Submitted:
22 May 2024
Posted:
05 June 2024
You are already at the latest version
Abstract
Dehydroepiandrosterone (DHEA) is considered an endogenous steroid hormone precursor, and 17-ß Estradiol (E2) is one of the estrogen steroid hormones. Of the thirteen known human cytosolic sulfotransferases (SULTs), SULT2B1a has been shown to be expressed in steroid hormone-responsive tissues such as the prostate, ovary, and placenta, as well as the fetal brain. Previous studies have demonstrated that SULT2B1a is capable of sulfating 3β-hydroxysteroids such as DHEA and pregnenolone. The present study aimed to investigate the effects of human SULT2B1 SNPs on the enzymatic characteristics of SULT2B1a allozymes in mediating the sulfation of DHEA and E2. To inspect the effects of single nucleotide polymorphisms of the SULT2B1 gene on the sulfation of DHEA and E2 by SULT2B1a allozymes, 13 recombinant SULT2B1a allozymes were produced, expressed, and purified using established procedures. 13 SULT 2B1a nonsynonymous missense coding SNPs (cSNPs) were selected among numerous identified human SULT 2B1a SNPs by a comprehensive database search. The corresponding cDNAs, packaged in pGEX-2TK expression vector, and encoding the selected 13 SULT2B1a allozymes, have been generated by performing site-directed mutagenesis. These were then bacterially expressed in BL21 E. coli cells and purified using glutathione-Sepharose affinity chromatography. The purified allozymes were tested for their ability to sulfonate DHEA and E2. In terms of the kinetic parameters, the wild-type SULT2B1a exhibited higher enzyme affinity towards DHEA than with E2. In comparison with the wild-type SULT2B1a, the purified allozymes displayed differential sulfating activities towards DHEA and E2. Accordingly, these findings indicate an apparent effect of SULT2B1 cSNPs on the sulfating activities of SULT2B1a allozymes toward DHEA and E2, and may provide for a better understanding of the pharmacokinetics of DHEA and E2 in individuals with differing SULT2B1a genotypes.
Keywords:
DHEA
; E2
; sulfation
; SULT2B1a
; SNPs
1. Introduction
In humans and other mammals, steroids such as dehydroepiandrosterone (DHEA) and 17-β estradiol (E2) are responsible for a variety of essential physiological processes such as reproductive function and the regulation of cell proliferation and apoptosis, particularly in endometrial, ovarian, and breast tissues [1,2,3,4]. DHEA is an endogenous steroid hormone that is abundantly present in the bloodstream compared to other steroids. In the body, DHEA is mainly produced in and secreted from the adrenal glands [1,2]. The pathway for its synthesis is well-established. Initially, cholesterol is converted to pregnenolone by the cytochrome P450 (CYP) side-chain cleavage enzyme (CYP11A1), which is present on the inner membranes of mitochondria; and then, DHEA is converted to pregnenolone under the action of CYP 17α-hydroxylase/17,20-lyase (CYP17A1), which performs two enzymatic reactions: hydroxylation of pregnenolone at C17 generating 17α-hydroxypregnenolone and cleavage of the C17-C20 bond generating DHEA. DHEA can then be used to synthesize testosterone and androstenedione, from which the CYPA19 aromatase enzyme mediates the biosynthesis of E2 and E1, respectively [2,5]. The pathway summary is illustrated in Figure 1. Notably, E2 is considered the most important estrogen that plays important physiological roles in the human body. It is specifically biosynthesized in and secreted from the reproductive organs [4,6].
Sulfation is considered an important Phase II metabolic pathway for the biotransformation and homeostasis of some endogenous compounds, as well as the detoxification of exogenous compounds [7,8,9,10]. Sulfation is mediated by cytosolic sulfotransferase (SULT) enzymes, which catalyze the transfer of a sulfonate group from 3’-phosphoadenosine 5’-phosphosulfate (PAPS) to the hydroxyl or amino group of an acceptor compound, thereby increasing the acceptor’s hydrophilicity and facilitating its urinary and biliary excretion from the body [10,11,12]. Human sulfotransferase enzymes (SULT2A1, SULT2B1a, and SULT2B1b) have been reported to be involved in the sulfation of DHEA to form sulfated DHEA (DHEA-S) [13]. Importantly, DHEA-S may prevent the conversion of DHEA to androgen hormones and is considered a neurosteroid since it is also synthesized in neural cells [14,15]. Interestingly, genomic studies have found that the single SULT2B1 gene encodes two distinct protein isoforms, SULT2B1a and SULT2B1b. These isoforms differ in their N-termini on account of alternative initiation and splicing [16]. SULT2B1 has been reported to contain single nucleotide polymorphisms (SNPs) [13,17,18,19,20,21,22,23]. For E2, there is precedent for effects from SULT1E1 SNPs, which have been investigated in the presence of E2 [24]. The interesting question is whether the SULT1B1a allozymes encoded by transcripts containing missense SNPs exhibit differential sulfating activities toward DHEA and E2 that impact physiology and pathology in different individuals.
This study was designed to investigate the effects of SULT2B1 SNPs on the sulfating activities of SULT2B1a allozymes toward DHEA and E2.
2. Materials and Methods
2.1. Materials
Dehydroepiandrosterone (DHEA), 17β-estradiol (E2), adenosine 5′-triphosphate (ATP), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), Trizma base, and dithiothreitol (DTT) were Sigma-Aldrich (St. Louis, MO, USA). PrimeSTAR® Max DNA polymerase was a product of Takara Bio (Mountain View, CA, USA). Oligonucleotide primers were synthesized by Eurofins Genomics (Louisville, KY, USA). Dpn I and the DNA ladder were products of New England Biolabs (Ipswich, MA, USA). QIAprep® Spin Miniprep Kit was from QIAGEN (Germantown, MD, USA). Glutathione Sepharose 4B GST-tagged protein purification resin was a product of GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). Cellulose TLC plates, and Ultrafree-MC 5000 NMWL filter units were from EMD Millipore (Billerica, MA, USA). X-ray films were sourced from Products International Corporation (Mt Prospect, IL, USA). Ecolume scintillation cocktail was purchased from MP Biomedicals, LLC. (Irvine, CA, USA). Carrier-free sodium [35S] sulfate was from American Radiolabeled Chemicals (St. Louis, MO, USA). Recombinant human bifunctional ATP sulfurylase/adenosine 5′-phosphosulfate kinase was prepared as previously described [25]. PAP[35S] was synthesized using recombinant human bifunctional PAPS synthase based on a previously established procedure [25]. All other chemicals were of the highest grade commercially available.
