Submitted:
24 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
. The Tau protein is associated with microtubule function in the neuron and is crucial for normal axonal transport. In several different neurodegenerative disorders, Tau misfolding leads to hyper-phosphorylation (p-Tau) and intracellular p-Tau aggregates known as neurofibrillary tangles (NFTs). This is particularly evident in individuals with Down syndrome (DS), who develop Alzheimer’s disease-like (AD) neuropathology early in life with almost complete penetrance, associated with the development of dementia symptoms in their 40s or 50s. Our previous findings have shown that certain forms of p-Tau are present already in childhood in individuals with DS in neuron-derived exosomes, suggesting an early phosphorylation profile in this population that could influence the development of AD pathology. Misfolded p-Tau isoforms are known to be seeding competent and may be responsible for spreading AD pathology in different regions of the brain. This review is focused on the accumulation of p-Tau in the brain of individuals with DS and potential consequences for brain function.
Keywords:
Down syndrome (DS)
; Alzheimer’s disease (AD)
; neurofibrillary tangles (NFTs)
; seeding competent p-Tau
; neuropathology.
1. Introduction
Individuals with Down syndrome (DS) exhibit Alzheimer’s disease (AD)-related pathology in the brain early in life, leading to the development of dementia in their 40s or 50s, with few exceptions [1-3]. The AD pathology that develops in the DS brain includes widespread amyloid plaques, neuroinflammation, cell death, and NFTs [1,4-6]. Amyloid pathology is naturally occurring in the DS brain because the amyloid precursor protein gene (APP) is located on Chromosome 21 (Chr. 21), leading to increased APP and amyloid production early in life, in humans with DS along with DS mouse models [3,7-10]. Neuroinflammation is also an inherent trait of the DS brain due to several different genes encoded on Chr. 21, including 4 of 6 interferon receptors [11,12] and the superoxide dismutase 1 gene (SOD-1)[13], among other factors. Therefore, cells obtained from the DS brain exhibit early signs of inflammatory activation as well as oxidative stress [3,4,6,14,15].
Associated with AD pathology is the spreading of misfolded Tau, including hyperphosphorylated Tau (p-Tau), from region to region in the brain, finally leading to a Braak stage of V-VI in the final stages of AD pathology [16,17]. We have demonstrated that neuron-derived exosomes (NDEs), isolated from the plasma of people with DS at different ages, contain unusual amounts of p-Tau already in childhood in some individuals with DS [18,19]. In addition, recent work from our group has shown that NDEs from individuals with DS contain seeding-competent p-Tau that can spread to mouse brain when NDEs are injected into the hippocampus via stereotactic injections [20].
Based on these findings, the current review will be focused on current findings regarding p-Tau and its potential to seed pathology as a “prion-like” spreader leading to AD, particularly in individuals with DS.
2. Microtubule-Associated Protein Tau (MAPT) Structure and Function
The Tau protein is a highly conserved protein within mammals and is known to have six isoforms, caused by alternate splicing of exons 2, 3, and 10 of the microtubule-associated protein tau (MAPT) gene [21-24]. Tau is a microtubule-associated protein that contributes to the stability of microtubules in neurites and is crucial for the maintenance of normal microtubule structure and function as well as axonal transport in the neuron [25]. The Tau protein interacts with microtubules via either 3 or 4 microtubule binding repeats (3R or 4R, respectively), and the balance between 3R and 4R Tau proteins is altered during pathological states. When Tau undergoes modifications, this can lead to de-stabilization of neuronal microtubules and, consequently, neuronal dysfunction and death [26]. Proteinaceous filaments of p-Tau are known to spread in a prion-like fashion in the human brain, leading to the detrimental formation of neurofibrillary tangles (NFTs) that contribute to AD pathology and correlate significantly with cognitive deficits in Alzheimer’s disease (AD) and also contribute to distinguishing differences between AD and other tauopathies [27,28]. A distinct set of modifications occur in different neurodegenerative conditions, of which AD is the most common [25]. A precise balance between 4-repeat (4R) and 3-repeat (3R) isoforms of Tau are found in normal conditions, while dysregulation of the 3R:4R ratio is associated with different forms of tauopathy [22,29-32].
2.1. Posttranslational Modifications of Tau
The larger 4R Tau isoform contains 441 amino acid residues and around 35 percent of 4R Tau isoforms undergo posttranslational modifications (PTMs) [33]. PTMs of Tau can occur at several different residues, including serine, threonine, tyrosine, lysine, arginine, asparagine, histidine, and cysteine [21]. PTMs at these sites include glycation, nitration, ubiquitination, and phosphorylation [25]. During pathological conditions caused either by environmental or genetic factors, Tau can undergo multiple different PTMs along with conformational changes, which can lead to Tau aggregation and tangle formations that can develop into hallmarks for specific tauopathies [21,25,28]. Phosphorylation is the most studied PTM of the Tau protein and alters the physiological function and Tau protein affinity to microtubules. Phosphorylation at 85 potential sites on the Tau molecule, located at the serine, tyrosine, or threonine sites [25>], can modulate intracellular interactions significantly and lead to aggregating isoforms that can be toxic, both in vivo and in vitro [34]. The phosphorylation of a protein changes the charge, adding a negatively charged hydrophilic group, which then results in a hydrophilic rather than a hydrophobic protein molecule. More than 20 different kinases and phosphatases are known to regulate the phosphorylation of the Tau protein and are thought to underlie imbalances.
The larger 4R Tau isoform contains 441 amino acid residues and around 35 percent of 4R Tau isoforms undergo posttranslational modifications (PTMs) [33]. PTMs of Tau can occur at several different residues, including serine, threonine, tyrosine, lysine, arginine, asparagine, histidine, and cysteine [21]. PTMs at these sites include glycation, nitration, ubiquitination, and phosphorylation [25]. During pathological conditions caused either by environmental or genetic factors, Tau can undergo multiple different PTMs along with conformational changes, which can lead to Tau aggregation and tangle formations that can develop into hallmarks for specific tauopathies [21,25,28]. Phosphorylation is the most studied PTM of the Tau protein and alters the physiological function and Tau protein affinity to microtubules. Phosphorylation at 85 potential sites on the Tau molecule, located at the serine, tyrosine, or threonine sites [25>], can modulate intracellular interactions significantly and lead to aggregating isoforms that can be toxic, both in vivo and in vitro [34]. The phosphorylation of a protein changes the charge, adding a negatively charged hydrophilic group, which then results in a hydrophilic rather than a hydrophobic protein molecule. More than 20 different kinases and phosphatases are known to regulate the phosphorylation of the Tau protein and are thought to underlie imbalances.
The complex relationship between Tau isoforms, phosphorylation and other PTMs at specific sites on the molecule is crucial information for determining the cause of different tauopathies as well as the development of novel and effective treatment paradigms. While Tau PTMs have been extensively studied in mouse models and in postmortem brains from donors with AD and other tauopathies, there is still little research examining how DS affects the occurrence of Tau PTMs. Further, the complex changes associated with trisomy 21 may drive unique molecular effects on Tau solubility, intermolecular interactions, protein localization and degradation in a manner that would make certain PTMs more likely to be altered and perhaps more amenable to selective treatments. For example, DYRK1A, a dual kinase that can phosphorylate Tau at multiple positions [35], is overexpressed in postmortem samples from brains with AD and particularly with Down syndrome (DS), where a 50% increase is measured due to having an extra copy of the gene [36-38]. Several strategies invoking DYRK1A inhibition improve cognitive function in mouse models of DS and have shown therapeutic benefits in young patients with DS in clinical trials [36,39]. However, the direct link between DYRK1A inhibition and suppression of Tau phosphorylation has only been demonstrated in fly and mouse models [40-42]. We have recently demonstrated overexpression of Dyrk1A in specific hippocampal layers in postmortem human tissue using spatial transcriptomics (personal communication, Granholm) correlated with Tau-related alterations in the DS-AD brain, suggesting a definite link between p-Tau and Dyrk1A. However, this remains to be demonstrated in vivo.
2.2. The role of truncation, RNA and DNA in Tau aggregation
Posttranslational modifications of Tau also include truncation of the protein, which can occur at different locations on the molecule by various proteases present in the cytosol. Proteolytic truncated pieces of Tau are highly prone to aggregation and contribute to progressive pathology in both AD and DS-AD via site-specific phosphorylation, self-aggregation, and binding to hyperphosphorylated and oligomeric Tau [43,44]. It has recently been shown that truncated forms of Tau from the C-terminal have seeding capacity and spread via axonal pathways to different brain regions, causing local toxicity and neuronal death, for example, by affecting the autophagy system [45]. Truncated Tau protein also contributes to microtubule instability, potentially leading to synaptic dysfunction as well.
