Submitted:
15 December 2023
Posted:
18 December 2023
You are already at the latest version
Abstract
African swine fever virus (ASFV) is the causative agent of African swine fever, an economically important disease of pigs, often with a high case fatality rate. ASFV has demonstrated low genetic diversity among isolates collected within Eurasia. To explore the influence of viral variants on clinical outcomes and infection-dynamics in pigs experimentally infected with ASFV, we have designed a deep sequencing strategy. Variant analysis revealed 3 distinct SNPs present in the virus inoculum (at 2.4%, 0.7%, and 13% frequency, respectively) that were maintained within all infected pigs (1-6 % frequency, and the latter at 16-21%). Several pigs displayed other unique SNPs at <10% frequency. In addition, a deletion of 10487 bp (resulting in the complete loss of 21 genes) was present at nearly 100% frequency in the ASFV DNA from one pig at position 6362-16849. This deletion was also found to be present at low levels in the virus inoculum and in two other infected pigs. The current methodology can be used for the currently circulating Eurasian ASFVs and also adapted to other ASFV strains and genotypes. Comprehensive deep sequencing is critical for following ASFV molecular evolution, especially for identification of modifications that affect virus virulence.
Keywords:
African swine fever virus (ASFV)
; next-generation sequencing (NGS)
; deep sequencing
; Nanopore sequencing
; minority variant analysis
1. Introduction
African swine fever virus (ASFV) is the causative agent of African swine fever (ASF), an economically important disease of domestic pigs, often with a high case fatality rate (up to 100%); the virus also infects wild boar and other members of the family Suidae [1,2,3,4]. It is the only known DNA arbovirus [2]. The ASFV genome is a large linear double stranded DNA molecule that ranges in size from 170 to 194 kb, depending on the strain, and includes almost 200 genes. The genome can be divided into three main regions, with a central conserved region of around 125 kb, flanked by two, more variable, terminal regions [5].
The ASFV genome has displayed low diversity among isolates collected over long time periods and from different geographical regions in Europe and Asia [3,6,7,8,9,10,11]. In contrast, greater genetic diversity has been found in ASFVs from East and Southern Africa, likely due to the sylvatic cycle of ASFV within soft ticks and warthogs [3,8,12,13]. While DNA viruses generally have more accurate replication mechanisms than RNA viruses due to the presence of proofreading enzymes, they can still generate genetic diversity through replication errors and recombination events [14,15,16,17,18]. An examination of ASFV samples collected over a period of 70 years revealed evolution rates of approximately 10-4 substitutions per nucleotide per year. This rate is closer to that observed in many RNA viruses than is typical for DNA viruses [19]. The DNA polymerase X together with the downstream DNA ligase, both exhibit low fidelity, suggesting a potential mutagenic role [20,21]. The quasispecies nature of viral populations has revealed a huge layer of minority genomes, which can modulate the behavior of the viral population or become prominent in response to selective forces or random events such as bottlenecks [14]. The diversity and quasispecies composition of ASFV has been insufficiently explored, with no known investigations conducted on the impact of quasispecies composition on virulence. In the present study, we have developed a long-range PCR method, which generates 17 overlapping PCR fragments covering almost the entirety of the ASFV genotype II POL/2015/Podlaskie genome. The PCR products, derived from samples from experimentally infected pigs, were sequenced and subsequent variant analysis revealed that the virus present in several pigs exhibited unique SNPs at 2-10% frequency. All samples were found to have a 10 bp insertion in the intergenic region (IGR) between genes I73R and I329L. The number of occurrences of a 10 bp tandem repeat in this region, has been used to further distinguish between ASFV genotype II viruses, by dividing them into 4 different IGR-variants: IGR-I with 2 repeats, IGR-II with 3 repeats, IGR-III with 4 repeats, IGR-IV with 5 repeats [22,23,24]. A large deletion > 10 kb was present at nearly 100% frequency in the virus from one infected pig; this was also found to be present at low levels in the virus inoculum and at higher levels in two other infected pigs. This methodology provides the necessary tools for comprehensive characterization of virus to explore the influence of viral variants on clinical outcomes and infection-dynamics in pigs.
2. Materials and Methods
2.1 Primers
Primers were designed using the updated ASFV/Georgia/2007 (Accession no. FR682468.2) [25] and ASFV/POL/2015/Podlaskie (Accession no. MH681419.1) [26] reference sequences, covering positions 300-177600 (in Podlaskie) in overlapping regions (Table 1). Two additional primer pairs were designed based on shot-gun sequencing results from DNA isolates from pig 8 to recover information from non-amplifying regions.
2.2 Infection Study
Twelve pigs were experimentally infected with the ASFV/POL/2015/Podlaskie, as previously described [27,28]. Briefly, 12 male Landrace x Large White pigs obtained from a conventional Spanish swine herd were challenged intranasally with 4 log10 TCID50 of the ASFV/POL/2015/Podlaskie (2nd passage) [28]. The pigs were euthanized when reaching the humane end points pre-set in the study, at 6 days post infection (dpi) [27]. During the study, EDTA-stabilized blood and serum were obtained from the twelve pigs at 0, 3, 5 and 6 (euthanasia) dpi [27]. At necropsy, spleen material and bone marrow were collected in TriReagent® (Sigma-Aldrich, St. Louis, MO, USA) or phosphate buffered saline, respectively. The tissue samples were then fast-frozen in liquid nitrogen.

Table 1.
Primer overview.
| Name | Fragment | Sequence |
|---|---|---|
| ASFV-300-Fα | 01_F | AGGTGGTTTGGCCGTATTCT |
| ASFV-11927-Rα | 01_R | ATGCGTAGGCCTCCTGAAAG |
| ASFV-10819-Fα | 02_F | ATAGGAGCGGCTTGAAGGAC |
| ASFV-22803-Rα | 02_R | TGCGGCAACATATGTCCAAAC |
| ASFV-22129-Fα | 03_F | CAAAGATGCCGTACCTCCGA |
| ASFV-33909-Rα | 03_R | TTTACGGCTTGGGTCAGGAC |
| ASFV-33237-Fα | 04_F | GCTCCCTTCAACGCATAGGA |
| ASFV-44936-Rα | 04_R | TGCGGGTCTTGGATTAAAGGA |
| ASFV-43757-Fα | 05a_F | TGGACCCAAAAAGGGTGGTC |
| ASFV-56602-Rα | 05a_R | GCGGCATTGAAAAACACCCT |
| ASFV-55750-Fα | 05b_F | TGAGCTGTTCCCAGGATTCG |
| ASFV-68434-Rα | 05b_R | AGCGCGCGTATTTATCAACG |
| ASFV-67660-Fβ | 06_F | GGACTGCGACACGATCACAGAGTC |
| ASFV-78076-Rβ | 06_R | GTCCTACGACACCATGCGAACCAAG |
| ASFV-77808-Fβ | 07_F | CTATATTGGCAAACTGTTTCACGTC |
| ASFV-87981-Rθ | 07_R | CAATCACAACGGTTCTCCTGTTAAG |
| ASFV-87000-Fα | 08_F | GCATAATCAATGGCAATCCCCC |
| ASFV-97970-Rα | 08_R | TGGCCTTAATCATTACAGCGGT |
| ASFV-97345-Fβ | 09_F | GTTACGTAGATCACTGAGTTGCAATC |
| ASFV-107683-Rβ | 09_R | GGCGCCCTCCTATACGATGAG |
| ASFV-107531-Fβ | 10_F | GTGTCCTCCATCGGATATACTATAC |
| ASFV-117620-Rθ | 10_R | AGTGTGCTGACCTATATCACGGAAC |
| ASFV-117410-Fβ | 11_F | CATTTCTGAACTGCGAGAGTTCTAG |
| ASFV-127433-Rβ | 11_R | TCGCTGTGCGTAATTTATCCCAATC |
| ASFV-126429-Fα | 12_F | AACACCTAACCTCGTCGTGC |
| ASFV-137952-Rα | 12_R | ACAGGTAAGGTCCGACTCGT |
| ASFV-137097-Fβ | 14_F | GAGAACAGGTCTTAGAATTACTTCATG |
| ASFV-147178-Rβ | 14_R | ACGCATCCGAAGGTGTTACAAGGAC |
| ASFV-146744-Fθ | 15_F | CTCTGAATGCGCAGAGCATCTTAC |
| ASFV-156426-Rβ | 15_R | GAACATGGGAATACGTGTGTCCAG |
| ASFV-155654-Fα | 17_F | AGGAACTGGACATGCAAGCAG |
| ASFV-166805-Rα | 17_R | ATGAGCTCGCCCACATAACC |
| ASFV-166089-Fα | 18_F | TATTGCCCGAGCCTCTGTATTC |
| ASFV-177600-Rα | 18_R | GGGGGAATCAACTCTCGCTTAA |
| ASFV-6188-Fα | del-PCR_F | GCTTCTAACTCTCTGTACAACA |
| ASFV-17145-Rα | del-PCR_R | CGGCATATCATAAGTAGGTTGGT |
| ASFV-6708-Fα | noDel-PCR_F | AAGTGGCTGCTCGTCAACAA |
| ASFV-7668-Rα | noDel-PCR_R | AGCCGTAGCAATGTTGGTGA |
2.3 Preparation of Long PCR Products from Viral DNA
DNA was extracted using a MagNA Pure 96 system (Roche, Basel, Switzerland) with the DNA/Viral NA 2.0 kit and the Viral NA Plasma external lysis S.V. 3.1. protocol, from serum, EDTA blood, spleen and bone marrow samples of individual pigs from the infection study, collected on the day of euthanasia, as well as from the virus used for inoculum (1st and 2nd passage) (Table 2). In short, spleen and bone marrow homogenate suspensions (25% w/v) were prepared in Minimum Essential Medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). The samples were homogenized using two 3 mm stainless steel beads (Dejay Distribution Ltd., Launceston, UK) in a TissueLyser II (QIAGEN, Hilden, Germany).
