
Submitted:
21 December 2023
Posted:
22 December 2023
You are already at the latest version
Abstract
Hydrogen sulfide (H2S) is an environmental toxicant of health concern. The brain is a major target in acute H2S poisoning. This study was to test the hypothesis that acute and subchronic ambient H2S exposures alter the brain metabolome. Male 7-8 week-old C57BL/6J mice were exposed by whole-body inhalation to 1000 ppm H2S for 45 min and euthanized at 5 min or 72 h for acute exposure. For subchronic study, mice were exposed to 5 ppm H2S 2 h/day, 5 days/week for 5 weeks. The brainstem was removed for metabolomic analysis. Enrichment analysis showed the metabolomic profiles in acute and subchronic H2S exposures matched with those of cerebral spinal fluid from patients with seizures or Alzheimer’s disease. Acute H2S exposure decreased excitatory neurotransmitters, aspartate and glutamate, while the inhibitory neurotransmitter, serotonin, was increased, which may explain H2S-induced loss of consciousness. Branched-chain amino acids and glucose were increased by acute H2S exposure. Subchronic H2S exposure within OSHA guideline surprisingly decreased serotonin altering central nervous system. In subchronic H2S exposure, glucose was decreased while polyunsaturated fatty acids, inosine, and hypoxanthine were increased. Collectively, these results provide important mechanistic clues of acute and subchronic ambient H2S poisonings and show that H2S alters brainstem metabolome.
Keywords:
Hydrogen sulfide
; Metabolomics
; Brainstem
; Brain
; Metabolism
; Biomarkers
; Neurotransmitters
; Branched-chain amino acids
; Polyunsaturated fatty acids
1. Introduction
The brain is a highly complex organ that regulates many body functions. To perform these roles, it relies on complex metabolic processes which are energy dependent [1,2]. Consequently, the brain is susceptible to toxic environmental chemicals that target mitochondria and impair energy metabolism. An example of such chemicals is hydrogen sulfide (H2S), a potent gas produced endogenously and in the environment. Endogenously produced H2S plays key roles in brain signaling, essential for normal brain physiologic functions. However, excessive amounts of H2S in the brain, either from aberrant endogenous metabolic pathways (genetic defects) or environmental exposures, leads to neuronal dysfunction, including neurodegeneration [3,4,5,6,7,8,9,10]. Ethylmalonic encephalopathy (OMIM 602473), a life-limiting disease, is caused by a genetic defect in H2S metabolism in mitochondria, resulting in chronic excessive H2S exposure in the brain [11].
Studies from our laboratory showed that acute exogenous H2S exposure induces transcriptomic and proteomic changes in the brainstem, thalamus, and other brain regions [3,5,6,7,8,9,12,13]. A wealth of literature show that H2S impairs mitochondrial function, specifically targeting cytochrome c oxidase (Complex IV) in the electron transport chain, thus inhibiting ATP production [14,15,16,17]. Chemical hypoxia and/or energy deficits are linked to loss of central respiratory drive, convulsions, and death following acute H2S exposures [18,19]. In cases without a fatal outcome, it is likely that deficits in brain metabolism may cause immediate, intermediate, delayed, and long-term neurotoxicity following acute or chronic H2S exposure. It is possible that some of these H2S-induced toxicological changes may predispose exposed individuals to other neurological conditions, including neurodegenerative conditions via gene-environment interactions. To our knowledge, despite the well-known impact of H2S on cellular metabolism, there are no metabolomics studies on the brainstem upon acute and chronic H2S exposures. This is a knowledge gap curtailing our understanding of toxic mechanisms of H2S -induced neurotoxicity.
We postulated that metabolic signatures of acute H2S poisoning were different from those following subchronic exposure to low level ambient H2S as clinical presentations are different [3,5,6,7,8,9,12,13,20]. We therefore conducted this study to understand brain metabolomic changes induced by a single acute exposure to a high concentration of H2S and that caused by subchronic exposure to ambient concentration of H2S. To do this, we used our well-established inhalation whole-mouse model because human exposure to H2S gas is typically via inhalation. For this project, we focused on the brainstem because this region has been previously shown to undergo neurodegeneration following acute H2S exposure and also because it controls vital autonomic functions such as breathing [3]. Impaired breathing is widely implicated as a cause of death following acute H2S poisoning [18]. It was also previously reported that H2S preferentially accumulates in the brainstem region [21]. Therefore, understanding the metabolomic changes in the brainstem following H2S exposure will advance our knowledge of the mechanisms underlying H2S-induced neurotoxicity, neuropathology, and death.
2. Materials and Methods
2.1. Animals
All animal studies were approved by the Institutional animal Care and Use Committee (IACUC) of Iowa State University (ISU) and University of California at Davis (UC Davis). IACUC-18-136, Feb. 2016 and IACUC-21819, Oct. 2020 were approved by ISU and UC Davis, respectively. Care and use of animals were performed in accordance with IACUC, and animals were treated humanely and handled with care. We also followed ARRIVE guidelines in the design and execution of this study [22].
Briefly, 7-8-week-old male C57BL/6J mice were purchased from the Jackson Laboratories (Sacramento, CA and Bar Harbor, ME). Only male mice were used because previous studies in our laboratory found male mice to be more sensitive to acute H2S poisoning than females [9]. Mice were randomly assigned to different groups and housed in Laboratory Animal Resources at the College of Veterinary Medicine, ISU and in the Teaching and Research Animal Care Services facility (TRACS) at the School of Veterinary Medicine, UC Davis with a 12:12 h light and dark cycle. Room temperature and relative humidity were maintained at 22 ℃ and 50 ± 10% respectively. Protein rodent maintenance diet (Teklad HSD Inc., WI) and drinking water were provided ad libitum. Mice were acclimated for 1 week before the start of the experiments.
2.2. Gas exposure
We conducted two separate experiments to mimic the two real world scenarios of H2S exposure. In the first scenario we conducted acute H2S exposure to mimic situations involving large scale industrial accidents or nefarious acts (Figure 1A). In the second scenario, we conducted subchronic exposure to low level ambient H2S to mimic occupational settings (Figure 1B). According to OSHA guidelines, workers can be exposed up to 10 ppm H2S for 8 hours a day (TWA), 5 days a week without negatively impacting their health [23]. For this reason we chose to use H2S at the concentration of 5 ppm.
2.2.1. Acute Exposure Experiment
For the acute study, we followed our previously published LCt50 model [3,5,6,7,8,9,12,13] (Figure 1A). Briefly, two groups male mice (n=10) were exposed to 1000 ppm of H2S for 45 min using a whole-body inhalation exposure chamber as reported previously [3,5,6,7,8,9,12,13]. This level and length of exposure results in 50% mortality (lethal concentration time 50 [LCt50]) during exposure and mice typically exhibit dyspnea, convulsions, loss of consciousness, and death in that order [9]. For the metabolomics analysis, we used mice that survived the LCt50 exposure. One cohort of surviving mice was sacrificed 5 min post exposure in order to determine the metabolic profile of mice immediately after exposure while the mice were still manifesting clinical effects of acute exposure. The other cohort was sacrificed 72 h post H2S exposure in order to determine metabolic changes at the time when neurodegenerative changes are typically initially observed in this model histologically [3,5,9]. For the controls, we used mice exposed once to normal room air (RA) for the same duration as H2S-exposed mice. As was the case for H2S exposed mice, one cohort of control mice (n=5) was euthanized at 5 min post RA exposure while the other was euthanized at 72 post RA exposure. All mice that received RA survived. Both H2S gas and RA were introduced to the exposure chamber from pressurized gas cylinders. The concentration of H2S in the exposure chamber was monitored in real-time using a H2S sensor (RKI Eagle, RKI Instrument, Union City, CA). Following exposure, mice were euthanized by decapitation and the brainstem (medulla and pons) was immediately further micro-dissected and the pontine reticular nucleus and the gigantocellular reticular nucleus isolated on ice. These tissue samples were stored at −80 °C until metabolomics analysis.
2.2.2. Ambient Exposure Experiment
In this experiment male mice (n=5) were exposed to 5 ppm H2S, 2 h/day, 5 days/week for 5 weeks while control group of mice (n=4) were exposed to normal room air (RA). This dosage was chosen based on our prior pilot studies and also because 5 ppm is the midpoint concentration of the OSHA guideline of a time weighted average of 10 ppm for H2S an 8 h work day, 5 days a week for the construction and shipyard industries [24]. Both H2S gas and RA were delivered to the exposure chambers from pressurized gas cylinders. None of the mice on the ambient H2S exposure experiment died. The experimental design is shown in Figure 1B.
2.3. Metabolomics analysis
The micro-dissected brainstem tissue samples were stored at -80 °C before submission to the West Coast Metabolomics Center, UC Davis for untargeted metabolomic analysis. These metabolomics analyses consisted of three assays, (1) primary metabolism by GC-TOF MS, (2) biogenic amines (hydrophilic compounds) by HILIC-MS/MS and (3) lipidomics by RPLC-MS/MS. Detailed procedures of metabolomic analysis was described in supplementary data.
2.4. Data analysis
Raw data peak heights were normalized to the sum of internal standards for primary amine and lipidomics data analyses. Data of primary metabolites were normalized by a vector normalization methods by calculating the sum of all peak heights of all identified metabolites. Data were further processed to identify outliers by using a boxplot with 1.5 x interquartile range (IQR). Outliers were removed for future analysis. The dataset of metabolomics analysis is publicly available in dryad, an open data publishing platform, (https://doi.org/10.5061/dryad.tdz08kq5n).
2.4. Enrichment analysis
Quantitative metabolite set enrichment analysis (MSEA) was performed using MetaboAnalyst 5.0 (https://www.metaboanalyst.ca). Metabolites that were significantly altered compared to the RA group were further analyzed by quantitative MSEA.
2.5. Heatmap
Metabolites that were significantly altered compared to the RA group were summarized and visualized in a heatmap. Concentration changes of metabolites were visualized as a log 2-fold change. Visualization was processed using Python version 3.0 (https://www.python.org).
2.6. Statistical analysis
Data are presented as mean and standard deviation of the mean. ANOVA with post-hoc Tukey HSD test was performed for metabolites of acute H2S exposure using the statsmodels module in Python (https://www.python.org, version 3.0.). Unpaired student’s t-test was performed for metabolites of ambient H2S exposure using Excel software of Microsoft 365. P-values of <=0.1 considered statistically significant for this explorative analysis. Statistical analysis of the metabolite set enrichment data was performed in MetaboAnalyst 5.0 (https://www.metaboanalyst.ca).
3. Results
3.1. Exposure to acute and ambient hydrogen sulfide
Mice exposed acutely to H2S for 45 minutes manifested seizures, dyspnea, ataxia, and knockdown during exposure as previously reported [3,5,6,7,9,12,13]. Those that survived the acute exposure were still weak, ataxic, and had labored breathing and were euthanized 5 min post exposure. Those euthanized at 72 h post acute H2S exposure were active and moved about normally. However, mice acutely exposed to H2S exhibited a reduction in body weight [3]. In contrast, mice from the 5 ppm exposure group did not exhibit any abnromal physiological symptoms or behavior albeit a reduction in body weight 30 days post ambient H2S exposure (data not shown).
3.2. Metabolomic changes in the brainstem following acute and ambient hydrogen sulfide
H2S induced metabolomic chnages. Metabolomic changes in brainstems from mice given a single high dose acute exposure were different from those of mice repeatedly administered low levels of H2S for over a month. Results of the changes from the metabolomics analyses included primary metabolites, biogenic amine metabolites, and lipidomic metabolites (sumarized in heatmaps for acute and ambient H2S exposures; Figure 2 and Figure 3). The Venn diagram shows the number of significantly altered identified metabolites which were increased more than 50 % or decreased 33 % compared to room air control group (Figure 4). Venn diagram represents the number of significantly altered metabolites in common or unique in 5 min and 72 h of acute H2S exposure and subchronic ambient H2S exposures. The significantly altered metabolites were listed in Table 1 and Table 2 for 5 min and 72 h of acute H2S exposure and subchronic ambient H2S exposure, respectively.
In the Venn diagram, immediate response reflects metabolic changes in brainstems of mice acutely exposed to H2S and euthanized 5 min post acute exposure, while early reflects changes at 72 h post acute exposure. Results show that both acute and subchronic H2S exposure significantly altered brainstem metabolites. Several metabolites including a phosphatidylinositol (PI 36:5), prostaglandin E1, thiazolidine-4-carboxylic acid, 3-methyhistidine, and phosphatidylethanol 18:1_18:1 (PEtOH 18:1_18:1) were increased both at 5 min post-acute H2S and subchronic ambient H2S while no metabolite was decreased in both acute and subchronic ambient H2S exposures relative to the RA group. 3-Methylhistidine and PEtOH 18:1_18:1 were increased at all three euthanasia time points. Metabolites increased in common by acute H2S exposure both at 5 min and 72 h included caffeic acid, N8-acetylspermidine, vanillin-4-sulfate, flavin adenine, 3-methylhistidine, and PEtOH 18:1_18:1, while those decreased by acute H2S exposure were N-α-methylhistamine, 2,8-quinolinediol, vanillin, a glycerophospholipid (Cardiolipin 40:4_42:7), adenosine, and behenic acid. Metabolites presented in the heatmap analyses are further listed in Table 1 and Table 2 with mass-to-charge ratio and fold change (log2 scale).
3.3. Enrichment analysis
We performed enrichment analysis using Metaboanalyst 5.0 to investigate whether H2S-induced metabolic profiles cause any biological dysregulation and whether H2S-induced metabolic dysregulations share any similarity with any existing pathological signature databases in metaboanalyst 5.0. While the database did not have brainstem as a tissue, we chose human cerebral spinal fluid (CSF) dataset as those most closely matching our experimenatl design. The results showed several matches with human neuro-pathological conditions which are summarized in Figure 5, Figure 6 and Figure 7. Briefly, the metabolomic profiles following acute H2S poisoning matched with several neurological disorders including seizures, epilepsy, Alzheimer’s disease, and Glut-1 deficiency syndrome among others (Figure 5 and Figure 6). The metabolomic profiles of subchronic ambient H2S exposure matched with several pathological conditions including Alzheimer’s disease, dementia, Parkinson’s disease, seizures, Glut-1 deficiency, and anoxia among others (Figure 7).