Another event occurring both with normal aging and AD is the accumulation of RNA and DNA quadruplexes [46], which have recently been shown to contribute to aging-related neuronal dysfunction and cell loss. G-Quadruplex (G4) DNA and RNA (RG4) are non-canonical secondary nucleic acid structures that have multiple roles in vital cellular processes both in health and disease [46,47]. G4s are involved in the regulation of both DNA and RNA processes, including replication, transcription, and translation, and RG4s have been shown to accumulate particularly during cellular stress conditions [47]. Our group has recently shown that RG4s accumulate particularly in the hippocampal formation with aging and in patients with AD, and that intracellular accumulation of RG4s correlates significantly both with NFTs and with Braak stage [48]. These findings are interesting and could lead to a better understanding of the role of DNA and RNA secondary structures for the development of Tau pathology in AD and DS-AD.
3. Tauopathy seeding and its spread in the DS brain
Dr. Stanely Prusiner and his research group have minted AD as a “double-prion” disease due to the prion-like activities conducted by both aggregating amyloid and Tau isoforms [49]. Interestingly, Condello and Prusiner examined prion activities in frozen brain tissue from individuals with DS of different ages by selectively precipitating Aβ and Tau from DS brain homogenates and measuring the number of prions using cellular bioassays. They discovered that while brain tissue from individuals with early-onset Alzheimer’s disease (EOAD) exhibit reduced prion activities with age, this is not the case for DS brain tissue, where the levels of Aβ and Tau seeds increased with age [50]. These findings suggest a partially different seeding mechanism for Tau aggregates in DS versus other forms of AD.
As mentioned above, truncated, fibrous, and oligomeric Tau are seeding competent and can seed and spread both in vivo and in vitro when given the opportunity to propagate in a prion-like manner [31,51]. Recent studies have implicated both fibrillar, truncated, and oligomeric Tau in terms of seeding, aggregation, and propagation of Tau pathology in the brain [22,31,32,52-54]. Interestingly, oligomeric and fibrillar Tau appear to be equivalent in potency in terms of seeding competency, and both are known to be taken up by local neurons after intracranial injection [55]. However, oligomeric Tau appears to drive a more potent glial response in the brain and a more rapid propagation of misfolded Tau to other brain regions [55].
3.1. P-Tau spreading in neurotransmitter systems with AD
Noradrenergic neurons of the locus coeruleus (LC-NE; [56,57]), basal forebrain cholinergic neurons (BFCNs; [58-61]), and hippocampal neurons [62-66] are particularly vulnerable to dysfunction and/or degeneration in the brain of persons with DS. Basal forebrain cholinergic neurons (BFCNs) are highly susceptible to degeneration in both DS-AD and AD and have crucial functions for learning and memory both in human cohorts and animal models [60,67]. BFCNs have been studied in detail in both DS-AD and AD, and the loss of these cells corresponds significantly with the decline in memory function in patients with dementia [67-70]. Because of the early findings in the 70s and 80s that acetylcholinesterase inhibitors could slow the progression of memory loss in patients with AD [67-69,71-73], this is the most used anti-AD class of drugs to this day, even though these drugs are not disease-modifying and can cause severe and uncomfortable side effects [73]. However, choline esterase inhibitors have not been used extensively in patients with DS-related AD due to side effects that might interfere with and contribute to comorbidities, even though a recent study suggested that also patients with DS-AD could benefit from cholinesterase inhibitors [74]. Interestingly, recent work has shown that pre-tangle pathology (with an antibody that detects phosphorylated Tau at S422) coincided with the loss of staining for the p75 neurotrophic factor receptor (NTR) in the basal forebrain of patients with no cognitive impairment (NCI) or mild cognitive impairment (MCI), and these changes correlated with cognitive decline and AD neuropathology, thus confirming earlier studies showing a connection between BFCN cell loss and Tau pathology [75]. Thus, it is strongly suggested that Tau misfolding and phosphorylation drive cholinergic cell loss in the basal forebrain, both in AD and in DS-AD.
LC-NE neurons degenerate prior to any other marked cell loss in the DS and AD brain [57,76]. Tau accumulates early in LC-NE neurons in AD, perhaps even decades prior to dementia symptoms, and from there, prion-like spread of oligomers or filamentous Tau spreads from the LC to the forebrain and on to other brain regions [77-79]. There is a direct innervation of LC-NE neurons to BFCNs [80], potentially suggesting that early Tau pathology observed in BFCNs could be the result of spreading Tau “seeds” from the LC-NE to BFCNs, although this pathogenic potential has not been examined as of yet. Others have shown that both Tau oligomers and Tau fibrils have seeding competency and can be secreted from neurons as well as taken up by other cells, including other neurons, astrocytes, microglia, or oligodendrocytes (Figure 1 above and [31,52,81,82]). However, the seeding competency of Tau extracted from the DS brain or from DS-AD NDEs had not been examined previously.
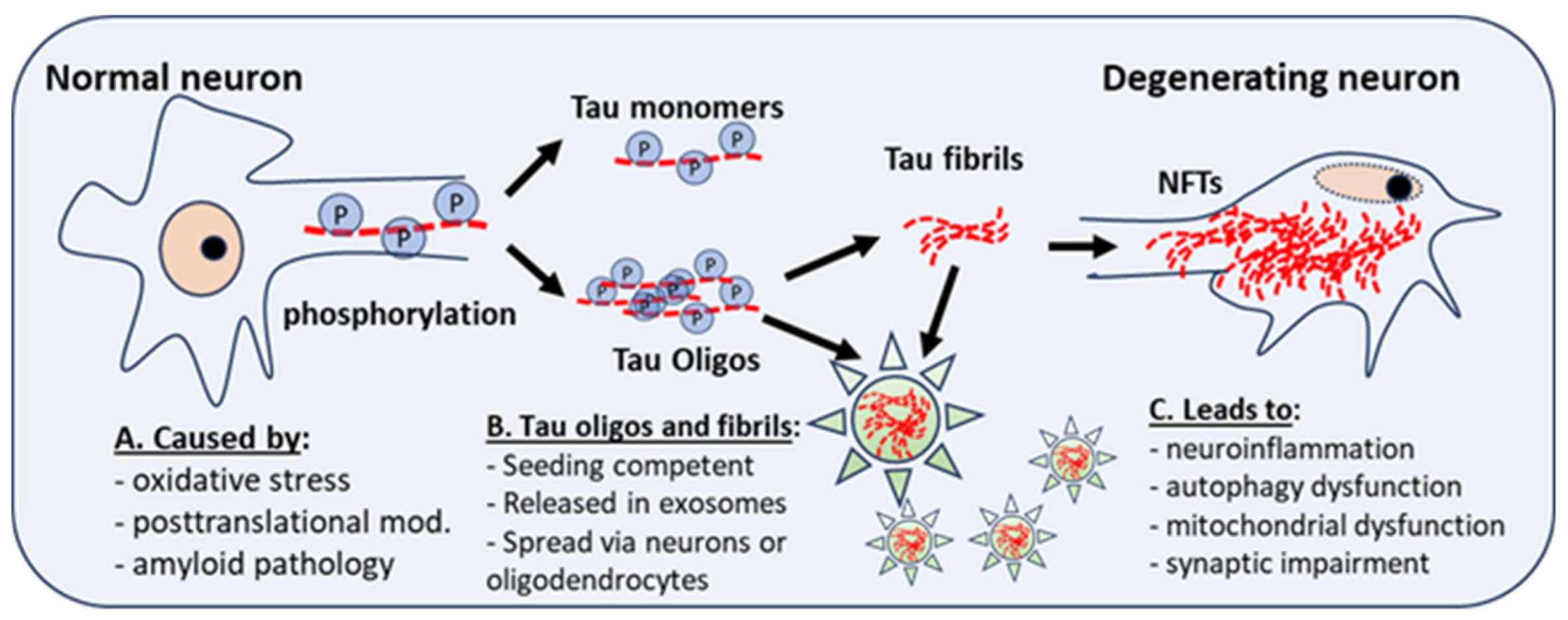
Figure 1.
Posttranslational modifications, neuroinflammation or oxidative stress, give rise to abnormal phosphorylation of Tau. This affects the stability of microtubules, and the development of Tau monomers and oligomers, which have aggregation potential and can form Tau fibrils, which aggregate into neurofibrillary tangles (NFTs), a hallmark for AD and DS-AD. Intra-neuronal NFT accumulation leads to neuroinflammation, autophagy dysfunction, and mitochondrial dysfunction. Tau fibrils and oligomers are seeding competent and are secreted in exosomes.spread to other neurons and to astrocytes, microglia, or oligodendrocytes.
Figure 1.
Posttranslational modifications, neuroinflammation or oxidative stress, give rise to abnormal phosphorylation of Tau. This affects the stability of microtubules, and the development of Tau monomers and oligomers, which have aggregation potential and can form Tau fibrils, which aggregate into neurofibrillary tangles (NFTs), a hallmark for AD and DS-AD. Intra-neuronal NFT accumulation leads to neuroinflammation, autophagy dysfunction, and mitochondrial dysfunction. Tau fibrils and oligomers are seeding competent and are secreted in exosomes.spread to other neurons and to astrocytes, microglia, or oligodendrocytes.
