Submitted:
27 February 2024
Posted:
26 March 2024
You are already at the latest version
Abstract
Understanding clinical significance of variants of unknown significance (VUS) reported in next-generation sequencing (NGS) has become essential in cancer treatment. Our study examined six widely used in silico tools: PolyPhen-2, Align-GVGD, MutationTaster2, CADD, REVEL, and Chat-GPT. We utilized a dataset of gene variants known to potentially affect immune therapy. No single tool could comprehensively determine mutation variant pathogenicity. MutationTaster2021 showed the highest overall accuracy and MCC among the tools. Notably, REVEL and Chat-GPT exhibited 100% specificity, suggesting their proficiency in accurately identifying pathogenic variants and minimizing false positives. In contrast, CADD displayed optimal sensitivity, making it suitable for effectively ruling out benign variants.
Keywords:
solid tumor
; Artificial intelligence
; Variant classification
; Pathogenicity
; Variants of uncertain significance
Introduction
Advancements in next-generation (NGS) sequencing technology have revealed a significant number of variants of unknown significance (VUS) in cancer, lacking a clear classification regarding their impact on cancer treatment [1]. With the integration of immunotherapy as a standard treatment, understanding the influence of these variants on immunotherapy outcomes has become increasingly imperative [2,3]. Numerous in silico tools have been developed to categorize these variants based on their pathogenicity and to provide insight into their clinical actionability [4,5,6]. This study evaluates the performance of six widely utilized in silico tools in predicting the pathogenicity of functional variants associated with differential immunotherapy outcome in cancer.
The in silico tools evaluated in this study have been developed utilizing AI (artificial intelligence)-based algorithms to predict whether specific variants of certain genes contribute to disease pathogenicity. ACMG Standards and Guidelines recommends researchers to exercise caution when using these in silico tools to predict the classification of genetic variants, and there is no standard method of using these tools [7]. Generally, these tools are used to predict where a given sequence variant falls along the spectrum of benign to pathogenic in its phenotypic effect.
It is suggested that the performance of the tools used to predict the pathogenicity of clinically actionable variants in solid tumor is not fully reliable. Further research needs to be done in order to increase the reliability of NGS in identifying the pathogenicity of clinically actionable variants. Among the tools available, broadly, there are three different AI-based approaches. One approach uses evolutionary principles, classifying variants based on multiple sequence alignments, preservation of the amino acid in the protein of interest, and allele frequency. DANN [8], PrimateAI [9], and PANTHER [10] are examples of tools that utilize such methods. Another approach analyzes the influence of variants on protein properties including polarity, charge, genomic location in relation to functional regions, and 3-D structure, and thus the protein function. Tools that utilize protein-structure/function analysis include iStable [11], SNPs&GO [12], and Mutpred2 [13]. Widely used in silico tools use a combined method for variant classification, as seen in PolyPhen-2 [14], Align-GVGD [15], and MutationTaster2021 [16]. For prediction of nsSNPs, recent advancements have begun to analyze changes in splice sites, chromatin effects, and patterns in regulatory motifs. Examples include DeepSEA [17], NetGene2 [18], and DanQ [19]. Meta-predictors including Revel [20], BayesDel [21], and CADD [22] integrate multiple classifiers and have outperformed traditional, individual in silico tools [23,24].
Attaining information regarding the performance and accuracy of in silico tools is critical as accuracy of these metrics allow direct implications on clinical management of patients with VUS. While studies have demonstrated variable results for performance of in silico tools, recent studies present a need for further research in a clinical context [25,26]. Of note, accuracy of in silico tools have not been evaluated for variants that impact immunotherapy. The aim of this study is to analyze the performance of six in silico tools for predicting the pathogenicity of 160 missense variants of immune-related genes associated with immunotherapy outcomes for solid tumors.
Materials and Methods
Variant Selection
We selected genes of interest for evaluation by conducting a literature search on PubMed. Our criteria for inclusion were genes that had evidence of clinical or preclinical significance in influencing immunotherapy response. The initial data set comprised 180 variants of POLE, STK11, PTEN, KEAP1, SMAD4, SMARC4, TP53, PTEN, and CDKN2A genes. Pathogenic variants were included based on a confirmed number of at least two annotations that specifically indicated pathogenicity in OncoKB, Cancer hotspots, CIViC, AACR Project GENIE and My Cancer Genome [27], MCG; mycancergenome.org]. The top ten variants with the highest frequencies were compared with ClinVar. Benign variants were curated with the same inclusion criteria and ClinVar assertion. Genes without 10 variants that had at least two annotations required variants with one annotation to be included based on their order listed in cBioPortal database. These sources were used to define a true classification for the variants in our dataset. A final dataset of 160 pathogenic (n=80) and benign (n=80) NSCLC variants was used for analysis.
