Submitted:
03 June 2024
Posted:
05 June 2024
You are already at the latest version
Abstract
Elevated levels of homocysteine (Hcy) and related metabolites are associated with Alzheimer’s disease (AD). Severe hyperhomocysteinemia causes neurological deficits and worsens behavioral and biochemical traits associated with AD. Although Hcy is precluded from entering the Genetic Code by proofreading mechanisms of aminoacyl-tRNA synthetases, and thus is a non-protein amino acid, it can be attached to proteins via an N-homocysteinylation reaction mediated by Hcy-thiolactone. Because N-homocysteinylation is detrimental to protein’s function and biological integrity, Hcy-thiolactone detoxifying enzymes – PON1, BLMH, BPHL – have evolved. This review provides an account of the biological function of these enzymes and of consequences of their impairments leading to phenotypes characteristic of AD.
Keywords:
BLMH
; BPHL
; PON1
; homocysteine thiolactone
; PHF8
; mTOR signaling
; autophagy
; Alzheimer’s disease
1. Introduction
Alzheimer’s disease (AD), the most common cause of dementia, is a major health problem in aging populations [1]. AD is characterized by the extracellular accumulation of amyloid β (Aβ) and the intracellular accumulation of neurofibrillary tangles of hyperphosphorylated tau protein leading to neuronal death. Mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) are responsible for the familial early-onset AD, which is relatively rare [2]. Although lifestyle and environmental factors have emerged as modulators of the susceptibility to AD [3], the causes of the most common sporadic late-onset AD are largely unknown, and no effective therapy is available. Thus, identification of novel risk factors and their mechanisms of action has important public health implications. Hyperhomocysteinemia (HHcy) is an emerging risk factor for AD [4]. However, mechanism(s) underlying the involvement of HHcy in AD are not fully understood. Specifically, it is not clear whether elevated levels of homocysteine (Hcy) itself or its downstream metabolites, such as Hcy-thiolactone (HTL) and N-homocysteinylated proteins, can be involved in AD.
Cystathionine β-synthase (CBS) deficiency, the most prevalent inborn error in the sulfur amino acid metabolism [5,6], is biochemically characterized by severe HHcy, i.e., severely elevated levels of Hcy and its metabolites, Hcy-thiolactone [7] and N-Hcy-protein [8,9]. CBS deficiency affects the central nervous system and causes severe learning and intellectual disability [5], reduced IQ [10], psychosis, obsessive-compulsive and behavior/personality disorders [11]. Accelerated brain atrophy associated with HHcy has been reported in healthy elderly individuals [12], alcoholic patients [13], and in AD patients [14] who also show upregulated brain mTOR signaling [15]. These phenotypes are also seen in an animal model of human CBS deficiency, the Cbs-/- mouse. Specifically, in the Cbs-/- mouse model, severe HHcy is accompanied by neurological impairments and cognitive deficiency characterized by attenuated problem-solving abilities, learning, short- and long-term memory [16,17]. The expression of the histone demethylase Phf8 was reduced, H4K20me1, mTOR signaling, and App were increased in brains of Cbs-/- mice compared to Cbs+/- sibling controls. Autophagy-related proteins Becn1, Atg5, and Atg7 were downregulated while neurodegeneration-related neurofilament-L (Nfl) and glial fibrillary acidic protein (Gfap) were upregulated in Cbs-/- brains. Treatments with Hcy-thiolactone, N-Hcy-protein or Hcy, or Cbs gene silencing by RNA interference significantly reduced Phf8 expression and increased total H4K20me1 as well as mTOR promoter-bound H4K20me1 in mouse neuroblastoma N2a and N2a-APPswe cells. This caused transcriptional mTOR upregulation, autophagy downregulation, and significantly elevated APP and Aβ levels. The Phf8 gene silencing increased Aβ, but not APP, levels. These findings show that Phf8 regulates Aβ synthesis and suggest that neuropathy seen in mouse Cbs deficiency is mediated by Hcy metabolites, which transcriptionally dysregulate the Phf8->H4K20me1->mTOR->autophagy pathway thus increasing Aβ accumulation [18]. Phospho-Tau level was also elevated in Cbs-/- mouse brain [19].
Because N-homocysteinylated by Hcy-thiolactone is detrimental to protein’s function and biological integrity [9,20,21,22,23], enzymes detoxifying Hcy-thiolactone have evolved: serum paraoxonase 1 (PON1) [24], cytoplasmic bleomycin hydrolase (BLMH) [25], and mitochondrial biphenyl hydrolase-like (BPHL) enzyme [26,27,28], all of which hydrolyze Hcy-thiolactone to Hcy. The enzymatic detoxification reaction protects proteins from N-homocysteinylation [24,29] because it eliminates Hcy-thiolactone, which would otherwise damage them [9,20,22,30,31].
Accumulating evidence suggests that Hcy-thiolactone hydrolyzing enzymes PON1, BLMH, and BPHL play an important role in the central nervous system. This review provides an overview of current understanding of the biological function of Hcy-thiolactone-detoxifying enzymes and of the consequences of their impairment leading to phenotypes characteristic of AD. To provide a context for the discussion of Hcy-thiolactone-detoxifying enzymes, Hcy metabolism, biogenesis, and chemical biology of Hcy-thiolactone and N-homocysteinylated proteins are also briefly summarized.
2. Homocysteine Metabolism
Hcy was synthesized in 1935 by the reduction with metallic sodium-ammonia [32] of the disulfide homocystine (Hcy-S-S-Hcy), obtained three years earlier in 1932 by boiling Met in sulfuric acid [33]. The article describing the first synthesis of Hcy also reported the synthesis of Hcy-thiolactone from Hcy in strongly acidic solutions [32]. Later studies clarified the role of Hcy formed as a product of Met metabolism in a reaction catalyzed by the enzyme AHCY [34] (Figure 1) and as a precursor of the sulfur amino acids Met (reaction (i)) and cysteine (reaction (ii)) [35], and of the thioester Hcy-thiolactone (reaction (iii)) [9,22] (Figure 1).
In humans and other mammals, Hcy is generated from Met as a byproduct of the S-adenosylmethionine (AdoMet)-mediated methylation reactions [35]. Met, an essential amino acid supplied with protein in a diet, is released in the digestive system, taken up by epithelium, and metabolized to Hcy via the Met->AdoMet->AdoHcy->Hcy pathway in various organs (Figure 1). Hcy is then metabolized to Hcy-thiolactone by methionyl-tRNA synthetase (MetRS or MARS) [21,36,37], remethylated back to Met, or transsulfurated to Cys [35] (Figure 1). The genetic or dietary deficiencies affecting transsulfuration (CBS, CSE) or remethylation (MS, MTHFR) enzymes (Figure 1) lead to the accumulation of Hcy, Hcy-thiolactone [7,21,37,38], and N-Hcy-protein [8,21,37,39], and are associated with various pathologies in humans [4,9].
3. Homocysteine Thiolactone
Hcy-thiolactone, an intramolecular thioester of Hcy, was first synthesized from methionine (Met) in 1934 [40] as a byproduct of an assay for the quantification of Met in proteins [40]. The assay involved demethylation of Met by boiling with hydriodic acid, which also produced methyl iodide, whose quantification provided the basis of the assay. A more recent study showed that the digestion of L-Met with hydriodic acid yields a D,L-Hcy-thiolactone racemate [41]. Because the recovery of Hcy-thiolactone was quantitative [40], the hydriodic acid digestion provided a convenient method for the preparation of D,L-[35S]Hcy-thiolactone [41], which facilitated the elucidation of Hcy-thiolactone metabolic pathways [9,20,21,22,23,37].
The enzymatic conversion of Hcy to Hcy thiolactone in an editing reaction of MARS, prevents access of Hcy to the Genetic Code [42,43], and is universal, occurring in all cell types and organisms investigated so far, from bacteria [44,45] and yeast [46], to plants [47], mice [7], and humans [21,36,37,48]. The Hcy editing reaction is the only known mechanism of Hcy-thiolactone biosynthesis [42,49,50] (Figure 1). The fundamental role of MARS in Hcy-thiolactone biosynthesis in mammalian cells has been established by showing that Chinese hamster ovary cells harboring a temperature-sensitive mutation in the gene encoding MARS are unable to synthesize Hcy-thiolactone at a non-permissive temperature [36] and that Hcy-thiolactone formation in human endothelial cells was inhibited by Met [21].
In mice, Hcy-thiolactone is quickly cleared from the body (t1/2 = 5.1 min) [51,52], about 6-times faster than Hcy (t1/2 = 31.8 min) [51,52], and 120-times faster than N-Hcy-protein (t1/2 = 10.2 h) [51] (Figure 2). Efficient Hcy-thiolactone clearance is responsible for its relatively low levels, compared to Hcy and N-Hcy-protein levels in humans and mice [9].
4. N-Homocysteinylated Proteins
Hcy-thiolactone is harmful because of its ability to chemically modify protein lysine residues, which impairs protein structure and function [20,37], as first shown for human N-homocysteinylated (N-Hcy)-albumin [53], whose K525Hcy modification increased the protein’s susceptibility to oxidation and proteolysis [53]. Two other N-homocysteinylated lysine residues were identified in human albumin in vivo: K137Hcy and K212Hcy [54,55]. Interestingly, N-homocysteinylation of K212 in mice was affected by sex: significantly more K212Hcy modification in male than in female mice [9].
Notably, albumin, a classical globular protein with predominantly α-helical secondary structures, was converted by N-homocysteinylation to amyloid-like aggregates with prevailing β-sheet secondary structures [56].
Subsequent studies showed that N-homocysteinylation of other proteins conferred on them immunogenic [57,58], atherogenic [59,60], thrombogenic [8,61,62], amyloidogenic [56], neuropathic [63,64,65,66], and oncogenic [23,67] properties [9,22,30,31].
In cell cultures, N-Hcy-protein biogenesis positively correlated with the concentrations of its precursors Hcy, Hcy-thiolactone, and with the MARS activity [21]. Vitamin B12 and folate, cofactors of Hcy-metabolizing enzymes, inhibited N-Hcy-protein biogenesis [68]. Methionine, which inhibits the MARS-dependent metabolic conversion of Hcy to Hcy-thiolactone, also inhibited N-Hcy-protein biogenesis [21,46]. The antifolate drug aminopterin, which prevents metabolic conversion of Hcy to Met, increased N-Hcy-protein biogenesis [37]. In humans, N-Hcy-protein biogenesis increased in CBS and MTHFR deficiencies [8] and was influenced by PON1 polymorphism [29] and PON1 arylesterase activity [69]. In mice, N-Hcy-protein biogenesis is affected by the diet and Cbs, Mthfr, Pcft, Pon1, and Blmh genotypes [22].
Additional evidence supporting the mechanism of N-Hcy-protein biogenesis comes from the identification by mass spectrometry of specific N-Hcy-lysine (KHcy) residues in proteins: K525Hcy, K212Hcy, and K137Hcy in human and mouse serum albumin [53,54,55,61]; αK562Hcy, βK344Hcy, and γK385Hcy in human fibrinogen [61,70]; K160Hcy in mouse collagen [71]; five KHcy residues (K14Hcy, K18Hcy, K23Hcy, K27Hcy, and K56Hcy) in histone H3 from HTL-treated HEK293 T cells [72]; five KHcy residues (K32Hcy, K121Hcy, K338Hcy, K1173, and K1812) in ATR from HCT116 cells [67], K1218Hcy in dynein from rat brain [63], 304 KHcy residues in proteins from HTL-treated HeLa cells [73]; 2,525 KHcy residues in 870 different proteins from NE4C cell [66]; H3K79Hcy and other histone KHcy residues in human fetal NTD brain [74]; K411Hcy in MAP1 from rat brain [75]; K80Hcy in α-synuclein from mouse brain [64]; K182Hcy in DJ-1 from HEK293 cells [65].
5. Hcy-Thiolactone Hydrolyzing Enzymes
5.1. Paraoxonase 1
Paraoxonase 1 (PON1), named for the ability of hydrolyze the organophosphate pesticide paraoxon (Table 1), is the first enzyme that was found to use Hcy-thiolactone as a physiological substrate [24] (Table 1). PON1, a monomeric enzyme of 43 kDa molecular weight, synthesized in the liver and carried in the blood attached to a minor subclass of high-density lipoprotein (HDL) that represents 5% of total HDL [76], is present in many organs, including the brain [77]. It protects from organophosphate toxicity in agricultural workers [78] and from major adverse cardiovascular events in patients with coronary artery disease [79,80] and chronic kidney disease [81]. Low PON1 Hcy-thiolactone hydrolytic activity predicted worse long-term mortality [82]. In a general population, PON1 arylesterase activity predicted major adverse cardiovascular events [83]. In mice, Pon1 protects from atherosclerosis induced by a high-fat diet [84] or ApoE depletion [85]. Cardio protection by PON1 can be due to its apparent antioxidative function mediated by interactions of PON1 with redox response-related proteins [86,87] and its ability to detoxify Hcy-thiolactone [24,52,88], which attenuate lipid/protein peroxidation [79,84,89], and protein N-homocysteinylation [29,52,69].