2.2. Methods
Preparation of Wild-Type and SULT2B1a Allozymes, Expression, and Purification of Recombinant SULT2B1a Allozymes
Wild-type and SULT2B1a allozymes were generated, expressed, and purified as previously reported [26].
Enzymatic Assay for the Sulfating Activity of the Purified Recombinant SULT2B1a Allozymes
The sulfating activity of the purified recombinant SULT2B1a allozymes towards DHEA and E2 was tested with [35S]-labeled PAPS. The standard assay mixture, performed with a final volume of 20 µL containing 50 mM HEPES buffer at pH 7.4, 1 mM DTT, the sulfonate donor (14 µM [35S]PAPS (15 Ci/mmol)), and either the substrates (DHEA and E2) or DMSO for the control. The reaction started with the addition of 0.8 µg of enzyme, ran for 10 minutes at 37°C, and was stopped by heating at 98°C for 3 minutes. After heating, precipitates were cleared by centrifugation at 13,000 × g for 3 minutes. 1 μl of the cleared reaction mixture was then spotted to a thin-layer chromatography (TLC) plate. Afterward, the TLC analysis was conducted with a solvent system of n-butanol/isopropanol/formic acid/water (3:1:1:1 by volume). Then, air drying was performed on the TLC plate to be autoradiographed using an X-ray film. The radioactive spot corresponding the sulfated product was located using the autoradiography film, excised from the TLC plate, eluted in 0.5 mL water, and mixed with 2 mL of Ecolume scintillation liquid. Radioactivity was then measured using a liquid scintillation counter. The obtained results were used to determine the specific activity as the amount of sulfated product formed per minute per mg of enzyme.
Kinetic Studies and Statistical Analysis
Following the previously described technique, the SULT assay was conducted using different concentrations of DHEA and E2 ( cf. Table 1) at pH 7.4 to determine the kinetic parameters for the wild-type SULT2B1a.
Data Analysis
GraphPad Prism® (RRID: SCR_002798) v 6.0 software was used to generate non-linear regression curves based on Michaelis-Menten kinetics to calculate the kinetic constants (Km and Vmax). For inter-group comparisons, one-way ANOVA was used, followed by Dunnett’s test to determine statistical differences between wt- SULT2B1a and individual SULT2B1a allozymes. P-values less than 0.05 were considered statistically significant.
3. Results
Kinetic Study on the Sulfation of DHEA and E2 by wild-Type SULT2B1a
Data obtained from the experiments performed in the kinetic study were processed using the GraphPad Prism® software program to generate the best fitting curves for the Michaelis-Menten equation with non-linear regression in order to calculate the values of the kinetic constants (Km and Vmax) for the WT-SULT2B1a in mediating the sulfation of DHEA and E2. Data on the sulfation of DHEA and E2 by the WT-SULT2B1a were fitted to hyperbolic kinetic curves (cf. Figure 2 and Figure 3). Calculated values of Km, and Vmax, are compiled in Table 2
Enzymatic Characterization of SULT2B1a Allozymes Using DHEA and E2
The activities of the recombinant SULT2B1a allozymes and wild-type SULT2B1a toward DHEA and E2 were determined and compared. Three different concentrations of DHEA (0.35 µM (about ten times below the Km), 3.5 µM (close to the Km), and 35 µM (about ten times the Km)) and 180 µM of E2 (about ten times higher than the Km) were selected for this part of the study based on the above-mentioned results from the kinetic assay of wild-type SULT2B1a. Figure 4 and Figure 5 show the results obtained.
At the tested concentrations of 35 µM DHEA and 180 µM E2, the highest concentrations, (cf. Figure 4C and Figure 5), eight of the allozymes (SULT2B1a-D46N, SULT2B1a-P54A, SULT2B1a-G57V, SULT2B1a-T58M SULT2B1a-R132H, SULT2B1a-R259Q, SULT2B1a-G261V, and SULT2B1a-G261W) displayed sulfating activities drastically lower than the wild-type, while one (SULT2B1a-P330L) exhibited approximately half of that of the wild-type. Two allozymes (SULT2B1a-P134L and SULT2B1a-D176N) displayed around (80 ± 5) % the activity of the wild-type. The remaining two allozymes (SULT2B1a-R215H and SULT2B1a-S229T) displayed sulfating activities that were higher than that of the wild-type (cf. Table 3).
4. Discussion
DHEA is a key metabolic intermediate in the biosynthesis of endogenous steroid hormones, with adrenally-secreted DHEA being converted to sex steroid hormones in peripheral tissues through an intracrinologic process [5]. DHEA is also produced in the adrenal glands, brain, gonads, and skin [2]. Some physiological effects of DHEA have been documented, such as reduced cardiovascular risk, alleviating insulin resistance, stimulating endothelial proliferation, and improving memory and cognitive function [1,27]. Previous studies have revealed that sulfation is involved in DHEA metabolism [10,11,28,29,30]. SULT2B1a has been shown to be capable of sulfating DHEA. Importantly, SULT2B1a is highly expressed in steroid hormone-responsive tissues such as the prostate, breast, placenta, and endometrium [16].
