Submitted:
03 February 2024
Posted:
05 February 2024
You are already at the latest version
Abstract
Porcine sapovirus (PoSaV) is a significant pathogen causing diarrhea with high morbidity and mortality rates. Given its zoonotic characteristics, this study conducts a comprehensive genetic evolutionary analysis of PoSaV. Results confirmed PoSaV as the primary RNA virus in piglet feces (27.54%). Phylogenetic analysis identifies the prevalent PoSaV strain SHCM/Mega2023 in the GIII genogroup, involving a recombinant event with MK962338 and KT922089. The time tree revealed that the GIII genogroup exhibits the widest divergence time span, indicating a high likelihood of viral recombination. Further selective pressure calculations demonstrate that PoSaV was under purifying selection (dN/dS < 1), with seven positively selected sites in VP1 protein which might be related to the antigencity. In conclusion, this study presents a novel genomic evolution of PoSaV, offering valuable insights into antigenicity and vaccine research.
Keywords:
Megagenomic sequencing
; porcine sapovirus
; genomic evolution
; recombination
; selective pressure
1. Introduction
Sapovirus (SaV), a member of the Caliciviridae family alongside Norovirus, Lagovirus, and Vesivirus, stands out as a leading cause of acute gastroenteritis in humans, especially among younger children[1]. SaVs, with a non-enveloped structure, possess a linear genome comprising polyadenylated, single-stranded, positive-sense RNA. The SaV genome features two primary open reading frames (ORFs): ORF1 encodes nonstructural proteins VP1, while ORF2 encodes the minor protein VP2 with an unknown function[2]. Genetically diverse, SaVs fall into 19 genogroups[3]. SaVs have been identified globally[4,5,6] and also in various species such as swine[7], mink[8], cows[9], dogs[10], and bats[11]. Therefore, it is essential to focus attention on this agent, given its potential for cross-species dissemination[12].
Porcine SaV (PoSaV) was first observed in fecal specimens from piglets in the USA using electron microscopy in 1980[13]. The initial gastroenteritis outbreak in Chinese piglets caused by PoSaV was reported in Shanghai in 2008. Despite its global presence, genomic characterization in China remains limited. Moreover, the elusive viral isolation poses a significant obstacle to understanding the mechanism and developing vaccines. A comprehensive investigation into the genetic evolution of PoSaV is pivotal for pathogenesis studies. Considering the potential for zoonosis, the continuous evolution of PoSaV variants poses a major concern for global porcine production. Phylogenetic and molecular evolutionary analyses can uncover codons under negative/purifying selection in a species. Thus, this study aims to genetically characterize prevalent PoSaV strains, determining their genetic divergence and selective pressure. These findings serve as a guide for effective defense and containment of PoSaV infections in the country.
2. Materials and Methods
2.1. Metagenomic Sequencing and Data Analysis
Twenty piglet diarrhea fecal samples were collected from three herds in Shanghai, divided into two groups (PCM1 and PCM2), and subjected to metagenomic sequencing. Genomic DNA/RNA was extracted from the pretreatment samples with MagPure Viral DNA/RNA Mino LQ Kit (Magen kitR6662-02). The viral RNA underwent reverse transcription by SuperScript III reverse transcriptase (Invitrogen). Subsequently, the SMARTer Ultra Low Input RNA kit was used to synthesize double-strand cDNA, and the quality of viral nucleic acids was assessed using the NanoDrop spectrophotometer (Thermo Fisher Scientific) and 1.5% agarose electrophoresis. The Next® UltraTM DNA Library Prep Kit for Illumina® (New England Biolabs) was employed for generating sequencing libraries. High-throughput sequencing occurred on an Illumina Novaseq 6,000, producing 150 bp paired-end reads by the Magigene Company (Guangzhou, China).
The raw sequencing reads were processed to obtain clean data using Soapnuke (v2.0.5) for further analysis. Quality-filtered reads were then de novo assembled to generate the metagenome. Different virus families were classified according to the annotation information from the NCBI taxonomy database.
2.2. Complete Genome Sequencing of PoSaV and Genomic Evolutionary Analysis
To analyze piglet-prevalent PoSaV’s genomic characteristics, the full-length sequence of a GIII genotype isolate was amplified. The PCM1 and PCM2 samples were then mixed, followed by extracting the total RNA using TRIzol reagent (TAKARA, Japan) followed by cDNA synthesis using the downstream primer (5’-CGGTACGCGTAACCAGGGAAAGA-3’). PCR, employing 2 × Phanta Max Master Mix (Vazyme, China), utilized prime pairs designed based on reference sequences KT922089. The amplification conditions involved initial denaturation at 98 ℃ for 2 min, followed by 30 cycles of 98 ℃ for 20 s, 60 ℃ for 20 s, and 72 ℃ for 1 min, with an additional extension step at 72 ℃ for 5 min. Each fragment underwent cloning into pEASY Blunt Zero Vector and sequencing by Sangon Biotech (Shanghai) Co., Ltd. (China). Furthermore, the full-length sequence was subsequently compared and spliced using Clustal and Lasergene 7.0 software.
For phylogenetic analysis, PoSaV ORF2 genes were obtained from GenBank, forming the basis for a neighbor-joining phylogenetic tree. This tree, annotated via the interactive Tree Of Life (iTOL) software (http://itol.embl.de/)[14], aimed at illustrating the evolutionary relationships. To assess PoSaV evolution rates, RdRp sequences from GenBank-derived PoSaVs underwent analysis. This involved constructing both a phylogenetic tree and a time tree, using the maximum likelihood method within MEGA 11 with 1000 bootstrap replicates. A 50% cut-off value was applied to condense the tree. The RelTime with Dated Tips (RTDT) method estimated divergence times for all tree branching points. This analysis referred to 50 PoSaV strains identified between 1998 and 2022.
2.3. Recombination and Selective Pressure Analysis
Recombination analysis utilized Recombination Detection Program (RDP) software version 4, examining the entire genome of PoSaVs[15].
