Submitted:
28 May 2024
Posted:
29 May 2024
You are already at the latest version
Abstract
Background: We evaluate the genetic and phenotypic features of a cohort of 12 Italian patients affected by Retinitis Pigmentosa (RP) associated with RP1 sequence variants.; Methods: Retrospective, single center study (Careggi Hospital, Florence), including a cohort of twelve Italian patients (Five male and seven female) affected by RP carrying pathogenic variants of the RP1 gene. A complete ophthalmic assessment and pedigree analysis was conducted with focus on the onset of disease symptoms, the patient’s age at the first diagnosis, the follow up duration and the possible presence of comorbidities; Results: The mean age at onset of symptoms was 31,0 ± 24,1 years old, with a mean follow-up period of 9,1± 2,4 years. The main symptoms at presentation were hemeralopia and visual field constriction. Fundus evaluation revealed a classic type of RP. Fundus autofluorescence (FAF), Optical Coherence Tomography (OCT), Electroretinogram (ERG), and visual field confirmed in most of the cases the typical features of classic retinitis pigmentosa.; (4) Conclusions: This single-center cohort of Italian patients provided information on the clinical and genetic features of RP1-associated RP. By comprehensively clarifying the genetic variations and their associated clinical manifestations, we can better tailor therapeutic interventions aimed at targeting specific genetic abnormalities. This ultimately promises to improve the prognosis and quality of life for individuals with RP-associated RP1.
Keywords:
RP1 gene
; Inherited Retinal Dystrophies
; Genotype
; Phenotype
; Retinitis Pigmentosa
1. Introduction
Inherited retinal dystrophies (IRD) consist of a clinical and genetically variable spectrum of conditions that affect photoreceptor cells, causing visual impairment. [1,2] Among retinal dystrophies, retinitis pigmentosa (RP) (OMIM # 268000), predominantly affects rod photoreceptors and has the highest worldwide prevalence of 1 in 4000 individuals. Mostly the condition exhibits three patterns of inheritance: autosomal recessive (ArRP), autosomal dominant (AdRP) and X linked RP (xLRP) with up to 50% of cases being isolated (simple or sporadic RP or sRP), with no family history. [3]
The onset, progression, retinal manifestations, and visual prognosis vary considerably among RP cases. Typically, degeneration of the rods begins, causing night blindness, loss of peripheral vision, and tunnel vision. Then, cone photoreceptors are affected in later stages, causing dyschromatopsia and progressive loss of central vision. [4]
Often IRD affects the eye without additional systemic manifestations (non-syndromic IRD), although in some cases retinal degeneration accompanies systemic disorders (syndromic IRD).
RP1 gene sequence variants have been identified as possible non syndromic RP determinants. [5]
RP1 gene is localized in the pericentric region of the chromosome 8 (Locus 8q11.23-q12.1) and encodes a protein 2156 amino acids long mainly located in photoreceptor connection cilia and axonema. The N-terminal segment includes a microtubule-binding domain (DCX), which is vital for photoreceptor longevity by stabilizing microtubules and regulating cilia length. [6]
RP1 mRNA is expressed only in the photoreceptor cell bodies and inner segments of the retinas. [7] The protein regulates the stability and length of the microtubule-based axoneme of photoreceptors, required for the differentiation of photoreceptor cells and plays a role in the organization of the outer segment of rod and cone photoreceptors ensuring the correct orientation and higher-order stacking of outer segment disks along the photoreceptor axoneme.
RP1 mutations can be associated with autosomal dominant retinitis pigmentosa [8,9,10] but can also cause autosomal recessive forms of the disease. [11,12] Moreover, RP1 sequence variants have been reported in association with other phenotypes like cone-rod dystrophy, and macular degeneration. [13]
Most RP1 gene mutations are single nucleotide substitutions that produce a premature stop codon or insertions/deletions, resulting in a truncated protein. [14,15]
This study investigates the genetic and phenotypic characteristics of a group of Italian RP patients carrying RP1 sequence variants.
