Submitted:
03 January 2024
Posted:
17 January 2024
You are already at the latest version
Abstract
Platelet-activating factor (PAF) is a phospholipid-derived inflammatory mediator, triggering various inflammatory conditions, including eosinophil activation and recruitment. Despite the unclear mechanism underlying the formation and maintenance of nasal polyp in chronic rhinosinusitis with nasal polyps (CRSwNP), there is strongly indication of its association with type 2 inflammation. This study aimed to quantify PAF-associated gene expression, examining enzymes involved in PAF synthesis (lysophosphatidylcholine acyltransferase 1 (LPCAT1), LPCAT2, LPCAT3, and LPCAT4), and PAF degradation (PAF acetylhydrolases 1 B2 (PAFAH1B2), PAFAH1B3, and PAF acetylhydrolases 2 (PAFAH2)), and PAF receptor (PTAFR). Transcriptomic analysis encompassed CRSwNP, including eosinophilic CRS (ECRS) (n=9), nonECRS (n=8), and a subgroup of ECRS with aspirin exacerbated respiratory disease (Asp) (n=3) based on the Japanese Epidemiological Survey of Refractory Eosinophilic Chronic Rhinosinusitis (JESREC) criteria. Gene expression profiles from nasal polyps in CRSwNP patients and the uncinate process in normal subjects (controls) (n=6) were analyzed using bulk RNA barcoding and sequencing (BRB-seq). Hierarchal analysis, utilizing the average linkage method with iDEP1.1, clarified the relationship between Differentially Expressed Genes (DEG) expressions and cluster formation. Two clusters (cluster 1 (n=11) and cluster 2 (n=9)) of CRSwNP divided by hierarchal analysis showed distinct gene expression profiles in the specific pathway of “cytokine-cytokine receptor interaction” in KEGG pathway and upregulated type 2 inflammatory gene expression in cluster 2. In CRSwNP subclassification of ECRS, nonECRS, and Ctrl, PTAFR was only upregulated in ECRS and nonECRS. In cluster analysis, cluster 1 showed significant downregulation of LPCAT2 and upregulation of PTAFR expression, while cluster 2 showed significant upregulation of LPCAT1, PAFAH1B2, and PTAFR and downregulation of PAFAH2 expression. These findings suggest that cluster 2, characterized by severe type 2 inflammatory gene expression, manifest a prominent PAF-associated pathophysiology. Understanding this high PAF-associated pathophysiology in cluster 2 could provide valuable insights into the treatment and management of CRSwNP.
Keywords:
Paranasal sinuses
; Chronic rhinosinusitis (CRS)
; BRB-seq
; Nasal polyps
; Differentially Expressed Genes (DEG) analysis
; Hierarchal analysis
; Pathway analysis
; Type 2 inflammation
; LPCAT1
; LPCAT2
; PAFAH1B2
; PAFAH1B3
; PAFAH2
; PTAFR
1. Introduction
Chronic rhinosinusitis (CRS) with nasal polyps (CRSwNP) is a complex inflammatory disorder characterized by diverse clinical presentations and underlying inflammatory profiles. It can be categorized into eosinophilic CRS (ECRS) and non-ECRS based on the extent of eosinophilic infiltration in sinonasal tissue [1]. Pathological association between the inflammatory endotype of type 1, 2, and 3 and phenotypes of CRSwNP are deeply associated with the pathophysiology of CRSwNP [2]. ECRS, a subtype characterized by Th2-dominant inflammation, often results in less favorable surgical outcomes [1,3], and cases of ECRS with aspirin exacerbated respiratory disease (AERD) mostly show more severe surgical outcome due to the severe type 2 inflammation [4].
The precise mechanism behind the formation and maintenance of nasal polyps in CRSwNP remains unclear, but there's strong evidence suggesting an association with type 2 inflammation. The pathophysiology of ECRS, especially in ECRS with AERD, involves the dysregulation in the synthesis of glycerophospholipid mediators including leukotrienes, prostaglandin, thromboxane, and PAF [5]. In the context of type 2 inflammatory reactions, IgE-mediated hypersensitivity triggers immune cells to process allergens, prompting a B-cell mediated immune response. This leads to the cross-linking of IgE antibodies on inflammatory cells, initiating mast cell degranulation and the release of various mediators like PAF, histamine, and pro-inflammatory cytokines [6,7]. These mediators induce both early and late-phase allergic responses, resulting in cellular inflammation by recruiting eosinophils, basophils, monocytes, and lymphocytes. PAF, a phospholipid-derived inflammatory mediator released by various cell types [8], plays a pivotal role in multiple inflammatory states, recruiting eosinophils and neutrophils, triggering leukocyte degranulation and adhesion, and generating free radicals like superoxide and hydroxyl anions in the nasal mucosa [9]. A previous report mentioned that a large amount of PAF was contained in nasal polyps from the patients of CRSwNP with AERD and it correlated with tissue eosinophilia [10]. Moreover, PAF contributes to mast cell activation, often linked to severe anaphylactic responses [9]. Its role in inducing vascular permeability leads to nasal hyperreactivity and congestion [11,12,13].
PAF is tightly regulated and produced by two distinct pathways 4 with 1) de novo pathway, which constitutively produce low levels of PAF and 2) remodeling pathway, which rapidly synthesize the majority of PAF removing fatty acids (typically arachidonic acid) from the sn-2 position of membrane phospholipid phosphatidylcholine (PC), which results in lyso-PAF, following to convert lyso-PAF into PAF by LPCAT1 and LPCAT2 [14,15]. The reverse reaction from lyso-PAF to PC is catalyzed by LPCAT1–4 acyltransferase activity [16]. PAF is synthesized by a variety of cells including endothelial cells, platelets, macrophages, monocytes, neutrophils, and eosinophils [17], and shows its biological effects at high-picomolar concentrations, such as the typical serum concentration of PAF of 400 pg/ml in healthy individuals [18].
Produced PAF is rapidly degraded to lyso-PAF by PAF acetylhydrolases (PAFAHs), a family of Ca2+- independent lipoprotein-associated phospholipase A2. Half-life of PAF ranges from 3–13 min because PAFAHs regulate PAF activity by hydrolyzing PAF to lyso-PAF as a biologically inactive form [17]. PAFAHs have three isoforms and one isoform is a secreted form (Phospholipase A2 Group VII(PLA2G7)) and two isoforms are intracellular forms (PAFAH Ib and PAFAH II) [19]. PLA2G7 and PAFAH II can non-specifically hydrolyze oxidatively fragmented phospholipids with potent biological activities, while PAF-AH Ib displays high specificity for the sn-2 acetyl group of glycerophosphocholine (GPC) including PAF [20]. On the other hand, there was no report about the association between allergic conditions and PLA2G7 activity, although increased PLA2G7 activity was reported in various pathologic conditions, including ischemic stroke, myocardial infarction, familial HDL deficiency, chronic cholestasis, diabetes mellitus, rheumatoid arthritis, essential hypertension, and peripheral vascular disease [20]. Therefore, currently PLA2G7 is thought to be separated from the PAF-metabolism associated genes in allergic conditions.
