Submitted:
08 January 2024
Posted:
09 January 2024
You are already at the latest version
Abstract
The major arboviruses belong mainly to the families Bunyaviridae, Togaviridae, and Flaviviridae amongst which chikungunya virus and dengue virus have emerged as a global public health problem. The main objective of the study was to develop a specific, sensitive, and cost-effective molecular multiplex RT-PCR and RT-qPCR assay for the rapid and simultaneous detection of CHIKV and the four serotypes of DENV for arbovirus surveillance. Specific primers of all virus were designed, one step multiplex RT-PCR (mRT-PCR) and RT-qPCR (mRT-qPCR) was developed using reference strains of CHIKV and DENV serotypes. The specificity of the test for all the viruses was confirmed by sequencing. The standard curves showed a high correlation coefficient, R2= 0.99, for DENV-2 and DENV-3; R2= 0.98, for DENV-4 and CHIKV; R2= 0.93, for DENV-1. The limits of detection were calculated to be 10-2 copies/µL for all viruses. The specificity and sensitivity of the newly developed mRT-PCR and mRT-qPCR were validated using positive serum samples collected in India and Burkina Faso. The sensitivity of mRT-PCR and mRT-qPCR are 91%, and 100% respectively. The specificity of both assays was 100% specific. mRT-PCR and mRT-qPCR assays are low cost and a combination of both will be a useful tool for arbovirus surveillance.
Keywords:
multiplex
; molecular
; detection
; dengue virus
; chikungunya virus
; arbovirus
1. Introduction
Arboviral diseases are potent global public health problems due to their significant negative health and socio-economic impacts on many countries, particularly those in resource-limited settings. The major arboviruses belong to the families Bunyaviridae, Togaviridae, and Flaviviridae [1] and are mainly transmitted to humans by hematophagous arthropods (such as mosquitoes, ticks, sandflies), from an animal reservoir or from an infected individual. Amongst the mosquito, the Aedes and Culex mosquitoes are considered the most medically important mosquito vectors with most of the arbovirues being transmitted by the Aedes mosquitoes, namely, Aedes aegypti and Aedes albopictus [2,3,4]. The mosquito-borne arboviruses, such as, yellow fever virus (YFV), Dengue virus (DENV), Zika virus (ZIKV), West Nile virus (WNV), and chikungunya virus (CHIKV), are globally distributed [5,6]; growing human populations, urbanization, climatic changes, international traveling, and trades contribute to the extensive spread of these viruses to areas where Aedes mosquitoes have been infested [2]. High mutation rates among RNA viruses such as these arboviruses provide conditions for adaptive evolution to new mosquito species and often gain a high degree of receptivity and infectivity which may facilitate arbovirus disease emergence [7]. DENV and CHIKV are arboviruses of the family Flaviviridae and Togaviridae respectively. DENV is the most prevalent arbovirus, present in more than 100 countries within tropical and subtropical regions of the world [8]. The virus is an enveloped single-stranded, positive-sense RNA virus, comprising four antigenically distinct serotypes, DENV-1 to DENV-4 that exhibit 65% to 70% sequence homology [2,3]. CHIKV has now been identified in over 110 countries in Asia, Africa, Europe and the Americas [9] and is also a positive-sense single-stranded RNA virus with a genome of approximately 11.8 kb and comprises of two open reading frames (ORFs), ORF1 and ORF2, flanked by a 5′ cap and a polyadenylated tail at the 3′UTR [10].
Moreover, when the multiple serotypes of DENV circulate concurrently with CHIKV, there is a higher risk for more severe forms of the disease such as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) [11]. This highlights the importance of accurate and early diagnosis in humans, determination of the circulating viral serotypes, and entomological surveillance at any given location [6].
Between 2014–2017, 28.4% of dengue cases were serologically confirmed in India [12]. In the year 2016, followed by 2017, and 2019 maximum number of chikungunya laboratory- confirmed cases were reported in India. Highest confirmed cases were reported in Karnataka, Delhi, and Maharashtra [13]. In Burkina Faso, DENV is the most widespread arbovirus with the highest prevalence, incidence, and significant morbidities and mortality. In 2016, more than 1061 cases of dengue were reported in Burkina Faso with a case fatality rate of 1.2% [14]. In 2017, a prevalence of 28.54% of dengue fever was reported among pregnant women in Ouagadougou [15]. Another sero-epidemiological study of CHIKV was conducted in Ouagadougou, Burkina Faso, with blood samples collected in 2015, and a seroprevalence of 29.1% was reported [16]. These consecutive reports in different populations (general and pregnant populations) suggest a considerable dengue burden in Burkina Faso. The risk of arbovirus transmission in the country is real because of its tropical climate, which favors the multiplication of Aedes. Almost 80% of the Burkinabe population engages in farming and/or other agricultural practices. This may constitute a major factor of arbovirus transmission because, during the rainy season, farmers settle in the fields close to the forest and are in closer contact with vectors, which increases the risk of arbovirus transmission [17,18]. In the absence of a vaccine and specific antiviral treatment against the DENV and CHIKV, the most effective means of disease control remain surveillance for early detection of cases in order to intervene with public health measures to contain the cases and thus control the infection.
Serological testing for these infections has well-documented limitations: antibodies immunoglobulin M (IgM) may not be detectable early in the course of infection, a rise inimmunoglobulin G (IgG) between acute and convalescent samples can only provide a retrospective diagnosis, and anti-flavivirus antibodies may cross-react with one another [19]. IgM and IgG ELISA tests are widely used for rapid serological diagnosis but have the limitation of the inability to identify the circulating viral serotypes [20]. Virus isolation and amplification in susceptible cell lines is the gold standard for the detection of serotypes, but it is not an appropriate clinical diagnostic assay in early infection since it is laborious and time consuming [21]. For the prevention and detection of local transmission of DENV and CHIKV via both human and mosquito surveillance, developing a highly specific, sensitive and less expensive detection method with rapid outcomes appear particularly important. Multiplexed PCR-based assays often failed because multiple primers presented in high concentrations interact with each other unless they are exquisitely designed. Non-specific interference of oligonucleotides (DNA and RNA) is also thought to limit further multiplexed PCR [22]. Many molecular methods have been developed to improve the simultaneous detection of several arboviruses around the world [11,23,24,25,26]. The present study was conducted to develop a one-step multiplex reverse transcription polymerase chain reaction (mRT-PCR) and Real-time polymerase chain reaction (mRT-qPCR) for the rapid and simultaneous detection and serotyping of DENV and CHIKV to be used in arbovirus surveillance in Burkina Faso.
2. Materials and Methods
2.1. Viruses
Isolates of DENV and CHIKV were used for the development of multiplex PCRs and their analytical performances determination. Reference strains of DENV1-4 (1 isolate per serotype) were obtained from ATCC (VR-1856 ™ DENV-1; VR-1584 ™ DENV-2; VR-1256_FD ™ DENV-3; VR-1490 ™ DENV-4) and CHIKV was isolated from a PCR positive human sera sample [27]. The viruses were propagated in C6/36 Aedes albopictus cells prior to viral RNA extraction. Briefly, 20 µl of isolated sera sample was mixed with DMEM with 2% FBS, filtered and inoculated in confluent 24 well plates of C6/36 cells followed by incubation for 6 days. 90-100% confluent 6-well plates of Vero cells were used for CHIKV and DENV amplifications. 50 µL of virus stocks prepared was added to the flask and homogenized. The flask was incubated at 37°C for 1h, the volume of the flask was made to 25 mL with DMEM 2% FBS supplemented with 1% Pen/Strep and incubated until cytopathic effect was 90%. Infected cell supernatants were harvested in a 15 mL Falcon tubes and centrifuged at 2500 rpm for 5 min and stored at -80 °C. All procedures were carried out using sterile techniques and in a biosafety cabinet.