3.4. H2S dysregulated brainstem neurotransmitters
We analyzed neurotransmitters following both acute and subchronic ambient H2S exposures in the brainstem region. Interestingly, excitatory neurotransmitters, namely aspartate and glutamate were decreased while serotonin, an inhibitory neurotransmitter, was increased at 5 min after a single acute H2S exposure (Figure 8A). Adenosine, which acts both as a central excitatory and as an inhibitory neurotransmitter in the brain, was decreased following both acute and subchronic H2S exposures. Subchronic ambient H2S decreased serotonin concentration in the brainstem (Figure 8B). Tyrosine, precursor of cathecolamines (excitatory), was also increased at 5 min post acute H2S exposure, while tryptophan, a precursor of serotonin, was increased at 72 h post acute H2S exposure. Tryptophan was also increased by subchronic ambient H2S exposure (Figure 9B).
3.5. H2S dysregulated energy homeostasis
We analyzed for changes in lipid metabolism in the brainstem following acute and subchronic H2S exposures. Immediately following acute H2S exposure, BCAA were significantly increased more than 2-fold (Figure 10A). Interestingly, subchronic ambient H2S also significantly increased BCAA albeit less than acute exposure (Figure 10B).
Behenic acid concentration, a very long-chain fatty acid, was significantly decreased more than 2 folds following acute H2S exposure (Figure 11A). Following subchronic ambient H2S exposure major fatty acids in brainstem including oleic acid, arachidonic acid (AA), and docosahexaenoic acid (DHA) were significantly increased (Figure 11B).
Cellular respiration was also dysregulated following both acute and subchronic ambient H2S exposures. Glucose concentration was significantly elevated at 5 min post acute H2S exposure, while it decreased following ambient H2S exposure (Figure 12). Elevated glucose concentration can affect multiple biological pathways. Polyol pathway converts glucose to sorbitol to fructose. Sorbitol and fructose concentrations were also significantly increased at 5 min post acute H2S exposure. Lactate is formed via glycolysis and was also increased following acute H2S exposure but it was decreased following subchronic ambient H2S exposure. However, ribulose 5-phosphate and UDP-GlcNAc, intermediates of pentose phosphate pathay (PPP) and hexosamine biosynthetic pathways (HBP), respectively, were not significantly altered (data not shown). Succinate, fumarate, and malate which are intermediates of the Krebs cycle were also increased at 5 min of acute H2S exposure.
3.6. H2S dysregulated inosine metabolism
Inosine and its metabolites (hypoxanthine, xanthine) were increased by subchronic ambient H2S exposure (Figure 12) indicating an increased catabolism of purines under these conditions. Allantoin, an oxidation product of urate and usually considered a marker of oxidative stress, was the only metabolite increased at 5 min post acute H2S exposure. In contrast, inosine, hypoxanthine, and xanthine were all increased following subchronic ambient H2S exposure along with guanine and guanosine which are converted to xanthine (Figure 13).
4. Discussion
Hydrogen sulfide is widely reported to have a broad spectrum of physiological functions. It is also reported to be a highly toxic compound with steep dose-response relationships. The brain is a primary target organ. Acute high dose exposures are linked to acute death and to permanent neurobehavioral abnormalities and neurodegeneration [25,26,27,28] while the impacts of chronic low level H2S exposure in humans remain ambiguous. Despite the significance of this compound in health and disease the lack of data on impact of H2S on brain metabolism is striking. This is the first comprehensive metabolomic study of acute high dose H2S exposure and subchronic low dose H2S exposure on the brain. For the subchronic low dose study, we used 5 ppm H2S to expose mice only for 2 h per day Monday through Friday for 5 weeks. As a reference, the OSHA TWA exposure guidelines for an 8 h workday 5 days a week is 10 ppm. Results showed that both acute and subchronic ambient H2S significantly altered many classes of metabolites including organic oxygen compounds, organic acids, phenylpropanoids, polyketides, organoheterocyclic compounds, benzenoids, nucleosides, nucleotides, organic nitrogen compounds, lipids, and lipid-like molecules in the brain. Importantly, metabolic changes induced by H2S exposure were different depending on whether it was a high dose exposure or subchronic low dose exposure. Some metabolites, however, were increased both by acute and by subchronic exposures suggesting such compounds are sensitive indicators of H2S exposure.
3-Methylhistidine, a derivative of histidine and methionine amino acids, and phosphatidylethanol 18:1_18:1 (PEtOH 18:1_18:1) a glycerophosphoethanol were both significantly increased following both a single H2S exposure and by subchronic ambient H2S exposure. The functions of these two metabolites in brainstem are not clear at the moment.Vanillin was decreased while vanillin-4-sulfate was increased by acute H2S exposure. Vanillin has been linked to colorectal cancer. Roles of vanillin and vanillin-4-sulfate and roles of H2S exposure in altering vanillin and vanillin-4-sulfate remain to be determined. However, these metabolites may serve as potential biomarkers of H2S-poisoning.
Several metabolites were altered at 5 min and 72 h following a single acute H2S exposure. N8-acetyl spermindine was increased by 0.8 fold following acute H2S exposure. It is a polyamine derived from spermidine by deacetylation and reported to play a role in regulating cell signaling and gene expression among other functions [29]. It was previously reported that increase of N8-acetyl spermidine is caused by by inhibiting N8-acetylspermidine deacetylase. This compound is linked to the differentiation of neurons and to elevated dopamine [29]. Interestingly, acute H2S exposure increased dopamine concentration in the brainstem [3]. N8-acetyl spermidine is also considered as a biomarker for ischemic cardiomyopathy [30]. Interestingly, H2S is a cytochrome c oxidase inhibitor and causes chemical ischemia [31]. The exact role of N8-acetyl spermidine remains to be studied, but it may signal brain ischemia.
Flavin adenine, a derivative of riboflavin or vitamin B2, is also known as flavin adenine dinucleotide (FAD), and it plays a crtical role as a cofactor of many enzymes. FAD was increased more than 1 fold at 5 min and 0.5 fold after 72 h following a single acute exposure and is therefore a potential biomarker of acute H2S exposure in the brainstem. Since FAD plays a significant role as a cofactor in brain energy metabolism, it is possible that sudden increase of FAD in the brainstem may indicate an imbalance in the redox cellular state (favoring FAD over FADH). FADH is non detectable by any of the mass spectrometers used in this study.
Adenosine and behenic acid were decreased 1 fold and 2 fold at 5 min and 72 h post acute H2S exposure, respectively. Adenosine is a nucleoside which plays many important roles in energy transfer, signal transduction, as a neurotransmitter, and as a potent vasodilator [32]. H2S is well known to reduce ATP generation by inhibiting cytochrome c oxidase in the electron transport chain. Adenosine 5’-monophosphate was significantly decreased by about 1.5 fold following subchronic ambient H2S exposure. A decrease of adenosine concentration may limit several biological functions in which it is involved. Behenic acid, in contrast, is a very long-chain saturated fatty acid (VLCFA) and was decreased by more than 1.5 fold at 5 min and 72 h post acute H2S exposure; but not by subchronic H2S exposure. The reduction in behenic acid concentration may negatively impact energy production and homeostasis among other effects. These results suggest that both acute and subchronic H2S exposure alter brainstem metabolism, but in different ways. More research is needed to undertand the implications of these results on brain function.
The brainstem has been reported to be particularly vulnerable to acute H2S poisoning. Following exogenous exposure, H2S was reportedly found in highest concentrations in this brain region [21]. This brain region is also recognized as the center of regulation for key autonomic functions [33,34]. We have also previously shown that the brainstem is susceptible to H2S -induced neurodegeneration [3]. In addition, we showed that the inhibition of central respiratory drive (from the brainstem) is a key cause of death in our mouse model {Santana Maldonado, 2023 #17406}. Clinical effects of acute H2S poisoning characterized by seizures, and apnea among others were not seen in mice exposed to 5 ppm H2S for 1 month, indicating mechanisms involved were different. In this study, the only clinical effects noted following subchronic H2S exposure in mice was a loss in body weight.
Results of metabolite enrichment analysis summarized in Figure 5, Figure 6 and Figure 7 are very interesting. When the metabolomic profiles of acute and subchronic H2S exposure in mice are compared to existing databases of metabolomics of the cerebrospinal fluid from patients suffering from different neurological conditions, a pattern of similarities and differences emerges. At 5 min post exposure, the metabolomics enrichment pattern of acute H2S poisoning matches with many human neurological disorders including early-onset encephalopathy and cortical myoclonus, leukemia, propionic acidemia, anoxia, Rett syndrome, epilepsy, Alzheimer’s disease (AD), Parkinson’s disease (PD), Glut-1 deficiency syndrome, different seizure disorders, and schizophrenia among others. In this mouse model we have reported convulsions, and neurodegeneration [3,7,8,9,12,13]. AD and PD are neurodegenerative diseases while epilepsy matched with convultions in our mouse model. We have also reported a significant increase in dopamine and serotonin concentrations in this model {Santana Maldonado, 2023 #17406}{Anantharam, 2017 #17414;Anantharam, 2017 #17413}. Dopamine is a modulatory neurotransmitter which is both excitatory or inhibitory depending on which receptors in binds to. Serotonin is predominantly inhibitory and has been linked to schizophrenia. The summary of neurotransmitter changes in Figure 8 suggests that acute and subchronic H2S exposure tilts towards inhibitory and excitatory effects, respectively.
H2S exposure is widely reported to cause hypoxic neuronal injury which is consistent with its mechanism of action of inhibiting cytochrome C oxidase activity [31]. Reduced cytochrome c oxidase activity is also reported in Alzheimer’s disease [35] and chronic neurodegenerative diseases are characterized by brain energy deficits [1,2]. The metabolomic profiles at 5 mins post acute H2S exposure matched a metabolic profile of neurological disorders observed at 72 h min postacute exposure, including epilepsy, aseptic menigitis, Alzheimer’s disease, shizophrenia, and different seizure disorders.
We were surprised to see that subchronic H2S exposure to 5 ppm, which is within the OSHA guidelines of 10 ppm for an 8 h work day, caused metabolic derangement in the brainstem. This is a significant finding which needs confirmation because of the health implications in the workplace. Strikingly, the metabolome of subchronic ambient H2S also matched with many human neurological disorders including Alzheimer’s disease, Parknson’s disease, and Schizophrenia, which was similar to what we observed after a single acute H2S exposure with some exceptions. Notably, the metabolome of subchronic ambient H2S exposure additionally matched that of CSF from patients with dementia, canavan disease, alcoholism, and multiple sclerosis. Common themes to both acute and subchronic low dose H2S exposure revolve around metabolomic similarities between the metabolome of H2S exposure and that of seizure disorders, neurodegenerative conditions especially Alzheimer’s disease, Parkinson’s disease, neuroinflammation, ataxia, and hypoxic-induced injury. This possibly signals that environmental H2S exposure, shifts brain metabolism which may predispose or increase susceptibility to these debilitating disease conditions. Indeed, this mouse model with acute H2S exposure exhibited seizure and loss of consiousness {Santana Maldonado, 2023 #17406;Kim, 2018 #17411;Anantharam, 2017 #17414;Anantharam, 2017 #17413;Kim, 2023 #17405;Anantharam, 2018 #17412;Kim, 2020 #17410}. These results signal that H2S-induced metabolomic changes play important roles in H2S-induced physiological symptoms including seizure and loss of consiousness. Notably, it is interesting that patients predisposed to seizures or neurofibromatosis are very sensitive to acute H2S poisoning [36]. Also, H2S aggravated seizure-like events in rat seizure model [37]. Regarding the discovery of similarities in the metabolomes of acute and subchronic H2S exposure to the of CSF of patients with Alzheimer’s disease, there is meagre literature on this topic. It is well known that the interaction of gene and environment plays important roles in many neurological disorders including Alzheimers disease and Parkinson’s disease. Considering that H2S is an environmental pollutant and of occupational concern and that H2S exposure may be closely linked to many nuerological disorders with big burden in the society, more work should be focused in this area to investigate the potential role of ambient H2S exposure in AD and/or other neurological conditions in Figure 5, Figure 6 and Figure 7.
The effects of acute H2S and subchronic ambient H2S exposures on neurotransmitters were investigated. Acute H2S exposure decreased aspartic acid, glutamic acid and adenosine while serotonin was increased. Serotonin was significantly decreased by subchronic ambient H2S exposure. Aspartic acid and glutamic acid are exatatory neurotransmitters while serotonin is inhibitory [38,39]. Adenosine acts both as excitatory and inhibitory neurotransmitter. We previously reported that dopamine and serotonin were significantly increased in brainstem at 5 min post acute H2S [3], which is consistent with findings in this study. Tryptophan is a precurosor to serotonin while phenylalanine and tyrosine are precursor to dopamine. In this study tyrosine was significanlty increased at 5 min post acute exposure in brainstem while tryptophan was increased by subchronic ambient H2S exposure. Other neurotransmitters including acetylcholine, histidine, serine, noradrenaline, γ-aminobutyric acid (GABA), glycine, and histamine were not significantly altered in this study (data not shown).
Glutamate and aspartate are main excitatory neurotransmitters in brain and are released in a Ca2+-dependent manner upon electrical stimulation. Serotonin is an inhibitory neurotransmitter and has impact on a wide range from behavioral effects such appetie, aggression, memory, fear, depression. It is also involved in other CNS effects such as respiration, body temperature, motor control, and bowel movement [40]. It is well known that acute exposure to high concentrations of H2S induces loss of consicousness in humans. However, the exact mechanisms behind H2S-induced loss of consiousness are not known. A decrease of glutamate and aspartate coupled with an increase of serotonin following acute H2S exposure may play important roles in H2S-induced loss of consciousness. Interestingly, we recently reportated that sodium sulfide, a H2S donor, suppressed neuronal activity in vitro by suppressing Ca2+ oscillation in primay cortical neurons [6]. Electrical stimulus releases glutamate from synaptic vesicles to synaptic cleft. The released glutmate is taken up to glial cells by glutamate-aspartate transporter and converted to glutamine which was transported back to the neuron. Glutaminase in mitochondria convertes glutamine to glutamate and aspartate. Acute H2S induced increased glutamine at 5 min post acute acute exposure indicating that glutamate-glutamine cycling may be dysregulated. Decrease of aspartate may influence in conversion of glutamine to glutamate leading to decrease of glutamate upon acute H2S exposure. Serotonin was also reported to inhibit glutamate release and the action of released glutamate [39]. Besides, an acute surge of serotonin induces focal seizures [41].
Interestingly, subchronic ambient H2S exposure significantly decreased serotonin concentration. Current OSHA guideline sets recommended airborne exposure limit (REL) to 10 ppm for hydrogen sulfide in the workplaces [23]. To the best of our knowledge, it has not been previously reported that subchronic ambient H2S exposure dysregulates serotonin. Therefore, this finding is novel. There are many serotonin receptors. It should be interesting to investigate the health impact of decreased serotonin in subchronic ambient H2S exposure.
