
Submitted:
12 June 2024
Posted:
18 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
This review investigates links between post-acute sequelae of SARS-CoV-2 infection (PASC), post-infection viral persistence, mitochondrial involvement and aberrant innate immune response and cellular metabolism during SARS-CoV-2 infection. Advancement of proteomic and metabolomic studies now allows deeper investigation of alterations to cellular metabolism, autophagic processes and mitochondrial dysfunction caused by SARS-CoV-2 infection while computational biology and machine learning have advanced methodologies of predicting virus-host gene and protein interactions. Particular focus is given to interaction between viral genes and proteins with mitochondrial function and that of the innate immune system. Finally, the authors hypothesise that viral persistence may be a function of mitochondrial involvement in sequestration of viral genetic material. While further work is necessary to understand the mechanisms definitively, a number of studies now point to resolution of questions regarding the pathogenesis of PASC.
Keywords:
SARS-CoV-2
; PASC
; long-Covid
; mitochondria
; mtDNA
; autophagy
; mitophagy
; innate immunity
; cell metabolism
; reactive oxygen species
1. Introduction
1.1. Post-Acute Sequelae Of SARS-CoV-2
Referred to variously as “long COVID”, “long COVID syndrome”, “post COVID-19 condition [1] , “post-acute COVID-19 syndrome” [2], or “post-acute sequelae of SARS-CoV-2 (PASC)” [3], PASC is defined as a multi-organ syndrome with a varying yet consistent constellation of symptoms including fatigue, shortness of breath, neurological symptoms of confusion or “brain fog”, stress, anxiety, joint pain, muscle pain, cough, nasal congestion, runny nose, “tightening” chest pain, palpitations, tachycardia and a variety of other symptoms not within 12 weeks of the start of COVID-19 [2,4].
It is estimated that PASC affects around 10% of patients following acute infection [5] with more than 65 million formerly infected individuals currently suffering at least one long-term sequela [3]. Much research has been undertaken among previously hospitalised patient cohorts with moderate to severe symptoms of acute infection, among whom estimates ranging between 32.6% to 87% suffered sequelae [6]. Risk factors for developing PASC include advanced age, female sex, history of smoking, obesity, presence of comorbidities and previous hospitalisation or admission to an intensive care unit [7].
1.2. Proposed Pathogenesis Of PASC
The exact pathogenesis of PASC syndrome has received much discussion, with long-term organ damage [8], immune dysfunction, microbiota disruption, autoimmune interactions, metabolic dysfunction, endothelial dysfunction and post-intensive care syndrome among the forefront of proposed theories [7]. The phenomenon of long-term retention of symptoms is not unique to SARS-COV-19; similar post-acute syndromes having been noted in association with previous coronavirus outbreaks such as severe acute respiratory syndrome (SARS) and Middle-East respiratory syndrome (MERS). Post-mortem, histologic patterns of damage in patients dying from acute COVID infection have some similarities to those of patients who die from acute respiratory distress syndrome (ARDS) secondary to H1N1 flu virus, SARS or MERS, having diffuse alveolar damage and perivascular T cell lymphocytic infiltration. In contrast, they also have characteristic endothelial disruption with the presence of intracellular viruses, widespread thrombosis with microangiopathy and sustained angiogenesis at 2.7 times that of patients from control groups [9]. Patients suffered symptoms including abnormalities in pulmonary function [10,11] chronic fatigue [12], significantly increased stress and psychological distress [13] manifesting as increased incidence of psychiatric disease [12], depression [14,15], and overall reduced quality of life. In the aftermath of the more localised SARS & MERS outbreaks some authors proposed that post-acute sequelae developed as a result of psychological distress [15].
Nalbandian et al. proposed three methods of long-COVID syndrome pathogenesis: pathophysiological changes mediated by the specific virus, immunological aberration and inflammatory damage in response to acute infection and expected sequelae of post-critical illness [2]. Venturelli et al. conducted large scale study attempting to separate features of long COVID syndrome by method of pathogenesis. Their proposed categorisation grouped symptoms into those explained by post-traumatic stress disorder (including anxiety, flashbacks, and nightmares, those from post-viral chronic fatigue syndrome (such as persistent fatigue, sleep disturbance and headache) and finally those from post-critical illness syndrome (including muscle weakness, respiratory difficulties and cognitive impairment) [16,17]. Surveying 2,050 individuals in Tyrol (Austria) and South Tyrol (Italy) Sahanic et al. characterised two distinct phenotypes of acute COVID syndrome, non-specific infection with generalised symptoms and a “multi-organ phenotype” (MOP), suffering multiple neurological, cardiopulmonary, gastrointestinal and dermatological complaints. They note that patients expressing MOP type acute infection suffered longer and more damaging post-acute symptoms compared to those with non-specific symptoms [18].
It was recognised in early stages of the global pandemic that encephalopathy presented as a rare but significant complication of acute infection [19], and Muccioli et al. propose that similar pathogenesis could be considered responsible for the neuropsychiatric PASC symptoms [20]. While there is little evidence to support the theory that SARS-CoV-2 or previous coronaviruses are directly neuropathogenic, neurological manifestation of disease have nevertheless been features of post-infectious subacute states associated with SARS, MERS and COVID-19 [21].
2. Coronavirus Replication & Persistence
2.1. Structure & Replication
Coronaviruses are positive sense single-stranded RNA (+ssRNA) viruses with a wide degree of variance capable of infecting both humans and a multitude of animal species. Noted for their large >30kb genomic RNA load, they consist of a number of protein subunits holding varied basic and specialised functions [22]. In total there are 14 open reading frames (ORFs), 16 non-structural proteins (NSPs) and 9 accessory and structural proteins. The major function of viral proteins are assisting the virus in binding to and penetrating host cells, participating in the viral genome replication process, assisting in the assembly and release of new viral particles, while some particles may also interfere with elements of the the host immune response [23]. In particular, the structural S “spike” protein forms homotrimers binding the cellular entry receptor and mediates infection, the E “envelope” and M “membrane” proteins form structural assembly components and the N “nucleocapsid” protein encapsulates the viral +ssRNA genetic material [23,24,25].
Initial infection results when the coronavirus S protein binds to cellular entry receptors – most importantly human angiotensin-converting enzyme 2 (ACE2) receptor and cell-surface serine protease TMPRSS2, both found extensively throughout respiratory tract tissues. After cell entry viral particles have structural proteins removed in endosomes and open reading frames (ORF1a & ORF1b) are immediately translated to NSPs which will go on to form the perinuclear replication and translation complex (RTC). The RTC produces genetic material within double membrane vesicles (DMVs) that are translated into more viral particles which will be exocytosed from the infected cell [22]. Cryo-electron microscopy indicates that perinuclear DMVs have a hexameric NSP3 constituted molecular pore which allows replicated viral genetic material to be exported for translation and packaging of new virions [26].
2.2. Persistence Of SARS-CoV-2 Proteins & Genetic Material
SARS-CoV-2 RNA and replicating viruses are not fully cleared after acute infection, indicating that reservoirs of viral material persist long after infection [27]. The great persistence of this viral RNA is surprising because endogenous mRNA is typically degraded in efficient manner by nuclease enzymes, with various studies estimating median half-life for all genes between 7 [28] and 10 hours [29]. Yang et al. showed that alternative transcripts have higher degradation rates owing to specific sequences within retained introns or alternatively spliced exons [29]. Sharova et al. demonstrated that genes involved in regulation of the cell cycle, transcription, apoptosis had the shortest half-lives while genes regulating cellular metabolism, protein biosynthesis and maintenance of the cytoskeleton and extracellular matrix were the most persistent [28].
Multiple studies have confirmed the presence of +ssRNA, the full S protein, spike S1 subunit and N protein long after acute symptoms have resolved [30,31]. Craddock et al. demonstrated persistence of both viral RNA and the S protein in both convalescent patients and patients with long term sequelae, the latter demonstrating significantly higher levels of both RNA and S protein than convalescent patients returned to health [31]. Multi-organ involvement seems highly likely with regards to both acute and post-acute symptoms, especially in light of viral RNA and spike proteins being localised throughout the blood serum [30], gastrointestinal tract [32], gallbladder [33], tonsils [34], and central nervous system [35].
A number of early pandemic autopsies in 2020 localised viral RNA and protein within the central nervous system of patients succumbing to acute infection [36], while Stein et al. localised viral RNA and protein to a number of locations, most significantly the cervical spinal cord, cerebellum, basal ganglia and hypothalamus [35] and others have determined the presence of viral genetic material in cerebrospinal fluid via PCR [37]. Autopsies undertaken in 2020 revealed that the presence of viral components accompanied profound histologic change including perivascular leukocytic infiltration, hypoxic change, focal haemorrhage, necrosis and oedema [9]. This correlates with a large scale longitudinal study conducted by the UK Biobank where two sessions of magnetic resonance imaging were conducted on 401 individuals who tested positive for SARS-CoV-2 after their first imaging, with the results compared with 384 control individuals who did not test positive. Overall, individuals contracting SARS-CoV-2 infection showed pathological change consistent with tissue damage to the primary olfactory cortex, greater degree of cortical atrophy and greater than expected loss of cognitive function than would be expected in this cohort [38].
While these studies provide ample evidence for neurovascular invasion and viral mediation of multi-systemic symptoms in acute infection, nothing provides better evidence for the persistence of neurological post-acute sequelae than the long-term retention of viral RNA and spike proteins in highly variable tissues around the body. Roden et al. demonstrated that post-mortem lung tissue retained viral RNA up to 174 days after acute infection [39], while Zollner et al. demonstrated viral RNA and nucleocapsid protein within the gut mucosa of 32 patients with irritable bowel syndrome averaging 219 days since acute infection diagnosis [40]. Viral RNA has been further localised in the skin and appendix of a patient 426 days after symptom onset [41].
Clearly, viral genetic material persists many orders of magnitude longer than endogenous mRNA. The long latent period suggests that patients with post-acute sequelae harbour actively replicating viral reservoirs, however the highly varying quantities of genetic material present indicates that the size or activity of these reservoirs is both variable among the patient cohort and can be expressed irregularly at different times of post-acute activity [27].
3. Autophagy, Mitochondrial Damage & Innate Immune Response
3.1. Autophagy & Mitophagy
Autophagy (or autophagocytosis) is a highly conserved cellular adaptation process seen in ageing, neurodegeneration, cancer and infection. This natural process of cell degradation eliminates unnecessary or dysfunctional components using a regulated mechanism that depends on lysosomes. Unstressed cells typically have minimal autophagy, yet it is rapidly enhanced in cases of nutrient deprivation, infection, and cellular damage. Initially, autophagy is characterised by the formation of a double-membrane phagophore (PG) vesicle within the cytosol allowing cellular structures to be identified for disposal. PG vesicles mature to fuse with lysosomes resulting in degradation and recycling of cellular structures [42]. Autophagy is essential in balancing innate immune inflammatory responses. The protective effect is typically exerted through regulation of inflammatory responses and maintenance of cellular homeostasis during stress. Dysfunctional autophagy is linked to infections, inflammations, neurodegeneration and tumorigenesis [43,44].
The inflammasome, consisting of cytoplasmic multiprotein complexes, is a critical component of the innate immune system's protection against pathogens. “Canonical” inflammasomes are capable of activating caspase-1 and are activated either by infection, endogenous proteins associated with mitochondrial damage caused by reactive oxygen species (ROS), other damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) [45,46]. Several types of inflammasomes have been discovered, such as NLRP1, NLRP3, NLRC4, and AIM2 [47]. Cleavage and pro-caspase 1 to caspase 1 activation results in inflammasome activation and the secretion of pro-inflammatory cytokines and IL-1β and IL-18. The degree of intracellular caspase 1 activation directly influences the extent of cytokines production [48].
Mitochondria damaged by endogenous or infective processes produce higher amounts of superoxide anions and other ROS and such damaged mitochondria must be cleared from the cell by autophagic processes or their presence will trigger inflammasome activation and production of pro-inflammatory cytokines [48]. Mitophagy is a specific form of autophagy where damaged mitochondria are degraded within autophagosomes and acts as a form of “quality control” over damaged mitochondria that inappropriately produce ROS or leak internal contents [42]. It is controlled within the cell by the PINK1-Parkin pathway and the accumulation of mitochondrial p62 also known as sequestosome-1 [49]. PTEN-induced kinase 1 (PINK1) is a serine/threonine kinase that phosphorylates both ubiquitin and Parkin (an E3 ubiquitin ligase). Active Parkin attaches serial ubiquitin chains to mitochondrial outer membrane proteins marking the defective organelle for destruction (Figure 1) [50,51]. The damaged organelle is then packaged into ATG9a integrated vesicles, upon which the “phagophore” can then be fused with lysosomes and degraded [52]. p62 is an autophagy receptor protein which aggregates around damaged or misfolded proteins, sensitising them to ubiquitin binding. Defective p62 activation is associated with proteinopathies such as Huntingdon disease [53] while the PINK-Parkin pathway of mitochondrial degradation relies on recruitment and activation of p62 [54].
3.2. Is Mitochondrial DNA A Warning Of An Ageing Immune System?
Mitochondrial dysfunction is vital in inflammatory responses in human disease. Mitochondria function as signalling hubs for antiviral response and enhance antimicrobial immunity via ROS production. However, they can also induce inflammation when there is cellular damage and stress. [55]. Mitochondria are an important source of DAMPs, among the most prominent of which are ROS and mitochondrial DNA. mtDNA is a circular, double-stranded DNA molecule found throughout the organelle that comprises 37 genes encoding 13 subunits of electron transport chain proteins, two ribosomal RNAs and 22 transfer RNAs [56]. At least three molecules sense mtDNA: Toll-like receptor 9 (TLR-9), NOD-, LRR -and pyrin domain containing protein 3 (NLRP3) that function as a pattern recognition receptor, and cyclic GMP–AMP synthase (cGAS) (Figure 1) [57]. The triggering of these sensors can result in the secretion of proinflammatory cytokines (i.e., TNF-α, IL-6, IL-1β) [58].
