
Submitted:
19 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
Cytomegalovirus infection contributes to 10-30% congenital hearing loss in children. Vertebrate peripheral auditory organs include the outer, middle and inner ear. Their development is regulated by multiple signaling pathways. However, most ear diseases due to viral infections are due to congenital infections and reactivation, and affect healthy adults to a lesser extent. This may be due to the fact that viral infections affect signaling pathways that are important for the development of peripheral hearing organs. Therefore, an in-depth understanding of the relationship between viral infections and the signaling pathways involved in the development of peripheral hearing organs is important for the prevention and treatment of ear diseases. In this review, we summaries the effects of viruses on signaling pathways and signaling molecules in development of peripheral auditory organ, with a focus on Human cytomegalovirus as an example.
Keywords:
virus
; hearing
; cytomegalovirus
; development
Introduction
The Vertebrate peripheral auditory organs (PAOs) of vertebrates include the outer, middle and inner ear. The outer ear is composed primarily of the auricle and external auditory canal, which serves to transmit sound to the eardrum. The middle ear is comprised of various structures, including the tympanic chamber, eustachian tube, sinus and mastoid, which contains the auditory ossicles. The middle ear transmits sound vibrations to the inner ear. The inner ear is located in the rocky part of the temporal bone and consists of two parts, the vestibule and the cochlea, which provide sensory information about sound, movement, balance and spatial orientation. Viral infections can affect the development of PAOs and/or damage PAOs, causing hearing loss and balance disorders. Wiertsema et al. [1] identified the presence of human rhinovirus, bocavirus, adenovirus, parainfluenza virus, and respiratory syncytial virus nucleic acids in the nasopharynx of patients with a history of acute otitis media episodes. Vestibular neuritis is believed to have a potential causal link with herpes simplex virus type 1 (HSV-1) infection and reactivation [2]. Pyykko and colleagues [3] conducted serological studies of virus-specific IgG in patients with Meniere's disease, recurrent vertigo of unknown etiology, and sensorineural deafness. Their findings revealed that these patients exhibited significantly higher titres of varicella zoster, influenza virus B, coxsackievirus B5, and respiratory syncytial virus. Additionally, researchers have identified the presence of parotid and herpes viruses in individuals diagnosed with idiopathic sensorineural sudden deafness [4,5]. Rosenthal et al. followed up 580 neonates with Congenital cytomegalovirus (cCMV) infection and found that 77 had hearing loss at birth, 38 children had delayed hearing loss at the end of follow-up, and that delayed hearing loss was strongly associated with symptomatic Human cytomegalovirus(HCMV) infection at birth [6]. cCMV infection occurs in approximately 1% of live births. The majority of infants are born asymptomatic, while approximately 11% present with microcephaly, mental retardation, seizures, hepatosplenomegaly, petechiae or jaundice [7,8]. Approximately 10% of infants born asymptomatic subsequently exhibit sensorineural hearing loss (SNHL), mental retardation, and learning disabilities [9,10]. These studies all indicate that viral infection is one of the important causes of hearing loss. However, the pathological mechanism of hearing loss caused by viral infection has not been fully elucidated.
cCMV s the most prevalent virus causing SNHL. SNHL induced by cCMV infection may be present at birth or may occur later in childhood, and the severity of cCMV related hearing loss ranges from unilateral high-frequency loss to severe bilateral loss. Interestingly, hearing loss is typically not apparent in healthy adults, likely due to their well-developed immunity, but It may also be attributed to the fact that mature PAOs are less prone to developmental abnormalities. As developmental defects or stagnation of the inner ear and its surrounding structures are frequently diagnosed in children with SNHL, researchers speculate that CMV infection may affect the formation of inner ear structure and function by regulating signaling pathways during the development of PAOs. The development of the inner ear is subtly regulated by several important signaling pathways. Cross-species microarrays have identified seven different known signaling pathways; TGFβ, PAX, Notch, Wnt, NFκB, insulin/IGF1 and AP1[11]. It is possible that viral infections may affect these pathways, thereby impairing inner ear function. Stevens et al employed a retroviral gene to stimulate the Wnt signaling pathway, with the aim of modifying the morphogenesis of the chicken inner ear [12]. By embryonic day nine, the resulting morphological defects were evident in the ear sac(ES) and membranous labyrinth. In a study conducted by Harding et al [13], human stem cell-derived ear progenitor cells infected with HCMV were observed to exhibit disrupted TGFβ signaling, which was found to be associated with hearing loss. The human immunodeficiency virus has been shown to downregulate the RBPJ protein in primary CD4+ T cells [14]. RBPJ is a regulator of the Notch signaling pathway, and its absence has been linked to sensory deficits. Therefore, it can be hypothesized that HIV infection may have a similar effect on structures derived from the developing ES. The measles virus (MV) is known to cause otosclerosis, a condition characterized by the abnormal growth of bones within the middle and inner ear, which can ultimately result in SNHL [15]. MV infection of human dendritic cells leads to PAX2 upregulation. MV-induced upregulation of PAX2 may lead to dysregulation of neuroblast genesis if it occurs at a critical pattern formation stage in the ES. Sox2 expression was significantly reduced in neural stem cells (NSCs) from HSV infected mice [16]. Sox2 is a key transcription factor controlling axial specification of the ES and cochlear canal formation, which is essential for the development of the inner ear. Defects in Sox2 can lead to severe inner ear malformations within the developing inner ear [17]. The infection of neonatal rats with the lymphocytic choroidal meningitis virus results in an elevation of ATOH1 mRNA levels in the cerebellum and a disruption of the normal hair cell production process [18]. These studies support the regulation of signaling pathways related to the development of PAOs by viral infection.
The exact cause of developmental disorders and hearing loss in PAOs caused by viral infections is unknown. There are few studies on the effects of viral infections on the development of the outer and middle ear. Therefore, in this review we focus on the interactions between viral infections and inner ear developmental pathways and take HCMV as an example to summaries the effects of HCMV infection on inner ear developmental signaling pathways. It is hoped that this knowledge will be useful in the treatment of developmental defects and hearing loss in PAOs and other related diseases caused by viral infections.
The Development Process of Inner Ear
During organogenesis, signaling pathways between embryonic tissues interact to build highly organized functional tissues and organs. The inner ear contains auditory and vestibular sensory organs. In mammals, the cochlea is responsible for hearing and contains the Corti apparatus, in which mechanosensory hair cells convert acoustic stimuli and generate electrochemical signals in response, which are transmitted to the brain by ear neurons [19]. The vestibular system contains balance receptors for mechanoreceptor hair cells [20]. The enlarged part at the base of the semicircular canals, designated the juxtaglomerular ridge, is responsible for balance perception. In contrast, the two "maculae" of the globus pallidus and the ellipsoid capsule detect linear and angular acceleration.
The sensory organs of the inner ear originate from the otic plate (OP), a thickening of the ectoderm that develops near the hindbrain from the cranial ectoderm immediately lateral to the neural crest (Figure 1). Before the visible morphology of the OP, four genes Pax2, Sox3, BMP7, and Notch are expressed in the ectoderm [21]. Subsequently, the ectodermal cell surface undergoes induction of transcription factors such as the Dlx family, Sox9a, and Foxi1[22,23]. Fibroblast growth factors produced by the underlying mesenchyme (e.g. FGF10, FGF19, or FGF15) and FGF3 secreted by the hindbrain are also induced [24,25,26]. Following induction, the OP invaginates, separates from the ectoderm, and develops into an ES through interactions with neighboring tissues and the addition of cells from the neural crest and mesoderm [27]. As development progresses, the otic capsule undergoes a transformation from a simple epithelial capsule to a complex fluid-filled labyrinth [28]. The epithelial cells in the anterior medial part of the OP/otic capsule differentiate into a proto-neurosensory epithelium, giving rise to neuromasts, which later form the auditory vestibular ganglion(AVG)[29]. The presensory domain of the OP develops into the Corti apparatus and vestibular sensory organs. The Corti apparatus is a highly specialized acoustic sensory epithelium consisting of sensory hair cells and supporting cells, including outer hair cells, inner hair cells, inner finger cells, Deiters cells, and column cells. The dorsal epithelium of the otic capsule expands to create a vertical capsule. In the vertical outer pouch, opposing epithelial cells come together to form two fusion plates, which subsequently merge and are absorbed to give rise to the two upper semicircular canals and the common peduncle [30,31]. The lateral semicircular canals develop from the lateral sac. Neuronal cells from the ES form the AVG, which contains the neural precursors of the auditory and vestibular ganglia [32]. These ganglia initially form a single ganglion in early development. Ear neurons connect the sensory epithelium to the nucleus accumbens via extensions of the eighth pair of cranial nerves [33]. Concurrently, nearby mesenchymal cells are recruited by the inner ear to form the bony capsule surrounding the labyrinth.