Glutamate not only acts as a neurotransmitter but also as a anaplerotic component in tricarboxylic acid (TCA) cycle in enegery balance. Glutamate is converted to α-ketoglutarate. In addition to the altered glutamate and glutamine, fumarate and malate were increased by 0.5 fold compared to the RA control mice at 5 min post acute H2S , which may indicate that acute H2S exposure dysregulates the TCA cycle in other ways besides inhibiting cytochrome c oxidase in the electron transport chain (ETC). Indeed, intermediates of the TCA cycle including succinate, fumarate, and malate were increased at 5 min post acute H2S exposure while succinate and fumarate were decreased following ambient H2S exposure. Glucose is a key compound in glycolysis. Glucose concentration was significantly increased at 5 min post acute H2S exposure while decreased following ambient H2S exposure. Increase in brain glucose during acute H2S exposure may suggest it is spared following inhibition of cytochrome c oxidase by H2S. Elevated glucose concentration can affect multiple biological pathways including polyol, glycolysis, PPP, and HBP. Intermediates of polyol and glycolysis were increased while PPP and HBP were not significantly altered at 5 min post acute H2S exposure, indicating acute H2S exposure significantly dysregulate energy balance. Accumulation of succinate may be caused by inverse activity of succinate dehydrogenase converting fumate to succinate {Zhang, 2020 #17728} indicating the TCA cycle was inhibited. Lactate concentration remained elevated following acute H2S exposure, which is in line with previous findings that treatment of sodium hydrosulfide induced elevation of lactate [42]. It was shown that acute H2S exposure-induced inhibition of cytochrome c oxidase in brain including brainstem remained upto 72 h [3]. It is plausible that inhibition of cytochrome c oxidase subsequently inhibit ETC and TCA cycle, which could not meet the energy demand in central nervous system. Accumulation of succinate was shown to produce ROS and induce static epielpticus in a kainic acid rat model [43].
Fatty acids have multiple important roles in nervous system; from serving as structural component of membranes, energy source, signaling molecule, cellular differentiation, apoptosis, to pathological conditions such as aging and neurodegeneartive diseases [44]. Regulation of fatty acid is tightly-controlled and differently in different brain regions. Behenic acid is a VLCFA. VLCFA are preferentially metabolized in the peroxisome. The statistically significant reduction of behenic acid observed in this study may indicate altered peroxisome fatty acid beta oxidation. Subchronic ambient H2S strikingly increased metabolites of both the mono-unsaturated fatty acid (MUFA) and poly-unsaturated fatty acid (PUFA) synthesis pathways in brainstem including oleic acid, arachidonic acid (AA), and docosahexaenoic acid (DHA) in brainstem. Arachidonic acid, which was also increased, is pro-inflammatory and is also linked to neurodegenerative diseases such as AD [45]. A deficiency of DHA has been been associated with neurodegenerative disorders [46]. We have also previously reported that acute H2S poisoning causes neuroinflammation and neurodegeneration [3,9,12,13].
Subchronic ambient H2S also dysregulated inosine metabolism and increased inosine, hypoxanthine, and xanthine. Inosine is metabolized to hypoxanthine, xanthine and finally to uric acid [47]. Inosine was shown to play important roles in neuroprotection among others, presumably via anti-inflammatory and antioxidant properties [48]. AMP was shown to be metabolised to IMP, hypoxanthine, xanthine and uric acid to enhance ATP production during acute energy consumption [47]. Administration of inosine was shown to induce hypoglycemicaemia [49]. Indeed, subchronic ambient H2S decreased glucose concentration more than 1 fold compared to the RA control group. This finding shows that subchronic ambient H2S dysregulate energy homeostasis. More work is needed on this topic to determine whether subchronic ambient H2S exposure causes a brain energy deficit. Brain energy deficit has been cited a contributing factor in neurodegeneration [1,2]. Potentially this suggests that ambient H2S exposure may be an environmental factor predisposing to neurodegeneration.
This study, though yielding interesting results had some limitations. For example, the study only consisted of male, adult mice. Future studies should include female mice as well so as to determine whether there may be sex differences in brain metabolism following H2S exposure. As has been reported [9], male mice were more sensitive to acute H2S poisoning than females. Therefore, including female mice in the study may shed light on toxic mechanisms of H2S . The other limitation of this study was that the acute study only lasted up to 72 h post H2S exposure. In previous studies, we have shown neurodegeneration to be fully manifested on day 7 in the brain stem and other brain regions [3,5,7,8,9]. Also, in human victims of single acute H2S poisoning accidents, a plethora of long-term debilitating neurological sequalae including neuropsychiatric disturbance, sleep disorders, headaches, memory and cognition deficits and persistent vegetative states are reported [25,26,27,28,50]. Therefore, future studies should investigate metabolomic changes in the brain-stem several months later to determine the metabolomic profiles of delayed neurotoxic effects.
5. Conclusions
In summary, results of this study have identified 3-methylhistidine and PEtOH18:1_18:1 as potential biomarkers of H2S exposure, because they were significantly increased in brainstems of mice exposed to H2S both acutely and subchronically. Twelve metabolites were altered by acute H2S exposure including N-acetylspermidine, adenosine, behenic acid, flavin adenine, vanillin, vanillin-4-sulfate, caffeic acid, and behenic acid which were identified as potential biomarkers of acute H2S exposure. Acute H2S decreased glutamate and aspartate while serotonin was increased, suggesting that overall acute H2S shifts neurotransmitters predominantntly towards inhibitory status. Serotonin was decreased by subchronic ambient H2S exposure and so were adenosine suggesting chronic H2S exposure may predispose neurological excitation. Glucose and fructose were increased in acute H2S poisoning, while glucose was significantly decreased in subchronic ambient H2S. MUFAs and PUFAs were increased in subchronic ambient H2S exposure. Overall, results in this study provide evidence that both acute and subchronic H2S exposures alter neurotransmitters and energy metabolism in the brainstem. This has opened novel mechanisms underlying H2S-induced neurotoxicty. Surprisingly and more importantly, this study showed that current OSHA guidelines (10 ppm for 40 h/work week) may not be adequately protecting worker health as a subchronic exposure of mice to 5 ppm H2S for only a few hours perday sigificantly altered neurotransmitters and energy metabolism.
Author Contributions
Conceptualization. W.K.R.; Data curation. D.K.; methodology. D.K., C.S.M. and W.K.R.; Software. D.K.; Investigation. D.K. and C.S.M.; Project administration. D.K.; Resources. W.K.R..; validation. D.K.; formal analysis D.K.; Supervision. D.K. and W.K.R.; Visualization. D.K.; writing—original draft preparation, D.K. and W.K.R.; writing—review and editing, D.K., C.G., W.K.R.; funding acquisition, W.K.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by ISU internal fundings for W.K.R. and start-up fundings by UC Davis for W.K.R.
Institutional Review Board Statement
The study was conducted in accordance with the Institutional animal Care and Use Committee (IACUC) of University of California at Davis (UC Davis). The animal study protocol was approved by IACUC with IACUC-18-136 at ISU and with IACUC-21819 at UC Davis.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in Dryad at [https://doi.org/10.5061/dryad.tdz08kq5n].
Acknowledgments
Authors are thankful to West Coast Metabolomics Center, UC Davis for their support.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nature reviews. Drug discovery 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radic Biol Med 2016, 100, 108–122. [Google Scholar] [CrossRef]
- Santana Maldonado, C.M.; Kim, D.S.; Purnell, B.; Li, R.; Buchanan, G.F.; Smith, J.; Thedens, D.R.; Gauger, P.; Rumbeiha, W.K. Acute hydrogen sulfide-induced neurochemical and morphological changes in the brainstem. Toxicology 2023, 485, 153424. [Google Scholar] [CrossRef]
- Santana Maldonado, C.; Weir, A.; Rumbeiha, W.K. A comprehensive review of treatments for hydrogen sulfide poisoning: past, present, and future. Toxicology mechanisms and methods 2023, 33, 183–196. [Google Scholar] [CrossRef]
- Rumbeiha, W.K.; Kim, D.S.; Min, A.; Nair, M.; Giulivi, C. Disrupted brain mitochondrial morphology after in vivo hydrogen sulfide exposure. Scientific reports 2023, 13, 18129. [Google Scholar] [CrossRef]
- Kim, D.S.; Pessah, I.N.; Santana, C.M.; Purnell, B.; Li, R.; Buchanan, G.F.; Rumbeiha, W.K. Investigations into hydrogen sulfide-induced suppression of neuronal activity in vivo and calcium dysregulation in vitro. Toxicol Sci 2023, 192, 247–264. [Google Scholar] [CrossRef]
- Kim, D.S.; Anantharam, P.; Padhi, P.; Thedens, D.R.; Li, G.; Gilbreath, E.; Rumbeiha, W.K. Transcriptomic profile analysis of brain inferior colliculus following acute hydrogen sulfide exposure. Toxicology 2020, 430, 152345. [Google Scholar] [CrossRef]
- Kim, D.S.; Anantharam, P.; Hoffmann, A.; Meade, M.L.; Grobe, N.; Gearhart, J.M.; Whitley, E.M.; Mahama, B.; Rumbeiha, W.K. Broad spectrum proteomics analysis of the inferior colliculus following acute hydrogen sulfide exposure. Toxicology and applied pharmacology 2018, 355, 28–42. [Google Scholar] [CrossRef]
- Anantharam, P.; Whitley, E.M.; Mahama, B.; Kim, D.S.; Imerman, P.M.; Shao, D.; Langley, M.R.; Kanthasamy, A.; Rumbeiha, W.K. Characterizing a mouse model for evaluation of countermeasures against hydrogen sulfide-induced neurotoxicity and neurological sequelae. Ann N Y Acad Sci 2017, 1400, 46–64. [Google Scholar] [CrossRef]
- Rumbeiha, W.; Whitley, E.; Anantharam, P.; Kim, D.S.; Kanthasamy, A. Acute hydrogen sulfide-induced neuropathology and neurological sequelae: challenges for translational neuroprotective research. Ann N Y Acad Sci 2016, 1378, 5–16. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 2009, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Anantharam, P.; Whitley, E.M.; Mahama, B.; Kim, D.S.; Sarkar, S.; Santana, C.; Chan, A.; Kanthasamy, A.G.; Kanthasamy, A.; Boss, G.R.; et al. Cobinamide is effective for treatment of hydrogen sulfide-induced neurological sequelae in a mouse model. Ann N Y Acad Sci 2017, 1408, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Anantharam, P.; Kim, D.S.; Whitley, E.M.; Mahama, B.; Imerman, P.; Padhi, P.; Rumbeiha, W.K. Midazolam Efficacy Against Acute Hydrogen Sulfide-Induced Mortality and Neurotoxicity. Journal of medical toxicology : official journal of the American College of Medical Toxicology 2018, 14, 79–90. [Google Scholar] [CrossRef]
- Guidotti, T.L. Hydrogen sulfide: advances in understanding human toxicity. International journal of toxicology 2010, 29, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, T.L. Hydrogen Sulfide intoxication. In Handbook of clinical neurology, 3rd ed.; Lotti, M., Bleecker, M.L., Eds.; 2015; Volume 131. [Google Scholar]
- Reiffenstein, R.J. Neurochemical and neurophysiological effects of high dose hydrogen sulphide. In Proceedings of international conference on hydrogen sulphide toxicity; Prior, M.G., Roth, S.H., Green, F.H.Y., Hulbert, W.C., Reiffenstein, R., Eds.; The Sulphide Research Network: Banff, Alberta, Canada, 1989. [Google Scholar]
- Reiffenstein, R.J.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annual review of pharmacology and toxicology 1992, 32, 109–134. [Google Scholar] [CrossRef]
- Beauchamp, R.O., Jr.; Bus, J.S.; Popp, J.A.; Boreiko, C.J.; Andjelkovich, D.A. A critical review of the literature on hydrogen sulfide toxicity. Critical reviews in toxicology 1984, 13, 25–97. [Google Scholar] [CrossRef]
- Vicas, I.M.O. Therapeutics and Management of H2S Poisoning. In Proceedings of International Conference on Hydrogen Sulphide Toxicity, Prior, M.G., Roth, S.H., Green, F.H.Y., Hulbert, W.C., Reiffenstein, R., Eds. Sulphide Research Network, in care of the Department of Pharmacology, University of Alberta: Banff, Alberta, 1989; pp. 217–219. [Google Scholar]
- Santana, C.M.; Gauger, P.; Vetger, A.; Magstadt, D.; Kim, D.S.; Shrestha, D.; Charavaryamath, C.; Rumbeiha, W.K. Ambient hydrogen sulfide exposure increases the severity of influenza A virus infection in swine. Archives of environmental & occupational health 2021, 76, 526–538. [Google Scholar] [CrossRef]
- warenycia, m.w.; Goodwin, l.R.; benishin, c.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute Hydrogen sulfide poisoning. Biochemical Pharmacology 1989, 38. [Google Scholar] [CrossRef]
- Sprague, W. Arrive Guidelines 2.0. Vet Clin Pathol 2020, 49, 378–379. [Google Scholar] [CrossRef]
- Occupational Safety and Health Administration. Hydrogen Sulfide. Available online: https://www.osha.gov/hydrogen-sulfide/hazards (accessed on Oct. 3).
- Occupational Safety and Health Administration. Hydrogen Sulfide. Available online: https://www.osha.gov/SLTC/hydrogensulfide/standards.html (accessed on 08/20).