PON1 has also been implicated in AD [90,91,92], which may not be unexpected given that AD has a significant vascular component [93]. For example, PON1 arylesterase activity, reflecting levels of the PON1 protein [94,95], was significantly reduced in AD and dementia patients compared to healthy controls [96,97,98,99] and negatively correlated with the extent of AD-related cognitive decline [100]. In mild cognitive impairment patients, PON1 arylesterase activity predicted global cognition, verbal episodic memory, and attention/processing speed [101]. ApoE-/-Pon1-/- mice with severe carotid atherosclerosis [85], also showed markers of AD and impaired brain vasculature at 14 months, although it was not clear whether brain pathology occurred due to ApoE-/-, Pon1-/-, or both knockouts [102]. In a Tg2576 mouse AD model, immunohistochemical signals for Pon1 surrounded Aβ plaques in various brain regions but could not be assigned to any cell type [103].
5.1.1. Consequences of Pon1 Gene Ablation
Pon1 gene deletion in mice diminished their Hcy-thiolactone hydrolyzing ability (Table 2), causing Hcy-thiolactone accumulation in the brain, kidney, and urine. Pon1-/- mice exhibited significantly increased neurotoxic response to Hcy-thiolactone injections compared to their Pon1+/+ siblings [52] (Figure 3). Pon1-/- mice were also more susceptible to organophosphates and other neurotoxic agents [78,84] and to atherosclerosis [84,85]. Exposure to organophosphates has been linked to neurological disorders including AD, Parkinson’s disease (PD), intellectual disability, attention deficit hyperactivity disorder (ADHD), autism, and other developmental neuropathies [104].
Studies of brain proteomes in Pon1-/- vs. Pon1+/+ mice showed that Pon1 interacts with diverse cellular processes, such as energy metabolism, anti-oxidative defenses, cell cycle, cytoskeleton dynamics, and synaptic plasticity, that are essential for brain homeostasis [105]. The findings that Pon1 depletion influenced the expression of oxidative stress-responsive proteins associated with AD, such as Sod1, Prdx2, and DJ-1 suggests that Pon1 involvement in oxidative stress is indirect [105].
Clusterin (CLU, APOJ), involved in transport of amyloid beta (Aβ) from plasma to brain in humans (reviewed in [2]), is carried on a distinct HDL subspecies that contains three major proteins: PON1, CLU, and APOA1 [106]. Notably, levels of Clu (ApoJ) were significantly elevated in plasma of Pon1-/- vs. Pon1+/+ mice [86]. Taken together, these findings suggest that PON1 plays an important role in the CNS.
5.1.2. Pon1 Depletion Downregulates Phf8, Upregulates mTOR Signaling, Inhibits Autophagy
That PON1 plays an important role in the CNS is further supported by a recent study using a new mouse model of AD, the Pon1-/-5xFAD mouse, which elucidated molecular mechanism by which Pon1 maintains CNS homeostasis and protects brain from accumulation of Aβ, a hallmark of AD [107]. The study showed that Pon1 depletion, which causes accumulation of Hcy-thiolactone and N-Hcy-protein in mice [52], downregulated histone demethylase Phf8 and upregulated the H4K20me1 epigenetic mark in brains of Pon1-/- mice and in Pon1-silenced mouse neuroblastoma N2a-APPswe cells [107]. The depletion of Pon1 increased H4K20me1 binding to the mTOR promoter, demonstrated in mouse neuroblastoma N2a-APPswe cells, and upregulated mTOR signaling, which in turn inhibited the autophagy flux.
5.1.3. Pon1 Depletion Upregulates App and Aβ
In mouse neuroblastoma N2a-APPswe cells and in brains of Pon1-/-5xFAD mice, Pon1 depletion upregulated amyloid precursor protein (App) and amyloid beta (Aβ) [107]. Treatments with N-Hcy-protein and Hcy-thiolactone induced similar biochemical changes App and Aβ levels in the mouse neuroblastoma cells.
These findings provide direct mechanistic evidence linking Hcy-thiolactone and N-Hcy-protein with dysregulated mTOR signaling and its downstream consequences such as downregulation of autophagy and upregulation of Aβ. This mechanism is further supported by findings showing that Phf8 depletion by RNA interference affected mTOR, autophagy, APP and Aβ, as did Pon1 depletion or treatments with Hcy-thiolactone or N-Hcy-protein. These findings also suggest that Pon1 is a negative regulator of mTOR signaling by controlling levels of Hcy metabolites that affect binding of H4K20me1 at the mTOR promotor and define a neuroprotective mechanism by which Pon1 protects from amyloidogenic App processing to Aβ in mouse brain [107].
5.1.4. Pon1 Interacts with App but Phf8 Does Not
Although depletion of Pon1 downregulated Phf8 and upregulated APP in brains of Pon1-/-5xFAD mice, Phf8 depletion did not change APP level [107], suggesting that Pon1 interacts with APP in the mouse brain while Phf8 does not. The nature of the Pon1-APP interaction, whether it is direct or indirect, remains to be elucidated.
Pon1 depletion downregulated Phf8 and upregulated Aβ in brains of Pon1-/-5xFAD mice and in mouse neuroblastoma N2a-APPswe cells. In contrast, Phf8 depletion upregulated Aβ, although it did not affect APP level [107]. This suggests that two pathways are involved in Aβ generation in Pon1-depleted mouse brain and neural cells. One pathway involves Hcy-thiolactone and N-Hcy-protein metabolites, which upregulate APP, while another pathway, mediated by Phf8, H4K20me1, and mTOR, involves impaired Aβ clearance due to downregulated autophagy (Figure 4).
5.1.5. Similar Effects of Pon1 Depletion and Hcy-Thiolactone/N-Hcy-Protein on Pathways Leading to Aβ
Interestingly, Pon1 depletion induced changes in the Phf8->H4K20me1->mTOR->autophagy pathway in Pon1-/-5xFAD mouse brain and in Pon1-silenced neuroblastoma cells that mimicked the changes induced by HHcy in Pon1+/-5xFAD mouse brain and in Hcy-thiolactone or N-Hcy-protein treated mouse neuroblastoma N2a-APPswe cells [107]. Pon1 depletion or HHcy similarly increased accumulation of Aβ in the brain. Earlier work has shown that biochemical outcomes of Pon1 depletion and HHcy were identical: HHcy elevated Hcy-thiolactone and N-Hcy-protein [39] as did Pon1 depletion [52,69]. Pon1 depletion by RNA interference or treatments with Hcy-thiolactone or N-Hcy-protein similarly elevated Aβ in mouse neuroblastoma cells. Taken together, these findings suggest that increased accumulation of Aβ in Pon1-depleted brain is mediated by effects of Hcy metabolites on mTOR signaling and autophagy. These findings also suggest that Pon1 is a negative regulator of mTOR signaling by controlling Hcy-related metabolite levels that influence the extent of H4K20me1 binding at the mTOR promoter.
5.2. Bleomycin Hydrolase
Human BLMH [108,109], named for the ability to hydrolyze the anticancer drug bleomycin, is the second enzyme that was found to use Hcy-thiolactone as a physiological substrate [25] (Table 2). BLMH, a cytoplasmic enzyme expressed in various organs, has a quaternary structure like the 20 S proteasome, and is a member of the self-compartmentalizing cysteine proteases family [110]. In addition to being studied in relation to Hcy toxicity [25,111] and Alzheimer’s disease [112,113,114,115], BLMH was also studied in the field of protein turnover [116,117], cancer therapy [109,118], and keratinization disorders [119,120]. The I443V polymorphic site located in the C-terminal domain important for the activity of the human BLMH is associated with a risk of AD in some [113,121] but not all studies [122,123,124].
A cytoplasmic Hcy-thiolactone hydrolyzing activity was originally purified from human placenta and identified by proteomic and biochemical analyses as BLMH [25]. Substrate specificity studies showed that the human BLMH exhibits absolute stereo-specificity for L-Hcy-thiolactone, the preferred natural substrate, and does not hydrolyze D-Hcy-thiolactone (Table 1). Methyl esters of sulfur-containing amino acids such as L-Cys and L-Met were also hydrolyzed, while D-Met methyl ester was not. L-homoserine lactone, γ-thiobutyrolactone, and other L-amino acids, were not hydrolyzed by human BLMH [25].
Recombinant human and yeast BLMH, expressed in E. coli, exhibit Hcy-thiolactone hydrolyzing activity like that of the corresponding native enzymes. Active site mutation C73A in human BLMH and H369A in yeast BLMH inactivate their Hcy-thiolactone hydrolyzing activity [25].
5.2.1. Consequences of Blmh Gene Ablation
In mice, the deletion of the Blmh gene [125] diminished the animals ability to detoxify Hcy-thiolactone, which led to its accumulation in the brain, kidney, and urine [51], and resulted in several brain-related phenotypes such as astrogliosis and behavioral changes [126], increased neurotoxic response to Hcy-thiolactone injections [51] (Figure 5), in addition to skin-related (tail dermatitis [125]) and immune response-related phenotypes (impaired antigen presentation [127]).
In brains of AD patients, the Hcy-thiolactonase and aminopeptidase activities of BLMH were significantly decreased compared to control brains, suggesting that the attenuated BLMH activity contributes to the pathology of AD [111]. Serum BLMH level was significantly reduced in Parkinsons disease (PD) patients who responded to the therapeutic deep brain stimulation [128], a treatment recommended for advanced stages of PD [129]. Levels of BLMH in extracellular vesicles from the cerebrospinal fluid were significantly lower in amyotrophic lateral sclerosis patients compared with healthy controls [130]. Proteomic studies of Blmh-/- mouse brain showed that Blmh affects various cellular processes, which are important for brain homeostasis, including synaptic plasticity, cytoskeleton dynamics, cell cycle, energy metabolism, and antioxidant defenses [131]. Taken together, these findings suggest that Blmh plays an important role in the CNS.
5.2.2. Blmh Depletion Downregulates Phf8, Upregulates mTOR Signaling, Inhibits Autophagy
To elucidate molecular mechanism by which Blmh maintains CNS homeostasis and protects brain from the accumulation of Aβ, a hallmark of AD, a recent study examined biochemical and behavioral traits related to AD in a new mouse model, the Blmh1-/-5xFAD mouse [132]. 5xFAD mice overexpress the K670N/M671L (Swedish), I716V (Florida), and V717I (London) mutations in human APP(695), and M146L and L286V mutations in human PS1 associated with familial early-onset AD and accumulate elevated levels of Aβ42 beginning around 2 months of age [133].
The study showed that Blmh depletion, which causes accumulation of Hcy-thiolactone and N-Hcy-protein in mice [51], downregulated histone demethylase Phf8 and upregulated the H4K20me1 epigenetic mark in brains of Blmh-/- and Blmh-/-5xFAD mice [132]. These findings were recapitulated in Blmh-silenced mouse neuroblastoma N2a-APPswe cells that harbor a human APP transgene with the K670N and M671L Swedish mutations associated with familial early-onset AD [134]. Blmh depletion increased H4K20me1 binding to the mTOR promoter (demonstrated in N2a-APPswe cells) and upregulated mTOR signaling, which in turn inhibited the autophagy flux in N2a-APPswe cells and in brains of Blmh-/-5xFAD mice.
5.2.3. Blmh Depletion Upregulates App and Aβ and Worsens Cognitive and Neuromotor Deficits
Blmh depletion upregulated App and Aβ in mouse neuroblastoma cells and in Blmh-/-5xFAD mouse brains [132]. Treatments with N-Hcy-protein and Hcy-thiolactone induced similar biochemical changes in mouse neuroblastoma cells. These biochemical changes were associated with cognitive and neuromotor deficits in Blmh-/- and Blmh-/-5xFAD mice. For example, one-year-old Blmh-/-5xFAD mice scored worse compared to Blmh+/+5xFAD animals in the ovel object recognition test, indicating impaired memory, and in the hindlimb and cylinder tests, indicating sensorimotor impairments. Four-month-old Blmh-/- mice, which did not accumulate Aβ, showed similar memory and sensorimotor impairments compared to Blmh+/+ animals. These findings show that the absence of the Blmh protein causes memory and sensorimotor impairments independently of the Aβ-producing transgene [51].