HCMV Regulates Sox2: A Transcription Factor Required for Inner Ear Growth and Cochlear Nonsensory Formation
Sox2, a member of the Sox B1 family of transcription factors, directs the differentiation of pluripotent stem cells into neural progenitor cells (NPCs) and the maintenance of neural progenitor stem cell identity [34]. Sox2 stabilizes embryonic stem cell cells in a pluripotent state by maintaining the necessary level of Oct 3/4 expression [35]. A small increase in Sox2 protein triggers embryonic stem cell differentiation, so embryonic stem cell self-renewal requires tight control of Sox2 levels [36]. Sox2 is widely expressed in developing neural precursor cells and NSCs in the adult brain [37,38]. In embryonic stem cells that differentiate into neural progenitors, Sox2 is directly regulated by signal transducer and activator of transcription 3 (STAT3) to promote NSCs fate commitment [39]. The forced expression of Sox2 in fibroblasts results in the generation of induced NSC [40]. These cells express NSCs markers and exhibit morphological, self-renewal, neurosphere formation and gene expression profiles comparable to those of wild type NSCs.
Sox2 missense or heterozygous loss-of-function mutations have been shown to cause bilateral anophthalmia, which can be accompanied by learning disabilities, seizures, brain malformations, or hearing loss [41]. In Sox2-deficient mice, inner ear neurons initially form normally, whereas late-differentiating neurons in the cochlear apex are never formed, and there is a complete absence of sensory epithelium formation in the apical part of the juxtaposition of the semicircular canals and in all three cristae [42]. In addition, the transcription factor Sox2 is a key gene for cochlear hair cell development. Sox2 activates the transcription factor Atoh1, which is important for hair cell differentiation, by interacting with the 3' enhancer of Atoh1[43]. Deletion of Sox2 results in impaired inner ear development with reduced and disorganized hair cells [17]. Hair cell development was found to cease even after progenitor cells were fully established in mice conditionally knocked out of Sox2, and silencing also inhibited postnatal differentiation of hair cells induced by inhibition of γ-secretase [44].
Genome wide analysis showed that HCMV infected NPCs exhibited downregulation of Sox2[45]. In patient-derived glioma stem cell-like cells endogenously infected with HCMV, the attenuation of immediate early protein expression using RNAi has been observed to suppress Sox2 expression [46]. Wu et al. found that the HCMV immediate early protein 1 (IE1) mediated Sox2 protein depletion in infected NPCs by promoting nuclear accumulation and inhibiting phosphorylation of STAT3 to cause Sox2 downregulation [47]. Sox2 also regulates the expression of HCMV viral genes. Wen et al. found that Sox2 downregulated the expression of promyelocytic leukaemia and Sp100, thereby promoting viral gene expression by reducing the number of PML nucleosomes in HCMV infected glioma cells [48].
HCMV Regulates the Wnt Signaling Pathway: Affecting Auditory Substrate Specialization, Otic Vesicle Formation and Hair Cell Differentiation
The Wnt signaling pathway is a highly conserved mechanism that plays a crucial role in various developmental processes, including cell fate determination, cell migration, neural patterning, and cell polarity [49]. Wnt proteins are known for their abundance of conserved cysteine residues. Wnt ligands have the ability to interact with the extracellular N-terminal cysteine-rich structural domain (CRD) of Frizzled (Fzd), thereby triggering the initiation of Wnt signaling [50]. This binding of Wnt proteins to the extracellular surface initiates the activation of intracellular signaling pathways, such as the classical Wnt pathway, the non-classical planar cell polarity (PCP) pathway, and the non-classical Wnt/calcium pathway [51]. The PCP pathway regulates the orientation of hair cell cilia bundles and the convergent extension of the cochlea during cochlear tube formation. The classical Wnt/β-catenin signaling pathway is particularly important for ear development. Cochlear development in selective knockout Wnt5a mice showed significant PCP defects, as evidenced by disorganized hair cell orientation and shorter cochlea formation compared to wild-type mice [52].
Activation of the classical Wnt pathway involves the binding of the ligand to the co-receptors of the Fzd receptor and the low-density lipoprotein receptor-related protein (LRP5/6). This pathway regulates intracellular β-catenin activity upon activation. In the absence of Wnt ligand, intracellular glycogen synthase kinase 3β (GSK3β), activated Axin, and adenomatous polyposis coli (APC) form a complex, leading to the phosphorylation and subsequent degradation of β-catenin by the proteasome system. This results in low intracellular levels of β-catenin and the suppression of downstream target gene transcription. Upon Wnt stimulation, the Fzd receptor recruits Disheveled (Dvl) proteins to the plasma membrane, promoting the phosphorylation of LRP5/6. This disrupts the Axin-APC-GSK3β complex, leading to the release of β-catenin from degradation and its accumulation in the cell. The accumulated β-catenin translocates to the nucleus, where it binds to transcription factors Tcf/Lef, directing the transcription of downstream target genes (Figure 2) [53].
The Wnt/β-catenin pathway plays a crucial role in various processes during inner ear development, such as the specialization of the auditory substrate, the formation of auditory vesicles, and the regulation of hair cell differentiation [53]. Jacques et al. [54] applied FH535, an inhibitor of the Wnt/ β-catenin signaling pathway, to cochlear explants at Embryonic day 12.5, resulting in inhibition of the Wnt/β-catenin signaling pathway, and the results showed that differentiation of sensory progenitor cells to hair cells was inhibited. Conversely, the addition of LiCl (a Wnt signaling activator that prevents the degradation of β-catenin by inhibiting GSK3β activity)[55], activated the Wnt/β-catenin pathway and led to an increase in the number of differentiated hair cells.
HCMV can target the Wnt pathway and regulate its activity. Teo et al. observed increased expression levels of Wnt11, Fzd7, GSK3β and β-catenin in the Wnt signaling pathway during HCMV infection in HCMV infected colorectal cells and their derived cells [56]. Zhou and colleagues found that Mouse cytomegalovirus(MCMV) infection significantly inhibits the expression of Wnt-1 in NSCs cultured in vitro, which affects the differentiation of NSCs [57]. Maussang et al. highlighted the Wnt/β-linker pathway in their transcriptional profiling of NIH-3T3 cells expressing the HCMV-encoded chemokine receptor US-28[58]. In the US28 transgenic mouse model, US28 acts as GSK3β in intestinal epithelial cells, promoting the accumulation of β-catenin protein and increasing the expression of Wnt target genes that control cell proliferation [59]. US28 transgenic mice developed intestinal adenomas and adenocarcinomas at 40 weeks of age. US28 does not activate β-catenin through the classical Wnt/Fzd signaling pathway. Langemeijer et al. suggest that the Rho-Rho kinase pathway is involved in β-catenin activation. Additionally, HCMV infected cells showed a significant increase in β-catenin stability and signaling, largely mediated by US28 expression [60]. HCMV can also negatively regulate the Wnt pathway. Angelova et al. demonstrated that HCMV disrupts Wnt/β-catenin signaling in dermal fibroblasts and human placental trophoblasts [61]. HCMV infection alters the subcellular distribution of β-catenin, with decreased levels of membrane-associated and cytoplasmic pool β-catenin, and accumulation in discrete nuclear regions, inhibiting Wnt/β-catenin signaling. Roy et al. demonstrated that HCMV infection inhibits the PARsylation activity of Tankyrase, leading to the accumulation of Axin1 (a negative regulator of the Wnt pathway) and reducing its PARylation, thereby inhibiting the β-catenin pathway [62]. It is not yet clear whether HCMV affects the non-canonical Wnt signaling pathway. However, Zuylen et al. demonstrated that HCMV infection can increase the expression of the non-canonical Wnt receptor ROR2 to alter Wnt5a-mediated signaling and modulate the migration of nourishing cells [63].
Many studies have confirmed that HCMV can regulate the Wnt pathway. However, the impact of HCMV infection on the regulation of the Wnt pathway in inner ear development remains unclear, and further research is needed to elucidate its potential mechanisms.
HCMV Regulates the Nonth Signaling Pathway: Affecting Cell Fate and Interfering with Inner Ear Development
The Notch pathway is highly conserved throughout evolution and is considered to be one of the major signaling pathways coordinating the developmental processes of most organs and tissues in all postnatal animals [64]. It is involved in the coordination between neighboring cells during development and homeostasis [65]. The core pathway of the Notch signaling pathway is the anchoring of transmembrane Notch receptors in one cell to transmembrane Notch ligands in neighboring cells (Figure 3). Mammals have four Notch receptors (Notch1-Notch4), and the transmembrane Notch receptor consists of the Notch extracellular structural domain (NECD) with multiple epidermal growth factor (EGF)-like repeats and a negative regulatory region (NRR), and the membrane-bound intracellular structural domain (NICD). The Notch receptor binds to type I transmembrane proteins collectively referred to as DSL proteins. Notch receptors bind to type I transmembrane proteins collectively known as DSL proteins. Mammals have five DSL ligands [three from the Delta-like family (Dll1, Dll3 and Dll4) and two Serrate orthologues known as the Jagged family (Jag1 and Jag2)]. Following the binding of the DSL ligand to the Notch receptor, the DSL ligand initiates a process of endocytosis that pulls on the Notch receptor [66]. The force generated by ligand endocytosis pulls on the Notch receptor, changing the conformation of the hinge region in the NRR structural domain and exposing the ADAM metalloproteinase cleavage site. Subsequently, the Notch receptor is cleaved first at the NRR by Adam10 and then at the intramembrane site by γ-secretase cleavage. These cleavages result in the release of NICD in the receptor cell. Upon release, NICD translocases to the nucleus and binds to nuclear proteins of the RBPJ-kappa family (also known as CSL or CBF1/SuH/Lag-1) and the cofactor MAML (Mastermind/Lag-3) to form the NICD-CSL-MAML transcriptional activator, which drives the transcriptional expression of downstream basic-helix-loop-helix transcription factors, such as Atoh1 and Hes/Hey family [67,68]. The Notch pathway can also target the regulation of Sox2. The Notch effector proteins Hes1 and Hes5 bind to Janus kinase 2 (JAK2) and STAT3, promoting the phosphorylation of STAT3 to directly regulate the Sox2 promoter and upregulate Sox2 expression [39].