2.2. Patient Sera Samples
A total of 130 human serum samples were used in the present study after obtaining informed consent from the patients in both India and Burkina Faso. Among the 130 serum samples, 32 were positive for DENV (n=16) and CHIKV (n=16) and 32 from uninfected individuals (negative for both viruses) were part of a study funded by government of India [28]. Also, 33 of the 130 serum samples were positive only for DENV and 33 serum samples were negative for DENV kindly offered by the National Institute of Public Health, Burkina Faso. The sera samples were well characterized by ELISA and singleplex RT-PCR methods prior to use for the validation (after the proof-of-concept step) of the new multiplex RT-PCR and RT-qPCR methods. The protocol of this study was reviewed and approved by ethical committees of the institutional ethical committees of the Health Science Research, Burkina Faso N° A026-2023/CEIRES/IRSS and ICGEB, New Delhi, ICGEB/IEC/2014/01 version 3.
2.3. Primer Design
Nucleotide sequences for the complete genome of each serotype of the Dengue virus were downloaded from the National Center for Biotechnology Information (NCBI) database and aligned to identify highly conserved regions using MEGA Software version 11. DENV1-4 primers were designed manually, forward conserved primer was designed for all serotypes, and reverse primers of specific serotypes were designed using only specific conserved regions of different serotypes of DENV. A new DENV-3 forward (DENV-3 q F) and the reverse DENV-3 R) have been designed by multi-alignment of another genome of four DENV serotypes for RT-qPCR use. The representative sequences of CHIKV were downloaded from Virus Pathogen Database and Analysis Resource (ViPR) and primers were designed to target the E1 gene using Snap Gene version 5.3.1. All RT-PCR primers were designed using similar parameters so that they would have similar melting temperatures (Tm). The potentials for dimerization and secondary structures were analyzed using Oligo EvaluatorTM online software (data not shown). The length of all amplicons was (200 bp DENV-1, 367 bp DENV-2, 1359 bp DENV-3, 118 bp DENV-4, and 574 bp CHIKV). The specificity of all primer sequences was further confirmed using primer BLAST (NCBI). Detailed information on primers is provided in Table 1 and genome nucleotide accession numbers are in provided in Supplementary information.
2.4. Viral RNA Extraction
Viral RNA was extracted from 150 µL cell culture supernatant and clinical samples using a NucleoSpin RNA Virus kit (MACHEREY-NAGEL Gmbh Co.KG. Germany), according to the manufacturer’s instructions and eluted in 50 µL of diethyl pyrocarbonate (DEPC) treated water. A purified RNA was quantified using Nanodrop 2000 and cryopreserved at -80 °C until further processing.
2.5. One-Step Multiplex RT-PCR Amplification
Multiplex RT-PCR assay was optimized for simultaneous detection and serotyping of DENV and CHIKV RNAs in cell culture supernatants and human sera. mRT-PCR was performed using PrimeScript One Step RT-PCR Kit (Takara bio-INC). A total volume of 15µL of reaction mixture consisting of 100 ng of extracted RNA, 7.5 µL of 2X RT buffer, 0.6 µL of the enzyme, and 0.45µL (10µM) of forward conserved primer (DENV-1, DENV-2, DENV-3, DENV-4), 0.11µL (10 µM) of forward primer CHIKV, 0.3µL (10 µM) of Reverse primer DENV-1 and DENV-4, 0.11µL (10 µM) of Reverse primer of DENV-2, DENV-3, and CHIKV, the reaction was complete to 15µL with DEPC water. DEPC water was used for negative control. The thermal cycling profile of this assay consists of a 30 min Reverse Transcriptase (RT) step which is performed at 50°C, 95°C for 5 min, and then 35 cycles of 95°C for 30 s, 62°C for 20 s, 72°C for 30s and final extension 72°C for 5 min. mRT-PCR was performed using Applied Biosystems ProFlex™ 3 x 32-well PCR System machine. The PCR products were then analyzed by gel electrophoresis. 15 µL of PCR product were loaded into a 1% (W/V) agarose gel in 1x Tris-Acetate-EDTA buffer with a 1Kbp ladder as molecular weight marker. To detect coinfections between DENV 1-4 and CHIKV, individual serotypes RNA of reference sample were mixed in equal quantities and 3µL was used in mRT-PCR detection.
2.6. Multiplex One-Step Real-Time RT-PCR
A real-time, one-step, multiplex SYBR Green I RT-PCR assay was also developed for the detection of DENV-1 to DENV-4 and CHIKV. This assay was performed in PikoReal 96 Real-Time PCR System machine (Thermo Scientific, United States) using a one-step QuantiTect SYBR Green kit (Qiagen, Hilden, Germany). All mRT-qPCR reactions were performed in 50 µL reactions with 25 µL of SYBR Green, 0.5 µL of the enzyme, 1.4 µL (10µM) of forward conserved primer (DENV-1, DENV-2, DENV-4), 0.46 µl (10 µM) of DENV-3 q forward and reverse primer, 0.47µL (10 µM) of CHIKV forward and reverse primer, 0.5 µL (10 µM) of reverse primer DENV-1 and DENV-4, 0.44 µL (10 µM) of DENV-2 reverse primer, and 100 ng of RNA template. 15µL of reaction was used in triplicate for all viruses. DEPC water was used for negative control. The RT-PCR conditions for the real-time RT-PCR consisted of a 30-minute RT step at 50°C and 10 min of Taq polymerase activation at 95°C, followed by 40 cycles of PCR at 95°C for 30s (denaturation), 62°C for 30s (annealing), 72°C for 30s (extension) and final extension at 60°C for 30s. The melting curve temperature ranged from 60°C to 95°C. The result was positive if cycle threshold (Ct) values is equal to or less than 33 cycles. If Ct is more than 33 cycles, the result was considered negative. The limit of sensitivity of the assay was carried out with known quantitative RNA standards prepared using the one-step QuantiTect SYBR Green method. Briefly, the RNA template of each virus was serially diluted ten-fold with the known concentration of DENV 1-4, CHIKV, and 4µL of the diluted RNA were added to the mRT-qPCR reaction tube and amplified in triplicate. Standard curves have been determined using dilution 102 to 100 Copie/µL, for detection limits determination 10-1 and 10-2 have been added. Melting-curve analysis was performed after PCR amplification to verify that the correct product was amplified by examining its specific Melting temperature (Tm) that was also used to serotype DENV.
After optimization, the positive RNA of DENV and CHIKV and negative RNA from pre-collected sera were assayed in the same condition to evaluate the multiplex RT-PCR and RT-qPCR for their diagnostic potential in a clinical sample and validation. In Burkina, the clinical sample was performed using Applied Biosystems (SimpliAmp Thermo Fisher Scientific) thermal cycler for mRT-PCR and CFX96 Real-Time System (BIO-RAD) for mRT-qPCR.