PAF effects are mediated by binding to the PAF receptor (PTAFR), leading to mobilization of intracellular calcium and activation of kinases. Activation of PTAFR leads to the activation of cytoplasmic PLA2 with subsequent formation of leukotrienes, prostaglandins, and thromboxane after the cleavage of arachidonic acid [19]. PTAFR is expressed in T lymphocytes, monocytes and macrophages, platelets, tracheal epithelium, vascular endothelium, lung alveolar wall, liver, small intestine, heart, skeletal and smooth muscle, brain microgalia and neurons, myometrium, spleen, and kidney [21,22]. PTAFR expression is regulated by intracellular cyclic AMP, which can downregulate PTAFR gene expression and reduce PAF-induced arachidonic acid release [23,24].
Stimulation of the PTAFR induces production of nitric oxide [25], histamine release from basophils, activation and degranulation of mast cells, chemotaxis of mast cells and eosinophils, recruitment of neutrophils, production of IL-4 by B lymphocytes, bronchial smooth muscle contraction, and mucus secretion as well as increase vascular permeability [19]. The role of PAF in the pathogenesis of allergic rhinitis was currently demonstrated [26], but the role of PAF in the pathogenesis of CRSwNP was underestimated. The intricate relationship between PAF and the pathophysiology of CRSwNP, which include ECRS with AERD often showing anaphylactic reaction after administration of NSAIDS, suggests a potential link between PAF metabolism and its pathophysiology including the formation and maintenance of nasal polyps derived from vascular permeability associated with plasma extravasation [11,12,13,27].
Current classification methods like the JESREC criteria attempt to stratify ECRS based on clinical findings and eosinophil count [1], transcriptome analyses have revealed that the expression level of type 2 inflammatory cytokine and chemokine do not align with these clinical classifications [28]. These analyses have unveiled significant differences between the clinical classification and the severity of type 2 inflammation. Therefore, this study aims to explore and elucidate the complexities of PAF metabolism-associated gene expressions of key enzymes involved in PAF synthesis (LPCAT1, LPCAT2, LPCAT3, LPCAT4) and degradation (PAFAH1B2, PAFAH1B3, PAFAH2), along with PAF receptor (PTAFR), across subtypes classified as nonECRS and ECRS including ECRS with AERD(Asp) by clinical classification and hierarchal analysis-based classification. Understanding the significance of PAF in orchestrating eosinophil recruitment and its association with type 2 inflammation sets the stage for investigating how variations in PAF metabolism-associated gene expressions align with these subtypes. Identifying disparities in PAF metabolism-associated gene expressions could be crucial in comprehending the heterogeneity of CRSwNP and potentially guiding tailored therapeutic interventions. This investigation seeks to bridge the gap between transcriptomic variations, inflammatory phenotypes, and PAF-related molecular signatures, aiming to illuminate the intricate interplay between PAF metabolism and CRSwNP subtypes and/or cluster-based classification. Ultimately, this study aims to unravel the diverse landscape of PAF-associated gene expressions across distinct CRSwNP subtypes and/or cluster-based classification providing deeper insights into the molecular foundations contributing to the varying inflammatory profiles observed in this condition.
2. Results
2.1. Gene expression profiles of NPs between ECRS including Asp and nonECRS.
2.1.1 Principal Component Analysis (PCA), heatmap of the correlation matrix, and hierarchical cluster analysis PCA was conducted across all groups, indicating distinct segregation only for the control group, while ECRS, Asp and nonECRS did not show clear separation from each other (Figure 1). These findings support the rationale for subsequent analyses of PAF metabolisms, considering ECRS and Asp as part of the same ECRS group.
2.2. Segregation of all CRSwNPs into two clusters using hierarchical cluster analysis
Hierarchical cluster analysis divided all CRSwNPs into two distinct clusters (referred to as cluster 1 and cluster 2) (Figure 2). However, no correlation was found between the clusters and the presence of asthma as a comorbidity. Notably, these two clusters exhibited significant variations in Differentially Expressed Genes (DEG) related to a wide array of cytokines and chemokines including interleukine (IL)-4 like cytokines, Chemokine (C-C motif) ligand (CCL26), and leukotriene B (LTB), indicating the differing severity of type 2 inflammation (Figure 3).
2.3. Gene expression analysis about PAF-metabolism associated gene based on the JESREC-based clinical classification and clusters reflecting the severity of type 2 inflammation.
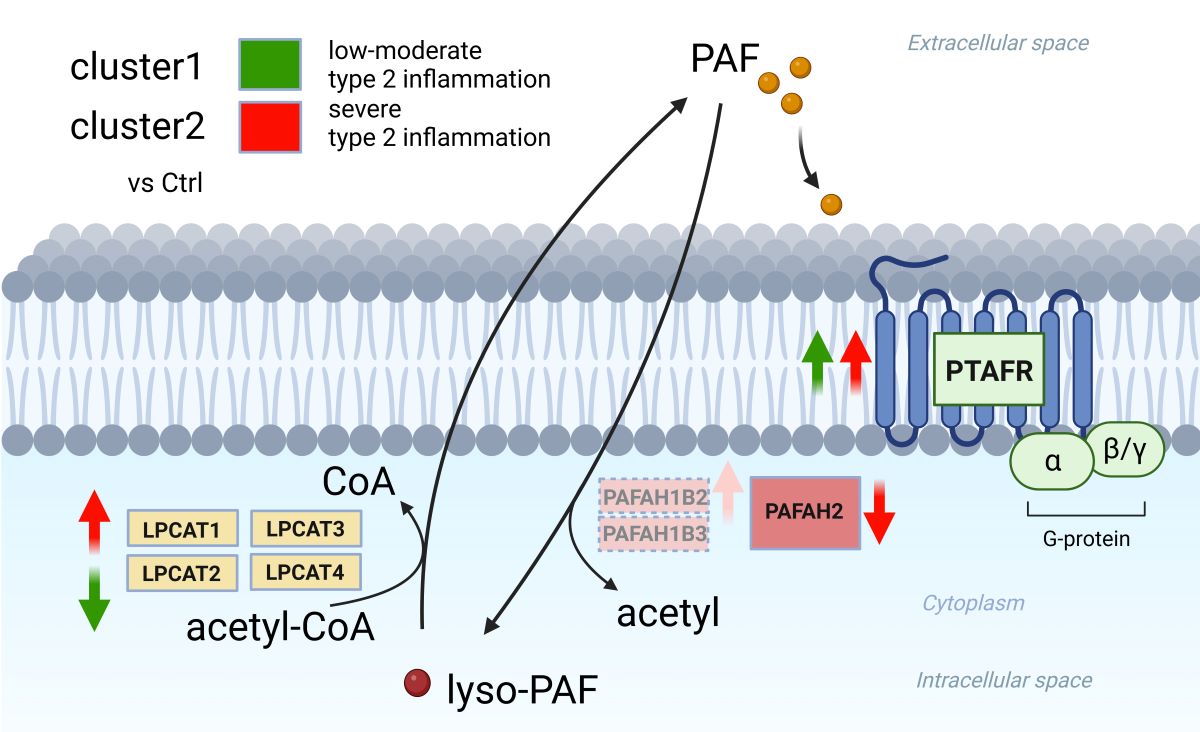
Each group of CRSwNP as nonECRS and ECRS including Asp and Ctrl showed no significant difference in LPCAT1, LPCAT2, LPCAT3, LPCAT4, PAFAH1B2,PAFAH1B3, and PAFAH2, but both groups of CRSwNP were upregulated in PTAFR compared to Ctrl (Figure 4). Each clusters reflecting the severity of type 2 inflammation showed no significant difference in LPCAT3, LPCAT4, and PAFAH1B3. Although cluster1 showed upregulated PTAFR and downregulated LPCAT2 compared to Ctrl, cluster2 showed upregulated LPCAT1, PAFAH1B2, and PTAFR, and downregulated PAFAH2. Compared between each clusters, cluster2 was upregulated in LPCAT1 and LPCAT2.