3.2. Neuronal exosomes harbor pathogenic Tau seeds
A few years ago, Goetzl et al. [83-85] developed a new biomarker method, in which exosomes derived from neurons (NDEs, [83]) or astrocytes (ADEs, [84,85]) were purified from plasma or serum samples and could predict dementia diagnoses and show both amyloid and Tau pathology cargo in exosomes decades prior to the onset of dementia [83]. We utilized this method and purified NDEs from plasma samples of individuals with DS at different ages, from childhood to older adults [86], and showed that NDEs from children with DS contained different p-Tau species already early in life, compared to typically developing children. Our group recently showed that NDEs, isolated from DS-AD plasma, contained seeding competent Tau conformers [20], using consecutive cycles of shear-induced fragmentation[24,27, 51,87]. As discussed below, NDEs from DS-AD patients injected into the hippocampus of wild-type mice gave rise to the spreading of Tau pathology to different portions of the mouse brain. The ability for misfolded Tau to spread from a patient with DS-AD to the mouse brain had not been demonstrated previously. Others have shown that toxic isoforms of Tau can be carried in exosomes and spread to other neurons via the normal exosomal expulsion/uptake mechanisms (Figure 2, see also [20,88,89]).
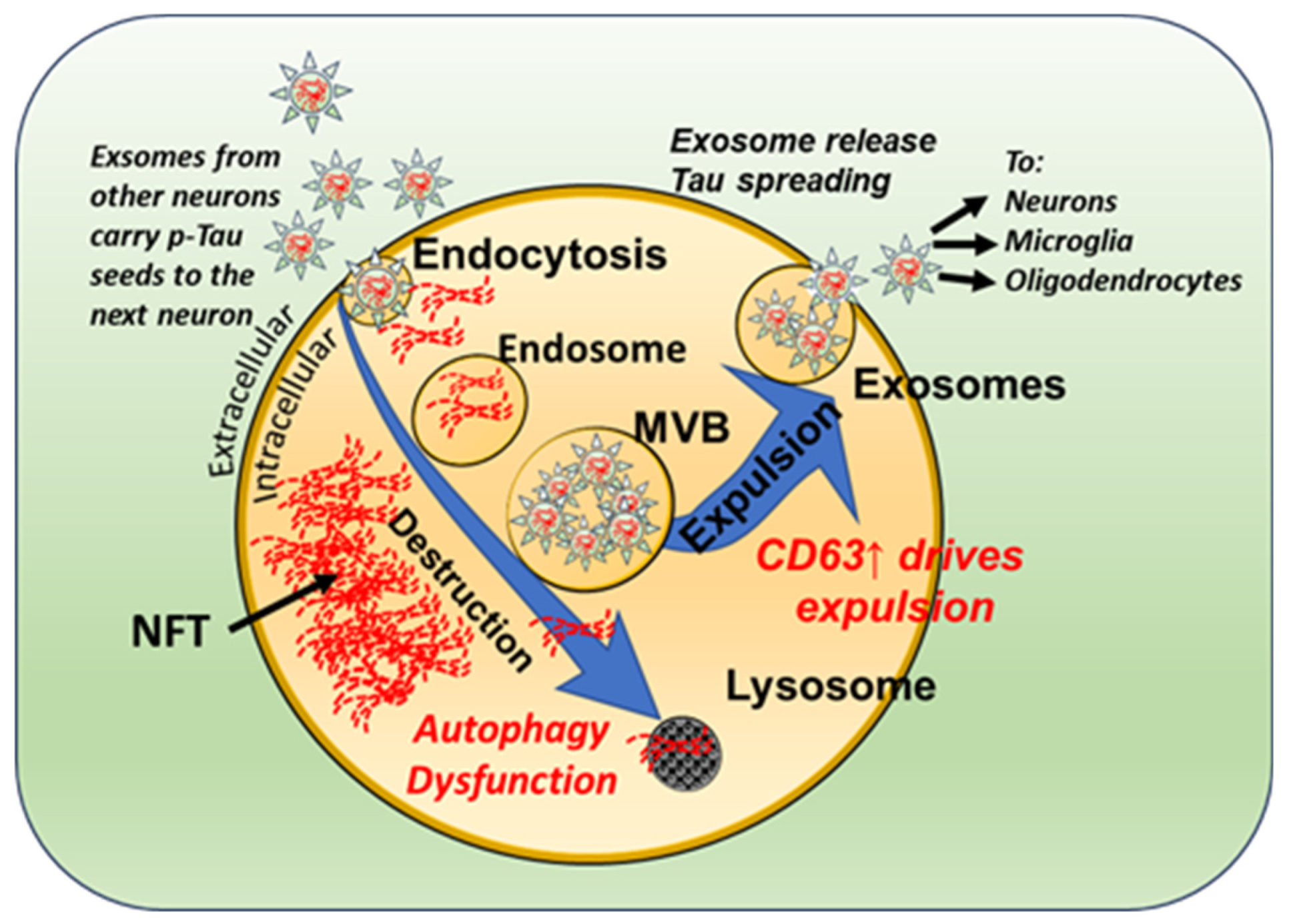
Figure 2.
Schematic drawing showing alterations in the exosomal pathways in DS. Exosomal secretion is increased in individuals with DS due to a reduced lysosomal/autophagy function and increased levels of the tetraspannin CD63. Endocytosis leads to the accumulation of misfolded Tau (oligomeric and/or fibrillar, Red threads) intracellularly into NFTs. Exosomes containing the toxic Tau species can either undergo autophagy or be expelled. It has been shown that exosomes released from neurons can be taken up by other neurons and glial cells in the vicinity.
Figure 2.
Schematic drawing showing alterations in the exosomal pathways in DS. Exosomal secretion is increased in individuals with DS due to a reduced lysosomal/autophagy function and increased levels of the tetraspannin CD63. Endocytosis leads to the accumulation of misfolded Tau (oligomeric and/or fibrillar, Red threads) intracellularly into NFTs. Exosomes containing the toxic Tau species can either undergo autophagy or be expelled. It has been shown that exosomes released from neurons can be taken up by other neurons and glial cells in the vicinity.
3.3. Tau seeds impart a bystander-spreading effect in the brain
Since p-Tau and other forms of toxic Tau are part of exosome cargo and can be spread in the brain via exosomes (Figure 2 and [88]), we conducted experiments in which neuron-derived exosomes (NDEs) were purified from DS-AD plasma and injected into the dorsal hippocampus of a wild-type mouse (WT) using stereotaxic techniques (Figure 3 and [20]). Control experiments using exosome-specific antibodies and Nanosight technology, demonstrated the purity of the NDE injection and confirmed that only NDEs and not other vesicles with different cellular origins were injected [20].
Further, in our studies, we observed a significant spreading of p-Tau (S396) immunoreactivity (i.r.) after the NDE injection after NDEs from patients with DS-AD, but not after control NDE injections. We saw no evidence of spreading of p-Tau (T231) i.r. beyond the cells that were stained with this different p-Tau isoform in the hippocampus. Double labeling with glial and neuronal stains revealed that most of the white matter cells stained with p-Tau (S396) antibodies also co-labeled with Olig-2 and SOX-10; two stains that are specific for oligodendrocytes in the CNS. These findings suggest that NDEs from DS-AD patients can give rise to p-Tau (S396) seeding in the WT mouse brain and lead to the spreading of Tau pathology from neurons in the hippocampus to oligodendrocytes in the white matter and on to other brain regions, at least after NDE injections into the mouse brain [20].