The amino acid homology of PoSaV ORF1 proteins was assessed using Larsergene 7.1 and MEGA 11 for selective pressure analysis. Synonymous nonsynonymous analysis was conducted through SNAP services (http://hcv.lanl.gov/content/sequence/SNAP/SNAP.html) to determine the nonsynonymous to synonymous substitutions ratio (dN/dS) at each ORF1 coding region amino acid site, indicating evidence of positive or negative selection[16].
3. Results
3.1. Sapovirus Is Highly Prevalent in the Piglet Diarrhea Feces
To perform a comprehensive search for piglet gut viruses, twenty diarrhea feces were collected and divided into two groups: PCM1 and PCM2. The sapovirus abundance in piglet diarrhea feces was estimated using metagenomics. Results showed that RNA viruses, excluding phages, constituted 55.47%, with Caliciviridae and Astroviridae accounting for 27.54% and 21.78%, respectively (Figure 1a). Sapovirus emerged as the sole Caliciviridae member in this metagenomic sequencing. Analyzing the sapovirus ratio among total RNA viruses confirmed infection rates of 55.36% and 29.89% in PCM1 and PCM2 samples, respectively (Figure 1a). These findings indicate sapovirus as one of the predominant viruses in piglet diarrhea guts.
3.2. Phylogenetic and Time Evolutionary Analysis
To understand the genomic evolution of sapovirus in piglet diarrhea, a full-length sequence of sapovirus was amplified and spliced, named as SHCM/Mega2023. The phylogenetic tree, based on the ORF2 genes of SHCM/Mega2023 and corresponding GenBank strains, identified its classification as GIII genotype. SHCM/Mega2023 demonstrated the closest relationship with the 2016 USA-isolated JJ259 strain (KT922089) (Figure 2).
Subsequently, employing the RTDT method based on the RdRp genes, a time tree was calculated to examine sapovirus divergence times (Figure 3). The GII genotype showed the earliest origin among eighteen genotypes, dating back to the 1980s. GIV emerged in 1987. GXVII, GVII, and GV shared a common divergence time in 2005. GVIII distinguished itself between 1998 and 2009, while GXI emerged between 2001 and 2014. Notably, the GIII genotype exhibited the most extended span of divergence times, ranging from 1998 to 2014. SHCM/Mega2023, along with Orf1 (MH490911), diverged around 2014.
3.3. Recombinant Analysis
A complete genome recombination analysis utilized the RDP5 software to examine SHCM/Meg2023 and 33 reference strains. Results indicated that SHCM/Meg2023 potentially resulted from recombination between MK962338 (major parent) and KT922089 (minor parent), supported by six detection algorithms (RDP 6.821×10-20, BootScan 6.750×10-18, MaxChi 1.314×10-14, Chimaera 6.32×10-14, SiScan 1.743×10-24, 3Seq 6.574×10-11) (Figure 4). SAV/Pig-wt/ESP/P452/2017 (MK962338) and JJ259 (KT922089) are GIII PoSaV isolates from Spain (2017) and the USA (2016), respectively. Further analysis identifies position 2969-5132 in the SHCM/Meg2023 genome as the predicted recombinant fragment, with the breakpoint within ORF1 (Figure 4).
3.4. Evolutionary Selective Pressures on the ORF1 Codons
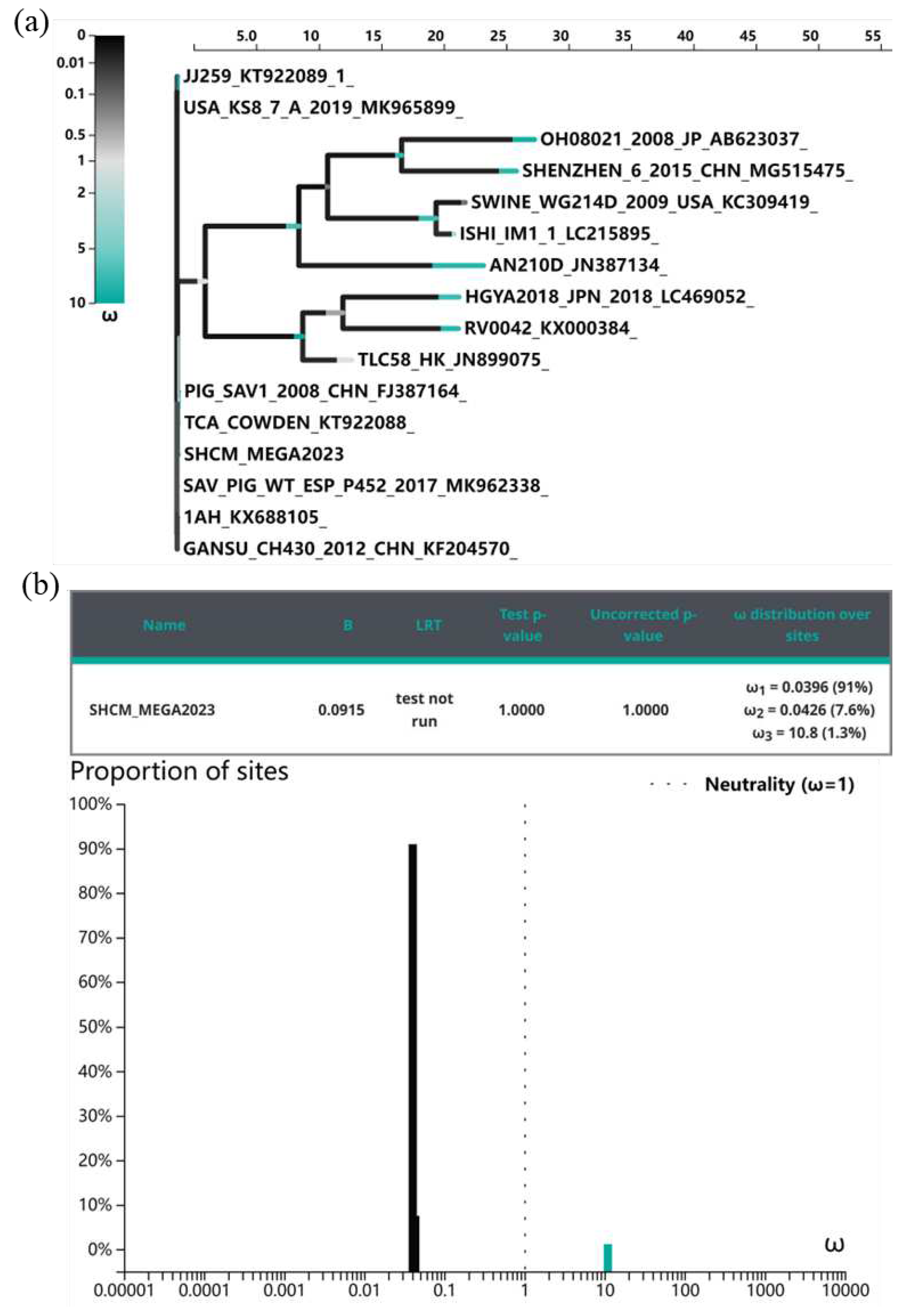
To further evaluate the extent of selective pressure on the ORF1 gene, the standardized differences in dN/dS were calculated for each position. Scores > 0 suggest heightened diversifying selection, highlighting the propensity for ORF1 protein regions to diversify. Purifying selection (dN/dS < 1) was indicated by BUSTED models for SHCM/Mega2023 (Figure 5a). In the ORF1 protein, seven amino acids (2 aa, 110 aa, 529 aa, 620 aa, 1668 aa, 1977 aa, and 2232 aa) were subject to diversifying positive selection (ω3 = 10.8), while 1314 amino acids experienced purifying selection (ω1 = 0.0396) (Figure 5b). Three positively selected sites (1668 aa, 1977 aa, and 2232 aa) were situated within ORF1 at capsid protein VP1.
4. Discussion