2.4. Gene expression analysis between PAF-metabolism associated gene and clinical features.
Significant correlation only between oral FeNO or total nasal FeNO and LPCAT2, LPCAT3, and PTAFR were recognized in CRSwNP (Figure 5), but there were no significant correlation between both FeNO and LPCAT1, LPCAT4, PAFAH1B2, and PAFAH1B3 (data not shown). There were also no correlation between tissue eosinophil, blood eosinophil, total IgE, and JESREC score and each genes expressions of LPCAT1, LPCAT2, LPCAT3, LPCAT4, PAFAH1B2,PAFAH1B3, PAFAH2, and PTAFR (data not shown).
3. Discussion
PAF-metabolism associated gene expression using transcriptome analysis revealed that clinical classification based on the JESREC study showed the significant upregulation of PTAFR gene expression in nonECRS and ECRS compared to Ctrl, but the other genes did not show any difference among nonECRS, ECRS and Ctrl. Additionally, there was no difference between nonECRS and ECRS in PTAFR gene expression. These results showing significant upregulation in nonECRS and ECRS compared to Ctrl and no difference between nonECRS and ECRS in PTAFR gene expression were also previously demonstrated [27] and these results suggested that clinical classification based on the JESREC criteria or the status with/without comorbid asthma and/or AERD may not detect the difference of PAF-metabolism in each clinical classification because PCA analysis with nonECRS, ECRS, and Asp could not segregate each other. On the other hand, upregulation of PTAFR gene expression in nasal polyp from both nonECRS and ECRS suggests that PTAFR upregulation enhancing the possibility of PAF-signaling upregulation associate with the formation or maintenance of nasal polyp.
However, two clusters from hierarchical analysis could clearly segregate from each other in PCA analysis and showed the clear difference in variety of cytokines expression in KEGG pathway analysis suggesting the reflection of the severity of type 2 inflammation. In the cluster analysis of PAF-associated gene expression, cluster 2 showed significant upregulation of LPCAT1, PAFAHB2, and PTAFR, and significant downregulation of PAFAH2 compared to Ctrl. In contrast, cluster 1 showed significant upregulation of PTAFR and significant downregulation of LPCAT2. Compared between cluster 1 and 2, cluster 2 was significantly upregulated in LPCAT1 and LPCAT2 expression.
LPCAT1 and LPCAT2 are main enzymes of PAF synthesis from lyso-PAF [14,15], and PAFAH2 is main degrading enzymes of PAF by hydrolyzing PAF to lyso-PAF as a biologically inactive form [19]. Although PAFAH1B2 and PAFAH1B3 are capable of hydrolyzing PAF, previous report demonstrated that RNAi-mediated knockdown of PAFAH1B2 or PAFAH1B3 does not alter PAF levels or PAF hydrolytic activity, indicating that these enzymes may possess alternate endogenous substrates [29]. These results suggest that cluster2, which can reflect the severity of type 2 inflammation, have high PAF-associated pathophysiology with upregulation of PAF synthesis and downregulation of PAF degrading leading to the local PAF accumulation and intensify the effects of PAF signaling via upregulation of PTAFR.
The result showed for the first time that severe type 2 inflammation induce the high PAF-associated pathophysiology and the main cause of the high PAF-associated pathophysiology assumingly comes from high lyso-PAF and PAF synthesis via overexpressed LPCAT1 and LPCAT2 compared to the cluster of moderate to low type 2 inflammatory type of CRSwNP (cluster 1). On the other hand, our result did not show any correlation between each gene expressions associating PAF metabolism and blood/tissue eosinophil count or the clinical classification. Only the oral FeNO and total nasal FeNO showed the relationship between LPCAT2, LPCAT3, and PTAFR, but could not exhibit the above differences of PAF-metabolism associated gene expression, especially in LPCAT1 and LPCAT2, shown in cluster analysis. The difference may come from the discrepancy between the severity of type 2 inflammation and blood/tissue eosinophilia, both FeNO level, or clinical classification because blood/tissue eosinophil count is used for the biomarker of eosinophilia [30] and FeNO is for airway inflammation or irritation [31] and clinical classification is for the phenotype diagnosis [1]. Although oral FeNO and total nasal FeNO reflect the severity of allergic inflammation [31], our result revealed that both FeNO can reflect the severity of allergic inflammation but not the severity of type 2 inflammation. Direct biomarker for the severity of type 2 inflammation, which have been underdeveloped yet, should be made for the further analysis of pathophysiology in CRSwNP.
Previous report mentioned that difference of lyso-PAF concentration between normal nasal polyp and nasal polyp having AERD suggested the association between airway inflammation and/or disease severity and lyso-PAF concentration [27]. Other paper also mentioned that human nasal polyp with severe eosinophil infiltration showed high PAF concentration [10]. Our result also clearly showed that the group of nasal polyps with severe type 2 inflammation had the increased PAF-metabolism, which can play an important role in the pathogenesis of CRSwNP inducing increased tissue neutrophilia and eosinophilia in nasal mucosa as a function of PAF signaling [19]. Therefore, these results may enhance that PAF-metabolism associated gene expression, especially in combination of LPCAT1 and LPCAT2 gene expression, can become a biomarker for the severity of type 2 inflammation in CRSwNP.
In summary, this study demonstrates the upregulation of PTAFR gene expression in nasal polyp and severe type 2 inflammation leads to increase high PAF-metabolism in nasal polyp, which may induce the proliferation and maintenance of nasal polyp. However, due to the classification using hierarchical analysis, small sample size, and only from Japanese patients, further research is needed to confirm our findings and the association between type 2 inflammation and the production of PAF-metabolism associated protein including PAF and/or lyso-PAF to reach the development of anti-PAF therapies for CRSwNP especially with severe type 2 inflammation. Additionally, the research about LPCAT1 and LPCAT2 gene expression in CRSwNP should be performed to confirm whether these gene expression become a good biomarker for the severity of type 2 inflammation or not.