During Drosophila external sensory organ development, the Notch signaling pathway restricts sensory organ precursor cell fates through lateral inhibition, which then gives rise to the entire sensory organ [69]. Similarly, in the vertebrate ear, the Notch signaling pathway regulates the proliferation of inner ear sensory precursor cells and maintains homeostasis of cochlear sensory epithelial cell number and structure through lateral inhibition and lateral induction [70]. In the initial stages of inner ear development, there is a positive feedback loop between the Wnt and Notch pathways, which together coordinate the refinement of the auricular plate boundary. The Wnt pathway regulates the early expression of Notch1, Jag1, Hes1 and Hey1 in the auricular plate. The Notch pathway also regulates the expression of ear markers such as Pax8 and the thickening of the ear plate. Inactivation of Notch1 reduces the size of the ear plate. Although Notch signaling does not regulate its own expression in the auricular plate and the onset of activation, it enhances Wnt activity, thereby maintaining Notch activity [71].
Notch signaling is also required for the early patterns of the ear basal plate, regulating neurogenesis and presensory norms. The Notch pathway regulates presensory norms through lateral induction, which promotes the formation of presensory cells in some cells of the ES. In lateral induction, the Notch pathway establishes a positive feedback loop through which Notch activation in one cell induces expression of Notch-activated ligands in the same cell [69]. Notch activation in the mouse inner ear not only autonomously induces Jag1-expressing cells, but also non-autonomously induces cells that propagate signals to neighboring cells [72]. The prosensory marker Notch ligands Jag1 and Sox2 were initially expressed at high levels in the ES. Neves et al. observed that Jag1-mediated Notch activity maintained Sox2 expression, rather than inducing Sox2 expression de novo [73]. he expression of Jag1 maintains the expression of Sox2, which in turn drives sensory capacity. Furthermore, Notch induces the expression of Hes and Hey factors, and Sox2 also prevents hair cell differentiation by promoting a discrete feed-forward loop activated by Atoh1 inhibitors [43]. The deletion of Jag1 results in developmental defects in the sensory epithelium of the inner ear, including truncation or absence of sensory organs and loss of hair cells [74]. The reduction in the number of hair cells is not due to a differentiation defect but to a loss of cell specification [75].
The Notch pathway also inhibits lateral inhibition, thus forming a mosaic pattern of hair/support cells. Activated Notch receptors inhibit the expression of Jag2, Dll1 and Atoh1 in signaling cells, resulting in the differentiation of signaling cells into supporting cells and adjacent cells into hair cells. In mammals, Dll1 expression predicts differentiation of ear neurons and hair cells, whereas Jag2 predicts differentiation of hair cells [76,77]. Hair cells express Dll1 and Jag2, and they activate the Notch pathway in neighboring cells to express genes such as Hes1, Hes5 and Hey1, which synergistically induce support for cell fate [78]. Inactivation of the γ-secretase inhibitors DAPT or MAML leads to neuronal and hair cell overproduction [79]. In addition, downregulation of Notch signaling pathway related genes including Dll1, Jag2 and Notch1 with siRNAs induced the production of large numbers of sensory hair cells [80]. Defects in the cochlear vestibular ganglion and deficits in the macular sensory epithelium occur in mice with conditional knockout of Dll1. This suggests that deletion of Dll1 disrupts lateral inhibition and leads to excessive neuron numbers and depletion of the sensory precursor pool [80]. Furthermore, there is functional redundancy between Notch ligands, and the cochlea of Dll3 mutant mice shows no abnormalities, suggesting that loss of one ligand can be at least partially compensated for by another [81].
HCMV infection can interfere with organ development by altering cell fate decisions through disruption of the Notch pathway. In HCMV infected NPCs, the expression of the periplasmid proteins pp71 and UL26, both of which are expressed endogenously and exogenously, results in a reduction in the levels of NICD1 and Jag1 proteins and an alteration in the subcellular localization of NICD1[82]. The mRNA levels of Notch related receptors and ligands were found to be significantly reduced in HCMV infected NSCs following 1 day of induced differentiation. In particular, the levels of Notch1, Notch2 and DLL1 were found to be significantly decreased. Furthermore, the intracellular levels of NICD were found to be significantly reduced 7 days after viral infection. HCMV infection also promotes the proliferation of U251 glioma cells by regulating the ATF5 signaling pathway to upregulate the expression of NICD and Notch1[83]. Hes1 serves as an important downstream effector of the Notch signaling pathway, and deletion of the Hes1 protein inhibits NPCs proliferation and neutrosphere formation, driving aberrant differentiation of NPCs. Liu et al. were the first to observe that HCMV infection disrupts the Hes1 rhythm. to interfere with NPCs cell fate [84]. Further studies revealed that IE1 of HCMV promotes Hes1 ubiquitination and degrades Hes1 through the proteasome, downregulating Hes1 expression [85].
Other Signaling Pathways Regulated by HCMV
HCMV can target Pax2, and in primary fibroblasts infected with HCMV, Browne et al. applied gene chip technology analysis to find that PAX2 was downregulated at 1- and 48-hours post-infection [86]. HCMV induced downregulation of PAX2 may hypothetically disrupt the otic medial–lateral specification toward the generation of neuroblasts within the epithelium of otocyst, which subsequently affects the segregation and migration of vestibular and auditory neuromasts from the neurosensory domain to the vestibular cochlear ganglion. Similarly, HCMV infection in fibroblasts also downregulates the transcription factor SIX1, which plays a role in the axial specification of ES [86]. MCMV infected differentiated mouse NSC significantly downregulate the transcription factor NGN1, which determines vestibulocochlear ganglion nerve fate [57,86]. HCMV major IE1 can interact with FGFR3 in astrocytoma cell lines, but the absence of FGFR3 leads to abnormal column cell development [87,88]. Furthermore, FGFR3 deficient mice exhibit deficiencies in supporting cell differentiation, and virus-induced excess of FGFR3 may alter the balance of sensory and non-sensory cells [89]. Another possibility for hearing damage from HCMV infection is reduced levels of CDKN1B, an enzyme inhibitor that regulates the G1 phase of the cell cycle [86]. HCMV infection of human embryonic lung cells leads to CDKN1B degradation [90]. The deletion of CDKN1B results in the overproduction of hair cells and supporting cells during cochlear development, which in turn causes severe hearing loss [91].
Viral Infections Affect the Formation of Ossicular Chains
The mammalian ossicular chains contains three auditory ossicles, the malleus, incus and stapes. The auditory ossicles are mainly derived from cranial neural crest cells (NCCs). NCCs migrating to the first pharyngeal arch (PA1) form the malleus-incus condensation [92]. In mouse PA1, the malleus-incus forms as a single cohesive mass attached to Meckel's cartilage at embryonic day(E) 10.5. Subsequently, the malleus and incus bones separate from each other at E13.5[93,94]. NCCs migrating to the second pharyngeal arch (PA2) form the stapedial condensation [95]. At E10.5 the stapedial condensation forms separately in the PA2 mesenchyme [96]. The stapes acquires its characteristic stirrup-like structure at E11.5. The stapes is attached to the inner ear by annular ligaments at E15.5.
Sonic Hedgehog (SHH), bone morphogenetic protein 4 (BMP4), FGF and Notch signaling molecules, which play a crucial role in middle ear development. Shh is expressed in the endoderm of PA1 at E10.5, Its receptor is expressed in the mesenchyme adjacent to the endoderm, forming the malleus-incus anlagen. Knockout of Shh in the pharyngeal endoderm results in loss of malleus-incus condensation in PA1[92]. The forced activation of the SHH signaling pathway in NCCs impairs the development of normal auditory ossicles [92]. BMP signaling mediates induction and migration of NCCs [97]. Mouse embryos with downregulated expression of BMP signaling in NCCs exhibit the loss of stapes and styloid process [98]. The knockdown of Bmp4 in the pharyngeal endoderm results in the disruption of NCC migration to the stapes region in PA2, which in turn affects the formation of the stapes [92]. Fgf8 is strongly expressed in arch ectoderm and the pharyngeal endoderm [99]. Fgf8 deficient mice exhibit severe deficits in the malleus and incus bones [100]. Heterozygotes with mutations in FGF receptor 1 show incus abnormalities and multiple malformations of the stapes bone [101]. Mice lacking Jag1 or Notch2 in NCCs show malformed stapes, manifesting as a monopodial structure lacking crural regions [102].