2. Materials and Methods
This retrospective single-center study conducted at Careggi Hospital in Florence followed the principles outlined in the Declaration of Helsinki. Through the Retinal Degeneration Referring Center of the Hospital, we enrolled 12 patients affected by RP, associated with the detection of pathogenic mutation in the RP1 gene.
Ophthalmic examinations included assessments of best-corrected visual acuity (BCVA), refraction, intraocular pressure measurement, anterior and posterior segment biomicroscopy, color and autofluorescence retinal imaging, OCT, electroretinography (ERG) and Goldmann perimetry. An accurate clinical history was collected in all patients included in the study, with particular focus on the onset of disease symptoms, the patient’s age at first diagnosis, the follow up duration, the possible presence of extraocular pathologies, and the presence of other affected family members.
Imaging modalities included wide-field ocular fundus photography (Optos PLC, Dunfermline, UK), spectral-domain Optical Coherence Tomography (Heidelberg Engineering, Heidelberg, Germany; Carl Zeiss Meditec, Germany) and Goldmann visual field test. Electroretinography (ERG) was performed following the International Society for Clinical Electrophysiology of Vision (ISCEV) standard protocol. [16] Following pupil dilation, ERGs were obtained using Retiscan 201 B4 (Roland Consult, Brandenburg, Germany). We compared the results with those obtained from a group of age-similar control patients (n = 15) with no significant ocular pathology who attended our clinic. An Excel database (Microsoft Excel 2010, Microsoft Office Professional Plus 2010) was used to record all the data.
2.1. Molecular Genetic Analysis
Following informed consent and a pre-test genetic counseling, 10 ml of peripheral blood were obtained from the antecubital vein using EDTA-containing vials. DNA was extracted from 200 μl of peripheral blood by using automated DNA extractors: BioRobot EZ1 (QIAGEN, Hilden, Germany) or QIAsymphony SP workstation (QIAGEN GmbH, Germany), according to the manufacturer protocols. The proband was the first patient with a clinical diagnosis of RP included in the study. In 11 probands from five independent pedigrees, genetic analysis was performed with targeted next-generation sequencing (NGS) at the Department of Genetic Diagnosis (Careggi Teaching Hospital, Florence, Italy) which is a certified UNI EN ISO 9001:2008 laboratory. A panel of 137 genes known to be associated with retinal dystrophies was used for targeted NGS: Exons of DNA samples were captured and investigated as shown previously with enrichment methodology SureSelect QXT Target Enrichment (Agilent Technologies, Santa Clara, CA), using the Illumina NextSeq TM500 platform (Illumina, San Diego, CA). All identified variants were confirmed with Sanger sequencing and further segregated in the respective families when other relatives were available. Variations were annotated using Alissa Interpret Rev. 5.4.2 Agilent Inc., by comparing with other databases (1000 Genome project, Exome aggregation consortium, Ensmble, dbSNP, ClinVar, Human Gene Mutation Database (HGMD), Variation Database (LOVD), RetinoGenetics, RetNet, Mutation Database of Retina International) and predicted for pathogenicity with online bioinformatic tools (SIFT, PolyPhen, Mutation taster, Mutation assessor, and Variant Effect Predictor). We evaluated allele frequencies (GnomAD), co-segregation analysis and published data. The variants were classified according to the current revised guidelines of ACMG. [17]
3. Results
3.1. Subsection
In this study we enrolled twelve Italian patients (five men, seven women) with a mean age of 59,2 ±15,4 years (range 32 – 79 years) affected by RP and carrying pathogenic mutations in the RP1 gene. Demographics and clinical data of patients included in the study are summarized in Table 1. Patients P10 and P11 are sisters. Of note visual acuity was very variable ranging from hand motion (HM) or light perception (LP) to almost normal values.