4. Materials and Methods
4.1. Patient recruitment
Patients with or without CRS, who underwent endoscopic sinus surgery in Hiroshima Medical University Hospital between October 2016 and October 2021, were enrolled. The diagnosis of CRS was based on computed tomography (CT) scanning, patient history, clinical symptoms, and endoscopic findings. The inclusion criteria for CRSwNP were as follows: treatment without oral/nasal steroids within 4 weeks before surgery, no improvement in continuous nasal drip, post-nasal drip and nasal congestion after medical treatment including low-dose macrolide therapy. Patients with CRSwNP were clinically diagnosed as ECRS or nonECRS based on the diagnostic criteria of the JESREC study [4]. The diagnostic criteria for ECRS (JESREC score) include 1) side affected: both sides; 3 points, 2) with nasal polyps; 2 points, 3) CT changes: ethmoid/maxillary ≥ 1; 2 points, and 4) peripheral blood eosinophil count (%): 2<and≤5%; 4 points, 5<and≤10%; 8 points, 10%<; 10 points. Eosinophil infiltration into the NPs was diagnosed by calculating the mean cell count of the 3 most dense areas of eosinophils in the hematoxylin and eosin (HE) stained sections under x400 magnification (ocular lens, field number 22). A diagnosis of ECRS was made by both the total JESREC score of 11 points or higher and eosinophil infiltration count into the NPs of 70 or more [4]. The group of ECRS with aspirin exacerbated respiratory disease was independently classified as Asp (N=3), and the others were classified as ECRS (N=9) and nonECRS (N=8). Control patients (Ctrl) (N=6) were also diagnosed based on the anatomical abnormalities but showed no inflammatory mucosal change or bacterial infection around the uncinate process. The exclusion criteria for CRSwNP were as follows: treatment with oral/nasal steroids within 4 weeks before surgery, fungal/allergic fungal rhinosinusitis, and primary ciliary dyskinesia. Demographics and clinical characteristics were obtained from the medical records. The detailed patient demography is shown in Table 1.

Table 1.
Patient demographics. BMI: body mass index, NP: nasal polyp.
 |
4.2. RNA-seq using BRB-seq
BRB-seq [32] was performed for library preparation with the following modifications. Barcoded oligo-dT based primer (5’-GCCGGTAATACGACTCACTATAGGGAGTTCTACAGTCCGACGATCNNNNNNNNNNCCCCCCCCCTTTTTTTTTTTTTTTTTTTTTTTTV -3’ ; 10bp “N”=UMI, 9bp “C”=cell barcode) was used for single-stranded synthesis and Second Strand Synthesis Module (NEB, #E6111) was used for double-stranded cDNA synthesis. In-house MEDS-B Tn5 transposase [27,28] was used for tagmentation and libraries were amplified for 10 cycles of PCR using Phusion High-Fidelity DNA Polymerase (Thermo Scientific, #M0530) with the following primers (5’ -AATGATACGGCGACCACCGAGATCTACACindexGTTCAGAGTTCTACAGTCCGA-3',5’-CAAGCAGAAGACGGCATACGAGATindex GTCTCGTGGGCTCGGAGATGT-3'). An Illumina NovaSeq6000 was used to obtain 15bp of barcode read (Read1) and 81bp of insert read (Read2).
4.3. Data processing of BRB-seq
Read1(barcode read) was extracted by using UMI-tools(ver.1.1.1) with the command “umi_tools extract -I read1.fastq --read2-in=read2.fastq --bc-pattern=NNNNNNNNNNCCCCCCCCC --read2-stdout ”. Adaptor sequences and low-quality sequence were removed and read length below 20 bp were discarded using Trim Galore (ver.0.6.7). Reads were mapped to the GRCh38 reference using HISAT2 (ver.2.2.1). Read counts for each gene were obtained by featureCounts (ver.2.0.1), and UMI duplication was removed by UMI-tools with the command “umi_tools count --method=unique --per-gene --per-cell --gene-tag=XT”. Normalized read count value was obtained by using DESeq2 (ver.1.34.0). Gene-level expression data (read counts) were processed by using the web portal for integrated differential expression and pathway analysis (iDEP.1.13; http://bioinformatics sdstate.edu/idep11). iDEP.1.13 was used for principal component analysis (PCA), hierarchical clustering with a heatmap by center genes, identification of differentially expressed genes (DEGs), and pathway analysis. DEGs were extracted with an FDR cutoff of 0.1 and min-fold change of 2. Pathway analysis was performed using GAGE with hsa04060 cytokine-cytokine receptor interaction of Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Geneset size was set at minimum 5 and maximum 2000, and pathway significance cutoff (FDR) was set at 0.2.
4.4. Data analysis for PAF metabolism associated gene expression
In the catergory of fold change in CRSwNP, PAF synthesis (LPCAT1, LPCAT2, LPCAT3, LPCAT4) and degradation (PAFAH1B2, PAFAH1B3, PAFAH2), along with the PAF receptor (PTAFR) were measured and classified with clinical classification and hierarchical classification. Statifitical analysis was performed using welch’s t test and Pearson correlation coefficient with Graphpad prism 8.4.3.
5. Conclusions
This study revealed and shed light on the PAF metabolism in endotype categorization of the severity of type 2 inflammation. Additionally, this study also revealed that hierarchical analysis-based classification as endotype categorization was useful to elucidate the differences of CRSwNP. This result may provide a valuable data for further studies to elucidate the differences of endotype of CRSwNPs, future therapy for CRSwNP using anti-PAF metabolism drug, and possible biomarker of LPCAT1 and LPCAT2 for severity of type 2 inflammation.
Author Contributions
Conceptualization, T.I.; methodology, T.I.; software, T.I.; validation, T.I., N.C. and T.K.; formal analysis, T.T.; investigation, S.T.; resources, Y.H.; data curation, K.T,M.N,T.O,T.H; Writing – Original Draft Preparation, T.I.; Writing – Review & Editing, T.I. and S.T.; visualization, T.I.; supervision, S.T.; project administration, T.U.; funding acquisition, S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded partly by the Japan Society for the Promotion of Science KAKENHI grant, the Health Labor Sciences Research grant, and Research Collaboration Fund with Universal Sound Design Inc.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and was approved by the Ethics Committee of Hiroshima University Hospital, Hiroshima University, Hiroshima, Japan (Hi-136; August.11.2014 as date of approval).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Not applicable.