Many reports describe the presence of MV RNA or protein in the ear bones of patients with otosclerosis [103,104,105]. Therefore, the researchers hypothesized that persistent MV infection of the ES is one of the causes of otosclerosis. The characteristic pathological change in otosclerosis is a disturbed reconstruction of the bone of the ear capsule. This abnormal bone reconstruction can lead to stapes fixation resulting in conductive hearing loss and can also be combined with SNHL [106]. Furthermore, immunohistochemical staining of fragments of osteosclerotic footplates revealed the expression of mumps,MV and rubella virus antigens in vascular connective tissue, osteoblasts, osteoclasts, and chondrocytes in or around the lesion area [107]. These results support the idea that otosclerosis is caused by a viral infection. However, how viral infection affects the development of the ossicles of the ear capsule remains unclear, and more research is needed to elucidate the underlying mechanisms.
3. Conclusions
Viral infections are considered a significant cause of congenital hearing loss, but little attention has been paid so far to the effects of viral infections on the molecular and signaling pathways involved in the development of the PAOs. The aim of this paper is to summaries the effects of viruses (especially HCMV) on the main signaling molecules and signaling pathways involved in PAOs development. By regulating the signaling pathways of PAOs development, it can play a therapeutic role in related diseases. Inhibitors of the Wnt pathway have been demonstrated to suppress the replication of HCMV and the expression of viral proteins IE2, UL44 and pp65[108]. Notch signaling blockers also show favorable effects on the recovery of hearing loss in mice [78]. Inhibitor of γ-secretase induces new hair cells and partially restores noise-induced hearing loss [109]. We hope that the study of abnormalities in the developmental pathway of PAOs caused by viral infection will provide new directions for intervention in virus-mediated developmental malformations of PAOs and related diseases.
Author Contributions
YS and L-XZ designed the study and reviewed and edited the manuscript. Z-YJ, and S-XY drew Figures. Z-DZ and L-XZ wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (Nos.2021YFF0702303, 2023YFE0203200), the Innovative Research Groups of Hubei Province (No.2023AFA038), the Basic Research Support Program of Huazhong University of Science and Technology(No.2024BRA019).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wiertsema, S.P.; Chidlow, G.R.; Kirkham, L.A.; Corscadden, K.J.; Mowe, E.N.; Vijayasekaran, S.; Coates, H.L.; Harnett, G.B.; Richmond, P.C. High detection rates of nucleic acids of a wide range of respiratory viruses in the nasopharynx and the middle ear of children with a history of recurrent acute otitis media. J Med Virol 2011, 83, 2008–17. [Google Scholar] [CrossRef]
- Rujescu, D.; Hartmann, A.M.; Giegling, I.; Konte, B.; Herrling, M.; Himmelein, S.; Strupp, M. Genome-Wide Association Study in Vestibular Neuritis: Involvement of the Host Factor for HSV-1 Replication. Front Neurol 2018, 9, 591. [Google Scholar] [CrossRef] [PubMed]
- Pyykko, I.; Zou, J. Do viruses cause inner ear disturbances? ORL J Otorhinolaryngol Relat Spec discussion 40-1. 2008, 70, 32–40. [Google Scholar] [CrossRef]
- Tsubota, M.; Shojaku, H.; Ishimaru, H.; Fujisaka, M.; Watanabe, Y. Mumps virus may damage the vestibular nerve as well as the inner ear. Acta Otolaryngol 2008, 128, 644–7. [Google Scholar] [CrossRef]
- Kikidis, D.; Nikolopoulos, T.P.; Kampessis, G.; Stamatiou, G.; Chrysovergis, A. Sudden sensorineural hearing loss: subclinical viral and toxoplasmosis infections as aetiology and how they alter the clinical course. ORL J Otorhinolaryngol Relat Spec 2011, 73, 110–5. [Google Scholar] [CrossRef]
- Rosenthal, L.S.; Fowler, K.B.; Boppana, S.B.; Britt, W.J.; Pass, R.F.; Schmid, S.D.; Stagno, S.; Cannon, M.J. Cytomegalovirus shedding and delayed sensorineural hearing loss: results from longitudinal follow-up of children with congenital infection. Pediatr Infect Dis J 2009, 28, 515–20. [Google Scholar] [CrossRef]
- Bale, J.F. Jr. Human cytomegalovirus infection and disorders of the nervous system. Arch Neurol 1984, 41, 310–20. [Google Scholar] [CrossRef]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol 2007, 17, 253–76. [Google Scholar] [CrossRef]
- Fowler, K.B.; Boppana, S.B. Congenital cytomegalovirus (CMV) infection and hearing deficit. J Clin Virol 2006, 35, 226–31. [Google Scholar] [CrossRef] [PubMed]
- Conboy, T.J.; Pass, R.F.; Stagno, S.; Britt, W.J.; Alford, C.A.; McFarland, C.E.; Boll, T.J. Intellectual development in school-aged children with asymptomatic congenital cytomegalovirus infection. Pediatrics 1986, 77, 801–6. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Bashiardes, S.; Powder, K.E.; Sajan, S.A.; Bhonagiri, V.; Alvarado, D.M.; Speck, J.; Warchol, M.E.; Lovett, M. Large scale gene expression profiles of regenerating inner ear sensory epithelia. PLoS One 2007, 2, e525. [Google Scholar] [CrossRef]
- Stevens, C.B.; Davies, A.L.; Battista, S.; Lewis, J.H.; Fekete, D.M. Forced activation of Wnt signaling alters morphogenesis and sensory organ identity in the chicken inner ear. Dev Biol 2003, 261, 149–64. [Google Scholar] [CrossRef]
- Harding, A.T.; Ocwieja, K.; Jeong, M.; Zhang, Y.; Leger, V.; Jhala, N.; Stankovic, K.M.; Gehrke, L. Human otic progenitor cell models of congenital hearing loss reveal potential pathophysiologic mechanisms of Zika virus and cytomegalovirus infections. mBio 2024, 15, e0019924. [Google Scholar] [CrossRef]
- Bradley, T.; Ferrari, G.; Haynes, B.F.; Margolis, D.M.; Browne, E.P. Single-Cell Analysis of Quiescent HIV Infection Reveals Host Transcriptional Profiles that Regulate Proviral Latency. Cell Rep 2018, 25, 107–117.e3. [Google Scholar] [CrossRef]
- Karosi, T.; Kónya, J.; Petkó, M.; Sziklai, I. Histologic otosclerosis is associated with the presence of measles virus in the stapes footplate. Otol Neurotol 2005, 26, 1128–33. [Google Scholar] [CrossRef]
- Ozaki, H.; Nakamura, K.; Funahashi, J.; Ikeda, K.; Yamada, G.; Tokano, H.; Okamura, H.O.; Kitamura, K.; Muto, S.; Kotaki, H.; Sudo, K.; Horai, R.; Iwakura, Y.; Kawakami, K. Six1 controls patterning of the mouse otic vesicle. Development 2004, 131, 551–62. [Google Scholar] [CrossRef]
- Kiernan, A.E.; Pelling, A.L.; Leung, K.K.; Tang, A.S.; Bell, D.M.; Tease, C.; Lovell-Badge, R.; Steel, K.P.; Cheah, K.S. Sox2 is required for sensory organ development in the mammalian inner ear. Nature 2005, 434, 1031–5. [Google Scholar] [CrossRef]
- Licona, K. d. a. H. W. Congenital LCM virus: Mechanism of brain disease in a rat model of congenital viral infection. 2010. [Google Scholar]
- Raphael, Y.; Altschuler, R.A. Structure and innervation of the cochlea. Brain Res Bull 2003, 60, 397–422. [Google Scholar] [CrossRef]
- Goldberg, J.M. The vestibular end organs: morphological and physiological diversity of afferents. Curr Opin Neurobiol 1991, 1, 229–35. [Google Scholar] [CrossRef]
- Groves, A.K.; Bronner-Fraser, M. Competence, specification and commitment in otic placode induction. Development 2000, 127, 3489–99. [Google Scholar] [CrossRef]
- Ekker, M.; Akimenko, M.A.; Bremiller, R.; Westerfield, M. Regional expression of three homeobox transcripts in the inner ear of zebrafish embryos. Neuron 1992, 9, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chu, H.; Maves, L.; Yan, Y.L.; Morcos, P.A.; Postlethwait, J.H.; Westerfield, M. Fgf3 and Fgf8 dependent and independent transcription factors are required for otic placode specification. Development 2003, 130, 2213–24. [Google Scholar] [CrossRef] [PubMed]
- Léger, S.; Brand, M. Fgf8 and Fgf3 are required for zebrafish ear placode induction, maintenance and inner ear patterning. Mech Dev 2002, 119, 91–108. [Google Scholar] [CrossRef]
- Maroon, H.; Walshe, J.; Mahmood, R.; Kiefer, P.; Dickson, C.; Mason, I. Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development 2002, 129, 2099–108. [Google Scholar] [CrossRef]
- Wright, T.J.; Mansour, S.L. FGF signaling in ear development and innervation. Curr Top Dev Biol 2003, 57, 225–59. [Google Scholar] [PubMed]
- Couly, G.F.; Coltey, P.M.; Le Douarin, N.M. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development 1993, 117, 409–29. [Google Scholar] [CrossRef]