The main age at which symptoms first appeared was 31,0± 24,1 years (range 2 – 74 years) and the mean follow up period was 9,1 ± 2,4 years (range 6 – 12 years). Generally, the main symptoms at onset were hemeralopia and visual field constriction.
Most of the patients showed a classic RP fundus appearance with diffused retinal dystrophy with bone spicules and some dark clumps of pigment in the midperiphery, attenuated retinal vessels and optic disc pallor. FAF showed hypo-autofluorescent areas corresponding to the areas of RPE atrophy and in some patients a perimacular hyperautofluorescent ring at the border between the peripheral atrophic retina and the surviving area at the posterior pole. Figure 1 illustrates fundus autofluorescence imaging of a patient of our group.
Out of twelve patients (twenty-four eyes), nine eyes were pseudophakic, nine had predominantly posterior cortical cataracts and six had a lens within normal limits. OCT images showed some abnormalities in the outer retina, e.g., disrupted ellipsoid zone (EZ) and thinning of the outer nuclear layer. OCT evaluation of the macular area detected cystoid macular oedema in five eyes, an epiretinal membrane in three eyes and a macular atrophy in three eyes. Cystoid macular edema in one of our patients is shown in Figure 2.
ERG findings showed reduced or no photopic and scotopic responses, while Goldmann visual perimetry always presented with a progressive concentric constriction and sometimes with a tubular visual field. The visual field was bilaterally not evaluable in five patients with very poor visual acuity (P2, P7, P9, P10,P12).
3.2. Genetics
The sequence variants identified in our series are summarized in Table II. Most of the mutations have already been reported in the literature [11,12,18,19,20] while one novel variant [c.5753G>A (p.Cys1918Tyr) ] could be classified as likely pathogenic through the evaluation by means of the dedicated software.

Table 2.
shows the genetic characteristic of our choort of patients.
| P1 | c.2029C>T p.(Arg677*) | |
| P2 | c.5962dup p.(Ile1988Asnfs*3) | |
| P3 | c.118C>T (p.Thr373Ile) | |
| P4 | c.2219C>G p.(Ser740*) | |
| P5 | c.2219C>G p.(Ser740*) | |
| P6 | c.965G>A p.(Ser322*) | |
| P7 | c.2029C>T p.(Arg677*) | |
| P8 | c.2029C>T p.(Arg677*) | |
| P9 | c.1234dup (p.Met412Asnfs*7) | |
| P10 | c.2029C>T p.(Arg677*) | |
| P11 | c.2029C>T p.(Arg677*) | |
| P12 | c.5962dup p.(Ile1988Asnfs*3) | |
Table 2: RP1 mutations spectrum in our series.
The most prevalent mutation was the variant c.2029C>T (p.Arg677*), which was already reported as to be quite common in other studies. [21] This type of mutation lead to the production of a truncated protein without 50–70% of the C-terminal. It was reported in different populations (European, American, Asian and African). It is a recurrent mutation in Caucasian RP patients and is present in approximately 3% of cases of dominant RP in North America. [7]
In our series from eleven independent pedigrees (the patients P10 and 11 were sisters) seven patients carried an heterozygous RP1 mutation and showed some affected relatives according a dominant inheritance fashion while other five patients showed RP1 biallelic mutations (three homozygous duplications and two compound heterozygous nucleotide changes) and a pedigree compatible with a recessive mode of inheritance.
4. Discussion
In this study, we investigated the genetic and phenotypic profiles of a cohort consisting of twelve Italian retinitis pigmentosa (RP) patients carrying RP1 mutations.
On the clinical point of view most of the patients show a classic RP fundus appearance with diffused retinal dystrophy and bone spicules, attenuated vessels and pale optic disc, as shown in Figure 3; only patient (P3) presented with a higher concentration of the pigment deposit ad midperiphery along the vascular arcades. Contrarily the functional situation was quite diversified with the coexistence of milder phenotypes and very severe clinical pictures with a more or less complete loss of the visual capacity.