2.7. Sequence Analysis
After gel electrophoresis, the amplicon of the different viruses was cut and purified using NucleoSpin® Gel and PCR Clean-up Kit (MACHEREY-NAGEL Gmbh Co.KG. Germany) according to the manufacturer’s instructions and eluted in 25 µl NE buffer. 20 𝜇L of purified PCR product was sent to Macrogen (Seoul, South Korea) for Deoxyribonucleic acid (DNA) sequencing by the Sanger method, using the amplification primers. Forward and reverse sequence was assembled by BioEdit Software version 7.2.5.0, to determine consensus sequence and blast using the NCBI BLAST tool to confirm the specificity and accuracy of the amplification of different viruses RNA fragments.
3. Results and Discussion
3.1. Primer Design and Their Specificity Assessment for Multiplex Detection of DENV1-4 and CHIKV:
For developing multiplex RT-PCR and RT-qPCR assay for the simultaneous detection of all DENV serotypes and CHIKV as co-infection, specificities of the primer were used. A set of primers was designed based on the optimal conserved regions revealed by multiple sequence alignments of three complete genomes of all DENV serotypes (DENV-1 to DENV-4) using MEGA software (Supplementary Figure 1). A universal forward primer was designed from a conserved region of the DENV-1 to DENV-4 genome to ensure the detection of all four DENV serotypes; the reverse primers of all serotypes were designed using only specific conserved regions of different serotypes of DENV, but for improving mRT-qPCR detection, another DENV-3 forward primer (DENV-3 q F) was designed to use in SYBR Green real-time PCR (Supplementary Figure 1). CHIKV primers were designed using SnapGene by targeting the E1 gene. Compatible melting temperatures (average Tm ranging from 60- 65ºC) were selected from the primer set. Of the different conserved regions selected for primer design, DENV conserved forward was located in the capsid (C) protein, Reverse DENV-1, and DENV-4 in the C protein, Reverse DENV-2 in precursor membrane prM protein, DENV-3 q forward and Reverse DENV-3 in envelope protein E (Figure 1). The oligonucleotide sequences and genome positions of all primers for the multiplex assay are listed in Table 1.

Table 1.
Oligonucleotide primers used in multiplex RT-PCR and RT-qPCR assay.
| Family/Genus | Virus | Primers | Sequence (5′ to 3′) | Target | Genome position |
|---|---|---|---|---|---|
| Flaviviridae/Flavivirus | DENV | DENVcon F | TCAATATGCTGAAACGCGAGAGAAACCG | C | 137-164 |
| DENV-3 q F | AGGAGCTACGTGGGTTGACGTG | E | 1219-1240 | ||
| DENV-1 R | TTTGATCGCTCCATTCTTCTTGAATGAG | C | 309-336 | ||
| DENV-2 R | TTCCCTTTCTCTTGTCTACTGACG | prM | 480-503 | ||
| DENV-3 R | GTGGTGAGCATTCTAGCCCAA | E | 1855-1875 | ||
| DENV-4 R | GTGATGAATGCTAGCACCATCCGTAAG | C | 228-254 | ||
| Togaviridae/Alphavirus | CHIKV | CHIKV F | ACACGTAAACAGTGATCCCGAACAC | E1 | 9998-10022 |
| CHIKV R | CCAAACGGCGGGTAGTCCATGT | 10550-10571 |
DENVconF: Dengue virus conserved Forward; R: Reverse; DENV-3 q F: DENV-3 forward for RT-qPCR using. GenBank access: DENV-1 (NC_001477.1, MN869914.1, MT929577.1, KC692515.1, KC692512.1, JN697057.1); DENV-2 (AY858035.2, AY858036.2, AB189124.1, FM210237.2, FM210232.2, FM210227.1); DENV-3(AB189127.1, AY858044.2, AY858048.2, KF954948.1, ON123669.1, KC762688.1); DENV-4(KF955510.1, HQ332172.1, HQ332176.1, JX024758, OK605599.1, KC762698.1); CHIKV (NC_004162.2).
DENV1-4 and CHIKV specific region were amplified successfully by multiplex RT-PCR and RT-qPCR assay and their amplicons were further observed on agarose gel electrophoresis for the confirmation based on the amplicon size and real-time detection based on Cycle threshold (Ct) value. Ct were DENV-1 (Ct = 17.28), DENV-2 (Ct = 23.5), DENV-3 (Ct = 22.06), DENV-4 (Ct = 21.73), and CHIKV (Ct = 22.75) (Table 2 and Figure 2). The expected size of the amplicons was 200 bp DENV-1, 367 bp DENV-2, 1359 bp DENV-3 (RT-PCR only), 118 bp DENV-4, and 574 bp CHIKV.
The amplicon size of DENV-3 used in real time RT-PCR was 509 bp. The primer pairs designed for the mRT-PCR revealed equally specific and sensitive to the singleplex RT-PCR. Furthermore, the specificity of all viruses was confirmed by subsequent DNA sequencing results and BLAST analysis (Supplementary Figure 2). Evaluation of coinfection of all possible combinations amongst DENV 1-4 and CHIKV are indicated that our mRT-PCR assay could simultaneously detect and serotype DENV and CHIKV in a single reaction (Figure 3).
Several studies have previously developed multiplex assays for detecting dengue serotypes as well as other flaviviruses [11,23,24]. Similarly, commercial rapid diagnostic tests (RDTs) such as SD BIOLINE Chikungunya IgM, RTK ProDetectTM, SD BIOLINE Dengue Duo® are available for detecting both CHIKV and the DENV serotypes [29,30] . However, reports of false positive results have dampened the enthusiasm of use of these kits by clinicians. These reports also emphasize the need of designing robust primer sets for efficient and stringent detection of the viruses, a feature employed in the current study. For instance, primer designing was performed according to several parameters in order to avoid heterodimer formation likelihood and thermal compatibility in order to increase the sensitivity and specificity of our assay. Further, DENV serotyping primers were designed based on the genome alignment of multiple strains of all four serotypes of DENV both globally as well as taking country specific sequences to study the variations at the genes critically. The alignment of several sequences assured the improvement of the universal primers, mainly by the elimination of potential mismatches, increasing the possibility of amplification of templates with a greater sequence diversity. Information on genes, such as C, prM and E domains, used in earlier similar studies was considered, for designing the primers [11,31,32].
3.2. Multiplex RT-qPCR Assessment and Sensitivity:
mRT-qPCR was performed with 100 ng of RNA and to evaluate sensitivity, 10-fold serial dilutions of RNA standards were used to draw a standard curve and estimate the limits of detection (LOD) of viral RNA copy load for the developed one-step real-time RT-PCR assay. Ct values obtained for serial 10-fold dilutions of known concentrations of DENV 1-4 and CHIKV RNA have been used to draw a linear curve against the amounts of standard RNA copy numbers and was used to calculate correlation coefficient (Supplementary Figure 3). The details are provided in Table 2.
Table 2.
Sensitivity of DENV 1-4 and CHIKV in mRT-qPCR.
| Parameters | mRT-qPCR | ||||
|---|---|---|---|---|---|
| DENV-1 | DENV-2 | DENV-3 | DENV-4 | CHIKV | |
| Ct value | 17.28 | 23.5 | 22.06 | 21.73 | 22.75 |
| R2 | 0.93 | 0.99 | 0.99 | 0.98 | 0.98 |
| LOD (Copies/µL) |
10-2 | 10-2 | 10-2 | 10-2 | 10-2 |
| Tm | 79.17 | 80.01 | 82.7 | 78.71 | 81.84 |
Tm: melting temperature, Ct: Cycle threshold, R2: coefficient of correlation, LOD: limit of detection.