Impaired quality control of mitochondrial genetic material and organelle homeostasis associated with ageing produces increased “leakage” of mtDNA into cytosol, extracellular space, and into plasma. Physiologically, this is seen in increasing circulating mtDNA levels in the elderly and has implications for age-related inflammation, known as “inflamm-ageing” [59]. This release can cause immunometabolic dysfunction, reduced health span, and accelerated ageing, underscoring the influence of mtDNA on immune responses and ageing [60]. Increased presence of mtDNA is associated with and exacerbates the intensity of cardiovascular diseases, inflammatory arthritis, and age-related inflammation [61,62]. Moreover, mutations in mtDNA that lead to defective mitophagy can result in innate immunity disorders, contributing to the chronicity of inflammation [63]. Additionally, the decline in mtDNA copy number as individuals age has been well studied and is associated with decreases in mitochondrial function and overall performance, especially in conditions such as peripheral artery disease [64].
3.3. Mitochondrial Fusion & Fission
It is common practice to represent mitochondria as static organelles enclosed within the cytoplasm of a cell, isolated from each other and individual in form and function. Under-represented in the literature is an understanding of the highly dynamic nature of the cellular mitochondrial mass. Individual mitochondria exist within a continuum of fusion and fission to form new organelles and split those that have become too large. This cycle functions to produce a well-controlled mass of mitochondrial material of regulated size and efficiency catering to the specific energy requirements of the cell which, when surplus to requirements or no longer functional can be catabolised to its component parts.
Fusion is mediated by highly conserved outer membrane GTPases mitofusin-1 and -2 and inner membrane protein optic atrophy 1 (OPA1). The role of OPA1 is to uphold the structure of the membrane and provide protection for mtDNA. Mitochondrial fragmentation occurs when any of these proteins are removed, thus allowing the clearance of damaged mitochondria via mitophagy [65,66]. Although the specific molecular triggers for OPA1 processing are not well understood, it is clear that both apoptosis activation and disruption of mitochondrial membrane potential result in the cleavage of OPA1 [67,68]. During fusion, there is a rapid exchange of components like metabolites and soluble proteins, whereas membrane embedded proteins and mtDNA spread at a slower rate. The fusion of mitochondria helps minimize heterogeneity of the content, making it the first line of defense against dysfunction [69,70].
Fission is mediated by the outer membrane protein mammalian homologue of yeast FIS1 (hFIS1) and dynamin-related protein 1 (DRP1) in the cytosol [71]. The process is well regulated as cells with a low energy balance undergo changes allowing mitochondria to become more granular, separate from each other and gain higher surface area [72]. When visualised using fluorescent biomarkers localised within mitochondria the process is seen to be highly dynamic, providing balance between the rate of fusion and fission allowing self-regulation of mitochondrial length and motility. The probability that a single mitochondrion will undergo fission is largely a product of length, while the probability that it will undergo fusion is largely a product of motility [73]. Imbalance in the fusion-fission cycle can lead to the pathological generation of excessively large “megamitochondria” [72], formation of which has been associated with various cardiac, renal, hepatic and neurodegenerative diseases [74].
Cellular mitochondria have a dynamic nature governed by repeated, cyclical fusion-fission episodes which must remain in balance for the proper function of the cell to continue. We can, therefore, dispel the orthodox view that singular organelles are largely separate in function and efficiency and, once no longer functional, are degraded by mitophagy. Instead, pathological disturbances resulting in damage to mitochondrial components have the potential to affect the whole mitochondrial compartment, while individual components of the mitochondrial compartment are degraded and recycled as necessary. Mitochondrial fusion-fission processes have been found to be disrupted in several diseases, including neurodegeneration, obesity, type II diabetes, and COVID-19 [75,76]. Disruption in mitochondrial dynamics can cause oxidative stress, mitochondrial disfunction and dysregulation of the innate immune response during infection by SARS-CoV-2. During the early stages of infection, cells exhibit modifications in mitochondria characterized by thinner and elongated structures, suggesting morphological and dynamic fusion-fission related change [77]. The virus induces a rise in mitochondrial transmembrane potential resulting in elongated mitochondria and increased ATP synthesis, potentially contributing to viral replication and disease progression [78]. The virus therefore exacerbates the severity of disease by promoting the fusion and elongation of mitochondria, more effectively replicating within the host cells [76].
3.4. Mitochondrial Control of Inflammation
Inflammation is a complex biological response to infection or tissue damage, involving the coordinated action of various mediators to defend pathogens, repair damaged tissues, and prevent further injury [79]. Mitochondria can act as signalling platforms for controlling inflammation through various mechanisms. The dysfunctional mitochondria can lead to oxidative stress, which subsequently triggers inflammation and tissue remodelling [80]. The release of mitochondrial constituent and metabolic products can act as DAMPs. Examples of DAMPs include N-formyl peptidase, mtDNA, the DNA derived from viral infections or single-stranded viral RNA [81].
Fragmented or oxidized mtDNA can trigger inflammation via the activation of three main pro-inflammatory mechanisms: (1) Toll-like receptors (TLR9 signalling pathway), and (2) cytosolic cyclic GMP/AMP synthase—stimulator of interferon genes DNA-sensing system (cGAS-STING pathway), and (3) nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome (NLRP3- mediated inflammation) (Figure 1).
Extracellularly, mtDNA activates inflammatory responses via neutrophil bound PRR TLR-9 [82], and intracellular, endosomal TLR-9 is an element of the important signalling pathway that recognizes unmethylated CpG motifs in DNA commonly found in bacteria, viruses [83], and also mtDNA [84]. Protein 88 (MYD88) facilitates the communication between mtDNA and TLR9, resulting in the activation of nuclear factor κB (NF-κB) and triggering inflammatory and antiviral responses (Figure 2). Intracellularly, mtDNA release into cytosol is sensed by the DNA sensor cGAS and triggers the cGAS-STING pathway, leading to the expression of type I interferon and inflammatory cytokines like TNF and interleukins [85]. STING recognises the presence of viral genetic material and, via a series of second messengers including TANK-binding kinase 1 (TBK1), causes phosphorylation of IRF3 to drive the transcription of type I IFNs such as IFNα and IFNβ and starts the process of autophagy (Figure 1) [86]. Not only IRF3, but the cGAS-STING pathway can also activate Nf-κB through alternative mechanisms [87]. There is significant evidence that suggests mtDNA acts as an endogenous activator of inflammasomes. Various receptor proteins, including NLRP3, NLRC4, and AIM-2 are involved in the recognition and activation of inflammasomes, specifically through the recognition of mtDNA [88,89]. There is a possibility that the process of autophagy is influenced by mtDNA, which can lead to the overproduction of inflammatory mediators and activation of apoptotic signal pathways, and the interaction between autophagy and apoptosis further supports this connection [90]. Defective autophagy (mitophagy) allows accumulation of damaged mitochondria and release of mtDNA, triggering inflammatory pathways like cGAS-STING and NLRP3 [91,92]. The cGAS-STING pathway activated by cytosolic mtDNA can induce apoptosis through various mechanisms including ER stress and NF-κB activation [93]. Alternatively, autophagy could act as a negative regulator of mtDNA-induced inflammatory responses by inhibiting TLR9 overexpression during inflammation. [94].
MAVS, also known as a mitochondrial antiviral signalling protein, is a key component in the innate immune system's response to viral infections [62]. Localised in the outer mitochondrial membrane, MAVS plays a role in activating type I interferon production during viral infections, acting downstream of the cytosolic RNA sensor RIG-I (Figure 2). Currently, RIG-I and MDA-5 are identified as members of RIG-I-like receptor family (RLR) [95]. In the resting state MAVS is bound to mitofusin-2 and located in the mitochondrial membrane. Activated RIG-1 and associated chaperone proteins form a complex with MAVS which, dissociating from mitofusin-2 and the outer mitochondrial membrane, is free to translocate and activate transcription of antiviral proteins [96]. MAVS-RIG-I (for cytosolic RNA sensing) and cGAS-STING (for cytosolic DNA sensing) pathways are regulated by a complex series of ubiquitination and de-ubiquitination reactions [97] and it is notable that both converge on IRF-3 [86].
Activation of Toll-like receptors (TLRs) occurs when they recognise pathogen-associated molecular patterns (PAMPs). These patterns are typically found in foreign organisms, such as bacteria and viruses. TLR localisation can occur on either the cell surface (TLR-1, -2, -4, -5, -6, -10) or in intracellular compartments such as endosomes (TLR-3, -7, -8, -9). The ability to recognise viral single-stranded RNA (ssRNA) implies its potential for SARS-CoV-2 clearance of SARS-CoV-2. These receptors detect signals and initiate NF-κB and IRFs activation. Activation can occur through the MyD88-dependent and MyD88-independent pathways, ultimately leading to the expression of cytokines and interferon (IFN-I). The binding of viral RNA to RIG-I or MDA5 prompts the creation of MAVS polymers in mitochondria, followed by the subsequent attachment of TRAFs. TRAFs activate the NF-κB, and IRFs, mainly IRF-3 and IRF-7. This process leads to the expression of antiviral ISGs and pro-inflammatory cytokines. Activation of the JAK-STAT pathway by IFN and cytokines initiates the innate immune response against viral infections. The role of this pathway is critical in various physiological and pathological processes like cancer development, inflammation, tissue damage, and viral infections. The binding of Type I IFN to IFNα receptors (IFNAR) activates JAKs, which in turn phosphorylates and moves STATs to the nucleus. This process leads to the expression of antiviral ISGs. Once the STAT1-STAT2 heterodimer is formed, it binds to IRF9 to create the transcriptionally active ISGF3. The early immune response to viral infections heavily relies on the activation of the Jak2/STAT3 signalling pathway by Il-6, which facilitates the virus clearance through neutrophils. The IL-6 protein binds to the IL-6R receptor, which is composed of the IL-6α receptor molecule and the gp130 signal transducer, allowing cell signalling. JAK2 activation induced by IL-6 through gp130 results in the activation of STATs, particularly STAT3.
ISGs - interferon-stimulated genes; GAS - interferon-activated site; IRF9 - interferon regulatory factor 9; TBK1 - TANK binding kinase, IKKα - IκB kinase α; TRAF - tumour necrosis factor receptor-related factor; Jak - Janus kinase; STAT - signal transducer and activator of transcription; IFNs - interferons; Tyk2 - tyrosine kinase 2; ISGF3 - INF-stimulated gene factor 3; ISRE - interferon-stimulated response element; RIG-I - retinoic acid-inducible gene I; MDA5 - melanoma differentiation-associated protein 5; MODS - Multiple Organ Dysfunction Syndrome; MyD88 - myeloid differentiation primary response 88; PAMPs - pathogen-associated molecular patterns; TRAF6 - tumour necrosis factor receptor-associated factor 6; NEMO - NF-κB essential modulator; TRAM - Trif-related adaptor molecule; TRIF - TIR domain-containing adaptor protein inducing interferon β. (Created with BioRender.com)
3.5. Viral Interference in Innate Immune Response
The SARS-CoV-2 virus employs a range of strategies to disrupt and avoid the innate immune response of the host, with a particular focus on the interferon (IFN) response. Proteins immediately translated from viral open reading frames interfere at multiple levels of anti-viral interferon responses [98]. It has now been recognised that SARS-CoV-2 and a variety of other coronaviruses are capable of de-ubiquitination and can produce dysregulation at multiple layers of interferon mediated innate immune response. In SARS-CoV-2 this is accomplished by the papain-like protease (PLpro) domain of NSP3 which acts as a deubiquitinating enzyme and is capable of a wide pattern of disruption to interferon production and response [99]. Indeed, Zhao et al. went as far as to characterise the entire interplay between SARS-CoV-2 and infected hosts as a battle for dominance over E3 ubiquitin ligase (one of a triad of ubiquitinating enzymes) and de-ubiquitinating enzymes [100]. Through deubiquitinating the proteins involved in IFN signalling pathways, PLpro has the ability to inhibit the synthesis of IFN-β and suppress the downstream antiviral effects of IFN-stimulated genes (ISGs).
A surprisingly large selection of viral proteins have secondary roles interfering with innate cellular antiviral processes. Immediately on cellular access NSP1 decreases cytoplasmic translation of type I (including IFN-α and IFN-β) and III interferons and favours cellular translation of viral mRNA over cellular mRNA [21], ORF-3 [101], ORF-6 and N protein all inhibit phosphorylation of interferon regulatory factor-3 (IRF-3), preventing its translocation from cytoplasm to nucleus where it should act as a transcription factor activating interferon production [98]. Similarly, the SARS-CoV-2 M protein inhibits production of IFN-β and other type I interferons by inhibiting IRF3 phosphorylation and nuclear translocation [24]. N protein, meanwhile, has been also been found to dramatically inhibit cellular response to interferon proteins. Cells with viral N protein have reduced expression of DNA binding nuclear factor protein NF-κB, here responsible for activating the expression of interferon anti-viral genes via interferon-stimulated response element (ISRE) [98]. Viral elements interfere with the cGAS-STING-TBK1 axis of IRF-3 activation at numerous levels, with ORF3a and ORF9b interfering with STING activation and ORF7a, NSP5, NSP6 and NSP13 interfering with TBK1 activation [86]. In a 2020 study Jiang et al. determined that viral accessory protein ORF9b interfered with MAVS in the outer mitochondrial membrane, also inhibiting type I IFN production [102].
Viral infections can change the shape and function of mitochondria and these changes can lead to cell death and affect how cells produce energy and defend against viruses. Mitochondria have a vital role defending the body against viruses, and understanding how viruses and mitochondria interact could assist in finding new ways to treat disease. Studies demonstrate that viral infection interferes with the anti-inflammatory effects of IL-6, a cytokine which, via the JAK/STAT pathway, activates genes involved in differentiation, survival, apoptosis and proliferation [103] and viral targeting of parts of the JAK/STAT pathway results in both interferon dysfunction and insensitivity to IL-6 [104]. This, accompanied by viral-mediated mitochondrial dysfunction leading to activation of TLR-9-NFκB-IL-6 axis, potentially explains why severe acute COVID-19 infection is characterised by high circulating IL-6, sustained cytokine production and hyper-inflammation [105]. Evidently, the SARS-CoV family has evolved to evade interferon mediated cell death by both inhibiting the production of IFNs and inhibiting cellular response to their activation at multiple levels.