Neurological impairments seen in Blmh-/- and Blmh-/-5xFAD mice are likely to be caused, at least partly, by Phf8 depletion, which does occur in Blmh-/- brains [132]. That Phf8 depletion could account for the neurological deficits in Blmh-/- and Blmh-/-5xFAD mice, is supported by findings showing that PHF8 depletion in humans causes neurological impairments such as intellectual disability, autism spectrum disorder, and attention deficit hyperactivity disorder [135,136] and that Phf8-/- mice also show similar neuropathies [137].
5.2.4. Treatments with Hcy-Thiolactone or N-Hcy-Protein Mimicked the Effects of Blmh Depletion
Notably, treatments with Hcy-thiolactone or N-Hcy-protein mimicked the effects of Blmh depletion by siRNA treatments in the mouse neuroblastoma cells [132]. For example, Hcy-thiolactone, N-Hcy-protein, or Blmh depletion inhibited Phf8 expression, elevated total H4K20me1 level, increased H4K20me1 bound at the mTOR promoter, upregulated mTOR signaling, and impaired autophagy. These findings suggest that Blmh is a negative regulator of mTOR signaling by controlling Hcy-related metabolite levels that influence the extent of H4K20me1 binding at the mTOR promoter [132].
Phf8 is also a mediator directly linking Hcy-thiolactone and N-Hcy-protein with dysregulated mTOR signaling and its downstream outcomes such as impaired autophagy flux and upregulated Aβ accumulation, thus supplying a plausible mechanism explaining neuropathy induced by Blmh deficiency [132] (Figure 6) and explaining an association of HHcy with Alzheimer’s disease [4]. This function of Phf8 is further supported by experiments showing that Phf8 gene silencing had the same impact on mTOR, autophagy, and Aβ as did Blmh gene silencing or the treatments with Hcy-thiolactone or N-Hcy-protein [132].
5.2.5. Blmh Interacts with App, but Phf8 Does Not
Importantly, Blmh gene deletion upregulated App in Blmh-/- in Blmh-/-5xFAD mice as did Blmh gene silencing in mouse neuroblastoma N2a-APPswe cells [132]. However, silencing the Phf8 gene had no effect on App expression, suggesting that the Blmh interacts with App in the CNS while Phf8 does not. The interaction Blmh-App interaction is most likely direct, as suggested by other investigators who found that human BLMH interacts with APP in vitro and that overexpressed BLMH processed human APP to Aβ in the 293-HEK and CHO cells [138]. Another report showed that rat Blmh has the ability to hydrolyze Aβ40 and Aβ42 in vitro, with fibrillar Aβ forms being more resistant than nonfibrillar Aβ [115]. Another possibility is that BLMH can regulate mTOR expression via binding to the mTOR promoter, supported by findings that BLMH can bind to DNA [139,140]. Further studies are needed to clarify the mechanism underlying the regulation of APP by BLMH.
Although Blmh depletion downregulated Phf8 and upregulated APP and Aβ, Phf8 depletion upregulated Aβ but not APP. These findings suggest that three pathways contribute to Aβ upregulation in Blmh-depleted mouse brain (Figure 6) [132]. In the first pathway (i, Figure 6A), Hcy metabolites upregulate APP (independently of Phf8), which leads to Aβ upregulation in Blmh-depleted or Hcy-thiolactone/N-Hcy-protein-treated mouse brain cells. In the second pathway, (ii-a, Figure 6A), Hcy metabolites downregulate Phf8, which upregulates mTOR signaling and thereby reduces autophagy flux resulting in Aβ upregulation due to impaired clearance. Direct depletion of Phf8 by RNA interference, independently of Hcy metabolites, also starts a similar pathway mediated by mTOR (ii-b, Figure 6A) that results in Aβ accumulation due to impaired autophagy. These pathways remain to be verified in future studies by testing effects of Phf8 overexpression or mTOR downregulation (by pharmacological inhibition using rapamycin or by RNA interference) on APP and Aβ accumulation in Blmh-depleted cells.
5.2.6. Becn1 Interacts with App
The findings that APP upregulation was accompanied by downregulation of Becn1, a protein with a central role in autophagy initiation, in the Blmh-/-5xFAD mouse brain and in mouse neuroblastoma cells suggest that a third pathway, involving an interaction between Bcln1 and APP, contributes to Aβ upregulation [132]. In this pathway, Becn1 is a negative regulator of APP expression and processing (iii, Figure 6B). This conclusion is supported by prior findings showing that level of Becn1 is significantly reduced in human AD brains compared with non-AD controls, and that reduction of Becn1 level in transgenic APP-overexpressing APP+Becn+/- mice increased Aβ accumulation in neuronal cells [141]. Becn1 was also reported to regulate APP processing and turnover. Depletion of Bcln1 by siRNA in rat neuroblastoma B103/hAPPwt cells expressing human APP transgene elevated APP, Lc3, and Aβ, while overexpression of Becn1 reduced APP level [142]. The involvement of autophagy in APP and Aβ accumulation Blmh-depleted cells needs to be confirmed in future studies, e.g., by enhancing autophagy (e.g., with TAT-Beclin1), which should rescue APP and Aβ accumulation in these cells.
5.2.7. Similar Effects of Blmh Depletion and Hcy-Thiolactone/N-Hcy-Protein on Pathways Leading to Aβ
Interestingly, Blmh gene deletion or HHcy induced by a high Met diet led to similar changes in the Phf8->H4K20me1->mTOR->autophagy pathway and Aβ accumulation [132]. These findings are consistent with the fact that the Blmh gene deletion and high Met diet lead to the same biochemical outcome: upregulation of Hcy-thiolactone and N-Hcy-protein levels [39,51]. Indeed, treatments of mouse neuroblastoma cells with individual metabolites that accumulate in HHcy, Hcy-thiolactone or N-Hcy-protein, recapitulated changes in the Phf8->H4K20me1->mTOR->autophagy pathway and Aβ accumulation seen in Blmh-depleted or Met-supplemented HHcy wild type mice [132]. These findings also suggest that dysregulation of Hcy metabolism in general would affect the Phf8->H4K20me1->mTOR->autophagy pathway in the CNS. Indeed, Hcy metabolites inhibit autophagy, elevate Aβ, and induce neuropathy by dysregulating Phf8/H4K20me1-dependent epigenetic regulation of mTOR in cystathionine β-synthase-deficient mice and Cbs-silenced mouse neuroblastoma cells [18].
Blmh deficiency or HHcy induced by a high Met diet elevated level of methylated histone H4K20me1 via downregulation of the histone demethylase Phf8 in mouse brain [132]. HHcy is also known to affect DNA and protein methylation via S-adenosylhomocysteine (AdoHcy, an inhibitor of cellular AdoMet-dependent methylation reactions), which underlies the pathology of HHcy-associated human disease (reviewed in ref. [143]). However, possible inhibition of H4K20 histone methylase by AdoHcy would have an opposing effect, i.e., it would reduce H4K20me1 level. The findings linking Blmh with the status of the histone H4K20me1 methylation, are reminiscent of findings showing that Pon1 deletion in mice elevated the H4K20me1 methylation level via downregulation of Phf8 [107] (Figure 4). Thus, these two Hcy-thiolactone-detoxifying enzymes exert similar effects on H4K20me1 levels. Although there is no evidence that Blmh or Pon1 are linked to DNA methylation, these findings provide the first evidence that Blmh and Pon1 influence histone methylation.
5.3. Biphenyl Hydrolase-Like Enzyme
Biphenyl hydrolase-like (BPHL) enzyme, also called valacyclovir hydrolase, is the third enzyme shown to use Hcy-thiolactone as a natural substrate [26,27,28]. It is a 32 kDa mitochondrial protein highly expressed in human liver and kidney [144,145]. BPHL hydrolyzes and activates the antiviral prodrug esters valacyclovir and valganciclovir, used in the treatment of herpes simplex, herpes zoster (shingles), and herpes B [146]. However, valacyclovir was rapidly hydrolyzed to acyclovir in Bphl-/- mice, which shows that BPHL is not obligatory for the conversion of valacyclovir to acyclovir [147].
First cloned from the breast carcinoma cells and expressed in E. coli, BPHL, a member of the alpha/beta hydrolase fold family, is a serine hydrolase distantly related to other members of the serine hydrolase family [144,145]. The BPHL gene is located on human chromosome 6p25 in a locus with other serine hydoxylases.
Crystallographic studies showed that human BPHL has the catalytic triad S122-D227-H255, a serine hydrolase consensus sequence GSXSG, and a unique binding mode and the specificity for esters of α-amino acids [148]. The α-amino acid specificity, including the ability of BPHL to hydrolyze L-Met methyl ester shared with the Hcy-thiolactone-hydrolyzing enzyme BLMH [25], suggested that BPHL could also hydrolyze Hcy-thiolactone. Indeed, this prediction, was substantiated by conference reports published in 2010-2011 [26,27] and a report published in a 2014 PlosOne article [28].
BPHL, BLMH, and PON1 differ in their specificities towards non-physiological substrates for which they have been originally named and in catalytic efficiencies towards Hcy-thiolactone (Table 1). Catalytic efficiency of BPHL in the Hcy-thiolactone hydrolytic reaction is higher than that of BLMH or PON1, suggesting that BPHL can have a significant contribution to Hcy-thiolactone detoxification in vivo [28].
5.3.1. Consequences of Bphl Ablation
A recent study found that BPHL gene is overexpressed in lung cancer and promotes lung carcinogenesis and that downregulation of BPHL expression by RNA interference inhibited tumor growth and metastasis by impairing the progression of cell cycle and inducing apoptosis in A549, NCI-H1975, and NCI-H-1299 human lung carcinoma cell lines [149]. Deletion of the Bphl gene in mice decreased circulating creatinine levels in males, suggesting a kidney function defect (http://www.informatics.jax.org/allele/allgenoviews/MGI:5548556).
A recent report has shown that the deletion of Bphl gene in mice significantly attenuated Hcy-thiolactone turnover in vivo [150], similar to the impairment of Hcy-thiolactone turnover in Blmh-/- mice [51]. Notably, silencing the Bphl gene by RNA interference in mouse neuroblastoma N2a-APPswe cells caused changes in the Phf8->H4K20me1->mTOR->autophagy pathway and APP/Aβ levels characteristic of AD [150], similar to the changes seen in mouse neuroblastoma N2a-APPswe cells in which Pon1 [107] or Blmh [132] was silenced by RNA interference.
6. Conclusions
Hcy-thiolactone, a product of an error-correcting reaction during protein biosynthesis, is generated in the human body when Hcy is selected in place of methionine by methionyl-tRNA synthetase. Hcy-thiolactone is a chemically reactive thioester metabolite that modifies protein lysine residues in a process called N-homocysteinylation. The modification causes protein damage/aggregation, a hallmark of many diseases, including Alzheimer’s. Hcy-thiolactone-detoxifying enzymes—serum paraoxonase PON1 carried in the circulation on high-density lipoprotein, cytoplasmic bleomycin hydrolase BLMH, and mitochondrial biphenyl hydrolase-like enzyme BPLH—protect the human body proteins from Hcy-thiolactone/N-homocysteinylation-associated damage. Depletion of any of these enzymes elevates Hcy-tiolactone and N-Hcy-protein, which dysregulate the Phf8->H4K20me1->mTOR->autophagy pathway and upregulate APP, causing Aβ accumulation, a hallmark of Alzheimer’s disease.
Funding
This research was funded by the National Science Center, Poland grants 2018/29/B/NZ4/00771, 2019/33/B/NZ4/01760, 2021/43/B/NZ4/00339 and the American Heart Association grant 17GRNT32910002.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available in the methods of this article.
Conflicts of Interest
No conflicts of interest, financial or otherwise, are declared by the author.
References
- Alzheimer's disease facts and figures. Alzheimers Dement 2021, 17, 327–406. [CrossRef] [PubMed]
- Tanzi, R. E. (2012) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2.
- Dorszewska, J., Prendecki, M., Oczkowska, A., Dezor, M., and Kozubski, W. (2016) Molecular Basis of Familial and Sporadic Alzheimer's Disease. Curr Alzheimer Res 13, 952-963.
- Smith, A. D., and Refsum, H. (2021) Homocysteine - from disease biomarker to disease prevention. J Intern Med 290, 826-854.
- Mudd, S.; Skovby, F.; Levy, H.; Pettigrew, K.; Wilcken, B.; Pyeritz, R.; Andria, G.; Boers, G.; Bromberg, I.; Cerone, R.; et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar] [PubMed]
- Kozich, V., Sokolova, J., Morris, A. A. M., Pavlikova, M., Gleich, F., Kolker, S., Krijt, J., Dionisi-Vici, C., Baumgartner, M. R., Blom, H. J., Huemer, M., and consortium, E. H. (2021) Cystathionine beta-synthase deficiency in the E-HOD registry-part I: pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis. J Inherit Metab Dis 44, 677-692.