Some very severe clinical pictures could be observed in elderly patients (P7, P10) so we cannot exclude that their severe phenotype could be related with a longer duration of the disease. On the other side some patients presented as well with a severe phenotype at a younger age (P2, P9, P12 at 58,32 and 57 years of age respectively). Interestingly some patients with a severe visual loss report an early onset and then a long duration of the disease (P2, P9, P12 with onset at 4,12, and 6 years of age respectively).
Our data confirm the possible association of RP1 sequence variants with both autosomal and recessive mode of disease transmission. However, we could not appreciate an evident difference of the clinical course between the dominant and recessive forms of the disease, because the patients of both groups generally show at fundus examination a classic RP phenotype, with milder or more severe functional involvement
On the other side among the five patients with bilateral almost complete vision loss (visual acuity bilaterally reduced to HM or LP) two carried a heterozygous mutation (P7, P10) while the other 3 ones (patients P2, P9 and P12) carried a biallelic mutation, in two cases the c.5962dup. variant. This genotype was previously reported in some Spanish patients [20] but without providing clinical details allowing a reliable evaluation of the associated clinical pictures. Then our data do not clearly support the results of previous studies reporting a significantly more severe disease progression associated with RP1 variants in autosomal recessive forms than in autosomal dominant cases. [12] However, we have to consider that the limited number of patients included in our study prevents a definite evaluation of the clinical difference between RP1 dominant and recessive RP phenotypes.
As previously reported in other studies [7,21,22] in our series c.2029C>T (p.Arg677*) is the most prevalent RP1 mutation; interestingly among our patients this mutation is not associated with a specific geographic origin of the patients who come from different and far Italian Regions (Lombardia, Tuscany, Marche, Sicily ).
5. Conclusions
In conclusion we reported the clinical and genetic data of a group of Italian patients with the clinical diagnosis of RP associated with mutations of the RP1 gene. Our results confirm the possible association of RP1 sequence variants with both a dominant and a recessive mode of inheritance, with a certain variability of the clinical pictures in both groups.
These findings may be useful to plan the development of future therapeutic trials, including gene- therapies for RP1-related phenotypes.
Author Contributions
Writing – original draft, V.S.; Investigation, V.M. and D.P.M.; Resources, M.P., I.P., M.M. and E.P.; Data curation, D.G. and M.P.; Visualization, V.S. and I.B.; Methodology, G.V.; Validation, G.V.; Supervision, F.G.; Project administration, A.S.; Writing – review & editing, A.S. and I.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the “Patients phenotyping and genotyping and innovative treatments for retinitis pigmentosa” network project (grant number NET-2016-02363765) funded by the Italian Ministry of Health, the Tuscany Region, and the Sicily Region.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author/s.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Retinitis pigmentosa. Hamel C. Orphanet Journal of Rare Diseases.
- Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. Nguyen X, Moekotte L, […] Boon C. International Journal of Molecular Sciences. [CrossRef]
- Retinitis pigmentosa. Hartong D, Berson E, Dryja T. Lancet.
- A missense mutation in the nuclear localization signal sequence of CERKL (p.R106S) causes autosomal recessive retinal degeneration. Ali M, Ramprasad L, […]Inglehearn C. Molecular Vision (2008) 14.
- Updating the Genetic Landscape of Inherited Retinal Dystrophies. García Bohórquez B, Aller E, […] Millán J. Frontiers in Cell and Developmental Biology (2021) 9.
- Novel variants in the hotspot region of RP1 in South African patients with retinitis pigmentosa. Roberts L, Bartmann L, […] Greenberg J. Molecular Vision (2006) 12.
- Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Pierce E, Quinn T, […] Dryja T. Nature Genetics (1999) 22(3). [CrossRef]
- Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Jacobson S, Cideciyan A, […] Stone E. Investigative Ophthalmology and Visual Science (2000) 41(7).
- Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1). Berson E, Grimsby J, […] Dryja T. Investigative Ophthalmology and Visual Science (2001) 42(10).
- Novel C2orf71 mutations account for ~1% of cases in a large French arRP cohort. Audo I, Lancelot M, […] Zeitz C. Human Mutation (2011) 32(4).
- Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. Khaliq S, Abid A, […] Mehdi S. Journal of Medical Genetics (2005) 42(5). [CrossRef]
- Retinitis Pigmentosa Due to Rp1 Biallelic Variants. Silva R, Salles M, […]. Sallum J. Scientific Reports (2020) 10(1).
- Macular dystrophy and cone-rod dystrophy caused by mutations in the RP1 gene: Extending the RP1 disease spectrum. Verbakel S, Van Huet R, […] Nishiguchi K. Investigative Ophthalmology and Visual Science (2019) 60(4).
- RP1 protein truncating mutations predominate at the RP1 adRP locus. Payne A, Vithana E, […] Bhattacharya S. Investigative Ophthalmology and Visual Science (2000) 41(13).
- Differential pattern of RP1 mutations in retinitis pigmentosa. Zhang X, Chen L, […] Pang C. Molecular Vision (2010) 16.
- ISCEV Standard for full-field clinical electroretinography (2022 update). Robson A, Frishman L, […] McCulloch D. Documenta Ophthalmologica.
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Richards S, Aziz N, […] Rehm H. Genetics in Medicine (2015) 17(5).
- Molecular epidemiology in 591 italian probands with nonsyndromic retinitis pigmentosa and usher syndrome. Colombo L, Maltese P, […] Rossetti L. Investigative Ophthalmology and Visual Science (2021) 62(2). [CrossRef]
- Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Karali M, Testa F, […] Banfi S. Scientific Reports (2022) 12(1).
- Novel RP1 mutations and a recurrent BBS1 variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. Méndez-Vidal C, Bravo-Gil N, […] Antiñolo GBMC Genetics (2014) 15(1).
- Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Bowne S, Daiger S, […] Sullivan L. Human Molecular Genetics (1999) 8(11).
- A novel missense RP1 mutation in retinitis pigmentosa. Chiang S, Wang D, […] Pang C. Eye (2006) 20(5).
Figure 1.
Fundus Autofluorescence (FAF) of right and left eyes, shows symmetric areas of mottled hypo autofluorescence corresponding to the dystrophic retina, starting from the optic disc. There is also an irregular ring of hyper autofluorescence delimitating the spared retina.
Figure 1.
Fundus Autofluorescence (FAF) of right and left eyes, shows symmetric areas of mottled hypo autofluorescence corresponding to the dystrophic retina, starting from the optic disc. There is also an irregular ring of hyper autofluorescence delimitating the spared retina.