4. Alteration To Cellular Metabolism In SARS-CoV-2 Infection
4.1. Functional Change In Energy Production In Infected Cells
Metabolomic studies have focused on acute COVID-19 biomarkers, the most successful including lactic acid, glutamate, aspartate, phenylalanine, β-alanine, ornithine, arachidonic acid, choline and hypoxanthine [106]. A key feature of acute infection by SARS-CoV-2 is dysregulation of hepatic carbon metabolism [107]. Using gas chromatography and mass spectrometry, metabolomic profiles of patients with mild and severe acute infection reveal dysregulation of energy production and amino acid catabolism strongly correlated with the degree of hypoxia suffered by the patient [106]. In mild cases, tricarboxylic acid cycle metabolites succinate, citrate, pyruvate and glutamate accumulate in the serum in increased quantities, indicating reduced acetyl-CoA oxidation and the removal of oxaloacetate from the mitochondria on failure to condense with acetyl-CoA. Nuclear magnetic resonance imaging of serum metabolites shows marked increase in ketone bodies (acetoacetate and 3-hydroxybutyrate) further indicating a failure of oxidative phosphorylation within hepatocytes [108]. In severe cases of acute infection citrate levels decrease and positively correlate with the level of hypoxaemia [109].
Codo et al. isolated and sequenced RNA from bronchoalveolar lavage (BAL) of patients with severe infection and determined that circulating monocytes reconfigured their energy producing metabolism to overwhelmingly glycolytic means while simultaneously upregulating expression of ACE2 receptors [110]. Similar levels of mitochondrial derangement were found by Ajaz et al. in 2021 [111]. These early studies support observations regarding the interesting changes in metabolic profile of classically and alternatively activated macrophages. Classically activated (M1) pro-inflammatory macrophages rely on aerobic glycolysis to provide rapid and efficient responses to microbes whereas alternatively activated (M2) anti-inflammatory macrophages have a metabolic profile based on oxidative phosphorylation [112]. Activation is determined by two mutually inhibitory arginine requiring pathways: M1 macrophages via the inducible nitric oxide (NO) synthase pathway involving PI3K/AKT/mTOR and M2 via arginase and AMPK signalling [113]. In M1 activated macrophages the presence of NO inhibits mitochondrial aconitase (ACO2) and inactivates isocitrate dehydrogenase 2 (IDH2) resulting in a curtailing of the TCA cycle, exfiltration of citrate from the mitochondria and stabilisation of hypoxia inducible factor-1α (HIF-1α) [114].
Changes in metabolites associated with catabolism of branched chain amino acids, glutamate and phenylalanine have been noted in patients with both milder and more severe acute infection and suggest a “Warburg-like” reconfiguration of cellular energy production to adapt to hypoxic conditions [109]. Hypoxaemic states cause adaptation of energy production away from TCA cycle and fatty acid β-oxidation and towards anaerobic glycolysis. Similar to the neoplastic Warburg effect, it has been hypothesised that “Warburg-like” reconfiguration of cellular glucose metabolism enables virus-invaded cells to produce more biosynthetic substrates enabling greater capacity for virion manufacture, viral replication in host cells likely being supported by alteration of cellular glucose metabolism [114].
Viruses can regulate host cell oxidative phosphorylation and mitochondrial function to facilitate their replication and survival [115] and alternative means of curtailing the role of oxidative phosphorylation exist in virally infected cells. C15orf48 is a mitochondrial protein translated from the gene known both as C15orf48 or NMES1. TLR signalling upregulates C15orf48 in M1 macrophages of patients with autoimmune disease and with severe SARS-CoV-2 infection and appears to both downregulate pro-inflammatory cytokines [116] and interfere with complexes I, III and IV of the electron transport chain (ETC), potentially modulating cytochrome c (ETC complex IV) in states of viral infection [116,117].
Nevertheless, cells in hypoxic conditions have been shown to maintain the TCA cycle even when there are profound reductions in glucose-dependent citrate production so long as a source of glutamine derived α-ketoglutarate remains able to be reductively carboxylated by mitochondrial IDH2 [118]. This situation, extensively described in cancer cells adapting to function in low-oxygen environments, occurs when HIF-1α becomes stabilised in hypoxic condition. HIF-1α stimulates glycolysis and represses mitochondrial function and O2 consumption by inducing pyruvate dehydrogenase kinase 1 (PDK1) to phosphorylate and inhibit pyruvate dehydrogenase (PDH) from processing pyruvate for use in the TCA cycle [119]. This active cessation of pyruvate metabolism results in NADPH dependent “reverse flux” through IDH2 allowing the continued reductive synthesis of citrate from glutamate and cellular proliferation even in states of severe hypoxia [118].
In the infected patient, alterations to energy metabolism and mitochondrial function can occur in brain tissue even in the absence of viral RNA. Guarnieri et al. hypothesise that this could be caused by the action of diffusible factors after integrated stress response (ISR) activation in infected tissue [120]. The ISR is an intracellular signalling response activated in response to proteostatic stress, regulated by four key “sentinel” kinases – PERK, PKR, GCN2, HRI [121,122]. The presence of misfolded or damaged proteins in the ER [123], relative amino acid starvation, oxidative stress and mitochondrial dysfunction [124] in the ER or cytosol activates sentinel kinases, the activity of which converges on eukaryotic translation initiation factor 2 subunit α (eIF2α) phosphorylation. Phosphorylation of eIF2α produces wide ranging downstream effects, most notably alterations in protein synthesis and a reduction in global rates of mRNA translation. When stress cannot be mitigated the cell undergoes apoptosis [122]. Of particular interest, PKR is an IFN-induced double stranded RNA-dependent protein kinase which, via two N terminal dsRNA binding motifs, is activated on contact with viral dsRNA [125]. ISR activation is well described in neural tissues, especially in Alzheimer’s disease [122]. Growth/differentiation factor 15 (GDF15) is a TGF-β related cytokine affecting systemic energy metabolism via GDNF-family receptor α-like (GFRAL) receptors and is produced in tissues in response to mitochondrial stress in a wide array of tissues [126]. GFRAL is highly expressed in hindbrain tissues of the area postrema, a region integrating a wide array of metabolic and physiological function [127]. It naturally follows that protein synthesis in this area is especially sensitive to biological stress during pathogenic infection. The suggestion by Guarnieri et al. that diffusible cytokines such as GDF15 produced after viral or mitochondrial ISR activation can influence distant metabolic function in otherwise unaffected tissue [120] remains not only convincing, but also merits further investigation.
4.2. Immune System Impacts Of SARS-CoV-2 On Lipid Metabolism
Viruses use their host’s lipid metabolism for replication and immune system evasion while viral mediated dysregulation of lipid metabolism results in heightened severity of infection [107]. Replication in perinuclear DMVs and trafficking through the ER and Golgi apparatus requires “hijacking” of the host’s intracellular lipid metabolism [128]. Post-replicative coronaviruses utilise the host’s endoplasmic reticulum-Golgi intermediate compartment (ERGIC) where new viruses are assembled before they undergo exocytosis from the trans-Golgi network (TGN) [129] allowing viral replication to bypass intracellular immune surveillance [128]. Similarly, SARS-CoV-2 exploits the presence of sphingomyelin derived ceramide lipid rafts in the cell membrane which cluster and organise a number of membrane-based receptor molecules including the ACE2 viral port of entry [130,131]. A 2022 study by Kornhuber et al. indicated that SARS-CoV-2 activates acid sphingomyelinase (ASM) inducing increases in ceramide and facilitating viral entry [130].
Patients with low levels of circulating high-density lipoprotein (HDL-C) had lower lymphocyte counts, higher C-reactive protein (CRP) and other acute phase proteins [132]. A number of studies have determined that patients with low serum HDL-C suffer a more severe course of COVID-19 infection [132,133]. High serum HDL correlates with good cardiovascular health [134], a large-scale meta-analysis of over a million subjects concluding that increased HDL is associated with reduced mortality [135]. Clearly patients with a greater level of overall health prior to infection are more capable of sustained immune response and should have better outcomes, but studies also indicate that acute infection and inflammation causes changes in lipid profile. Decreased HDL-C and increased low-density lipoprotein (LDL-C) and very-low density lipoprotein (VLDL-C) are seen in patients with acute infection [108], and HDL-C, LDL-C and apolipoprotein-A1 (Apo-A1) have been used as prognostic markers in a variety of disease states [136]. High density lipoprotein appears to confer protection during inflammation and sepsis and acts as a component of the innate immune system [137]. Reduced formation of Apo-A1 by the liver in inflammation, reduced ester formation owing to decreased lecithin-cholesterol acyltransferase (LCAT) activity and increased clearance owing to overproduction of serum amyloid A (SAA), which displaces Apo-A1 in HDL-C are all hypothesised as causing the observed reduction [136]. Increased SAA is associated with increased cardiovascular mortality and appears to be incorporated into HDL-C, rendering the protective effects dysfunctional [138].
Multiple studies now support the hypothesis that acute infection depletes serum cholesterol, activating sterol regulatory element binding protein-2 (SREBP2) [139,140]. This regulatory protein controls lipid cholesterol and fatty acid gene expressions via the MAPK signalling pathway [141]. As the amount of cholesterol within the cell decreases SREBP2 is increasingly activated, resulting in increased cholesterol [142]. Indeed, the presence of the C terminal end of SREBP in blood correlates with severity of disease progression and can be used as an indicator of acute disease prognosis [139]. It is further hypothesised that the reduction in cellular and serum cholesterol acts as an innate immune mechanism preventing viral access to cells – dramatic depletion of cholesterol results in less bond formation between viral S protein and cellular ACE2 receptors and decreased expression of ACE2 receptors overall [143].
4.3. Oxidative Stress
Oxidative stress occurs when the rate of ROS production exceeds the capacity of the anti-oxidant defence system [144] and signs of cellular metabolic and oxidative stress are evident both in acute and post-acute SARS-CoV-2 infection [145]. SARS-CoV-2 both directly and indirectly impairs mitochondrial function resulting in increased production of ROS and oxidative stress [146], and involvement of the ACE2 receptor, NADPH oxidases and inflammatory pathways are potential factors in the induction of oxidative stress by SARS-CoV-2 [147]. Reduction of ACE2 receptor expression in infected cells causes upregulation of angiotensin II (ATII). When bound to ACE1 receptors, ATII directly upregulates NADPH oxidase (isoform 2) causing increased ROS production throughout the organism. In physiological conditions excessive ROS activates Bcl-2-family pro-apoptotic proteins increasing mitochondrial permeability and releasing caspases, mtDNA and cytochrome c which can result in programmed death of the cell. Infection by SARS-CoV-2 also interferes with anti-oxidant pathways such as nuclear factor (erythroid-derived 2) -like 2 (NRF2) transcription factor which controls expression of genes that protect against cellular stress, indirectly resulting in increased intra- and extracellular oxidative stress and overall systemic inflammation [146].
Increased production of ketone bodies within hepatocytes and a failure of oxidative phosphorylation of acetyl-CoA suggests increased metabolic stress throughout the acute phase of SARS-CoV-2 infection and is characterised by increased production of ROS, as are reduced serum levels of essential amino acids, tyrosine and glutamine [108]. Reduced ability to oxidise acetyl-CoA in hepatic mitochondria may also result in significantly increased accumulation of VLDL-C and TAG in serum and indicate further metabolic stress [107]. Similarly, increased expression of the phosphorylated form of H2A histone family member X (γ-H2AX), an intracellular marker of ROS damage to DNA, is seen in tissues expressing viral S protein [148].
A small study of 50 patients in North-West Nigeria found that patients with severe acute infection have relative deficiency in antioxidant trace elements, vitamins, reduced glutathione and enzymatic antioxidants while expressing increased markers of oxidative stress [149]. Patients with increased oxidative stress from prior comorbidities also have a higher rate of severe complication and death upon infection [145] and in an early study in Italy patients with chronic obstructive pulmonary disease, acute respiratory distress syndrome and severe SARS-CoV-2 infection all patients were hypoxaemic and displayed an acute imbalance in blood redox state, indicating excess production of ROS [150].
Little noted but of major interest to the study of PASC is micro-circulatory dysfunction resulting from viral invasion of endothelial tissue contributing to widespread thrombosis in severe disease states [9]. Expression of the S protein in endothelial cells lines caused increased generation of ROS and increased expression of senescence markers indicating metabolic dysfunction, macromolecular damage and cell-cycle arrest [148]. Nevertheless, it remains to be seen whether endothelial dysfunction is caused by direct invasion of cells or by action of ROS, cytokines and unbalanced immune response to severe infection [151,152]. What has been determined is that hypoxia in endothelial cells results in increased production of ROS, HIF-1α stabilisation, activation of the PI3K signalling pathway with “Warburg-like” reconfiguration of cellular glucose metabolism and extensive thromboxane activation and micro-circulatory thrombosis [114].
During acute SARS-CoV-2 infection ROS mediated damage to the endothelial system results in thrombosis and hyperinflammation which in turn causes damage to the respiratory, cardiovascular and neurological systems. Increased oxidative stress results in disruption of the immune system, specifically in neutrophil extracellular trap (NET) formation. Neutrophils produce NETs (consisting of DNA and globular proteins) in response to inflammatory cytokines and their function is to provide a matrix for anti-microbial activity. Balance between NET formation and degradation is essential in the prosecution of disease. Excessive NET production results in immuno-thrombotic states, increased susceptibility to sepsis, acute respiratory distress syndrome (ARDS) and acute lung injury. Owing to the high glycaemic burden and the presence of advanced glycation end products, diabetic patients already maintain increased extracellular oxidative stress. Their concurrent, heightened, systemic inflammation explains the more severe course of disease within this cohort and similar effects are noted in other groups of patients with inflammatory conditions such as obesity, atherosclerosis and cancer [153]. Meanwhile, the total effect of increased ROS on the immune system remains elusive even though effects of increased ROS can be seen in certain aspects of immune function. Studies demonstrate the key role of surfactant protein-A (SP-A) on macrophage activation [112,145] and increased presence of ROS causes oxidation of SP-A and a consequent decrease in classical activation of macrophages, resulting in reduced classical immune response and phagocytic activity [154]. Excessive oxygen exposure over a prolonged period is a recognised factor that could increase oxidative stress and potentially play a part in the persistent inflammation that leads to the pulmonary fibrosis development post-COVID-19 infection [155].