- Chwatko, G., Boers, G. H., Strauss, K. A., Shih, D. M., and Jakubowski, H. (2007) Mutations in methylenetetrahydrofolate reductase or cystathionine beta-synthase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. Faseb J 21, 1707-1713.
- Jakubowski, H., Boers, G. H., and Strauss, K. A. (2008) Mutations in cystathionine {beta}-synthase or methylenetetrahydrofolate reductase gene increase N-homocysteinylated protein levels in humans. FASEB J 22, 4071-4076.
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef] [PubMed]
- El Bashir, H., Dekair, L., Mahmoud, Y., and Ben-Omran, T. (2015) Neurodevelopmental and Cognitive Outcomes of Classical Homocystinuria: Experience from Qatar. JIMD Rep 21, 89-95.
- Abbott, M. H., Folstein, S. E., Abbey, H., and Pyeritz, R. E. (1987) Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am J Med Genet 26, 959-969.
- Sachdev, P.S.; Valenzuela, M.; Wang, X.L.; Looi, J.C.; Brodaty, H. Relationship between plasma homocysteine levels and brain atrophy in healthy elderly individuals. Neurology 2002, 58, 1539–1541. [Google Scholar] [CrossRef]
- Bleich, S.; Bandelow, B.; Javaheripour, K.; Müller, A.; Degner, D.; Wilhelm, J.; Havemann-Reinecke, U.; Sperling, W.; Rüther, E.; Kornhuber, J. Hyperhomocysteinemia as a new risk factor for brain shrinkage in patients with alcoholism. Neurosci. Lett. 2003, 335, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, Vitamin B12, and Serum Total Homocysteine Levels in Confirmed Alzheimer Disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.C.; Zafar, A.; Hubbard, P.; Nagy, S.; Durant, S.; Bicknell, R.; Wilcock, G.; Christie, S.; Esiri, M.M.; Smith, A.D.; et al. Dysfunction of the mTOR pathway is a risk factor for Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Majtan, T., Park, I., Cox, A., Branchford, B. R., di Paola, J., Bublil, E. M., and Kraus, J. P. (2019) Behavior, body composition, and vascular phenotype of homocystinuric mice on methionine-restricted diet or enzyme replacement therapy. FASEB J 33, 12477-12486.
- Akahoshi, N., Kobayashi, C., Ishizaki, Y., Izumi, T., Himi, T., Suematsu, M., and Ishii, I. (2008) Genetic background conversion ameliorates semi-lethality and permits behavioral analyses in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. Hum Mol Genet 17, 1994-2005.
- Witucki, L., and Jakubowski, H. (2023) Homocysteine metabolites inhibit autophagy, elevate amyloid beta, and induce neuropathy by impairing Phf8/H4K20me1-dependent epigenetic regulation of mTOR in cystathionine beta-synthase-deficient mice. J Inherit Metab Dis.
- Khayati, K.; Antikainen, H.; Bonder, E.M.; Weber, G.F.; Kruger, W.D.; Jakubowski, H.; Dobrowolski, R. The amino acid metabolite homocysteine activates mTORC1 to inhibit autophagy and form abnormal proteins in human neurons and mice. FASEB J. 2017, 31, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [CrossRef]
- Jakubowski, H., Zhang, L., Bardeguez, A., and Aviv, A. (2000) Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ Res 87, 45-51.
- Jakubowski, H. (2013) Homocysteine in Protein Structure/Function and Human Disease - Chemical Biology Of Homocysteine-containing Proteins, Springer, Wien.
- Jakubowski, H. Protein N-Homocysteinylation and Colorectal Cancer. Trends Cancer 2019, 5, 7–10. [Google Scholar] [CrossRef]
- Jakubowski, H. Calcium-dependent Human Serum Homocysteine Thiolactone Hydrolase. A protective mechanism against protein N-homocysteinylation. J Biol Chem. 2000, 275, 3957–3962. [Google Scholar] [CrossRef] [PubMed]
- Zimny, J., Sikora, M., Guranowski, A., and Jakubowski, H. (2006) Protective mechanisms against homocysteine toxicity: the role of bleomycin hydrolase. J Biol Chem 281, 22485-22492.
- Zimny, J., Bretes, E., and Guranowski, A. (2010) Novel mammalian homocysteine thiolactone hydrolase: Purification and characterization. Acta Biochimica Polonica 57 Suppl 4, 134.
- Zimny, J., Bretes, E., Grygiel, D., Guranowski, A. (2011) Human mitochondrial homocysteine thiolactone hydrolase; overexpression and purification. Acta Biochimica Polonica 58 Suppl 4, 57.
- Marsillach, J.; Suzuki, S.M.; Richter, R.J.; McDonald, M.G.; Rademacher, P.M.; MacCoss, M.J.; Hsieh, E.J.; Rettie, A.E.; Furlong, C.E. Human Valacyclovir Hydrolase/Biphenyl Hydrolase-Like Protein Is a Highly Efficient Homocysteine Thiolactonase. PLOS ONE 2014, 9, e110054. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H.; Ambrosius, W.T.; Pratt, J. Genetic determinants of homocysteine thiolactonase activity in humans: implications for atherosclerosis. FEBS Lett. 2001, 491, 35–39. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Pathophysiological Consequences of Homocysteine Excess. J. Nutr. 2006, 136, 1741S–1749S. [Google Scholar] [CrossRef]
- Riegel, B., Du Vigneaud, V. (1935) The isolation of homocysteine and its conversion to a thiolactone. J. Biol. Chem. 112, 149-154.
- Butz, L. W., du Vigneaud, V. (1932) The formation of homologue of cysteine by the decomposition of methionine with sulfuric acid. J Biol Chem 99, 135-142.
- de la Haba, G.; Cantoni, G. The Enzymatic Synthesis of S-Adenosyl-l-homocysteine from Adenosine and Homocysteine. J. Biol. Chem. 1959, 234, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D. Homocysteine: A History in Progress. Nutr. Rev. 2000, 58, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H., and Goldman, E. (1993) Synthesis of homocysteine thiolactone by methionyl-tRNA synthetase in cultured mammalian cells. FEBS Lett 317, 237-240.
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. J Biol Chem. 1997, 272, 1935–1942. [Google Scholar] [CrossRef]
- Jakubowski, H. (1997) Synthesis of homocysteine thiolactone in normal and malignant cells. In Homocysteine Metabolism: From Basic Science to Clinical Medicine (Rosenberg, I. H., Graham, I., Ueland, P. M., and Refsum, H., eds) pp. 157-165, Kluwer Academic Publishers, Norwell, MA.
- Jakubowski, H.; Perła-Kaján, J.; Finnell, R.H.; Cabrera, R.M.; Wang, H.; Gupta, S.; Kruger, W.D.; Kraus, J.P.; Shih, D.M. Genetic or nutritional disorders in homocysteine or folate metabolism increase proteinN-homocysteinylation in mice. FASEB J. 2009, 23, 1721–1727. [Google Scholar] [CrossRef]
- Baernstein, H.D. A modification of the method for determining methionine in proteins. J Biol Chem. 1934, 106, 451–456. [Google Scholar] [CrossRef]
- Jakubowski, H. (2007) Facile syntheses of [35S]homocysteine-thiolactone, [35S]homocystine, [35S]homocysteine, and [S-nitroso-35S]homocysteine Analytical biochemistry 370, 124-126.
- Jakubowski, H. Quality control in tRNA charging. Wiley Interdiscip. Rev. RNA 2012, 3, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. (2017) Homocysteine Editing, Thioester Chemistry, Coenzyme A, and the Origin of Coded Peptide Synthesis dagger. Life (Basel) 7.
- Jakubowski, H. (1990) Proofreading in vivo: editing of homocysteine by methionyl-tRNA synthetase in Escherichia coli Proc Natl Acad Sci U S A 87, 4504-4508.
- Jakubowski, H. (1995) Proofreading in vivo. Editing of homocysteine by aminoacyl-tRNA synthetases in Escherichia coli. J Biol Chem 270, 17672-17673.
- Jakubowski, H. (1991) Proofreading in vivo: editing of homocysteine by methionyl-tRNA synthetase in the yeast Saccharomyces cerevisiae Embo J 10, 593-598.
- Jakubowski, H.; Guranowski, A. Metabolism of Homocysteine-thiolactone in Plants. J Biol Chem 2003, 278, 6765–6770. [Google Scholar] [CrossRef] [PubMed]
- Borowczyk, K.; Piechocka, J.; Głowacki, R.; Dhar, I.; Midtun, Ø.; Tell, G.S.; Ueland, P.M.; Nygård, O.; Jakubowski, H. Urinary excretion of homocysteine thiolactone and the risk of acute myocardial infarction in coronary artery disease patients: the WENBIT trial. J. Intern. Med. 2019, 285, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Quality control in tRNA charging -- editing of homocysteine. Acta Biochim. Pol. 2011, 58, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. (2015) Transfer RNA Synthetase Editing of Errors in Amino Acid Selection. In eLS pp. 1-18, John Wiley & Sons, Ltd, Chichester.
- Borowczyk, K.; Tisończyk, J.; Jakubowski, H. Metabolism and neurotoxicity of homocysteine thiolactone in mice: protective role of bleomycin hydrolase. Amino Acids 2012, 43, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Borowczyk, K.; Shih, D.M.; Jakubowski, H. Metabolism and Neurotoxicity of Homocysteine Thiolactone in Mice: Evidence for a Protective Role of Paraoxonase 1. J. Alzheimer's Dis. 2012, 30, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Głowacki, R.; Jakubowski, H. Cross-talk between Cys34 and Lysine Residues in Human Serum Albumin Revealed by N-Homocysteinylation. J. Biol. Chem. 2004, 279, 10864–10871. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Marczak, L.; Twardowski, T.; Stobiecki, M.; Jakubowski, H. Direct monitoring of albumin lysine-525 N-homocysteinylation in human serum by liquid chromatography/mass spectrometry. Anal. Biochem. 2010, 405, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Marczak, L., Sikora, M., Stobiecki, M., and Jakubowski, H. (2011) Analysis of site-specific N-homocysteinylation of human serum albumin in vitro and in vivo using MALDI-ToF and LC-MS/MS mass spectrometry. J Proteomics 74, 967-974.
- Paoli, P.; Sbrana, F.; Tiribilli, B.; Caselli, A.; Pantera, B.; Cirri, P.; De Donatis, A.; Formigli, L.; Nosi, D.; Manao, G.; et al. Protein N-Homocysteinylation Induces the Formation of Toxic Amyloid-Like Protofibrils. J. Mol. Biol. 2010, 400, 889–907. [Google Scholar] [CrossRef]
- Undas, A., Perla, J., Lacinski, M., Trzeciak, W., Kazmierski, R., and Jakubowski, H. (2004) Autoantibodies against N-homocysteinylated proteins in humans: implications for atherosclerosis. Stroke 35, 1299-1304.
- Perla, J., Undas, A., Twardowski, T., and Jakubowski, H. (2004) Purification of antibodies against N-homocysteinylated proteins by affinity chromatography on Nomega-homocysteinyl-aminohexyl-Agarose. J Chromatogr B Analyt Technol Biomed Life Sci 807, 257-261.
- Perła-Kaján, J., Stanger, O., Ziółkowska, A., Melandowicz, L. K., Twardowski, T., and Jakubowski, H. (2007) Immunohistochemical detection of N-homocysteinylated proteins in cardiac surgery patients. Clin Chem Lab Med 45, A36.
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, L.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids 2013, 46, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of Fibrinogen by Homocysteine Thiolactone Increases Resistance to Fibrinolysis: A Potential Mechanism of the Thrombotic Tendency in Hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Akchiche, N., Bossenmeyer-Pourie, C., Kerek, R., Martin, N., Pourie, G., Koziel, V., Helle, D., Alberto, J. M., Ortiou, S., Camadro, J. M., Leger, T., Gueant, J. L., and Daval, J. L. (2012) Homocysteinylation of neuronal proteins contributes to folate deficiency-associated alterations of differentiation, vesicular transport, and plasticity in hippocampal neuronal cells. FASEB J 26, 3980-3992.
- Zhou, L., Guo, T., Meng, L., Zhang, X., Tian, Y., Dai, L., Niu, X., Li, Y., Liu, C., Chen, G., Liu, C., Ke, W., Zhang, Z., Bao, A., and Zhang, Z. (2023) N-homocysteinylation of alpha-synuclein promotes its aggregation and neurotoxicity. Aging Cell 22, e13745.