- Tvedt, B.; Edland, A.; Skyberg, K.; Forberg, O. Delayed neuropsychiatric sequelae after acute hydrogen sulfide poisoning: affection of motor function, memory, vision and hearing. Acta neurologica Scandinavica 1991, 84, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Nam, B.; Kim, H.; Choi, Y.; Lee, H.; Hong, E.S.; Park, J.K.; Lee, K.M.; Kim, Y. Neurologic sequela of hydrogen sulfide poisoning. Industrial health 2004, 42, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, F.; Cummins, J.W.; Anderson, R.E. Neurological sequelae of massive hydrogen sulfide inhalation. Archives of neurology 1979, 36, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.W.; Safir, E.F.; Summerville, G.P.; Middleberg, R.A. Occupational fatality and persistent neurological sequelae after mass exposure to hydrogen sulfide. Am J Emerg Med 1995, 13, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Mudumba, S.; Menezes, A.; Fries, D.; Blankenship, J. Differentiation of PC12 cells induced by N8-acetylspermidine and by N8-acetylspermidine deacetylase inhibition. Biochem Pharmacol 2002, 63, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Liu, C.; Mehta, A.; Ko, Y.A.; Tahhan, A.S.; Dhindsa, D.S.; Uppal, K.; Jones, D.P.; Butler, J.; Morris, A.A.; et al. N8-Acetylspermidine: A Polyamine Biomarker in Ischemic Cardiomyopathy With Reduced Ejection Fraction. Journal of the American Heart Association 2020, 9, e016055. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, R.J.; Green, F.H.; Auer, R.N. Sulfide toxicity: mechanical ventilation and hypotension determine survival rate and brain necrosis. Journal of applied physiology 1993, 75, 1348–1353. [Google Scholar] [CrossRef]
- Boison, D. Adenosine as a neuromodulator in neurological diseases. Current opinion in pharmacology 2008, 8, 2–7. [Google Scholar] [CrossRef]
- Faraguna, U.; Ferrucci, M.; Giorgi, F.S.; Fornai, F. Editorial: The Functional Anatomy of the Reticular Formation. Frontiers in neuroanatomy 2019, 13, 55. [Google Scholar] [CrossRef]
- Mangold, S.A.; Das, J.M. Neuroanatomy, Reticular Formation; StatPearls: StatPearls, 2023. [Google Scholar]
- Cardoso, S.M.; Proenca, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Shivanthan, M.C.; Perera, H.; Jayasinghe, S.; Karunanayake, P.; Chang, T.; Ruwanpathirana, S.; Jayasinghe, N.; De Silva, Y.; Jayaweerabandara, D. Hydrogen sulphide inhalational toxicity at a petroleum refinery in Sri Lanka: a case series of seven survivors following an industrial accident and a brief review of medical literature. Journal of occupational medicine and toxicology 2013, 8, 9. [Google Scholar] [CrossRef]
- Luo, Y.; Wu, P.F.; Zhou, J.; Xiao, W.; He, J.G.; Guan, X.L.; Zhang, J.T.; Hu, Z.L.; Wang, F.; Chen, J.G. Aggravation of seizure-like events by hydrogen sulfide: involvement of multiple targets that control neuronal excitability. CNS neuroscience & therapeutics 2014, 20, 411–419. [Google Scholar] [CrossRef]
- Dingledine, R.; McBain, C.J. Glutamate and Aspartate Are the Major Excitatory Transmitters in the Brain. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition; Lippincott: Lippincott, 1999. [Google Scholar]
- Maura, G.; Marcoli, M.; Pepicelli, O.; Rosu, C.; Viola, C.; Raiteri, M. Serotonin inhibition of the NMDA receptor/nitric oxide/cyclic GMP pathway in human neocortex slices: involvement of 5-HT(2C) and 5-HT(1A) receptors. British journal of pharmacology 2000, 130, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu Rev Med 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Almallouhi, E.; Rahwan, M.; Dainton, H.; Bonilha, L. Focal seizure as a manifestation of serotonin syndrome: case report. Avicenna J Med 2019, 9, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, P.; Chenuel, B.; Sonobe, T. High-dose hydroxocobalamin administered after H2S exposure counteracts sulfide-poisoning-induced cardiac depression in sheep. Clinical toxicology 2015, 53, 28–36. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Zhu, W.; Yu, J.; Wang, Q.; Zhang, J.; Cui, Y.; Pan, X.; Gao, X.; Sun, H. Succinate accumulation induces mitochondrial reactive oxygen species generation and promotes status epilepticus in the kainic acid rat model. Redox biology 2020, 28, 101365. [Google Scholar] [CrossRef] [PubMed]
- Hussain, G.; Schmitt, F.; Loeffler, J.P.; Gonzalez de Aguilar, J.L. Fatting the brain: a brief of recent research. Frontiers in cellular neuroscience 2013, 7, 144. [Google Scholar] [CrossRef]
- Thomas, M.H.; Pelleieux, S.; Vitale, N.; Olivier, J.L. Arachidonic acid in Alzheimer’s disease. J Neurol Neuromedicine 2016, 1, 1–6. [Google Scholar]
- Kerdiles, O.; Laye, S.; Calon, F. Omega-3 polyunsaturated fatty acids and brain health: Preclinical evidence for the prevention of neurodegenerative diseases. Trends in Food Science & Technology 2017, 69B, 203–213. [Google Scholar]
- Johnson, T.A.; Jinnah, H.A.; Kamatani, N. Shortage of Cellular ATP as a Cause of Diseases and Strategies to Enhance ATP. Frontiers in pharmacology 2019, 10, 98. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Macedo-Junior, S.J.; Lapa-Costa, F.R.; Cezar-Dos-Santos, F.; Santos, A.R.S. Inosine as a Tool to Understand and Treat Central Nervous System Disorders: A Neglected Actor? Frontiers in neuroscience 2021, 15, 703783. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, N.F.; Ibrahim, S.M.; Moustafa, P.E.; Elbaset, M.A. Inosine mitigated diabetic peripheral neuropathy via modulating GLO1/AGEs/RAGE/NF-kappaB/Nrf2 and TGF-beta/PKC/TRPV1 signaling pathways. Biomed Pharmacother 2022, 145, 112395. [Google Scholar] [CrossRef] [PubMed]
- Wasch, H.H.; Estrin, W.J.; Yip, P.; Bowler, R.; Cone, J.E. Prolongation of the P-300 latency associated with hydrogen sulfide exposure. Archives of neurology 1989, 46, 902–904. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A summary of experimental exposure paradigms. A) Exposure paradigm of single acute exposure to 1000 ppm H2S. This exposure model induces 50 % mortality (LCt50). B) Exposure paradigm of subchronic ambient exposure to 5 ppm H2S. In this model mice were exposed to 5 ppm H2S for 2 h/day, 5 days/week for 5 weeks.
Figure 1.
A summary of experimental exposure paradigms. A) Exposure paradigm of single acute exposure to 1000 ppm H2S. This exposure model induces 50 % mortality (LCt50). B) Exposure paradigm of subchronic ambient exposure to 5 ppm H2S. In this model mice were exposed to 5 ppm H2S for 2 h/day, 5 days/week for 5 weeks.
Figure 2.
Heatmap analysis of significantly altered metabolites following a single acute H2S exposure. Concentrations of significantly altered metabolites from primary metabolites, biogenic amines, and lipidomic metabolites are visualized in a heatmap by fold-change (log2 scale). Classification of individual metabolites is shown on the right side of the heatmap.
Figure 2.
Heatmap analysis of significantly altered metabolites following a single acute H2S exposure. Concentrations of significantly altered metabolites from primary metabolites, biogenic amines, and lipidomic metabolites are visualized in a heatmap by fold-change (log2 scale). Classification of individual metabolites is shown on the right side of the heatmap.
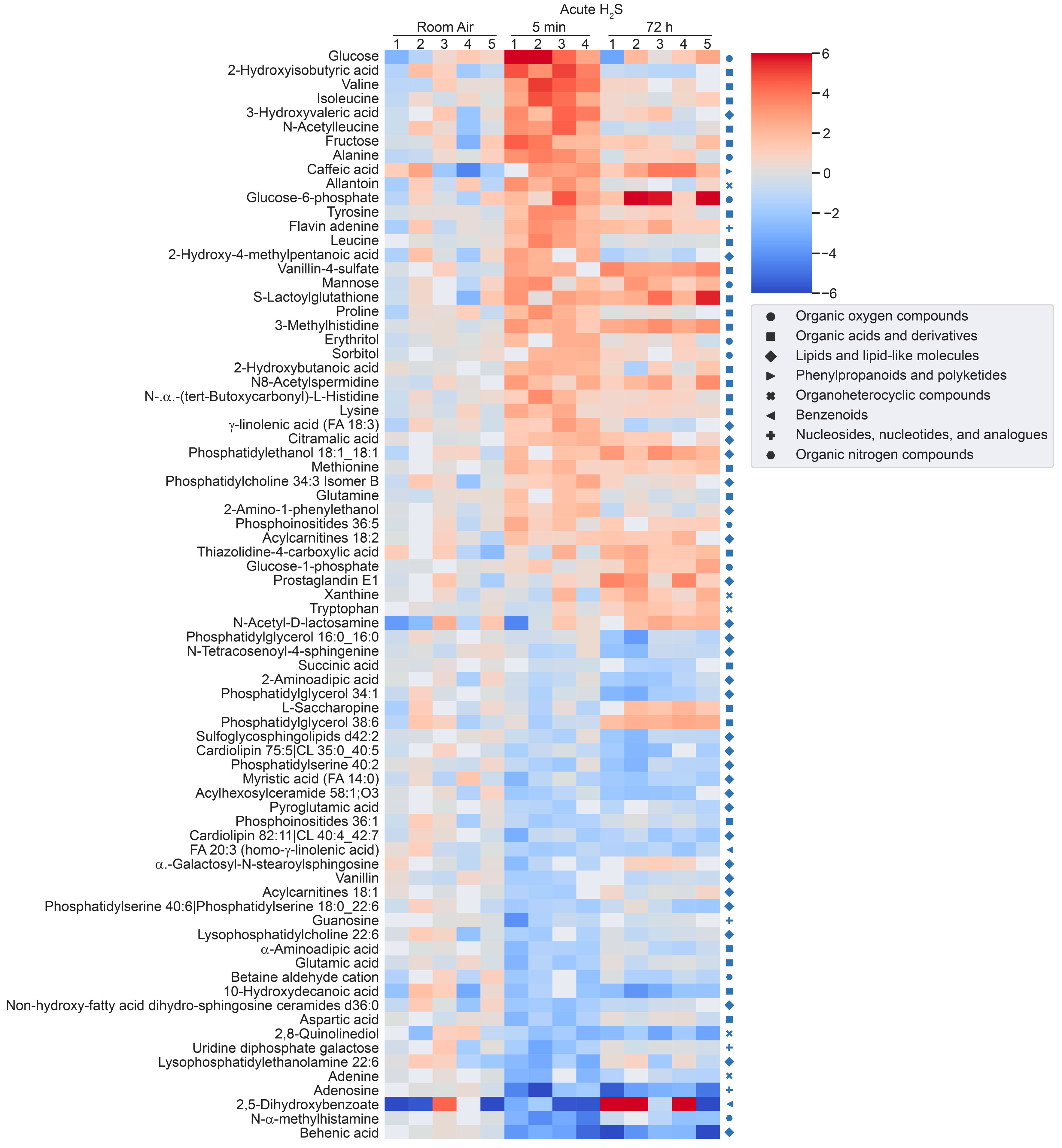
Figure 3.
Heatmap analysis of significantly altered metabolites following subchronic ambient H2S exposure. Concentrations of significantly altered primary organic metabolites, hydrophilic compounds including biogenic amines, and lipidomic metabolites are visualized in a heatmap by fold-change (log2 scale). Classification of individual metabolites is shown on the right side of the heatmap.
Figure 3.
Heatmap analysis of significantly altered metabolites following subchronic ambient H2S exposure. Concentrations of significantly altered primary organic metabolites, hydrophilic compounds including biogenic amines, and lipidomic metabolites are visualized in a heatmap by fold-change (log2 scale). Classification of individual metabolites is shown on the right side of the heatmap.
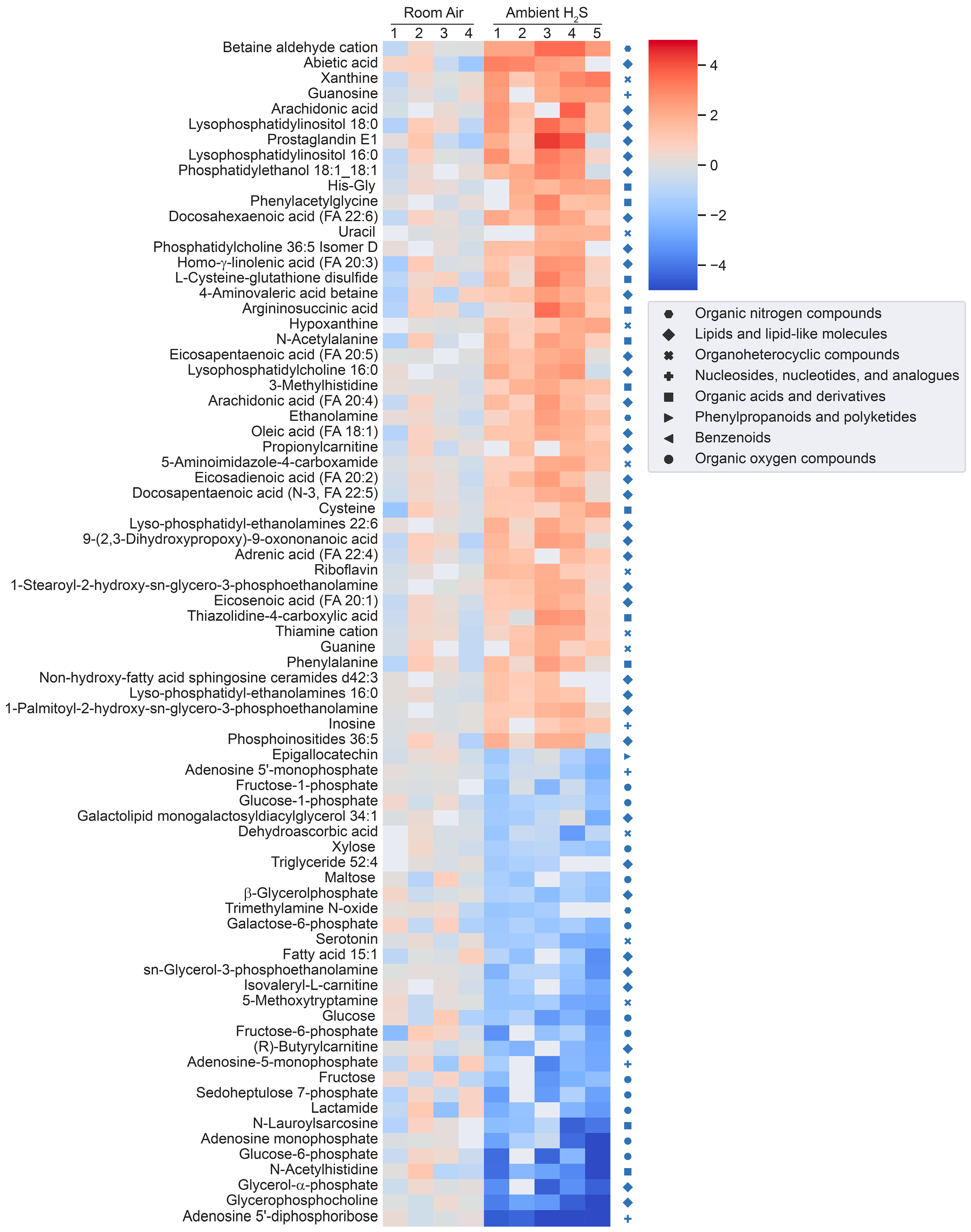
Figure 4.