5. Mitochondrial Interference & mtDNA
5.1. Computational Modelling
Pioneering work in computational biology conducted with the RNA-GPS machine learning tool in 2020 predicted that SARS-CoV-2 genetic material had a high affinity for residence within host cell mitochondria [156]. Similarly, in 2020 Gordon et al. Cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins and used affinity purification mass spectrometry (AP-MS) to predict high-confidence interactions between viral and host proteins, determining that viral NSP5 had a high likelihood of interacting with mitochondrial tRNA [157]. A 2014 study from SARS-CoV-1-host interaction pointed to the high possibility of host mitochondrial involvement in viral attempts to evade the innate immune system [158] and it is apparent that SARS-CoV-2 has continued this pattern of behaviour.
SARS-CoV-2 mitochondrial interaction was further demonstrated by Medini et al. in 2021, finding that mitochondrial genes were drastically downregulated in circulating blood cells and immune cells during acute infection but not in respiratory tract tissue [159]. This study highlights the crucial role of mitochondrial-nuclear co-regulation in the COVID-19 immune response. The decrease in mtDNA gene expression in patients' blood cells indicates a shift towards glycolysis, promoting virus replication [159].
Miller et al. reported after computational analysis of RNA transcriptome data from cell lines, BAL and clinical lung samples that while expression of mitochondrial genes, especially those responsible for proteins of complex I of the electron transport chain and other TCA cycle enzymes, were downregulated there was no significant effect on MAVS related innate antiviral defence [160]. The evidence indicates that the reduced expression of key mitochondrial genes might be the reason behind energy production difficulties and tissue damage seen in COVID-19 patients.
In contrast to this, during their 2022 study Li et al. found that expression of viral accessory protein ORF10 caused inhibition of MAVS-RIG1 pathway and inactivation of IFN type I production [161]. Confusion exists within the literature regarding this topic and more work is required to resolve the question of whether MAVS is inactivated in live, in vivo infection.
5.2. Breakthroughs With Fluorescence Microscopy & Multi-Omics
Remarkably, a 2022 study conducted by Shang et al. used fluorescence microscopy to localise SARS-CoV dsRNA (a genetic product of viral replication) within mitochondria, providing a seminal moment in understanding the complex interplay between autophagy, SARS-CoV-2 mitochondrial involvement and the efforts of the virus to evade the innate immune response. The authors noted the association of DMV sites of replication alongside damaged mitochondria and the role of outer membrane transport protein translocase of outer membrane 20 (TOM20) in internalising SARS-CoV-2 dsRNA genetic material, along with the failure of PINK1-Parkin pathway activation to result in successful mitophagy [162]. TOM20 is a component of the translocase of the outer membrane (TOM) complex, responsible for recognition, import and segregation of most the of the precursor proteins required within the organelle [163]. Confirming this, Mozzi et al. determined that the viral accessory protein ORF3c localises itself within the mitochondrion increasing production of mitochondrial ROS yet blocking mitophagy [164]. These studies indicate that SARS-CoV-2 appears capable of sequestration of its genetic material within mitochondria, ensuring that aberrant organelles evade destruction by mitophagy and this is one hypothesis to explain the greatly increased persistence of viral genetic material and proteins within post-infected patients.
In a wide-ranging 2023 study, Guarnieri et al. used nasopharyngeal biopsies, tissue from autopsies and infected rodent tissue to investigate the effects of SARS-CoV-2 infection on mitochondrial gene transcription [120]. They confirmed RNA-GPS predictions by Wu et al. [156] and Stukalov et al. [165] predicting viral polypeptide binding and interaction with mitochondrial proteins, especially those of ETC complex I, III & IV. Even when the virus was cleared cardiac, renal, hepatic and lymph node mitochondria remained injured. Mitochondrial bioenergetic gene function was depressed in autopsy samples from the heart, kidneys, liver, lungs and lymph nodes. The degree of mitochondrial gene modulation was proportional to viral load, while post-infection lung samples showed a return to normal expression of oxidative phosphorylation gene expression. Key mitochondrial inner membrane transport enzymes were downregulated in the heart, including SLC25AC phosphate carrier, SLC25A4 and SLC25A6 adenine nucleotide translocases, SLC25A1 citrate carrier and SLC25A12 Ca2+ binding aspartate-glutamate carrier, all of which are critical for the correct function of the TCA cycle and oxidative phosphorylation [120].
5.3. mtDNA As A Prognostic Marker Of Acute COVID Infection?
Increased circulating plasma levels of mtDNA have been demonstrated to determine poor outcomes in disease states. Quantitatively analysing systemic circulating mtDNA, Schneck et al. determined that patients suffering from septic shock showed consistently elevated levels when compared with patients suffering from post-operative inflammation [166]. Scozzi et al. demonstrated that high circulating levels of mtDNA were an early indicator of poor outcome in COVID-19 [91]. Edinger et al. conducted a study of 29 critically ill patients aged between 59 – 80 and found that significantly increased levels of peak plasma mtDNA could be used as a predictive marker of mortality [167]. While the full mechanism remains unclear, circulating mtDNA induces expression of inflammatory mediators TNF-α and IL-1β in mouse models [168]. Valdés-Aguayo et al. determined that damage to the mitochondrial membrane system during severe acute infection resulted in the release of mtDNA into circulation, triggering innate immune responses. They further conclude that high levels of mtDNA in circulation were useful as markers of infection [169].
According to recent studies, there is evidence to suggest that variations in mtDNA can play a role in determining the risk and severity of SARS-CoV-2 infection (for more information on haplogroups, see the mitomap database at www.mitomap.org). Studies in Slovak COVID-19 patients have shown that specific mtDNA haplogroups were associated with decreased risk of severe COVID-19 (J1, and clusters H+U5b, and T2b+U5b) while other haplogroups (T1, H11 and K (K1a)) were associated with an increased risk of severe COVID-19 [170]. This study recognizes that the limited number of samples and regional differences in mtDNA studies can create biases and impact the findings. The variations in haplogroups within the Han Chinese population of Hubei, China can be attributed to the presence of different mtDNA variants. Some of these variants, such as C5178a (in NADH dehydrogenase subunit 2), A249d (in the D-loop), T6392C (in the cytochrome C oxidase I gene) and G10310A, have been linked to a decreased risk of severe infection outcomes. On the other hand, variants like A4833G and T3394C have been associated with an increased risk [171]. Based on the results, it can be inferred that specific mtDNA variants have a substantial influence on the functioning of certain OXPHOS enzymes. It is suggested that these mtDNA variations may play a role in determining the severity of COVID-19 and the response to treatments targeting mitochondrial functions or OXPHOS.
Mitochondrial microRNAs (mitomiRs) have vital functions in regulating different aspects of mitochondrial homeostasis, such as controlling fission and fusion, managing mitophagy, regulating mitochondrial calcium levels, influencing OXPHOS, and safeguarding mtDNA integrity [172]. Furthermore, mitomiRs also play a role in regulating mtDNA translation by affecting the translational activity of mitochondrial genomes. A 2019 study of mitomiR-2392 showed that this non-coding RNA regulated chemoresistance in squamous cell carcinomas of the tongue by reprogramming cellular metabolism, downregulating electron transport chain complexes I, III and IV [173]. This fast-evolving field suggests that non-coding and micro-RNA (ncRNA and miRNA) interact at numerous levels to provide immediate control of cellular metabolism and protein function and further study can elucidate their interaction in virally mediated mitochondrial dysfunction [174]. Assessing the functionality of immune cells in the blood through mtDNA and mitomiR monitoring could serve as a non-intrusive approach to diagnose and predict outcomes of SARS-CoV-2 and infection by other viruses.
5.4. Mitochondrial Dysfunction In COVID-19 Patients & Therapeutic Implications
The replication of SARS-CoV-2 heavily depends on mitochondria, and the virus-cell interactions lead to disturbances in mitochondrial homeostasis. The investigation of potential treatments for mitochondrial dysfunction in COVID-19 is an active area of research.
The study by Guarnieri et al. highlights suppression of transcription of certain nuclear DNA-encoded mitochondrial oxidative phosphorylation genes in nasopharyngeal samples, inducing glycolysis and activating immune defence [120]. The decrease in expression of some genes, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and phosphoglycerate kinase 1 (PGK1), in human nasopharyngeal samples, can result in the accumulation of substrates in the initial steps of glycolysis. This accumulation promotes increased production of NADPH, fatty acids, and consequently supports viral biogenesis. In contrast, an increase in the expression of the HIF-1α gene, glycolysis genes, and mTOR signalling genes was observed in the autopsy tissue of patients with COVID-19. Mitochondrial gene expression was recovered in the lungs, yet mitochondrial function remained suppressed in the heart, kidney, and liver. The fact that organs outside the pulmonary system continue to malfunction implies that mitochondrial dysfunction might have a long-term impact on the internal organs of these patients. A significant finding of this study is that impaired mitochondrial gene expression persists in specific tissues even after the virus is cleared, potentially playing a role in the severity of COVID-19 pathology [120]. These findings suggest that therapies aimed at improving mitochondrial function, decreasing excessive mitochondrial reactive oxygen species (mROS), and preventing the release of mitochondrial DNA could potentially be effective in reducing the severity of acute infection by SARS-CoV-2 and relieving symptoms associated with long-covid.
A noteworthy finding in the research conducted by Guarnieri et al. was the detection of a potential therapeutic focus in microRNA 2392, also known as miR-2392. The study revealed that this particular microRNA controls the functioning of mitochondria in the human tissue samples examined [120]. Blocking miR-2392 is suggested as a way to improve mitochondrial function, decrease viral replication, and alleviate the severity of COVID-19 symptoms (Tab.1) [175]. microRNA can be also involved in the regulation of inflammatory responses to viral infections [176]. miR-146a and miR-21 are commonly referred to as "inflammamiRs" because they have the capability to control NF-κB-driven inflammatory pathways. They are upregulated in acute and chronic viral infections, and specifically target molecules involved in the NF-κB/NLRP3 inflammatory pathways [177]. Modulating their expression could potentially help control inflammation and protect mitochondrial function in COVID-19 patients.
Enhancing our comprehension of the function of mitochondria in SARS-CoV-2 infection could enhance intervention treatments and provide better protection for patients against pathogens. Many potential therapeutic drugs that reduce the severity of human infections with SARS-CoV-2 can be found in the Reactome database (https://reactome.org/content/detail/R-HSA-9679191.1), and therapeutic implications for mitochondrial dysfunction are in Table 1.
6. Conclusions
Recent studies suggest clear and significant links between SARS-CoV-2 infection and mitochondrial dysfunction, autophagy, innate immune response and cellular metabolic function. Circulating mtDNA clearly plays a role in acute COVID-19 pathogenesis yet it’s role in the pathogenesis of post-acute sequelae and “long COVID” syndrome nevertheless remains elusive. That the PASC-mtDNA association is still not fully understood highlights the need to continue to identify current gaps in our knowledge.
Complex computational multi-omics studies allow metabolic and proteomic profiling of infected tissues, providing deep understanding of the changes wrought to cellular metabolism by viral infection. In turn, this provides greater access to underlying genetic processes. Understanding the interplay of related processes underlying SARS-CoV-2 replication and pathogenesis allows investigation of the virally-induced mitochondrial dysfunction that determines the clinical events of disease progression. New evidence of SARS-CoV-2 RNA sequestration within the host mitochondrial matrix in addition to the nucleolus, predicted by computational models and confirmed in laboratory settings, provides tantalising hints to possible links between mitochondrial and innate immune system dysfunction, viral persistence and post-acute sequelae of COVID-19.
Author Contributions
Concept: BS & CW; writing and original draft presentation: CW; supervision: BS; review and commentary: BS. All authors have read and agreed to the published version of the manuscript.
Funding
This review was supported by Medical University of Gdańsk, grant no: 71-01415/0004604.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Post COVID-19 Condition (Long COVID) Available online:. Available online: https://www.who.int/europe/news-room/fact-sheets/item/post-covid-19-condition (accessed on 30 January 2024).
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-Acute COVID-19 Syndrome. Nat Med 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.L.; Catalfamo, C.J.; Farland, L.V.; Ernst, K.C.; Jacobs, E.T.; Klimentidis, Y.C.; Jehn, M.; Pogreba-Brown, K. Post-Acute Sequelae of COVID-19 in a Non-Hospitalized Cohort: Results from the Arizona CoVHORT. PLOS ONE 2021, 16, e0254347. [Google Scholar] [CrossRef] [PubMed]
- Overview | COVID-19 Rapid Guideline: Managing the Long-Term Effects of COVID-19 | Guidance | NICE Available online:. Available online: https://www.nice.org.uk/guidance/NG188 (accessed on 30 January 2024).