In Silico Classification Tool Selection
Chat-GPT; PolyPhen-2 [14], Align-GVGD [15], MutationTaster2021 [16], CADD [23], and REVEL [20] were the in silico classification tools evaluated in this study. These tools except Chat-GPT were selected primarily based on inclusion in the ACMG Standard and Guidelines [6] and common use based on literature reviews. CADD and REVEL are meta-predictors that incorporate a combination of individual scores to classify variants, while Chat-GTP;Polyphen-2, Align-GVGD, and MutationTaster2021 are individual tools for pathogenicity prediction. CADD combines 60 distinct annotations including Ensembl Variant Effect Predictor (VEP), phyloP, phastCons, GERP++, Grantham, SIFT and Polyphen-2. REVEL integrates scores from MutPred. FATHMM v2.3, VEST 3.0, PolyPhen-2, SIFT, PROVEAN, MutationAssessor, MutationTaster, LFT, GERP++, SiPhy, phyloP, and phastCOns–included in this study based on its exceptional performance in a recent study by Tian et al. [28]. MutationTaster2021, the latest update to MutationTaster, was employed for this study due to its improved prediction model that attains higher accuracy [16].
Parameter Setting
Default thresholds suggested by tools’ authors were implemented for classification of variants. Tools determining pathogenicity that present numerical scores were differentiated as score <0.05 for Polyphen-2, ≥C35 for Align-GVGD, >15 for CADD, and >0.05 for REVEL. MutationTaster provided categorical outputs for classified variants represented as Deleterious, Deleterious (ClinVar), Benign, or Benign (auto).
Evaluation of Performance
Each in silico tool classified each missense variant in the final dataset. True positive (TP) results refer to the correct prediction of pathogenicity, and true negative (TN) results refer to the correct prediction of benign variants. These were defined in our final dataset using the criteria aforementioned. The following measures were obtained for each tool: overall accuracy (OA = ), sensitivity (Sn = ), specificity (Sp = ), and Matthews correlation coefficient (MCC = ) were calculated.
Sensitivity (true positive rate) measured the ability of each in silico tool to account for true pathogenic variants. Specificity (true negative rate) was reflective of each tool’s ability to account for true benign variants. Some results were excluded from the study and recorded as errors due to reasons including yielding error in calling predictions and conflicting predictions within the same tool. MCC contributed a metric for fair comparison between different sample sizes per tool and the disproportionate samples between pathogenic and benign variants in the dataset. MCC values range from -1 (always false) to +1 (always correct), with a value of 0 indicating total random classification.
A quality check was performed for variants yielding no results, “error” results, or had conflicting interpretations within the same tool. An additional 10 variants were randomly selected for quality check and analyzed using each in silico tool. All variants were assessed again for PolyPhen-2 (HumDiv) and PolyPhen-2 (HumVar) due to discrepancies identified in the randomized quality check.
Characteristics of Selected Variants
The final dataset of 160 variants (80 pathogenic, 80 benign) was generated by removing variants with algorithmic interpretation errors or conflict. Notably, the number of variants for SMARCA4 and KEAP1 was fewer compared to other genes, due to a lack of references regarding their pathogenicity and errors in the computational tools. We selected pathogenic variants by focusing on single nucleotide variants with high frequencies for each gene in Cbioportal AACR genie. The same method was used to select benign variants.
Result
Overall Performance
The overall performances of the in silico tools are highlighted in Table 1. To assess the overall performance of each algorithm, we examined metrics including accuracy, sensitivity, specificity, PPV, NPV, and MCC using the 160 variants from our study. Out of the tools assessed, Mutation Taster 2021 displayed the best accuracy (0.83) and MCC (0.69). On the other hand, Align-GVGD reported the least accuracy (0.52) and the lowest MCC (0.06), while other tools recorded scores between 0.36 and 0.69. Interestingly, both Chat-GPT and REVEL had a perfect specificity and positive predictive value but lagged in negative predictive value, scoring 0.22. Conversely, CADD had the highest sensitivity and negative predictive value, both at 1.00, but its specificity was the lowest at 27.50%. Out of 8 genes, REVEL showed highest accuracy in 6 genes (STK11, TP53, PTEN, POLE, EZH2, CDKN2A).