- Anniko, M. Cytodifferentiation of cochlear hair cells. Am J Otolaryngol 1983, 4, 375–88. [Google Scholar] [CrossRef]
- Satoh, T.; Fekete, D.M. Clonal analysis of the relationships between mechanosensory cells and the neurons that innervate them in the chicken ear. Development 2005, 132, 1687–97. [Google Scholar] [CrossRef]
- Martin, P.; Swanson, G.J. Descriptive and experimental analysis of the epithelial remodellings that control semicircular canal formation in the developing mouse inner ear. Dev Biol 1993, 159, 549–58. [Google Scholar] [CrossRef]
- Chang, W.; Brigande, J.V.; Fekete, D.M.; Wu, D.K. The development of semicircular canals in the inner ear: role of FGFs in sensory cristae. Development 2004, 131, 4201–11. [Google Scholar] [CrossRef]
- Adam, J.; Myat, A.; Le Roux, I.; Eddison, M.; Henrique, D.; Ish-Horowicz, D.; Lewis, J. Cell fate choices and the expression of Notch, Delta and Serrate homologues in the chick inner ear: parallels with Drosophila sense-organ development. Development 1998, 125, 4645–54. [Google Scholar] [CrossRef] [PubMed]
- Hemond, S.G.; Morest, D.K. Ganglion formation from the otic placode and the otic crest in the chick embryo: mitosis, migration, and the basal lamina. Anat Embryol (Berl) 1991, 184, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J Stem Cells 2014, 6, 305–11. [Google Scholar] [CrossRef]
- Masui, S.; Nakatake, Y.; Toyooka, Y.; Shimosato, D.; Yagi, R.; Takahashi, K.; Okochi, H.; Okuda, A.; Matoba, R.; Sharov, A.A.; Ko, M.S.; Niwa, H. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 2007, 9, 625–35. [Google Scholar] [CrossRef]
- Kopp, J.L.; Ormsbee, B.D.; Desler, M.; Rizzino, A. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 2008, 26, 903–11. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M. SOX after SOX: SOXession regulates neurogenesis. Genes Dev 2011, 25, 2423–8. [Google Scholar] [CrossRef] [PubMed]
- Brazel, C.Y.; Limke, T.L.; Osborne, J.K.; Miura, T.; Cai, J.; Pevny, L.; Rao, M.S. Sox2 expression defines a heterogeneous population of neurosphere-forming cells in the adult murine brain. Aging Cell 2005, 4, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Foshay, K.M.; Gallicano, G.I. Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev 2008, 17, 269–78. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; Tong, L.M.; Balestra, M.E.; Javier, R.; Andrews-Zwilling, Y.; Li, G.; Walker, D.; Zhang, W.R.; Kreitzer, A.C.; Huang, Y. Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 2012, 11, 100–9. [Google Scholar] [CrossRef]
- Ragge, N.K.; Lorenz, B.; Schneider, A.; Bushby, K.; de Sanctis, L.; de Sanctis, U.; Salt, A.; Collin, J.R.; Vivian, A.J.; Free, S.L.; Thompson, P.; Williamson, K.A.; Sisodiya, S.M.; van Heyningen, V.; Fitzpatrick, D.R. SOX2 anophthalmia syndrome. Am J Med Genet A discussion 8. 2005, 135, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, M.; Jahan, I.; Macova, I.; Chumak, T.; Bohuslavova, R.; Syka, J.; Fritzsch, B.; Pavlinkova, G. Incomplete and delayed Sox2 deletion defines residual ear neurosensory development and maintenance. Sci Rep 2016, 6, 38253. [Google Scholar] [CrossRef]
- Neves, J.; Uchikawa, M.; Bigas, A.; Giraldez, F. The prosensory function of Sox2 in the chicken inner ear relies on the direct regulation of Atoh1. PLoS One 2012, 7, e30871. [Google Scholar] [CrossRef] [PubMed]
- Kempfle, J.S.; Turban, J.L.; Edge, A.S. Sox2 in the differentiation of cochlear progenitor cells. Sci Rep 2016, 6, 23293. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Hannemann, H.; Kulkarni, A.S.; Schwartz, P.H.; O'Dowd, J.M.; Fortunato, E.A. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol 2010, 84, 3528–41. [Google Scholar] [CrossRef]
- Soroceanu, L.; Matlaf, L.; Khan, S.; Akhavan, A.; Singer, E.; Bezrookove, V.; Decker, S.; Ghanny, S.; Hadaczek, P.; Bengtsson, H.; Ohlfest, J.; Luciani-Torres, M.G.; Harkins, L.; Perry, A.; Guo, H.; Soteropoulos, P.; Cobbs, C.S. Cytomegalovirus Immediate-Early Proteins Promote Stemness Properties in Glioblastoma. Cancer Res 2015, 75, 3065–76. [Google Scholar] [CrossRef]
- Wu, C.C.; Jiang, X.; Wang, X.Z.; Liu, X.J.; Li, X.J.; Yang, B.; Ye, H.Q.; Harwardt, T.; Jiang, M.; Xia, H.M.; Wang, W.; Britt, W.J.; Paulus, C.; Nevels, M.; Luo, M.H. Human Cytomegalovirus Immediate Early 1 Protein Causes Loss of SOX2 from Neural Progenitor Cells by Trapping Unphosphorylated STAT3 in the Nucleus. J Virol 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Wang, X.Z.; Qiu, Y.; Zhou, Y.P.; Zhang, Q.Y.; Cheng, S.; Sun, J.Y.; Jiang, X.J.; Rayner, S.; Britt, W.J.; Chen, J.; Hu, F.; Li, F.C.; Luo, M.H.; Cheng, H. SOX2 downregulation of PML increases HCMV gene expression and growth of glioma cells. PLoS Pathog 2023, 19, e1011316. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382, 225–30. [Google Scholar] [CrossRef]
- Habas, R.; Dawid, I.B. Dishevelled and Wnt signaling: is the nucleus the final frontier? J Biol 2005, 4, 2. [Google Scholar] [CrossRef]
- Qian, D.; Jones, C.; Rzadzinska, A.; Mark, S.; Zhang, X.; Steel, K.P.; Dai, X.; Chen, P. Wnt5a functions in planar cell polarity regulation in mice. Dev Biol 2007, 306, 121–33. [Google Scholar] [CrossRef] [PubMed]
- Munnamalai, V.; Fekete, D.M. Wnt signaling during cochlear development. Semin Cell Dev Biol 2013, 24, 480–9. [Google Scholar] [CrossRef] [PubMed]
- Jacques, B.E.; Puligilla, C.; Weichert, R.M.; Ferrer-Vaquer, A.; Hadjantonakis, A.K.; Kelley, M.W.; Dabdoub, A. A dual function for canonical Wnt/β-catenin signaling in the developing mammalian cochlea. Development 2012, 139, 4395–404. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 1996, 93, 8455–9. [Google Scholar] [CrossRef]
- Teo, W.H.; Chen, H.P.; Huang, J.C.; Chan, Y.J. Human cytomegalovirus infection enhances cell proliferation, migration and upregulation of EMT markers in colorectal cancer-derived stem cell-like cells. Int J Oncol 2017, 51, 1415–1426. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Fang, F.; Dong, Y.S.; Zhou, H.; Zhen, H.; Liu, J.; Li, G. [Inhibitory effect of murine cytomegalovirus infection on neural stem cells' differentiation and its mechanisms]. Zhonghua Er Ke Za Zhi 2006, 44, 505–8. [Google Scholar] [PubMed]
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Stigter-van Walsum, M.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; van Dongen, G.A.; Leurs, R.; Smit, M.J. The human cytomegalovirus-encoded chemokine receptor US28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res 2009, 69, 2861–9. [Google Scholar] [CrossRef]
- Bongers, G.; Maussang, D.; Muniz, L.R.; Noriega, V.M.; Fraile-Ramos, A.; Barker, N.; Marchesi, F.; Thirunarayanan, N.; Vischer, H.F.; Qin, L.; Mayer, L.; Harpaz, N.; Leurs, R.; Furtado, G.C.; Clevers, H.; Tortorella, D.; Smit, M.J.; Lira, S.A. The cytomegalovirus-encoded chemokine receptor US28 promotes intestinal neoplasia in transgenic mice. J Clin Invest 2010, 120, 3969–78. [Google Scholar] [CrossRef]
- Langemeijer, E.V.; Slinger, E.; de Munnik, S.; Schreiber, A.; Maussang, D.; Vischer, H.; Verkaar, F.; Leurs, R.; Siderius, M.; Smit, M.J. Constitutive β-catenin signaling by the viral chemokine receptor US28. PLoS One 2012, 7, e48935. [Google Scholar] [CrossRef]
- Angelova, M.; Zwezdaryk, K.; Ferris, M.; Shan, B.; Morris, C.A.; Sullivan, D.E. Human cytomegalovirus infection dysregulates the canonical Wnt/β-catenin signaling pathway. PLoS Pathog 2012, 8, e1002959. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Liu, F.; Arav-Boger, R. Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase--A Potential Strategy for Suppression of the Wnt Pathway. Viruses 2015, 8. [Google Scholar] [CrossRef]
- van Zuylen, W.J.; Ford, C.E.; Wong, D.D.; Rawlinson, W.D. Human Cytomegalovirus Modulates Expression of Noncanonical Wnt Receptor ROR2 To Alter Trophoblast Migration. J Virol 2016, 90, 1108–15. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: cell fate control and signal integration in development. Science 1999, 284, 770–6. [Google Scholar] [CrossRef] [PubMed]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev Cell 2017, 41, 228–241. [Google Scholar] [CrossRef]
- Seib, E.; Klein, T. The role of ligand endocytosis in notch signalling. Biol Cell 2021, 113, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zang, C.; Taing, L.; Arnett, K.L.; Wong, Y.J.; Pear, W.S.; Blacklow, S.C.; Liu, X.S.; Aster, J.C. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A 2014, 111, 705–10. [Google Scholar] [CrossRef]