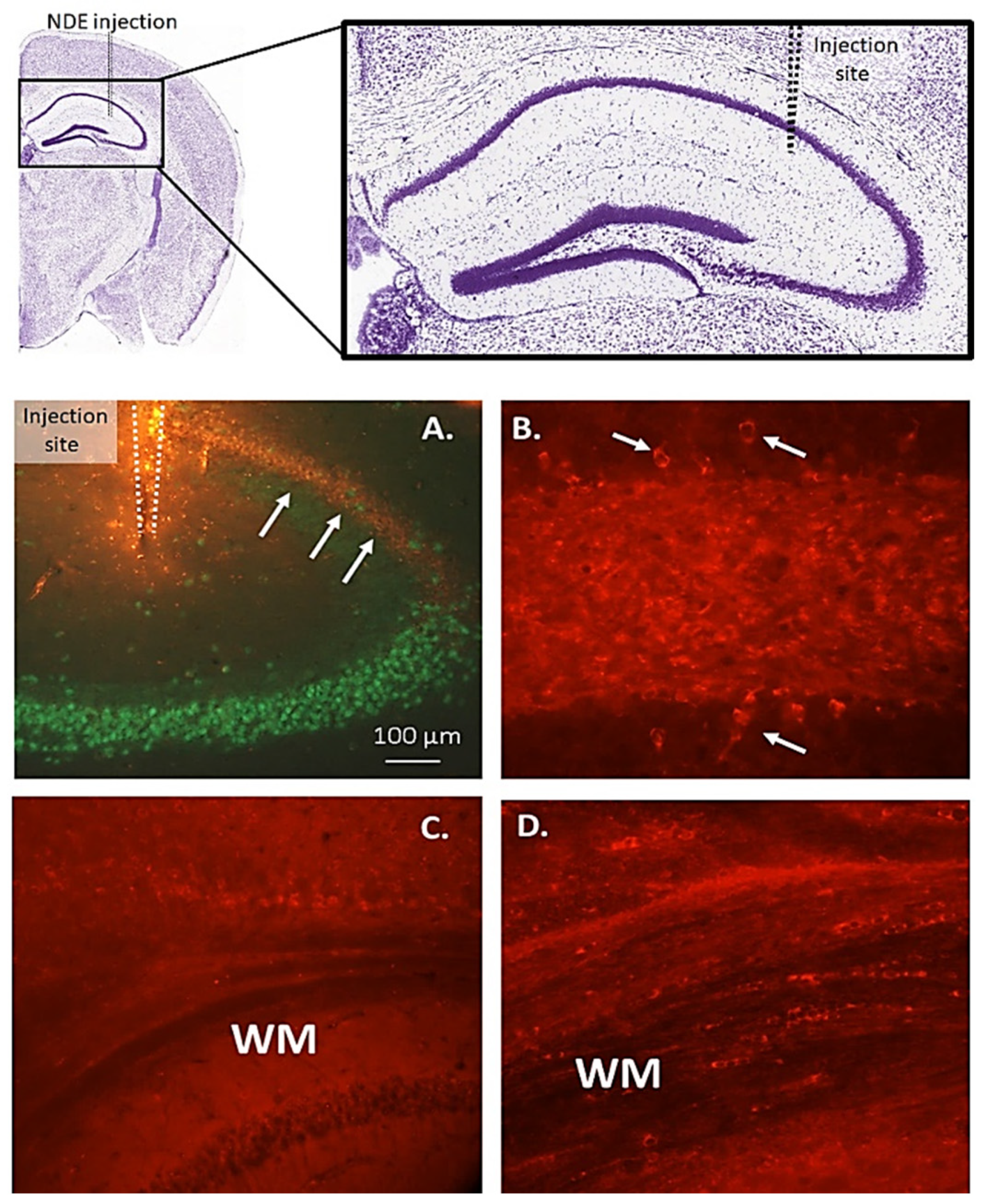
Figure 3.
Stereotaxic injection of DS-AD neuron-derived exosomes (NDEs) into the dorsal hippocampus gave rise to a spreading of p-Tau (S396) immunostaining along the CA1 pyramidal cell layer (A, arrows) to reach distances several millimeters away from the injection site after several months. Within the hippocampal dentate gyrus (B), multiple flame-like neuronal inclusions of p-Tau (S396) i.r. could be seen 1-month post-injection (B, small white arrows), and a more diffuse, granular staining was observed within the hilar region (B). The NDE injection gave rise to a plexus of small cell bodies and fibers in the white matter (WM) that stained for p-Tau (S396) (D) but not for p-Tau (T231, C), suggesting that certain isoforms of p-Tau are more involved in seeding from neurons to oligodendrocytes in the white matter.
Figure 3.
Stereotaxic injection of DS-AD neuron-derived exosomes (NDEs) into the dorsal hippocampus gave rise to a spreading of p-Tau (S396) immunostaining along the CA1 pyramidal cell layer (A, arrows) to reach distances several millimeters away from the injection site after several months. Within the hippocampal dentate gyrus (B), multiple flame-like neuronal inclusions of p-Tau (S396) i.r. could be seen 1-month post-injection (B, small white arrows), and a more diffuse, granular staining was observed within the hilar region (B). The NDE injection gave rise to a plexus of small cell bodies and fibers in the white matter (WM) that stained for p-Tau (S396) (D) but not for p-Tau (T231, C), suggesting that certain isoforms of p-Tau are more involved in seeding from neurons to oligodendrocytes in the white matter.
These results are corroborated by findings by Ferrer et al. [90], who recently published an interesting study where they showed that oligodendrocytes are highly involved in Tau spreading in the brain after brain homogenate injections from patients with different tauopathies, including AD, primary age-related tauopathy (PART), aging-related tau astrogliopathy (ARTAG) and globular glial tauopathy (GGT). After inoculation into the mouse brain, they reported that p-Tau aggregates were found in oligodendrocytes along with neurons to a greater degree than in other glial populations [90]. They also discovered that Tau seeding from human brain gave rise to a response in mouse neurons, with increased levels of active Tau kinases, including p38 and ERK 1/2, suggesting that human misfolded Tau could give rise to active Tau phosphorylation of murine Tau [90]. Further, the seeding effect of pathogenic Tau seeds from AD brains has been shown to trigger Tau spread in macaque brain brains [91]. Thus, to date, misfolded Tau isoforms can spread as seeds between different species and give rise to Tau pathology in the recipient brain – causing alterations in the conformation of the recipient Tau. Such seeding competency has not been described yet for amyloid, which is another prion-like protein in the AD brain. However, it has recently been shown that amyloid can promote seeding capacity and spreading of both Tau and alpha-synuclein in neurodegenerative disorders [92].
We do not yet know whether the injections into mouse hippocampus of DS-AD NDEs give rise to murine Tau inclusions in neurons or oligodendrocytes in our studies, but this important concept is the topic for future studies. We also have not seen other studies investigating the seeding potency of p-Tau (S396) versus other isoforms of p-Tau, and this will be an interesting topic for future experiments. It is also possible that human tau mRNA transcript isoforms with differences in the predominance of 3R tau versus 4R tau may exhibit different seeding potencies and/or different morphological consequences in the host mouse brain, as suggested by some investigators [93]. Ferrer and collaborators showed that Tau strains produce different patterns of active neuronal seeding, which also depend on the host Tau [90,93]. These studies could have a significant bearing on the pathological cascade in the brain caused by Tau misfolding and seeding and can lead to better and more targeted drugs that could stop the progression of dementia in several different tauopathies.
4. Tau biomarkers in biofluids, including exosomes.
Individuals with DS develop dementia and AD pathology at a variable age, but earlier than that seen in late-onset AD (LOAD) and perhaps earlier than seen with EOAD. Given that approximately 40–80% of individuals with DS develop AD-like dementia by the fifth decade of life [94], one might hypothesize that Tau seeding and spread within the DS brain may occur well within the fourth decade. Recent improvements in measuring P-Tau by immunoassays, seeding assays and Positron Emission tomography (PET) have enabled testing of this hypothesis and led to several breakthroughs and new questions about when Tau gains seeding “prion-like” capacities.
4.1. Ultrasensitive Immunoassays
With the recent development of new sensitive assays such as the Single Molecule Array (Simoa [95]), investigators are now capable of measuring even minute amounts of biomarkers in plasma during different neurological conditions. Using these novel sensitive measures, we performed a study focused on exosome biomarkers in individuals with DS, showing increased amyloid exosomal levels already in early childhood, and levels increased through adulthood [18]. However, we found an unexpected increase also in p-Tau (S396 and T181) in comparisons between all DS and all control participants at an early age, and with an increase with age in p-Tau (T181) [86]. These findings suggested that misfolding and phosphorylation of Tau may be an early event in DS and could be used to predict the onset of dementia as well as treatment efficacy also in this population. Measuring biomarkers in exosomes proved to be a sensitive and possibly less variable assay of dementia-related biomarkers than that previously shown for the same biomarkers in plasma due to the relatively low levels of AD-related proteins observed in plasma.
In older adults with DS, investigators in the Alzheimer Biomarkers Consortium — Down Syndrome (ABC-DS) have demonstrated early changes in persons with DS in different p-Tau isoforms in plasma [96]. The Petersen et al. group [96] showed, studying more than 300 participants with DS at different ages, that plasma levels of total Tau and neurofilament light (NfL) were highly predictable of both AD pathology and clinical status in those with DS at different ages. In another recent biomarker study from the European DS clinical network Horizon 21, Carmona and collaborators examined neurofilament light (NfL) [97] and found that NfL plasma levels had excellent diagnostic performance and a highly similar temporal distribution of change compared to that seen in autosomal dominant AD [97]. Phospho-Tau biomarkers in plasma have also been examined using ultrasensitive methods [98,99]. Plasma p-Tau (T217), glial fibrillary acidic protein (GFAP), amyloid beta peptides 42 and 40 (Aβ42/Aβ40), NfL, and total Tau (t-Tau) were assayed. The study included 300 participants with DS and 37 non-DS siblings. They found that higher p-Tau (T217) levels but no other biomarkers were associated with worse performance on DS Mental Status Examination and Cued Recall Test, thus suggesting that plasma p-Tau (T217) is an accurate blood-based biomarker of both Tau and Aβ pathological brain changes in DS that could be used for inclusion of individuals with DS in AD clinical trials, especially when combined with age as a covariate [98]. Since plasma levels of AD-related pathology is easier to conduct experimentally than NDE assessments, this newly developed measure may lend itself better to large clinical studies than the more cumbersome exosomal biomarker studies [19,100].