Single-Gene Analysis
<TP53>
Mutation Taster 2021, Chat-GPT, and Polyphen showed the highest accuracy (90%). Align-GVGD and Chat-GPT demonstrated 100% specificity. (Figure 1, Table 2)
<PTEN>
Mutation Taster 2021 and REVEL showed 100% accuracy. All the tools except Chat-GPT and Ailgn-GVGD demonstrated 100% sensitivity for PTEN variants. Chat-GPT and Align-GVGD showed 100% specificity. (Figure 1, Table 2)
<SMARCA4>
100% sensitivity was shown for CADD and Align-GVGD. Notably, Chat-GPT showed 100% specificity. (Figure 1, Table 2)
<SMAD4>
All the tools except Chat-GPT and Ailgn-GVGD demonstrated 100% sensitivity for SMAD4 variants. Chat-GPT and Align-GVGD showed 100% specificity. (Figure 1, Table 2)
<KEAP1>
Study only included 3 variants in KEAP1 gene due to insufficient number of references on Clinvar and Cbioportal. (Figure 1, Table 2)
<STK11>
Polyphen, CADD, REVEL and Mutation Tater had sensitivity of 100%. REVEL and Mutation Taster 2021 showed 100% Accuracy. (Figure 1, Table 2)
<POLE>
REVEL, Chat-GPT, Mutation Taster 2021, and Align-GVGD performed with 100% specificity. CADD showed 100% sensitivity. (Figure 1, Table 2)
<EZH2>
REVEL and Mutation Taster 2021 showed 100% Accuracy while Chat-GPT showed 100% specificity. (Figure 1, Table 2)
<CDKN2A>
CADD and Mutation Taster 2021 showed 100% sensitivity. 100% specificity was shown in REVEL and Chat-GPT. REVEL showed the highest accuracy of 95% (Figure 1, Table 2)
In this study, we analyzed the performance of 6 in-silico tools including Chat-GPT for determining the pathogenicity of 160 unidentified missense variants present in immune-related genes associated with immunotherapy outcomes for solid tumors. To the best of our understanding, it is the first study to evaluate the potential clinical impacts of in-silico tools in the field of immuno-oncology. Furthermore, we integrated Chat-GPT into our in-silico toolkit, leveraging its capabilities for extensive analysis.
Single Algorithm Predictors: Polyphen-HumDiv, CADD, Mutation Taster 2021, Align-GVGD
Interestingly, CADD, MutationTaster 2021, and Polyphen-HumDiv showcased potential to identify non-pathogenic variants with sensitivity values of 1.00, 0.9375, and 0.8625, and negative predictive values of 1.00, 0.9219, and 0.8429, respectively. Polyphen-HumDiv and MutationTaster 2021 obtained high score for specificity and positive predictive value as well. Their specificities of 0.7375 and positive predictive value of 0.7667 and 0.7813 respectively were comparably higher than those for CADD. Previous studies agree with these findings. In past studies, PolyPhen-2 version demonstrated high sensitivity and specificity values, with one study presenting 100% sensitivity in prediction of pathogenicity of BRCA2 variants [29,30]. In other studies, MutationTaster 2021 performed exceptionally well in identifying pathogenicity and was ranked with better accuracy for sensitivity and specificity compared to PolyPhen2, which concur with aforementioned data [31,32].
Moreover, while CADD performs exceptionally well in ruling out benign ones, it may not be the best at identifying pathogenic variants. CADD's superior performance to identify nonpathogenic variants due to its large training set was explained in previous studies [33]. Furthermore, past studies also are consistent with CADD’s limitations. In one study, an unknown proportion of proxy-deleterious variants were concluded as neutral, indicating probable low positive rate. Another study concluded that CADD had the lowest specificity relative to other in silico tools [34].
Align-GVGD had the lowest overall accuracy of 0.5250 of the six predictors evaluated. It is worth mentioning that our findings resonate with a prior study which also underscored the subpar performance of Align-GVGD in comparison to other predictors, though they stand in opposition to the conclusions drawn in other research [26,27]. Some studies indicated high sensitivity, compared to one study that suggested significantly low sensitivity with greatest variability in Align-GVGD compared to PolyPhen-2 and SIFT [35,36,37]. Further study may provide more solid evidence on these tools’ performances to identify pathogenic and benign variants relative to each other.