Acknowledgments
We can acknowledge Y.O. for the technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tokunaga, T.; Sakashita, M.; Haruna, T.; Asaka, D.; Takeno, S.; Ikeda, H.; Nakayama, T.; Seki, N.; Ito, S.; Murata, J.; et al. Novel scoring system and algorithm for classifying chronic rhinosinusitis: the JESREC Study. Allergy. 2015, 70, 995–1003. [Google Scholar] [CrossRef]
- Grayson, JW.; Hopkins, C.; Mori, E.; Senior, B.; Harvey, RJ. Contemporary Classification of Chronic Rhinosinusitis Beyond Polyps vs No Polyps: A Review. JAMA. Otolaryngol. Head. Neck. Surg. 2020, 146, 831–838. [Google Scholar] [CrossRef]
- Matsuwaki, Y.; Ookushi, T.; Asaka, D.; Mori, E.; Nakajima, T.; Yoshida, T.; Kojima, J.; Chiba, S.; Ootori, N.; Moriyama, H. Chronic rhinosinusitis: risk factors for the recurrence of chronic rhinosinusitis based on 5-year follow-up after endoscopic sinus surgery. Int. Arch. Allergy. Immunol. 2008, 146 (Suppl), 77–81. [Google Scholar] [CrossRef]
- Walgama, ES.; Hwang, PH. Aspirin-exacerbated respiratory disease. Otolaryngol. Clin. North. Am. 2017, 50, 83–94. [Google Scholar] [CrossRef]
- Laidlaw, TM.; Boyce, JA. Pathogenesis of aspirin-exacerbated respiratory disease and reactions. Immunol. Allergy. Clinics. North. America. 2013, 33, 195–210. [Google Scholar] [CrossRef]
- Min, YG. The pathophysiology, diagnosis and treatment of allergic rhinitis. Allergy Asthma Immunol Res. 2010, 2, 65–76. https://doi.org/10.4168/aair.2010.2.2.65. Epub 2010 Mar 24. [PubMed]
- Sin, B.; Togias. A. Pathophysiology of allergic and nonallergic rhinitis. Proc. Am. Thorac. Soc. 2011, 8:106–14. 1: 8. [CrossRef] [PubMed]
- Small P, Keith PK, Kim H. Allergic rhinitis. Allergy asthma. Clin. Immunol. 2018,14(s2),51. [CrossRef] [PubMed]
- Muñoz-Cano, RM.; Casas-Saucedo, R.; Valero Santiago. A.; Bobolea, I.; Ribó, P.; Mullol, J. Platelet-Activating Factor (PAF) in allergic rhinitis: clinical and therapeutic implications. J. Clin. Med. 2019,8,1338. [CrossRef] [PubMed]
- Furukawa, M.; Ogura, M.; Tsutsumi, T.; Tsuji, H.; Yamashita, T. Presence of platelet-activating factor in nasal polyps and eosinophils. Acta. Otolaryngol. 2002, 122, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Leggieri, E.; Tedeschi, A.; Lorini, M.; Bianco, A.; Miadonna, A. Study of the effects of paf-acether on human nasal airways. Allergy. 1991, 46, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, V. Role of histamine and platelet-activating factor in allergic rhinitis. J. Physiol. Biochem. 2004, 60, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cano, R.; Valero, A.; Roca-Ferrer, J.; Bartra, J.; Sanchez-Lopez, J.; Mullol, J. Platelet-activating factor nasal challenge induces nasal congestion and reduces nasal volume in both healthy volunteers and allergic rhinitis patients. Am. J. Rhinol. Allergy. 2013,27:e48-52. [CrossRef] [PubMed]
- Shindou, H.; Hishikawa, D.; Nakanishi, H.; Harayama, T.; Ishii, S.; Taguchi, R. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J. Biol. Chem. 2007,282,6532–6539. https://doi.org/10.1074/jbc.M609641200. Epub 2006 Dec 20. [PubMed]
- Harayama, T.; Shindou, H.; Shimizu, T. Biosynthesis of phosphatidylcholine by human lysophosphatidylcholine acyltransferase 1. J. Lipid. Res. 2009,50,1824–1831. https://doi.org/10.1194/jlr.M800500-JLR200. Epub 2009 Apr 21. [PubMed] [PubMed Central]
- Saito. RF.; Rangel, MC.; Halman, JR.; Chandler, M.; de Sousa Andrade, LN.; Odete-Bustos, S.; Furuya, TK.; Carrasco, AGM.; Chaves-Filho, AB.; Yoshinaga, MY.; Miyamoto, S.; Afonin, KA.; Chammas, R. Simultaneous silencing of lysophosphatidylcholine acyltransferases 1-4 by nucleic acid nanoparticles (NANPs) improves radiation response of melanoma cells. Nanomedicine. 2021,36,102418. https://doi.org/10.1016/j.nano.2021.102418. Epub 2021 Jun 24. [PubMed]
- Palgan, K.; Bartuzi, Z. Platelet activating factor in allergies. Int. J. Immunopathol. Pharmacol. 2015,28,584–589. https://doi.org/10.1177/0394632015600598. Epub 2015 Oct 20. [PubMed]
- Vadas, P.; Gold, M.; Perelman, B.; Liss, GM.; Lack, G.; Blyth, T. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N. Engl. J. Med. 2008, 358, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Upton, JEM.; Grunebaum, E.; Sussman, G.; Vadas, P. Platelet Activating Factor (PAF): A Mediator of Inflammation. Biofactors. 2022,48,1189-1202. https://doi.org/10.1002/biof.1883. Epub 2022 Aug 27. [PubMed]