Venn diagrams summarising numbers of significantly altered metabolites following acute and ambient H2S exposure. Immediate (5 min) and early (72 h) responses to acute H2S and response to ambient H2S exposure in brainstem are shown. A) Increased metabolites compared to RA control group. B) Decreased metabolites compared to RA control group.
Figure 4.
Venn diagrams summarising numbers of significantly altered metabolites following acute and ambient H2S exposure. Immediate (5 min) and early (72 h) responses to acute H2S and response to ambient H2S exposure in brainstem are shown. A) Increased metabolites compared to RA control group. B) Decreased metabolites compared to RA control group.
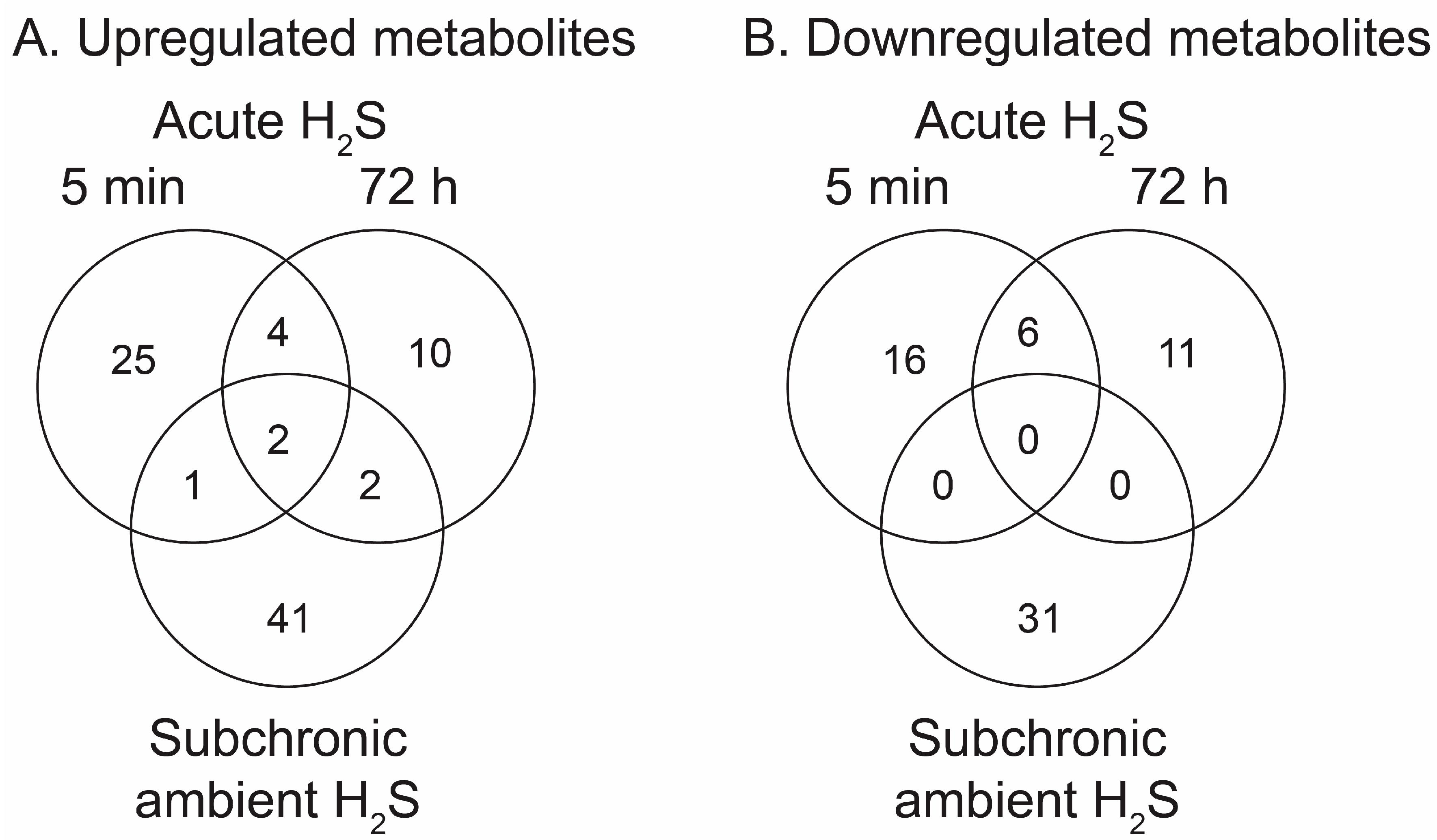
Figure 5.
Disease-based enrichment analysis of signficantly altered metabolites of immediate response to H2S exposure (5 min post a single acute H2S exposure) for primary metabolites, biogenic amines, and lipidomic metabolites.
Figure 5.
Disease-based enrichment analysis of signficantly altered metabolites of immediate response to H2S exposure (5 min post a single acute H2S exposure) for primary metabolites, biogenic amines, and lipidomic metabolites.
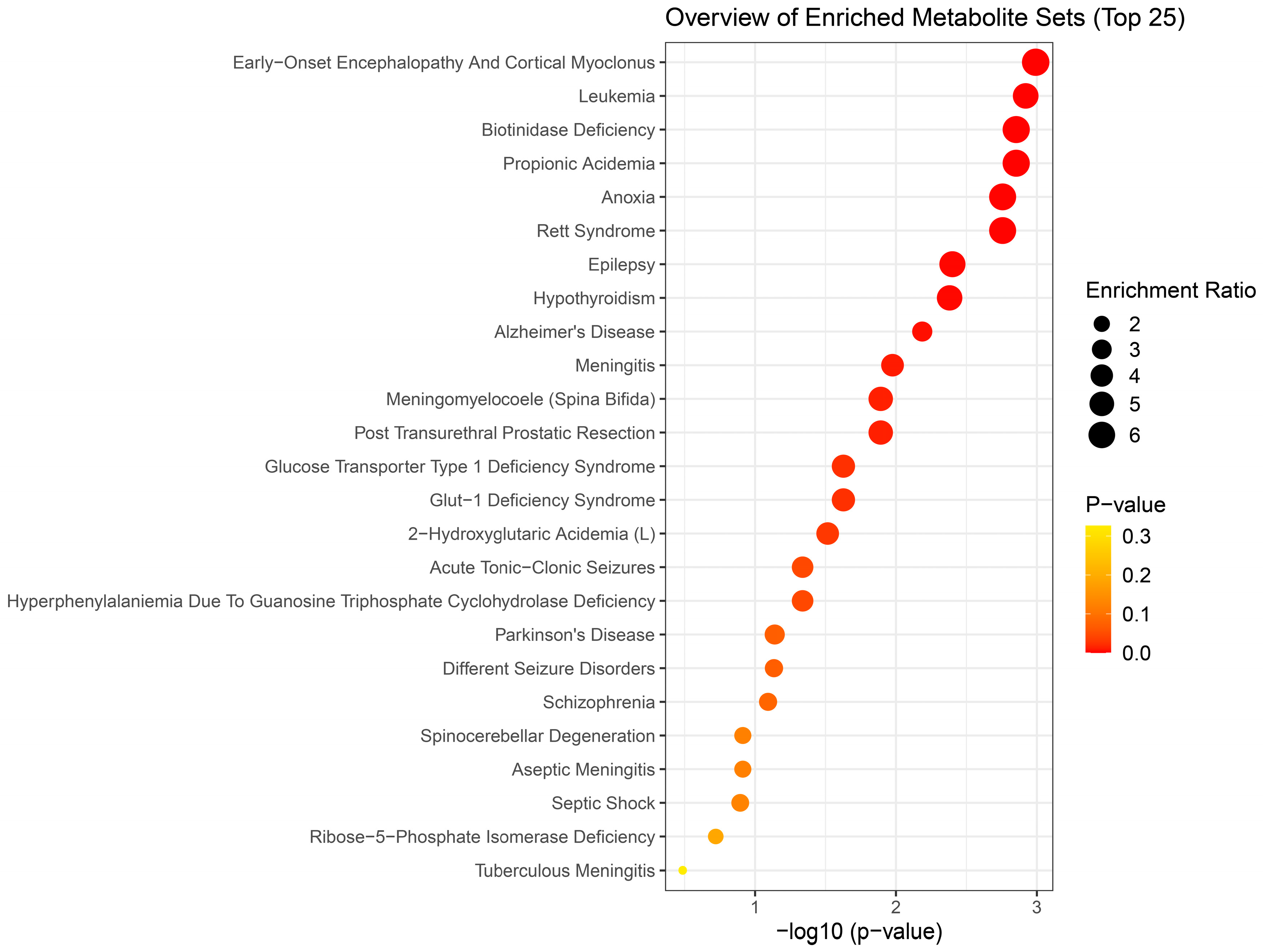
Figure 6.
Disease-based enrichment analysis of signficantly altered metabolites of early response 72 h post a single acute H2S exposure for primary metabolites, biogenic amines, and lipidomic metabolites.
Figure 6.
Disease-based enrichment analysis of signficantly altered metabolites of early response 72 h post a single acute H2S exposure for primary metabolites, biogenic amines, and lipidomic metabolites.
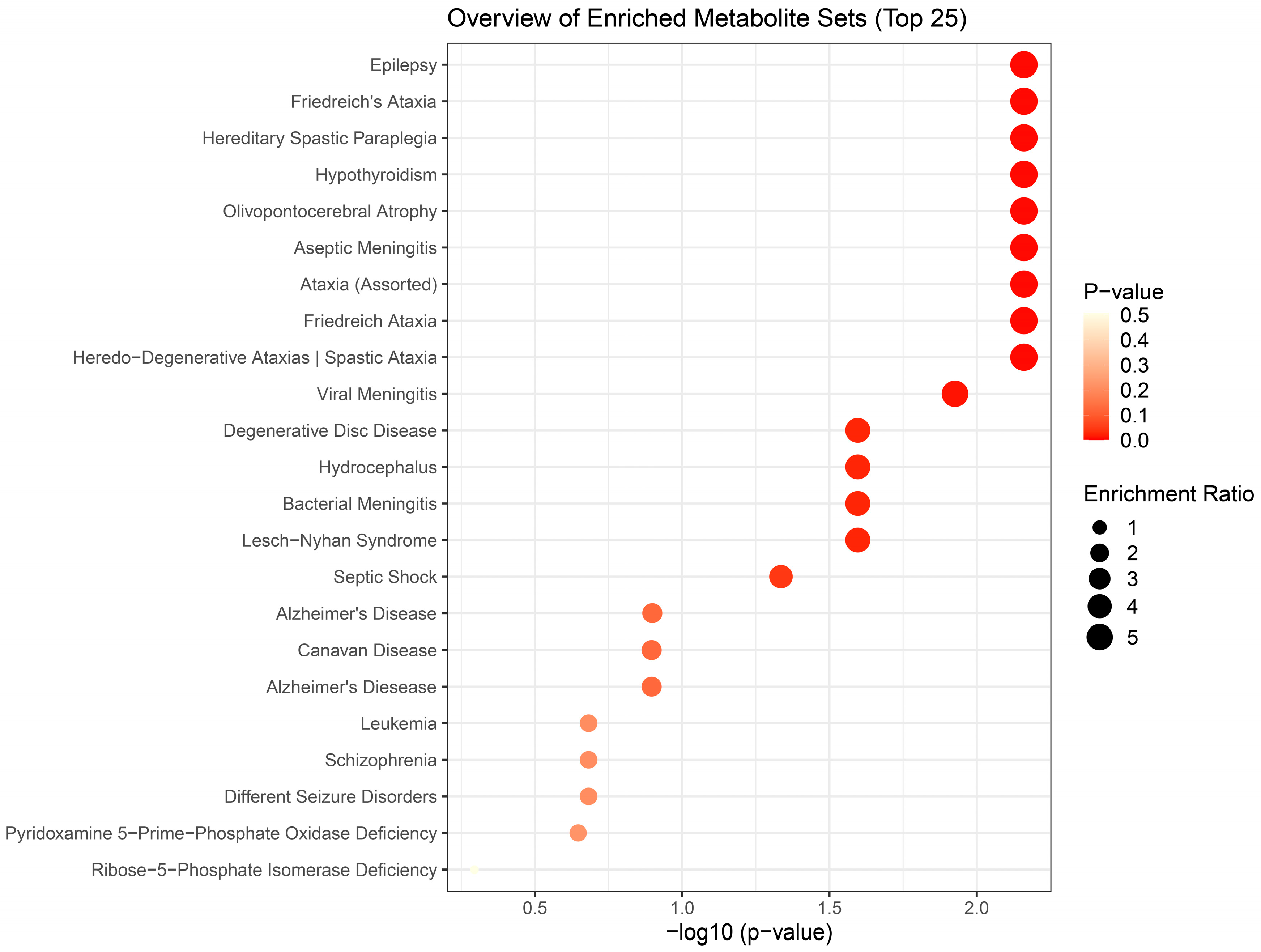
Figure 7.
Disease-based enrichment analysis of signficantly altered metabolites of subchronic response to repeated H2S exposure for 5 weeks for primary metabolites, biogenic amines, and lipidomic metabolites.
Figure 7.
Disease-based enrichment analysis of signficantly altered metabolites of subchronic response to repeated H2S exposure for 5 weeks for primary metabolites, biogenic amines, and lipidomic metabolites.