Meta-Predictor: REVEL
Our investigation revealed the compelling performance of REVEL, demonstrating high specificity and PPV. This indicates its ability to minimize the likelihood of misclassifying pathogenic variants. REVEL surpassed all other meta-predictors, correctly identifying the variants as disease-causing with a high level of accuracy. This result is consistent with previous studies as REVEL utilizes an optimized amalgamation of 13 distinct in silico tools, trained using newly identified pathogenic and rare neutral missense variants [38,39,40]. However, certain disparities emerged in terms of REVEL's overall accuracy in our findings, compared to another study. While previous investigations have consistently shown REVEL's highest overall performance over tools including for CADD, our result suggests that REVEL might not be as accurate in predicting pathogenesis of the specific genes in this study.
Furthermore, another study indicated that REVEL exhibited its ability to predict gain of function (GoF) variants with greater accuracy than loss of function (LoF), which was manifested by 98% of LoF and 100% of GoF meeting the recommended REVEL threshold of 0.5 for pathogenicity [41]. Therefore, further investigation in the distinctive attributes of these mechanisms and their influence on REVEL’s predictions could provide invaluable insights for enhancing the precision of its predictive functionalities.
Generative AI: Chat-GPT
Chat-GPT is a relatively recent chatbot that was developed using a huge amount of textual data from the internet. The evaluation of ChatGPT among other tools in this study carries significance, as this is the first study to use ChatGPT for variant classification. In our study, Chat-GPT demonstrated perfect specificity and PPV, both attaining a score of 1.00; however, the model displayed a lower sensitivity of 0.2250 and NPV of 0.5634. This indicates that ChatGPT may erroneously classify some certain pathogenic variants as benign, yet underscores its usefulness in confirming the pathogenicity of variants categorized as of unknown significance. This result highlights the ability of ChatGPT to assist in the identification of potentially pathogenic genetic changes.
Although Chat-GPT, originally a language model, was not intended for the assessment of variant classification, its impressive performance in this research opens up a new possibility. The generative AI’s ability to exclude certain pathogenic variants as benign and confirm the pathogenicity of VUS showcases its utility in the field of molecular genetics. It also suggests the possibility of more machine learning models being trained to predict the pathogenicity of genetic variants, perhaps with superior sensitivity and specificity. Further research needs to be done to fully assess the clinical applicability of ChatGPT in variant classification.
Limitations
Several variants were omitted from our study due to limitations in certain algorithms' assessment capabilities. Errors in variant assessment may arise from a variety of reasons. Tools are often limited in the types of mutations they can evaluate.
While prediction software commonly utilizes machine-learning algorithms and is validated with variants from public databases including ExAC/gnomAD, ClinVar10, and SwissProt21, inherent biases from standardized datasets can lead to overfitting and false concordance. It has been previously shown that prediction algorithms have variable performance when applied to different datasets. Therefore the use of variant datasets derived from online public databases may not be representative of the performance of tools when applied in a clinical setting.
Variant predictors frequently rely on diverse external data, necessitating cross-referencing gene identifiers across databases. For example, for Mutation Taster, external data sometimes draws from NCBI and at other times from Ensembl; both are essential for pathogenicity predictions in Mutation Taster. This dichotomy can introduce assessment errors.
Our study also encounters certain limitations owing to the varied update timelines of each tool involved. The Chat-GPT tool received its latest update in September 2021. Meanwhile, The MutationTaster2021, which was launched on 07/02/2021, serves as a recent upgrade to the preceding MutationTaster2, and is claimed to exhibit enhanced accuracy compared to its predecessor, according to its developers [16]. On the other hand, as mentioned on their official website, Align-GVGD has not been updated since September 8, 2014.
Our study focused primarily on missense mutations. Some in silico tools are capable of analyzing mutations beyond missense mutations - DANN and DeepSEA. Presently, no algorithm can evaluate all mutation classes, highlighting the need for a versatile in silico tool that accurately classifies pathogenicity across different mutation types.
Conclusion
Despite its limitations, our study offers insights that have the potential to shape the future of immunotherapy. To begin with, our research stands out due to its exclusive focus on missense mutations within immunotherapy-related genes. Moreover, our study provides evidence regarding the accuracy of in silico tools in assessing the pathogenicity of variants classified as of unknown significance (VUS). In the evaluation, MutationTaster2021 surpassed CADD, Align-GVGD, REVEL, and Chat-GPT in terms of overall precision, negative predictive valuation, and Matthew's Correlation Coefficient. Conversely, when focusing on the analysis of individual genes, depicted in Table 2 and Figure 1, REVEL emerged as the most accurate tool in predicting six out of the eight scrutinized genes (STK11, SMAD4, PTEN, POLE, EZH2, CDKN2A). In stark contrast, Align-GVGD lagged, recording the lowest efficacy both generally and in six out of the eight analyzed genes (TP53, SMARCA4, SMAD4, PTEN, POLE, EZH2, CDKN2A). Furthermore, our study introduces an innovative application of ChatGPT in variant classification, introducing its potential role in aiding immunotherapy efforts. Our findings indicate that ChatGPT achieves notable specificity and positive predictive value (PPV). Consequently, we can suggest which tools to employ when confirming or ruling out the pathogenicity of VUS. ChatGPT excels in confirming positive results, while CADD proves valuable in affirming negative outcomes. In conclusion, our study suggests that utilizing in silico algorithms can aid in making treatment decisions including in the field of immuno-oncology.