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol 2003, 194, 237–55. [Google Scholar] [CrossRef] [PubMed]
- Bray, S. Notch signalling in Drosophila: three ways to use a pathway. Semin Cell Dev Biol 1998, 9, 591–7. [Google Scholar] [CrossRef]
- Brown, R.; Groves, A.K. Hear, Hear for Notch: Control of Cell Fates in the Inner Ear by Notch Signaling. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Jayasena, C.S.; Ohyama, T.; Segil, N.; Groves, A.K. Notch signaling augments the canonical Wnt pathway to specify the size of the otic placode. Development 2008, 135, 2251–61. [Google Scholar] [CrossRef] [PubMed]
- Hartman, B.H.; Reh, T.A.; Bermingham-McDonogh, O. Notch signaling specifies prosensory domains via lateral induction in the developing mammalian inner ear. Proc Natl Acad Sci U S A 2010, 107, 15792–7. [Google Scholar] [CrossRef]
- Neves, J.; Parada, C.; Chamizo, M.; Giráldez, F. Jagged 1 regulates the restriction of Sox2 expression in the developing chicken inner ear: a mechanism for sensory organ specification. Development 2011, 138, 735–44. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, A.E.; Xu, J.; Gridley, T. The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet 2006, 2, e4. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Jin, Y.; Stanger, B.; Kiernan, A.E. Notch signaling is required for the generation of hair cells and supporting cells in the mammalian inner ear. Proc Natl Acad Sci U S A 2010, 107, 15798–803. [Google Scholar] [CrossRef] [PubMed]
- Lanford, P.J.; Lan, Y.; Jiang, R.; Lindsell, C.; Weinmaster, G.; Gridley, T.; Kelley, M.W. Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet 1999, 21, 289–92. [Google Scholar] [CrossRef]
- Daudet, N.; Lewis, J. Two contrasting roles for Notch activity in chick inner ear development: specification of prosensory patches and lateral inhibition of hair-cell differentiation. Development 2005, 132, 541–51. [Google Scholar] [CrossRef]
- Tateya, T.; Imayoshi, I.; Tateya, I.; Ito, J.; Kageyama, R. Cooperative functions of Hes/Hey genes in auditory hair cell and supporting cell development. Dev Biol 2011, 352, 329–40. [Google Scholar] [CrossRef] [PubMed]
- Abelló, G.; Khatri, S.; Giráldez, F.; Alsina, B. Early regionalization of the otic placode and its regulation by the Notch signaling pathway. Mech Dev 2007, 124, 631–45. [Google Scholar] [CrossRef]
- Jung, J.Y.; Avenarius, M.R.; Adamsky, S.; Alpert, E.; Feinstein, E.; Raphael, Y. siRNA targeting Hes5 augments hair cell regeneration in aminoglycoside-damaged mouse utricle. Mol Ther 2013, 21, 834–41. [Google Scholar] [CrossRef]
- Hartman, B.H.; Hayashi, T.; Nelson, B.R.; Bermingham-McDonogh, O.; Reh, T.A. Dll3 is expressed in developing hair cells in the mammalian cochlea. Dev Dyn 2007, 236, 2875–83. [Google Scholar] [CrossRef]
- Li, X.J.; Liu, X.J.; Yang, B.; Fu, Y.R.; Zhao, F.; Shen, Z.Z.; Miao, L.F.; Rayner, S.; Chavanas, S.; Zhu, H.; Britt, W.J.; Tang, Q.; McVoy, M.A.; Luo, M.H. Human Cytomegalovirus Infection Dysregulates the Localization and Stability of NICD1 and Jag1 in Neural Progenitor Cells. J Virol 2015, 89, 6792–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, M.; Xing, F.; Wang, M.; Wang, B.; Qian, D. Human cytomegalovirus infection promotes the stemness of U251 glioma cells. J Med Virol 2017, 89, 878–886. [Google Scholar] [CrossRef]
- Liu, X.J.; Jiang, X.; Huang, S.N.; Sun, J.Y.; Zhao, F.; Zeng, W.B.; Luo, M.H. Human cytomegalovirus infection dysregulates neural progenitor cell fate by disrupting Hes1 rhythm and down-regulating its expression. Virol Sin 2017, 32, 188–198. [Google Scholar] [CrossRef]
- Liu, X.J.; Yang, B.; Huang, S.N.; Wu, C.C.; Li, X.J.; Cheng, S.; Jiang, X.; Hu, F.; Ming, Y.Z.; Nevels, M.; Britt, W.J.; Rayner, S.; Tang, Q.; Zeng, W.B.; Zhao, F.; Luo, M.H. Human cytomegalovirus IE1 downregulates Hes1 in neural progenitor cells as a potential E3 ubiquitin ligase. PLoS Pathog 2017, 13, e1006542. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J Virol 2001, 75, 12319–30. [Google Scholar] [CrossRef] [PubMed]
- Martínez, F.P.; Tang, Q. Identification of cellular proteins that interact with human cytomegalovirus immediate-early protein 1 by protein array assay. Viruses 2013, 6, 89–105. [Google Scholar] [CrossRef]
- Shim, K.; Minowada, G.; Coling, D.E.; Martin, G.R. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev Cell 2005, 8, 553–64. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Cunningham, D.; Bermingham-McDonogh, O. Loss of Fgfr3 leads to excess hair cell development in the mouse organ of Corti. Dev Dyn 2007, 236, 525–33. [Google Scholar] [CrossRef]
- Chen, Z.; Knutson, E.; Kurosky, A.; Albrecht, T. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J Virol 2001, 75, 3613–25. [Google Scholar] [CrossRef]
- Chen, P.; Segil, N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development 1999, 126, 1581–90. [Google Scholar] [CrossRef]
- Ankamreddy, H.; Min, H.; Kim, J.Y.; Yang, X.; Cho, E.S.; Kim, U.K.; Bok, J. Region-specific endodermal signals direct neural crest cells to form the three middle ear ossicles. Development 2019, 146. [Google Scholar]
- Miyake, T.; Cameron, A.M.; Hall, B.K. Stage-specific onset of condensation and matrix deposition for Meckel's and other first arch cartilages in inbred C57BL/6 mice. J Craniofac Genet Dev Biol 1996, 16, 32–47. [Google Scholar] [PubMed]
- Amin, S.; Matalova, E.; Simpson, C.; Yoshida, H.; Tucker, A.S. Incudomalleal joint formation: the roles of apoptosis, migration and downregulation. BMC Dev Biol 2007, 7, 134. [Google Scholar] [CrossRef]
- Chapman, S.C. Can you hear me now? Understanding vertebrate middle ear development. Front Biosci (Landmark Ed) 2011, 16, 1675–1692. [Google Scholar] [CrossRef]
- Santagati, F.; Rijli, F.M. Cranial neural crest and the building of the vertebrate head. Nat Rev Neurosci 2003, 4, 806–18. [Google Scholar] [CrossRef]
- Nie, X.; Luukko, K.; Kettunen, P. BMP signalling in craniofacial development. Int J Dev Biol 2006, 50, 511–21. [Google Scholar] [CrossRef]
- Kanzler, B.; Foreman, R.K.; Labosky, P.A.; Mallo, M. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development 2000, 127, 1095–104. [Google Scholar] [CrossRef] [PubMed]
- Crossley, P.H.; Martin, G.R. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 1995, 121, 439–51. [Google Scholar] [CrossRef]
- Trumpp, A.; Depew, M.J.; Rubenstein, J.L.; Bishop, J.M.; Martin, G.R. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev 1999, 13, 3136–48. [Google Scholar] [CrossRef]
- Pau, H.; Fuchs, H.; de Angelis, M.H.; Steel, K.P. Hush puppy: a new mouse mutant with pinna, ossicle, and inner ear defects. Laryngoscope 2005, 115, 116–24. [Google Scholar] [CrossRef]
- Teng, C.S.; Yen, H.Y.; Barske, L.; Smith, B.; Llamas, J.; Segil, N.; Go, J.; Sanchez-Lara, P.A.; Maxson, R.E.; Crump, J.G. Requirement for Jagged1-Notch2 signaling in patterning the bones of the mouse and human middle ear. Sci Rep 2017, 7, 2497. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.; Friedmann, I. [Detection of measles and rubella-specific antigens in the endochondral ossification zone in otosclerosis]. Laryngol Rhinol Otol (Stuttg) 1987, 66, 167–71. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, H.; Arnold, W.; Neubert, W.J.; Höfler, H. Evidence of measles virus RNA in otosclerotic tissue. ORL J Otorhinolaryngol Relat Spec 1994, 56, 130–2. [Google Scholar] [CrossRef]
- McKenna, M.J.; Kristiansen, A.G.; Haines, J. Polymerase chain reaction amplification of a measles virus sequence from human temporal bone sections with active otosclerosis. Am J Otol 1996, 17, 827–30. [Google Scholar]
- Browning, G.G.; Gatehouse, S. Sensorineural hearing loss in stapedial otosclerosis. Ann Otol Rhinol Laryngol 1984, 93, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.; Friedmann, I. Otosclerosis--an inflammatory disease of the otic capsule of viral aetiology? J Laryngol Otol 1988, 102, 865–71. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; He, R.; Venkatadri, R.; Forman, M.; Arav-Boger, R. Wnt modulating agents inhibit human cytomegalovirus replication. Antimicrob Agents Chemother 2013, 57, 2761–7. [Google Scholar] [CrossRef]
- Mizutari, K.; Fujioka, M.; Hosoya, M.; Bramhall, N.; Okano, H.J.; Okano, H.; Edge, A.S. Notch inhibition induces cochlear hair cell regeneration and recovery of hearing after acoustic trauma. Neuron 2013, 77, 58–69. [Google Scholar] [CrossRef]
Figure 1.