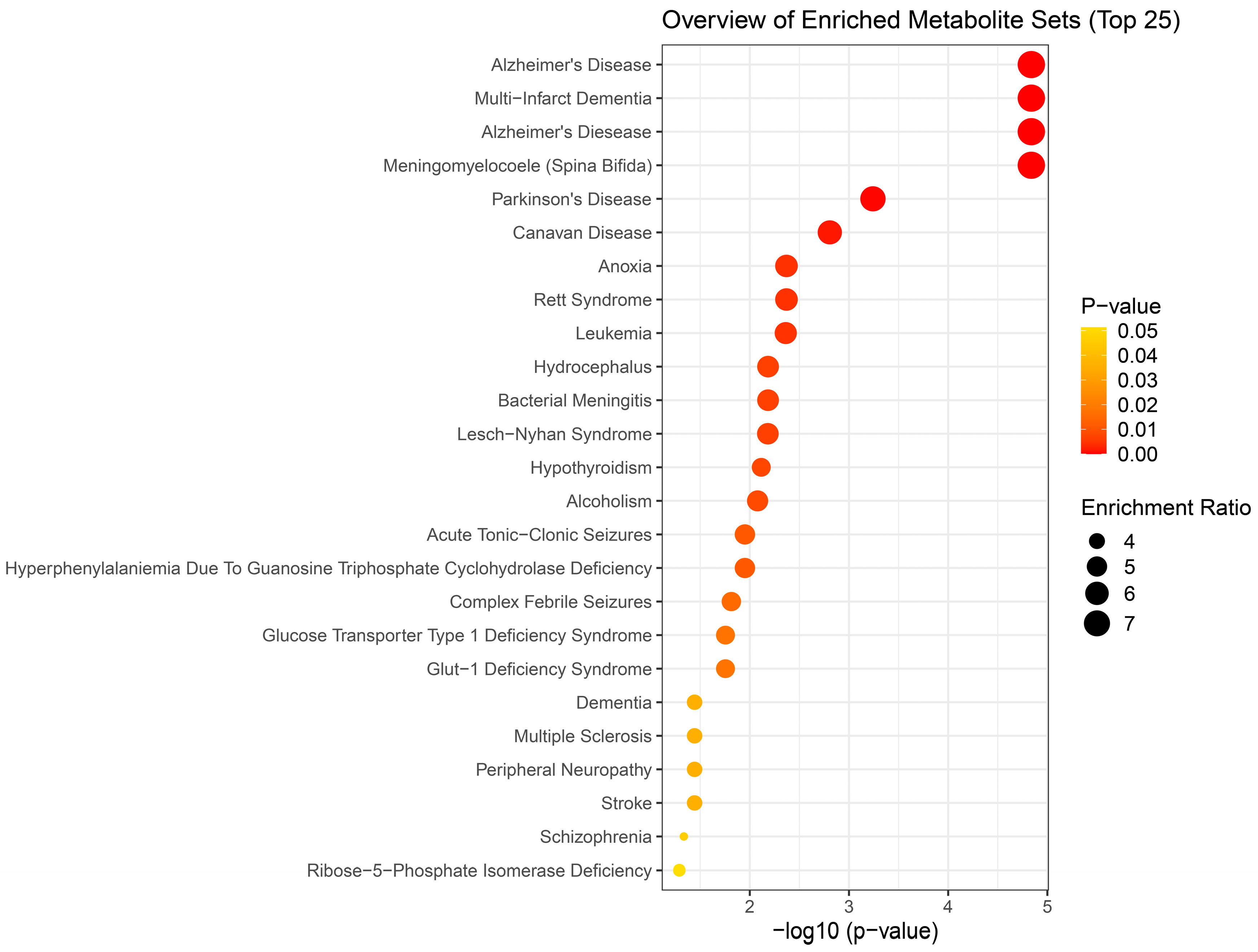
Figure 8.
Neurotransmitter changes following H2S exposure. A) Fold change of neurotransmitters following acute 1000 ppm H2S exposure. B) Fold change of neurotransmitters following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups to test for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, ** < 0.05, and ****<0.001.
Figure 8.
Neurotransmitter changes following H2S exposure. A) Fold change of neurotransmitters following acute 1000 ppm H2S exposure. B) Fold change of neurotransmitters following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups to test for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, ** < 0.05, and ****<0.001.
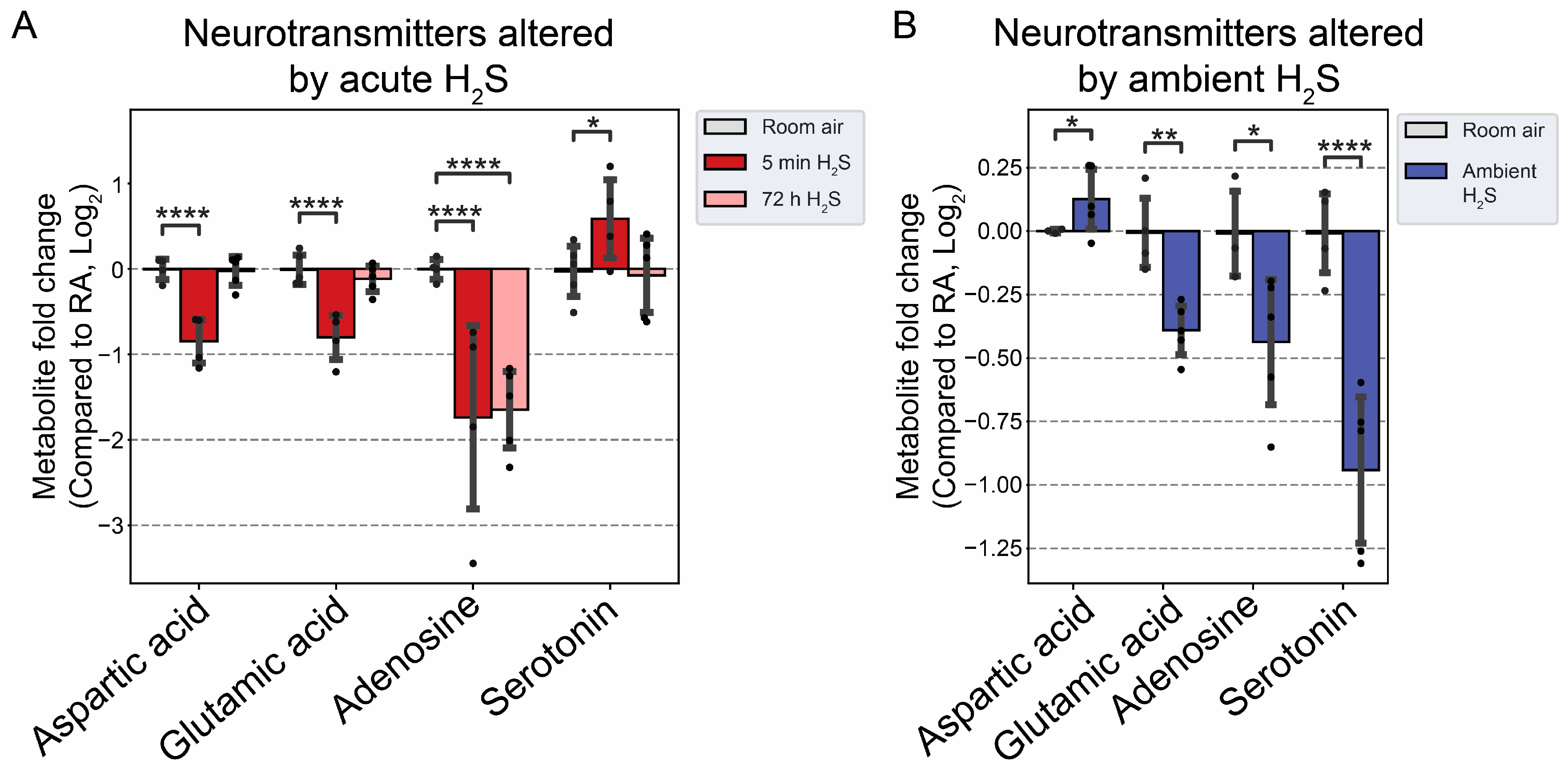
Figure 9.
Altered aromatic amino acids following H2S exposure. A) Fold change of aromatic amino acids following acute 1000 ppm H2S exposure. B) Fold change of aromatic amino acids following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups to test for statistical significance. Asterisks indicate statistically significant differences compared to RA group. P ** < 0.05 and ***<0.01.
Figure 9.
Altered aromatic amino acids following H2S exposure. A) Fold change of aromatic amino acids following acute 1000 ppm H2S exposure. B) Fold change of aromatic amino acids following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups to test for statistical significance. Asterisks indicate statistically significant differences compared to RA group. P ** < 0.05 and ***<0.01.
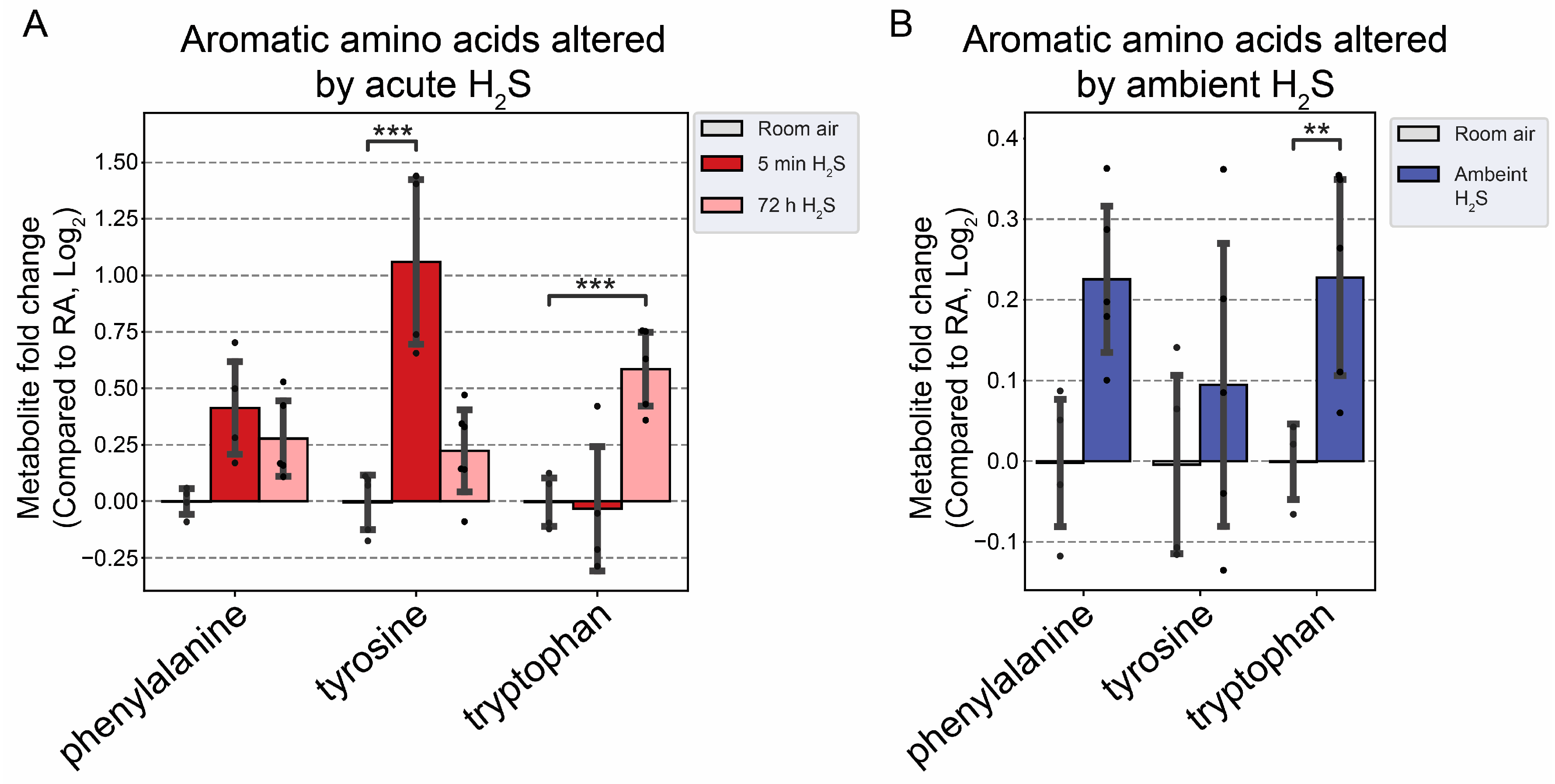
Figure 10.
Fold change of branched chain amino acid following H2S exposure. A) Fold change of BCAA following acute H2S exposure. B) Fold change of BCAA following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, ***<0.01, and ****<0.001.
Figure 10.
Fold change of branched chain amino acid following H2S exposure. A) Fold change of BCAA following acute H2S exposure. B) Fold change of BCAA following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, ***<0.01, and ****<0.001.
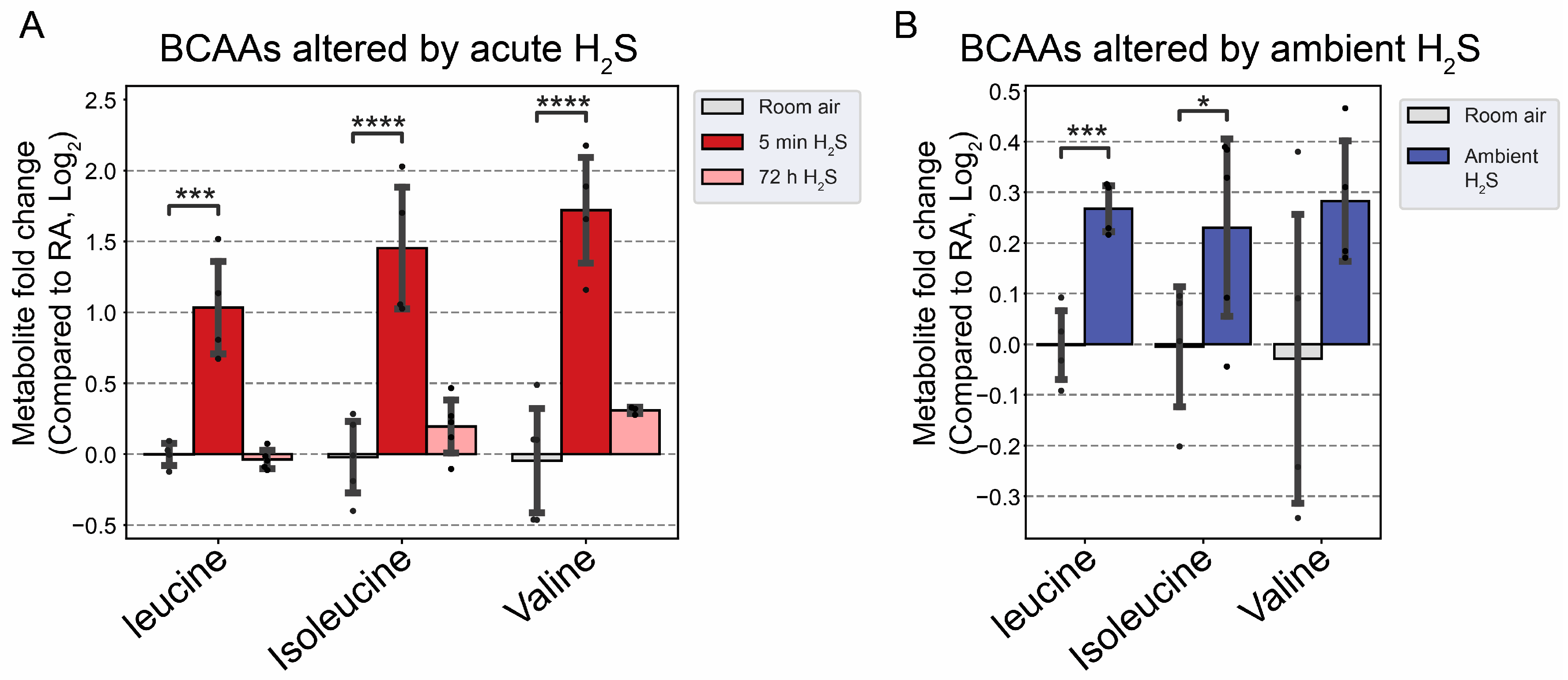
Figure 11.