Pigs in all growth stages can contract PoSaVs, with a higher infection rate observed in post-weaning pigs[17,18]. No significant differences in SaV prevalence were found between age-matched groups of pigs with and without diarrhea in the field. However, PoSaVs frequently co-infect with other enteric pathogens, such as porcine epidemic diarrhea virus, porcine astrovirus, porcine kobuvirus, porcine deltacornavirus, etc.[19]. While most PoSaV genogroups remain unknown[20], a genomic evolutionary investigation of PoSaVs is necessary to provide effective clues for further research on cellular adaptability, pathogenic mechanisms, and prevention and control strategies.
To date, SaV has been categorized into 19 genogroups and 52 subtypes based on VP1 gene sequences, revealing four genogroups (GI, GII, GIV, and GV) and eight genogroups (GIII and GV-GXI) in humans and pigs, respectively[21]. GIII predominantly circulates in global swine herds worldwide[14], consistent with our study's findings. Our metagenomic data confirmed porcine sapovirus as the primary pathogen in piglet diarrhea feces, warranting attention. Phylogenetic analysis identified the prevalent PoSaV within the GIII genogroup. Given sapovirus's current division into 19 genogroups, our study pioneered the calculation and comparison of their evolutionary rates. Results affirmed GIII's widest span of divergence among genogroups, spanning from 1998 to 2014. We hypothesize that GIII exhibits the highest rates of transmission and variation.
SaV exhibits a broad host range with the potential for zoonotic transmission[22]. Recombination, acknowledged as an evolutionary force, can yield new viruses with distinct pathogenesis and virulence[23]. Intra- and inter-genogroup recombinant strains have been reported[7,24,25]. Here, we present the novel PoSaV isolate SHCM/Mega2023, characterized by recombination with MK962338 (major parent) and KT922089 (minor parent), with the breakpoint identified at 2969–5132. This differs from the intra-recombinant PoSaV (p2 strain) reported in Shanghai in 2015[26], which featured a breakpoint in the RaRp-capsid junction region, aligning with most SaV recombination events[27]. Although both SHCM/Mega2023 and p2 strains were reported in Shanghai, they exhibited distinct recombinant events, with the event in SHCM/Mega2023 being novel. The relationship between the novel recombinant event and pathogenesis remains unknown and requires further exploration.
Investigation into the selective pressures acting on a gene is crucial for comprehending its evolutionary dynamics and may unveil potential drug-targeting regions, as amino acid substitutions at negatively selected sites are likely intolerable[28,29]. Positively selected sites aid in identifying epitope-contacting residues[30]. Meanwhile, the presence of positive selection signatures in the genome is a characteristic indication of adaptation, revealing ongoing, recent, or ancient responses to environmental changes throughout a population's evolution[31]. In SaV, the polyprotein encoded by ORF1 undergoes cleavage to form various viral proteins, including 2C-like NTPase, 3C-like protease, VPg, 3D-like RNA-dependent RNA polymerase, and the major capsid protein (VP1). In this study, the selective pressure force of the ORF1 protein of PoSaVs was initially calculated, revealing seven positively selected sites in the ORF1 protein of SHCM/Mega2023. Notably, three positively selected sites (1668 aa, 1977 aa, and 2232 aa) within the capsid protein VP1 of ORF1 are potentially associated with antigenicity. VP1, the main structural protein of PoSaV, safeguards the viral nucleic acid and features multiple immunogenic epitopes that elicit both humoral and cellular immunity.
Author Contributions
Conceptualization, H.L. (Huili Liu) and J.T. (Jie Tao); Methodology, J.C. (Jinghua Cheng) and Y.S. (Ying Shi); Investigation, B.L. (Benqiang Li); resources, B.L. and P.T. (Pan Tang); Data curation, J.J. (Jiajie Jiao) and P.T.; Writing-original draft preparation, H.L. and J.T.; Writing-review and editing, J.C. and Y.S.; Project administration, H.L. and J.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Shanghai Agricultural Technology Development Program, China (Grant NO. 2022-02-08-00-12-F01167), SAAS Program for Excellent Research Team (Grant No. [2022]012).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Conflicts of Interest
The authors do not have any conflict of interest.