3.3. Performance of mRT-PCR and mRT-qPCR Assay on Clinical Samples and Validation:
To determine the performance of multiplex RT-PCR and real-time RT-PCR assays, 32 human sera from the patients infected with DENV and CHIKV, and 32 human sera samples from individuals negative for these viruses from India was used. Also, 33 clinical samples positive for DENV and 33 samples from uninfected serum pre-collected in the National Institute of public health, Burkina Faso were used. All clinical samples were already tested for their infection status through RT-PCR and serology and were used for multiplex detection with mRT-PCR and mRT-qPCR. Among India clinical samples, the mRT-PCR assay was able to detect 87.5% (14/16) samples for DENV and 93.7% (15/16) for CHIKV, whereas the mRT-qPCR assay was able to detect 100% (16/16) samples for DENV and 100% (16/16) for CHIKV; serotyping showed that 43.7% (7/16) sample were positive for DENV-1, 12.5% (2/16) were positive for DENV-2, 37.5% (6/16) were positive for DENV-3 and 6.3% (1/16) were positive for DENV-4. In Burkina, clinical sample positives for DENV, 90.9% (30/33) were positives by mRT-PCR and 100% (33/33) by mRT-qPCR (Table 3), according to serotyping 57.5 % (19/33) sample were DENV-1 and 42.5 (14/33) for DENV-3. The sensitivity of mRT-PCR and mRT-qPCR is 91%, and 100% respectively. All positive samples tested by mRT-PCR were also positive for real-time RT-PCR. mRT-qPCR assay was seen to be more sensitive than the conventional multiplex RT-PCR. The results and the comparison of detection rates between both assays are summarised in Table 3. The specificity of the assay was evaluated using negative clinical samples, and the assay showed that none of them were detected by the assay, leading to a conclusion that both conventional multiplex RT-PCR and SYBR Green real-time RT-PCR assays were 100% specific (Table 3).
Two multiplex assays were validated using positive clinical samples pre-collected in India and Burkina Faso, the sensitivity and specificity of the conventional multiplex RT-PCR assays are 91% and 100% respectively, and for the real-time SYBR Green assay, 100% and 100% respectively, mRT-qPCR is more sensitive than mRT-PCR. In the study of Chen et al, evaluation of SYBR Green-I based one-step multiplex real-time RT-PCR assay was done according to DENV serotype, sensitivity for DENV-1, DENV-2, DENV-3, DENV-4, and CHIKV was 89.66%, 96.67%, 96.67%, 94.12%, and 95.74%, respectively, with 100% specificity [24]. In our study, evaluations were done according to the sample positive for DENV, the sensitivity of mRT-PCR (91%) is similar to the study of Chen et al, but the sensitivity of our real-time SYBR Green assay is better (100%). These results show that mRT-PCR and mRT-qPCR can be used in Burkina Faso for DENV and CHIKV surveillance. In Burkina Faso, DENV is the main endemic arbovirus included in the diagnostic and surveillance protocol, which explains the difficulty of obtaining CHIKV-positive samples. Only one seroprevalence study on CHIKV has been documented [16]. These multiplexed methods will help to improve early detection and effective surveillance of CHIKV in both humans and vectors.
4. Conclusion
Differential diagnosis is key to effective disease and patient management. Dengue serotyping gives evidence of displacement of the dominant circulating serotypes across time, and plays an important role in predicting severity of future outbreaks [34]. From an international perspective, accurate serotyping will allow a better understanding of traveling waves in dengue fever transmission by identifying related outbreaks across borders [35]. In addition, serotyping data can inform research into the multi-annual cross-country periodicity of dengue, thought to be related to the cycling of host immunity to different serotypes [36]. The present study is a proof of concept of successfully employing multiplex PCR for efficient detection of CHIKV and DENV serotypes using SYBR Green PCR chemistry and serves to be a cost-effective option for use in resource constraint countries in Africa and Asia both for diagnosis and epidemiologic surveillance of CHIKV and DENV.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, L.R.W.B., M.K.G., I.S., and S.S.; methodology, L.R.W.B., S.A.I., D.K.V. and A.K.; validation, L.R.W.B., S.A.I. and S.S.; writing-Original Draft, L.R.W.B., S.A.I., M.K.G., I.S., and S.S; supervision, S.A.I. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by UNTBLDC-TWAS-ICGEB South-South Programme for Exchanges and Collaborations (n° 4500459676).
Acknowledgments
We sincerely thank UNTBLDC-TWAS-ICGEB, ICGEB New Delhi, CEA/ITECH-MTV, and FONRID of Burkina Faso.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huang, Y.J.S.; Higgs, S.; Vanlandingham, D.L. Biological Control Strategies for Mosquito Vectors of Arboviruses. Insects 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Gao, X.; Gould, E.A. Factors Responsible for the Emergence of Arboviruses; Strategies, Challenges and Limitations for Their Control. Emerg. Microbes Infect. 2015, 4, e18. [Google Scholar] [CrossRef]
- Carrington, L.B.; Simmons, C.P. Human to Mosquito Transmission of Dengue Viruses. Front. Immunol. 2014, 5, 290. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.G.; Martinez, N.; Abdalla, L.; Duarte dos Santos, C.N.; Chame, M. Animals in the Zika Virus Life Cycle: What to Expect from Megadiverse Latin American Countries. Plos Neglect. Trop. Dis. 2016, 10, e0005073. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging Arboviruses: Why Today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The Global Ecology and Epidemiology of West Nile Virus. BioMed Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, L.G.; Alto, B.W.; Kim, M.S.; Hutter, D.; Bradley, A.; Bradley, K.M.; Burkett-Cadena, N.D.; Benner, S.A. Multiplexed Kit Based on Luminex Technology and Achievements in Synthetic Biology Discriminates Zika, Chikungunya, and Dengue Viruses in Mosquitoes. BMC Infect. Dis. 2019, 19, 418. [Google Scholar] [CrossRef] [PubMed]
- Sekaran, S.D.; Artsob, H. Molecular Diagnostics for the Detection of Human Flavivirus Infections. Expert Opin. Med. Diagn. 2007, 1, 521–530. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Chikungunya. Available online: https://www.who.int/health topics/chikungunya#tab=tab_1 (accessed on 15 September 2023).