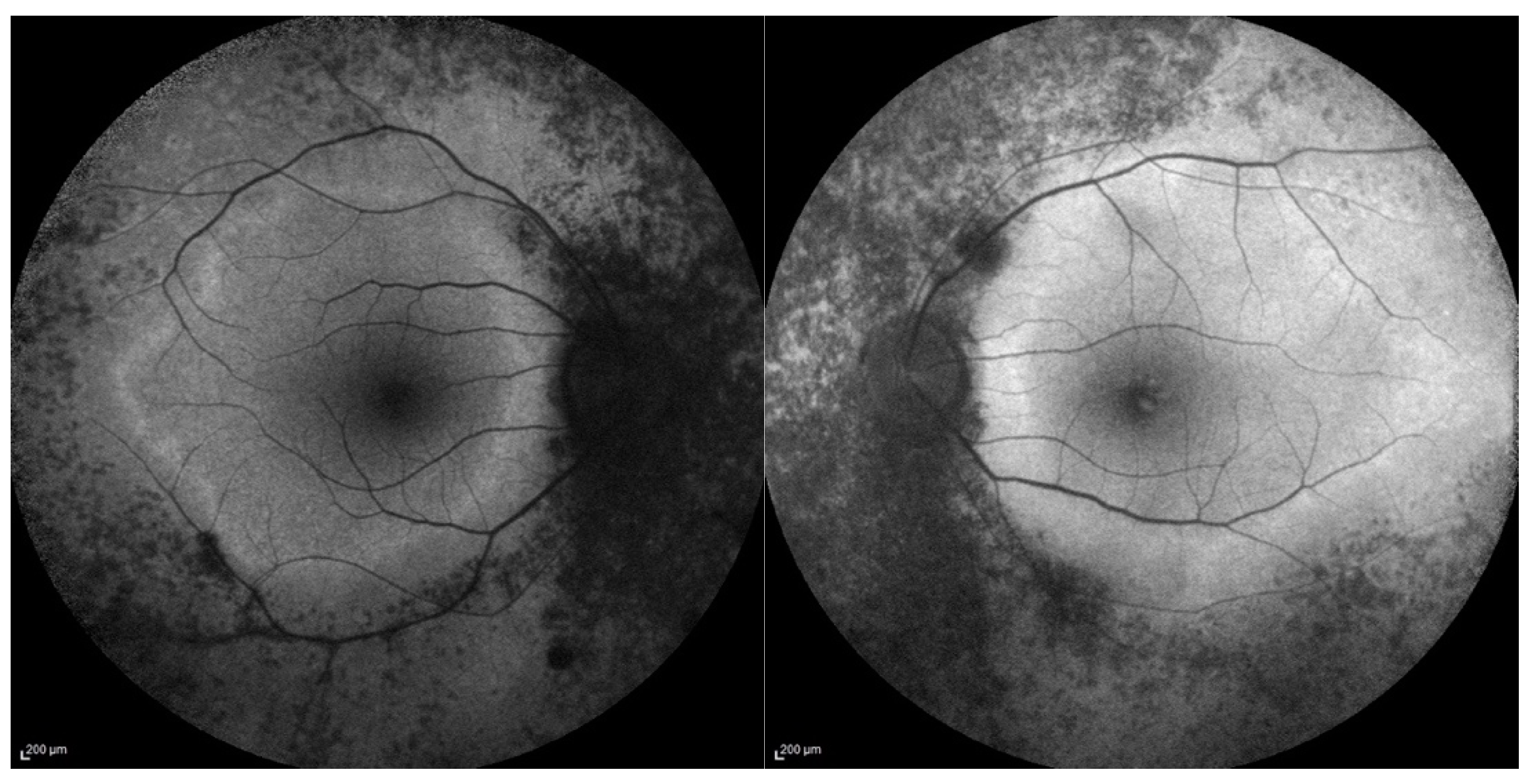
Figure 2.
SD-OCT showing a case of RP complicated by cystic macular oedema. It is possible to observe the absence of the foveal depression and IS-OS disruption with outer layer atrophy sparing only of a small subfoveal area.
Figure 2.
SD-OCT showing a case of RP complicated by cystic macular oedema. It is possible to observe the absence of the foveal depression and IS-OS disruption with outer layer atrophy sparing only of a small subfoveal area.