- Guo, T., Zhou, L., Xiong, M., Xiong, J., Huang, J., Li, Y., Zhang, G., Chen, G., Wang, Z. H., Xiao, T., Hu, D., Bao, A., and Zhang, Z. (2024) N-homocysteinylation of DJ-1 promotes neurodegeneration in Parkinson's disease. Aging Cell, e14124.
- Mei, X.; Qi, D.; Zhang, T.; Zhao, Y.; Jin, L.; Hou, J.; Wang, J.; Lin, Y.; Xue, Y.; Zhu, P.; et al. Inhibiting MARSs reduces hyperhomocysteinemia-associated neural tube and congenital heart defects. EMBO Mol. Med. 2020, 12, e9469. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhao, R.; Qu, Y.-Y.; Mei, X.-Y.; Zhang, X.; Zhou, Q.; Li, Y.; Yang, S.-B.; Zuo, Z.-G.; Chen, Y.-M.; et al. Colonic Lysine Homocysteinylation Induced by High-Fat Diet Suppresses DNA Damage Repair. Cell Rep. 2018, 25, 398–412.e6. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Thiolactone: Metabolic Origin and Protein Homocysteinylation in Humans. J. Nutr. 2000, 130, 377S–381S. [Google Scholar] [CrossRef] [PubMed]
- Perla-Kajan, J., and Jakubowski, H. (2010) Paraoxonase 1 protects against protein N-homocysteinylation in humans. FASEB J 24, 931-936.
- Sikora, M., Marczak, L., Suszynska-Zajczyk, J., Jakubowski, H. (2012) Monitoring site-specific N-homocysteinylation in fibrinogen in vitro and in vivo as a potential marker of thrombosis in CBS-deficient patients. In 22nd International Fibrinogen Workshop Vol. Abstract Book, P018 p. 94, International Fibrinogen Research Society (IFRS), Brighton, UK.
- Perla-Kajan, J., Utyro, O., Rusek, M., Malinowska, A., Sitkiewicz, E., and Jakubowski, H. (2016) N-Homocysteinylation impairs collagen cross-linking in cystathionine beta-synthase-deficient mice: a novel mechanism of connective tissue abnormalities. FASEB J 30, 3810-3821.
- Xu, L.; Chen, J.; Gao, J.; Yu, H.; Yang, P. Crosstalk of homocysteinylation, methylation and acetylation on histone H3. Anal. 2015, 140, 3057–3063. [Google Scholar] [CrossRef]
- Chen, N.; Liu, J.; Qiao, Z.; Liu, Y.; Yang, Y.; Jiang, C.; Wang, X.; Wang, C. Chemical proteomic profiling of proteinN-homocysteinylation with a thioester probe. Chem. Sci. 2018, 9, 2826–2830. [Google Scholar] [CrossRef]
- Zhang, Q.; Bai, B.; Mei, X.; Wan, C.; Cao, H.; Li, D.; Wang, S.; Zhang, M.; Wang, Z.; Wu, J.; et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nat. Commun. 2018, 9, 3436. [Google Scholar] [CrossRef]
- Bossenmeyer-Pourie, C., Smith, A. D., Lehmann, S., Deramecourt, V., Sablonniere, B., Camadro, J. M., Pourie, G., Kerek, R., Helle, D., Umoret, R., Gueant-Rodriguez, R. M., Rigau, V., Gabelle, A., Sequeira, J. M., Quadros, E. V., Daval, J. L., and Gueant, J. L. (2019) N-homocysteinylation of tau and MAP1 is increased in autopsy specimens of Alzheimer's disease and vascular dementia. J Pathol 248, 291-303.
- Perła-Kaján, J.; Jakubowski, H. Paraoxonase 1 and homocysteine metabolism. Amino Acids 2012, 43, 1405–1417. [Google Scholar] [CrossRef]
- Marsillach, J., Mackness, B., Mackness, M., Riu, F., Beltran, R., Joven, J., and Camps, J. (2008) Immunohistochemical analysis of paraoxonases-1, 2, and 3 expression in normal mouse tissues. Free Radic Biol Med 45, 146-157.
- Costa, L.G.; Giordano, G.; Cole, T.B.; Marsillach, J.; Furlong, C.E. Paraoxonase 1 (PON1) as a genetic determinant of susceptibility to organophosphate toxicity. Toxicology 2012, 307, 115–122. [Google Scholar] [CrossRef]
- Bhattacharyya, T.; Nicholls, S.J.; Topol, E.J.; Zhang, R.; Yang, X.; Schmitt, D.; Fu, X.; Shao, M.; Brennan, D.M.; Ellis, S.G.; et al. Relationship of Paraoxonase 1 (PON1) Gene Polymorphisms and Functional Activity With Systemic Oxidative Stress and Cardiovascular Risk. JAMA 2008, 299, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Hartiala, J.; Fan, Y.; Wu, Y.; Stewart, A.F.; Erdmann, J.; Kathiresan, S.; Roberts, R.; McPherson, R.; Allayee, H.; et al. Clinical and Genetic Association of Serum Paraoxonase and Arylesterase Activities With Cardiovascular Risk. Arter. Thromb. Vasc. Biol. 2012, 32, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Tang, W.H.W.; Fan, Y.; Wu, Y.; Mann, S.; Pepoy, M.; Hazen, S.L. Diminished Antioxidant Activity of High-Density Lipoprotein-Associated Proteins in Chronic Kidney Disease. J. Am. Hear. Assoc. 2013, 2, e000104. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Dohi, T.; Miyauchi, K.; Ogita, M.; Kurano, M.; Ohkawa, R.; Nakamura, K.; Tamura, H.; Isoda, K.; Okazaki, S.; et al. Prognostic impact of homocysteine levels and homocysteine thiolactonase activity on long-term clinical outcomes in patients undergoing percutaneous coronary intervention. J. Cardiol. 2017, 69, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Bakker, S.J.; James, R.W.; Dullaart, R.P. Serum paraoxonase-1 activity and risk of incident cardiovascular disease: The PREVEND study and meta-analysis of prospective population studies. Atherosclerosis 2015, 245, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.M.; Gu, L.; Xia, Y.-R.; Navab, M.; Li, W.-F.; Hama, S.; Castellani, L.W.; Furlong, C.E.; Costa, L.G.; Fogelman, A.M.; et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 1998, 394, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.M.; Xia, Y.-R.; Wang, X.-P.; Miller, E.; Castellani, L.W.; Subbanagounder, G.; Cheroutre, H.; Faull, K.F.; Berliner, J.A.; Witztum, J.L.; et al. Combined Serum Paraoxonase Knockout/Apolipoprotein E Knockout Mice Exhibit Increased Lipoprotein Oxidation and Atherosclerosis. J. Biol. Chem. 2000, 275, 17527–17535. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Bretes, E.; Perła-Kaján, J.; Lewandowska, I.; Marczak, Ł.; Jakubowski, H. Genetic Attenuation of Paraoxonase 1 Activity Induces Proatherogenic Changes in Plasma Proteomes of Mice and Humans. Antioxidants 2020, 9, 1198. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M., and Jakubowski, H. (2021) Changes in redox plasma proteome of Pon1-/- mice are exacerbated by a hyperhomocysteinemic diet. Free Radic Biol Med 169, 169-180.
- Perla-Kajan, J.; Borowczyk, K.; Glowacki, R.; Nygard, O.; Jakubowski, H. Paraoxonase 1 Q192R genotype and activity affect homocysteine thiolactone levels in humans. FASEB J. 2018, 32, 6019–6024. [Google Scholar] [CrossRef]
- Tward, A.; Xia, Y.-R.; Wang, X.-P.; Shi, Y.-S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M.; A, R.; M, W.; et al. Decreased Atherosclerotic Lesion Formation in Human Serum Paraoxonase Transgenic Mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef]
- Menini, T.; Gugliucci, A. Paraoxonase 1 in neurological disorders. Redox Rep. 2013, 19, 49–58. [Google Scholar] [CrossRef]
- Marsillach, J.; Adorni, M.P.; Zimetti, F.; Papotti, B.; Zuliani, G.; Cervellati, C. HDL Proteome and Alzheimer’s Disease: Evidence of a Link. Antioxidants 2020, 9, 1224. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C. Valacchi, G., Tisato, V., Zuliani, G., and Marsillach, J. (2019) Evaluating the link between Paraoxonase-1 levels and Alzheimer's disease development. Minerva Med 110, 238-250.
- de la Torre, J.C. (2002) Alzheimer disease as a vascular disorder: nosological evidence. Stroke 33, 1152-1162.
- Costa, L. G., Cole, T. B., Jarvik, G. P., and Furlong, C. E. (2003) Functional genomic of the paraoxonase (PON1) polymorphisms: effects on pesticide sensitivity, cardiovascular disease, and drug metabolism. Annu Rev Med 54, 371-392.
- Moren, X.; Lhomme, M.; Bulla, A.; Sanchez, J.-C.; Kontush, A.; James, R.W. Proteomic and lipidomic analyses of paraoxonase defined high density lipoprotein particles: Association of paraoxonase with the anti-coagulant, protein S. Proteom. – Clin. Appl. 2015, 10, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Erlich, P.M.; Lunetta, K.L.; Cupples, L.A.; Abraham, C.R.; Green, R.C.; Baldwin, C.T.; Farrer, L.A. Serum paraoxonase activity is associated with variants in the PON gene cluster and risk of Alzheimer disease. Neurobiol. Aging 2012, 33, 1015.e7–1015.e23. [Google Scholar] [CrossRef] [PubMed]
- Bednarska-Makaruk, M.E.; Krzywkowski, T.; Graban, A.; Lipczyńska-Łojkowska, W.; Bochyńska, A.; Rodo, M.; Wehr, H.; Ryglewicz, D.K. Original article Paraoxonase 1 (PON1) gene -108C>T and p.Q192R polymorphisms and arylesterase activity of the enzyme in patients with dementia. Folia Neuropathol. 2013, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Dantoine, T. F., Debord, J., Merle, L., Lacroix-Ramiandrisoa, H., Bourzeix, L., and Charmes, J. P. (2002) Paraoxonase 1 activity: a new vascular marker of dementia? Ann N Y Acad Sci 977, 96-101.
- Paragh, G., Balla, P., Katona, E., Seres, I., Egerhazi, A., and Degrell, I. (2002) Serum paraoxonase activity changes in patients with Alzheimer's disease and vascular dementia. Eur Arch Psychiatry Clin Neurosci 252, 63-67.
- Bednarz-Misa, I.; Berdowska, I.; Zboch, M.; Misiak, B.; Zieliński, B.; Płaczkowska, S.; Fleszar, M.; Wiśniewski, J.; Gamian, A.; Krzystek-Korpacka, M. Paraoxonase 1 decline and lipid peroxidation rise reflect a degree of brain atrophy and vascular impairment in dementia. Adv. Clin. Exp. Med. 2020, 29, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Perla-Kajan, J., Wloczkowska, O., Ziola-Frankowska, A., Frankowski, M., Smith, A. D., de Jager, C. A., Refsum, H., and Jakubowski, H. (2021) Paraoxonase 1, B Vitamins Supplementation, and Mild Cognitive Impairment. J Alzheimers Dis 81, 1211-1229.
- Aluganti Narasimhulu, C., Mitra, C., Bhardwaj, D., Burge, K. Y., and Parthasarathy, S. (2019) Alzheimer's Disease Markers in Aged ApoE-PON1 Deficient Mice. J Alzheimers Dis 67, 1353-1365.
- Salazar, J.G.; Marsillach, J.; Reverte, I.; Mackness, B.; Mackness, M.; Joven, J.; Camps, J.; Colomina, M.T. Paraoxonase-1 and -3 Protein Expression in the Brain of the Tg2576 Mouse Model of Alzheimer’s Disease. Antioxidants 2021, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Mostafalou, S.; Abdollahi, M. The susceptibility of humans to neurodegenerative and neurodevelopmental toxicities caused by organophosphorus pesticides. Arch. Toxicol. 2023, 97, 3037–3060. [Google Scholar] [CrossRef] [PubMed]
- Suszyńska-Zajczyk, J.; Łuczak, M.; Marczak, Ł.; Jakubowski, H. Inactivation of the Paraoxonase 1 Gene Affects the Expression of Mouse Brain Proteins Involved in Neurodegeneration. J. Alzheimer's Dis. 2014, 42, 247–260. [Google Scholar] [CrossRef]
- Blatter, M. C., James, R. W., Messmer, S., Barja, F., and Pometta, D. (1993) Identification of a distinct human high-density lipoprotein subspecies defined by a lipoprotein-associated protein, K-45. Identity of K-45 with paraoxonase. Eur J Biochem 211, 871-879.
- Witucki, L., and Jakubowski, H. (2023) Depletion of Paraoxonase 1 (Pon1) Dysregulates mTOR, Autophagy, and Accelerates Amyloid Beta Accumulation in Mice. Cells 12.