Estrogen sulfation is considered a key detoxification process for preventing estrogen-mediated mitogenicity and genotoxicity [3]. Of the thirteen known SULTs, SULT1E1 has been shown to display the most significant efficiency in catalyzing estrogen sulfation [31,32,33]. The present study investigated the capability of SULT2B1a to catalyze the sulfation of E2, which is the most potent member among all estrogens [34].
Previous studies have linked the genetic polymorphisms of SULT2B1 to the progression and proliferation of some types of cancer, including prostate cancer, esophageal squamous cell carcinoma, hepatocellular carcinoma, gastric cancer, and colorectal cancer [10,11,16,28,29,30,35,36,37,38,39,40,41]. This raises a question concerning the effects of genetic polymorphisms on the sulfating activity of SULT2B1a allozymes towards DHEA and E2.
In this study, a comprehensive database search was performed to identify missense cSNPs in the human SULT2B1 gene, which codes for SULT2B1a allozymes. Site-directed mutagenesis was performed to synthesize the corresponding cDNAs. Subsequently, the thirteen coded SULT2B1a allozymes were expressed, purified, and characterized in regard to their sulfating activity toward DHEA and E2.
Two allozymes, SULT2B1a-R215H, and SULT2B1a-S229T, exhibited higher sulfating activities than the wild-type enzyme. Based on the crystal structure report of SULT2B1, Arg215 is neither involved in nor close to regions of catalytic center, PAPS binding, or substrate binding region. However, substituting the arginine residue (a basic amino acid) with histidine (heterocyclic amino acid) could be the reason for the increased sulfating activity. In contrast, S229 is within the PAPS/PAP binding region; the higher sulfating activity of SULT2B1a-S229T is because the original serine was substituted with a highly similar residue, threonine (both being polar and hydroxylic amino acids).
In the cases of SULT2B1a-P134L and SULT2B1a-D176N, their sulfating activities were around 20 - 25% lower than the wild-type. In P134L, the replacement of proline (a turn-inducing residue) with leucine (a non-turn-inducing residue) could explain the decreased sulfating activity by impacting the conformation of the protein backbone, consequently impairing hydrogen bonding between the NH2 of R132 and the oxygen atom of the 3’-phosphate in PAPS [42,43]. On the other hand, the activity of SULT2B1a-D176N was found to be the same as that in a previous report using DHEA as a substrate, even though the preparations were different (purified recombinant enzymes vs. enzymes expressed in COS-1 cells) [44].
A previous mutational analysis study on SULT2B1a revealed that the removing 53 amino acids from the long carboxyl-terminal end (including P330) had no effect on sulfating activity towards pregnenolone [45]. The allozyme SULT2B1a-P330L exhibited pregnenolone sulfating activity comparable to that of the wild-type [26]. In the present study, SULT2B1a-P330L displayed sulfating activity approximately 45 - 50% lower than the wild-type enzyme. This is lower than the sulfating activity previously reported for the same allozyme using DHEA as a substrate [44].
In the cases of SULT2B1a-D46N, SULT2B1a-P54A, SULT2B1a-G57V, SULT2B1a-T58M, SULT2B1a-R132H, SULT2B1a-R259Q, SULT2B1a-G261V, and SULT2B1a-G261W, the amino acid substitutions may have completely abolished or dramatically decreased their sulfating activity toward DHEA and E2 as follows:
The substitution of glycine (a small amino acid residue with more conformational flexibility) with valine or tryptophan (both being branched amino acid residues with more restricted freedom in conformation) in SULT2B1a-G57V, SULT2B1a-G261V, and SULT2B1a-G261W might have weakened the hydrogen-bonding of PAPS with the O4P and O2P phosphate oxygens, respectively, resulting in decreased sulfating activity toward DHEA and E2 [43,46].
In the case of SULT2B1a-T58M, SULT2B1a-R132H, and SULT2B1a-R259Q, the substituted residues are all located in the PAP/PAPS-binding pocket [43]. Therefore, the substitution of threonine (a polar residue) with methionine (a nonpolar residue), arginine (a basic amino acid) with histidine (a heterocyclic amino acid), and arginine (a basic amino acid) with glutamine (an uncharged amino acid) might have affected the interaction of SULT2B1a and its essential cofactor PAPS/PAP, thus causing significant decrease in or complete abolishment of sulfating activity [43,46].
In the case of SULT2B1a-P54A, the altered amino acid residue is located in the PSB loop, involving the substitution of a turn-inducing amino acid (proline) with a non-turn inducing residue (alanine). This replacement likely has an indirect effect on PAPS by causing displacement of K55, a residue that might be involved in a polar interaction with the O6P oxygen atom of the 5’-phosphate of PAPS [43]. Thus, SULT2B1a-P54A exhibited lower sulfating activity toward DHEA and E2 than the wild-type enzyme.
In the case of SULT2B1a-D46N, although D46 is not located in any of the critical components of the SULT2B1a structure known to be required for sulfation, this allozyme exhibited extremely low sulfating activity relative to the wild-type. The reason might be due to the substituted amino acid being totally different in terms of chemical characteristics.
5. Conclusion
The current study aimed to insight into the effects of human SULT2B1a single nucleotide polymorphisms on the enzyme’s sulfating activity towards DHEA and E2. Enzymatic assays revealed that human SULT2B1a allozymes displayed differential sulfating activities. Of the thirteen examined allozymes, no sulfating activity was detected for two (SULT2B1a-R132H/, and SULT2B1a-R259Q), while differential sulfating activities were found for the other eleven (SULT2B1a-D46N, SULT2B1a-P54A, SULT2B1a-G57V, SULT2B1a-T58M, SULT2B1a-P134L, SULT2B1a-D176N, SULT2B1a-R215H, SULT2B1a-S229T, SULT2B1a-G261V, SULT2B1a-G261W, and SULT2B1a-P330L). The data obtained from this study confirmed a significant effect of SULT2B1a genetic polymorphisms on the DHEA- and E2-sulfating activities of the encoded allozymes, which may underscore the differential metabolism of DHEA and E2 metabolism in individuals with different SULT2B1a genotypes. Further work is needed in order to clarify the role of genetic polymorphisms in the risk of some SULT2B1a-associated diseases, in which may shed light on the design of personalized dosages of impacted drugs for individuals with different SULT2B1 genotypes.