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major Findings, Mechanisms and Recommendations. Nat Rev Microbiol 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Lechner-Scott, J.; Levy, M.; Hawkes, C.; Yeh, A.; Giovannoni, G. Long COVID or Post COVID-19 Syndrome. Multiple Sclerosis and Related Disorders 2021, 55. [Google Scholar] [CrossRef] [PubMed]
- Tziolos, N.-R.; Ioannou, P.; Baliou, S.; Kofteridis, D.P. Long COVID-19 Pathophysiology: What Do We Know So Far? Microorganisms 2023, 11, 2458. [Google Scholar] [CrossRef] [PubMed]
- Haunhorst, S.; Bloch, W.; Wagner, H.; Ellert, C.; Krüger, K.; Vilser, D.C.; Finke, K.; Reuken, P.; Pletz, M.W.; Stallmach, A.; et al. Long COVID: A Narrative Review of the Clinical Aftermaths of COVID-19 with a Focus on the Putative Pathophysiology and Aspects of Physical Activity. Oxf Open Immunol 2022, 3. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Patel, K.; Greenwood, D.C.; Halpin, S.; Lewthwaite, P.; Salawu, A.; Eyre, L.; Breen, A.; O’Connor, R.; Jones, A.; et al. Long-Term Clinical Outcomes in Survivors of Severe Acute Respiratory Syndrome and Middle East Respiratory Syndrome Coronavirus Outbreaks after Hospitalisation or ICU Admission: A Systematic Review and Meta-Analysis. J Rehabil Med 2020, 52, jrm00063. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Joynt, G.M.; Wong, K.T.; Gomersall, C.D.; Li, T.S.; Antonio, G.; Ko, F.W.; Chan, M.C.; Chan, D.P.; Tong, M.W.; et al. Impact of Severe Acute Respiratory Syndrome (SARS) on Pulmonary Function, Functional Capacity and Quality of Life in a Cohort of Survivors. Thorax 2005, 60, 401–409. [Google Scholar] [CrossRef]
- Lam, M.H.-B.; Wing, Y.-K.; Yu, M.W.-M.; Leung, C.-M.; Ma, R.C.W.; Kong, A.P.S.; So, W.Y.; Fong, S.Y.-Y.; Lam, S.-P. Mental Morbidities and Chronic Fatigue in Severe Acute Respiratory Syndrome Survivors: Long-Term Follow-Up. Arch Intern Med 2009, 169, 2142–2147. [Google Scholar] [CrossRef]
- Lee, A.M.; Wong, J.G.W.S.; McAlonan, G.M.; Cheung, V.; Cheung, C.; Sham, P.C.; Chu, C.-M.; Wong, P.-C.; Tsang, K.W.T.; Chua, S.E. Stress and Psychological Distress among SARS Survivors 1 Year after the Outbreak. Can J Psychiatry 2007, 52, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lancee, W.J.; Maunder, R.G.; Goldbloom, D.S. ; Coauthors for the Impact of SARS Study Prevalence of Psychiatric Disorders among Toronto Hospital Workers One to Two Years after the SARS Outbreak. Psychiatr Serv 2008, 59, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Shin, H.-S.; Park, H.Y.; Kim, J.L.; Lee, J.J.; Lee, H.; Won, S.-D.; Han, W. Depression as a Mediator of Chronic Fatigue and Post-Traumatic Stress Symptoms in Middle East Respiratory Syndrome Survivors. Psychiatry Investig 2019, 16, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Benatti, S.V.; Casati, M.; Binda, F.; Zuglian, G.; Imeri, G.; Conti, C.; Biffi, A.M.; Spada, M.S.; Bondi, E.; et al. Surviving COVID-19 in Bergamo Province: A Post-Acute Outpatient Re-Evaluation. Epidemiol Infect 2021, 149, e32. [Google Scholar] [CrossRef] [PubMed]
- Diem, L.; Fregolente-Gomes, L.; Warncke, J.D.; Hammer, H.; Friedli, C.; Kamber, N.; Jung, S.; Bigi, S.; Funke-Chambour, M.; Chan, A.; et al. Fatigue in Post-COVID-19 Syndrome: Clinical Phenomenology, Comorbidities and Association With Initial Course of COVID-19. J Cent Nerv Syst Dis 2022, 14, 11795735221102727. [Google Scholar] [CrossRef] [PubMed]
- Sahanic, S.; Tymoszuk, P.; Ausserhofer, D.; Rass, V.; Pizzini, A.; Nordmeyer, G.; Hüfner, K.; Kurz, K.; Weber, P.M.; Sonnweber, T.; et al. Phenotyping of Acute and Persistent Coronavirus Disease 2019 Features in the Outpatient Setting: Exploratory Analysis of an International Cross-Sectional Online Survey. Clin Infect Dis 2022, 75, e418–e431. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Ren, Y.; Lv, T. Encephalitis as a Clinical Manifestation of COVID-19. Brain Behav Immun 2020, 88, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, L.; Pensato, U.; Cani, I.; Guarino, M.; Cortelli, P.; Bisulli, F. COVID-19-Associated Encephalopathy and Cytokine-Mediated Neuroinflammation. Ann Neurol 2020, 88, 860–861. [Google Scholar] [CrossRef] [PubMed]
- Maury, A.; Lyoubi, A.; Peiffer-Smadja, N.; de Broucker, T.; Meppiel, E. Neurological Manifestations Associated with SARS-CoV-2 and Other Coronaviruses: A Narrative Review for Clinicians. Rev Neurol (Paris) 2021, 177, 51–64. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat Rev Microbiol 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 Genome-Encoded Proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci 2021, 30, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Sui, L.; Zhao, Y.; Wang, W.; Wu, P.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q. SARS-CoV-2 Membrane Protein Inhibits Type I Interferon Production Through Ubiquitin-Mediated Degradation of TBK1. Front Immunol 2021, 12, 662989. [Google Scholar] [CrossRef]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; De Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A Molecular Pore Spans the Double Membrane of the Coronavirus Replication Organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef]
- Proal, A.D.; VanElzakker, M.B.; Aleman, S.; Bach, K.; Boribong, B.P.; Buggert, M.; Cherry, S.; Chertow, D.S.; Davies, H.E.; Dupont, C.L.; et al. SARS-CoV-2 Reservoir in Post-Acute Sequelae of COVID-19 (PASC). Nature Immunology 2023, 24, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Sharova, L.V.; Sharov, A.A.; Nedorezov, T.; Piao, Y.; Shaik, N.; Ko, M.S.H. Database for mRNA Half-Life of 19 977 Genes Obtained by DNA Microarray Analysis of Pluripotent and Differentiating Mouse Embryonic Stem Cells. DNA Res 2009, 16, 45–58. [Google Scholar] [CrossRef]
- Yang, E.; van Nimwegen, E.; Zavolan, M.; Rajewsky, N.; Schroeder, M.; Magnasco, M.; Darnell, J.E. Decay Rates of Human mRNAs: Correlation With Functional Characteristics and Sequence Attributes. Genome Res 2003, 13, 1863–1872. [Google Scholar] [CrossRef]
- Swank, Z.; Senussi, Y.; Manickas-Hill, Z.; Yu, X.G.; Li, J.Z.; Alter, G.; Walt, D.R. Persistent Circulating Severe Acute Respiratory Syndrome Coronavirus 2 Spike Is Associated With Post-Acute Coronavirus Disease 2019 Sequelae. Clin Infect Dis 2022, 76, e487–e490. [Google Scholar] [CrossRef] [PubMed]
- Craddock, V.; Mahajan, A.; Spikes, L.; Krishnamachary, B.; Ram, A.K.; Kumar, A.; Chen, L.; Chalise, P.; Dhillon, N.K. Persistent Circulation of Soluble and Extracellular Vesicle-linked Spike Protein in Individuals with Postacute Sequelae of COVID-19. J. Med. Virol. 2023, 95. [Google Scholar] [CrossRef]
- Chun Chau Lawrence Cheung; Denise Goh; Xinru Lim; Tracy Zhijun Tien; Jeffrey Chun Tatt Lim; Justina Nadia Lee; Benedict Tan; Zhi En Amos Tay; Wei Yee Wan; Eileen Xueqin Chen; et al. Residual SARS-CoV-2 Viral Antigens Detected in GI and Hepatic Tissues from Five Recovered Patients with COVID-19. Gut 2022, 71, 226. [Google Scholar] [CrossRef]
- Hany, M.; Zidan, A.; Gaballa, M.; Ibrahim, M.; Agayby, A.S.S.; Abouelnasr, A.A.; Sheta, E.; Torensma, B. Lingering SARS-CoV-2 in Gastric and Gallbladder Tissues of Patients with Previous COVID-19 Infection Undergoing Bariatric Surgery. Obes Surg 2023, 33, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Miura, C.S.; Lima, T.M.; Martins, R.B.; Jorge, D.M.M.; Tamashiro, E.; Anselmo-Lima, W.T.; Arruda, E.; Valera, F.C.P. Asymptomatic SARS-COV-2 Infection in Children’s Tonsils. Braz J Otorhinolaryngol 2022, 88, 9. [Google Scholar] [CrossRef]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.-Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 Infection and Persistence in the Human Body and Brain at Autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Mukerji, S.S.; Solomon, I.H. What Can We Learn from Brain Autopsies in COVID-19? Neurosci Lett 2021, 742, 135528. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Tan, B. What Can Cerebrospinal Fluid Testing and Brain Autopsies Tell Us about Viral Neuroinvasion of SARS-CoV-2. J Med Virol 2021, 93, 4247–4257. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 Is Associated with Changes in Brain Structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Roden, A.C.; Boland, J.M.; Johnson, T.F.; Aubry, M.C.; Lo, Y.-C.; Butt, Y.M.; Maleszewski, J.J.; Larsen, B.T.; Tazelaar, H.D.; Khoor, A.; et al. Late Complications of COVID-19A Morphologic, Imaging, and Droplet Digital Polymerase Chain Reaction Study of Lung Tissue. Arch Pathol Lab Med 2022, 146, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Zollner, A.; Koch, R.; Jukic, A.; Pfister, A.; Meyer, M.; Rössler, A.; Kimpel, J.; Adolph, T.E.; Tilg, H. Postacute COVID-19 Is Characterized by Gut Viral Antigen Persistence in Inflammatory Bowel Diseases. Gastroenterology 2022, 163, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Goh, D.; Lim, J.C.T.; Fernaíndez, S.B.; Joseph, C.R.; Edwards, S.G.; Neo, Z.W.; Lee, J.N.; Caballero, S.G.; Lau, M.C.; Yeong, J.P.S. Case Report: Persistence of Residual Antigen and RNA of the SARS-CoV-2 Virus in Tissues of Two Patients with Long COVID. Frontiers in Immunology 2022, 13. [Google Scholar]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Huo, Y.; Sawant, A.; Tan, Y.; Mahdi, A.H.; Li, T.; Ma, H.; Bhatt, V.; Yan, R.; Coleman, J.; Dreyfus, C.F.; et al. Tumor Suppressor PALB2 Maintains Redox and Mitochondrial Homeostasis in the Brain and Cooperates with ATG7/Autophagy to Suppress Neurodegeneration. PLOS Genetics 2022, 18, e1010138. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.D.; Galle-Treger, L.; Akbari, O. Role of Autophagy in Lung Inflammation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy Balances Inflammation in Innate Immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-C.; Jha, S.; Linhoff, M.W.; Ting, J.P.-Y. The NLR Gene Family: From Discovery to Present Day. Nat Rev Immunol 2023, 23, 635–654. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Response by Inhibiting NALP3 Inflammasome-Mediated Mitochondrial DNA Release. Nat Immunol 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Poon, A.; Saini, H.; Sethi, S.; O’Sullivan, G.A.; Plun-Favreau, H.; Wray, S.; Dawson, L.A.; McCarthy, J.M. The Role of SQSTM1 (P62) in Mitochondrial Function and Clearance in Human Cortical Neurons. Stem Cell Reports 2021, 16, 1276–1289. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin Is Phosphorylated by PINK1 to Activate Parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Münch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab Cycles Regulate Parkin-Mediated Mitophagy. eLife 7. [CrossRef]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic Stress Induces Phosphorylation of P62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates. PLoS Genet 2015, 11, e1004987. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-Mediated Mitophagy Is Dependent on VDAC1 and P62/SQSTM1. Nat Cell Biol 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in Innate Immune Responses and Inflammatory Pathology. Nat Rev Immunol 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Hisata, S.; Choi, A.M.K. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid Redox Signal 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating Mitochondrial DNA Increases with Age and Is a Familiar Trait: Implications for “Inflamm-Aging. ” Eur J Immunol 2014, 44, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Guerra Martinez, C.; Torres-Odio, S.; Bell, S.L.; Birdwell, C.E.; Bryant, J.D.; Tong, C.W.; Watson, R.O.; West, L.C.; West, A.P. Elevated Type I Interferon Responses Potentiate Metabolic Dysfunction, Inflammation, and Accelerated Aging in mtDNA Mutator Mice. Science Advances 2021, 7, eabe7548. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an Inflammatory Mediator in Cardiovascular Diseases. Biochem J 2018, 475, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial Control of Inflammation. Nat Rev Immunol 2023, 23, 159–173. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J Clin Med 2020, 9, 978. [Google Scholar] [CrossRef]
- Peripheral Artery Disease, Calf Skeletal Muscle Mitochondrial DNA Copy Number, and Functional Performance - Mary M McDermott, Charlotte A Peterson, Robert Sufit, Luigi Ferrucci, Jack M Guralnik, Melina R Kibbe, Tamar S Polonsky, Lu Tian, Michael H Criqui, Lihui Zhao, James H Stein, Lingyu Li, Christiaan Leeuwenburgh, 2018 Available online:. Available online: https://journals.sagepub.com/doi/10.1177/1358863X18765667 (accessed on 7 May 2024).
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J Cell Biol 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Scorrano, L. A Cut Short to Death: Parl and Opa1 in the Regulation of Mitochondrial Morphology and Apoptosis. Cell Death Differ 2007, 14, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E. OPA1 and PARL Keep a Lid on Apoptosis. Cell 2006, 126, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeong, S.-Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the Mammalian Mitochondrial Fission and Fusion Mediators Fis1, Drp1, and Opa1 in Apoptosis. Mol Biol Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Emerging Functions of Mammalian Mitochondrial Fusion and Fission. Hum Mol Genet 2005, 14 Spec No. 2, R283–289. [Google Scholar] [CrossRef]
- Griffin, E.E.; Detmer, S.A.; Chan, D.C. Molecular Mechanism of Mitochondrial Membrane Fusion. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2006, 1763, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial Fragmentation in Neurodegeneration. Nat Rev Neurosci 2008, 9, 505–518. [Google Scholar] [CrossRef]
- Wakabayashi, T. Megamitochondria Formation - Physiology and Pathology. J Cell Mol Med 2002, 6, 497–538. [Google Scholar] [CrossRef] [PubMed]
- Principles of the Mitochondrial Fusion and Fission Cycle in Neurons - PubMed Available online:. Available online: https://pubmed.ncbi.nlm.nih.gov/23525002/ (accessed on 13 March 2024).