References
- Umair, M.; Rehman, Z.; Haider, S.A.; Usman, M.; Rana, M.S.; Ikram, A.; Salman, M. First report of coinfection and whole-genome sequencing of norovirus and sapovirus in an acute gastroenteritis patient from Pakistan. J. Med Virol. 2023, 95, e28458. [Google Scholar] [CrossRef]
- Choi, H.L.; Suh, C.-I.; Park, S.-W.; Jin, J.-Y.; Cho, H.-G.; Paik, S.-Y. Whole-Genome Sequencing Analysis of Sapovirus Detected in South Korea. PLOS ONE 2015, 10, e0132328. [Google Scholar] [CrossRef]
- Yinda, C.K.; Conceição-Neto, N.; Zeller, M.; Heylen, E.; Maes, P.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent sapoviruses detected by metagenomics analysis in straw-colored fruit bats in Cameroon. Emerg. Microbes Infect. 2017, 6, e38. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, R.; Ding, X.; Freedman, S.B.; Lee, B.E.; Ali, S.; Luong, J.; Xie, J.; Chui, L.; Wu, Y.; Pang, X.; Alberta Provincial Pediatric EnTeric Infection, T. Molecular Epidemiology of Human Sapovirus among Children with Acute Gastroenteritis in Western Canada. J. Clin. Microbiol. 2021, 59, JCM0098621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ullman, K.; Chowdry, V.; Reining, M.; Benyeda, Z.; Baule, C.; Juremalm, M.; Wallgren, P.; Schwarz, L.; Zhou, E.; et al. Molecular investigations on the prevalence and viral load of enteric viruses in pigs from five European countries. Veter- Microbiol. 2015, 182, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Hoque, S.A.; Pham, N.T.K.; Onda-Shimizu, Y.; Nishimura, S.; Sugita, K.; Kobayashi, M.; Islam, M.T.; Okitsu, S.; Khamrin, P.; Maneekarn, N.; et al. Sapovirus infections in Japan before and after the emergence of the COVID-19 pandemic: An alarming update. J. Med Virol. 2023, 95, e29023. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Shang, R.; Cheng, K.; Wang, S.; Wu, J.; Yu, Z. A novel recombinant porcine sapovirus infection in piglets with diarrhea in Shandong Province, China, 2022. Braz. J. Microbiol. 2023, 54, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- 8. Birch, J.M.; Leijon, M.; Nielsen, S.S.; Struve, T.; Jensen, H.E. Visualization of intestinal infections with astro- and sapovirus in mink (Neovison vison) kits by in situ hybridization. FEMS Microbes 2011, 2, xtab005. [Google Scholar] [CrossRef] [PubMed]
- Mijovski, J.Z.; Poljšak-Prijatelj, M.; Steyer, A.; Barlič-Maganja, D.; Koren, S. Detection and molecular characterisation of noroviruses and sapoviruses in asymptomatic swine and cattle in Slovenian farms. Infect. Genet. Evol. 2010, 10, 413–420. [Google Scholar] [CrossRef]
- Stamelou, E.; Giantsis, I.A.; Papageorgiou, K.V.; Petridou, E.; Davidson, I.; Polizopοulou, Z.S.; Papa, A.; Kritas, S.K. First report of canine Astrovirus and Sapovirus in Greece, hosting both asymptomatic and gastroenteritis symptomatic dogs. Virol. J. 2022, 19, 1–9. [Google Scholar] [CrossRef]
- Tse, H.; Chan, W.M.; Li, K.S.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Discovery and Genomic Characterization of a Novel Bat Sapovirus with Unusual Genomic Features and Phylogenetic Position. PLOS ONE 2012, 7, e34987. [Google Scholar] [CrossRef]
- dos Anjos, K.; Lima, L.M.; Silva, P.A.; Inoue-Nagata, A.K.; Nagata, T. The possible molecular evolution of sapoviruses by inter- and intra-genogroup recombination. Arch. Virol. 2011, 156, 1953–1959. [Google Scholar] [CrossRef]
- Saif, L.J.; Bohl, E.H.; Theil, K.W.; Cross, R.F.; House, J.A. Rotavirus-Like, Calicivirus-Like, and 23-nm Virus-Like Particles Associated with Diarrhea in Young Pigs. J. Clin. Microbiol. 1980, 12, 105–111. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Tao, J.; Li, B.; Cheng, J.; Shi, Y.; Qiao, C.; Lin, Z.; Liu, H. Genomic Divergence Characterization and Quantitative Proteomics Exploration of Type 4 Porcine Astrovirus. Viruses 2022, 14, 1383. [Google Scholar] [CrossRef] [PubMed]
- Wisotsky, S.R.; Pond, S.L.K.; Shank, S.D.; Muse, S.V. Synonymous Site-to-Site Substitution Rate Variation Dramatically Inflates False Positive Rates of Selection Analyses: Ignore at Your Own Peril. Mol. Biol. Evol. 2020, 37, 2430–2439. [Google Scholar] [CrossRef] [PubMed]