Data Availability
The data generated in this study are available upon request from the corresponding author.
Financial Support
Not applicable.
Conflicts of Interest
The authors declare no potential conflicts of interest. Acknowledgements: Not applicable.
References
- Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nature Reviews Genetics. 2011;12(9):628-40.
- Lier A, Penzel R, Heining C, Horak P, Fröhlich M, Uhrig S, et al. Validating Comprehensive Next-Generation Sequencing Results for Precision Oncology: The NCT/DKTK Molecularly Aided Stratification for Tumor Eradication Research Experience. JCO Precision Oncology. 2018(2):1-13.
- Volckmar A-L, Leichsenring J, Kirchner M, Christopoulos P, Neumann O, Budczies J, et al. Combined targeted DNA and RNA sequencing of advanced NSCLC in routine molecular diagnostics: Analysis of the first 3,000 Heidelberg cases. International Journal of Cancer. 2019;145(3):649-61.
- Casey RT, McLean MA, Madhu B, Challis BG, Ten Hoopen R, Roberts T, et al. Translating in vivo metabolomic analysis of succinate dehydrogenase deficient tumours into clinical utility. JCO precision oncology. 2018;2:1-12.
- van der Velden DL, van Herpen CML, van Laarhoven HWM, Smit EF, Groen HJM, Willems SM, et al. Molecular Tumor Boards: current practice and future needs. Annals of Oncology. 2017;28(12):3070-5.
- Volckmar A-L, Christopoulos P, Kirchner M, Allgäuer M, Neumann O, Budczies J, et al. Targeting rare and non-canonical driver variants in NSCLC – An uncharted clinical field. Lung Cancer. 2021;154:131-41.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015;17(5):405-23.
- Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2014;31(5):761-3.
- Sundaram L, Gao H, Padigepati SR, McRae JF, Li Y, Kosmicki JA, et al. Predicting the clinical impact of human mutation with deep neural networks. Nature Genetics. 2018;50(8):1161-70.
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nature Protocols. 2019;14(3):703-21.
- Chen C-W, Lin M-H, Liao C-C, Chang H-P, Chu Y-W. iStable 2.0: Predicting protein thermal stability changes by integrating various characteristic modules. Computational and Structural Biotechnology Journal. 2020;18:622-30.
- Capriotti E, Calabrese R, Fariselli P, Martelli PL, Altman RB, Casadio R. WS- SNPs&GO: a web server for predicting the deleterious effect of human protein variants using functional annotation. BMC Genomics. 2013;14(3):S6.
- Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, et al. MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv. 2017:134981.
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248-9.
- Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics. 2006;43(4):295-305.
- Steinhaus R, Proft S, Schuelke M, Cooper DN, Schwarz Jana M, Seelow D. MutationTaster2021. Nucleic Acids Res. 2021;49(W1):W446-W51.
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning–based sequence model. Nature Methods. 2015;12(10):931-4.
- Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24(17):3439-52.
- Quang D, Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016;44(11):e107-e.
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99(4):877-85.
- Feng B-J. PERCH: A Unified Framework for Disease Gene Prioritization. Human Mutation. 2017;38(3):243-51.
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics. 2014;46(3):310-5.
- Gunning AC, Fryer V, Fasham J, Crosby AH, Ellard S, Baple E, et al. Assessing performance of pathogenicity predictors using clinically-relevant variant datasets. bioRxiv. 2020:2020.02.06.937169.
- Leong IUS, Stuckey A, Lai D, Skinner JR, Love DR. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med Genet. 2015;16:34-.
- Ernst C, Hahnen E, Engel C, Nothnagel M, Weber J, Schmutzler RK, et al. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med Genomics. 2018;11(1):35-.
- Kerr ID, Cox HC, Moyes K, Evans B, Burdett BC, van Kan A, et al. Assessment of in silico protein sequence analysis in the clinical classification of variants in cancer risk genes. Journal of Community Genetics. 2017;8(2):87-95.
- Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precision Oncology. 2017(1):1-16.
- Tian Y, Pesaran T, Chamberlin A, Fenwick RB, Li S, Gau C-L, et al. REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Scientific Reports. 2019;9(1):12752.
- Zimbru CG, Nicoleta Andreescu, Albu A, Chirita-Emandi A, Stanciu A, Puiu M. Performance Evaluation of in Silico Predictors for the Classification of ClinVar Variants. 2019 Nov 1;
- Poon KS. In silico analysis of BRCA1 and BRCA2 missense variants and the relevance in molecular genetic testing. Scientific Reports. 2021 May 27;11(1).
- Li J, Zhao T, Zhang Y, Zhang K, Shi L, Chen Y, et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Research. 2018 Jul 8;46(15):7793–804.
- Chen Q, Dai C, Zhang Q, Du J, Li W. [Evaluation of performance of five bioinformatics software for the prediction of missense mutations]. PubMed. 2016 Oct 1;33(5):625–8.
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research [Internet]. 2018 Oct 29 [cited 2019 Mar 20];47(D1):D886–94. Available from: https://academic.oup.com/nar/article/47/D1/D886/5146191.
- Niroula A, Vihinen M. How good are pathogenicity predictors in detecting benign variants? Panchenko ARR, editor. PLOS Computational Biology. 2019 Feb 11;15(2):e1006481.
- Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Human Mutation. 2011 Apr 7;32(6):661–8.
- Fortuno C, James PA, Young EL, Feng B, Olivier M, Pesaran T, et al. Improved, ACMG-compliant, in silico prediction of pathogenicity for missense substitutions encoded by TP53 variants. Human Mutation. 2018 Jun 5;39(8):1061–9.
- Ernst C, Hahnen E, Engel C, Nothnagel M, Weber J, Schmutzler RK, et al. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Medical Genomics. 2018 Mar 27;11(1).
- Tian Y, Pesaran T, Chamberlin A, Fenwick RB, Li S, Gau C-L, et al. REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Scientific Reports. 2019;9(1):12752.
- Fao G, Es de A, Ei P. Insights on variant analysis in silico tools for pathogenicity prediction. Frontiers in genetics [Internet]. 2022 Nov 29;13. Available from: https://pubmed.ncbi.nlm.nih.gov/36568376/.