- Shang, Y.; Li, Z.; Cai, P.; Li, W.; Xu, Y.; Zhao, Y.; Xia, S.; Shao, Q.; Wang, H. Megamitochondria Plasticity: Function Transition from Adaption to Disease. Mitochondrion 2023, 71, 64–75. [Google Scholar] [CrossRef]
- Grel, H.; Woznica, D.; Ratajczak, K.; Kalwarczyk, E.; Anchimowicz, J.; Switlik, W.; Olejnik, P.; Zielonka, P.; Stobiecka, M.; Jakiela, S. Mitochondrial Dynamics in Neurodegenerative Diseases: Unraveling the Role of Fusion and Fission Processes. International Journal of Molecular Sciences 2023, 24, 13033. [Google Scholar] [CrossRef]
- Holder, K.; Reddy, P.H. The COVID-19 Effect on the Immune System and Mitochondrial Dynamics in Diabetes, Obesity, and Dementia. The Neuroscientist 2020. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Lee, W.; Ku, K.B.; Yoon, G.Y.; Moon, H.-W.; Kim, C.; Kim, M.-H.; Yi, Y.-S.; Jun, S.; Kim, B.-T.; et al. SARS-CoV-2 Aberrantly Elevates Mitochondrial Bioenergetics to Induce Robust Virus Propagation. Sig Transduct Target Ther 2024, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Liu, J.-J.; Ma, S.-L. Analysis of Mitochondrial Function in Lymphocytes Obtained from COVID-19 Patients. Int J Immunopathol Pharmacol 2023, 37, 03946320231210736. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Chen, Y.-F.; Chen, Y.-F.; Chung, L.-C.; Tamilselvi, S.; Shen, C.-Y.; Day, C.H.; Chen, R.-J.; Viswanadha, V.P.; Kuo, W.-W.; et al. The Multifaceted Link between Inflammation and Human Diseases. Journal of Cellular Physiology 2018, 233, 6458–6471. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune Sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating Mitochondrial DAMPs Cause Inflammatory Responses to Injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. Cytoplasmic DNA Innate Immune Pathways. Immunological Reviews 2011, 243, 99–108. [Google Scholar] [CrossRef] [PubMed]
- T, O.; S, H.; O, Y.; M, T.; T, T.; T, T.; J, O.; T, M.; H, N.; K, N.; et al. Mitochondrial DNA That Escapes from Autophagy Causes Inflammation and Heart Failure. Nature 2012, 485. [Google Scholar] [CrossRef]
- Hu, M.-M.; Shu, H.-B. Mitochondrial DNA-Triggered Innate Immune Response: Mechanisms and Diseases. Cell Mol Immunol 2023, 20, 1403–1412. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, M.; Yuan, C.; Ma, Z.; Li, W.; Zhang, Y.; Su, L.; Xu, J.; Liu, W. Progress of cGAS-STING Signaling in Response to SARS-CoV-2 Infection. Front Immunol 2022, 13, 1010911. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, C.; Liao, Z.; Xu, P. cGAS-STING Signaling in Cell Death: Mechanisms of Action and Implications in Pathologies. European Journal of Immunology 2023, 53, 2350386. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Chen, J.; Zhao, L.; Wu, S.; Chen, X.; Chen, H. Mitochondrial DNA Leakage Triggers Inflammation in Age-Related Cardiovascular Diseases. Front. Cell Dev. Biol. 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Jabir, M.S.; Hopkins, L.; Ritchie, N.D.; Ullah, I.; Bayes, H.K.; Li, D.; Tourlomousis, P.; Lupton, A.; Puleston, D.; Simon, A.K.; et al. Mitochondrial Damage Contributes to Pseudomonas Aeruginosa Activation of the Inflammasome and Is Downregulated by Autophagy. Autophagy 2015, 11, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Wang, W.; Lu, L.; Wang, L.; Chen, X.; Guo, R.; Li, S.; Jiang, J. Inactivation of Beclin-1-Dependent Autophagy Promotes Ursolic Acid-Induced Apoptosis in Hypertrophic Scar Fibroblasts. Experimental Dermatology 2018, 27, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.; Rauseo, A.M.; Liu, Z.; Sahu, S.K.; et al. Circulating Mitochondrial DNA Is an Early Indicator of Severe Illness and Mortality from COVID-19. JCI Insight 2021, 6, e143299–143299. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; De Vries, R.L.A.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial Autophagy in Cells with mtDNA Mutations Results from Synergistic Loss of Transmembrane Potential and mTORC1 Inhibition. Hum Mol Genet 2012, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, J.; Zhang, X.; Li, X.; Wu, X.; Zhao, Y.; Ren, J. Circulating Mitochondrial DNA-Triggered Autophagy Dysfunction via STING Underlies Sepsis-Related Acute Lung Injury. Cell Death Dis 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Rodgers, M.A.; Bowman, J.W.; Liang, Q.; Jung, J.U. Regulation Where Autophagy Intersects the Inflammasome. Antioxid Redox Signal 2014, 20, 495–506. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, Y.; Wu, J.; Qi, N. MAVS: A Two-Sided CARD Mediating Antiviral Innate Immune Signaling and Regulating Immune Homeostasis. Front Microbiol 2021, 12, 744348. [Google Scholar] [CrossRef]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J Virol 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Q.; Pan, Y.; Wang, C. Sensing and Responding to Cytosolic Viruses Invasions: An Orchestra of Kaleidoscopic Ubiquitinations. Cytokine & Growth Factor Reviews 2015, 26, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe Acute Respiratory Syndrome Coronavirus Open Reading Frame (ORF) 3b, ORF 6, and Nucleocapsid Proteins Function as Interferon Antagonists. J Virol 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Duan, L.; Huang, B.; Xiong, Y.; Zhang, G.; Huang, H. The SARS-CoV-2 Papain-like Protease Suppresses Type I Interferon Responses by Deubiquitinating STING. Sci Signal 2023, 16, eadd0082. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, M.; Yang, Z.; Zhou, Z.; Huang, J.; Zhao, B. Role of E3 Ubiquitin Ligases and Deubiquitinating Enzymes in SARS-CoV-2 Infection. Front Cell Infect Microbiol 2023, 13, 1217383. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yang, X.; Chang, M.; Xue, Z.; Wang, W.; Bai, L.; Zhao, S.; Liu, E. ORF3a Protein of Severe Acute Respiratory Syndrome Coronavirus 2 Inhibits Interferon-Activated Janus Kinase/Signal Transducer and Activator of Transcription Signaling via Elevating Suppressor of Cytokine Signaling 1. Front Microbiol 2021, 12, 752597. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, H.; Meng, Q.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.; Wang, X.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b Suppresses Type I Interferon Responses by Targeting TOM70. Cell Mol Immunol 2020, 17, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of Interleukin (IL)-6-Type Cytokine Signalling and Its Regulation. Biochem J 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-Y.; Khan, N.; Close, B.J.; Goel, R.K.; Blum, B.; Tavares, A.H.; Kenney, D.; Conway, H.L.; Ewoldt, J.K.; Chitalia, V.C.; et al. SARS-CoV-2 Disrupts Proximal Elements in the JAK-STAT Pathway. J Virol 95. [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef]
- Onoja, A.; von Gerichten, J.; Lewis, H.-M.; Bailey, M.J.; Skene, D.J.; Geifman, N.; Spick, M. Meta-Analysis of COVID-19 Metabolomics Identifies Variations in Robustness of Biomarkers. International Journal of Molecular Sciences 2023, 24, 14371. [Google Scholar] [CrossRef]
- Casari, I.; Manfredi, M.; Metharom, P.; Falasca, M. Dissecting Lipid Metabolism Alterations in SARS-CoV-2. Prog Lipid Res 2021, 82, 101092. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, C.; Bizkarguenaga, M.; Gil-Redondo, R.; Diercks, T.; Arana, E.; García de Vicuña, A.; Seco, M.; Bosch, A.; Palazón, A.; San Juan, I.; et al. SARS-CoV-2 Infection Dysregulates the Metabolomic and Lipidomic Profiles of Serum. iScience 2020, 23, 101645. [Google Scholar] [CrossRef] [PubMed]
- Páez-Franco, J.C.; Torres-Ruiz, J.; Sosa-Hernández, V.A.; Cervantes-Díaz, R.; Romero-Ramírez, S.; Pérez-Fragoso, A.; Meza-Sánchez, D.E.; Germán-Acacio, J.M.; Maravillas-Montero, J.L.; Mejía-Domínguez, N.R.; et al. Metabolomics Analysis Reveals a Modified Amino Acid Metabolism That Correlates with Altered Oxygen Homeostasis in COVID-19 Patients. Sci Rep 2021, 11, 6350. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L. de B.; Souza, G.F. de; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; Junior, C.A.O. de B.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metabolism 2020, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial Metabolic Manipulation by SARS-CoV-2 in Peripheral Blood Mononuclear Cells of Patients with COVID-19. Am J Physiol Cell Physiol 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Shibata, M.; Horng, T. Metabolism Supports Macrophage Activation. Front Immunol 2017, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The Key Role of Warburg Effect in SARS-CoV-2 Replication and Associated Inflammatory Response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef]
- Moreno-Altamirano, M.M.B.; Kolstoe, S.E.; Sánchez-García, F.J. Virus Control of Cell Metabolism for Replication and Evasion of Host Immune Responses. Front Cell Infect Microbiol 2019, 9, 95. [Google Scholar] [CrossRef]
- Clayton, S.A.; Daley, K.K.; MacDonald, L.; Fernandez-Vizarra, E.; Bottegoni, G.; O’Neil, J.D.; Major, T.; Griffin, D.; Zhuang, Q.; Adewoye, A.B.; et al. Inflammation Causes Remodeling of Mitochondrial Cytochrome c Oxidase Mediated by the Bifunctional Gene C15orf48. Sci Adv 7. [CrossRef]
- Floyd, B.J.; Wilkerson, E.M.; Veling, M.T.; Minogue, C.E.; Xia, C.; Beebe, E.T.; Wrobel, R.L.; Cho, H.; Kremer, L.S.; Alston, C.L.; et al. Mitochondrial Protein Interaction Mapping Identifies New Regulators of Respiratory Chain Function. Mol Cell 2016, 63, 621–632. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia Promotes Isocitrate Dehydrogenase-Dependent Carboxylation of α-Ketoglutarate to Citrate to Support Cell Growth and Viability. Proc Natl Acad Sci U S A 2011, 108, 19611–19616. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab 2006, 3, 187–197. [Google Scholar] [CrossRef]
- Guarnieri, J.W.; Dybas, J.M.; Fazelinia, H.; Kim, M.S.; Frere, J.; Zhang, Y.; Soto Albrecht, Y.; Murdock, D.G.; Angelin, A.; Singh, L.N.; et al. Core Mitochondrial Genes Are Down-Regulated during SARS-CoV-2 Infection of Rodent and Human Hosts. Sci Transl Med 2023, 15, eabq1533. [Google Scholar] [CrossRef]
- Lu, H.; Koju, N.; Sheng, R. Mammalian Integrated Stress Responses in Stressed Organelles and Their Functions. Acta Pharmacol Sin 2024, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The Integrated Stress Response: From Mechanism to Disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial Dysfunction Is Signaled to the Integrated Stress Response by OMA1, DELE1 and HRI 2019, 715896.