The homogenates were centrifuged and supernatants collected for DNA purification. The extracted samples were analyzed for the presence of ASFV DNA by qPCR using the CFX Opus Real-Time PCR System (Bio-Rad, Hercules, CA, USA), essentially as described by Tignon et al. [30].
Table 2.
Sample overview.
| Sample IDα | Sample name | Matrix | Ctβ |
|---|---|---|---|
| Spleen, 1/50 2. passage LPPAM 02-07-2019 | Inoc | Cell culture supernatant | 25.2 |
| CReSA_2020_pig_1_eut. | S1 | Serum | 20.2 |
| CReSA_2020_pig_1_eut. | E1 | EDTA-blood | 15.4 |
| CReSA_2020_pig_1_eut. | B1 | Bone-marrowθ | 19.0 |
| CReSA_2020_pig_1_eut. | SP1 | Spleen θ | 15.6 |
| CReSA_2020_pig_2_eut. | S2 | Serum | 19.3 |
| CReSA_2020_pig_2_eut. | E2 | EDTA-blood | 17.2 |
| CReSA_2020_pig_2_eut. | B2 | Bone-marrow θ | 16.8 |
| CReSA_2020_pig_2_eut. | SP2 | Spleen θ | 15.7 |
| CReSA_2020_pig_3_eut. | S3 | Serum | No Ct |
| CReSA_2020_pig_3_eut. | E3 | EDTA-blood | No Ct |
| CReSA_2020_pig_3_eut. | B3 | Bone-marrow θ | 38.3 |
| CReSA_2020_pig_3_eut. | SP3 | Spleen θ | 39.5 |
| CReSA_2020_pig_4_eut. | S4 | Serum | 19.2 |
| CReSA_2020_pig_4_eut. | E4 | EDTA-blood | 16.3 |
| CReSA_2020_pig_4_eut. | B4 | Bone-marrow θ | 19.1 |
| CReSA_2020_pig_4_eut. | SP4 | Spleen θ | 16.2 |
| CReSA_2020_pig_5_eut. | S5 | Serum | No Ct |
| CReSA_2020_pig_5_eut. | E5 | EDTA-blood | No Ct |
| CReSA_2020_pig_5_eut. | B5 | Bone-marrow θ | No Ct |
| CReSA_2020_pig_5_eut. | SP5 | Spleen θ | 39.8 |
| CReSA_2020_pig_6_eut. | S6 | Serum | 29.0 |
| CReSA_2020_pig_6_eut. | E6 | EDTA-blood | 22.0 |
| CReSA_2020_pig_6_eut. | B6 | Bone-marrow θ | 22.3 |
| CReSA_2020_pig_6_eut. | SP6 | Spleen θ | 16.5 |
| CReSA_2020_pig_7_eut. | S7 | Serum | 20.5 |
| CReSA_2020_pig_7_eut. | E7 | EDTA-blood | 17.3 |
| CReSA_2020_pig_7_eut. | B7 | Bone-marrow θ | 19.3 |
| CReSA_2020_pig_7_eut. | SP7 | Spleen θ | 16.5 |
| CReSA_2020_pig_8_eut. | S8 | Serum | 19.4 |
| CReSA_2020_pig_8_eut. | E8 | EDTA-blood | 15.4 |
| CReSA_2020_pig_8_eut. | B8 | Bone-marrow θ | 19.1 |
| CReSA_2020_pig_8_eut. | SP8 | Spleen θ | 15.8 |
| CReSA_2020_pig_9_eut. | S9 | Serum | 19.7 |
| CReSA_2020_pig_9_eut. | E9 | EDTA-blood | 16.7 |
| CReSA_2020_pig_9_eut. | B9 | Bone-marrow θ | 19.2 |
| CReSA_2020_pig_9_eut. | SP9 | Spleen θ | 17.3 |
| CReSA_2020_pig_10_eut. | S10 | Serum | 19.3 |
| CReSA_2020_pig_10_eut. | E10 | EDTA-blood | 14.8 |
| CReSA_2020_pig_10_eut. | B10 | Bone-marrow θ | 18.2 |
| CReSA_2020_pig_10_eut. | SP10 | Spleen θ | 15.5 |
| CReSA_2020_pig_11_eut. | S11 | Serum | 18.6 |
| CReSA_2020_pig_11_eut. | E11 | EDTA-blood | 16.2 |
| CReSA_2020_pig_11_eut. | B11 | Bone-marrow θ | 18.8 |
| CReSA_2020_pig_11_eut. | SP11 | Spleen θ | 17.0 |
| CReSA_2020_pig_12_eut. | S12 | Serum | 19.1 |
| CReSA_2020_pig_12_eut. | E12 | EDTA-blood | 15.5 |
| CReSA_2020_pig_12_eut. | B12 | Bone-marrow θ | 17.4 |
| CReSA_2020_pig_12_eut. | SP12 | Spleen θ | 15.8 |
| Spleen, 1st passage LPPAM 13-05-2019 1/50 | 1p19 | Cell culture supernatant | 27.2 |
The extracted DNA preparations from ASFV positive samples were used to generate overlapping long PCRs, ranging in size from approx. 9600 bp to 12800 bp, employing a modified version of the long PCR method described previously [31]. Briefly, the samples, except for those from serum, were diluted 1:10 in UltraPure™ DNase/RNase-Free Distilled Water (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). The products were amplified by long PCR using AccuPrime high-fidelity DNA polymerase (Thermo Scientific, Thermo Fisher Scientific, Waltham, MA, USA) in a final volume of 50 μl using 94°C for 30 s followed by 35 cycles of 94°C for 15 s, 55°C for 30 s and 68°C for 12 min, and a final extension of 68°C for 12 min. As a positive control we used extracted DNA from the spleen of an ASFV-infected pig, derived from a previous study [32]. The PCR products were analyzed using the Genomic DNA ScreenTape on a 4200 TapeStation (Agilent Technologies, Santa Clara, CA, USA) together with GeneRuler 1 kb Plus DNA Ladder (Thermo Scientific), and their concentrations were estimated using either the Qubit™ 1x dsDNA broad-range kit (Invitrogen) with the Qubit™ Fluorometer (Invitrogen) or Quant-iT™ 1X dsDNA broad-range kit (Invitrogen) on a FLUOstar® Omega (BMG LABTECH, Mornington, VIC, Australia) instrument.
2.4 Next Generation Sequencing (NGS)
PCR products were pooled for each sample and sequenced by NGS using MiSeq (Illumina, San Diego, CA, USA) with a modified Nextera XT DNA library protocol and also sequenced on the Nanopore (Oxford Nanopore Technologies, Oxford, UK) using a modified SQK-LSK109 protocol with native barcoding on a R9.4.1 flow-cell. MiSeq reads were trimmed using AdapterRemoval [33] by at least 30 bp at both the 5’ and 3’ ends, to ensure primer removal, as well as for quality (q3) using a sliding window. Some samples were sequenced directly (no PCR-amplification) using the MiSeq as described above.
2.5 PCR Deletion Screening and Sanger Sequencing
Long PCRs were performed as described above on ASFV positive samples with primers spanning the deletion. The PCR products were analyzed as above using the Genomic DNA ScreenTape or D5000 ScreenTape and their concentrations were estimated as above. PCR products were purified using the GeneJET PCR Purification Kit (Thermo Scientific) according to the manufacturer’s instructions, and were then sequenced using the Sanger system with a combination of BigDye Terminator v. 1.1. Cycle Sequencing Kit (Applied BioSystems, Waltham, MA, USA) with 10 µM primers, purification using SigmaSpin Post-Reaction columns (Sigma-Aldrich, St. Louis, MO, USA) and an ABI3500 Genetic Analyzer (Applied BioSystems). Sequences were analyzed using Geneious (Biomatters INC., Boston, MA, USA).