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. American Journal of Human Genetics [Internet]. 2016 Oct 6;99(4):877–85. Available from: https://pubmed.ncbi.nlm.nih.gov/27666373.
- Hopkins JJ, Wakeling MN, Johnson M, Flanagan SE, Laver TW. REVEL is better at predicting pathogenicity of loss-of-function than gain-of-function variants. medRxiv (Cold Spring Harbor Laboratory). 2023 Jun 7;
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. American Journal of Human Genetics [Internet]. 2016 Oct 6;99(4):877–85. Available from: https://pubmed.ncbi.nlm.nih.gov/27666373.
Figure 1.
Single-gene analysis in graphics.
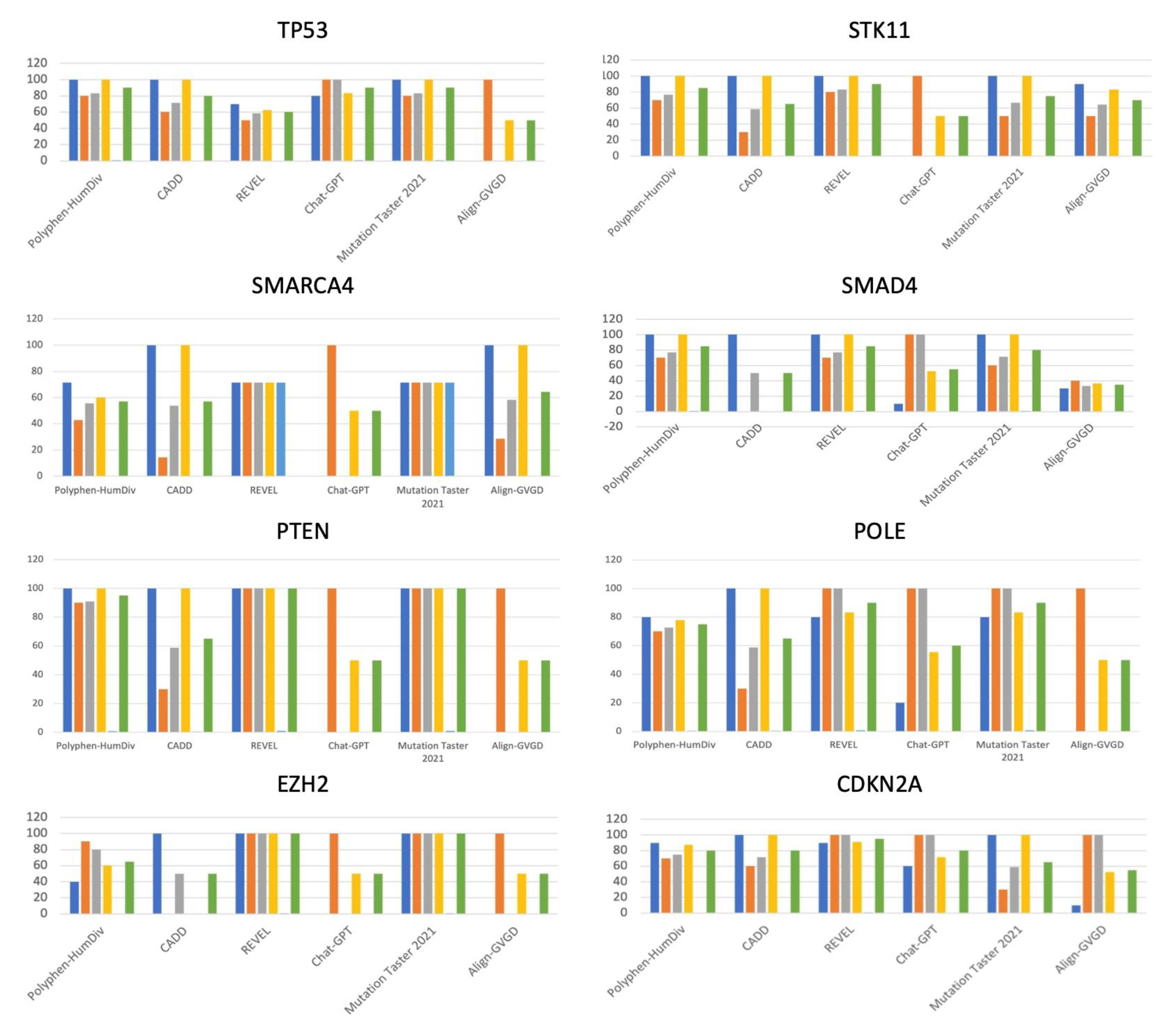

Table 1.
Integrated Gene Analysis. We have calculated six indicators (sensitivity, specificity, positive predictive value, negative predictive value, Matthew's correlation coefficient, overall accuracy) using the entire variants of all genes as samples, and displayed them in this table.
Table 1.
Integrated Gene Analysis. We have calculated six indicators (sensitivity, specificity, positive predictive value, negative predictive value, Matthew's correlation coefficient, overall accuracy) using the entire variants of all genes as samples, and displayed them in this table.
| Polyphen-HumDiv | CADD | Mutation Taster 2021 | Align-GVGD | REVEL | Chat-GPT | |
|---|---|---|---|---|---|---|
| Sensitivity | 86.25 | 100.00 | 93.75 | 28.75 | 22.50 | 22.50 |
| Specificity | 73.75 | 27.50 | 73.75 | 76.25 | 100.00 | 100.00 |
| Positive Predictive Value | 76.67 | 57.97 | 78.13 | 54.76 | 100.00 | 100.00 |
| Negative Predictive Value | 84.29 | 100.00 | 92.19 | 51.69 | 56.34 | 56.34 |
| MCC | 0.60 | 0.40 | 0.69 | 0.06 | 0.36 | 0.36 |
| Overall accuracy | 80.00 | 63.75 | 83.75 | 52.50 | 61.25 | 61.25 |
Table 2.
Single Gene Analysis. We have separately calculated six indicators (sensitivity, specificity, positive predictive value, negative predictive value, Matthew's correlation coefficient, overall accuracy) to evaluate each gene and have recorded their respective values in this table.
Table 2.