- García, M.A.; Meurs, E.F.; Esteban, M. The dsRNA Protein Kinase PKR: Virus and Cell Control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The Metabolic Effects of GDF15 Are Mediated by the Orphan Receptor GFRAL. Nat Med 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Johann, K.; Kleinert, M.; Klaus, S. The Role of GDF15 as a Myomitokine. Cells 2021, 10, 2990. [Google Scholar] [CrossRef]
- Song, J.-W.; Lam, S.M.; Fan, X.; Cao, W.-J.; Wang, S.-Y.; Tian, H.; Chua, G.H.; Zhang, C.; Meng, F.-P.; Xu, Z.; et al. Omics-Driven Systems Interrogation of Metabolic Dysregulation in COVID-19 Pathogenesis. Cell Metab 2020, 32, 188–202. [Google Scholar] [CrossRef]
- Hong, W. Combating COVID-19 with Chloroquine. J Mol Cell Biol 2020, 12, 249–250. [Google Scholar] [CrossRef]
- Kornhuber, J.; Hoertel, N.; Gulbins, E. The Acid Sphingomyelinase/Ceramide System in COVID-19. Mol Psychiatry 2022, 27, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Carpinteiro, A.; Gripp, B.; Hoffmann, M.; Pöhlmann, S.; Hoertel, N.; Edwards, M.J.; Kamler, M.; Kornhuber, J.; Becker, K.A.; Gulbins, E. Inhibition of Acid Sphingomyelinase by Ambroxol Prevents SARS-CoV-2 Entry into Epithelial Cells. Journal of Biological Chemistry 2021, 296. [Google Scholar] [CrossRef]
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Declined Serum High Density Lipoprotein Cholesterol Is Associated with the Severity of COVID-19 Infection. Clinica Chimica Acta 2020, 510, 105–110. [Google Scholar] [CrossRef]
- Masana, L.; Correig, E.; Ibarretxe, D.; Anoro, E.; Arroyo, J.A.; Jericó, C.; Guerrero, C.; Miret, M.; Näf, S.; Pardo, A.; et al. Low HDL and High Triglycerides Predict COVID-19 Severity. Sci Rep 2021, 11, 7217. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-H. The Current Status of Research on High-Density Lipoproteins (HDL): A Paradigm Shift from HDL Quantity to HDL Quality and HDL Functionality. Int J Mol Sci 2022, 23, 3967. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Kong, S.Y.; Ro, Y.S.; Ryu, H.H.; Shin, S.D. Serum Cholesterol Levels and Risk of Cardiovascular Death: A Systematic Review and a Dose-Response Meta-Analysis of Prospective Cohort Studies. Int J Environ Res Public Health 2022, 19, 8272. [Google Scholar] [CrossRef]
- Filippas-Ntekouan, S.; Liberopoulos, E.; Elisaf, M. Lipid Testing in Infectious Diseases: Possible Role in Diagnosis and Prognosis. Infection 2017, 45, 575–588. [Google Scholar] [CrossRef]
- Trinder, M.; Boyd, J.H.; Brunham, L.R. Molecular Regulation of Plasma Lipid Levels during Systemic Inflammation and Sepsis. Current Opinion in Lipidology 2019, 30, 108. [Google Scholar] [CrossRef]
- Zewinger, S.; Drechsler, C.; Kleber, M.E.; Dressel, A.; Riffel, J.; Triem, S.; Lehmann, M.; Kopecky, C.; Säemann, M.D.; Lepper, P.M.; et al. Serum Amyloid A: High-Density Lipoproteins Interaction and Cardiovascular Risk. Eur Heart J 2015, 36, 3007–3016. [Google Scholar] [CrossRef]
- Lee, W.; Ahn, J.H.; Park, H.H.; Kim, H.N.; Kim, H.; Yoo, Y.; Shin, H.; Hong, K.S.; Jang, J.G.; Park, C.G.; et al. COVID-19-Activated SREBP2 Disturbs Cholesterol Biosynthesis and Leads to Cytokine Storm. Signal Transduct Target Ther 2020, 5, 186. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zeng, W.; Su, J.; Wan, H.; Yu, X.; Cao, X.; Tan, W.; Wang, H. Hypolipidemia Is Associated with the Severity of COVID-19. J Clin Lipidol 2020, 14, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An Aberrant SREBP-Dependent Lipogenic Program Promotes Metastatic Prostate Cancer. Nat Genet 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; Chen, S.; Zhang, L.; Liu, Q.; Guo, X.; et al. Cholesterol Homeostatic Regulator SCAP-SREBP2 Integrates NLRP3 Inflammasome Activation and Cholesterol Biosynthetic Signaling in Macrophages. Immunity 2018, 49, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Abu-Farha, M.; Thanaraj, T.A.; Qaddoumi, M.G.; Hashem, A.; Abubaker, J.; Al-Mulla, F. The Role of Lipid Metabolism in COVID-19 Virus Infection and as a Drug Target. Int J Mol Sci 2020, 21, 3544. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Czajkowsky, D.M. SARS-CoV-2 Infection and Oxidative Stress: Pathophysiological Insight into Thrombosis and Therapeutic Opportunities. Cytokine Growth Factor Rev 2022, 63, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Tekos, F.; Skaperda, Z.; Goutzourelas, N.; Phelps, D.S.; Floros, J.; Kouretas, D. The Importance of Redox Status in the Frame of Lifestyle Approaches and the Genetics of the Lung Innate Immune Molecules, SP-A1 and SP-A2, on Differential Outcomes of COVID-19 Infection. Antioxidants (Basel) 2020, 9, 784. [Google Scholar] [CrossRef] [PubMed]
- Gain, C.; Song, S.; Angtuaco, T.; Satta, S.; Kelesidis, T. The Role of Oxidative Stress in the Pathogenesis of Infections with Coronaviruses. Front. Microbiol. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Forcados, G.E.; Muhammad, A.; Oladipo, O.O.; Makama, S.; Meseko, C.A. Metabolic Implications of Oxidative Stress and Inflammatory Process in SARS-CoV-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front. Cell. Infect. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Meyer, K.; Patra, T.; Vijayamahantesh, null; Ray, R. SARS-CoV-2 Spike Protein Induces Paracrine Senescence and Leukocyte Adhesion in Endothelial Cells. J Virol 2021, 95, e0079421. [Google Scholar] [CrossRef]
- Muhammad, Y.; Kani, Y.A.; Iliya, S.; Muhammad, J.B.; Binji, A.; El-Fulaty Ahmad, A.; Kabir, M.B.; Umar Bindawa, K.; Ahmed, A. Deficiency of Antioxidants and Increased Oxidative Stress in COVID-19 Patients: A Cross-Sectional Comparative Study in Jigawa, Northwestern Nigeria. SAGE Open Med 2021, 9, 2050312121991246. [Google Scholar] [CrossRef] [PubMed]
- Duca, L.; Ottolenghi, S.; Coppola, S.; Rinaldo, R.; Dei Cas, M.; Rubino, F.M.; Paroni, R.; Samaja, M.; Chiumello, D.A.; Motta, I. Differential Redox State and Iron Regulation in Chronic Obstructive Pulmonary Disease, Acute Respiratory Distress Syndrome and Coronavirus Disease 2019. Antioxidants (Basel) 2021, 10, 1460. [Google Scholar] [CrossRef] [PubMed]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mäe, M.; Muhl, L.; et al. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Levy, J.H. COVID-19 and Its Implications for Thrombosis and Anticoagulation. Blood 2020, 135, 2033–2040. [Google Scholar] [CrossRef]
- Vollbracht, C.; Kraft, K. Oxidative Stress and Hyper-Inflammation as Major Drivers of Severe COVID-19 and Long COVID: Implications for the Benefit of High-Dose Intravenous Vitamin C. Front Pharmacol 2022, 13, 899198. [Google Scholar] [CrossRef] [PubMed]
- Mikerov, A.N.; Umstead, T.M.; Gan, X.; Huang, W.; Guo, X.; Wang, G.; Phelps, D.S.; Floros, J. Impact of Ozone Exposure on the Phagocytic Activity of Human Surfactant Protein A (SP-A) and SP-A Variants. Am J Physiol Lung Cell Mol Physiol 2008, 294, L121–L130. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary Fibrosis and COVID-19: The Potential Role for Antifibrotic Therapy. The Lancet Respiratory Medicine 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Wu, K.E.; Fazal, F.M.; Parker, K.R.; Zou, J.; Chang, H.Y. RNA-GPS Predicts SARS-CoV-2 RNA Residency to Host Mitochondria and Nucleolus. Cell Syst 2020, 11, 102–108. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug-Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J Immunol 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Medini, H.; Zirman, A.; Mishmar, D. Immune System Cells from COVID-19 Patients Display Compromised Mitochondrial-Nuclear Expression Co-Regulation and Rewiring toward Glycolysis. iScience 2021, 24, 103471. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.; Silverstein, A.; Flores, M.; Cao, K.; Kumagai, H.; Mehta, H.H.; Yen, K.; Kim, S.-J.; Cohen, P. Host Mitochondrial Transcriptome Response to SARS-CoV-2 in Multiple Cell Models and Clinical Samples. Sci Rep 2021, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 Suppresses the Antiviral Innate Immune Response by Degrading MAVS through Mitophagy. Cell Mol Immunol 2022, 19, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front Microbiol 2021, 12, 780768. [Google Scholar] [CrossRef] [PubMed]
- Budzińska, M.; Gałgańska, H.; Karachitos, A.; Wojtkowska, M.; Kmita, H. The TOM Complex Is Involved in the Release of Superoxide Anion from Mitochondria. J Bioenerg Biomembr 2009, 41, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Mozzi, A.; Oldani, M.; Forcella, M.E.; Vantaggiato, C.; Cappelletti, G.; Pontremoli, C.; Valenti, F.; Forni, D.; Saresella, M.; Biasin, M.; et al. SARS-CoV-2 ORF3c Impairs Mitochondrial Respiratory Metabolism, Oxidative Stress, and Autophagic Flux. iScience 2023, 26, 107118. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel Proteomics Reveals Host Perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Schneck, E.; Edinger, F.; Hecker, M.; Sommer, N.; Pak, O.; Weissmann, N.; Hecker, A.; Reichert, M.; Markmann, M.; Sander, M.; et al. Blood Levels of Free-Circulating Mitochondrial DNA in Septic Shock and Postsurgical Systemic Inflammation and Its Influence on Coagulation: A Secondary Analysis of a Prospective Observational Study. J Clin Med 2020, 9, 2056. [Google Scholar] [CrossRef] [PubMed]
- Edinger, F.; Edinger, S.; Koch, C.; Markmann, M.; Hecker, M.; Sander, M.; Schneck, E. Peak Plasma Levels of mtDNA Serve as a Predictive Biomarker for COVID-19 in-Hospital Mortality. J Clin Med 2022, 11, 7161. [Google Scholar] [CrossRef]
- Denning, N.-L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front Immunol 2019, 10, 2536. [Google Scholar] [CrossRef]
- Valdés-Aguayo, J.J.; Garza-Veloz, I.; Vargas-Rodríguez, J.R.; Martinez-Vazquez, M.C.; Avila-Carrasco, L.; Bernal-Silva, S.; González-Fuentes, C.; Comas-García, A.; Alvarado-Hernández, D.E.; Centeno-Ramirez, A.S.H.; et al. Peripheral Blood Mitochondrial DNA Levels Were Modulated by SARS-CoV-2 Infection Severity and Its Lessening Was Associated With Mortality Among Hospitalized Patients With COVID-19. Front Cell Infect Microbiol 2021, 11, 754708. [Google Scholar] [CrossRef] [PubMed]
- Bľandová, G.; Janoštiaková, N.; Kodada, D.; Pastorek, M.; Lipták, R.; Hodosy, J.; Šebeková, K.; Celec, P.; Krasňanská, G.; Eliaš, V.; et al. Mitochondrial DNA Variability and Covid-19 in the Slovak Population. Mitochondrion 2024, 75, 101827. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, X.-H.; Li, X.-H.; Song, L.-Y.; Yu, S.-L.; Fang, Z.-C.; Liu, Y.-Q.; Yuan, L.-Y.; Peng, C.-Y.; Zhang, S.-Y.; et al. Common mtDNA Variations at C5178a and A249d/T6392C/G10310A Decrease the Risk of Severe COVID-19 in a Han Chinese Population from Central China. Military Med Res 2021, 8, 57. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chu, P.-Y. Advances in Understanding Mitochondrial MicroRNAs (mitomiRs) on the Pathogenesis of Triple-Negative Breast Cancer (TNBC). Oxid Med Cell Longev 2021, 2021, 5517777. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA Determines Chemoresistance by Reprogramming Metabolism and Regulating Mitochondrial Transcription. Cancer Research 2019, 79, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Lu, Y.; Zhang, H.; Zhang, J.; Fang, X.; Wang, J.; Li, M. Mitochondrial Non-Coding RNAs Are Potential Mediators of Mitochondrial Homeostasis. Biomolecules 2022, 12, 1863. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Enguita, F.J.; Taylor, D.; Griffin, R.J.; Priebe, W.; Emmett, M.R.; Sajadi, M.M.; Harris, A.D.; Clement, J.; Dybas, J.M.; et al. Role of miR-2392 in Driving SARS-CoV-2 Infection. Cell Rep 2021, 37, 109839. [Google Scholar] [CrossRef]
- Olivieri, F.; Prattichizzo, F.; Giuliani, A.; Matacchione, G.; Rippo, M.R.; Sabbatinelli, J.; Bonafè, M. miR-21 and miR-146a: The microRNAs of Inflammaging and Age-Related Diseases. Ageing Research Reviews 2021, 70, 101374. [Google Scholar] [CrossRef]
- Ballinas-Verdugo, M.A.; Jiménez-Ortega, R.F.; Martínez-Martínez, E.; Rivas, N.; Contreras-López, E.A.; Carbó, R.; Sánchez, F.; Bojalil, R.; Márquez-Velasco, R.; Sánchez-Muñoz, F.; et al. Circulating miR-146a as a Possible Candidate Biomarker in the Indeterminate Phase of Chagas Disease. Biological Research 2021, 54. [Google Scholar] [CrossRef]
- Ouyang, L.; Gong, J. Mitochondrial-Targeted Ubiquinone: A Potential Treatment for COVID-19. Medical Hypotheses 2020, 144, 110161. [Google Scholar] [CrossRef]
- Finnigan, L.E.M.; Cassar, M.P.; Koziel, M.J.; Pradines, J.; Lamlum, H.; Azer, K.; Kirby, D.; Montgomery, H.; Neubauer, S.; Valkovič, L.; et al. Efficacy and Tolerability of an Endogenous Metabolic Modulator (AXA1125) in Fatigue-Predominant Long COVID: A Single-Centre, Double-Blind, Randomised Controlled Phase 2a Pilot Study. eClinicalMedicine 2023, 59. [Google Scholar] [CrossRef] [PubMed]
- Gendrot, M.; Andreani, J.; Duflot, I.; Boxberger, M.; Le Bideau, M.; Mosnier, J.; Jardot, P.; Fonta, I.; Rolland, C.; Bogreau, H.; et al. Methylene Blue Inhibits Replication of SARS-CoV-2 in Vitro. Int J Antimicrob Agents 2020, 56, 106202. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J. da S.; Leão, A.H.F.F.; Erustes, A.G.; Morais, I.B. de M.; Vrechi, T.A. de M.; Zamarioli, L. dos S.; Pereira, C.A.S.; Marchioro, L. de O.; Sperandio, L.P.; Lins, Í.V.F.; et al. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. Int J Mol Sci 2021, 22, 4067. [Google Scholar] [CrossRef] [PubMed]
- Babajani, A.; Hosseini-Monfared, P.; Abbaspour, S.; Jamshidi, E.; Niknejad, H. Targeted Mitochondrial Therapy With Over-Expressed MAVS Protein From Mesenchymal Stem Cells: A New Therapeutic Approach for COVID-19. Front Cell Dev Biol 2021, 9, 695362. [Google Scholar] [CrossRef]
- Bramante, C.T.; Buse, J.B.; Liebovitz, D.M.; Nicklas, J.M.; Puskarich, M.A.; Cohen, K.; Belani, H.K.; Anderson, B.J.; Huling, J.D.; Tignanelli, C.J.; et al. Outpatient Treatment of COVID-19 and Incidence of Post-COVID-19 Condition over 10 Months (COVID-OUT): A Multicentre, Randomised, Quadruple-Blind, Parallel-Group, Phase 3 Trial. Lancet Infect Dis 2023, 23, 1119–1129. [Google Scholar] [CrossRef]
Figure 1.
Mechanism of SARS-CoV-2 entry, mitochondrial damage sensing and innate immune dysregulation. Viral particles enter cells via the angiotensin converting enzyme 2 (ACE2) receptor and TMPRSS2 protease. In normally functioning cells various mechanisms exist to warn of mitochondrial damage and viral infection, most notably via retinoic acid inducible gene-1 and mitochondrial antivirus signalling protein (RIG-1-MAVS), cyclic GMP–AMP synthase and stimulator of interferon genes (cGAS-STING) signalling, Toll-like receptor-9 and activation of the NOD-, LRR -and pyrin domain containing protein 3 (NLRP3) inflammasome. Damage to mitochondria resulting in the production of ROS initiates mitophagy via the PTEN-induced kinase 1 (PINK1)-Parkin E3 ubiquitin ligase pathway. Infection of cells by SARS-CoV-2 disrupts these axes at a number of control points and causes widespread aberration to systems of innate immunity. (Created with BioRender.com).
Figure 1.
Mechanism of SARS-CoV-2 entry, mitochondrial damage sensing and innate immune dysregulation. Viral particles enter cells via the angiotensin converting enzyme 2 (ACE2) receptor and TMPRSS2 protease. In normally functioning cells various mechanisms exist to warn of mitochondrial damage and viral infection, most notably via retinoic acid inducible gene-1 and mitochondrial antivirus signalling protein (RIG-1-MAVS), cyclic GMP–AMP synthase and stimulator of interferon genes (cGAS-STING) signalling, Toll-like receptor-9 and activation of the NOD-, LRR -and pyrin domain containing protein 3 (NLRP3) inflammasome. Damage to mitochondria resulting in the production of ROS initiates mitophagy via the PTEN-induced kinase 1 (PINK1)-Parkin E3 ubiquitin ligase pathway. Infection of cells by SARS-CoV-2 disrupts these axes at a number of control points and causes widespread aberration to systems of innate immunity. (Created with BioRender.com).