2.6 Variant and Indel Calling
MiSeq reads were mapped to the ASFV/POL/2015/Podlaskie reference sequence using BWA-MEM [34], and aligned reads were filtered for mapping quality (mapq) 60, secondary and supplementary reads were removed using Samtools [35]. Variant calling was performed using a combination of Lo-Freq [36] and SnpEFF [37] as described previously [38]. Briefly, variants were filtered for a minimum coverage of 100, frequency above 2% and strand-bias. A custom script was made to scan variants for homopolymer runs; a homopolymer was defined as 4 or more consecutive repeated nucleotides. Insertions and deletions (indels) were called for each sample by mapping the Nanopore reads to the ASFV/POL/2015/Podlaskie reference sequence using a combination of minimap2 [39] and Samtools. The alignment was first filtered to select reads that spanned the entirety of each PCR region. Up to 1000 reads were extracted for each PCR product from the alignment. The Compact Idiosyncratic Gapped Alignment Report (CIGAR) strings, which consist of matches (M), insertions (I) and deletions (D) [40], of the alignment files were parsed with a custom script to detect indels above 5 bp, and filtered for a minimum of 5 reads present on both forward and reverse strands, as well as a frequency ≥2%. Effects were called using SnpEFF and indels were scanned for homopolymer runs as described above.
3. Results
3.1 Infection Study
Twelve pigs had been experimentally infected with the ASFV/POL/2015/Podlaskie isolate as previously described [27]. Pigs 3 and 5 showed no clinical signs of disease; furthermore, the qPCRs to detect ASFV DNA in whole blood and serum were negative, and hence samples from these pigs were excluded from this study. Pig 6 displayed delayed onset of symptoms, consistent with the higher Ct-value of 22 for the EDTA-blood sample, compared to the other pigs, which all displayed Ct-values between approx. 15-17 (Figure 1).
3.2 Overlapping long PCRs
We designed 17 overlapping primer sets covering 93.6% of the ASFV/POL/2015/Podlaskie reference genome (Figure 2). We initially used serum samples for long PCR amplification, however the yield of products was low compared to our positive control. Therefore, we performed a dilution series on the four sample types (EDTA-blood, bone marrow, spleen and serum) to determine the optimum DNA input for the long PCRs (Table 3). Serum samples generated less product overall than the other sample types, whereas DNA isolated from EDTA-blood was able to generate the expected products at different yields, corresponding to the dilution. Bone-marrow and spleen samples both failed to generate products when used undiluted. Hence, the EDTA-blood samples collected at euthanasia and the virus inoculum were chosen for the initial analyses. Long PCRs were performed on the inoculum and 10x diluted EDTA-blood samples, and the products were sequenced by MiSeq, except for those from pig 6 which performed poorly in the PCR amplification, likely due to its lower concentration (higher Ct-value), and was therefore excluded from further study. In contrast to other samples, the samples from pig 8 consistently failed to yield PCR products for fragments 01 and 02, in all tissue types, although the other fragments were successfully generated from these samples.
3.3 Identification of a Large Deletion Event in the ASFV Genomes within Pig 8
Due to the failure of DNA isolated samples from pig 8 to yield PCR products for fragments 01 and 02, EDTA-blood, bone-marrow and spleen samples of pig 8 were sequenced directly by Illumina MiSeq. No reads were present that mapped to pos. 6364-16878 of the ASFV Podlaskie reference sequence, therefore we designed primers that spanned across this region, from pos. 6188-17145 (del-PCR). Use of these primers generated a PCR product of ~500 bp in all of the pig 8 samples (Figure 3B). The EDTA-blood samples from the other pigs and from the inoculum were screened for the presence of this deletion. Samples from pigs 1 and 7 also displayed prominent products of ~500 bp, consistent with the presence of the deletion, while the inoculum displayed a very weak product of this size (Figure 3A). All samples, except those from pig 7 and pig 8, also yielded products at ~11 kb, consistent with the expected full-length PCR products (without the deletion). The ca. 500bp PCR product from pig 8 was sequenced using the Nanopore and Sanger systems, which each revealed that the deletion occurred between positions 6362 and 16849, a total of 10487 bp. This deletion results in the loss of 21 complete genes, the majority are members of the MGF 110 (2L-14L) family, with a 3’ truncation of the MGF 110-1L gene, and ASFV G ACD 00090, 00120, 00160, 00190, 00240, and 00270, as well as MGF 360-4L. To confirm the deletion of nucleotides 6362-16849, we designed primers located within this region at pos. 6708-7668 (noDel-PCR), and screened all samples as above. All samples, except those from pig 8, yielded the expected product at ~1000 bp (Figure 3C), consistent with the ability to produce full-length PCR fragments 01 and 02. Screening of the other samples from pig 8, revealed a very weak ~1000 bp product in the bone-marrow and serum samples (Figure 3D).
In order to elucidate when this deletion variant arose in the inoculum, the first passage sample of the ASFV stock was also screened using these PCRs. They yielded only a full-length PCR product for the del-PCR, and the expected ~1000 bp product for the noDel-PCR (Figure S1), indicating that the deletion event took place during the production of the second passage virus stock, in porcine pulmonary alveolar macrophages (PPAM), which was used as the inoculum in the infection study.
3.4 SNP Analysis of EDTA-Blood Samples
In the virus inoculum a total of 3 SNPs, 2 silent, were present at 2.4% and 0.7% frequency, located at G3319A and A44769G, located in MGF 505-2R and MGF 505-10R, respectively (Figure 4 and Table S1). These 2 SNPs were shared among EDTA-blood samples from all the ASFV-infected pigs, ranging in frequencies between 1-6%. The third SNP in the inoculum was located in a non-coding region at A172451G with a frequency of 13%, and was found in all EDTA-blood samples at 16-21% frequency. The virus in EDTA-blood obtained from pig 4 had 2 unique silent SNPs, C136624T and C137323T located in NP868R, at 9.5% and 5% frequency, respectively. The ASFV in blood obtained from pig 8 had a unique missense SNP at A156198T (L265H) affecting the R298L gene present at 8.5% frequency. In EDTA-blood from pig 10, the ASFV DNA had 2 unique SNPs, a silent one (A11952G) at 2% frequency located in MGF 110-9L, and a missense change at G117050A (P46S) at 6.5% frequency, affecting CP123L. The virus in blood from pig 11 had a single missense SNP at C11870T (D213N) at 9.3% frequency, affecting MGF 110-9L while in pig 12 there were 3 SNPs, two silent at A33205T and T33212C, and one missense change at G33210A (R26Q), all located in MGF 505-2R, and these occurred at 2.3-2.4% frequency.
3.5 Samples from Pig 2
To explore virus tropism across various tissues, pig 2 was randomly chosen for further investigation. Only two SNPs were detected in samples from pig 2, above the 2% frequency cut-off, one silent change is located at G3319A with 2.4% frequency, and one in a non-coding region (A172451G) with a frequency of 13% (Figure 5 and Table S2). These SNPs were found in all samples from this pig, ranging in frequency from 1%-9% and 18.9-21.8%, respectively. The bone-marrow sample from this pig had a missense SNP at T146850C (S47P) affecting S273R, which was also found in the serum, with frequencies at 3.4% and 7.5% frequency, respectively. The serum sample from pig 2 had an additional 6 SNPs, 2 were missense at A36456G (M216V) and G144569A (G51R) affecting MGF 505-4R and D205R, at 2.6% and 2.2% frequency, respectively. The rest were silent SNPs ranging from 3.4-10% frequency.
3.6 Indel Analysis of EDTA Blood Samples
All samples were screened for the presence of indels above 5 bp in the full-length Nanopore reads derived from the various PCR products in order to detect larger deletions. As Nanopore reads contain a lot of noise, indels below 5 bp were ignored.
The identified 10487 bp deletion in the ASFV DNA from pig 8, which spans the region 6362-16849, results in the failure of the PCRs that generate fragments 01 and 02. This is due to the locations of their reverse and forward primers, respectively, within the deleted region. Consequently, only full-length versions of this region of the genome are successfully amplified. This means that the large deletion in the virus from pig 8 (and pigs 1 and 7) does not appear in this indel analysis.