- Karasawa, K.; Harada, A.; Satoh, N.; Inoue, K.; Setaka, M. Plasma platelet activating factor-acetylhydrolase (PAF-AH). Prog Lipid. Res. 2003, 42, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Honda, Z.; Ishii, S.; Shimizu, T. Platelet-activating factor receptor. J. Biochem. 2002,131,773–779. [CrossRef] [PubMed]
- Howard, KM. Platelet-activating factor receptor. In Encyclopedia of Biological Chemistry II. 2nd ed; Lennarz, WJ., Lane, MD., Eds.; Academic Press.: San Diego, CA, 2013; pp. 533–537. [Google Scholar]
- Vadas, P.; Perelman, B. Effect of epinephrine on platelet-activating factorstimulated human vascular smooth muscle cells. J. Allergy. Clin. Immunol 2012,129,1329-1333. https://doi.org/10.1016/j.jaci.2012.02.027. Epub 2012 Mar 27. [PubMed]
- Thivierge, M.; Parent, JL.; Stankova, J.; Rola-Pleszczynski, M. Modulation of human platelet-activating factor receptor gene expression by protein kinase C activation. J. Immunol. 1996, 157, 4681–4687. [Google Scholar] [CrossRef] [PubMed]
- Moritoki, H.; Hisayama, T.; Takeuchi, S.; Miyano, H.; Kondoh, W. Involvement of nitric oxide pathway in the PAF-induced relaxation of rat thoracic aorta. Br. J. Pharmacol. 1992, 107, 196–201. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Teo, CWL.; Png, SJY.; Ung, YW.; Yap, WN. Therapeutic effects of intranasal tocotrienol-rich fraction on rhinitis symptoms in platelet-activating factor induced allergic rhinitis. Allergy. Asthma. Clin. Immunol. 2022,18,52. [CrossRef] [PubMed]
- Roca-Ferrer, J.; Pérez-González, M.; Alobid, I.; Tubita, V.; Fuentes, M.; Bantulà, M.; Muñoz-Cano, R.; Valero, A.; Izquierdo, I.; Mullol, J. Upregulation of Platelet-Activating Factor Receptor Expression and Lyso-Platelet-Activating Factor Isoforms in Human Nasal Polyp Tissues. J. Clin. Med. 2023, 12, 7357. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ishino, T.; Takeno, S.; Takemoto, K.; Yamato, K.; Oda, T.; Nishida, M.; Horibe, Y.; Chikuie, N.; Kono, T.; Taruya, T.; Hamamoto, T.; Ueda, T. Distinct Gene Set Enrichment Profiles in Eosinophilic and Non-Eosinophilic Chronic Rhinosinusitis with Nasal Polyps by Bulk RNA Barcoding and Sequencing. Int. J. Mol. Sci. 2022, 23, 5653. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kohnz, RA.; Mulvihill, MM.; Chang, JW.; Hsu, KL.; Sorrentino, A.; Cravatt, BF.; Bandyopadhyay, S.; Goga, A.; Nomura, DK. Activity-Based Protein Profiling of Oncogene-Driven Changes in Metabolism Reveals Broad Dysregulation of PAFAH1B2 and 1B3 in Cancer. ACS. Chem. Biol. 2015,10,1624-1630. https://doi.org/10.1021/acschembio.5b00053. Epub 2015 May 7. [PubMed] [PubMed Central]
- Fulkerson, PC.; Rothenberg, ME. Targeting eosinophils in allergy, inflammation and beyond. Nat. Rev. Drug. Discov. 2013,12,117-129. https://doi.org/10.1038/nrd3838. Epub 2013 Jan 21. [PubMed] [PubMed Central]
- Ricciardolo, FL. Multiple roles of nitric oxide in the airways. Thorax. 2003, 58, 175–182. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alpern, D.; Gardeux, V.; Russeil, J.; Mangeat, B.; Meireles-Filho, ACA.; Breysse, R.; Hacker, D.; Deplancke, B. BRB-seq: ultra-affordable high-throughput transcriptomics enabled by bulk RNA barcoding and sequencing.Genome Biol 2019,20, 71. [CrossRef]
Figure 1.
PCA performed among all groups. The control group was clearly segregated, but ECRS, nonECRS, and Asp were not segregated from each other.
Figure 1.
PCA performed among all groups. The control group was clearly segregated, but ECRS, nonECRS, and Asp were not segregated from each other.
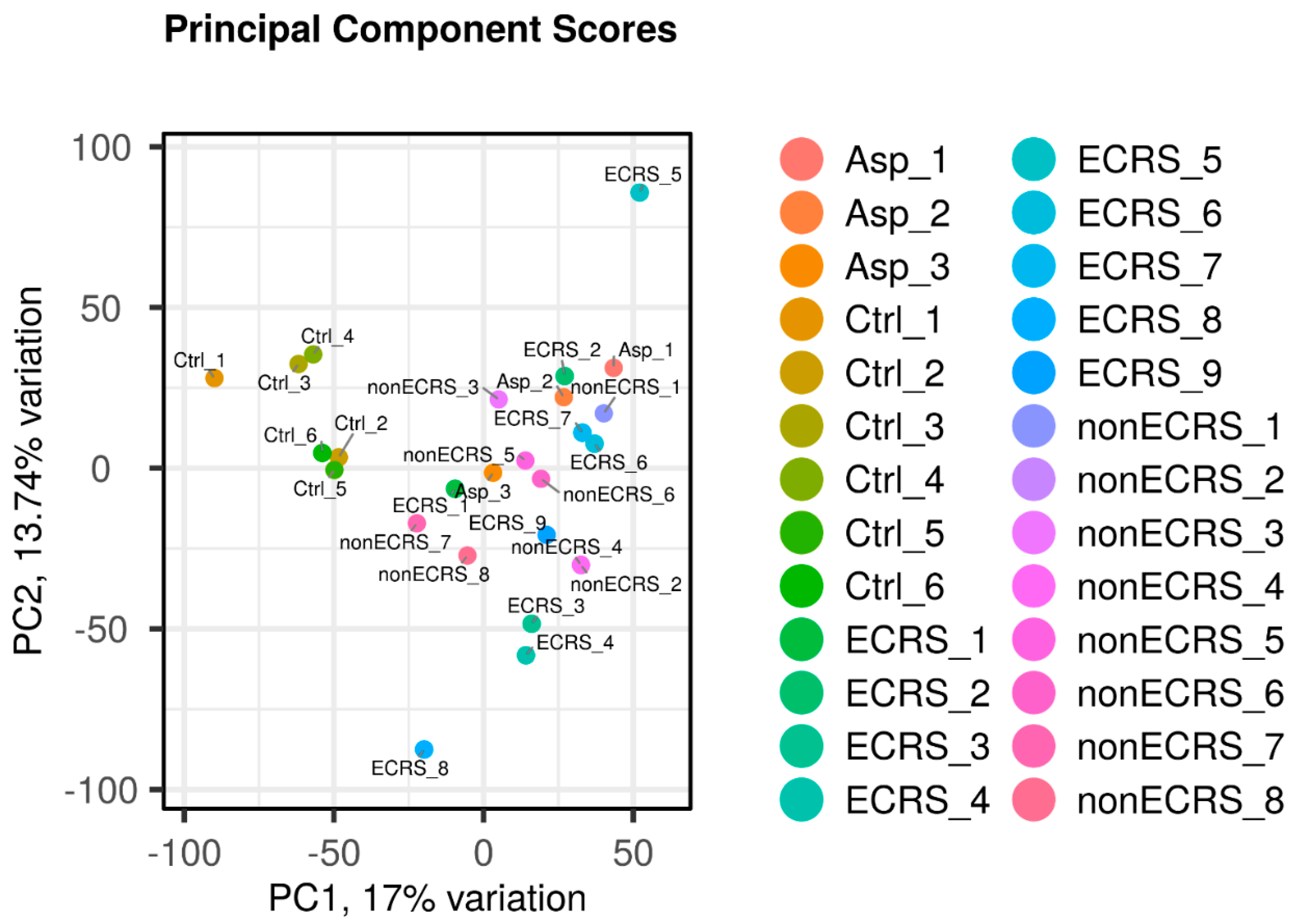
Figure 2.
Hierarchic cluster analysis with all CRSwNPs segregated into two cluster (cluster1 and 2) among these CRSwNPs (A). Segregation of two clusters and comorbidity of asthma were not correlated. The new clusters and control showed clear separation in the PCA analysis (B).
Figure 2.
Hierarchic cluster analysis with all CRSwNPs segregated into two cluster (cluster1 and 2) among these CRSwNPs (A). Segregation of two clusters and comorbidity of asthma were not correlated. The new clusters and control showed clear separation in the PCA analysis (B).