- Kamata, Y.; Itoh, Y.; Kajiya, A.; Karasawa, S.; Sakatani, C.; Takekoshi, S.; Osamura, R.Y.; Takeda, A. Quantification of Neutral Cysteine Protease Bleomycin Hydrolase and its Localization in Rat Tissues. J. Biochem. 2006, 141, 69–76. [Google Scholar] [CrossRef]
- Bromme, D., Rossi, A. B., Smeekens, S. P., Anderson, D. C., and Payan, D. G. (1996) Human bleomycin hydrolase: molecular cloning, sequencing, functional expression, and enzymatic characterization. Biochemistry 35, 6706-6714.
- O'Farrell, P. A., Gonzalez, F., Zheng, W., Johnston, S. A., and Joshua-Tor, L. (1999) Crystal structure of human bleomycin hydrolase, a self-compartmentalizing cysteine protease. Structure 7, 619-627.
- Suszynska, J., Tisonczyk, J., Lee, H. G., Smith, M. A., and Jakubowski, H. (2010) Reduced homocysteine-thiolactonase activity in Alzheimer's disease. J Alzheimers Dis 19, 1177-1183.
- Kajiya, A., Kaji, H., Isobe, T., and Takeda, A. (2006) Processing of amyloid beta-peptides by neutral cysteine protease bleomycin hydrolase. Protein and peptide letters 13, 119-123.
- Papassotiropoulos, A., Bagli, M., Jessen, F., Frahnert, C., Rao, M. L., Maier, W., and Heun, R. (2000) Confirmation of the association between bleomycin hydrolase genotype and Alzheimer's disease. Molecular psychiatry 5, 213-215.
- Lefterov, I.M.; Koldamova, R.P.; Lefterova, M.I.; Schwartz, D.R.; Lazo, J.S. Cysteine 73 in Bleomycin Hydrolase Is Critical for Amyloid Precursor Protein Processing. Biochem. Biophys. Res. Commun. 2001, 283, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Kajiya, A., Kaji, H., Isobe, T., and Takeda, A. (2006) Processing of amyloid beta-peptides by neutral cysteine protease bleomycin hydrolase. Protein and Peptide Letters 13, 119-123.
- Kamata, Y.; Taniguchi, A.; Yamamoto, M.; Nomura, J.; Ishihara, K.; Takahara, H.; Hibino, T.; Takeda, A. Neutral Cysteine Protease Bleomycin Hydrolase Is Essential for the Breakdown of Deiminated Filaggrin into Amino Acids. 2009, 284, 12829–12836. [CrossRef]
- Ratovitski, T.; Chighladze, E.; Waldron, E.; Hirschhorn, R.R.; Ross, C.A. Cysteine Proteases Bleomycin Hydrolase and Cathepsin Z Mediate N-terminal Proteolysis and Toxicity of Mutant Huntingtin. 2011, 286, 12578–12589. [CrossRef]
- Okamura, Y.; Nomoto, S.; Hayashi, M.; Hishida, M.; Nishikawa, Y.; Yamada, S.; Fujii, T.; Sugimoto, H.; Takeda, S.; Kodera, Y.; et al. Identification of the bleomycin hydrolase gene as a methylated tumor suppressor gene in hepatocellular carcinoma using a novel triple-combination array method. Cancer Lett. 2011, 312, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kamata, Y.; Maejima, H.; Watarai, A.; Saito, N.; Katsuoka, K.; Takeda, A.; Ishihara, K. Expression of bleomycin hydrolase in keratinization disorders. Arch. Dermatol. Res. 2011, 304, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, J.P.; Jakasa, I.; Riethmüller, C.; Schön, M.P.; Braun, A.; Haftek, M.; Fallon, P.G.; Wróblewski, J.; Jakubowski, H.; Eckhart, L.; et al. Filaggrin Expression and Processing Deficiencies Impair Corneocyte Surface Texture and Stiffness in Mice. J. Investig. Dermatol. 2020, 140, 615–623.e5. [Google Scholar] [CrossRef] [PubMed]
- Montoya, S. E., Aston, C. E., DeKosky, S. T., Kamboh, M. I., Lazo, J. S., and Ferrell, R. E. (1998) Bleomycin hydrolase is associated with risk of sporadic Alzheimer's disease. Nat Genet 18, 211-212.
- Namba, Y., Ouchi, Y., Asada, T., Hattori, H., Ueki, A., and Ikeda, K. (1999) Lack of association between bleomycin hydrolase gene polymorphism and Alzheimer's disease in Japanese people. Ann Neurol 46, 136-137.
- Farrer, L. A., Abraham, C. R., Haines, J. L., Rogaeva, E. A., Song, Y., McGraw, W. T., Brindle, N., Premkumar, S., Scott, W. K., Yamaoka, L. H., Saunders, A. M., Roses, A. D., Auerbach, S. A., Sorbi, S., Duara, R., Pericak-Vance, M. A., and St George-Hyslop, P. H. (1998) Association between bleomycin hydrolase and Alzheimer's disease in caucasians. Ann Neurol 44, 808-811.
- Thome, J., Gewirtz, J. C., Sakai, N., Zachariou, V., Retz-Junginger, P., Retz, W., Duman, R. S., and Rosler, M. (1999) Polymorphisms of the human apolipoprotein E promoter and bleomycin hydrolase gene: risk factors for Alzheimer's dementia? Neurosci Lett 274, 37-40.
- Schwartz, D.R.; Homanics, G.E.; Hoyt, D.G.; Klein, E.; Abernethy, J.; Lazo, J.S. The neutral cysteine protease bleomycin hydrolase is essential for epidermal integrity and bleomycin resistance. Proc. Natl. Acad. Sci. USA 1999, 96, 4680–4685. [Google Scholar] [CrossRef] [PubMed]
- Montoya, S.; Thiels, E.; Card, J.; Lazo, J. Astrogliosis and behavioral changes in mice lacking the neutral cysteine protease bleomycin hydrolase. Neuroscience 2007, 146, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Towne, C.F.; York, I.A.; Watkin, L.B.; Lazo, J.S.; Rock, K.L. Analysis of the Role of Bleomycin Hydrolase in Antigen Presentation and the Generation of CD8 T Cell Responses. J. Immunol. 2007, 178, 6923–6930. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y., Qian, S., Chen, D., Ye, M., Wu, J., and Wang, Y. L. (2023) Serum BLMH and CKM as Potential Biomarkers for Predicting Therapeutic Effects of Deep Brain Stimulation in Parkinson's Disease: A Proteomics Study. J Integr Neurosci 22, 163.
- Okun, M. S. (2012) Deep-brain stimulation for Parkinson's disease. N Engl J Med 367, 1529-1538.
- Thompson, A.G.; Gray, E.; Mäger, I.; Thézénas, M.-L.; Charles, P.D.; Talbot, K.; Fischer, R.; Kessler, B.M.; Wood, M.; Turner, M.R. CSF extracellular vesicle proteomics demonstrates altered protein homeostasis in amyotrophic lateral sclerosis. Clin. Proteom. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Suszyńska-Zajczyk, J.; Łuczak, M.; Marczak, Ł.; Jakubowski, H. Hyperhomocysteinemia and Bleomycin Hydrolase Modulate the Expression of Mouse Brain Proteins Involved in Neurodegeneration. J. Alzheimer's Dis. 2014, 40, 713–726. [Google Scholar] [CrossRef]
- Witucki, L., Borowczyk, K., Suszynska-Zajczyk, J., Warzych, E., Pawlak, P., and Jakubowski, H. (2023) Deletion of the Homocysteine Thiolactone Detoxifying Enzyme Bleomycin Hydrolase, in Mice, Causes Memory and Neurological Deficits and Worsens Alzheimer's Disease-Related Behavioral and Biochemical Traits in the 5xFAD Model of Alzheimer's Disease. J Alzheimers Dis.
- Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., Guillozet-Bongaarts, A., Ohno, M., Disterhoft, J., Van Eldik, L., Berry, R., and Vassar, R. (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129-10140.
- Thinakaran, G., Teplow, D. B., Siman, R., Greenberg, B., and Sisodia, S. S. (1996) Metabolism of the "Swedish" amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the "beta-secretase" site occurs in the golgi apparatus. J Biol Chem 271, 9390-9397.
- Sobering, A.K.; Bryant, L.M.; Li, D.; McGaughran, J.; Maystadt, I.; Moortgat, S.; Graham, J.M.; van Haeringen, A.; Ruivenkamp, C.; Cuperus, R.; et al. Variants in PHF8 cause a spectrum of X-linked neurodevelopmental disorders and facial dysmorphology. Hum. Genet. Genom. Adv. 2022, 3, 100102. [Google Scholar] [CrossRef]
- Laumonnier, F.; Holbert, S.; Ronce, N.; Faravelli, F.; Lenzner, S.; E Schwartz, C.; Lespinasse, J.; Van Esch, H.; Lacombe, D.; Goizet, C.; et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J. Med Genet. 2005, 42, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L.; et al. Phf8 histone demethylase deficiency causes cognitive impairments through the mTOR pathway. Nat. Commun. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Lefterov, I.M.; Koldamova, R.P.; Lazo, J.S. Human bleomycin hydrolase regulates the secretion of amyloid precursor protein. FASEB J. 2000, 14, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Joshua-Tor, L.; Xu, H.E.; Johnston, S.A.; Rees, D.C. Crystal Structure of a Conserved Protease That Binds DNA: the Bleomycin Hydrolase, Gal6. Science 1995, 269, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A., Higuchi, D., Yamamoto, T., Nakamura, Y., Masuda, Y., Hirabayashi, T., and Nakaya, K. (1996) Purification and characterization of bleomycin hydrolase, which represents a new family of cysteine proteases, from rat skin. Journal of biochemistry 119, 29-36.
- Pickford, F. Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., Small, S., Spencer, B., Rockenstein, E., Levine, B., and Wyss-Coray, T. (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118, 2190-2199.
- Jaeger, P.A.; Pickford, F.; Sun, C.-H.; Lucin, K.M.; Masliah, E.; Wyss-Coray, T. Regulation of Amyloid Precursor Protein Processing by the Beclin 1 Complex. PLOS ONE 2010, 5, e11102. [Google Scholar] [CrossRef] [PubMed]
- Perła-Kaján, J.; Jakubowski, H. Dysregulation of Epigenetic Mechanisms of Gene Expression in the Pathologies of Hyperhomocysteinemia. Int. J. Mol. Sci. 2019, 20, 3140. [Google Scholar] [CrossRef] [PubMed]
- Puente, X. S., and Lopez-Otin, C. (1995) Cloning and expression analysis of a novel human serine hydrolase with sequence similarity to prokaryotic enzymes involved in the degradation of aromatic compounds. J Biol Chem 270, 12926-12932.
- Puente, X.S.; Pendás, A.M.; López-Otı́n, C. Structural Characterization and Chromosomal Localization of the Gene Encoding Human Biphenyl Hydrolase-Related Protein (BPHL). Genomics 1998, 51, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Doh, M.J.; Kim, I.H.; Kong, H.S.; Lee, J.S.; Kim, Y.M. Prednisolone 21-sulfate sodium: a colon-specific pro-drug of prednisolone. J. Pharm. Pharmacol. 2003, 55, 1075–1082. [Google Scholar] [CrossRef]
- Hu, Y., Epling, D., Shi, J., Song, F., Tsume, Y., Zhu, H. J., Amidon, G. L., and Smith, D. E. (2018) Effect of biphenyl hydrolase-like (BPHL) gene disruption on the intestinal stability, permeability and absorption of valacyclovir in wildtype and Bphl knockout mice. Biochem Pharmacol 156, 147-156.
- Lai, L., Xu, Z., Zhou, J., Lee, K. D., and Amidon, G. L. (2008) Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase. J Biol Chem 283, 9318-9327.
- Ren, P.; Zhai, J.; Wang, X.; Yin, Y.; Lin, Z.; Cai, K.; Wang, H. Inhibition of BPHL inhibits proliferation in lung carcinoma cell lines. Transl. Lung Cancer Res. 2023, 12, 1051–1061. [Google Scholar] [CrossRef]
- Witucki, L., Suszyńska-Zajczyk, J., Perła-Kajan, J., Bretes, E., Włoczkowska, O., Jakubowski, H. . (2023) Deletion of the Homocysteine Thiolactone Detoxifying Enzyme Biphenyl Hydrolase-like (Bphl), in Mice, Induces Biochemical and Behavioral Hallmarks of Alzheimer’s Disease. In 14th International Conference One Carbon Metabolism, B Vitamins and Homocysteine & 2nd CluB-12 Annual Symposium (Hcy2023) (Cambridge University, U., ed), Cambridge University, Cambridge, UK.