The inoculum had a total of 4 indels above the cut-off, where 3 of these were 5 bp at 3.1-7.4% frequency and located in coding homopolymer regions, causing frameshifts in EP1242L, C717R, and G1340L (Figure 6 and Table S3). Each of these deletions were found in the EDTA-blood samples from at least one of the infected pigs. The 10 bp insertion, a tandem repeat ‘TATATAGGAA’, at 172423 was located in a non-coding region between the I73R and I329L genes with a frequency of 43.6% and was shared at similar frequencies (49-55%) in all the other samples. This insertion was also found in the aligned Miseq data, but at lower apparent frequencies. Due to this discrepancy, we counted the number of tandem repeats in the trimmed reads (pre-alignment), which revealed that that 96.3%-100% of the reads in both the Nanopore and MiSeq reads corresponded to the IGR-II variant. The disparity between the raw reads and alignment is due to reference bias (see Discussion). The published ASFV/POL/2015/Podlaskie reference strain sequence [26] does not contain this tandem repeat, however the results from this current study demonstrate that the ASFV/POL/2015/Podlaskie strain is actually an IGR-II variant. This disparity is discussed below.
In pig 1, the virus population exhibited 5 deletions, including a unique deletion of 5 bp with a frequency of 6%, located at position 115012 in a homopolymer region. This particular deletion caused a frameshift within the G1211R coding sequence. Within pig 2, the ASFV genomes displayed 2 deletions. One was a unique 5 bp deletion at position 116281 in a homopolymer region with a frequency of 3%, which also results in a frameshift within the G1211R coding sequence. The second deletion, of 5bp, was shared with pigs 4, 10, and 12 at position 85921, and ranged in frequency between 3.2% to 7%. This shared deletion caused a frameshift in the C475L gene coding sequence.
In the sample from pig 4, there were an additional 6 deletions. Two of these deletions were unique, with frequencies of 6.8% and 2.1%, and sizes of 6 bp and 5 bp, respectively. The 5 bp deletion led to a frameshift in the C717R gene, while the 6 bp deletion was situated in a non-coding homopolymer region. The remaining 4 deletions were 5 bp in size, found at frequencies of 2.2% to 2.5%, and were shared with at least one other pig, albeit sometimes at frequencies below the designated cut-off value.
The virus population in the sample from pig 7 exhibited 6 additional deletions, 3 of which were unique. The 6 bp deletion at pos. 167679 resulted in an in-frame deletion within the E248R gene at a 2.1% frequency. Another 5 bp deletion at pos. 139760, located in a homopolymeric region within the D339L ORF, and a 5 bp deletion at pos. 173667 in a non-coding region were also identified. Additionally, a 6 bp deletion at 170815 in a homopolymeric region caused an in-frame deletion in the I226R ORF at a 5.8% frequency and was present in all samples, albeit some below the cut-off level. The 5 bp deletion at 93795 at a 5.8% frequency caused a frameshift in the B962L ORF and was also detected below the cut-off level in pig 1.
The sample from pig 8 had 3 additional deletions, which were also detected in the sample from pig 12, though some below the cut-off level. Two of these deletions caused frameshifts within Q706L, in homopolymeric regions, present at frequencies of 2.3-2.5%
and 1.4-2.3%, respectively, and the third was in a non-coding homopolymer region. In contrast to all other samples, the virus in the sample from pig 9 did not have any unique indels.
The virus population in pig 10 showed 3 unique deletions, all resulting in frameshifts within the F1055L, G1340L, and R298L ORFs, in homopolymeric regions, at frequencies of 3%, 2.1%, and 4.3%, respectively. Another 4 deletions (5-6 bp) were identified in at least one other pig, though some were below the designated cut-off value. Two of these deletions caused in-frame deletions located in homopolymeric regions, in the C257L gene (pos. 85015), and the D1133L gene (pos. 141628), with frequencies of 2% and 4%, respectively. Another deletion occurred at a 2.3% frequency, and was situated in a homopolymeric region, and caused a frameshift in the B962L ORF.
The ASFV genomes within pig 11, displayed 4 unique 5 bp deletions, all leading to frameshifts within the C475L, G1211R , CP2475L, and H108R ORFs. There was also a 5 bp deletion in a non-coding homopolymeric region and an additional 6bp deletion in G1211R. These 6 deletions occurred at frequencies of 2-5.2%. The remaining 11 deletions (5-7 bp) were present in at least one other sample, although below the cut-off value. Of these deletions, 5 caused frameshifts in the MGF 505-2R, B962L, B169L, B602L, and H339R genes. Another 5 were located in homopolymeric regions within genes F165R, C962R, B175L, G1211R, and D1133L, with the four former changes causing frameshifts, and the latter being an in-frame deletion.
The virus in EDTA-blood from pig 12 had 1 unique deletion located at pos. 424 in a homopolymeric non-coding region with a frequency of 77 %. Another 5 bp frameshift deletion located at nt 158292 affecting the G706L ORF with a 2.1% frequency, was also detected in the inoculum but at below the designated cut-off level.
3.7 Samples from Pig 2
As mentioned above, the virus inoculum had a total for 4 indels present at above the cut-off frequency of 2%, where 3 of these were 5 bp in length at 3.1-7.4% frequency and located in coding regions, causing frameshifts in EP1242L, C717R, and G1340L (Figure 7 and Table S4). Two of these deletions were located in homopolymeric regions. The deletion at pos. 112741 (G1340L) was not present in any of the samples from pig 2. The 10 bp tandem repeat insertion (IGR-II) was present in each of the different samples from pig 2 at 46.7%-53.3% in the aligned Nanopore reads, and subsequent screening of the trimmed Nanopore and MiSeq reads also revealed a 99.9%-100% frequency. The 5 bp deletion at 83946 in the C717R gene, as found in the inoculum, was only detected in the serum sample, but was present below the cut-off level, and the 5 bp deletion at 69772 in the EP1242L ORF in the inoculum was only detected in the spleen sample, but at below the designated cut-off level.
In the EDTA-blood sample from pig 2, a distinctive 5 bp deletion at nt 85921 in the C475L gene, occurring at a frequency of 3.19%, was identified. This deletion was not found in other samples from pig 2, but was also present in the EDTA-blood of pigs 4, 10, and 12. Additionally, the 5 bp deletion at pos. 116281 in the G1211R ORF was observed at a 3.1% frequency in the EDTA-blood sample and at 2.5% in the bone-marrow sample.
In the bone marrow sample from pig 2, a unique 5 bp deletion causing a frameshift within the MGF 505-4R ORF, in a homopolymeric region, at a frequency of 2.3% was identified. Additionally, an in-frame deletion of 6 bp affecting the I226R gene, in a homopolymeric region, at a 2.4% frequency was present in the inoculum and in other samples from pig 2, although below the cut-off value.
Figure 6.
Indel frequency plots of insertions (red) and deletions (blue). Light grey background indicates areas with no PCR coverage, and dark grey background indicates areas where with failed PCRs (due to the deletion). Genes on the forward strand are indicated in blue, and genes on the reverse strand are indicated in gold, with affected genes in dark grey. (A) Indel frequencies in Inoculum. (B) Indel frequencies in sample E1. (C) Indel frequencies in sample E2. (D) Indel frequencies in sample E4. (E) Indel frequencies in sample E7. (F) Indel frequencies in sample E8. (G) Indel frequencies in sample E9. (H) Indel frequencies in sample E10. (I) Indel frequencies in sample E11. (J) Indel frequencies in sample E12.
Figure 6.
Indel frequency plots of insertions (red) and deletions (blue). Light grey background indicates areas with no PCR coverage, and dark grey background indicates areas where with failed PCRs (due to the deletion). Genes on the forward strand are indicated in blue, and genes on the reverse strand are indicated in gold, with affected genes in dark grey. (A) Indel frequencies in Inoculum. (B) Indel frequencies in sample E1. (C) Indel frequencies in sample E2. (D) Indel frequencies in sample E4. (E) Indel frequencies in sample E7. (F) Indel frequencies in sample E8. (G) Indel frequencies in sample E9. (H) Indel frequencies in sample E10. (I) Indel frequencies in sample E11. (J) Indel frequencies in sample E12.