References
- Labrie, F., DHEA, important source of sex steroids in men and even more in women, in Progress in brain research. 2010, Elsevier. p. 97-148.
- Davis, S., M. Panjari, and F. Stanczyk, DHEA replacement for postmenopausal women. The Journal of Clinical Endocrinology & Metabolism, 2011. 96(6): p. 1642-1653.
- Raftogianis, R., et al., Estrogen metabolism by conjugation. JNCI Monographs, 2000. 2000(27): p. 113-124. [CrossRef]
- Ruiz-Cortés, Z.T., Gonadal sex steroids: production, action and interactions in mammals. Steroids-From Physiology to Clinical Medicine, 2012: p. 3-44.
- Schiffer, L., W. Arlt, and K.-H. Storbeck, Intracrine androgen biosynthesis, metabolism and action revisited. Molecular and Cellular Endocrinology, 2018. 465: p. 4-26.
- Stocco, C., Tissue physiology and pathology of aromatase. Steroids, 2012. 77(1-2): p. 27-35. [CrossRef]
- Falany, C.N., Enzymology of human cytosolic sulfotransferases. The FASEB Journal, 1997. 11(4): p. 206-216. [CrossRef]
- Falany, C. and J. Roth, Human Drug Metabolism; From Molecular Biology to Man. by Jeffery EH, CRC Press, Boca Raton, 1993: p. 101-115.
- Mulder, G. and W. Jakoby, Sulfation. Conjugation reactions in drug metabolism. Mulder GJ, 1990.
- Weinshilboum, R. and D. Otterness, Sulfotransferase enzymes, in Conjugation—Deconjugation Reactions in Drug Metabolism and Toxicity. 1994, Springer. p. 45-78.
- Falany, C., Properties of human cytosolic sulfotransferases involved in drug matabolism. Human Drug Metabolism: From Molecular Biology to Man, 1993: p. 101-115.
- Lipmann, F., Biological sulfate activation and transfer. Science, 1958. 128(3324): p. 575-580. [CrossRef]
- Falany, C.N. and K.J. Rohn-Glowacki, SULT2B1: unique properties and characteristics of a hydroxysteroid sulfotransferase family. Drug metabolism reviews, 2013. 45(4): p. 388-400. [CrossRef]
- Noordam, C., et al., Inactivating PAPSS2 mutations in a patient with premature pubarche. New England Journal of Medicine, 2009. 360(22): p. 2310-2318. [CrossRef]
- Vallée, M., W. Mayo, and M. Le Moal, Role of pregnenolone, dehydroepiandrosterone and their sulfate esters on learning and memory in cognitive aging. Brain Research Reviews, 2001. 37(1-3): p. 301-312. [CrossRef]
- Falany, C., et al., Human cytosolic sulfotransferase 2B1: isoform expression, tissue specificity and subcellular localization. The Journal of steroid biochemistry and molecular biology, 2006. 102(1-5): p. 214-221.
- Freimuth, R.R., et al., Human cytosolic sulfotransferase database mining: identification of seven novel genes and pseudogenes. Pharmacogenomics J, 2004. 4(1): p. 54-65. [CrossRef]
- HENDERSON, E., M. WEINBERG, and W.A. WRIGHT, Pregnenolone. The Journal of Clinical Endocrinology, 1950. 10(4): p. 455-474.
- Schumacher, M., et al., Pregnenolone sulfate in the brain: a controversial neurosteroid. Neurochemistry international, 2008. 52(4-5): p. 522-540. [CrossRef]
- Li, Y., et al., Structure, function and polymorphism of human cytosolic sulfotransferases. Current drug metabolism, 2008. 9(2): p. 99-105. [CrossRef]
- Eagle, K., ADHD impacted by sulfotransferase (SULT1A) inhibition from artificial food colors and plant-based foods. Physiology & behavior, 2014. 135: p. 174-179. [CrossRef]
- Eagle, K., Toxicological effects of red wine, orange juice, and other dietary SULT1A inhibitors via excess catecholamines. Food and chemical toxicology, 2012. 50(6): p. 2243-2249. [CrossRef]
- Han, D.-F., et al., Sulfotransferase 1A1 (SULT1A1) polymorphism and breast cancer risk in Chinese women. Toxicology letters, 2004. 150(2): p. 167-177. [CrossRef]
- El Daibani, A.A., et al., Impact of Human SULT1E1 Polymorphisms on the Sulfation of 17β-Estradiol, 4-Hydroxytamoxifen, and Diethylstilbestrol by SULT1E1 Allozymes. Eur J Drug Metab Pharmacokinet, 2021. 46(1): p. 105-118.
- Yanagisawa, K., et al., cDNA cloning, expression, and characterization of the human bifunctional ATP sulfurylase/adenosine 5′-phosphosulfate kinase enzyme. Bioscience, biotechnology, and biochemistry, 1998. 62(5): p. 1037-1040.
- Alatwi, E. and A.F. Bairam, The role of genetic polymorphisms in the sulfation of pregnenolone by human cytosolic sulfotransferase SULT2B1a. Scientific Reports, 2024. 14(1): p. 8050. [CrossRef]
- Traish, A.M., et al., Dehydroepiandrosterone (DHEA)—a precursor steroid or an active hormone in human physiology (CME). The journal of sexual medicine, 2011. 8(11): p. 2960-2982. [CrossRef]
- Glatt, H., et al., Human cytosolic sulphotransferases: genetics, characteristics, toxicological aspects. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2001. 482(1-2): p. 27-40.