4.2. Tau seeding and aggregation assays
Promising techniques have been developed for the quantitation of Tau seeding activity in human biofluid. Holme et al. (2014) developed an in-cell-based assay where a Tau-containing biosample is incubated with a cell line overexpressing tau linked to a fluorescent protein [81]. Upon aggregation, energy transfers between fluorescent proteins (FRET) allowed for the detection of seeding capacity signals using flow cytometry. Using this FRET method, robust Tau seeding activity can be 1 month before histopathological stains show NFTs, suggesting that tau seeding is an early signature of tauopathy. Cell-free assays have been developed Real-Time Quaking-Induced Conversion (RT-QuIC) to measure prion seeding [101] and, more recently, Tau seeding [102]. Tau seed-containing material from biofluids or brain samples is incubated with recombinant tau substrate and thioflavin T in optimized conditions. Using RT-QuIC, seed-competent material induces the aggregation of the substrate, which generates a fluorescent signal.
Jin et al. (2022) recently developed a Tau seeding activity assay using truncated HA-tagged Tau151-391 peptide and cellular transformation [103]. Cells transiently express the HA-tagged Tau151-391 peptide, which is readily captured and aggregated by oligomeric Tau derived from postmortem AD brain samples. The captured Tau is then quantified using traditional immunoblot methods. They employed these assays on AD and DS brain samples in two different studies [104] and discovered that only brain extracts from AD or DS brain samples that contained hyperphosphorylated Tau seeded Tau aggregation in cultured cells. Interestingly, Tau extracts of DS corpus callosum showed low tau seeding activity, and no detectable tau seeding activity was observed in DS cerebellar cortex. Again, Tau seeding ability was found to be highly correlated with phosphorylation, but the group did not study effects of other Tau PTMs in this assay.
4.3. Tau binding studies using positron emission tomography (PET) ligands
Tau PET imaging is emerging as an important clinical tool for early diagnosis of AD and other tauopathies since p-Tau appears to correlate more closely than amyloid with symptomatic dementia progression. In the last ten years, multiple Tau PET ligands have been developed. Tau-specific ligands for use in PET include the first-generation ligands (e.g., [18F]THK5317, [18F]THK5351, [18F]AV1451, and [11C]PBB3) and, consequently, a set of second-generation ligands (e.g. [18F]MK-6240, [18F]RO-948, [18F]PI-2620, [18F]GTP1, [18F]PM-PBB3, and [18F]JNJ64349311 ([18F]JNJ311), see Leuzy et al. [105]. Several of these Tau PET ligands have been tested in patients with DS and AD, for example, the [18F]-AV-1451 Tau PET ligand [106]. Rafii et al. demonstrated that the amyloid-negative participants with DS imaged were all Tau-negative and that both amyloid and Tau burden correlated with age [106]. They also found that Tau binding in the brain correlated well with cognitive decline, suggesting that this clinical measure can be used to predict the onset or progression of dementia in DS-AD. Recently, Dr. Bradley Christian and his research group have performed longitudinal PET analyses in the DS population to better define a timeline of the progression of amyloid and Tau burden through the conventional Braak stages [107-109]. Based on PET scans, they revealed early and rapid Tau elevation following the onset of amyloid-positive PET imaging. While Tau elevation is highly variable in AD cohorts, individuals with DS displayed uniform increases in Tau within 2.5 years of the onset of amyloid binding in the brain.
To further quantify the binding of Tau PET ligands in specific brain regions, we performed an autoradiographic binding study of Tau PET ligand binding in postmortem, fixed and paraffin-embedded brain tissue sections from individuals that received a neuropathological diagnosis of either LOAD, EOAD, or DS-AD, and age-matched controls [110]. Interestingly, we found that binding of both the first generation (THK5117) and the second generation (MK6240) Tau PET ligand exhibited a significant increase in frontal cortex (middle frontal gyrus) in DS-AD postmortem cases compared to both EOAD and LOAD cases. In addition, autoradiographic binding of both of these Tau ligands correlated significantly with AT8 p-Tau immunostaining on adjacent sections, strongly suggesting that on-target binding was more prevalent than off-target binding, at least in these fixed and paraffin-embedded sections [110]. Other studies have suggested that in fresh frozen materials and in vivo, there is off-target binding of Tau ligands to, for example, monoamine oxidase B (MAO-B)[111], which did not appear to be the case in our study using fixed materials. Investigators have identified a binding site for all the Tau tracers on MAO-B [111], which could make clinical studies difficult to undertake unless a better understanding of off-target binding is first achieved. However, it was reported in the Murugan et al. manuscript [111] that a second-generation Tau PET ligand, MK6240, has a lower affinity for MAO-B than the first-generation tracers. Nevertheless, in our binding study, we found a significant correlation between p-Tau immunostaining (AT8 antibodies) with both the first and second-generation Tau ligands used in that study [110], suggesting less off-target binding in fixed tissues than what was previously reported for in vivo studies.
5. Discussion
Tau pathology is an important component of AD research but also carries a significant pathological burden in other tauopathies [112]. Tauopathies can be classified clinically by a range of symptoms that involve cognition, behavior, mental health, motor function, language disabilities and non-specific amnestic or executive symptoms. Pathologically, tauopathies can be classified based on the predominant Tau isoforms and/or phosphorylation sites that are present in inclusion bodies or cytoskeletal fibrils, including the ratio of 3R:4R and truncation of the Tau protein at specific sites [112]. The most recently classified tauopathy is Chronic Traumatic Encephalopathy (CTE), which pathologically is classified by accumulation of Tau inclusions, particularly in the sulci [29]. Recent work has shown that repeated traumatic brain injuries (TBIs) in mouse or rat brains give rise to an accumulation of cis-phosphorylated Tau [113] as well as Tau oligomers in CSF or brain tissue. However, the specific form and inclusion pattern of Tau appears to be significantly different in different forms of tauopathy [21,45].
In individuals with DS and AD (DS-AD), Condello and collaborators found a significant difference in prion activity compared to other forms of AD, where DS brains showed a continuously increasing prion activity of Tau and amyloid with age; this is contrary to the reduced prion activity of these proteins observed in brains from patients with LOAD or EOAD [50]. This would suggest that DS brains contain a particularly violent form of Tau seeds and may, at least partially, explain the early onset of AD pathology and dementia seen in this population [5].
Our binding studies using Tau tracers suggested that the frontal cortex was more affected in terms of Tau binding in DS-AD compared to LOAD or EOAD [110], potentially due to the developmental detriments in frontal cortex organization that have been reported for children with DS, with hypoconnectivity [114], reduced neuronal numbers and brain volume [115], and underdeveloped neuronal migration in the frontal cortex [70,116]. This could contribute to the relative vulnerability of the frontal cortex to Tau and amyloid pathology as the individual with DS gets older compared to the frontal cortex in typically developing individuals.
The fact that the gene that encodes amyloid precursor protein (APP) is located on Chr. 21 [117] can also contribute to the vulnerability of individuals with DS to AD pathology. Since APP in the fetal brain is highly involved in neurogenesis, neuronal differentiation and synaptogenesis during neurotypical development [118,119], an overproduction of amyloid could lead to a loss of normal APP function during development – hence, a delayed migration of neurons in the cortical plate and/or disturbance in synaptic development. The Tau protein is also directly involved in neural and synaptic development. Studies have shown that human phosphorylated fetal Tau (3R, containing exon 0) is found in the distal portions of cortical growth cones [120] and contributes to axonal growth and maturation [120]. This isoform of p-Tau does not generate aggregates in the brain but is altered early in the maturation of the DS brain [121], potentially also contributing to a reduced maturation, particularly of cortical regions. Thus, although we know that aberrant prenatal brain development in individuals with DS can disrupt the function and structure of cortical areas during adulthood and aging, further studies are needed in order to design appropriate interventions that can halt or reduce the impact of AD pathology in individuals with DS.
6. Conclusions
Tau misfolding and aggregation play a major role in AD pathology as well as in DS-AD pathology. This review demonstrates recent work focused on understanding the role of NFTs and monomeric/oligomeric Tau for the prion-like spreading of Tau pathology in the human brain to come up with novel treatment paradigms for this debilitating and early onset AD form that occurs in those with DS. This review sheds light on the underpinnings of Tau pathology in DS-AD and demonstrates the central role that misfolding of the Tau protein plays in this pathological process.
Author Contributions
ACG and EH contributed substantially and equally to the conceptualization and writing of this review. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful for funding from the BrightFocus Foundation (Grant no. CA2018010) and individual research grants from the NIH. ACG is funded by NIH grants R01AG071228-02, R01AG070153, and R01AG061566. EDH is funded by the Alzheimer’s Association (Grant no. AARG-22-974669) and the Infectious Diseases Society of America (Grant no. 00128).
Institutional Review Board Statement
This review did not require ethical approval.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the studies upon which this review is based.
Data Availability Statement
Data pertaining to this review are available via the DSBC website (https://medschool.cuanschutz.edu/neurosurgery/research-and-innovation/services/down-syndrome-biobank) or via our previously published work as indicated in the publication list.