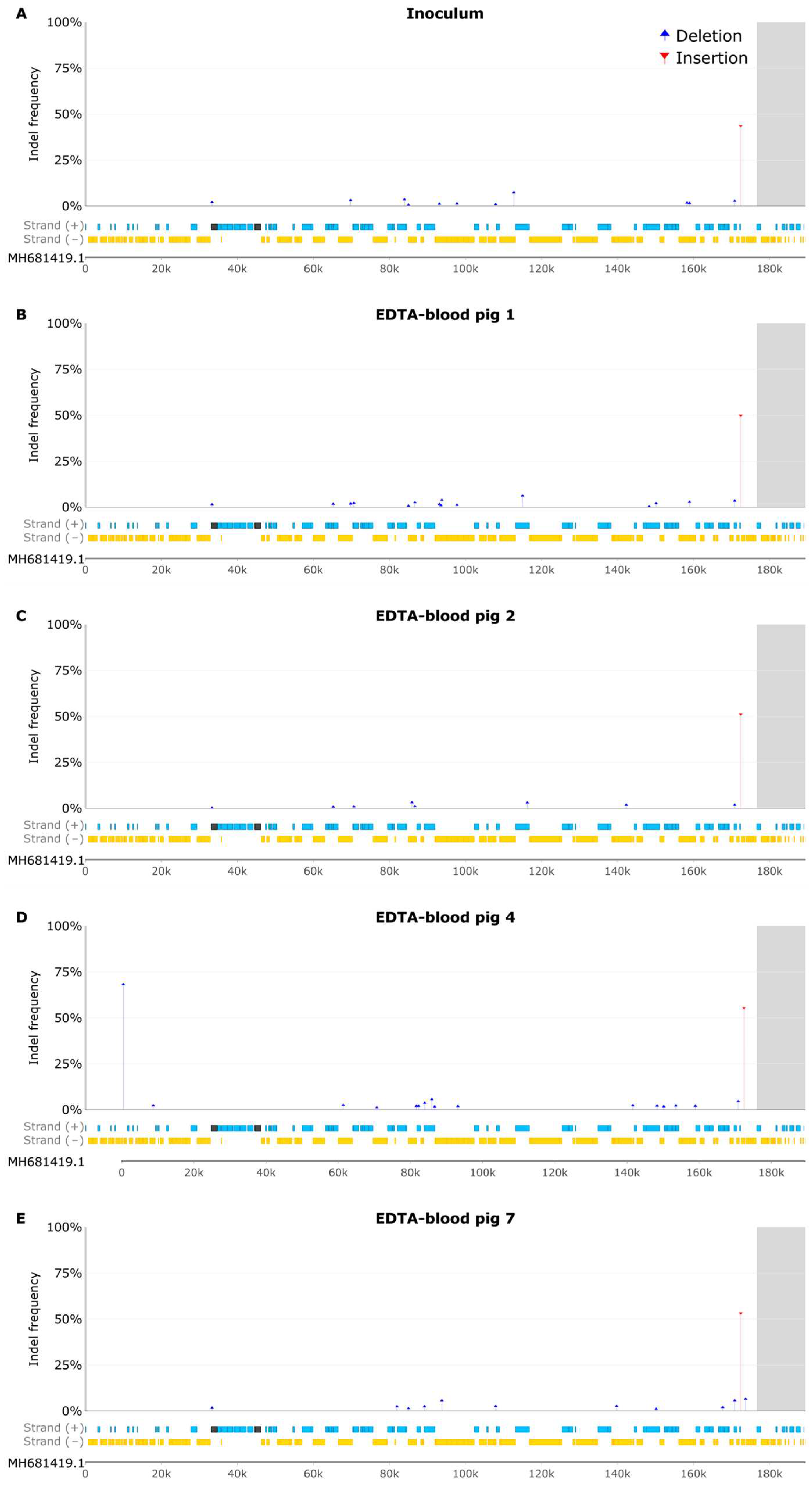
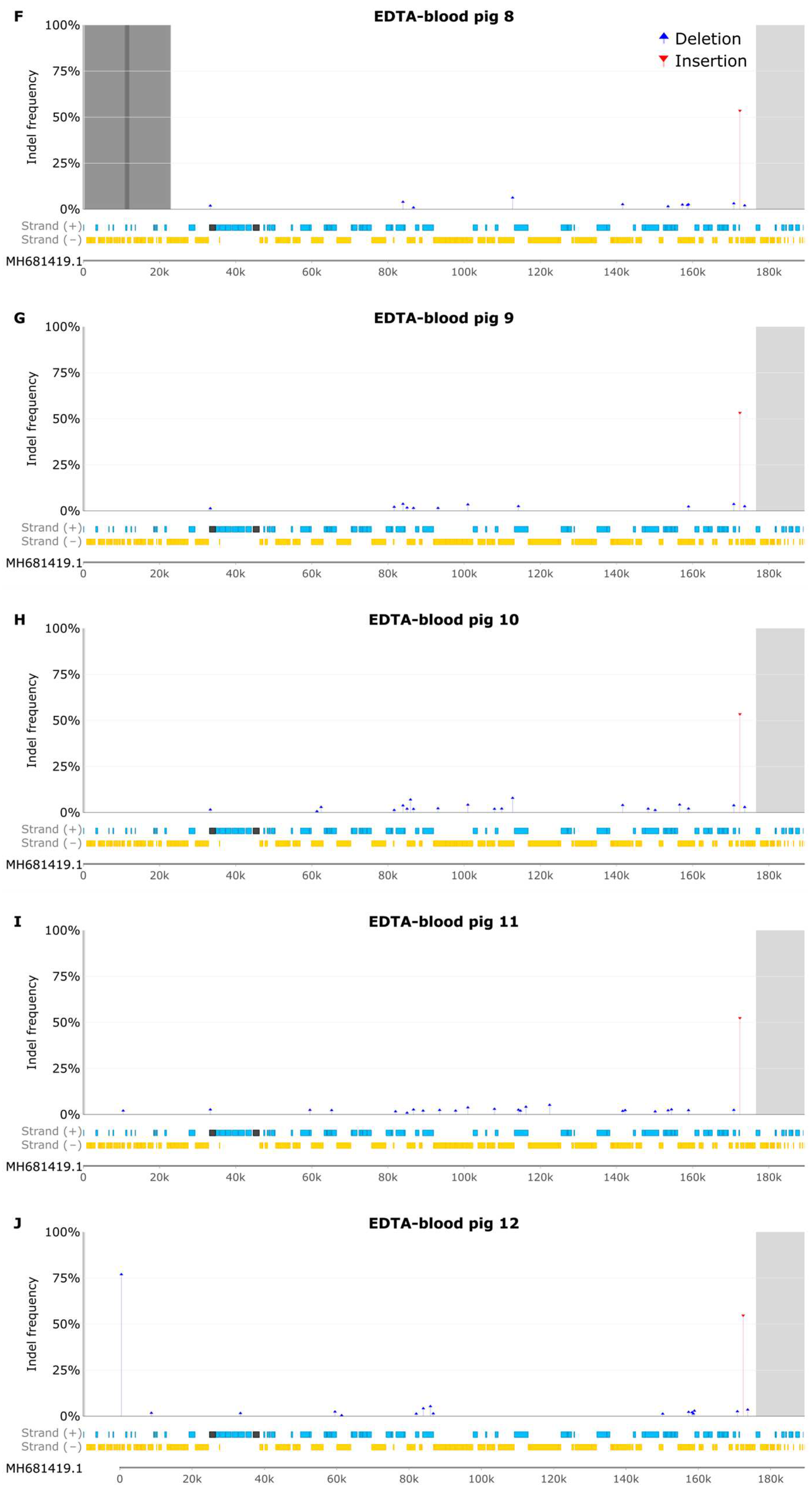
Figure 7.
Indel frequency plots of silent (green) and missense (red) mutations. Light grey background indicates areas with no PCR coverage. Genes on forward strand are indicated in blue, and genes on reverse strand in gold, with affected genes in dark grey. (A) Indel frequencies in the Inoculum. (B) Indel frequencies in sample E2. (C) Indel frequencies in sample B2. (D) Indel frequencies in sample SP2. (E) Indel frequencies in sample S2.
Figure 7.
Indel frequency plots of silent (green) and missense (red) mutations. Light grey background indicates areas with no PCR coverage. Genes on forward strand are indicated in blue, and genes on reverse strand in gold, with affected genes in dark grey. (A) Indel frequencies in the Inoculum. (B) Indel frequencies in sample E2. (C) Indel frequencies in sample B2. (D) Indel frequencies in sample SP2. (E) Indel frequencies in sample S2.