- 17. Valente, C.S.; Alfieri, A.F.; Barry, A.F.; Leme, R.A.; Lorenzetti, E.; Alfieri, A.A. Age distribution of porcine sapovirus asymptomatic infection and molecular evidence of genogroups GIII and GIX? circulation in distinct Brazilian pig production systems. Trop Anim Health Prod 2016, 48, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, S.; Yori, P.P.; Olortegui, M.P.; A Lima, A.; Ahmed, T.; Mduma, E.R.; George, A.; Samie, A.; Svensen, E.; Lima, I.; et al. The Epidemiology of Sapovirus in the Etiology, Risk Factors, and Interactions of Enteric Infection and Malnutrition and the Consequences for Child Health and Development Study: Evidence of Protection Following Natural Infection. Clin. Infect. Dis. 2022, 75, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Dufkova, L.; Scigalkova, I.; Moutelikova, R.; Malenovska, H.; Prodelalova, J. Genetic diversity of porcine sapoviruses, kobuviruses, and astroviruses in asymptomatic pigs: an emerging new sapovirus GIII genotype. Arch. Virol. 2012, 158, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Wang, Q.; Oka, T.; Saif, L.J. Porcine sapoviruses: Pathogenesis, epidemiology, genetic diversity, and diagnosis. Virus Res. 2020, 286, 198025–198025. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, F.; Masuda, T.; Aoki, H.; Ito, M.; Sano, K.; Naoi, Y.; Katayama, Y.; Omatsu, T.; Oba, M.; Furuya, T.; et al. Complete genome sequencing and genetic characterization of porcine sapovirus genogroup (G) X and GXI: GVI, GVII, GX, and GXI sapoviruses share common genomic features and form a unique porcine SaV clade. Infect. Genet. Evol. 2019, 75, 103959. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Saga, Y.; Iwai, M.; Obara, M.; Horimoto, E.; Hasegawa, S.; Kurata, T.; Okumura, H.; Nagoshi, M.; Takizawa, T. Frequent Detection of Noroviruses and Sapoviruses in Swine and High Genetic Diversity of Porcine Sapovirus in Japan during Fiscal Year 2008. J. Clin. Microbiol. 2010, 48, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, E.; Muylkens, B.; Mauroy, A.; Ziant, D.; Delwiche, T.; Thiry, E. Experimental evidence of recombination in murine noroviruses. J. Gen. Virol. 2010, 91, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Supadej, K.; Khamrin, P.; Kumthip, K.; Malasao, R.; Chaimongkol, N.; Saito, M.; Oshitani, H.; Ushijima, H.; Maneekarn, N. Distribution of norovirus and sapovirus genotypes with emergence of NoV GII.P16/GII.2 recombinant strains in Chiang Mai, Thailand. J. Med Virol. 2018, 91, 215–224. [Google Scholar] [CrossRef]
- Dey, S.K.; Mizuguchi, M.; Okitsu, S.; Ushijima, H. Novel recombinant sapovirus in Bangladesh. Clin Lab 2011, 57, 91–94. [Google Scholar] [PubMed]
- Li, J.; Shen, Q.; Zhang, W.; Zhao, T.; Li, Y.; Jiang, J.; Yu, X.; Guo, Z.; Cui, L.; Hua, X. Genomic organization and recombination analysis of a porcine sapovirus identified from a piglet with diarrhea in China. Virol. J. 2017, 14, 1–8. [Google Scholar] [CrossRef]
- Oka, T.; Wang, Q.; Katayama, K.; Saif, L.J. Comprehensive Review of Human Sapoviruses. Clin. Microbiol. Rev. 2015, 28, 32–53. [Google Scholar] [CrossRef]
- Suzuki, Y. Natural Selection on the Influenza Virus Genome. Mol. Biol. Evol. 2006, 23, 1902–1911. [Google Scholar] [CrossRef]
- Correia, V.; Abecasis, A.B.; Rebelo-De-Andrade, H. Molecular footprints of selective pressure in the neuraminidase gene of currently circulating human influenza subtypes and lineages. Virology 2018, 522, 122–130. [Google Scholar] [CrossRef]
- Air, G.M.; Laver, W.G.; Luo, M.; Stray, S.J.; Legrone, G.; Webster, R.G. Antigenic, sequence, and crystal variation in influenza B neuraminidase. Virology 1990, 177, 578–587. [Google Scholar] [CrossRef]
- Abondio, P.; Cilli, E.; Luiselli, D. Inferring Signatures of Positive Selection in Whole-Genome Sequencing Data: An Overview of Haplotype-Based Methods. Genes 2022, 13, 926. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sapovirus infection rate in the porcine diarrhea faces. (a) Taxonomic distributions of the phages, RNA virus, DNA virus, and unclassified viruses. (b) Sapovirus ratios in the two sequenced samples.
Figure 1.
Sapovirus infection rate in the porcine diarrhea faces. (a) Taxonomic distributions of the phages, RNA virus, DNA virus, and unclassified viruses. (b) Sapovirus ratios in the two sequenced samples.