Acknowledgments
The authors would like to acknowledge the patients and their families who donated their brains for research as well as the autopsy personnel and clinical coordinators who worked diligently with the brain donation network. We would also like to acknowledge the expert technical assistance provided by Ms. Anah Gilmore.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Alldred, M.J.; Martini, A.C.; Patterson, D.; Hendrix, J.; Granholm, A.C. Aging with Down Syndrome-Where Are We Now and Where Are We Going? J Clin Med 2021, 10. [Google Scholar] [CrossRef]
- Delabar, J.M.; Allinquant, B.; Bianchi, D.; Blumenthal, T.; Dekker, A.; Edgin, J.; O'Bryan, J.; Dierssen, M.; Potier, M.C.; Wiseman, F.; et al. Changing Paradigms in Down Syndrome: The First International Conference of the Trisomy 21 Research Society. Mol Syndromol 2016, 7, 251–261. [Google Scholar] [CrossRef]
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer's Disease Neuropathology. Curr Alzheimer Res 2016, 13, 18–29. [Google Scholar] [CrossRef]
- Flores-Aguilar, L.; Iulita, M.F.; Kovecses, O.; Torres, M.D.; Levi, S.M.; Zhang, Y.; Askenazi, M.; Wisniewski, T.; Busciglio, J.; Cuello, A.C. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 2020, 143, 3653–3671. [Google Scholar] [CrossRef]
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer's disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol 2021, 20, 930–942. [Google Scholar] [CrossRef]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer's disease: Common pathways, common goals. Alzheimers Dement 2015, 11, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Bartley, M.G.; Marquardt, K.; Kirchhof, D.; Wilkins, H.M.; Patterson, D.; Linseman, D.A. Overexpression of amyloid-beta protein precursor induces mitochondrial oxidative stress and activates the intrinsic apoptotic cascade. J Alzheimers Dis 2012, 28, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Helman, A.M.; Siever, M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Abner, E.L.; Schmitt, F.A.; Head, E. Microbleeds and Cerebral Amyloid Angiopathy in the Brains of People with Down Syndrome with Alzheimer's Disease. J Alzheimers Dis 2019, 67, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.L.; Isacson, O.; Nelson, M.; Bimonte-Nelson, H.; Seo, H.; Lin, L.; Ford, K.; Kindy, M.S.; Granholm, A.C. Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down's syndrome. Neurosci Res 2003, 45, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Millan Sanchez, M.; Heyn, S.N.; Das, D.; Moghadam, S.; Martin, K.J.; Salehi, A. Neurobiological elements of cognitive dysfunction in down syndrome: exploring the role of APP. Biol Psychiatry 2012, 71, 403–409. [Google Scholar] [CrossRef]
- Chung, H.; Green, P.H.R.; Wang, T.C.; Kong, X.F. Interferon-Driven Immune Dysregulation in Down Syndrome: A Review of the Evidence. J Inflamm Res 2021, 14, 5187–5200. [Google Scholar] [CrossRef]
- Powers, R.K.; Culp-Hill, R.; Ludwig, M.P.; Smith, K.P.; Waugh, K.A.; Minter, R.; Tuttle, K.D.; Lewis, H.C.; Rachubinski, A.L.; Granrath, R.E.; et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun 2019, 10, 4766. [Google Scholar] [CrossRef] [PubMed]
- Ram, G.; Chinen, J. Infections and immunodeficiency in Down syndrome. Clin Exp Immunol 2011, 164, 9–16. [Google Scholar] [CrossRef]
- Martini, A.C.; Helman, A.M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Schmitt, F.A.; Head, E. Distribution of microglial phenotypes as a function of age and Alzheimer's disease neuropathology in the brains of people with Down syndrome. Alzheimers Dement (Amst) 2020, 12, e12113. [Google Scholar] [CrossRef]
- Barone, E.; Arena, A.; Head, E.; Butterfield, D.A.; Perluigi, M. Disturbance of redox homeostasis in Down Syndrome: Role of iron dysmetabolism. Free Radic Biol Med 2018, 114, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, J.A.; Amon, A.; Abbeduto, L.; Agiovlasitis, S.; Alsaied, T.; Anderson, H.A.; Bain, L.J.; Baumer, N.; Bhattacharyya, A.; Bogunovic, D.; et al. Opportunities, barriers, and recommendations in down syndrome research. Transl Sci Rare Dis 2021, 5, 99–129. [Google Scholar] [CrossRef]
- Snyder, H.M.; Bain, L.J.; Brickman, A.M.; Carrillo, M.C.; Esbensen, A.J.; Espinosa, J.M.; Fernandez, F.; Fortea, J.; Hartley, S.L.; Head, E.; et al. Further understanding the connection between Alzheimer's disease and Down syndrome. Alzheimers Dement 2020, 16, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer's disease biomarkers in Down syndrome. Alzheimers Dement 2017, 13, 541–549. [Google Scholar] [CrossRef]
- Hamlett, E.D.; LaRosa, A.; Mufson, E.J.; Fortea, J.; Ledreux, A.; Granholm, A.C. Exosome release and cargo in Down syndrome. Dev Neurobiol 2019, 79, 639–655. [Google Scholar] [CrossRef]
- Ledreux, A.; Thomas, S.; Hamlett, E.D.; Trautman, C.; Gilmore, A.; Rickman Hager, E.; Paredes, D.A.; Margittai, M.; Fortea, J.; Granholm, A.C. Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain. J Clin Med 2021, 10. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer's Disease and Other Tauopathies. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Meyer, V.; Dinkel, P.D.; Luo, Y.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; Eaton, S.S.; et al. Single mutations in tau modulate the populations of fibril conformers through seed selection. Angew Chem Int Ed Engl 2014, 53, 1590–1593. [Google Scholar] [CrossRef]
- Siddiqua, A.; Luo, Y.; Meyer, V.; Swanson, M.A.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; et al. Conformational basis for asymmetric seeding barrier in filaments of three- and four-repeat tau. J Am Chem Soc 2012, 134, 10271–10278. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol Rev 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O.; et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J 2015, 34, 3028–3041. [Google Scholar] [CrossRef] [PubMed]
- Weismiller, H.A.; Murphy, R.; Wei, G.; Ma, B.; Nussinov, R.; Margittai, M. Structural disorder in four-repeat Tau fibrils reveals a new mechanism for barriers to cross-seeding of Tau isoforms. J Biol Chem 2018, 293, 17336–17348. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med 2020, 26, 1256–1263. [Google Scholar] [CrossRef]
- Armstrong, R.A.; McKee, A.C.; Alvarez, V.E.; Cairns, N.J. Clustering of tau-immunoreactive pathology in chronic traumatic encephalopathy. J Neural Transm (Vienna) 2017, 124, 185–192. [Google Scholar] [CrossRef]
- Capano, L.S.; Sato, C.; Ficulle, E.; Yu, A.; Horie, K.; Kwon, J.S.; Burbach, K.F.; Barthelemy, N.R.; Fox, S.G.; Karch, C.M.; et al. Recapitulation of endogenous 4R tau expression and formation of insoluble tau in directly reprogrammed human neurons. Cell Stem Cell 2022, 29, 918–932 e918. [Google Scholar] [CrossRef]
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L., 3rd; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol 2017, 133, 91–100. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 2011, 286, 15317–15331. [Google Scholar] [CrossRef] [PubMed]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front Neurol 2020, 11, 595532. [Google Scholar] [CrossRef] [PubMed]
- Trushina, N.I.; Bakota, L.; Mulkidjanian, A.Y.; Brandt, R. The Evolution of Tau Phosphorylation and Interactions. Front Aging Neurosci 2019, 11, 256. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. The Journal of biological chemistry 2007, 282, 34850–34857. [Google Scholar] [CrossRef]
- Neumann, F.; Gourdain, S.; Albac, C.; Dekker, A.D.; Bui, L.C.; Dairou, J.; Schmitz-Afonso, I.; Hue, N.; Rodrigues-Lima, F.; Delabar, J.M.; et al. DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci Rep 2018, 8, 2859. [Google Scholar] [CrossRef]
- Laham, A.J.; Saber-Ayad, M.; El-Awady, R. DYRK1A: a down syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cell Mol Life Sci 2021, 78, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J 2008, 22, 3224–3233. [Google Scholar] [CrossRef]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci 2018, 8. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.S.; Kim, A.K.; Choi, M.; Choi, K.; Kang, M.; Chi, S.W.; Lee, M.S.; Lee, J.S.; Lee, S.Y.; et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Disease models & mechanisms 2016, 9, 839–848. [Google Scholar] [CrossRef]