- Freimuth, R., et al., Human cytosolic sulfotransferase database mining: identification of seven novel genes and pseudogenes. The pharmacogenomics journal, 2004. 4(1): p. 54-65. [CrossRef]
- Her, C., et al., Human hydroxysteroid sulfotransferase SULT2B1: two enzymes encoded by a single chromosome 19 gene. Genomics, 1998. 53(3): p. 284-295. [CrossRef]
- Falany, C.N., V. Krasnykh, and J.L. Falany, Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. The Journal of steroid biochemistry and molecular biology, 1995. 52(6): p. 529-539. [CrossRef]
- Schrag, M.L., et al., Sulfotransferase 1E1 is a low km isoform mediating the 3-O-sulfation of ethinyl estradiol. Drug metabolism and disposition, 2004. 32(11): p. 1299-1303.
- Zhang, H., et al., Sulfuryl transfer: the catalytic mechanism of human estrogen sulfotransferase. Journal of Biological Chemistry, 1998. 273(18): p. 10888-10892. [CrossRef]
- Cui, J., Y. Shen, and R. Li, Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends in molecular medicine, 2013. 19(3): p. 197-209. [CrossRef]
- Hyland, P.L., et al., Genetic variants in sex hormone metabolic pathway genes and risk of esophageal squamous cell carcinoma. Carcinogenesis, 2013. 34(5): p. 1062-1068. [CrossRef]
- Lévesque, É., et al., Steroidogenic germline polymorphism predictors of prostate cancer progression in the estradiol pathway. Clinical Cancer Research, 2014. 20(11): p. 2971-2983. [CrossRef]
- Mostaghel, E.A., Steroid hormone synthetic pathways in prostate cancer. Translational andrology and urology, 2013. 2(3): p. 212.
- Yang, X., et al., Hydroxysteroid sulfotransferase SULT2B1b promotes hepatocellular carcinoma cells proliferation in vitro and in vivo. PLoS One, 2013. 8(4): p. e60853. [CrossRef]
- Vickman, R.E., et al., Cholesterol sulfonation enzyme, SULT2B1b, modulates AR and cell growth properties in prostate cancer. Molecular Cancer Research, 2016. 14(9): p. 776-786. [CrossRef]
- Chen, W., et al., Overexpression of SULT2B1b promotes angiogenesis in human gastric cancer. Cellular Physiology and Biochemistry, 2016. 38(3): p. 1040-1054. [CrossRef]