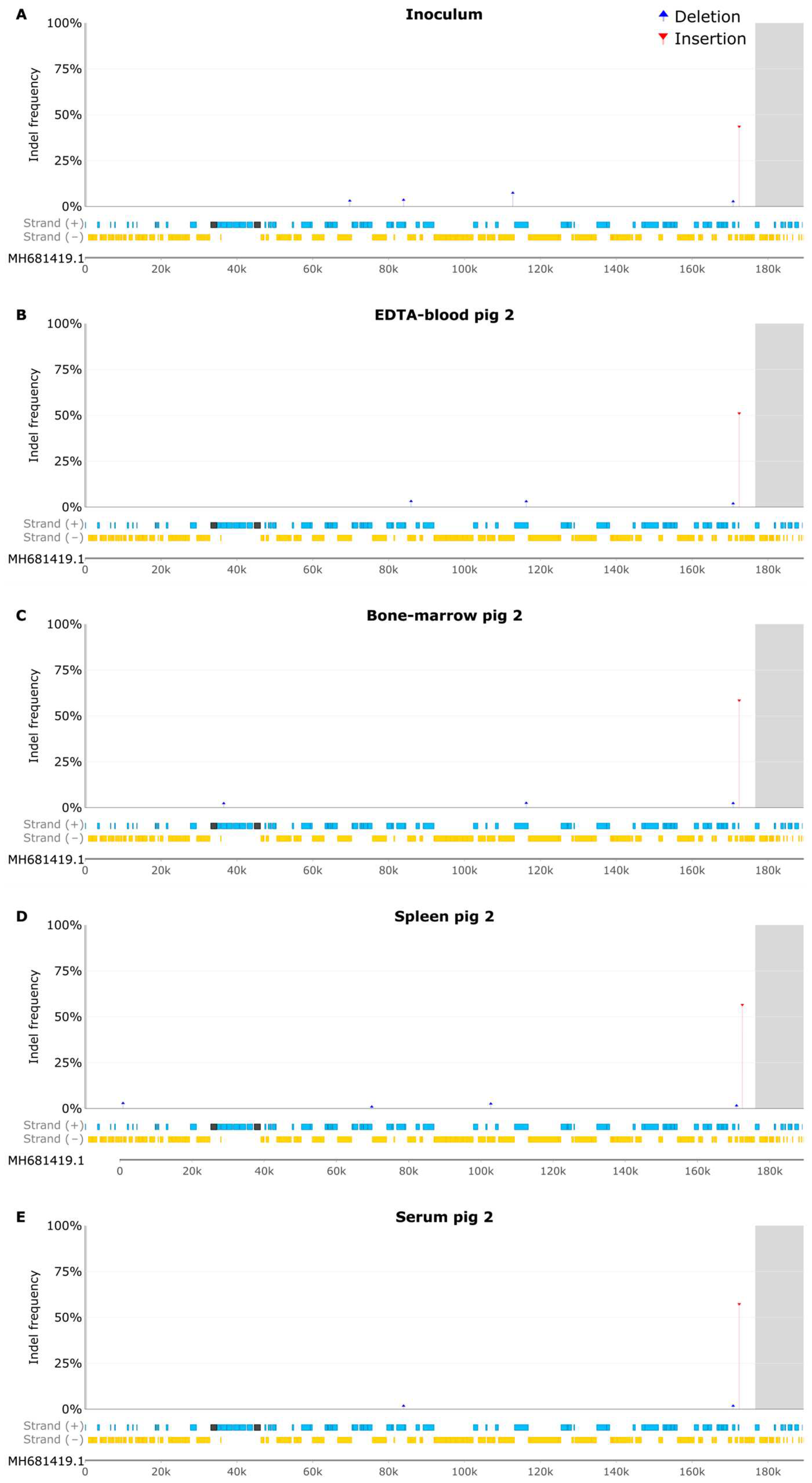
The spleen sample from pig 2 exhibited two unique 5 bp deletions causing frameshifts in the MGF 360-1La and B385R ORFs, in homopolymeric regions, with frequencies of 2.9% and 2.6%, respectively. The serum sample from pig 2 had no unique indels above the cut-off value.
4. Discussion
Using a deep-sequencing strategy on ASFV DNA samples derived from experimentally infected pigs, we have identified a variety of changes within the viral genome that have occurred during replication in the pigs. The largest change was a deletion of over 10 kb, which initially arose in cell culture in generating the inoculum, but became dominant in pig 8 and was present in pigs 1 and 7. This large deletion (pos. 6362 to 16849) in the ASFV DNA in pig 8 completely removes 21 genes, and truncates another gene. The deletion did not prevent the production of severe disease, as the pig was still clinically ill [27], and high levels of fragmented cell-free DNA were still released into the serum [41]. However, the effect of such deletions on transmission to a new host or its ability to grow in arthropod vectors is unknown, but there must have been a significant advantage for the deletion to have taken over nearly the entire virus population within this pig. Deletions in this part of the genome have also happened in the field, e.g. the majority of the lost genes in pig 8 are shared with the Estonia2014 strain (see [42]) except for ASFV G ACD 00270, MGF 110-13Lb and MGF 360-4L which are still present, however, MGF 110-13La is truncated at the 3’-end. Estonia2014 is characterized as having reduced virulence, it additionally lacks KP177R, L83L, L60L and MGF 360 (1L-3L) compared to the deletion within the ASFV DNA in pig 8. The MGF360-1L and MGF110-1L genes do not seem to be involved in virulence in swine [43,44], and similarly, KP177R, which encodes for the structural protein p22, does not seem to be involved in determining virulence in swine [45]. L83L is a nonessential protein, and does not affect pathogenicity in vitro or in vivo, however, it seems to play a role in the inhibition of the type I interferon production [46,47]. Results suggest that the L60L gene could serve as a virulence determinant [48].
Limited information is available regarding the majority of the genes deleted in the ASFVs in samples from pig 8. The MGF110-5L-6L gene appears not to play a role in virus replication or virulence in swine [49], whereas MGF110-9L does seem to play such a role [50]. The MGF110-7L is implicated in subverting the host protein synthesis machinery, inducing phosphorylation of eIF2α through protein kinase R (PKR) and PKR-like endoplasmic reticulum (ER) kinase (PERK). This process was found to be essential for host translation repression and stress granule (SG) formation [51]. While MGF-11L appears crucial for pathogenicity and lethality in pigs, its significance in in vitro replication seems limited. Certain members of the MGF360 family have been identified as essential for replication in pigs, ticks and macrophages [52,53,54,55,56,57,58].
The complete switch in pig 8 from the deletion being a small component of the inoculum, as evident by the generation of mostly full-length product, to being the dominant virus indicates that a minority variant can become dominant in just a single round of infection within pigs. Thus, potentially a minor variant can change into a major source of infection from a pig and hence the properties of the minority variant virus can become the properties of the circulating virus and the nature of the disease will reflect the properties of the new variant. Transmission of virus from one host to another will often involve some sort of bottleneck event and a minority variant can then become a major component within newly infected animals even without a major selection advantage.
Recently 24 genetic groups have been identified within the genotype II ASFVs, that have been circulating in Europe, by using a multi-gene-approach to subtyping [23]. These variants differ by a variety of changes including: SNPs and deletions in the central variable region (CVR variants), tandem repeat sequences (TRS) in the intergenic region (IGR) between genes I73R/I329L (IGR variants), a 14 nt insertion in the O174L gene (O174 variants), a SNP in the K145R gene (K145R variants), SNPs in the IGR between the I329L and I215L genes and the I215L gene itself (ECO2 variants). In addition, there are differences in the TRS in the IGR between the MGF505-9R and the MGF505-10R genes (MGF variants). Using this nomenclature system, the published sequence for ASFV/POL/2015/Podlaskie would be classified as an CVR-I, IGR-I, O174L-I, K145R-I, MGF-I and ECO2-I variant. However, the IGR-II variant has been found in the majority of genotype II strains circulating in Europe [23]. In the studies presented here, it is apparent that the ASFV/POL/2015/Podlaskie in the inoculum is also an IGR-II variant, and that the published ASFV/POL/2015/Podlaskie reference strain, should be re-classified as an IGR-II variant, which places the ASFV/POL/2015/Podlaskie isolate in genetic group 3 [23].
The complete genome sequence of ASFV/POL/2015/Podlaskie was originally generated using a reference-based alignment to the ASFV/Georgia/2007 reference strain [26]. The results of our study emphasize the importance of the awareness of reference bias, where reads harboring non-reference genomic variations, can be inaccurately aligned or overlooked by the aligner [59,60].
Nanopore sequencers are known to struggle to accurately sequence homopolymers, due to the lack of signal change [61]. Previous studies have found that about 47% of errors were linked to homopolymers [62]. The new Nanopore R10.4 flow cells are more accurate in calling homopolymers in the 4-9 bp range than the R9.4.1 flow cells [63], which we have used in this current study. The majority of the indels reported in this study are associated with homopolymers, and it is difficult to determine their veracity by other means, as most sequencing technologies have difficulties with homopolymers [64,65,66,67].
Comprehensive deep sequencing plays a pivotal role in following ASFV molecular evolution and pinpointing modifications that may influence virulence. This current methodology facilitates variant calling and indel detection, with the flexibility to be tailored for other ASFV strains and genotypes by modifying primers in regions with low compatibility. This approach is suitable for the current ASFVs circulating in Eurasia and provides a foundation for understanding ASFV evolution.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Deletion screening; Table S1: Variant analysis generated by LoFreq and SnpEff for Inoculum and E1-E12; Table S2: Variant analysis generated by LoFreq and SnpEff for the Inoculum and E2, B2, SP2 and S2; Table S3: Indels detected in ONT data by parsing the CIGAR string of Inoculum and E1-E12; Table S4: Indels detected in ONT data by parsing the CIGAR string of Inoculum and E2, B2, SP2 and S2.