- Melchior, B.; Mittapalli, G.K.; Lai, C.; Duong-Polk, K.; Stewart, J.; Guner, B.; Hofilena, B.; Tjitro, A.; Anderson, S.D.; Herman, D.S.; et al. Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for Alzheimer's disease. Aging Cell 2019, 18, e13000. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Parsons, T.; Foley, C.; Shaw, Y.; Dunckley, T.; Hulme, C.; Hodge, J.J.L. DYRK1A antagonists rescue degeneration and behavioural deficits of in vivo models based on amyloid-beta, Tau and DYRK1A neurotoxicity. Sci Rep 2022, 12, 15847. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sierra, F.; Mondragon-Rodriguez, S.; Basurto-Islas, G. Truncation of tau protein and its pathological significance in Alzheimer's disease. J Alzheimers Dis 2008, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.X.; Iqbal, K.; Liu, F. Truncation of Tau selectively facilitates its pathological activities. J Biol Chem 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.H.; Nakamoto, Y. Autophagy and Tau Protein. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Antcliff, A.; McCullough, L.D.; Tsvetkov, A.S. G-Quadruplexes and the DNA/RNA helicase DHX36 in health, disease, and aging. Aging (Albany NY) 2021, 13, 25578–25587. [Google Scholar] [CrossRef]
- Vijay Kumar, M.J.; Morales, R.; Tsvetkov, A.S. G-quadruplexes and associated proteins in aging and Alzheimer's disease. Front Aging 2023, 4, 1164057. [Google Scholar] [CrossRef]
- Kallweit, L.; Hamlett, E.D.; Saternos, H.; Gilmore, A.; Granholm, A.C.; Horowitz, S. A New Role for RNA G-quadruplexes in Aging and Alzheimer's Disease. bioRxiv 2023. [Google Scholar] [CrossRef]
- Condello, C.; Merz, G.E.; Aoyagi, A.; DeGrado, W.F.; Prusiner, S.B. Abeta and Tau Prions Causing Alzheimer's Disease. Methods Mol Biol 2023, 2561, 293–337. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Maxwell, A.M.; Castillo, E.; Aoyagi, A.; Graff, C.; Ingelsson, M.; Lannfelt, L.; Bird, T.D.; Keene, C.D.; Seeley, W.W.; et al. Abeta and tau prions feature in the neuropathogenesis of Down syndrome. Proc Natl Acad Sci U S A 2022, 119, e2212954119. [Google Scholar] [CrossRef]
- Dinkel, P.D.; Siddiqua, A.; Huynh, H.; Shah, M.; Margittai, M. Variations in filament conformation dictate seeding barrier beween three- and four-repeat tau. Biochemistry 2011, 50, 4330–4336. [Google Scholar] [CrossRef]
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau Oligomers as Pathogenic Seeds: Preparation and Propagation In Vitro and In Vivo. Methods Mol Biol 2017, 1523, 141–157. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu Rev Neurosci 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated microglia mitigate Abeta-associated tau seeding and spreading. J Exp Med 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Mate De Gerando, A.; Welikovitch, L.A.; Khasnavis, A.; Commins, C.; Glynn, C.; Chun, J.E.; Perbet, R.; Hyman, B.T. Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau. Acta Neuropathol 2023, 146, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Phillips, C.; Hsieh, W.; Sumanth, K.; Dang, V.; Salehi, A. Neurotransmitter-based strategies for the treatment of cognitive dysfunction in Down syndrome. Prog Neuropsychopharmacol Biol Psychiatry 2014, 54, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, R.; McNerney, M.W.; Moghadam, S.; Salehi, A. Assessing disease-modifying effects of norepinephrine in Down syndrome and Alzheimer's disease. Brain Res 2019, 1702, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Granholm, A.C.; Sanders, L.A.; Crnic, L.S. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down's syndrome. Exp Neurol 2000, 161, 647–663. [Google Scholar] [CrossRef]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Flores Aguilar, L.; Hanna, M.; Wisniewski, T.; Granholm, A.C.; Buhusi, M.; Busciglio, J.; et al. Nerve growth factor metabolic dysfunction in Down's syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef]
- Martinez, J.L.; Zammit, M.D.; West, N.R.; Christian, B.T.; Bhattacharyya, A. Basal Forebrain Cholinergic Neurons: Linking Down Syndrome and Alzheimer's Disease. Front Aging Neurosci 2021, 13, 703876. [Google Scholar] [CrossRef]
- Sendera, T.J.; Ma, S.Y.; Jaffar, S.; Kozlowski, P.B.; Kordower, J.H.; Mawal, Y.; Saragovi, H.U.; Mufson, E.J. Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down's syndrome. Journal of neurochemistry 2000, 74, 1185–1196. [Google Scholar] [CrossRef]
- Ball, M.J.; Nuttall, K. Neurofibrillary tangles, granulovacuolar degeneration, and neuron loss in Down Syndrome: quantitative comparison with Alzheimer dementia. Ann Neurol 1980, 7, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. mTOR Hyperactivation in down syndrome hippocampus appears early during development. J Neuropathol Exp Neurol 2014, 73, 671–683. [Google Scholar] [CrossRef]
- Milenkovic, I.; Stojanovic, T.; Aronica, E.; Fulop, L.; Bozso, Z.; Mate, Z.; Yanagawa, Y.; Adle-Biassette, H.; Lubec, G.; Szabo, G.; et al. GABAA receptor subunit deregulation in the hippocampus of human foetuses with Down syndrome. Brain Struct Funct 2018, 223, 1501–1518. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Wisniewski, T.; de Leon, M.J.; Reisberg, B.; Pirttila, T.; Kivimaki, T.; Lehtimaki, T. Vascular fibrosis and calcification in the hippocampus in aging, Alzheimer disease, and Down syndrome. Acta Neuropathol 2002, 103, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Liu, C.; Belichenko, P.; Clapcote, S.J.; Li, S.; Pao, A.; Kleschevnikov, A.; Bechard, A.R.; Asrar, S.; Chen, R.; et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res 2010, 1366, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer's disease: Targeting the Cholinergic System. Curr Neuropharmacol 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer's Disease: Emerging Evidence from Translational and Clinical Research. J Prev Alzheimers Dis 2019, 6, 2–15. [Google Scholar] [CrossRef]
- Perez, S.E.; Miguel, J.C.; He, B.; Malek-Ahmadi, M.; Abrahamson, E.E.; Ikonomovic, M.D.; Lott, I.; Doran, E.; Alldred, M.J.; Ginsberg, S.D.; et al. Frontal cortex and striatal cellular and molecular pathobiology in individuals with Down syndrome with and without dementia. Acta neuropathologica 2019, 137, 413–436. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Ringman, J.M. Cognitive symptoms of Alzheimer's disease: clinical management and prevention. BMJ 2019, 367, l6217. [Google Scholar] [CrossRef]
- Martorana, A.; Esposito, Z.; Koch, G. Beyond the cholinergic hypothesis: do current drugs work in Alzheimer's disease? CNS Neurosci Ther 2010, 16, 235–245. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer's therapeutics (Review). Mol Med Rep 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Eady, N.; Sheehan, R.; Rantell, K.; Sinai, A.; Bernal, J.; Bohnen, I.; Bonell, S.; Courtenay, K.; Dodd, K.; Gazizova, D.; et al. Impact of cholinesterase inhibitors or memantine on survival in adults with Down syndrome and dementia: clinical cohort study. Br J Psychiatry 2018, 212, 155–160. [Google Scholar] [CrossRef]
- Vana, L.; Kanaan, N.M.; Ugwu, I.C.; Wuu, J.; Mufson, E.J.; Binder, L.I. Progression of tau pathology in cholinergic Basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. Am J Pathol 2011, 179, 2533–2550. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Fahimi, A.; Das, D.; Mojabi, F.S.; Ponnusamy, R.; Salehi, A. Noradrenergic System in Down Syndrome and Alzheimer's Disease A Target for Therapy. Curr Alzheimer Res 2016, 13, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J Neurosci 2018, 38, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Chalermpalanupap, T.; Weinshenker, D.; Rorabaugh, J.M. Down but Not Out: The Consequences of Pretangle Tau in the Locus Coeruleus. Neural Plast 2017, 2017, 7829507. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci 2018, 41, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Szabadi, E. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol 2008, 6, 235–253. [Google Scholar] [CrossRef]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 2014, 111, E4376–E4385. [Google Scholar] [CrossRef]
- Reilly, P.; Winston, C.N.; Baron, K.R.; Trejo, M.; Rockenstein, E.M.; Akers, J.C.; Kfoury, N.; Diamond, M.; Masliah, E.; Rissman, R.A.; et al. Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol Dis 2017, 106, 222–234. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer's disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement 2015, 11, 600–607. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer's disease. FASEB J 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann Neurol 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer's disease biomarkers in Down syndrome. Alzheimers Dement 2016. [Google Scholar] [CrossRef] [PubMed]