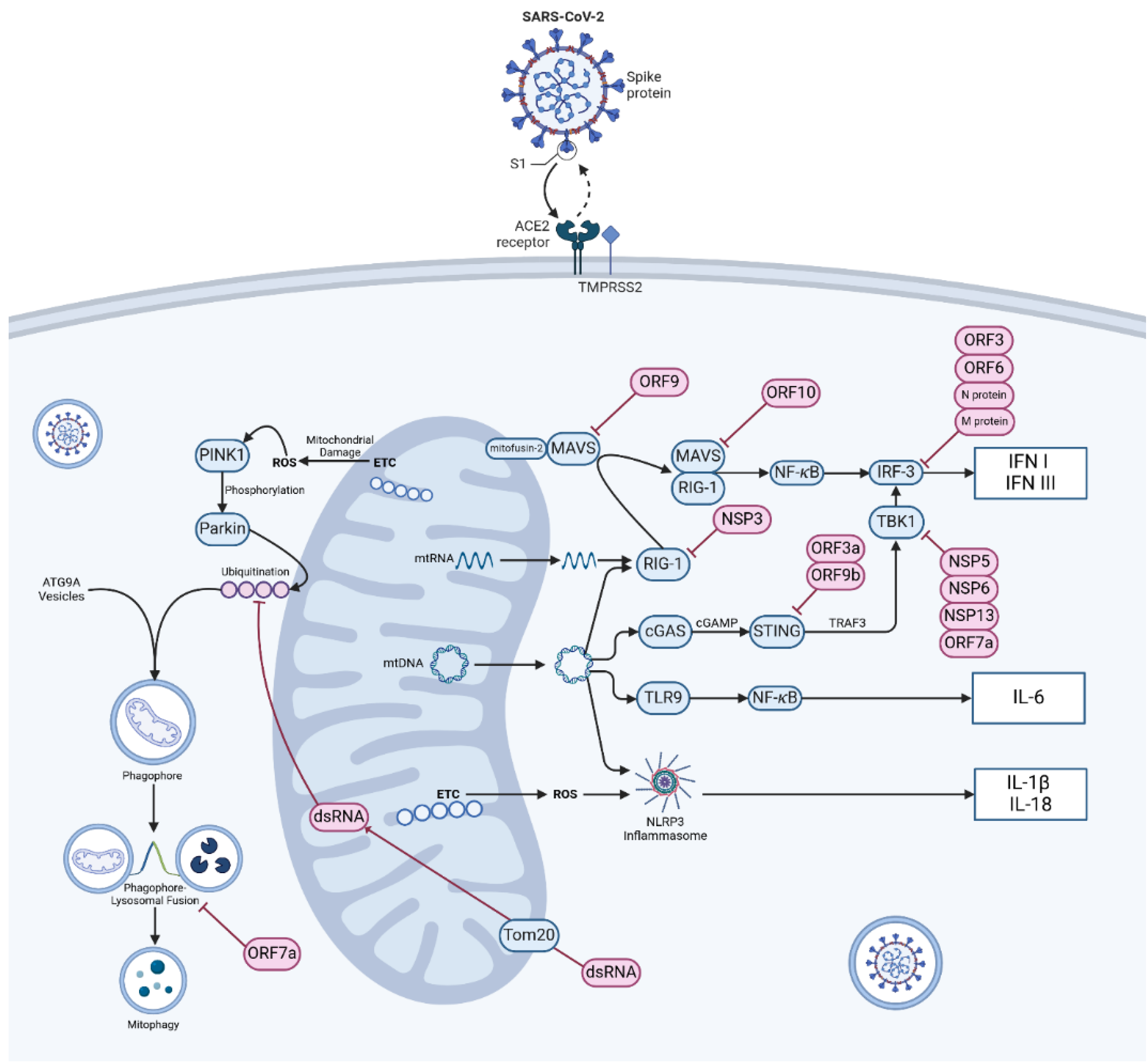
Figure 2.
Inflammation triggered by macrophage after SARS-CoV-2 infection.
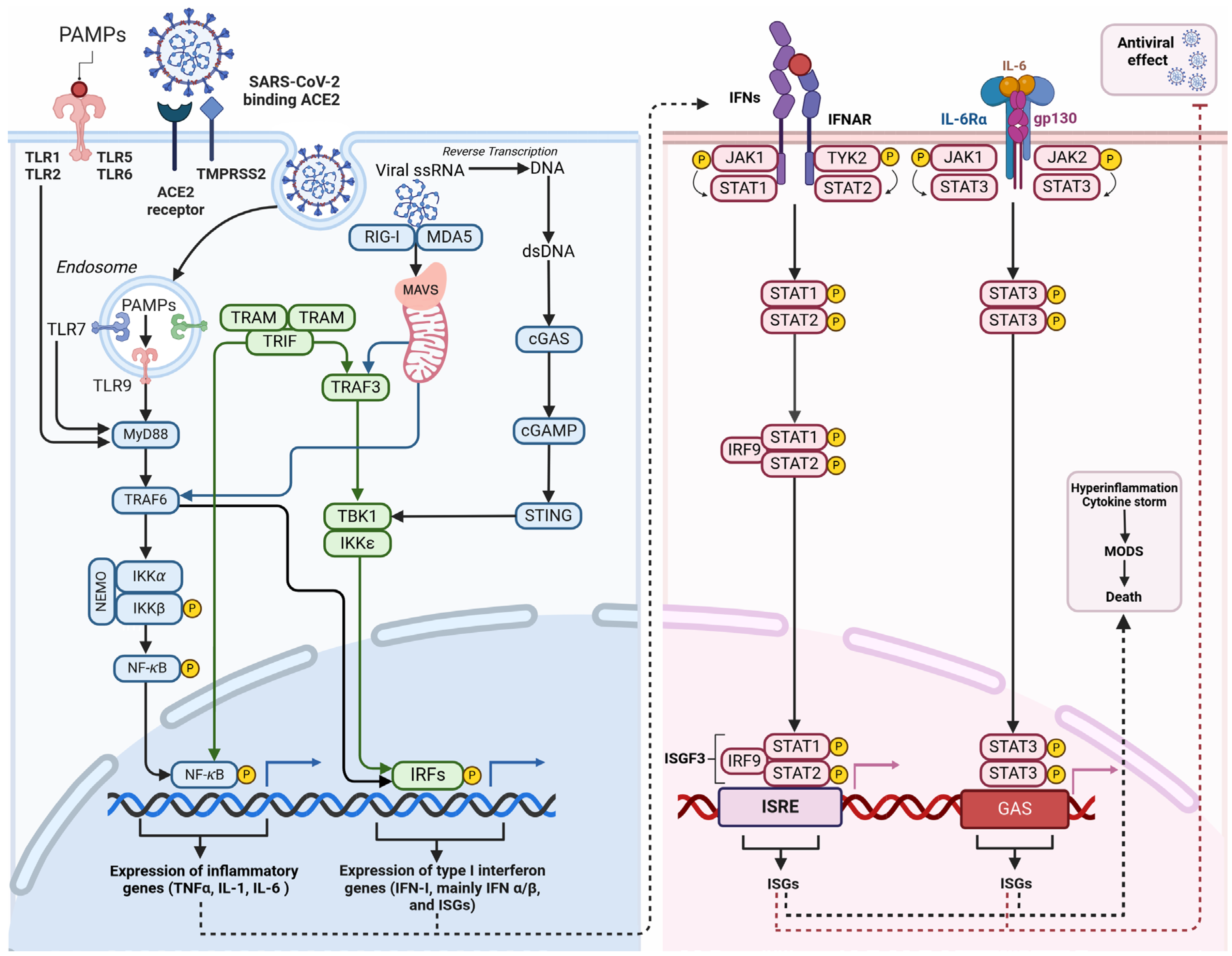

Table 1.
Therapeutic implications for mitochondrial dysfunction in COVID-19 patients including clinical trials related to assessment and improvement of mitochondrial function in patients with COVID-19 and PASC (from ClinicalTrials.gov, National Library of Medicine, Bethesda MD, USA).
Table 1.
Therapeutic implications for mitochondrial dysfunction in COVID-19 patients including clinical trials related to assessment and improvement of mitochondrial function in patients with COVID-19 and PASC (from ClinicalTrials.gov, National Library of Medicine, Bethesda MD, USA).
| Target/strategy | Effect/hypothesis | Intervention/treatment | ClinicalTrials.gov identifier (NCT number) | Ref. | |
| Antioxidants | N-acetylcysteine (NAC) | By supplementing with glycine and cysteine amino acids (in the form of N-acetylcysteine), it is possible to enhance GSH levels and improve mitochondrial function. As a result, this approach may help to lower oxidative stress, inflammation, and endothelial dysfunction. | Glycine N-acetylcysteine Alanine (placebo) |
NCT04703036 | |
| Quercetin Bromelain Zinc Vitamin C |
Zinc ionophore may act as antiviral agent, Bromelain is an anti-inflammatory agent, Vitamin C and Quercetin are antioxidants that also stimulate mitochondrial biogenesis |
Quercetin Bromelain Zinc Vitamin C |
NCT04468139 | ||
| Ubiquinol | Unique spa rehabilitation programmes in the High Tatras mountains can help patients with post-COVID 19 syndrome to restore impaired mitochondrial metabolism. The study has the potential to enhance physical and mental activity, boost immunity, reduce oxidative stress, and expedite the recovery process. | Ubiquinol (reduced coenzyme Q10) Mountain spa rehabilitation |
NCT05178225 | ||
| Mitochondrial-targeted ubiquinone (MitoQ) | MitoQ may help treat COVID-19 by reducing cytokine storms and restoring T cell function through improving mitochondrial dysfunction, which is linked to severe COVID-19 cases. Using MitoQ in the early stages could effectively slow down or postpone the progression of the disease in elderly COVID-19 patients or those with other comorbidities. |
MitoQ | - | [178] | |
| Mitoquinone | The overall objective of this study is to determine whether the daily administration of mito-MES at a dosage of 20 mg is effective in preventing confirmed SARS-CoV-2 infection. The study aims to compare the treatment to a placebo over a 14-day period and will focus on high-risk individuals who have had close contact with confirmed COVID-19 cases. | Mitoquinone/mitoquinol mesylate (mito-MES) Placebo |
NCT05886816 | ||
| Vitamin C Vitamin E Melatonin N-acetyl cysteine Pentoxifylline |
Inclusion of antioxidants like N-acetylcysteine (NAC), vitamin C, melatonin, and vitamin E in the treatment helps enhance intracellular GSH levels, sequester ROS, safeguard cell membrane lipids, cytosol proteins, nuclear DNA, and mitochondria. | Vitamin C Vitamin E Melatonin N-acetyl cysteine Pentoxifylline |
NCT04570254 | ||
| α-Lipoic acid | ALA has both antioxidant properties and the ability to suppress the NF-kB transcription factor, resulting in the inhibition of cytokine and pro-inflammatory factor production. | NAC (N-acetyl cysteine) α-lipoic acid (ALA) Liposomal glutathione (GSH) |
NCT05371288 | ||
| Mitochondria-targeted therapies | Biogenesis enhancers | Both adults and children with severe COVID-19 and multisystem inflammatory syndrome in children (MIS-C) have been found to have a significant lack of arginine. The limited availability of arginine in the plasma has been suggested as a factor contributing to problems with endothelial function, immune regulation, and excessive blood clotting. |
Arginine Hydrochloride | NCT05855330 | |
| The hypothesis proposes that supplementing with L-citrulline (CIT) is superior to ARG administration in correcting hypoargininemia, alleviating lymphocyte dysfunction, rectifying immunosuppression, and improving organ function in septic patients admitted to intensive care. | L-citrulline Placebo (water) |
NCT04404426 | |||
| Assessment of the effect of drug AXA1135 on improving bioenergetic function (measured via phosphocreatine recovery rate) in patients with fatigue-predominant PASC | AXA1125 | NCT05152849 | [179] | ||
| Mitochondria-protective agents | The study assesses the effect of methylene blue as a broad-spectrum antiviral agent and its stabilising impact on mitochondria. | Methylene mlue | - | [180] | |
| Autophagy modulation | Blocking autophagy in the early phase of COVID-19 infection could potentially control the body’s antiviral IFN response and suppress viral reproduction. |
Lysosomotropic agents (e.g., chloroquine, hydroxychloroquine, azithromycin, artemisinin, and imatinib),
Protease inhibitors/activating agents (camostat mesylate, lopinavir, ritonavir, umifenovir and teicoplanin), PI3K/AKT/mTOR modulators (e.g., rapamycin, wortmannin) |
[181] | ||
| Modulation of the mitochondrial function | The study builds on previous research suggesting that exogenous ketone supplementation can increase mitochondrial respiration in various tissues, including skeletal muscle and adipose tissue | Agilent Seahorse XF Cell Mito Stress Test Ketoneaid (Ketone monoester) |
NCT05798260 | ||
| MicroRNA targeting | miR-2392 | SBCov207 aims to mitigate the negative impacts of miR-2392 upregulation observed in COVID-19 patients, including mitochondrial dysfunction, heightened inflammation, increased glycolysis, and hypoxia, by inhibiting miR-2392. | SBCov207- antisense-based therapeutic against human miR-2392 | - | [175] |
| Enhancing the MAVS pathway | Overexpression of MAVS | The use of mesenchymal stem cells to deliver targeted mitochondrial therapy, specifically with an over-expressed MAVS protein, is being explored as a promising new treatment approach for COVID-19. The aim of this approach is to selectively enhance interferon (IFN) production and innate immune responses against SARS-CoV-2. |
Mesenchymal stem cells | - | [182] |
| Immune boosting action | Upregulation of TLR 3 | The presence of 13-cis retinoic acid led to a time-dependent upregulation of TLR3, MAVS, and IFN regulatory factor 1. Isotretinoin as “the Immunity passport" |
Isotretinoin (13 cis retinoic acid) |
NCT04353180 | |
| Drug repurposing | Metformin | By modulating mitochondrial function and reducing oxidative stress, metformin may offer potential relief for the cytokine storm and hyperinflammatory response observed in severe cases of COVID-19. Metformin can enhance the function of immune cells by optimizing their energy metabolism. | Metformin Placebo Fluvoxamine Ivermectin |
NCT04510194 | [183] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