Single Gene Analysis. We have separately calculated six indicators (sensitivity, specificity, positive predictive value, negative predictive value, Matthew's correlation coefficient, overall accuracy) to evaluate each gene and have recorded their respective values in this table.
| TP53 | ||||||
|---|---|---|---|---|---|---|
| Polyphen-HumDiv | CADD | REVEL | Chat-GPT | Mutation Taster 2021 | Align-GVGD | |
| Sensitivity | 100 | 100 | 70 | 80 | 100 | 0 |
| Specificity | 80 | 60 | 50 | 100 | 80 | 100 |
| Positive Predictive Value | 83.3 | 71.43 | 58.33 | 100 | 83.3 | N/A |
| Negative Predictive Value | 100 | 100 | 62.5 | 83.3 | 100 | 50 |
| MCC | 0.82 | 0.65 | 0.2 | 0.82 | 0.82 | N/A |
| Overall accuracy | 90 | 80 | 60 | 90 | 90 | 50 |
| STK11 | ||||||
| Sensitivity | 100 | 100 | 100 | 0 | 100 | 90 |
| Specificity | 70 | 30 | 80 | 100 | 50 | 50 |
| Positive Predictive Value | 76.92 | 58.82 | 83.33 | N/A | 66.67 | 64.29 |
| Negative Predictive Value | 100 | 100 | 100 | 50 | 100 | 83.33 |
| MCC | 0.73 | 0.42 | 0.82 | N/A | 0.58 | 0.44 |
| Overall accuracy | 85 | 65 | 90 | 50 | 75 | 70 |
| SMARCA4 | ||||||
| Sensitivity | 71.43 | 100 | 71.43 | 0 | 71.43 | 100 |
| Specificity | 42.86 | 14.29 | 71.43 | 100 | 71.43 | 28.57 |
| Positive Predictive Value | 55.56 | 53.85 | 71.43 | N/A | 71.43 | 58.33 |
| Negative Predictive Value | 60 | 100 | 71.43 | 50 | 71.43 | 100 |
| MCC | 0.15 | 0.28 | 71.43 | N/A | 71.43 | 0.41 |
| Overall accuracy | 57.14 | 57.14 | 0.43 | 50 | 0.43 | 64.29 |
| SMAD4 | ||||||
| Sensitivity | 100 | 100 | 100 | 10 | 100 | 30 |
| Specificity | 70 | 0 | 70 | 100 | 60 | 40 |
| Positive Predictive Value | 76.92 | 50 | 76.92 | 100 | 71.43 | 33.33 |
| Negative Predictive Value | 100 | N/A | 100 | 52.63 | 100 | 36.36 |
| MCC | 0.73 | N/A | 0.73 | 0.23 | 0.65 | -0.3 |
| Overall accuracy | 85 | 50 | 85 | 55 | 80 | 35 |
| PTEN | ||||||
| Sensitivity | 100 | 100 | 100 | 0 | 100 | 0 |
| Specificity | 90 | 30 | 100 | 100 | 100 | 100 |
| Positive Predictive Value | 90.91 | 58.82 | 100 | N/A | 100 | N/A |
| Negative Predictive Value | 100 | 100 | 100 | 50 | 100 | 50 |
| MCC | 0.9 | 0.42 | 1 | N/A | 1 | N/A |
| Overall accuracy | 95 | 65 | 100 | 50 | 100 | 50 |
| POLE | ||||||
| Sensitivity | 80 | 100 | 80 | 20 | 80 | 0 |
| Specificity | 70 | 30 | 100 | 100 | 100 | 100 |
| Positive Predictive Value | 72.73 | 58.82 | 100 | 100 | 100 | N/A |
| Negative Predictive Value | 77.78 | 100 | 83.33 | 55.56 | 83.33 | 50 |
| MCC | 0.5 | 0.42 | 0.82 | 0.33 | 0.82 | N/A |
| Overall accuracy | 75 | 65 | 90 | 60 | 90 | 50 |
| EZH2 | ||||||
| Sensitivity | 40 | 100 | 100 | 0 | 100 | 0 |
| Specificity | 90 | 0 | 100 | 100 | 100 | 100 |
| Positive Predictive Value | 80 | 50 | 100 | N/A | 100 | N/A |
| Negative Predictive Value | 60 | N/A | 100 | 50 | 100 | 50 |
| MCC | 0.35 | N/A | 1 | N/A | 1 | N/A |
| Overall accuracy | 65 | 50 | 100 | 50 | 100 | 50 |
| CDKN2A | ||||||
| Sensitivity | 90 | 100 | 90 | 60 | 100 | 10 |
| Specificity | 70 | 60 | 100 | 100 | 30 | 100 |
| Positive Predictive Value | 75 | 71.43 | 100 | 100 | 58.82 | 100 |
| Negative Predictive Value | 87.5 | 100 | 90.91 | 71.43 | 100 | 52.63 |
| MCC | 0.61 | 0.65 | 0.9 | 0.65 | 0.42 | 0.23 |
| Overall accuracy | 80 | 80 | 95 | 80 | 65 | 55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