Figure 1.
Schematic representation of homocysteine metabolism in humans and mice: the remethylation (i), transsulfuration (ii), and homocysteine (Hcy)-thiolactone (iii) pathways. Protein metabolism-related reactions involving Hcy are highlighted by blue arrows. The rectangle symbolizes the cell, the outside area is plasma, and the oval labelled “Dietary protein” represents the digestive tract. See text for description. AdoMet, adenosylmethionine; BPHL, biphenyl hydrolase-like; CBS, cystathionine β-synthase; MAT, Met S-adenosyltransferase; Met, methionine; MetRS, methionyl-tRNA synthetase; MS, Met synthase; MTHFR, methylenetetrahydrofolate reductase; THF, tetrahydrofolate. Reproduced with permission from Jakubowski [9].
Figure 1.
Schematic representation of homocysteine metabolism in humans and mice: the remethylation (i), transsulfuration (ii), and homocysteine (Hcy)-thiolactone (iii) pathways. Protein metabolism-related reactions involving Hcy are highlighted by blue arrows. The rectangle symbolizes the cell, the outside area is plasma, and the oval labelled “Dietary protein” represents the digestive tract. See text for description. AdoMet, adenosylmethionine; BPHL, biphenyl hydrolase-like; CBS, cystathionine β-synthase; MAT, Met S-adenosyltransferase; Met, methionine; MetRS, methionyl-tRNA synthetase; MS, Met synthase; MTHFR, methylenetetrahydrofolate reductase; THF, tetrahydrofolate. Reproduced with permission from Jakubowski [9].
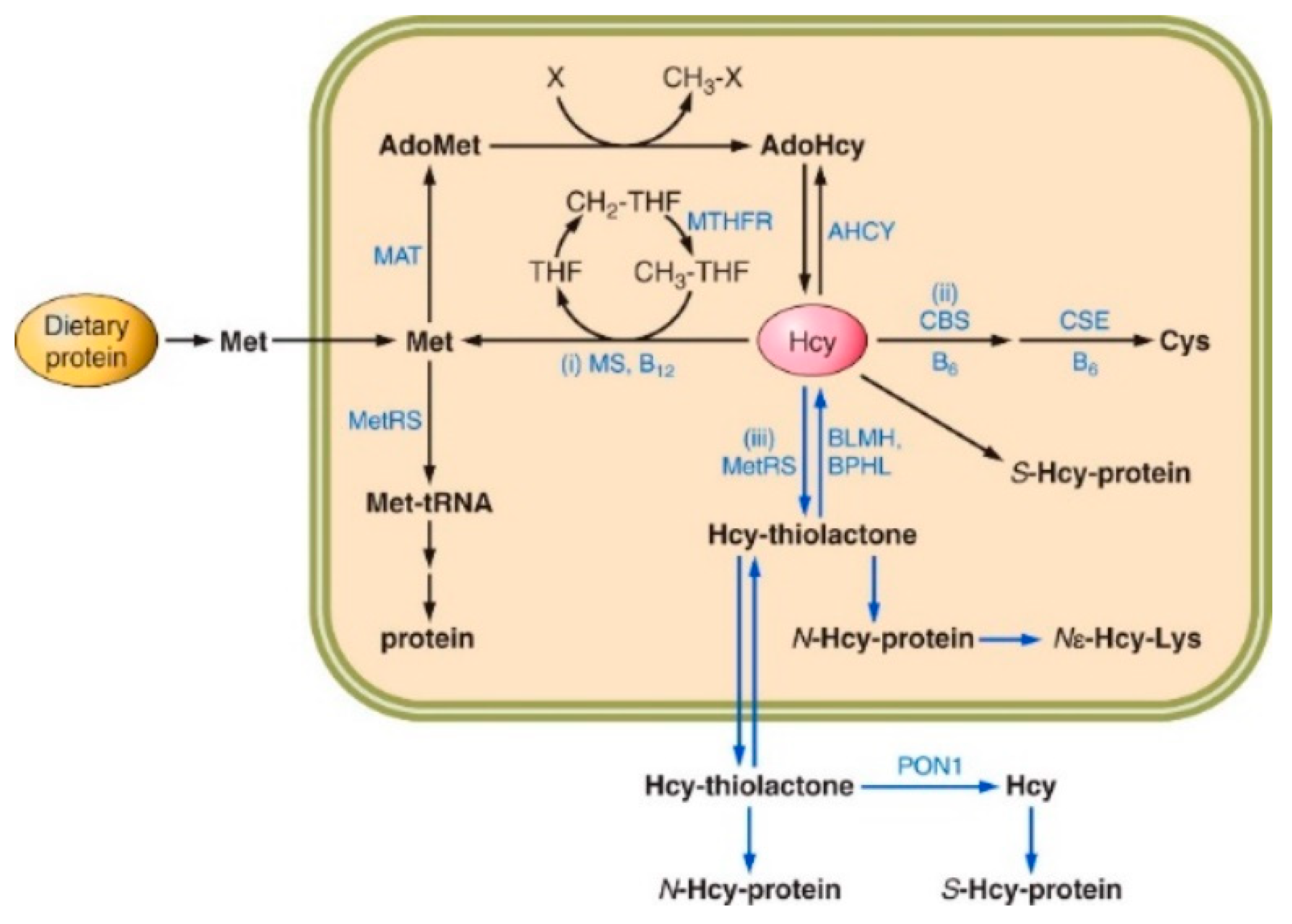
Figure 2.
Kinetics of plasma Hcy-thiolactone (A), total Hcy (B), and N-Hcy-protein (C) turnover in mice. For Hcy-thiolactone (A) and total Hcy (B) turnover experiments, mice were injected i.p. with 600 nmol L-Hcy-thiolactone/g body weight. For N-Hcy-protein (C) turnover experiments, 2,850 nmol L-Hcy-thiolactone/g body weight L-Hcy-thiolactone was used. Metabolites were analyzed at indicated times post-injection and data points were fitted to an exponential equation [At] = [A0]·e−k·t, where k is a first-order rate constant, [At] is metabolite concentration measured at time t, and [A0] is metabolite concentration extrapolated to time zero. Representative kinetics obtained for individual knockout Blmh−/− (•) and wild-type Blmh+/+ (x) mice are shown. Reproduced with permission from Borowczyk et al. [51].
Figure 2.
Kinetics of plasma Hcy-thiolactone (A), total Hcy (B), and N-Hcy-protein (C) turnover in mice. For Hcy-thiolactone (A) and total Hcy (B) turnover experiments, mice were injected i.p. with 600 nmol L-Hcy-thiolactone/g body weight. For N-Hcy-protein (C) turnover experiments, 2,850 nmol L-Hcy-thiolactone/g body weight L-Hcy-thiolactone was used. Metabolites were analyzed at indicated times post-injection and data points were fitted to an exponential equation [At] = [A0]·e−k·t, where k is a first-order rate constant, [At] is metabolite concentration measured at time t, and [A0] is metabolite concentration extrapolated to time zero. Representative kinetics obtained for individual knockout Blmh−/− (•) and wild-type Blmh+/+ (x) mice are shown. Reproduced with permission from Borowczyk et al. [51].
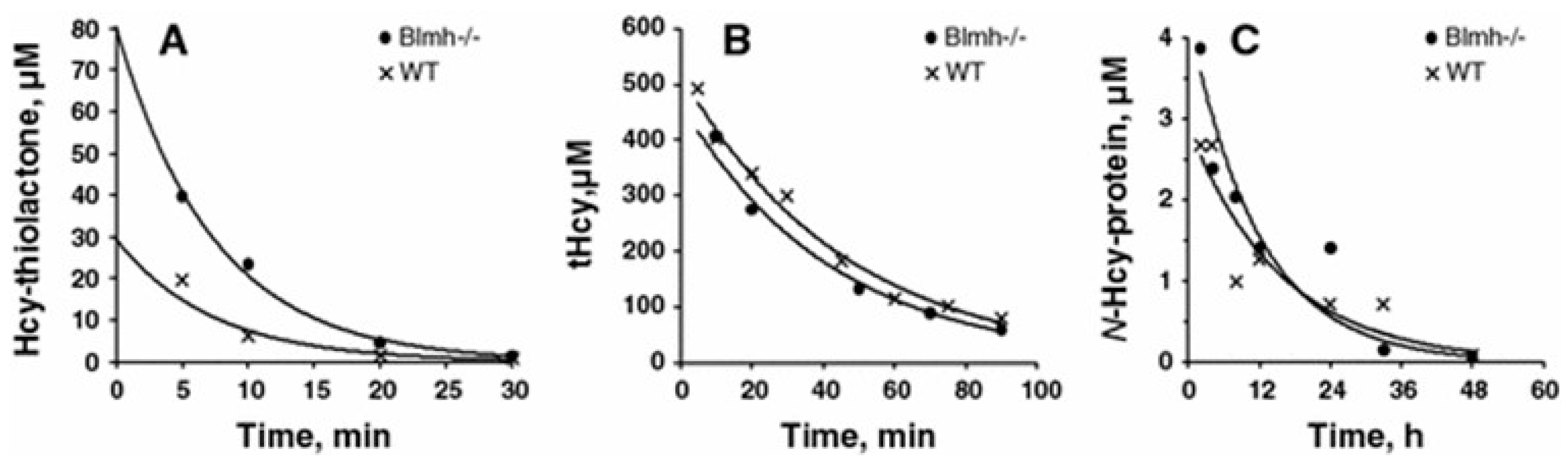
Figure 3.
Increased incidence of L-Hcy-thiolactone-induced seizures in Pon1-/- mice relative to their Pon1+/+ siblings (WT). L-Hcy-thiolactone was injected i.p. into Pon1-/- (n=19) and Pon1+/+ (WT, n=13) mice (3.7 μmol/g body weight); the animals were monitored for 90 min. Data from Borowczyk et al. [52].
Figure 3.
Increased incidence of L-Hcy-thiolactone-induced seizures in Pon1-/- mice relative to their Pon1+/+ siblings (WT). L-Hcy-thiolactone was injected i.p. into Pon1-/- (n=19) and Pon1+/+ (WT, n=13) mice (3.7 μmol/g body weight); the animals were monitored for 90 min. Data from Borowczyk et al. [52].
Figure 4.
Hypothetical pathways leading to Aβ generation in Pon1-/-5xFAD mice. Up and down arrows show direction of changes. Blmh, bleomycin hydrolase; Hcy, homocysteine; HTL, Hcy-thiolactone; APP, amyloid beta precursor protein; mTOR, mammalian target of rapamycin; pmTOR, phospho-mTOR; Phf8, Plant Homeodomain Finger protein 8. [H4K20me1-mTOR] represents H4K20me1 bound at the mTOR promoter.
Figure 4.
Hypothetical pathways leading to Aβ generation in Pon1-/-5xFAD mice. Up and down arrows show direction of changes. Blmh, bleomycin hydrolase; Hcy, homocysteine; HTL, Hcy-thiolactone; APP, amyloid beta precursor protein; mTOR, mammalian target of rapamycin; pmTOR, phospho-mTOR; Phf8, Plant Homeodomain Finger protein 8. [H4K20me1-mTOR] represents H4K20me1 bound at the mTOR promoter.