- Meyer, V.; Dinkel, P.D.; Rickman Hager, E.; Margittai, M. Amplification of Tau fibrils from minute quantities of seeds. Biochemistry 2014, 53, 5804–5809. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Gotz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J Biol Chem 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Aguilo Garcia, M.; Carmona, M.; Andres-Benito, P.; Torrejon-Escribano, B.; Garcia-Esparcia, P.; Del Rio, J.A. Involvement of Oligodendrocytes in Tau Seeding and Spreading in Tauopathies. Front Aging Neurosci 2019, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Darricau, M.; Dou, C.; Kinet, R.; Zhu, T.; Zhou, L.; Li, X.; Bedel, A.; Claverol, S.; Tokarski, C.; Katsinelos, T.; et al. Tau seeds from Alzheimer's disease brains trigger tau spread in macaques while oligomeric-Abeta mediates pathology maturation. Alzheimers Dement 2023. [Google Scholar] [CrossRef]
- Bassil, F.; Brown, H.J.; Pattabhiraman, S.; Iwasyk, J.E.; Maghames, C.M.; Meymand, E.S.; Cox, T.O.; Riddle, D.M.; Zhang, B.; Trojanowski, J.Q.; et al. Amyloid-Beta (Abeta) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Abeta Pathology. Neuron 2020, 105, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Zelaya, M.V.; Aguilo Garcia, M.; Carmona, M.; Lopez-Gonzalez, I.; Andres-Benito, P.; Lidon, L.; Gavin, R.; Garcia-Esparcia, P.; Del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol 2020, 30, 298–318. [Google Scholar] [CrossRef]
- Salehi, A.; Ashford, J.W.; Mufson, E.J. The Link between Alzheimer's Disease and Down Syndrome. A Historical Perspective. Current Alzheimer research 2016, 13, 2–6. [Google Scholar] [CrossRef]
- McCrea, M.; Broglio, S.P.; McAllister, T.W.; Gill, J.; Giza, C.C.; Huber, D.L.; Harezlak, J.; Cameron, K.L.; Houston, M.N.; McGinty, G.; et al. Association of Blood Biomarkers With Acute Sport-Related Concussion in Collegiate Athletes: Findings From the NCAA and Department of Defense CARE Consortium. JAMA Netw Open 2020, 3, e1919771. [Google Scholar] [CrossRef]
- Petersen, M.E.; Rafii, M.S.; Zhang, F.; Hall, J.; Julovich, D.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; et al. Plasma Total-Tau and Neurofilament Light Chain as Diagnostic Biomarkers of Alzheimer's Disease Dementia and Mild Cognitive Impairment in Adults with Down Syndrome. J Alzheimers Dis 2021, 79, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Iragui, M.; Alcolea, D.; Barroeta, I.; Videla, L.; Munoz, L.; Van Pelt, K.L.; Schmitt, F.A.; Lightner, D.D.; Koehl, L.M.; Jicha, G.; et al. Diagnostic and prognostic performance and longitudinal changes in plasma neurofilament light chain concentrations in adults with Down syndrome: a cohort study. Lancet Neurol 2021, 20, 605–614. [Google Scholar] [CrossRef]
- Janelidze, S.; Christian, B.T.; Price, J.; Laymon, C.; Schupf, N.; Klunk, W.E.; Lott, I.; Silverman, W.; Rosas, H.D.; Zaman, S.; et al. Detection of Brain Tau Pathology in Down Syndrome Using Plasma Biomarkers. JAMA Neurol 2022, 79, 797–807. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef]
- Hamlett, E.D.; Ledreux, A.; Potter, H.; Chial, H.J.; Patterson, D.; Espinosa, J.M.; Bettcher, B.M.; Granholm, A.C. Exosomal biomarkers in Down syndrome and Alzheimer's disease. Free Radic Biol Med 2018, 114, 110–121. [Google Scholar] [CrossRef]
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474. [Google Scholar] [CrossRef]
- Kraus, A.; Saijo, E.; Metrick, M.A., 2nd; Newell, K.; Sigurdson, C.J.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta neuropathologica 2019, 137, 585–598. [Google Scholar] [CrossRef]
- Liu, F.; Wu, R.; Jin, N.; Chu, D.; Gu, J.; Tung, Y.C.; Hu, Z.; Gong, C.X.; Iqbal, K. Two simple assays for assessing the seeding activity of proteopathic tau. Front Aging Neurosci 2023, 15, 1073774. [Google Scholar] [CrossRef]
- Jin, N.; Gu, J.; Wu, R.; Chu, D.; Tung, Y.C.; Wegiel, J.; Wisniewski, T.; Gong, C.X.; Iqbal, K.; Liu, F. Tau seeding activity in various regions of down syndrome brain assessed by two novel assays. Acta Neuropathol Commun 2022, 10, 132. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Rafii, M.S.; Lukic, A.S.; Andrews, R.D.; Brewer, J.; Rissman, R.A.; Strother, S.C.; Wernick, M.N.; Pennington, C.; Mobley, W.C.; Ness, S.; et al. PET Imaging of Tau Pathology and Relationship to Amyloid, Longitudinal MRI, and Cognitive Change in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J Alzheimers Dis 2017, 60, 439–450. [Google Scholar] [CrossRef]
- Lao, P.J.; Handen, B.L.; Betthauser, T.J.; Mihaila, I.; Hartley, S.L.; Cohen, A.D.; Tudorascu, D.L.; Bulova, P.D.; Lopresti, B.J.; Tumuluru, R.V.; et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down syndrome population. Alzheimers Dement (Amst) 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Zammit, M.D.; Tudorascu, D.L.; Laymon, C.M.; Hartley, S.L.; Zaman, S.H.; Ances, B.M.; Johnson, S.C.; Stone, C.K.; Mathis, C.A.; Klunk, W.E.; et al. PET measurement of longitudinal amyloid load identifies the earliest stages of amyloid-beta accumulation during Alzheimer's disease progression in Down syndrome. Neuroimage 2021, 228, 117728. [Google Scholar] [CrossRef]
- Zammit, M.D.; Betthauser, T.J.; McVea, A.K.; Laymon, C.M.; Tudorascu, D.L.; Johnson, S.C.; Hartley, S.L.; Converse, A.K.; Minhas, D.S.; Zaman, S.H.; et al. Characterizing the emergence of amyloid and tau burden in Down syndrome. Alzheimers Dement 2023. [Google Scholar] [CrossRef]
- Lemoine, L.; Ledreux, A.; Mufson, E.J.; Perez, S.E.; Simic, G.; Doran, E.; Lott, I.; Carroll, S.; Bharani, K.; Thomas, S.; et al. Regional binding of tau and amyloid PET tracers in Down syndrome autopsy brain tissue. Mol Neurodegener 2020, 15, 68. [Google Scholar] [CrossRef]
- Murugan, N.A.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Agren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: evidence from in silico modelling and in vivo imaging. Eur J Nucl Med Mol Imaging 2019, 46, 1369–1382. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, K.M.; Yang, L.; Dong, Q.; Yu, J.T. Tauopathies: new perspectives and challenges. Mol Neurodegener 2022, 17, 28. [Google Scholar] [CrossRef]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.Y.; Li, C.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 2017, 8, 1000. [Google Scholar] [CrossRef]
- Canete-Masse, C.; Carbo-Carrete, M.; Pero-Cebollero, M.; Cui, S.X.; Yan, C.G.; Guardia-Olmos, J. Abnormal degree centrality and functional connectivity in Down syndrome: A resting-state fMRI study. Int J Clin Health Psychol 2023, 23, 100341. [Google Scholar] [CrossRef]
- Hamadelseed, O.; Chan, M.K.S.; Wong, M.B.F.; Skutella, T. Distinct neuroanatomical and neuropsychological features of Down syndrome compared to related neurodevelopmental disorders: a systematic review. Front Neurosci 2023, 17, 1225228. [Google Scholar] [CrossRef] [PubMed]
- Utagawa, E.C.; Moreno, D.G.; Schafernak, K.T.; Arva, N.C.; Malek-Ahmadi, M.H.; Mufson, E.J.; Perez, S.E. Neurogenesis and neuronal differentiation in the postnatal frontal cortex in Down syndrome. Acta Neuropathol Commun 2022, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Kalil, K. The Microtubule-Associated Protein Tau Mediates the Organization of Microtubules and Their Dynamic Exploration of Actin-Rich Lamellipodia and Filopodia of Cortical Growth Cones. J Neurosci 2018, 38, 291–307. [Google Scholar] [CrossRef]
- Nicolas, M.; Hassan, B.A. Amyloid precursor protein and neural development. Development 2014, 141, 2543–2548. [Google Scholar] [CrossRef]
- Octave, J.N.; Pierrot, N.; Ferao Santos, S.; Nalivaeva, N.N.; Turner, A.J. From synaptic spines to nuclear signaling: nuclear and synaptic actions of the amyloid precursor protein. J Neurochem 2013, 126, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Waheed, Z.; Choudhary, J.; Jatala, F.H.; Fatimah; Noor, A.; Zerr, I.; Zafar, S. The Role of Tau Proteoforms in Health and Disease. Mol Neurobiol 2023, 60, 5155–5166. [Google Scholar] [CrossRef]
- Milenkovic, I.; Jarc, J.; Dassler, E.; Aronica, E.; Iyer, A.; Adle-Biassette, H.; Scharrer, A.; Reischer, T.; Hainfellner, J.A.; Kovacs, G.G. The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down syndrome. Neuropathol Appl Neurobiol 2018, 44, 314–327. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