- Khongwichit, S.; Chansaenroj, J.; Chirathaworn, C.; Poovorawan, Y. Chikungunya Virus Infection: Molecular Biology, Clinical Characteristics, and Epidemiology in Asian Countries. J. Biomed. Sci. 2021, 28, 84. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; el Zowalaty, M.E.; Islam, S.; Sharif, M.; Rahman, M.R.; Amin, M.R.; Ali, M.M.; Rahman, M.T.; Morita, K.; Ashour, H.M.A. Novel Multiplex RT-PCR Assay for Simultaneous Detection of Dengue and Chikungunya Viruses. Int. J. Mol. Sci. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Murhekar, M.; Joshua, V.; Kanagasabai, K.; Shete, V.; Ravi, M.; Ramachandran, R.; Sabarinathan, R.; Kirubakaran, B.; Gupta, N.; Mehendale, S. Epidemiology of Dengue Fever in India, Based on Laboratory Surveillance Data, 2014–2017. Int. J. Infect. Dis. 2019, 84, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Jagadesh, A.; Jayaram, A.; Babu, N.; Mudgal, P.P.; Sudandiradas, R.; Sheik, S.; Shetty, U.; Verma, D.K.; Mahilkar, S.; Sunil, S.; et al. Current Status of Chikungunya in India. Front. Microbiol. 2021, 12. [Google Scholar]
- Tarnagda, Z.; Cissé, A.; Bicaba, B.W.; Diagbouga, S.; Sagna, T.; Ilboudo, A.K.; Tialla, D.; Lingani, M.; Sondo, K.A.; Yougbaré, I.; et al. Dengue Fever in Burkina Faso, 2016. Emerg. Infect. Dis. 2018, 24, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Tougma, S.A.; Yaméogo, W.N.Z.; Dahourou, D.L.; Salou Kagoné, I.A.; Compaoré, T.R.; Kaboré, A.; Kagoné, T.; Drabo, M.K.; Meda, N. Dengue Virus Infection and Pregnancy Outcomes during the 2017 Outbreak in Ouagadougou, Burkina Faso: A Retrospective Cohort Study. PLoS One 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.; Ridde, V.; Agnandji, S.T.; Lell, B.; Yaro, S.; Yang, J.S.; Hoinard, D.; Weaver, S.C.; Vanhomwegen, J.; Salje, H.; et al. Seroepidemiological Reconstruction of Long-Term Chikungunya Virus Circulation in Burkina Faso and Gabon. J. Infect. Dis. 2022, 227, 261–267. [Google Scholar] [CrossRef]
- Bob, N.S.; Bâ, H.; Fall, G.; Ishagh, E.; Diallo, M.Y.; Sow, A.; Sembene, P.M.; Faye, O.; el Kouri, B.; Sidi, M.L.; et al. Detection of the Northeastern African Rift Valley Fever Virus Lineage during the 2015 Outbreak in Mauritania. Open Forum Infect. Dis. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Kwagonza, L.; Masiira, B.; Kyobe-Bosa, H.; Kadobera, D.; Atuheire, E.B.; Lubwama, B.; Kagirita, A.; Katushabe, E.; Kayiwa, J.T.; Lutwama, J.J.; et al. Outbreak of Yellow Fever in Central and Southwestern Uganda, February-May 2016. BMC Infect. Dis. 2018, 18, 548. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, L.G.; Bradley, A.; Bradley, K.M.; Alto, B.W.; Hoshika, S.; Hutter, D.; Sharma, N.; Yang, Z.; Kim, M.J.; Benner, S.A. High-Throughput Multiplexed XMAP Luminex Array Panel for Detection of Twenty-Two Medically Important Mosquito-Borne Arboviruses Based on Innovations in Synthetic Biology. J. Virol. Methods 2015, 214, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Prat, C.M.; Flusin, O.; Panella, A.; Tenebray, B.; Lanciotti, R.; Leparc-Goffart, I. Evaluation of Commercially Available Serologic Diagnostic Tests for Chikungunya Virus. Emerg. Infect. Dis. 2014, 20, 2129–2132. [Google Scholar] [CrossRef]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika Virus: History, Emergence, Biology, and Prospects for Control. Antiviral Res. 2016, 130, 69–80. [Google Scholar] [CrossRef]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and Application in Diagnostic Virology. Clin. Microbial. Rev. 2000, 13, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, J.; Yu, N.; Yan, J.; Zhuo, Z.; Chen, M.; Su, X.; Fang, M.; He, S.; Zhang, S.; et al. Development of Multiplex Real-Time Reverse-Transcriptase Polymerase Chain Reaction Assay for Simultaneous Detection of Zika, Dengue, Yellow Fever, and Chikungunya Viruses in a Single Tube. J. Med. Virol. 2018, 90, 1681–1686. [Google Scholar] [CrossRef]
- Chen, H.; Parimelalagan, M.; Lai, Y.L.; Lee, K.S.; Koay, E.S.C.; Hapuarachchi, H.C.; Ng, L.C.; Ho, P.S.; Chu, J.J.H. Development and Evaluation of a SYBR Green-Based Real-Time Multiplex RT-PCR Assay for Simultaneous Detection and Serotyping of Dengue and Chikungunya Viruses. J. Mol. Diagn. 2015, 17, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Yaren, O.; Alto, B.W.; Bradley, K.M.; Moussatche, P.; Glushakova, L.; Benner, S.A. Multiplexed Isothermal Amplification Based Diagnostic Platform to Detect Zika, Chikungunya, and Dengue 1. J. Vis. Exp. 2018, 133, 57051. [Google Scholar] [CrossRef]
- Priye, A.; Bird, S.W.; Light, Y.K.; Ball, C.S.; Negrete, O.A.; Meagher, R.J. A Smartphone-Based Diagnostic Platform for Rapid Detection of Zika, Chikungunya, and Dengue Viruses. Sci. Rep. 2017, 7, 44778. [Google Scholar] [CrossRef] [PubMed]
- Shrinet, J.; Jain, S.; Sharma, A.; Singh, S.S.; Mathur, K.; Rana, V.; Bhatnagar, R.K.; Gupta, B.; Gaind, R.; Deb, M.; Sunil, S. Genetic Characterization of Chikungunya Virus from New Delhi Reveal Emergence of a New Molecular Signature in Indian Isolates. Virol. J. 2012, 9, 100. [Google Scholar] [CrossRef]
- Ibemgbo, S.A.; Nyodu, R.; Chaudhary, S.; Verma, D.K.; Dixit, K.; Nayak, K.; Rani, V.; Gaind, R.; Chandele, A.; Sunil, S. Short Communication: Virological and B Cell Profiles of Chikungunya and Dengue Virus Co-Infections in Delhi during 2017–2019. Virus Res. 2022, 320, 198888. [Google Scholar] [CrossRef]
- Johnson, B.W.; Goodman, C.H.; Holloway, K.; De Salazar, P.M.; Valadere, A.M.; Drebot, M.A. Evaluation of Commercially Available Chikungunya Virus Immunoglobulin M Detection Assays. Am. J. Trop. Med. Hyg. 2016, 95, 182–192. [Google Scholar] [CrossRef]
- Mat Jusoh, T.N.A.; Shueb, R.H. Performance Evaluation of Commercial Dengue Diagnostic Tests for Early Detection of Dengue in Clinical Samples. J. Trop. Med. 2017, 2017. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Calisher, C.H.; Gubler, D.J.; Chang, G.-J.; Vorndamt, A.V. Rapid Detection and Typing of Dengue Viruses from Clinical Samples by Using Reverse Transcriptase-Polymerase Chain Reaction. J. Clin. Microbiol. 1992, 30, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Yenchitsomanus, P.T.; Sricharoen, P.; Jaruthasana, I.; Pattanakitsakul, S.N.; Nitayaphan, S.; Mongkolsapaya, J.; Malasit, P. Rapid Detection and Identification of Dengue Viruses by Polymerase Chain Reaction (PCR). Southeast Asian J. Trop. Med. Public Health 1996, 27, 228–236. [Google Scholar] [PubMed]
- Yong, Y.K.; Thayan, R.; Chong, H.T.; Tan, C.T.; Sekaran, S.D. Rapid Detection and Serotyping of Dengue Virus by Multiplex RT-PCR and Real-Time SYBR Green RT-PCR. Singapore Med. J. 2007, 1, 662–668. [Google Scholar]
- Li, D.S.; Liu, W.; Guigon, A.; Mostyn, C.; Grant, R.; Aaskov, J. Rapid Displacement of Dengue Virus Type 1 by Type 4, Pacific Region, 2007-2009. Emerg. Infect. Dis. 2010, 16, 123–125. [Google Scholar] [CrossRef]
- Cummings, D.A.T.; Irizarry, R.A.; Huang, N.E.; Endy, T.P.; Nisalak, A.; Ungchusak, K.; Burke, D.S. Travelling Waves in the Occurrence of Dengue Haemorrhagic Fever in Thailand. Nature 2004, 427, 344–347. [Google Scholar] [CrossRef] [PubMed]
- van Panhuis, W.G.; Choisy, M.; Xiong, X.; Chok, N.S.; Akarasewi, P.; Iamsirithaworn, S.; Lam, S.K.; Chong, C.K.; Lam, F.C.; Phommasak, B.; et al. Region-Wide Synchrony and Traveling Waves of Dengue across Eight Countries in Southeast Asia. Proc. Natl. Acad. Sci. U S A 2015, 112, 13069–13074. [Google Scholar] [CrossRef]
Figure 1.