Altered fatty acids following H2S exposure. A) Decreased behenic acid, a very long-chain fatty acid (VLCFA), following acute H2S exposure. B) Major unsaturated fatty acids in brain were increased following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups for statistical significance. Asterisks indicate significant differences compared to RA group. P ** < 0.05, ***<0.01, and ****<0.001.
Figure 11.
Altered fatty acids following H2S exposure. A) Decreased behenic acid, a very long-chain fatty acid (VLCFA), following acute H2S exposure. B) Major unsaturated fatty acids in brain were increased following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups for statistical significance. Asterisks indicate significant differences compared to RA group. P ** < 0.05, ***<0.01, and ****<0.001.
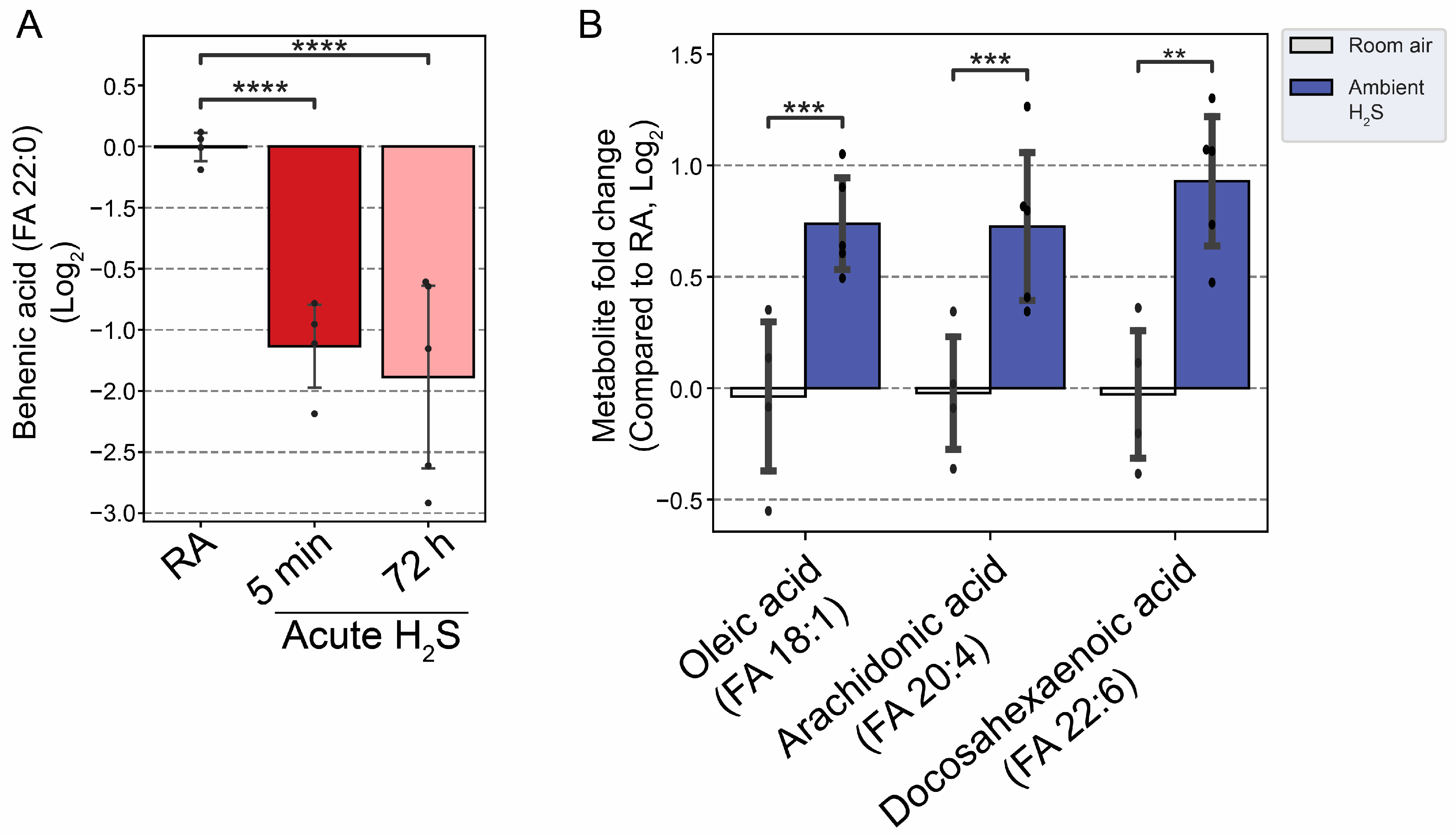
Figure 12.
Schematic pathway of glycolysis and TCA cycle. Responses at 5 min and 72 post acute H2S exposure and also following subchronic ambient H2S are presented in a heatmap analysis. Fructose, sorbitol, and glucose were significantly increased by acute H2S exposure but decreased by subchronic ambient H2S exposures compared to RA control. Changes in lactic acid are shown in a similar fashion. Succinate, fumarate, and malate concentrations were increased at 5 min of acute H2S exposure. Differentially altered metabolites are presented in fold changes (log 2 scale). Abbreviation: DHAP: Dihydroxyacetone phosphate, Fructose-1-P: Fructose 1-phosphate, Fructose-1,6-BP: Fructose 1,6-bisphosphate, Fructose-6-P: Fructose 6-phosphate, G3P: Glycerol-3-phosphate, GA3P: Glyceraldehyde 3-phosphate, Glucose-1-P: Glucose 1-phosphate, Glucose-6-P: Glucose 6-phosphate, and TCA cycle: Tricarboxylic acid cycle.
Figure 12.
Schematic pathway of glycolysis and TCA cycle. Responses at 5 min and 72 post acute H2S exposure and also following subchronic ambient H2S are presented in a heatmap analysis. Fructose, sorbitol, and glucose were significantly increased by acute H2S exposure but decreased by subchronic ambient H2S exposures compared to RA control. Changes in lactic acid are shown in a similar fashion. Succinate, fumarate, and malate concentrations were increased at 5 min of acute H2S exposure. Differentially altered metabolites are presented in fold changes (log 2 scale). Abbreviation: DHAP: Dihydroxyacetone phosphate, Fructose-1-P: Fructose 1-phosphate, Fructose-1,6-BP: Fructose 1,6-bisphosphate, Fructose-6-P: Fructose 6-phosphate, G3P: Glycerol-3-phosphate, GA3P: Glyceraldehyde 3-phosphate, Glucose-1-P: Glucose 1-phosphate, Glucose-6-P: Glucose 6-phosphate, and TCA cycle: Tricarboxylic acid cycle.
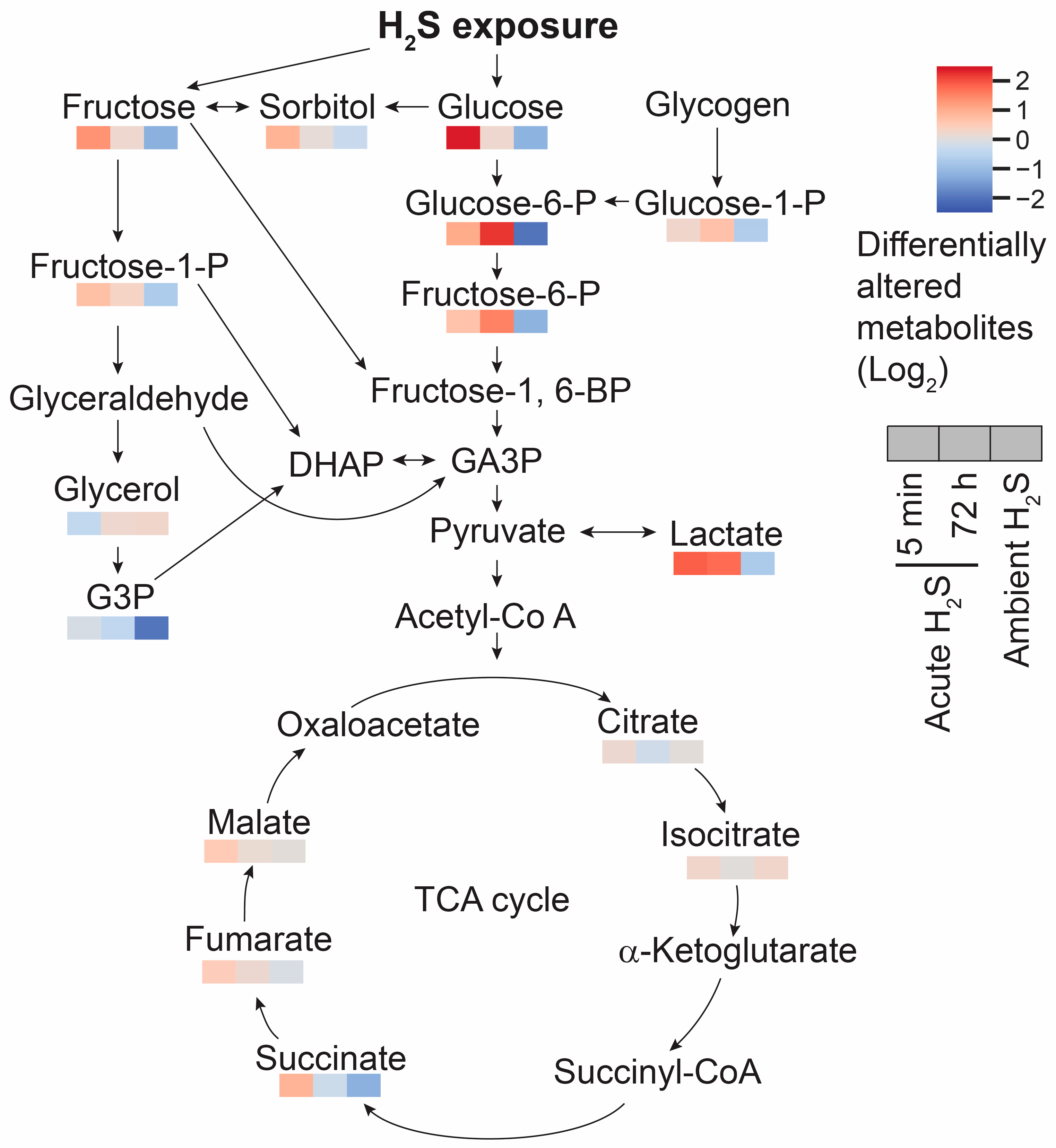
Figure 13.
Altered inosine and metabolites following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups for statistical significance. Asterisks indicate significant differences compared to RA group. P ***<0.01 and ****<0.001.
Figure 13.
Altered inosine and metabolites following subchronic ambient H2S exposure. Values are presented as mean ± standard deviation. ANOVA with post-hoc Tukey HSD test was performed for acute H2S exposure groups while unpaired Students’ t-test was performed for subchronic ambient H2S exposure groups for statistical significance. Asterisks indicate significant differences compared to RA group. P ***<0.01 and ****<0.001.
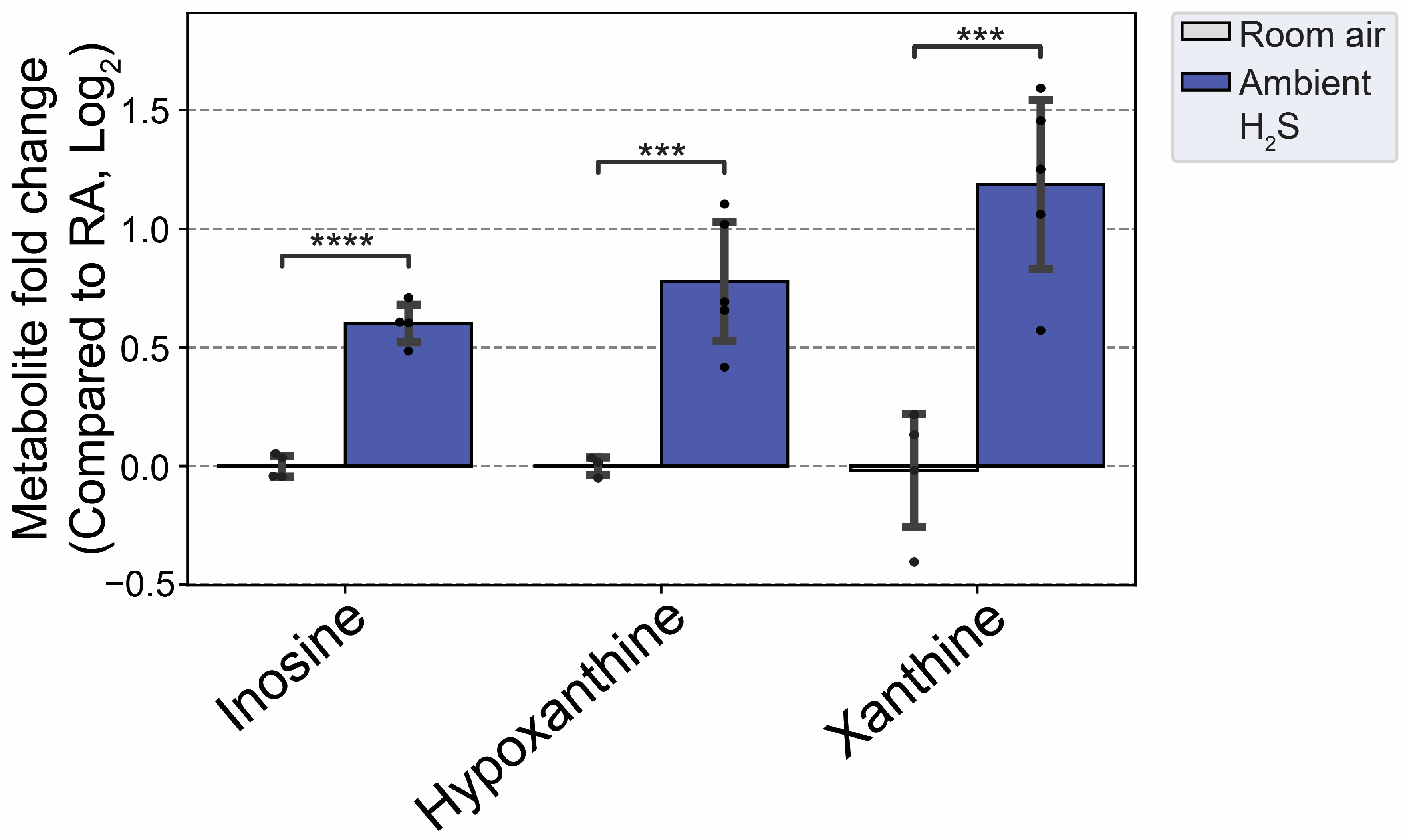

Table 1.