- Hu, L., et al., Overexpression of SULT2B1b is an independent prognostic indicator and promotes cell growth and invasion in colorectal carcinoma. Laboratory investigation, 2015. 95(9): p. 1005-1018. [CrossRef]
- Heinz, L., et al., Mutations in SULT2B1 cause autosomal-recessive congenital ichthyosis in humans. The American Journal of Human Genetics, 2017. 100(6): p. 926-939. [CrossRef]
- Lee, K.A., et al., Crystal Structure of Human Cholesterol Sulfotransferase (SULT2B1b) in the Presence of Pregnenolone and 3′-Phosphoadenosine 5′-Phosphate RATIONALE FOR SPECIFICITY DIFFERENCES BETWEEN PROTOTYPICAL SULT2A1 AND THE SULT2B1 ISOFORMS. Journal of Biological Chemistry, 2003. 278(45): p. 44593-44599.
- Ji, Y., et al., Human hydroxysteroid sulfotransferase SULT2B1 pharmacogenomics: gene sequence variation and functional genomics. Journal of Pharmacology and Experimental Therapeutics, 2007. 322(2): p. 529-540. [CrossRef]
- Fuda, H., et al., Mutational analysis of human hydroxysteroid sulfotransferase SULT2B1 isoforms reveals that exon 1B of the SULT2B1 gene produces cholesterol sulfotransferase, whereas exon 1A yields pregnenolone sulfotransferase. Journal of Biological Chemistry, 2002. 277(39): p. 36161-36166. [CrossRef]
- Betts, M.J. and R.B. Russell, Amino acid properties and consequences of substitutions. Bioinformatics for geneticists, 2003. 317: p. 289.
Figure 1.
Biosynthesis of steroidal hormones.
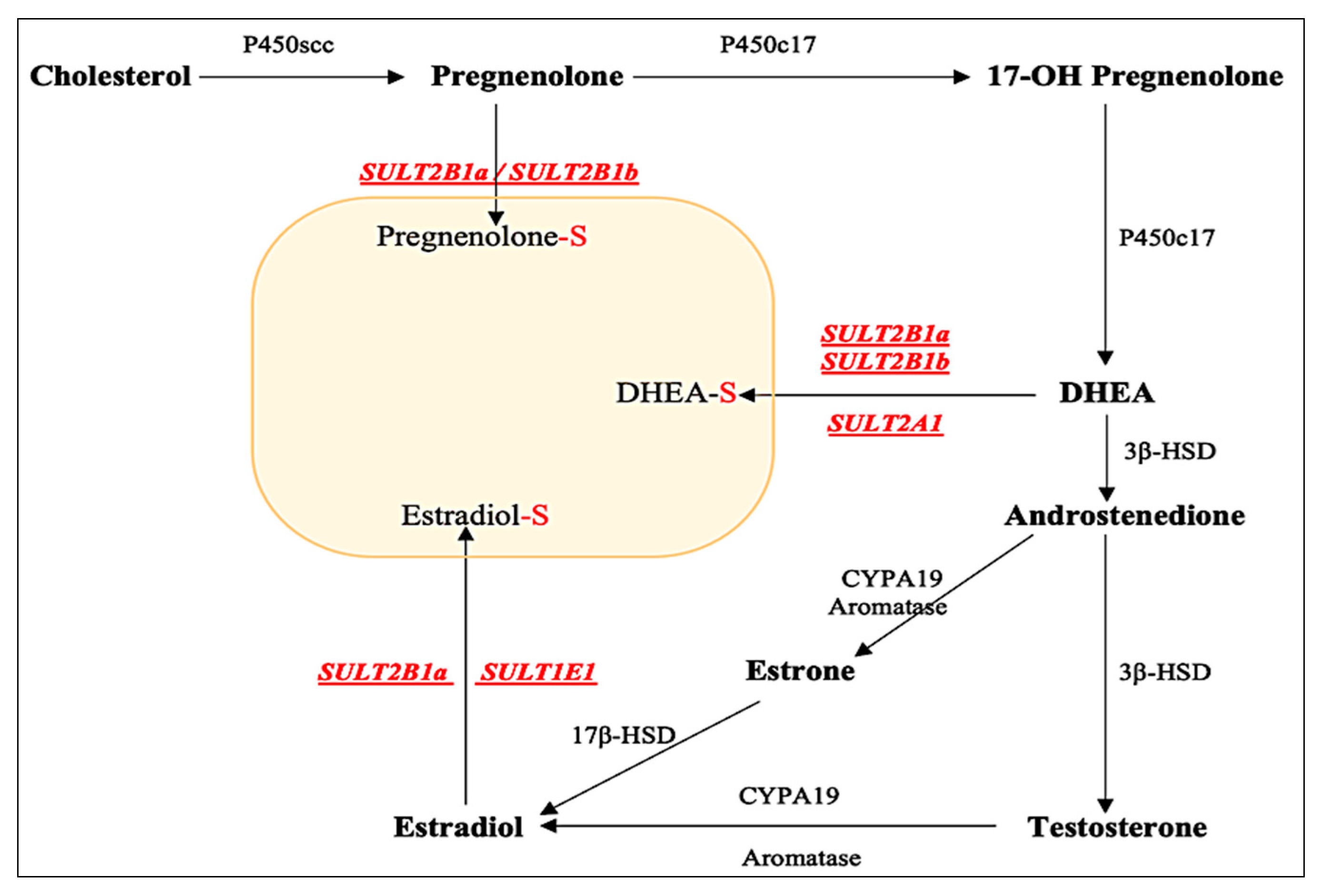
Figure 2.
Concentration-dependent sulfation of DHEA by wild-type SULT2B1a. Data obtained were processed using GraphPad Prism® 5.04 for the determination of kinetic constants Km and Vmax (3.616 ± 0.18 µM, 0.299 ± 0.004 nmol/min/mg). Data shown represent calculated mean ± standard deviation derived from three independent experiments.
Figure 2.
Concentration-dependent sulfation of DHEA by wild-type SULT2B1a. Data obtained were processed using GraphPad Prism® 5.04 for the determination of kinetic constants Km and Vmax (3.616 ± 0.18 µM, 0.299 ± 0.004 nmol/min/mg). Data shown represent calculated mean ± standard deviation derived from three independent experiments.
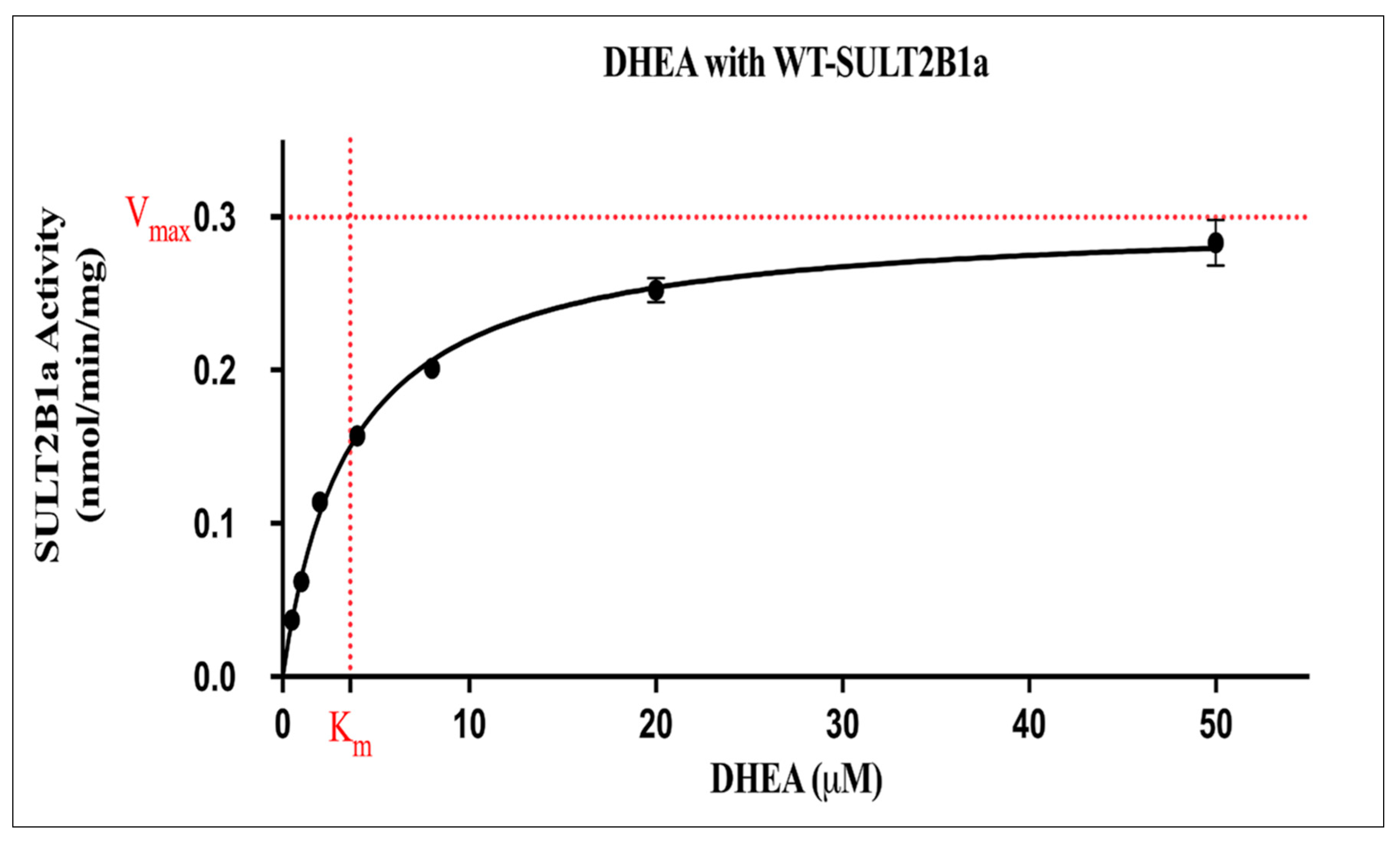
Figure 3.