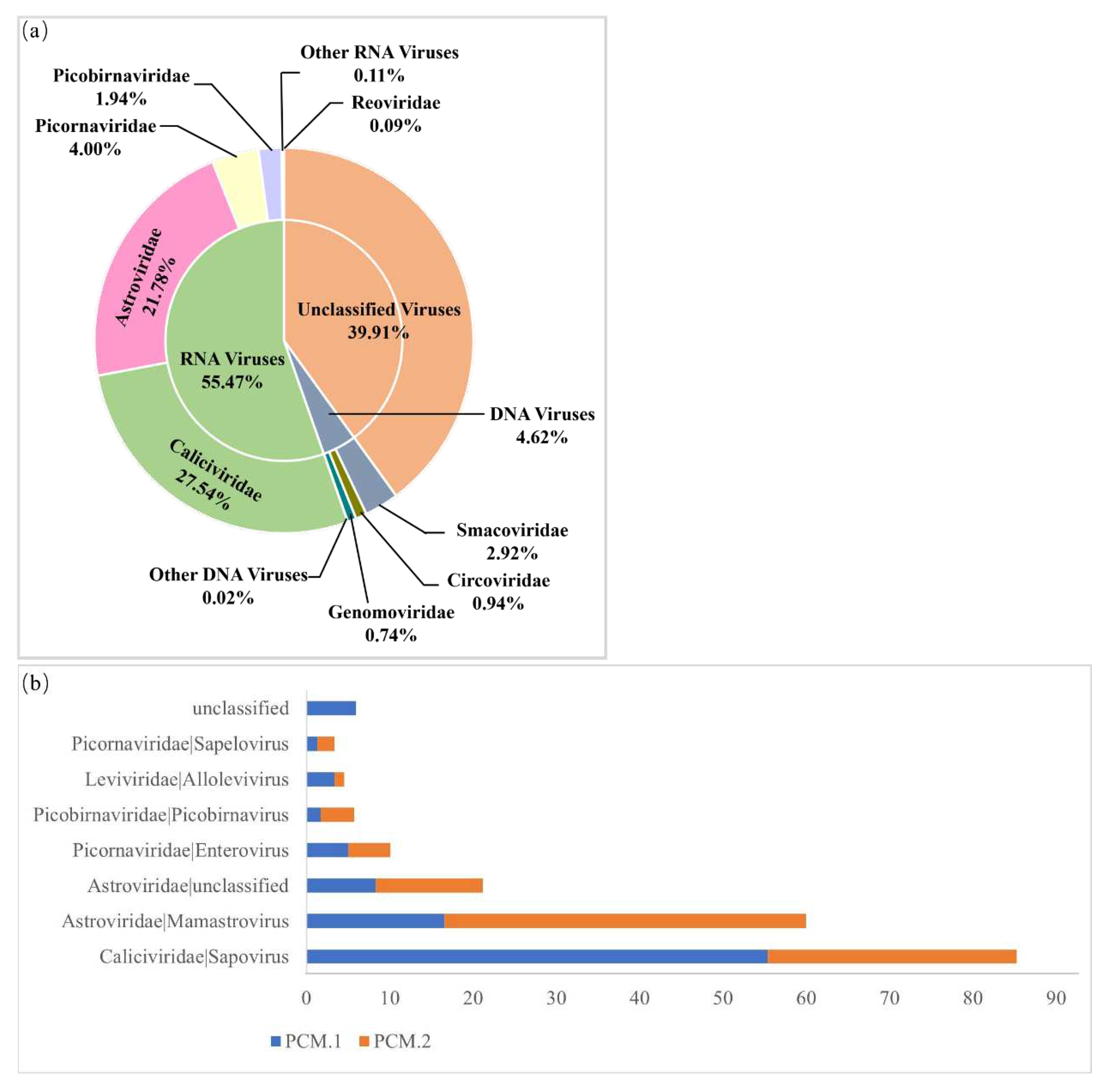
Figure 2.
Phylogenetic analysis of the ORF2 genes of PoSaVs. Phylogenetic tree constructed from the ORF2 genes of SHCM/Mega2023 and other 35 PoSaVs retrieved from GenBank using neighbour-joining method, which was annotated using the interactive Tree Of Life (iTOL) software. The outside layer denotes the genogroups. The green box (SHCM/Mega2023) indicates the PoSaV sequenced in this study. The purple dots indicate the bootstrap.
Figure 2.
Phylogenetic analysis of the ORF2 genes of PoSaVs. Phylogenetic tree constructed from the ORF2 genes of SHCM/Mega2023 and other 35 PoSaVs retrieved from GenBank using neighbour-joining method, which was annotated using the interactive Tree Of Life (iTOL) software. The outside layer denotes the genogroups. The green box (SHCM/Mega2023) indicates the PoSaV sequenced in this study. The purple dots indicate the bootstrap.
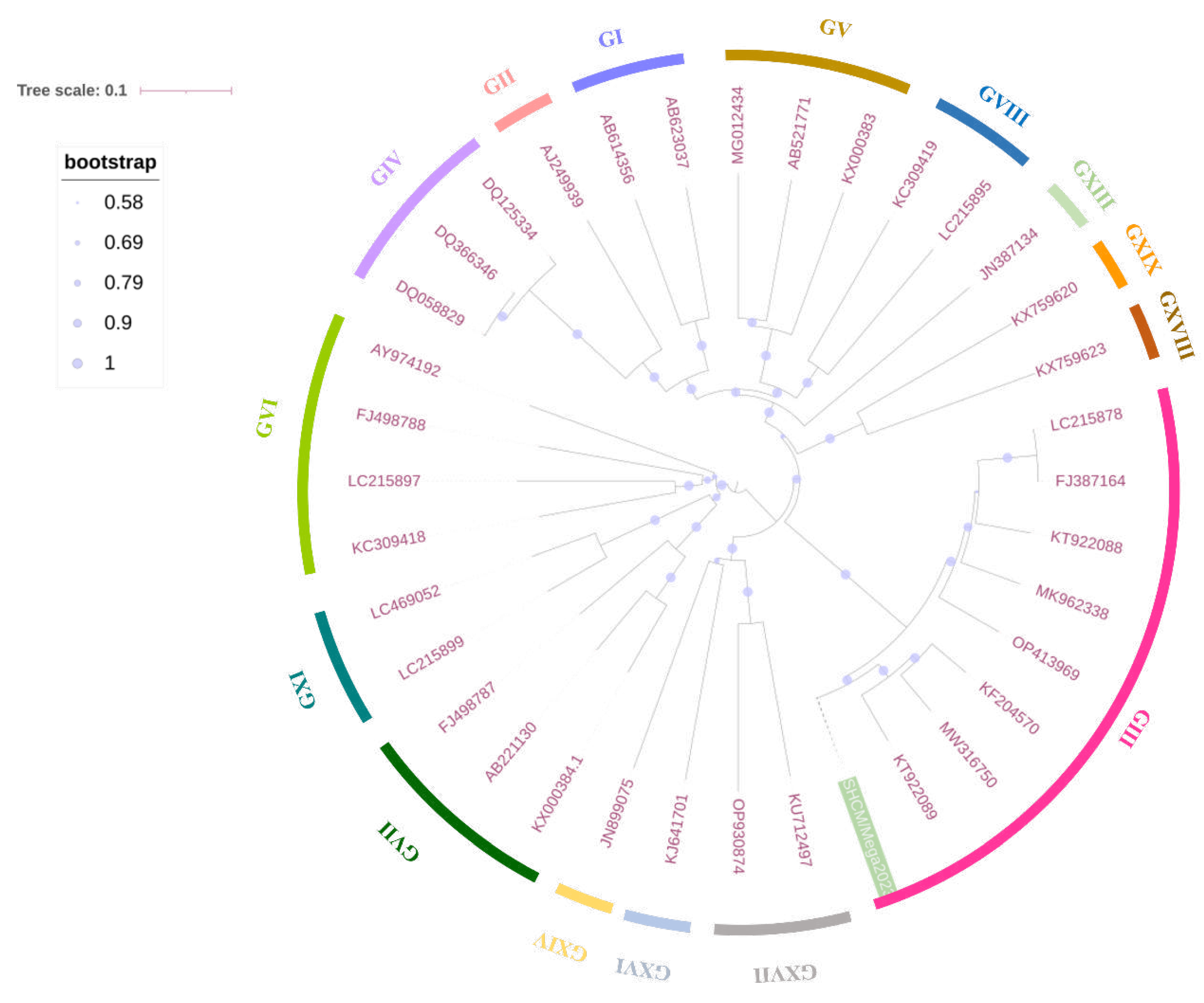
Figure 3.