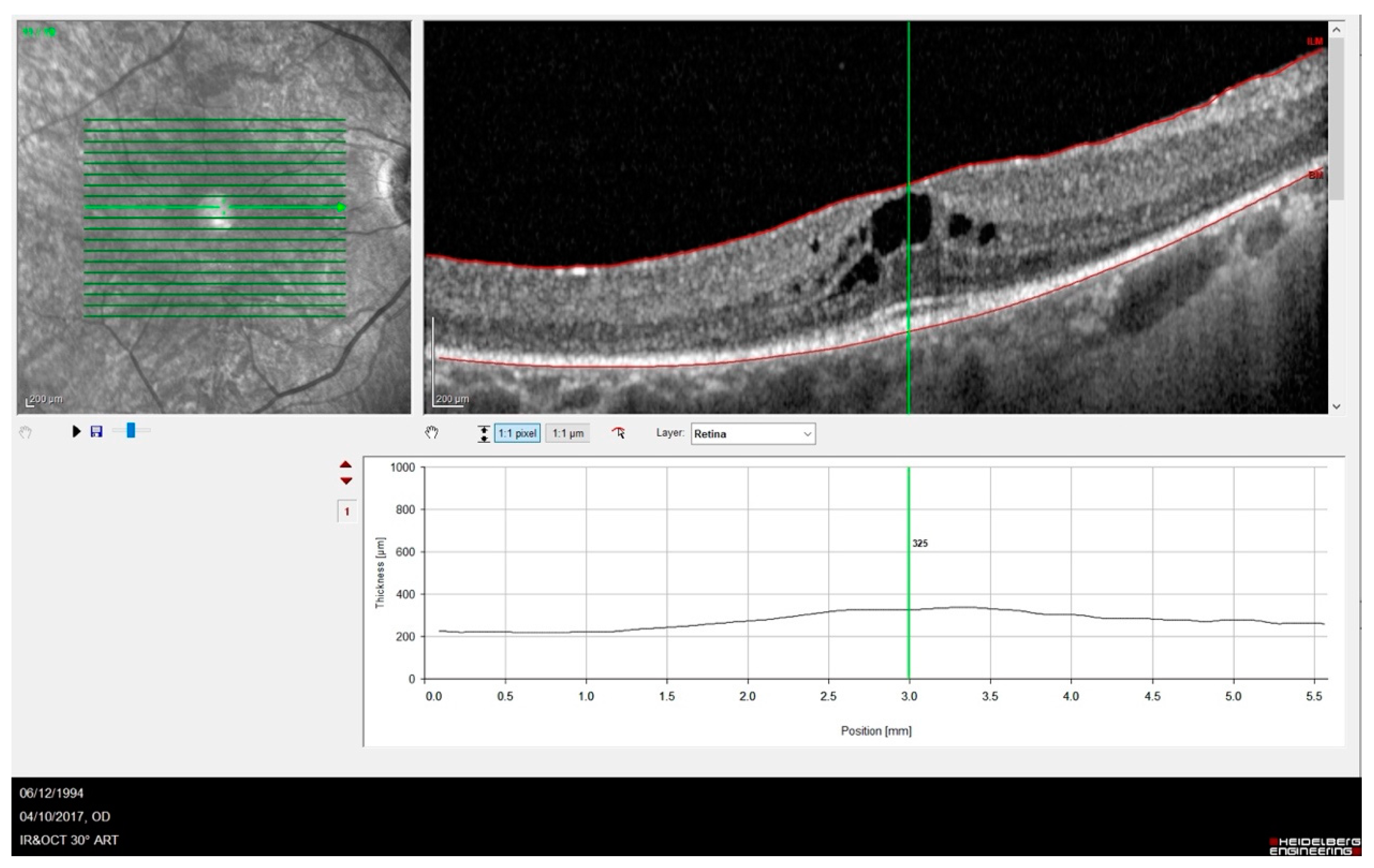
Figure 3.
RP fundus appearance of P9 with RP1 mutation.