Author Contributions
Conceptualization, C.M.J., T.B.R., and L.L.; methodology, C.M.J., A.l.M.M., and T.B.R; software, C.M.J.; validation, C.M.J.; formal analysis, C.M.J; investigation, C.M.J.; resources, A.S.O.; data curation, C.M.J., and T.B.R.; writing—original draft preparation, C.M.J.; writing—review and editing, all authors; visualization, C.M.J., and T.B.R.; supervision, T.B.R., L.L, A.B., G.J.B., and A.S.O.; project administration, T.B.R., and L.L.; funding acquisition, T.B.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Horizon 2020 ERA-NET Cofund International Coordination of Research of Infectious Animal Diseases (ICRAD), project ASF-RASH, and also from internal resources of the University of Copenhagen and the Statens Serum Institut.
Institutional Review Board Statement
Not applicable, no new animal studies were performed for this study.
Informed Consent Statement
Not applicable.
Data Availability Statement
Sequencing data and custom scripts are available upon request.
Acknowledgments
We thank Marie Hornstrup Christensen, Rasmus Oskar Rask Hansen, and Fie Fisker Brønnum Christensen for their invaluable technical assistance during this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Costard, S.; Jones, B.A.; Martínez-López, B.; Mur, L.; de la Torre, A.; Martínez, M.; Sánchez-Vizcaíno, F.; Sánchez-Vizcaíno, J.M.; Pfeiffer, D.U.; Wieland, B. Introduction of African Swine Fever into the European Union through Illegal Importation of Pork and Pork Products. PLoS ONE 2013, 8, e61104. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Sun, H.; Roberts, H. African Swine Fever. Antiviral Res. 2019, 165, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Stahl, K.; Jori, F.; Vial, L.; Pfeiffer, D.U. African Swine Fever Epidemiology and Control. Annu. Rev. Anim. Biosci. 2020, 8, 221–246. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cordón, P.J.P.J.; Montoya, M.; Reis, A.L.; Dixon, L.K.L.K. African Swine Fever: A Re-Emerging Viral Disease Threatening the Global Pig Industry. Vet. J. 2018, 233, 41–48. [Google Scholar] [CrossRef]
- Yáñez, R.J.; Rodrı́guez, J.M.; Nogal, M.L.; Yuste, L.; Enrı́quez, C.; Rodriguez, J.F.; Viñuela, E. Analysis of the Complete Nucleotide Sequence of African Swine Fever Virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef]
- Alonso, C.; Borca, M. V.; Dixon, L.K.; Revilla, Y.; Rodríguez, F.; Escribano, J.M. ICTV Virus Taxonomy Profile: Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef]
- Forth, J.H.; Forth, L.F.; Blome, S.; Höper, D.; Beer, M. African Swine Fever Whole-Genome Sequencing—Quantity Wanted but Quality Needed. PLOS Pathog. 2020, 16, e1008779. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Penrith, M.L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; R. Thomson, G. Genotyping Field Strains of African Swine Fever Virus by Partial P72 Gene Characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.A.L.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African Swine Fever Virus Isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Forth, J.H.; Tignon, M.; Cay, A.B.; Forth, L.F.; Höper, D.; Blome, S.; Beer, M. Comparative Analysis of Whole-Genome Sequence of African Swine Fever Virus Belgium 2018/1. Emerg. Infect. Dis. 2019, 25, 1249–1252. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrín, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.S.; Gerber, L.J.; Vosloo, W. Genetic Characterisation of African Swine Fever Viruses from Outbreaks in Southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef]
- Domingo, E.; García-Crespo, C.; Perales, C. Historical Perspective on the Discovery of the Quasispecies Concept. Annu. Rev. Virol. 2021, 8, 51–72. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Genetic Diversity and Evolution of Viral Populations. Encycl. Virol. 2021, 53–61. [Google Scholar] [CrossRef]
- Sanjuán, R.; Pereira-Gómez, M.; Risso, J. Genome Instability in DNA Viruses. In Genome Stability; Elsevier, 2016; pp. 37–47.
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Michaud, V.; Randriamparany, T.; Albina, E. Comprehensive Phylogenetic Reconstructions of African Swine Fever Virus: Proposal for a New Classification and Molecular Dating of the Virus. PLoS ONE 2013, 8, e69662. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, B.J.; Kumar, S.; Tsai, M.-D. ASFV DNA Polymerase X Is Extremely Error-Prone under Diverse Assay Conditions and within Multiple DNA Sequence Contexts. Biochemistry 2006, 45, 14826–14833. [Google Scholar] [CrossRef] [PubMed]
- García-Escudero, R.; Garcı́a-Dı́az, M.; Salas, M.L.; Blanco, L.; Salas, J. DNA Polymerase X of African Swine Fever Virus: Insertion Fidelity on Gapped DNA Substrates and AP Lyase Activity Support a Role in Base Excision Repair of Viral DNA. J. Mol. Biol. 2003, 326, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Cho, K.; Mai, N.T.A.; Park, J.-Y.; Trinh, T.B.N.; Jang, M.-K.; Nguyen, T.T.H.; Vu, X.D.; Nguyen, T.L.; Nguyen, V.D.; et al. Multiple Variants of African Swine Fever Virus Circulating in Vietnam. Arch. Virol. 2022, 167, 1137–1140. [Google Scholar] [CrossRef]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Nieto, R.; Perez, C.; Simon, A.; Ivanova, E.; et al. A Multi Gene-Approach Genotyping Method Identifies 24 Genetic Clusters within the Genotype II-European African Swine Fever Viruses Circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Woźniakowski, G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses 2020, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hübner, A.; Olesen, A.S.; Höper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Olesen, A.S.; Lohse, L.; Dalgaard, M.D.; Woźniakowski, G.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Complete Genome Sequence of an African Swine Fever Virus (ASFV POL/2015/Podlaskie) Determined Directly from Pig Erythrocyte-Associated Nucleic Acid. J. Virol. Methods 2018, 261, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Olesen, A.S.; Lohse, L.; Accensi, F.; Goldswain, H.; Belsham, G.J.; Bøtner, A.; Netherton, C.L.; Dixon, L.K.; Portugal, R. Inefficient Transmission of African Swine Fever Virus to Sentinel Pigs from Environmental Contamination under Experimental Conditions. Transbound. Emerg. Dis. (in press), preprint available at. [CrossRef]
- Olesen, A.S.; Kodama, M.; Lohse, L.; Accensi, F.; Rasmussen, T.B.; Lazov, C.; Limborg, M.T.; Gilbert, M.T.P.; Bøtner, A.; Belsham, G.J. Identification of African Swine Fever Virus Transcription within Peripheral Blood Mononuclear Cells of Acutely Infected Pigs. Viruses 2021, 13, 2333. [Google Scholar] [CrossRef] [PubMed]
- Portugal, R.; Coelho, J.; Höper, D.; Little, N.S.; Smithson, C.; Upton, C.; Martins, C.; Leitão, A.; Keil, G.M. Related Strains of African Swine Fever Virus with Different Virulence: Genome Comparison and Analysis. J. Gen. Virol. 2015, 96, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tignon, M.; Gallardo, C.; Iscaro, C.; Hutet, E.; Van der Stede, Y.; Kolbasov, D.; De Mia, G.M.; Le Potier, M.-F.; Bishop, R.P.; Arias, M.; et al. Development and Inter-Laboratory Validation Study of an Improved New Real-Time PCR Assay with Internal Control for Detection and Laboratory Diagnosis of African Swine Fever Virus. J. Virol. Methods 2011, 178, 161–170. [Google Scholar] [CrossRef]
- Rasmussen, T.B.; Reimann, I.; Uttenthal, Å.; Leifer, I.; Depner, K.; Schirrmeier, H.; Beer, M. Generation of Recombinant Pestiviruses Using a Full-Genome Amplification Strategy. Vet. Microbiol. 2010, 142, 13–17. [Google Scholar] [CrossRef]
- Olesen, A.S.; Lohse, L.; Hansen, M.F.; Boklund, A.; Halasa, T.H.B.; Belsham, G.J.; Rasmussen, T.B.; Bøtner, A.; Bødker, R. Infection of Pigs with African Swine Fever Virus via Ingestion of Stable Flies (Stomoxys Calcitrans). Transbound. Emerg. Dis. 2018, 65, 1152–1157. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid Adapter Trimming, Identification, and Read Merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. 2013.