Evolutionary time tree generated based on the RdRp genes of PoSaVs. The time tree was calculated in MEGA X where the divergence time was inferred by the RelTime with Dated Tips (RTDT) method. The GII genogroup strain SLV Bristol 98UK (AJ249939) (grey color) was designated as an outgroup taxon, with all sequences using the year of sampling as the tip dates for calibration constraints. The divergence time of each branch is marked. Different PoSaV genogroups are circled in differently colored dotted box.
Figure 3.
Evolutionary time tree generated based on the RdRp genes of PoSaVs. The time tree was calculated in MEGA X where the divergence time was inferred by the RelTime with Dated Tips (RTDT) method. The GII genogroup strain SLV Bristol 98UK (AJ249939) (grey color) was designated as an outgroup taxon, with all sequences using the year of sampling as the tip dates for calibration constraints. The divergence time of each branch is marked. Different PoSaV genogroups are circled in differently colored dotted box.
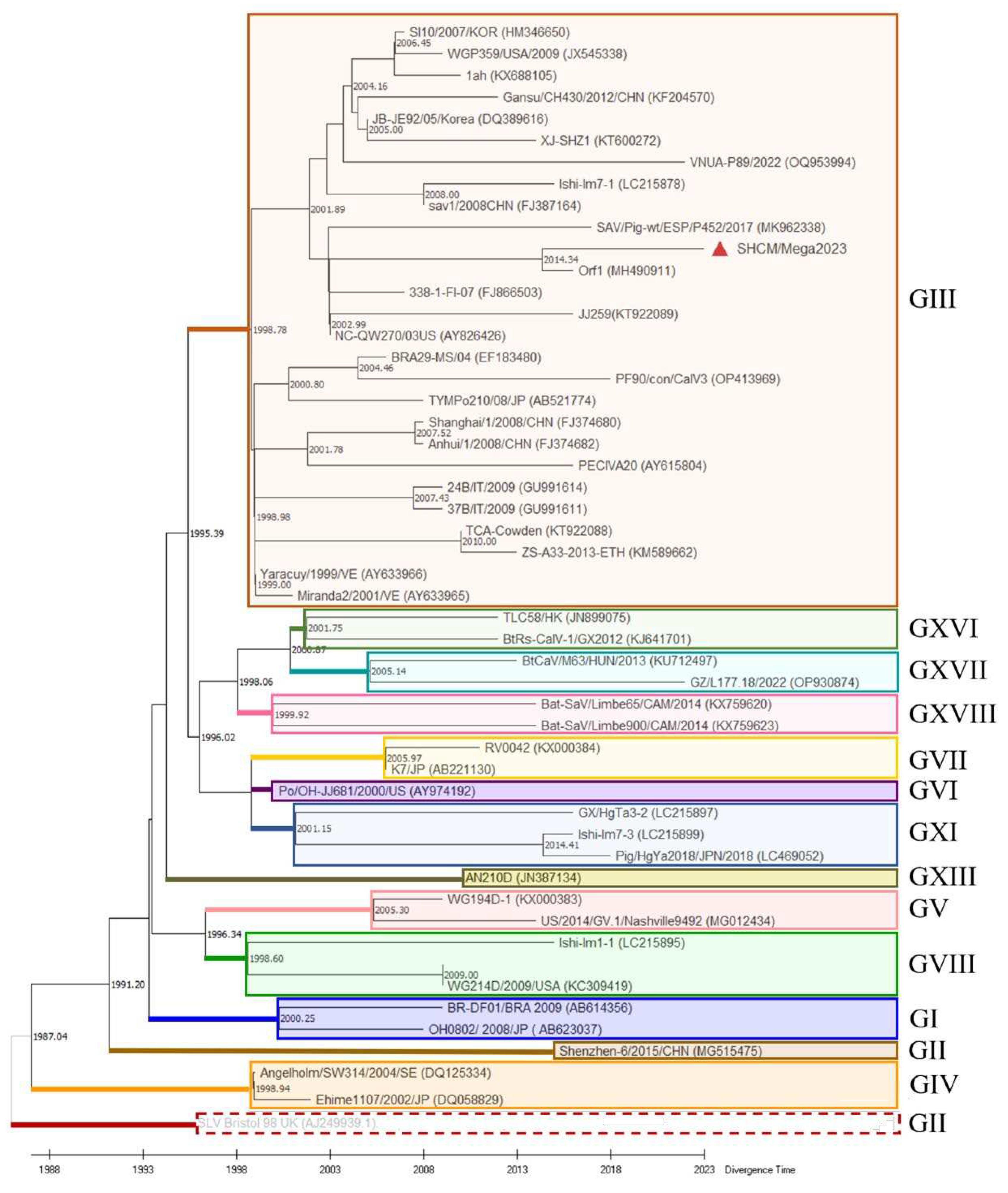
Figure 4.