Concentration-dependent sulfation of E2 by wild-type SULT2B1a. Data obtained were processed using GraphPad Prism® 5.04 for the determination of kinetic constants Km and Vmax (18.12 ± 2.46 µM, 4.092 ± 0.081 pmol/min/mg). Data shown represent calculated mean ± standard deviation derived from three independent experiments.
Figure 3.
Concentration-dependent sulfation of E2 by wild-type SULT2B1a. Data obtained were processed using GraphPad Prism® 5.04 for the determination of kinetic constants Km and Vmax (18.12 ± 2.46 µM, 4.092 ± 0.081 pmol/min/mg). Data shown represent calculated mean ± standard deviation derived from three independent experiments.
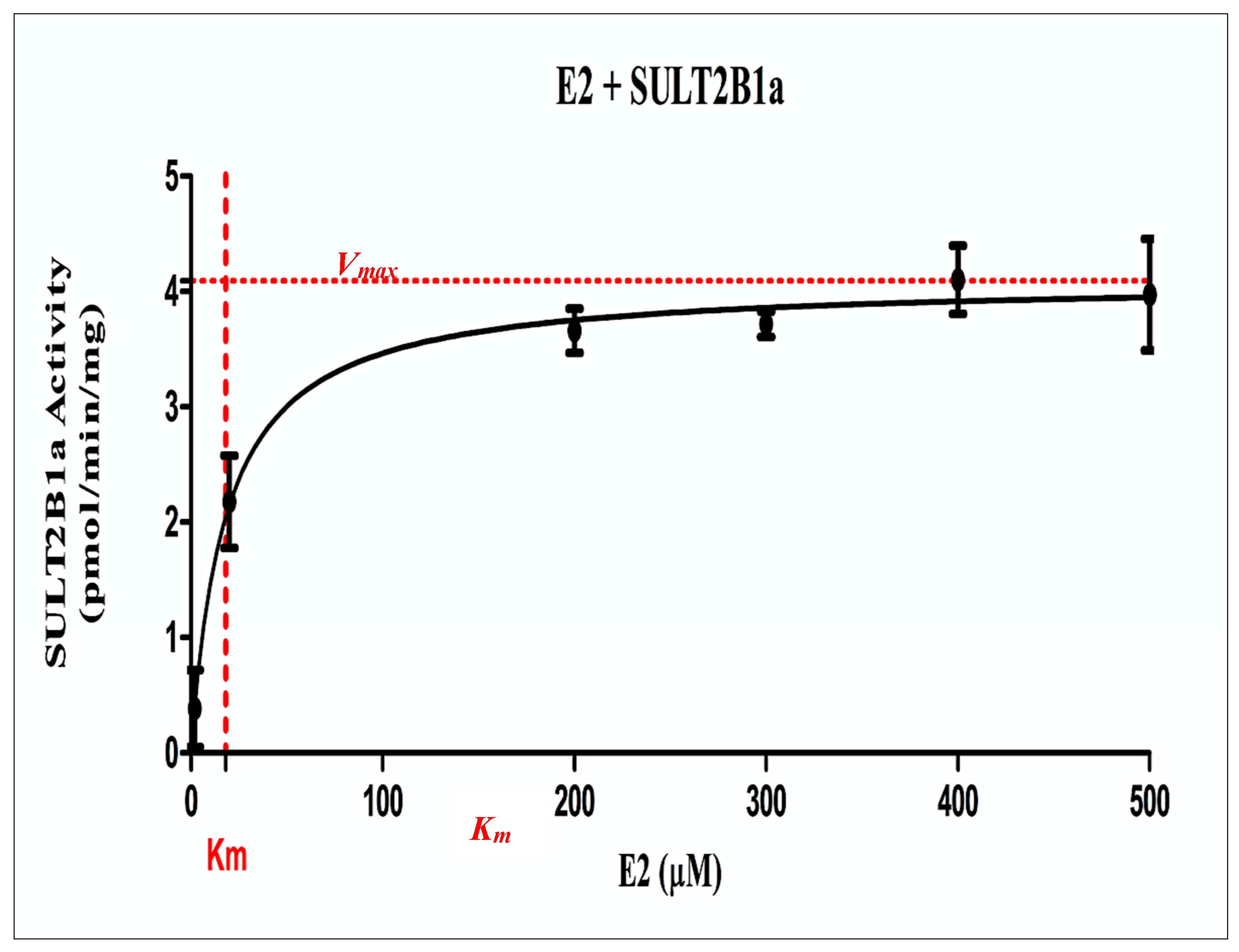
Figure 4.
Specific activities of human SULT2B1a allozymes in mediating the sulfation of DHEA at concentrations of 0.35 µM (A), 3.5 µM (B), and 35 µM (C). Data shown represent mean ± standard deviation, derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test.
Figure 4.
Specific activities of human SULT2B1a allozymes in mediating the sulfation of DHEA at concentrations of 0.35 µM (A), 3.5 µM (B), and 35 µM (C). Data shown represent mean ± standard deviation, derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test.
Figure 5.
Specific activities of human SULT2B1a allozymes in mediating the sulfation of E2 at a concentration of 180 µM. Data shown represent mean ± standard deviation, derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test.
Figure 5.
Specific activities of human SULT2B1a allozymes in mediating the sulfation of E2 at a concentration of 180 µM. Data shown represent mean ± standard deviation, derived from three determinations. One-way ANOVA was performed in combination with Dunnett’s multiple comparison test.