The DENV genome and primers targeted regions used in this multiplex detection study. Structural proteins are targeted in this study for DENV: DENV conserve forward is located in the capsid (C) protein, Reverse DENV-1 and DENV-4 in the C protein, and Reverse DENV-2 in precursor membrane prM protein. DENV-3 q forward and Reverse DENV-3 in envelope protein E; UTR: untranslated region.
Figure 1.
The DENV genome and primers targeted regions used in this multiplex detection study. Structural proteins are targeted in this study for DENV: DENV conserve forward is located in the capsid (C) protein, Reverse DENV-1 and DENV-4 in the C protein, and Reverse DENV-2 in precursor membrane prM protein. DENV-3 q forward and Reverse DENV-3 in envelope protein E; UTR: untranslated region.
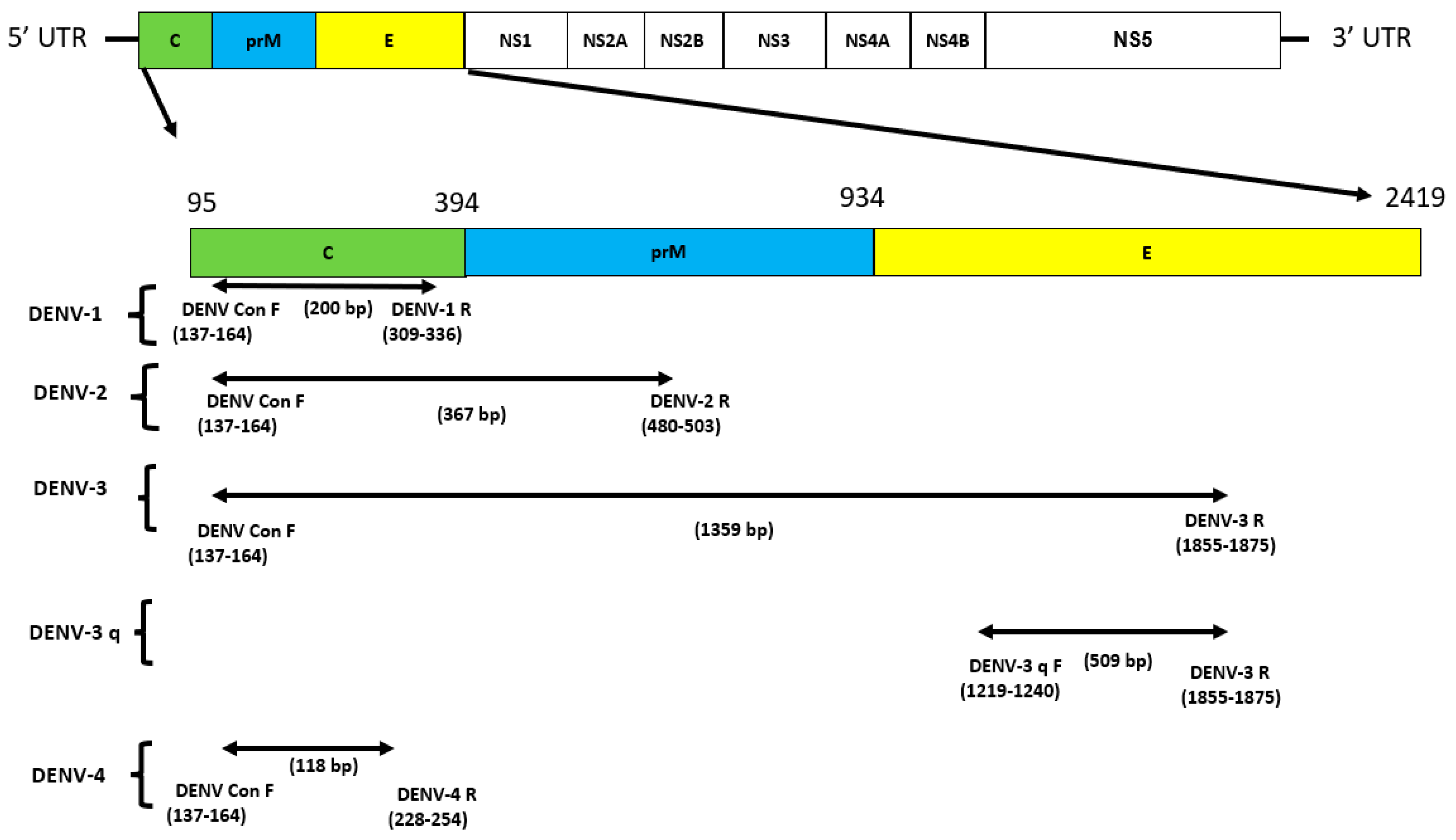
Figure 2.
Comparison sensibility and specificity of singleplex and multiplex RT-PCR. Electrophoresis of 1% agarose gel loaded with 15µL of singleplex RT-PCR and multiplex RT-PCR product showing the specificity of the primers. (A) Singleplex RT-PCR: Lane 1, M (1kb DNA marker); Lane 2, DENV-1; Lane 3, DENV-2; Lane 4, DEN-3; Lane 5, DENV-4; Lane 6, CHIKV; Lane 7, Negative Control (NC). (B) Multiplex RT-PCR: Lane 1, M (1kb DNA marker); Lane 2, mDENV-1; Lane 3, mDENV-2; Lane 4, mDEN-3; Lane 5, mDENV-4; Lane 6, mCHIKV; Lane 7, NC. m: multiplex, bp: basis pair, NC: negative control.
Figure 2.
Comparison sensibility and specificity of singleplex and multiplex RT-PCR. Electrophoresis of 1% agarose gel loaded with 15µL of singleplex RT-PCR and multiplex RT-PCR product showing the specificity of the primers. (A) Singleplex RT-PCR: Lane 1, M (1kb DNA marker); Lane 2, DENV-1; Lane 3, DENV-2; Lane 4, DEN-3; Lane 5, DENV-4; Lane 6, CHIKV; Lane 7, Negative Control (NC). (B) Multiplex RT-PCR: Lane 1, M (1kb DNA marker); Lane 2, mDENV-1; Lane 3, mDENV-2; Lane 4, mDEN-3; Lane 5, mDENV-4; Lane 6, mCHIKV; Lane 7, NC. m: multiplex, bp: basis pair, NC: negative control.