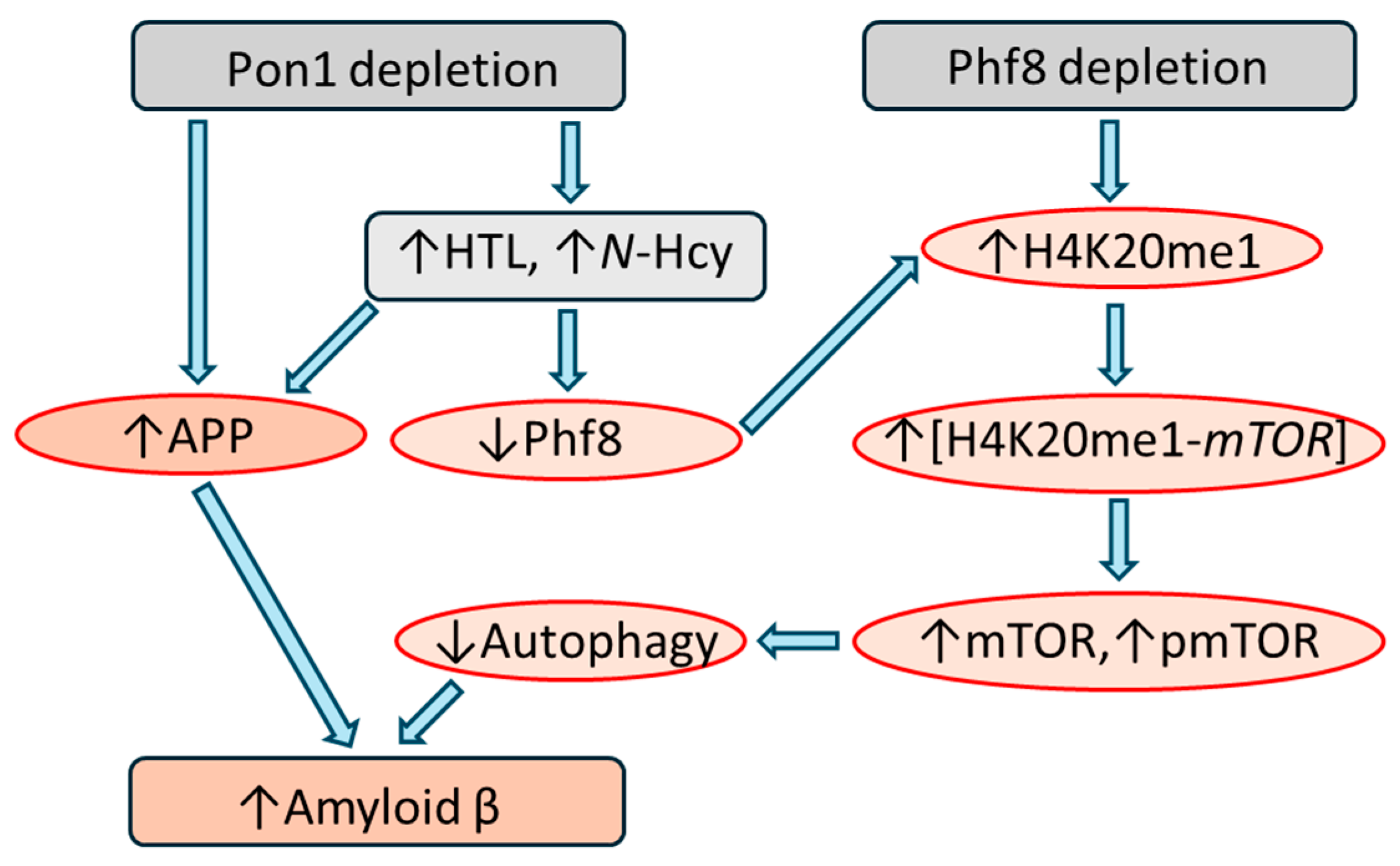
Figure 5.
Increased incidence of L-Hcy-thiolactone-induced seizures in Blmh1-/- mice relative to their Blmh1+/+ littermates (WT). L-Hcy-thiolactone was injected i.p. into Blmh1-/- (n = 32) and Blmh1+/+ (WT, n=44) mice (3.7 μmol/g body weight); the animals were monitored for 90 min. Data from Borowczyk et al. [51].
Figure 5.
Increased incidence of L-Hcy-thiolactone-induced seizures in Blmh1-/- mice relative to their Blmh1+/+ littermates (WT). L-Hcy-thiolactone was injected i.p. into Blmh1-/- (n = 32) and Blmh1+/+ (WT, n=44) mice (3.7 μmol/g body weight); the animals were monitored for 90 min. Data from Borowczyk et al. [51].
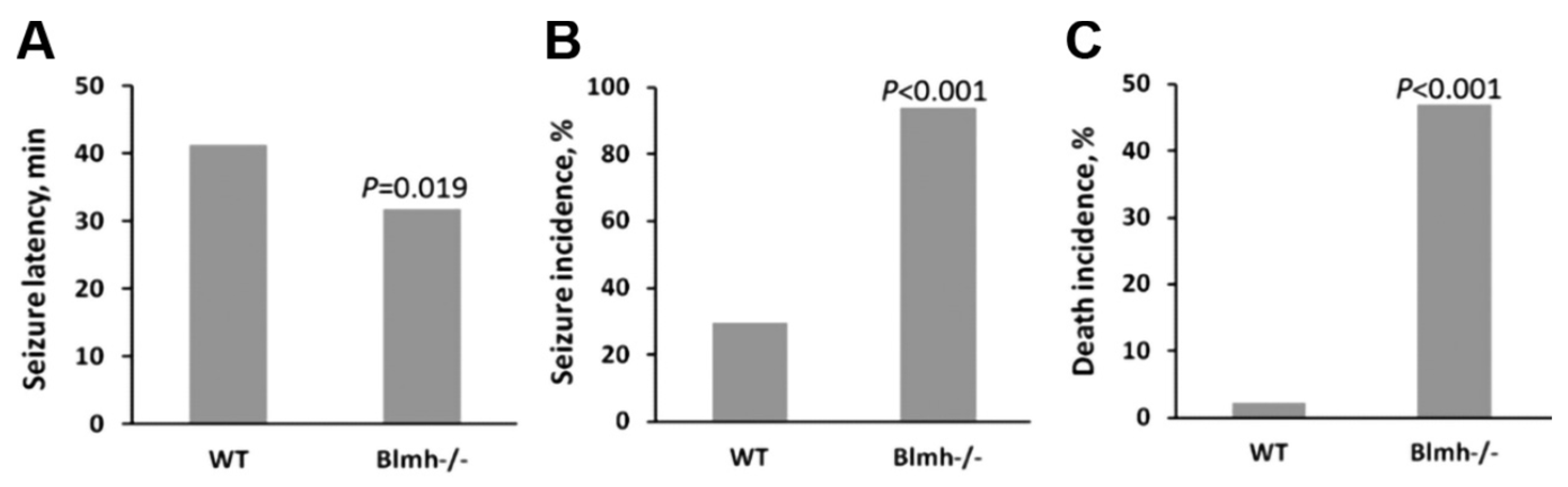
Figure 6.
Hypothetical pathways leading to Aβ upregulation in Blmh-/-5xFAD mice. Panel (A) illustrates the APP (i) and Phf8 (ii-a) pathways. Panel (B) highlights the interaction (iii) between autophagy (Becn1) and APP pathways. Up and down arrows show direction of changes. Blmh, bleomycin hydrolase; Hcy, homocysteine; HTL, Hcy-thiolactone; APP, amyloid beta precursor protein; mTOR, mammalian target of rapamycin; pmTOR, phospho-mTOR; Phf8, Plant Homeodomain Finger protein 8. [H4K20me1-mTOR] represents H4K20me1 bound at the mTOR promoter. Modified from Witucki et al. [132].
Figure 6.
Hypothetical pathways leading to Aβ upregulation in Blmh-/-5xFAD mice. Panel (A) illustrates the APP (i) and Phf8 (ii-a) pathways. Panel (B) highlights the interaction (iii) between autophagy (Becn1) and APP pathways. Up and down arrows show direction of changes. Blmh, bleomycin hydrolase; Hcy, homocysteine; HTL, Hcy-thiolactone; APP, amyloid beta precursor protein; mTOR, mammalian target of rapamycin; pmTOR, phospho-mTOR; Phf8, Plant Homeodomain Finger protein 8. [H4K20me1-mTOR] represents H4K20me1 bound at the mTOR promoter. Modified from Witucki et al. [132].
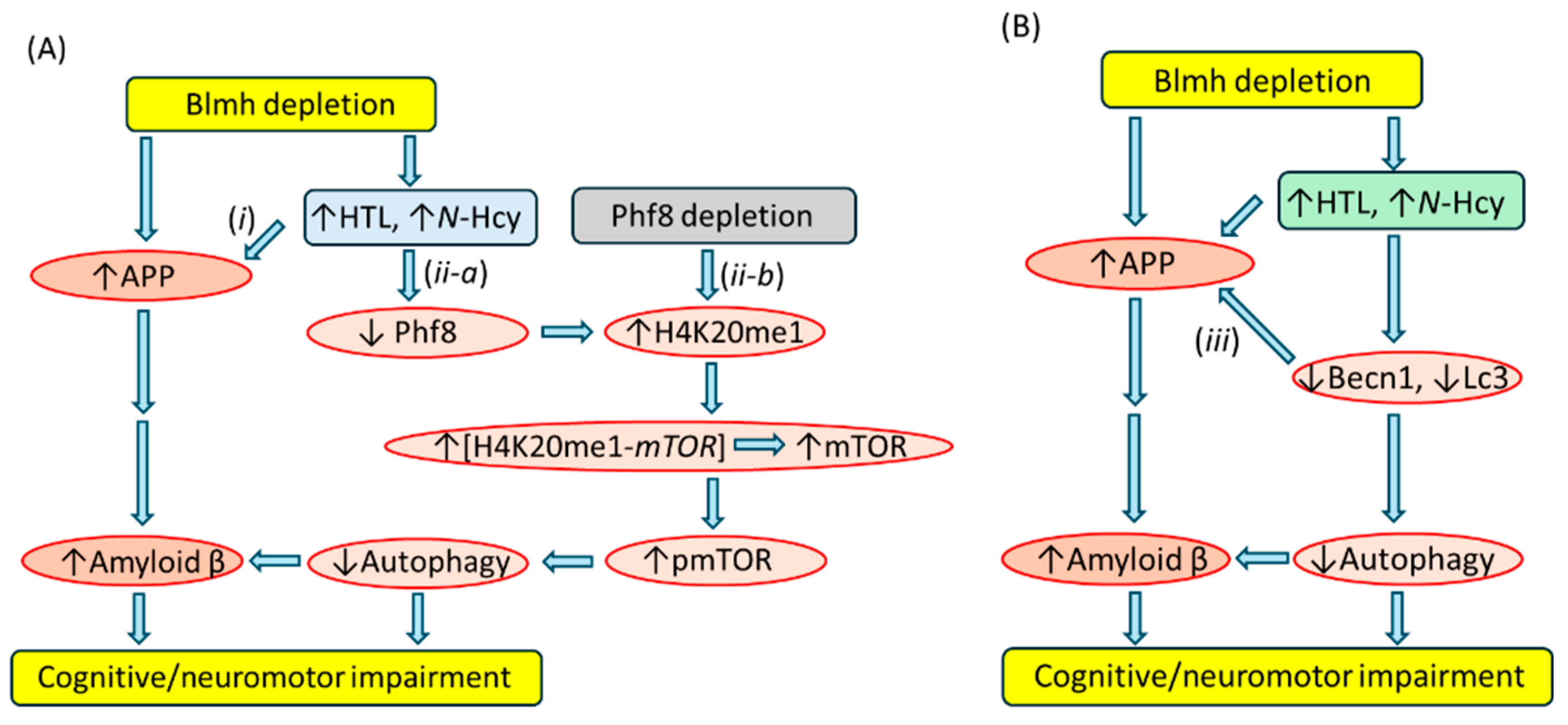

Table 1.
Substrate specificities of human Hcy-thiolactone hydrolyzing enzymes *.
| Substrate | PON1, % | BLMH, % | BPHL, % |
|---|---|---|---|
|
L-Hcy-thiolactone (kcat/Km) |
100 (10 M-1s-1) |
100 (103 M-1s-1) |
100 (7.7x104 M-1s-1) |
| D-Hcy-thiolactone | 24 | <1 | ND |
| γ-Thiobutyrolactone | 545 | <1 | <0.001 |
| N-Acetyl-D, L-HTL | <1 | <1 | <0.001 |
| L-Hse-lactone | ++++ | – | +++ |
| L-Met methyl ester | <1 | ++ | 30 |
| L-Cys methyl ester | <1 | ++ | ++ |
| L-Lys methyl ester | ND | – | – |
| L-Phe ethyl ester | 0 | ND | 16 |
| Nε-Hcy-aminocaproate | ND | ++++ | ND |
| Val(Nε-Hcy-Lys) | ND | ++++ | ND |
| HcyLeuAla | ND | ++++ | ND |
| Bleomycin | ND | 500 | ND |
| Paraoxon | 330 | – | ND |
| Phenyl acetate | 280,000 | – | <0.001 |
| Valacyclovir | – | ND | 22 |
* Data for PON1 and BLMH are from refs [24,25], respectively, and for BPHL from refs [26,27,28]. Multiple ‘+’ symbols indicate relative enzymatic activity for indicated substrates; a ‘–‘ symbol means no activity. Hcy, homocysteine; Hse, homoserine; ND, not determined. Reproduced with permission from Jakubowski [9].
Table 2.
Hcy-thiolactone hydrolyzing activity is absent in serum from Pon1-/- mice.
| Genotype | Hcy-thiolactone hydrolase activity*, % | Paraoxonase activity#, % | ||
| Male | Female | Male | Female | |
| Pon1-/- | 0 | 0 | 0 | 0 |
| Pon1+/- | 51 | 30 | 50 | 40 |
| Pon1+/+ | 100 | 73 | 100 | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