Significantly altered metabolites at 5 min post acute H2S exposure compared to room air control group. Common metabolite name, mass-to-charge ratio, fold change (log2 scale), and statistical significance were listed. ANOVA with post-hoc Tukey HSD test was performed for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, * * < 0.05, ***<0.01, and ****<0.001.
Table 1.
Significantly altered metabolites at 5 min post acute H2S exposure compared to room air control group. Common metabolite name, mass-to-charge ratio, fold change (log2 scale), and statistical significance were listed. ANOVA with post-hoc Tukey HSD test was performed for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, * * < 0.05, ***<0.01, and ****<0.001.
| Metabolites | m/z | 5min | 72h | ||
| Fold Change | Significance | Fold Change | Significance | ||
| 10-hydroxydecanoic acid | 187.135 | 1.77 | **** | 0.31 | |
| 2,5-dihydroxybenzoate | 329.025 | 1.52 | **** | 0.21 | |
| 2,8-quinolinediol | 162.057 | 1.46 | *** | 0.32 | |
| 2-amino-1-phenylethanol | 120.08 | 1.41 | **** | -0.2 | |
| 2-aminoadipic acid | 162.076 | 1.36 | *** | 0.48 | |
| 2-hydroxy-4-methylpentanoic acid | 131.071 | 1.35 | **** | 0.23 | |
| 2-hydroxybutanoic acid | 131 | 1.22 | * | 1.18 | ** |
| 2-hydroxyisobutyric acid | 103.041 | 1.15 | *** | 0.02 | |
| 3-hydroxyvaleric acid | 117.056 | 1.15 | 2.88 | * | |
| 3-methylhistidine | 170.091 | 1.11 | *** | 0.25 | |
| Acylcarnitines 18:1 | 426.358 | 0.9 | **** | 0.31 | *** |
| Acylcarnitines 18:2 | 424.344 | 0.87 | *** | 0.19 | |
| Acylhexosylceramide 58:1;O3 | 1048.91 | 1.07 | *** | -0.04 | |
| Adenine | 136.062 | 1.1 | 1.33 | * | |
| Adenosine | 236 | 1.07 | *** | 0.68 | ** |
| Alanine | 116 | 1.01 | *** | -0.44 | |
| Allantoin | 157.038 | 1 | *** | 1.29 | **** |
| α-galactosyl-N-stearoylsphingosine | 728.598 | 1.79 | **** | -0.43 | |
| α-aminoadipic acid | 260 | 0.97 | * | 0.81 | |
| Aspartic acid | 232 | 0.96 | 1.49 | ** | |
| Behenic acid | 117 | 0.93 | *** | 0.08 | |
| Betaine aldehyde cation | 120.1019_102.0893 | 0.93 | *** | 1.19 | **** |
| Caffeic acid | 181.057 | 0.91 | **** | 0.13 | |
| Cardiolipin 75:5|Cardiolipin 35:0_40:5 | 1496.04 | 0.79 | *** | 0.29 | |
| Cardiolipin 82:11|Cardiolipin 40:4_42:7 | 1582.09 | 0.78 | ** | -0.24 | |
| Citramalic acid | 247 | 0.83 | ** | 0.26 | |
| Erythritol | 217 | 0.78 | * | 0.76 | ** |
| Flavin adenine | 246 | 0.66 | ** | 0.2 | |
| Fructose | 307 | 0.63 | **** | -0.06 | |
| γ-Linolenic acid (FA 18:3) | 277.216 | 0.69 | * | 1.06 | *** |
| Glucose | 319 | 0.62 | *** | -0.02 | |
| Glucose-1-phosphate | 217 | 0.59 | * | 0.45 | |
| Glucose-6-phosphate | 387 | 0.47 | * | 0.65 | ** |
| Glutamic acid | 246 | 0.27 | 0.8 | * | |
| Glutamine | 156 | 0.26 | 0.7 | ** | |
| Guanosine | 324 | 0.12 | 1.07 | ** | |
| Homo-gamma-linolenic acid (FA 20:3) | 305.249 | 0.68 | *** | 0.48 | ** |
| Isoleucine | 158 | 0.09 | 0.71 | * | |
| Leucine | 158 | -0.06 | 0.87 | * | |
| L-Saccharopine | 277.139 | -0.23 | *** | -0.58 | **** |
| Lysine | 317 | -0.24 | -0.62 | ** | |
| Lysophosphatidylcholine 22:6 | 568.339 | -0.07 | -0.61 | ** | |
| Lysophosphatidylethanolamine 22:6 | 526.288 | -0.18 | -0.58 | ** | |
| Mannose | 205 | -0.24 | -0.8 | *** | |
| Methionine | 150.056 | -0.26 | 0.71 | ** | |
| Myristic acid (FA 14:0) | 227.202 | 0.75 | *** | 0.47 | ** |
| N-(Octadecanoyl)-sphinganine (Cer-NDS d36:0) | 566.547 | 0.86 | * | 0.88 | * |
| N-α-(Tert-Butoxycarbonyl)-L-histidine | 110.069 | -0.27 | 0.93 | **** | |
| N8-Acetylspermidine | 188.175 | -0.28 | -0.6 | ** | |
| N-Acetyl-D-lactosamine | 406.13 | -0.35 | -0.9 | *** | |
| N-Acetylleucine | 172.096 | -0.4 | -0.66 | ** | |
| N-α-Methylhistamine | 126.101 | -0.49 | -0.68 | * | |
| N-tetracosenoyl-4-sphingenine | 648.6272_630.6146 | -0.5 | * | -0.89 | ** |
| Phosphatidylcholine 34:3 Isomer B | 756.5538_778.5289 | -0.58 | *** | -0.39 | *** |
| Phosphatidylglycerol 16:0_16:0 | 721.502 | -0.6 | ** | -0.6 | ** |
| Phosphatidylserine 40:2 | 844.603 | -0.69 | * | -0.19 | |
| Phosphatidylserine 40:6|PS 18:0_22:6 | 836.547 | -0.74 | *** | -0.29 | * |
| Phosphoinositides 34:1 | 835.536 | -0.61 | ** | -0.59 | ** |
| Phosphoinositides 36:1 | 863.56 | -0.65 | ** | 0.38 | * |
| Phosphoinositides 36:5 | 855.5 | -0.66 | *** | -0.3 | |
| Phosphoinositides 38:6 | 881.512 | -0.66 | *** | 0.07 | |
| Phostatidylethanol 18:1_18:1 | 727.527 | -0.6 | * | -0.19 | |
| Proline | 142 | -0.66 | * | -0.37 | |
| Prostaglandin E1 | 353.232 | -0.67 | ** | -0.16 | |
| Pyroglutamic acid | 130.05 | -0.78 | **** | -0.11 | |
| S-Lactoylglutathione | 380.111 | -0.82 | -1.13 | * | |
| Sorbitol | 217 | -0.83 | ** | -0.3 | |
| Succinic acid | 117.019 | -0.83 | **** | -0.02 | |
| Sulfoglycosphingolipids d42:2 | 890.639 | -0.8 | * | -0.41 | |
| Thiazolidine-4-carboxylic acid | 134.025 | -0.84 | * | -1.05 | ** |
| Tryptophan | 202 | -0.87 | ** | -0.08 | |
| Tyrosine | 218 | -1.06 | * | -0.05 | |
| Uridine diphosphate galactose | 565.044 | -1.18 | **** | -0.49 | **** |
| Valine | 144 | -1.42 | **** | -1.58 | **** |
| Vanillin | 151.04 | -1.53 | 4.94 | * | |
| Vanillin-4-sulfate | 230.996 | -1.54 | **** | -0.64 | *** |
| Xanthine | 153.037 | -1.6 | **** | -1.71 | **** |
Table 2.
Significantly altered metabolites following subchronic ambient H2S exposure compared to RA control group. Common metabolite name, mass-to-charge ratio, fold change (log2 scale), and statistical significance were listed. Unpaired Students’ t-test was performed for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, * * < 0.05, ***<0.01, and ****<0.001.
Table 2.
Significantly altered metabolites following subchronic ambient H2S exposure compared to RA control group. Common metabolite name, mass-to-charge ratio, fold change (log2 scale), and statistical significance were listed. Unpaired Students’ t-test was performed for statistical significance. Asterisks indicate significant differences compared to RA group. P *< 0.1, * * < 0.05, ***<0.01, and ****<0.001.
| Metabolites | m/z | Fold Change | Significance |
| (R)-Butyrylcarnitine | 232.1547 | -1.18 | *** |
| 1-Palmitoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine | 454.2927 | 0.61 | ** |
| 1-Stearoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine | 482.3181 | 0.68 | ** |
| 3-Methylhistidine | 170.0913 | 0.77 | *** |
| 4-Aminovaleric acid betaine | 160.133 | 0.8 | ** |
| 5-Aminoimidazole-4-carboxamide | 110.0322 | 0.72 | ** |
| 5-Methoxytryptamine | 174 | -1.03 | *** |
| 9-(2,3-Dihydroxypropoxy)-9-oxononanoic acid | 261.1345 | 0.7 | ** |
| Abietic acid | 301.216 | 1.34 | *** |
| Adenosine 5'-diphosphoribose | 560.0761_582.0571 | -3.46 | **** |
| Adenosine 5'-monophosphate | 370.0487 | -0.61 | ** |
| Adenosine monophosphate | 695.1267 | -1.48 | ** |
| Adenosine-5-monophosphate | 315 | -1.2 | * |
| Adrenic acid (FA 22:4) | 331.2637 | 0.69 | *** |
| Arachidonic acid | 303.2328 | 1.13 | * |
| Arachidonic acid (FA 20:4) | 303.2333 | 0.77 | ** |
| Argininosuccinic acid | 291.127 | 0.8 | * |
| β-Glycerolphosphate | 243 | -0.76 | *** |
| Betaine aldehyde cation | 120.1019_102.0893 | 1.4 | *** |
| Cysteine | 220 | 0.71 | ** |
| Dehydroascorbic acid | 173 | -0.65 | ** |
| Docosahexaenoic acid (FA 22:6) | 327.2328 | 0.96 | *** |
| Docosapentaenoic acid (n-3, FA 22:5) | 329.2473 | 0.71 | ** |
| Eicosadienoic acid (FA 20:2) | 307.2637 | 0.72 | ** |
| Eicosapentaenoic acid (FA 20:5) | 301.2167 | 0.79 | ** |
| Eicosenoic acid (FA 20:1) | 309.2798 | 0.68 | ** |
| Epigallocatechin | 307.0835 | -0.59 | ** |
| Ethanolamine | 62.0596 | 0.76 | ** |
| Fatty acid 15:1 | 239.2009 | -0.92 | ** |
| Fructose | 307 | -1.21 | ** |
| Fructose-1-phosphate | 387 | -0.62 | ** |
| Fructose-6-phosphate | 315 | -1.13 | * |
| Galactolipid monogalactosyldiacylglycerol 34:1 | 774.605 | -0.64 | ** |
| Galactose-6-phosphate | 387 | -0.82 | * |
| Glucose | 319 | -1.11 | ** |
| Glucose-1-phosphate | 217 | -0.63 | ** |
| Glucose-6-phosphate | 387 | -1.9 | *** |
| Glycerol-alpha-phosphate | 357 | -1.97 | **** |
| Glycerophosphocholine | 515.2116_772.3096_280.0915_258.1106_296.0648 | -2.04 | **** |
| Guanine | 152.0564 | 0.64 | * |
| Guanosine | 324 | 1.16 | **** |
| His-Gly | 213.0949 | 1.01 | **** |
| Homo-γ-linolenic acid (FA 20:3) | 305.2487 | 0.86 | ** |
| Hypoxanthine | 265 | 0.8 | *** |
| Inosine | 230 | 0.6 | **** |
| Isovaleryl-L-carnitine | 246.17 | -0.98 | *** |
| Lactamide | 90.0544 | -1.25 | * |
| L-Cysteine-glutathione disulfide | 427.0924 | 0.81 | * |
| Lysophosphatidylcholine 16:0 | 540.3289 | 0.78 | ** |
| Lyso-phosphatidyl-ethanolamines 16:0 | 452.2776 | 0.62 | **** |
| Lyso-phosphatidyl-ethanolamines 22:6 | 526.2883 | 0.7 | ** |
| Lysophosphatidylinositol 16:0 | 571.2842 | 1.03 | ** |
| Lysophosphatidylinositol 18:0 | 599.3185 | 1.11 | ** |
| Maltose | 361 | -0.76 | * |
| N-Acetylalanine | 130.0515 | 0.79 | * |
| N-Acetylhistidine | 198.0888 | -1.96 | ** |
| N-Lauroylsarcosine | 272.2221 | -1.35 | * |
| Non-hydroxy-fatty acid sphingosine ceramides d42:3 | 704.6138 | 0.62 | *** |
| Oleic acid (FA 18:1) | 281.2491 | 0.75 | *** |
| Phenylacetylglycine | 192.0664 | 0.97 | ** |
| Phenylalanine | 164.0721 | 0.62 | * |
| Phosphatidylcholine 36:5 Isomer D | 780.555 | 0.88 | *** |
| Phosphoinositides 36:5 | 855.5001 | 0.6 | * |
| Phosphatidylethanol 18:1_18:1 | 727.5266 | 1.01 | ** |
| Propionylcarnitine | 218.1366 | 0.75 | *** |
| Prostaglandin E1 | 353.2316 | 1.07 | * |
| Riboflavin | 377.1473 | 0.69 | *** |
| Sedoheptulose 7-phosphate | 387 | -1.24 | ** |
| Serotonin | 174 | -0.91 | **** |
| sn-glycerol-3-phosphoethanolamine | 238.0433 | -0.96 | *** |
| Thiamine cation | 265.1075 | 0.65 | ** |
| Thiazolidine-4-carboxylic acid | 134.0252 | 0.66 | * |
| Triglyceride 52:4 | 872.7704 | -0.67 | *** |
| Trimethylamine N-oxide | 76.0748 | -0.8 | ** |
| Uracil | 241 | 0.89 | **** |
| Xanthine | 353 | 1.23 | *** |
| Xylose | 103 | -0.66 | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