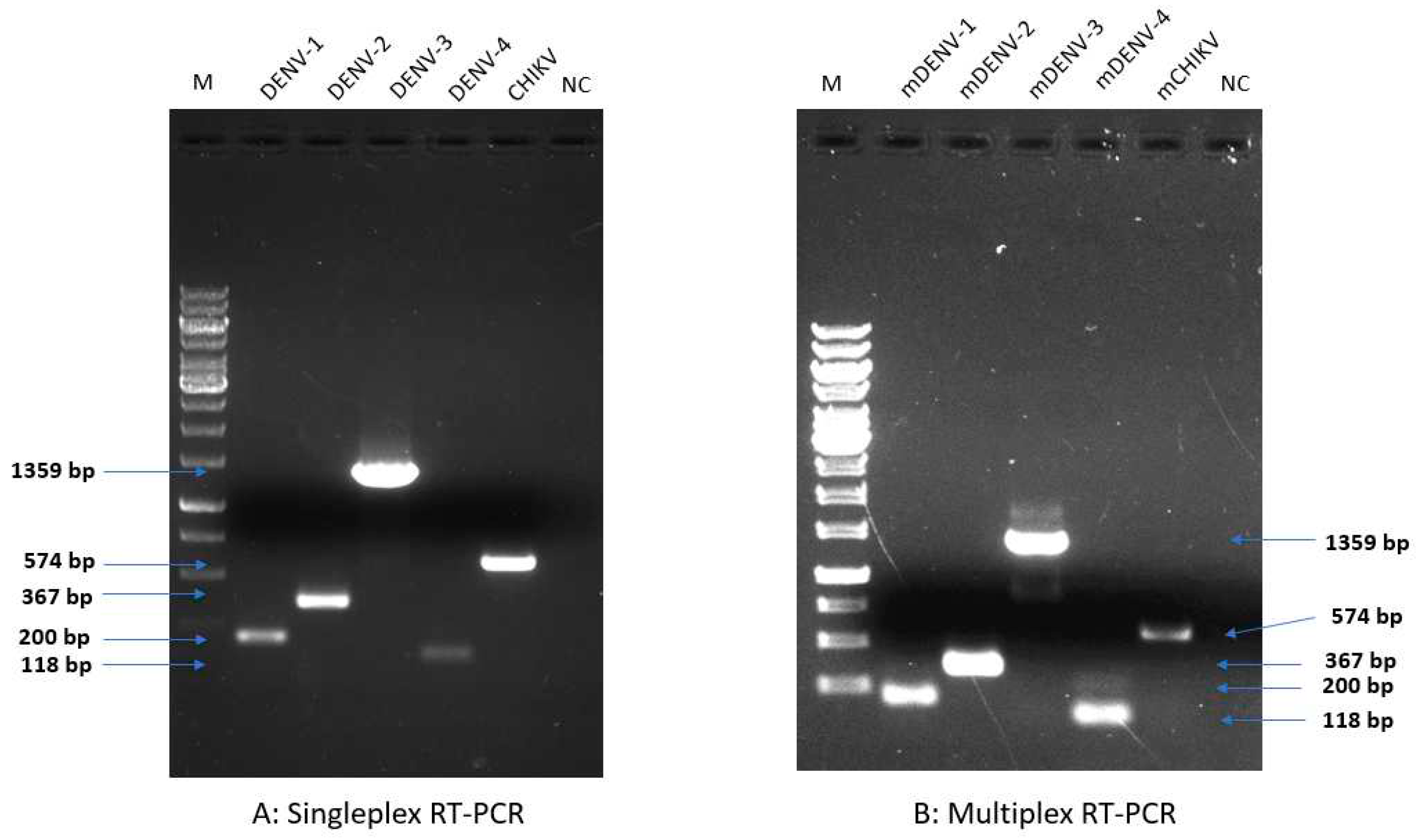
Figure 3.
Coinfection and concurrent infection detection between DENV 1-4 and CHIKV. (A) concurrent infection using RNA mixture of DENV-1 + DENV-2, lane 2; DENV-1 + DENV-3, lane 3; DENV-1 + DENV-4, lane 4; M is 1kb DNA marker. (B) coinfection using RNA mixture of DENV-1 + CHIKV, lane 1; DENV-2 + DENV-3, lane 2; DENV-2 +DENV-4, lane 3; DENV-2 + CHIKV, lane 4; DENV-3 +DENV-4, lane 5; DENV-3 + CHIKV, lane 6; DENV-4 + CHIKV, lane 7; DENV-1 +DENV-3 + CHIKV, lane 8; M is 1kb DNA marker. Electrophoresis of 1% agarose gel loaded with 15µL product showing the specificity of the primers in coinfection detection with mRT-PCR.
Figure 3.
Coinfection and concurrent infection detection between DENV 1-4 and CHIKV. (A) concurrent infection using RNA mixture of DENV-1 + DENV-2, lane 2; DENV-1 + DENV-3, lane 3; DENV-1 + DENV-4, lane 4; M is 1kb DNA marker. (B) coinfection using RNA mixture of DENV-1 + CHIKV, lane 1; DENV-2 + DENV-3, lane 2; DENV-2 +DENV-4, lane 3; DENV-2 + CHIKV, lane 4; DENV-3 +DENV-4, lane 5; DENV-3 + CHIKV, lane 6; DENV-4 + CHIKV, lane 7; DENV-1 +DENV-3 + CHIKV, lane 8; M is 1kb DNA marker. Electrophoresis of 1% agarose gel loaded with 15µL product showing the specificity of the primers in coinfection detection with mRT-PCR.