The recombination analysis of the complete SHCM/Meg2023 genome. The results were described using the RDP method, which was supported by ≥ 6 programs to further characterize the potential recombination events. The pink box indicates the regions for the recombinant and its putative parents. The Y-axis represents the pairwise identity between the recombinant and its putative parents. The X-axis represents the position when aligned with a 30 nt sliding window. The comparison of the recombinant-major parent, recombinant-minor parent, and major-minor parent was indicated by cyan, purple, and yellow lines, respectively.
Figure 4.
The recombination analysis of the complete SHCM/Meg2023 genome. The results were described using the RDP method, which was supported by ≥ 6 programs to further characterize the potential recombination events. The pink box indicates the regions for the recombinant and its putative parents. The Y-axis represents the pairwise identity between the recombinant and its putative parents. The X-axis represents the position when aligned with a 30 nt sliding window. The comparison of the recombinant-major parent, recombinant-minor parent, and major-minor parent was indicated by cyan, purple, and yellow lines, respectively.
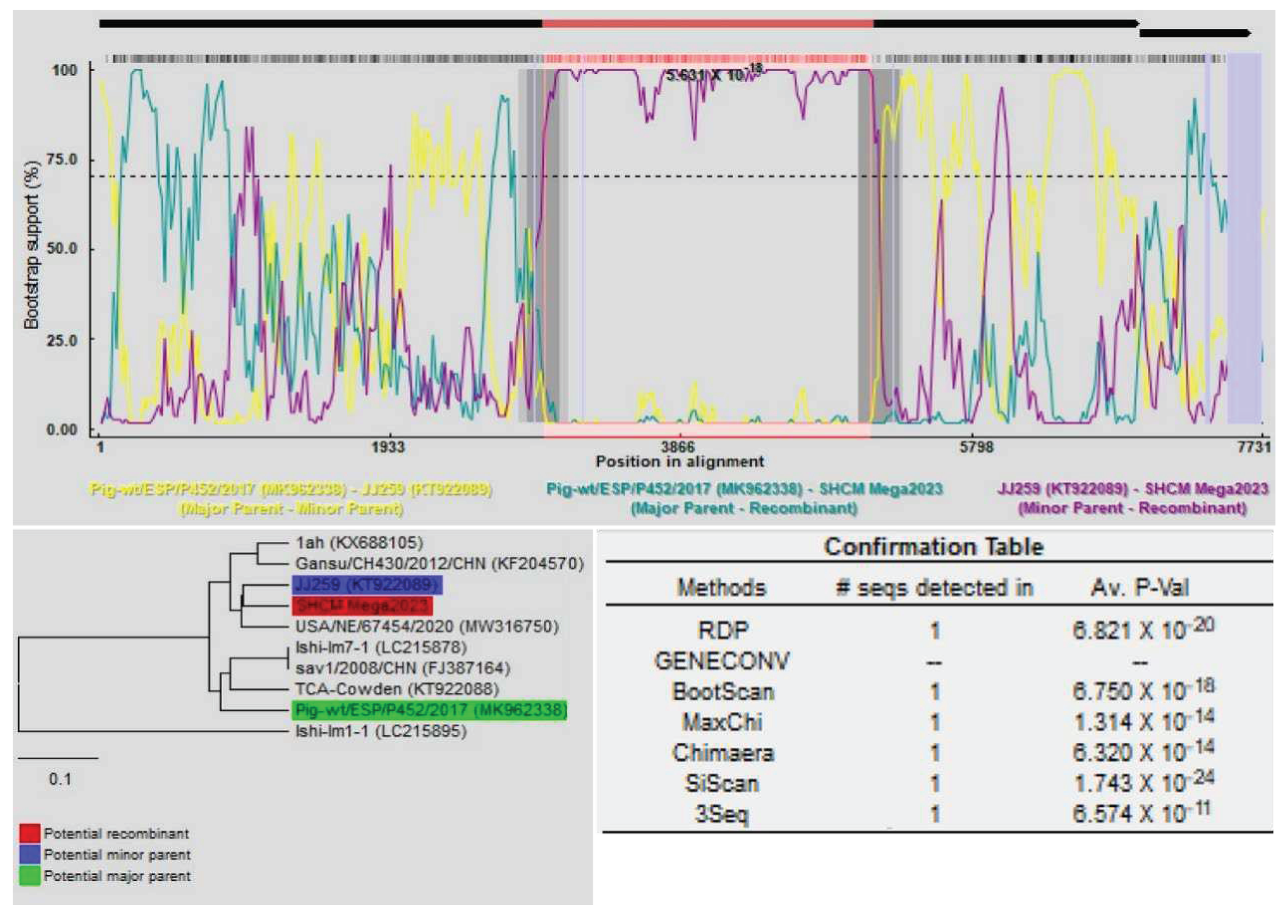
Figure 5.
Selective pressure analysis of the ORF1 protein of PoSaVs. The genetic evolutionary of ORF1 protein was first analyzed using Lasergene 7.1 and MEGA 11. Selection pressure was calculated using the FEL and BUSTED methods of Datamonkey online software (http://www.datamonkey.org/). The dN (non-synonymous replacement rate)/dS (synonymous replacement rate) > 1 represents positive selection, and vice versa, and is considered as the purification selection.
Figure 5.
Selective pressure analysis of the ORF1 protein of PoSaVs. The genetic evolutionary of ORF1 protein was first analyzed using Lasergene 7.1 and MEGA 11. Selection pressure was calculated using the FEL and BUSTED methods of Datamonkey online software (http://www.datamonkey.org/). The dN (non-synonymous replacement rate)/dS (synonymous replacement rate) > 1 represents positive selection, and vice versa, and is considered as the purification selection.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