Signals that regulate otic plate (OP) formation. Secretion of FGF8 by the pharyngeal endoderm induces secretion of FGF by the mesoderm near the hindbrain, and FGF defines the posterior PPR (blue) and induces expression of Wnt8a and FGF in the hindbrain. Anterior and posterior hindbrain-derived Wnt8a and FGF-mediated signaling designates the PPR as OP (green).
Figure 1.
Signals that regulate otic plate (OP) formation. Secretion of FGF8 by the pharyngeal endoderm induces secretion of FGF by the mesoderm near the hindbrain, and FGF defines the posterior PPR (blue) and induces expression of Wnt8a and FGF in the hindbrain. Anterior and posterior hindbrain-derived Wnt8a and FGF-mediated signaling designates the PPR as OP (green).
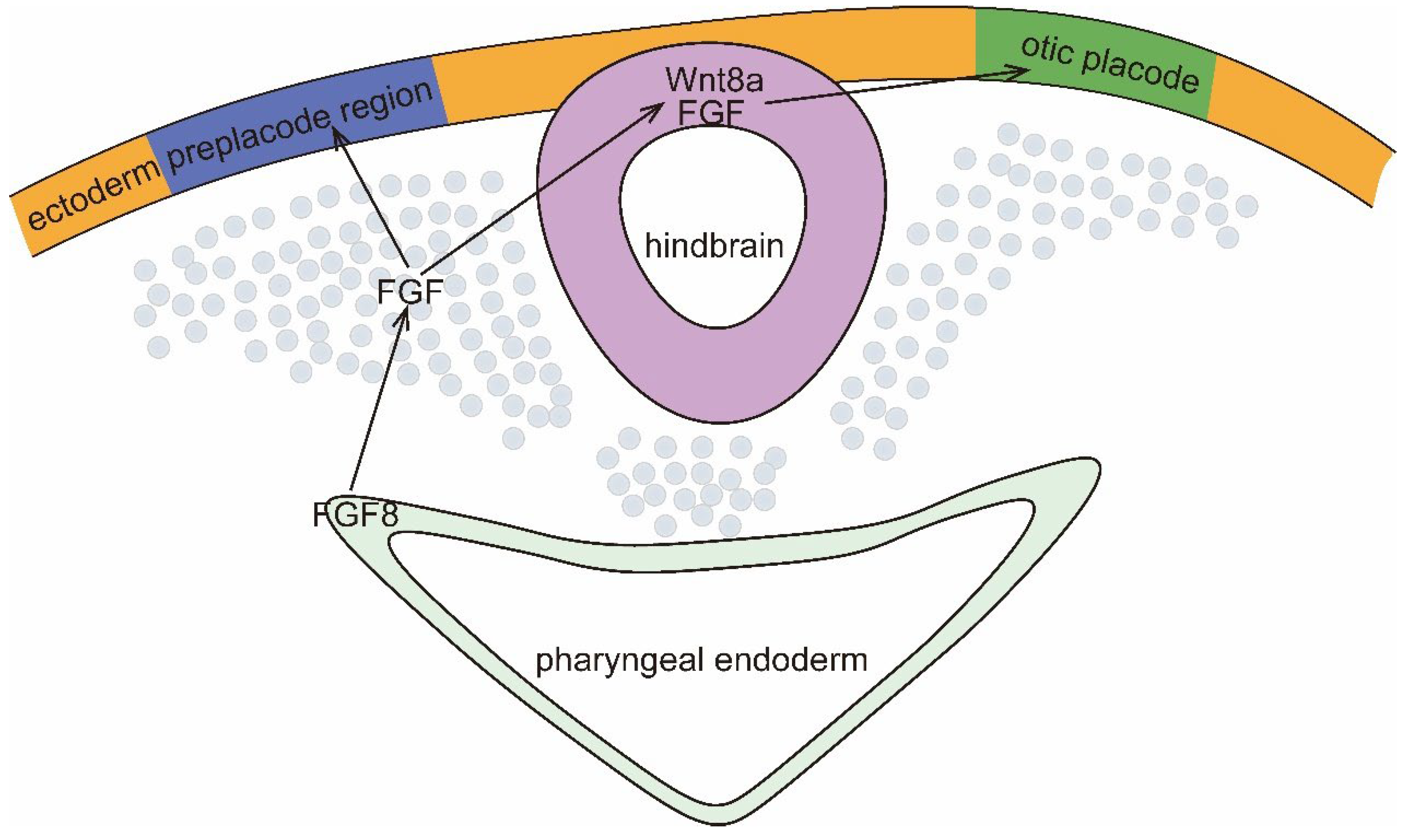
Figure 2.
HCMV regulates the classical Wnt/β-catenin signaling pathway. When the Wnt pathway is switched off (the Wnt receptor complex is not bound to the ligand), GSK3α/β phosphorylates β-catenin, which is then ubiquitinated and rapidly destroyed by proteasome targeting. In the nucleus, binding of Groucho to TCF/LEF inhibits transcription of Wnt target genes. When the Wnt signaling pathway is opened, the Fzd/LRP receptor complex activates the classical signaling pathway. Receptor recruitment to Dvl serves as a docking platform for components of the β-catenin disruption complex. Wnt induces GSK3β and CK1/CK2 to phosphorylate LRP5/6, which regulates Axin docking and releases β-catenin. In the nucleus, β-catenin replaces Groucho in Tcf/Lef, thereby promoting transcription of Wnt target genes. HCMV reduces the poly-ADP-ribosylation activity of end-anchor polymerase, thereby stabilizing Axin and inhibiting the Wnt pathway. HCMV US28 inhibits GSK3 activity via the ROCK pathway, β-catenin cannot be phosphorylated for degradation HCMV US28 inhibits GSK3 activity through the ROCK pathway, and β-catenin cannot be phosphorylated and degraded.
Figure 2.
HCMV regulates the classical Wnt/β-catenin signaling pathway. When the Wnt pathway is switched off (the Wnt receptor complex is not bound to the ligand), GSK3α/β phosphorylates β-catenin, which is then ubiquitinated and rapidly destroyed by proteasome targeting. In the nucleus, binding of Groucho to TCF/LEF inhibits transcription of Wnt target genes. When the Wnt signaling pathway is opened, the Fzd/LRP receptor complex activates the classical signaling pathway. Receptor recruitment to Dvl serves as a docking platform for components of the β-catenin disruption complex. Wnt induces GSK3β and CK1/CK2 to phosphorylate LRP5/6, which regulates Axin docking and releases β-catenin. In the nucleus, β-catenin replaces Groucho in Tcf/Lef, thereby promoting transcription of Wnt target genes. HCMV reduces the poly-ADP-ribosylation activity of end-anchor polymerase, thereby stabilizing Axin and inhibiting the Wnt pathway. HCMV US28 inhibits GSK3 activity via the ROCK pathway, β-catenin cannot be phosphorylated for degradation HCMV US28 inhibits GSK3 activity through the ROCK pathway, and β-catenin cannot be phosphorylated and degraded.