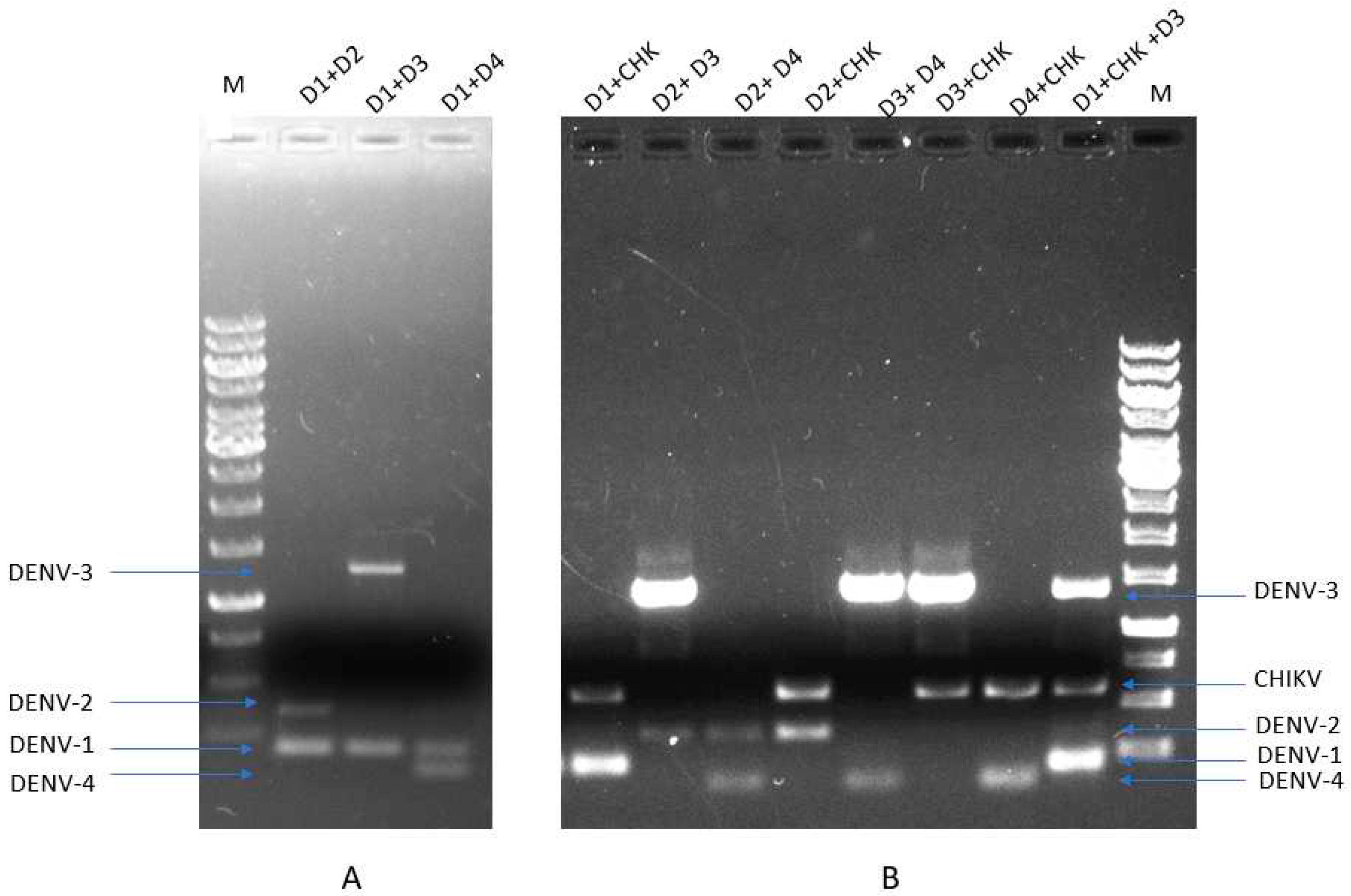
Figure 4.
Amplification of dengue virus DENV 1-4 and CHIKV in mRT-qPCR. (A) Real-time RT-PCR amplification. (B) Melt curve analysis. Serial 10-fold dilutions were used in amplification by mRT-qPCR in triplicate. The results are positive if the Ct value is equal to or less than 33 cycles, otherwise negative if the Ct value is > 33 cycles. The melting curve temperature ranged from 60°C to 95°C. The color curve represents different virus dilutions in triplicate, The detection limit of this assay for DENV-1, DENV-2, DENV-3, DENV-4, and CHIKV are 10-2 copies/µL. DEPC water was used for negative control.
Figure 4.
Amplification of dengue virus DENV 1-4 and CHIKV in mRT-qPCR. (A) Real-time RT-PCR amplification. (B) Melt curve analysis. Serial 10-fold dilutions were used in amplification by mRT-qPCR in triplicate. The results are positive if the Ct value is equal to or less than 33 cycles, otherwise negative if the Ct value is > 33 cycles. The melting curve temperature ranged from 60°C to 95°C. The color curve represents different virus dilutions in triplicate, The detection limit of this assay for DENV-1, DENV-2, DENV-3, DENV-4, and CHIKV are 10-2 copies/µL. DEPC water was used for negative control.
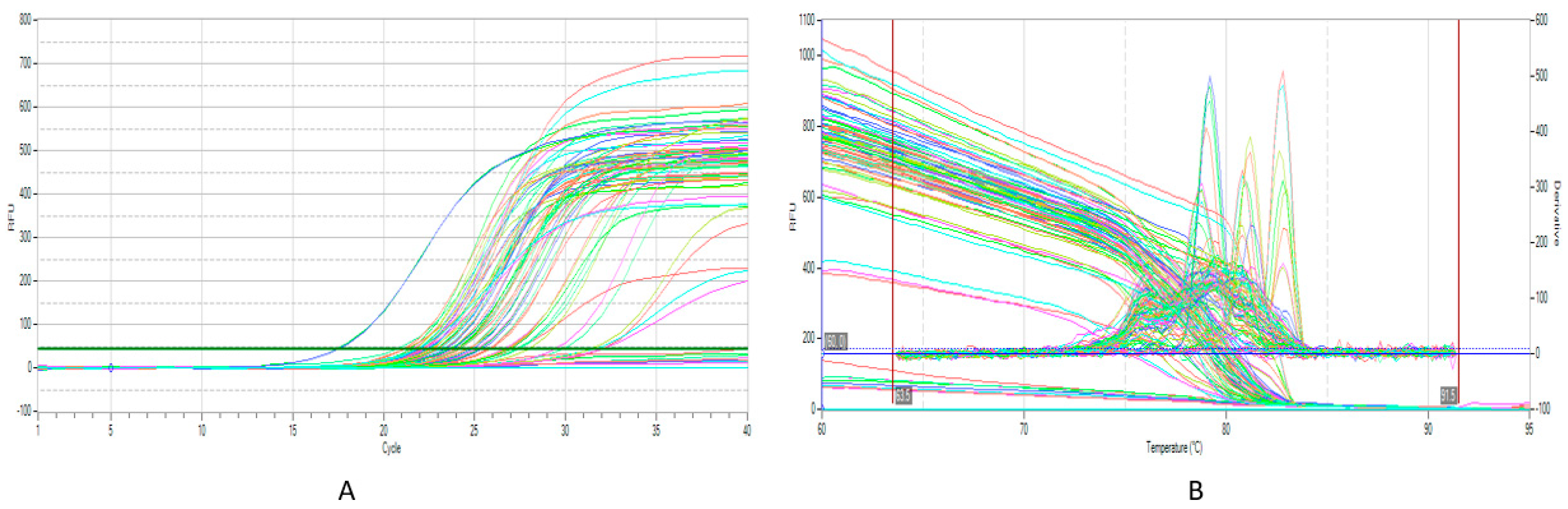
Table 3.
Diagnostic performance of mRT-qPCR and mRT-PCR in clinical samples.
| Country | Sample | Total | mRT-qPCR + N (%) | mRT-qPCR - N (%) | mRT-PCR + N (%) | mRT-PCR - N (%) |
|---|---|---|---|---|---|---|
| India | DENV positive | 16 | 16 (100.0) | 0 (0.0) | 14 (87.5) | 2 (12.5) |
| DENV negative | 16 | 0 (0.0) | 16 (100.0) | 0 (0.0) | 16 (100.0) | |
| CHIKV positive | 16 | 16 (100.0) | 0 (0.0) | 15 (93.7) | 1 (6.3) | |
| CHIKV negative | 16 | 0 (0.0) | 16 (100.0) | 0 (0.0) | 16 (100.0) | |
| Burkina Faso | DENV positive | 33 | 33 (100.0) | 0 (0.0) | 30 (90.9) | 3 (9.1) |
| DENV negative | 33 | 0 (0.0) | 33 (100.0) | 0 (0.0) | 33 (100.0) | |
| Sensitivity | 100% | 91 % | ||||
| Specificity | 100% | 100% | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