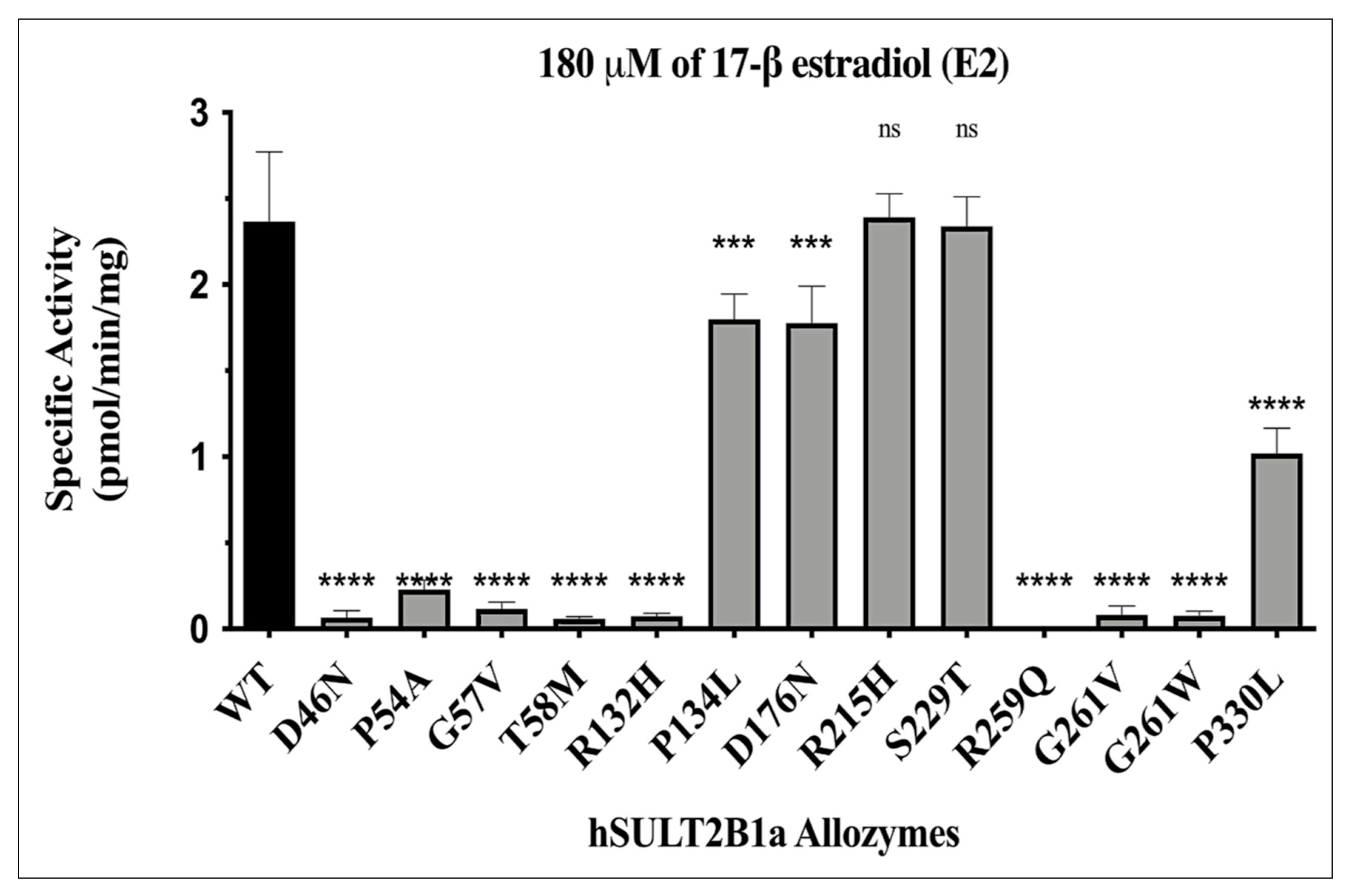

Table 1.
Concentrations of the substrates (DHEA and E2) used in the kinetic study on the wild-type SULT2B1a.
Table 1.
Concentrations of the substrates (DHEA and E2) used in the kinetic study on the wild-type SULT2B1a.
| Substrate | Final Concentrations (µM) |
|---|---|
| Dehydroepiandrosterone$(DHEA) | 0.5, 1, 2, 4, 8, 20, 50 |
| 17-β estradiol$(E2) | 2, 20, 200, 300, 400, 500 |
Table 2.
Kinetic parameters of DHEA and E2 sulfation by WT- SULT2B1a.
| Substrate | Vmax | Km (µM) |
|---|---|---|
| Dehydroepiandrosterone (DHEA) | 0.299 ± 0.004 (nmol/min/mg). | 3.616 ± 0.18 |
| 17-β estradiol$(E2) | 4.092 ± 0.081 (pmol/min/mg). | 18.12 ± 2.46 |
Table 3.
Relative activity of the SULT2B1a allozymes when using 35 µM DHEA and 180 µM E2 as substrates.
Table 3.
Relative activity of the SULT2B1a allozymes when using 35 µM DHEA and 180 µM E2 as substrates.
| Allozyme | Relative activity (% of wild-type SULT2B1a) |
|---|---|
| SULT2B1a-D46N | ≈2-3 |
| SULT2B1a-P54A | ≈10-20 |
| SULT2B1a-G57V | ≈5 |
| SULT2B1a-T58M | ≈3-2 |
| SULT2B1a-R132H | ≈1-3 |
| SULT2B1a-P134L | ≈75-85 |
| SULT2B1a-D176N | ≈75-80 |
| SULT2B1a-R215H | ≈100-130 |
| SULT2B1a-S229T | ≈100-130 |
| SULT2B1a-R259Q | ND |
| SULT2B1a-G261V | ≈1-3 |
| SULT2B1a-G261W | ≈2-3 |
| SULT2B1a-P330L | ≈45-50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