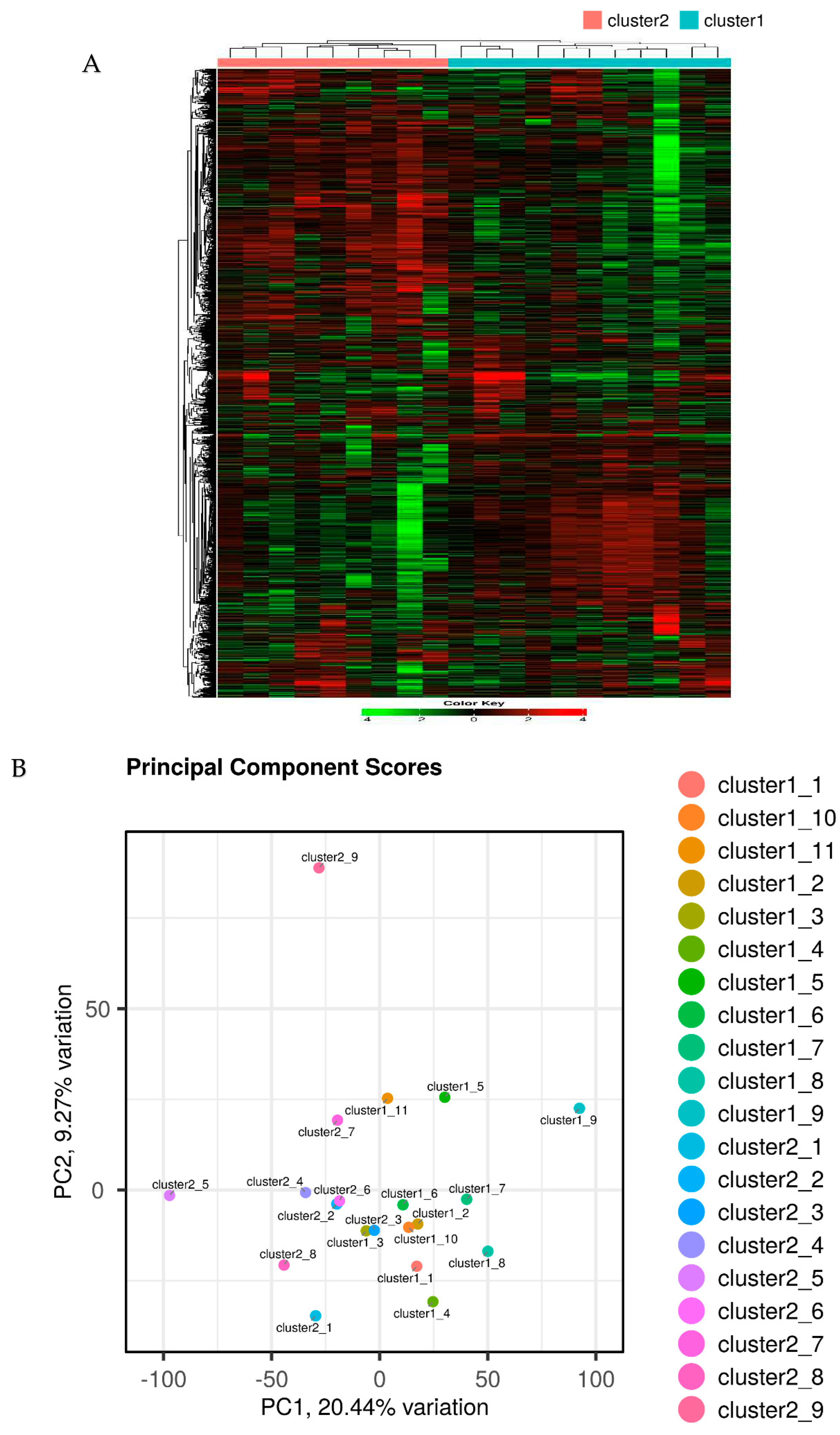
Figure 3.
KEGG pathway analysis of cluster 1 vs cluster 2 in “cytokine-cytokine receptor interaction” by iDEP1.13. Red represent the upregulated DEGs and Green represent the downregulated DEGs in cluster 2 compared to cluster 1. Intense result of DEGs including upregulation of interleukine (IL)-4 like cytokine, Chemokine (C-C motif) ligand (CCL26), and leukotriene B (LTB) and suggested that cluster 2 is a severe group of type 2 inflammation.
Figure 3.
KEGG pathway analysis of cluster 1 vs cluster 2 in “cytokine-cytokine receptor interaction” by iDEP1.13. Red represent the upregulated DEGs and Green represent the downregulated DEGs in cluster 2 compared to cluster 1. Intense result of DEGs including upregulation of interleukine (IL)-4 like cytokine, Chemokine (C-C motif) ligand (CCL26), and leukotriene B (LTB) and suggested that cluster 2 is a severe group of type 2 inflammation.
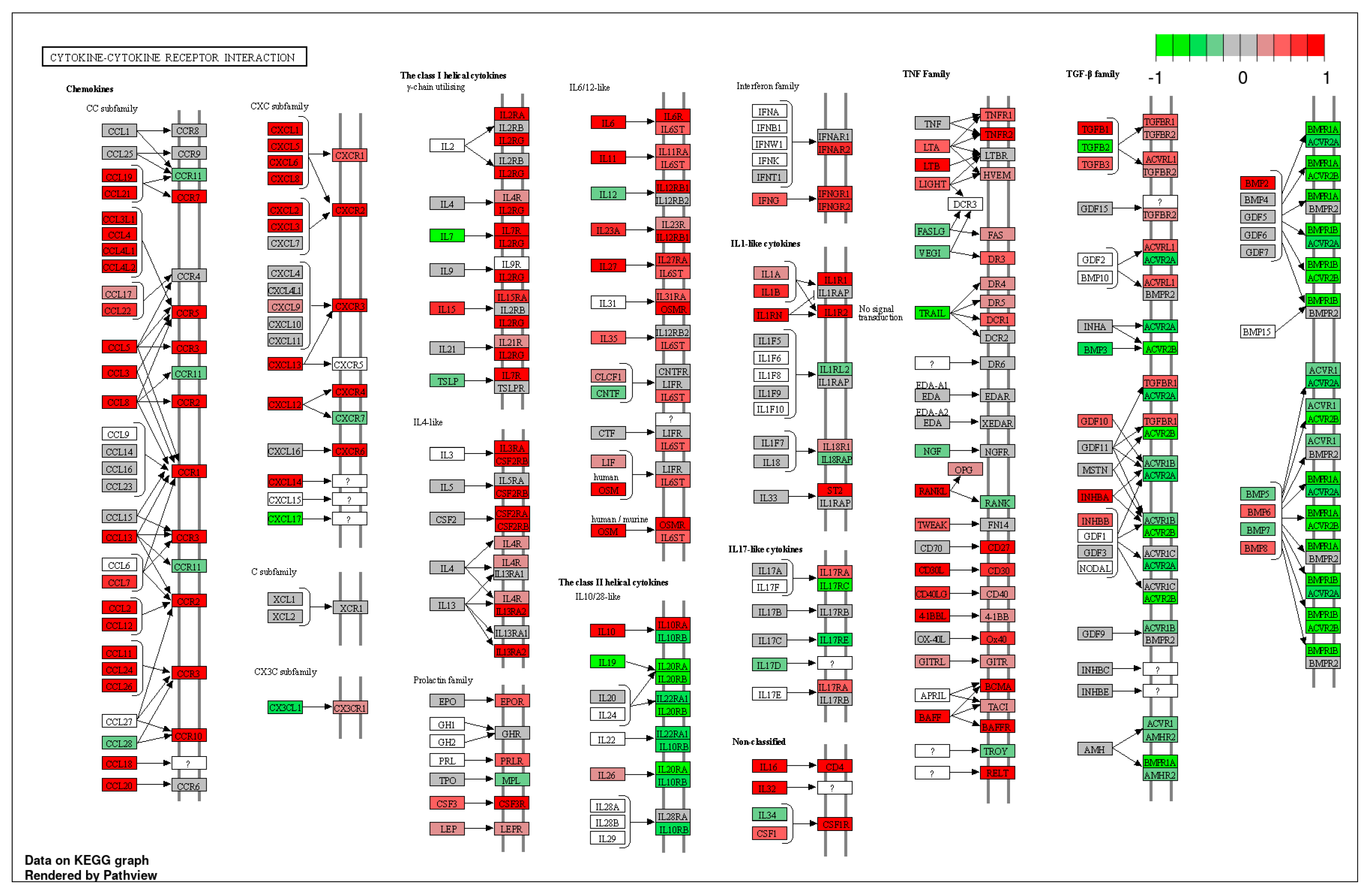
Figure 4.