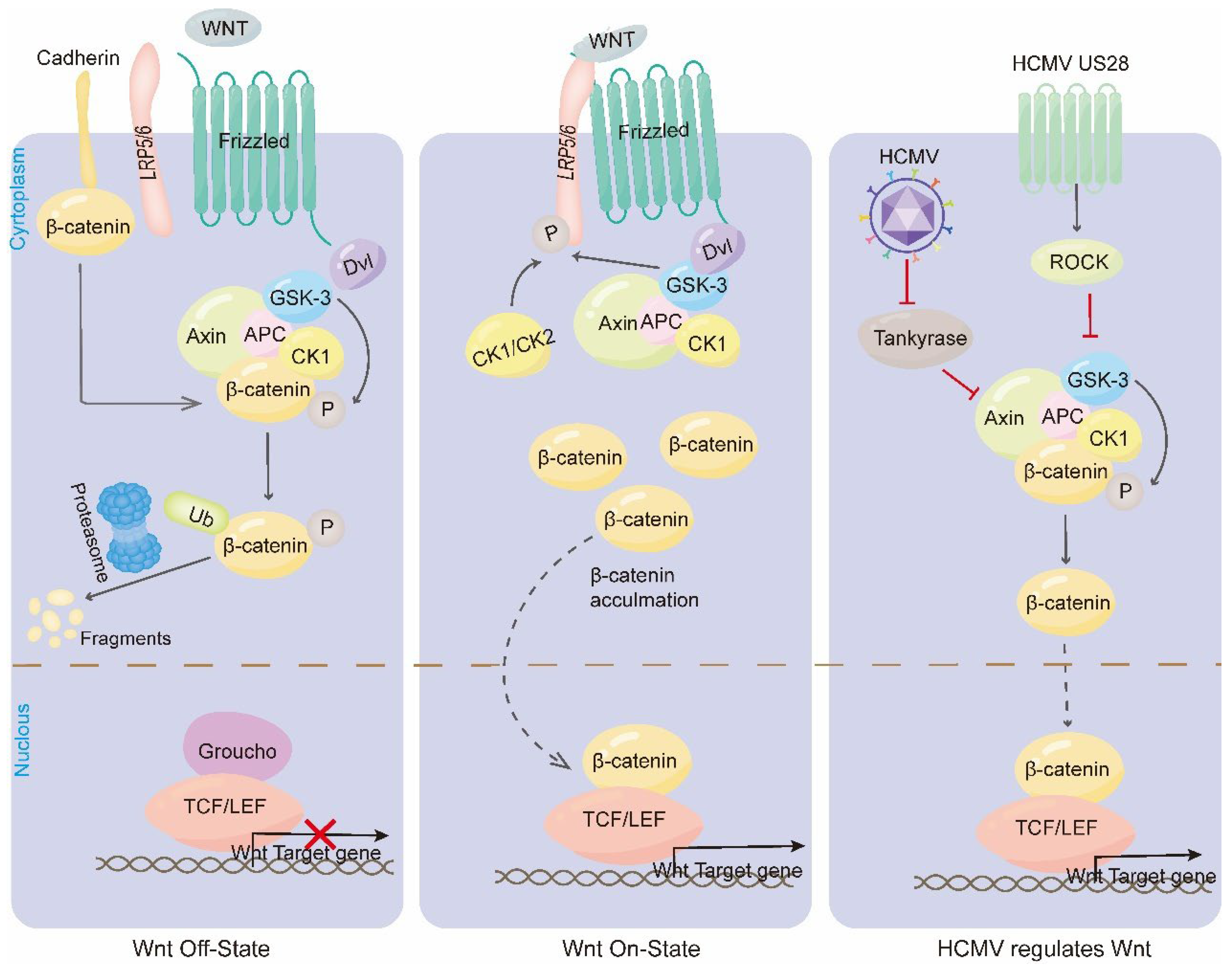
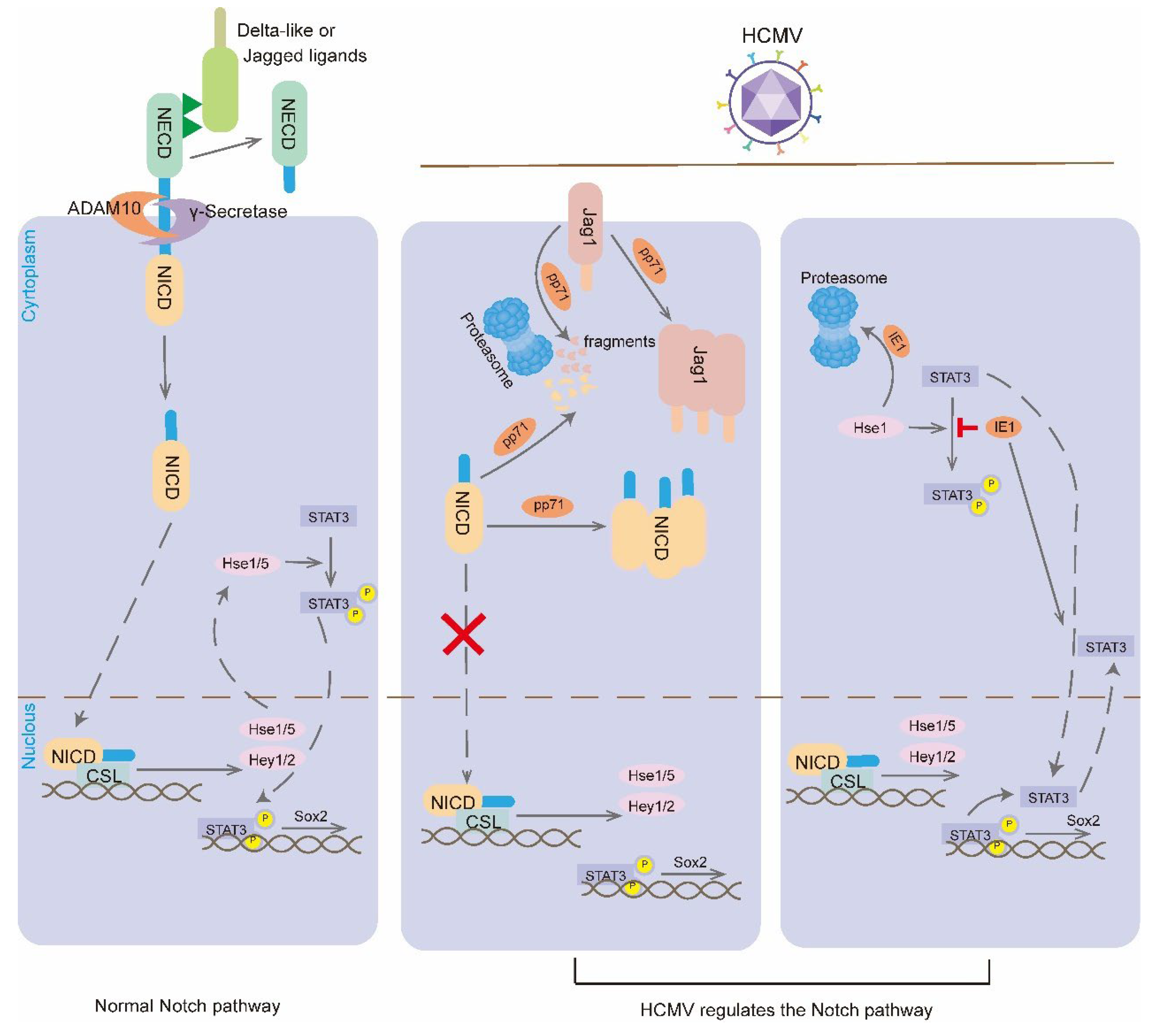
Figure 3.
HCMV regulates the Notch signaling pathway. When Notch receptor binds to Notch ligand, Notch receptor is cleaved by ADAM10 and γ-secretase, leading to the release of Notch intracellular structural domain (NICD).NICD fragment is the active form of the receptor that binds to the CSL transcription factor to trigger the transcription of the Notch target genes. Hes proteins also promote the phosphorylation and activation of STAT3, and the activated STAT3 shuttles to the nucleus to bind the Sox2 promoter and induce its expression. HCMV is a membrane protein that regulates Notch signaling pathway. phosphorylation and activation, and activated STAT3 shuttles to the nucleus to bind the Sox2 promoter and induce its expression. HCMV periplasmic protein pp71 can alter the cellular localization of Jag1 and NICD and inhibit Notch signaling by promoting their degradation via the proteasome. HCMV IE1 promotes proteasomal degradation of Hes1 to inhibit the phosphorylation of STAT3, and promotes the accumulation of unphosphorylated STAT3. accumulation of unphosphorylated STAT3 in the nucleus and inhibits Sox2 transcription.
Figure 3.
HCMV regulates the Notch signaling pathway. When Notch receptor binds to Notch ligand, Notch receptor is cleaved by ADAM10 and γ-secretase, leading to the release of Notch intracellular structural domain (NICD).NICD fragment is the active form of the receptor that binds to the CSL transcription factor to trigger the transcription of the Notch target genes. Hes proteins also promote the phosphorylation and activation of STAT3, and the activated STAT3 shuttles to the nucleus to bind the Sox2 promoter and induce its expression. HCMV is a membrane protein that regulates Notch signaling pathway. phosphorylation and activation, and activated STAT3 shuttles to the nucleus to bind the Sox2 promoter and induce its expression. HCMV periplasmic protein pp71 can alter the cellular localization of Jag1 and NICD and inhibit Notch signaling by promoting their degradation via the proteasome. HCMV IE1 promotes proteasomal degradation of Hes1 to inhibit the phosphorylation of STAT3, and promotes the accumulation of unphosphorylated STAT3. accumulation of unphosphorylated STAT3 in the nucleus and inhibits Sox2 transcription.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