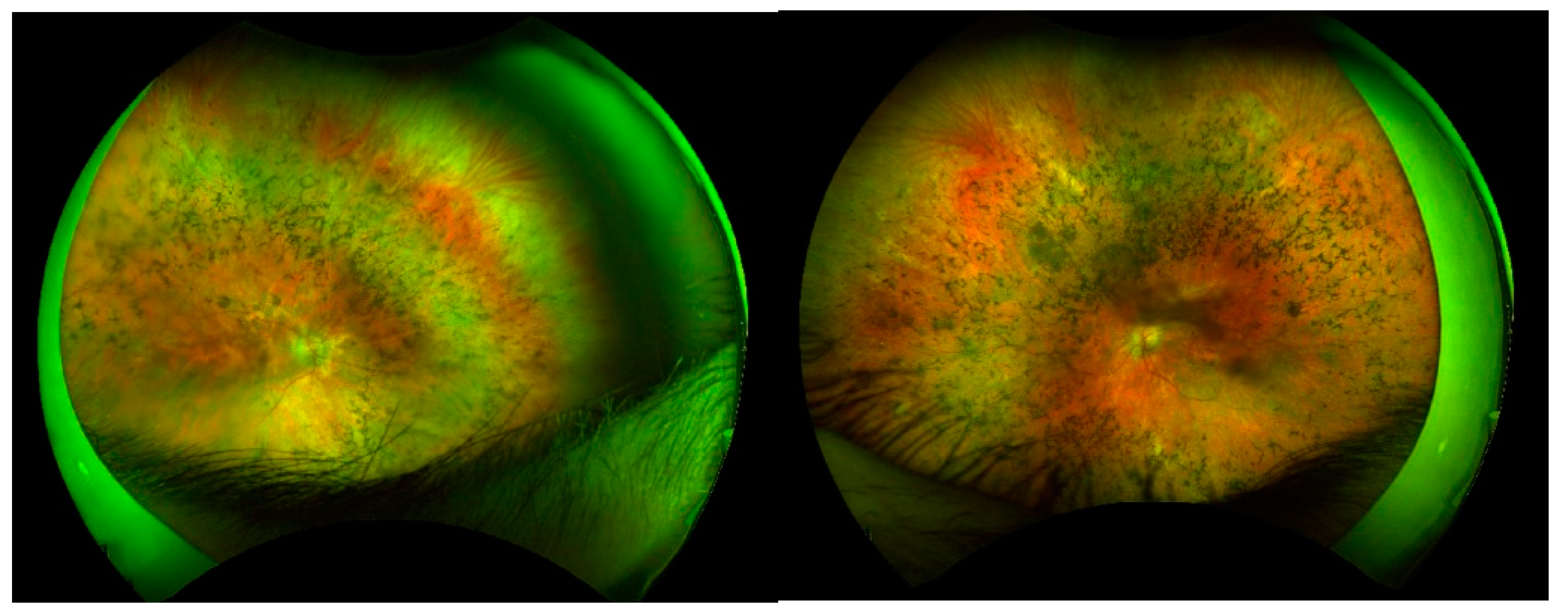
Table 1.
patients. LP (light perception), HM (hand motion).
| PATIENT | GENDER | AGE | SYMPTOMS AT TIME OF DIAGNOSIS | AGE OF SYMPTOM ONSET | BCVA LOGMAR (RE; LE) |
|---|---|---|---|---|---|
|
1 |
F | 66 | Reduced visual field | 55 | 0,1 ; 0,1 |
|
2 |
F | 58 | Hemeralopia |
4 | HM ; HM |
|
3 |
M | 62 | Hemeralopia | 30 | 1 ; 1 |
|
4 |
F | 32 | Nystagmus | 2 | HM ; 0,7 |
|
5 |
M | 60 | Hemeralopia | 43 | 0,2 ; 0,5 |
|
6 |
F | 58 | Reduced visual field | 20 | 0,15 ; 0,15 |
|
7 |
F | 77 | Hemeralopia | 47 | LP ; LP |
|
8 |
M | 52 | Reduced visual field | 48 | 0,1 ; 0,1 |
|
9 |
M | 32 | Hemeralopia | 12 | HM ; HM |
|
10 |
F | 79 | Reduced visual field | 74 | LP ; no LP |
|
11 |
F | 77 | Hemeralopia | 30 | 0,8 ; 1,0 |
|
12 |
M | 57 | Reduced visual field | 6 | LP ; LP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