PAF metabolism-associated gene expression(FC: fold change) in clinical classification with JESREC criteria (A) and clusters enhancing the severity of type 2 inflammation (B). Clinical classification did not show a clear difference between the phenotype of CRSwNP but the clusters enhancing endotype of CRSwNP showed clear difference in PAF metabolism-aassociated gene expression.
Figure 4.
PAF metabolism-associated gene expression(FC: fold change) in clinical classification with JESREC criteria (A) and clusters enhancing the severity of type 2 inflammation (B). Clinical classification did not show a clear difference between the phenotype of CRSwNP but the clusters enhancing endotype of CRSwNP showed clear difference in PAF metabolism-aassociated gene expression.
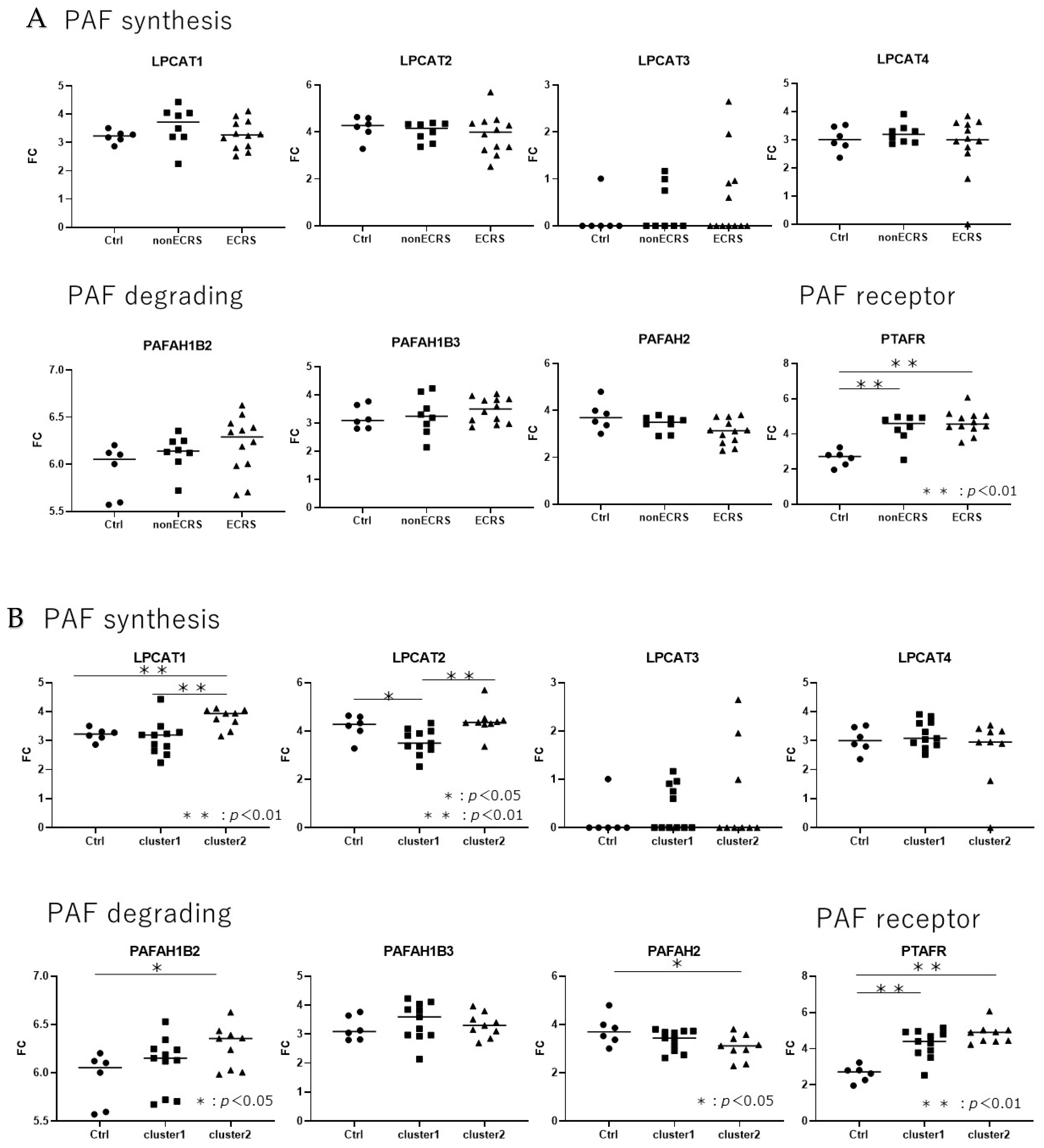
Figure 5.
PAF metabolism-associated gene expression (FC: fold change) in CRSwNP based on the test of oral FeNO and total nasal FeNO. Only oral FeNO and total nasal FeNO collerated with LPCAT2, LPCAT3 and PTAFR, but all of the data showed low R2 in linear regression analysis.
Figure 5.
PAF metabolism-associated gene expression (FC: fold change) in CRSwNP based on the test of oral FeNO and total nasal FeNO. Only oral FeNO and total nasal FeNO collerated with LPCAT2, LPCAT3 and PTAFR, but all of the data showed low R2 in linear regression analysis.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