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly (Austin). 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Johnston, C.M.; Fahnøe, U.; Lohse, L.; Bukh, J.; Belsham, G.J.; Rasmussen, T.B. Analysis of Virus Population Profiles within Pigs Infected with Virulent Classical Swine Fever Viruses: Evidence for Bottlenecks in Transmission but Absence of Tissue-Specific Virus Variants. J. Virol. 2020, 94, 1–20. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Olesen, A.S.; Lohse, L.; Johnston, C.M.; Rasmussen, T.B.; Bøtner, A.; Belsham, G.J. Increased Presence of Circulating Cell-Free, Fragmented, Host DNA in Pigs Infected with Virulent African Swine Fever Virus. Viruses 2023, 15, 2133. [Google Scholar] [CrossRef] [PubMed]
- Zani, L.; Forth, J.H.; Forth, L.F.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Höper, D.; et al. Deletion at the 5’-End of Estonian ASFV Strains Associated with an Attenuated Phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Medina, E.; Vuono, E.A.; Rai, A.; Pruitt, S.; Silva, E.; Velazquez-Salinas, L.; Zhu, J.; Gladue, D.P.; Borca, M. V. Evaluation in Swine of a Recombinant African Swine Fever Virus Lacking the MGF-360-1L Gene. Viruses 2020, 12, 1193. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Medina, E.; Vuono, E.; Pruitt, S.; Rai, A.; Silva, E.; Espinoza, N.; Zhu, J.; Velazquez-Salinas, L.; Borca, M. V.; Gladue, D.P. Development and In Vivo Evaluation of a MGF110-1L Deletion Mutant in African Swine Fever Strain Georgia. Viruses 2021, 13, 286. [Google Scholar] [CrossRef]
- Vuono, E.A.; Ramirez-Medina, E.; Pruitt, S.; Rai, A.; Espinoza, N.; Velazquez-Salinas, L.; Gladue, D.P.; Borca, M. V. Evaluation of the Function of the ASFV KP177R Gene, Encoding for Structural Protein P22, in the Process of Virus Replication and in Swine Virulence. Viruses 2021, 13, 986. [Google Scholar] [CrossRef] [PubMed]
- Borca, M. V.; O’Donnell, V.; Holinka, L.G.; Ramírez-Medina, E.; Clark, B.A.; Vuono, E.A.; Berggren, K.; Alfano, M.; Carey, L.B.; Richt, J.A.; et al. The L83L ORF of African Swine Fever Virus Strain Georgia Encodes for a Non-Essential Gene That Interacts with the Host Protein IL-1β. Virus Res. 2018, 249, 116–123. [Google Scholar] [CrossRef]
- Cheng, M.; Kanyema, M.M.; Sun, Y.; Zhao, W.; Lu, Y.; Wang, J.; Li, X.; Shi, C.; Wang, J.; Wang, N.; et al. African Swine Fever Virus L83L Negatively Regulates the CGAS-STING-Mediated IFN-I Pathway by Recruiting Tollip To Promote STING Autophagic Degradation. J. Virol. 2023, 97. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, R.; Zhang, Y.; Fan, J.; Zhou, X.; Yue, H.; Li, Q.; Miao, F.; Chen, T.; Mi, L.; et al. SY18ΔL60L: A New Recombinant Live Attenuated African Swine Fever Virus with Protection against Homologous Challenge. Front. Microbiol. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Medina, E.; Vuono, E.; Silva, E.; Rai, A.; Valladares, A.; Pruitt, S.; Espinoza, N.; Velazquez-Salinas, L.; Borca, M. V.; Gladue, D.P. Evaluation of the Deletion of MGF110-5L-6L on Swine Virulence from the Pandemic Strain of African Swine Fever Virus and Use as a DIVA Marker in Vaccine Candidate ASFV-G-ΔI177L. J. Virol. 2022, 96. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Qi, X.; Wen, Y.; Li, P.; Ma, Z.; Liu, Y.; Zheng, H.; Liu, Z. African Swine Fever Virus MGF-110-9L-Deficient Mutant Has Attenuated Virulence in Pigs. Virol. Sin. 2021, 36, 187–195. [Google Scholar] [CrossRef]
- Zhong, H.; Fan, S.; Du, Y.; Zhang, Y.; Zhang, A.; Jiang, D.; Han, S.; Wan, B.; Zhang, G. African Swine Fever Virus MGF110-7L Induces Host Cell Translation Suppression and Stress Granule Formation by Activating the PERK/PKR-EIF2α Pathway. Microbiol. Spectr. 2022, 10. [Google Scholar] [CrossRef]
- Burrage, T.G.; Lu, Z.; Neilan, J.G.; Rock, D.L.; Zsak, L. African Swine Fever Virus Multigene Family 360 Genes Affect Virus Replication and Generalization of Infection in Ornithodoros Porcinus Ticks. J. Virol. 2004, 78, 2445–2453. [Google Scholar] [CrossRef]
- Zsak, L.; Lu, Z.; Burrage, T.G.; Neilan, J.G.; Kutish, G.F.; Moore, D.M.; Rock, D.L. African Swine Fever Virus Multigene Family 360 and 530 Genes Are Novel Macrophage Host Range Determinants. J. Virol. 2001, 75, 3066–3076. [Google Scholar] [CrossRef]
- Neilan, J.G.; Zsak, L.; Lu, Z.; Kutish, G.F.; Afonso, C.L.; Rock, D.L. Novel Swine Virulence Determinant in the Left Variable Region of the African Swine Fever Virus Genome. J. Virol. 2002, 76, 3095–3104. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.L.; Abrams, C.C.; Goatley, L.C.; Netherton, C.; Chapman, D.G.; Sanchez-Cordon, P.; Dixon, L.K. Deletion of African Swine Fever Virus Interferon Inhibitors from the Genome of a Virulent Isolate Reduces Virulence in Domestic Pigs and Induces a Protective Response. Vaccine 2016, 34, 4698–4705. [Google Scholar] [CrossRef] [PubMed]
- Golding, J.P.; Goatley, L.; Goodbourn, S.; Dixon, L.K.; Taylor, G.; Netherton, C.L. Sensitivity of African Swine Fever Virus to Type I Interferon Is Linked to Genes within Multigene Families 360 and 505. Virology 2016, 493, 154–161. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Holinka, L.G.; Gladue, D.P.; Sanford, B.; Krug, P.W.; Lu, X.; Arzt, J.; Reese, B.; Carrillo, C.; Risatti, G.R.; et al. African Swine Fever Virus Georgia Isolate Harboring Deletions of MGF360 and MGF505 Genes Is Attenuated in Swine and Confers Protection against Challenge with Virulent Parental Virus. J. Virol. 2015, 89, 6048–6056. [Google Scholar] [CrossRef]
- O’Donnell, V.; Holinka, L.G.; Sanford, B.; Krug, P.W.; Carlson, J.; Pacheco, J.M.; Reese, B.; Risatti, G.R.; Gladue, D.P.; Borca, M. V. African Swine Fever Virus Georgia Isolate Harboring Deletions of 9GL and MGF360/505 Genes Is Highly Attenuated in Swine but Does Not Confer Protection against Parental Virus Challenge. Virus Res. 2016, 221, 8–14. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A Statistical Algorithm for Sensitive and Fast Mapping of Illumina Sequence Reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Brandt, D.Y.C.; Aguiar, V.R.C.; Bitarello, B.D.; Nunes, K.; Goudet, J.; Meyer, D. Mapping Bias Overestimates Reference Allele Frequencies at the HLA Genes in the 1000 Genomes Project Phase I Data. G3 Genes|Genomes|Genetics 2015, 5, 931–941. [Google Scholar] [CrossRef]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From Squiggle to Basepair: Computational Approaches for Improving Nanopore Sequencing Read Accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Delahaye, C.; Nicolas, J. Sequencing DNA with Nanopores: Troubles and Biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput. Struct. Biotechnol. J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef]
- Kieleczawa, J. Fundamentals of Sequencing of Difficult Templates--an Overview. J. Biomol. Tech. 2006, 17, 207–217. [Google Scholar] [PubMed]
- Zeng, F.; Jiang, R.; Chen, T. PyroHMMsnp: An SNP Caller for Ion Torrent and 454 Sequencing Data. Nucleic Acids Res. 2013, 41, e136. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.W.; Middendorf, L.R.; Benner, S.A.; Church, G.M.; Harris, T.; Huang, X.; Jovanovich, S.B.; Nelson, J.R.; Schloss, J.A.; Schwartz, D.C.; et al. The Challenges of Sequencing by Synthesis. Nat. Biotechnol. 2009, 27, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, F.; Gröber, C.; Blank, M.; Händler, K.; Beyer, M.; Schultze, J.L.; Mayer, G. Systematic Evaluation of Error Rates and Causes in Short Samples in Next-Generation Sequencing. Sci. Rep. 2018, 8, 10950. [Google Scholar] [CrossRef]
Figure 1.
Detection of ASFV DNA by qPCR [30] in EDTA blood of the different pigs collected on day of euthanasia (PID 6).
Figure 1.
Detection of ASFV DNA by qPCR [30] in EDTA blood of the different pigs collected on day of euthanasia (PID 6).
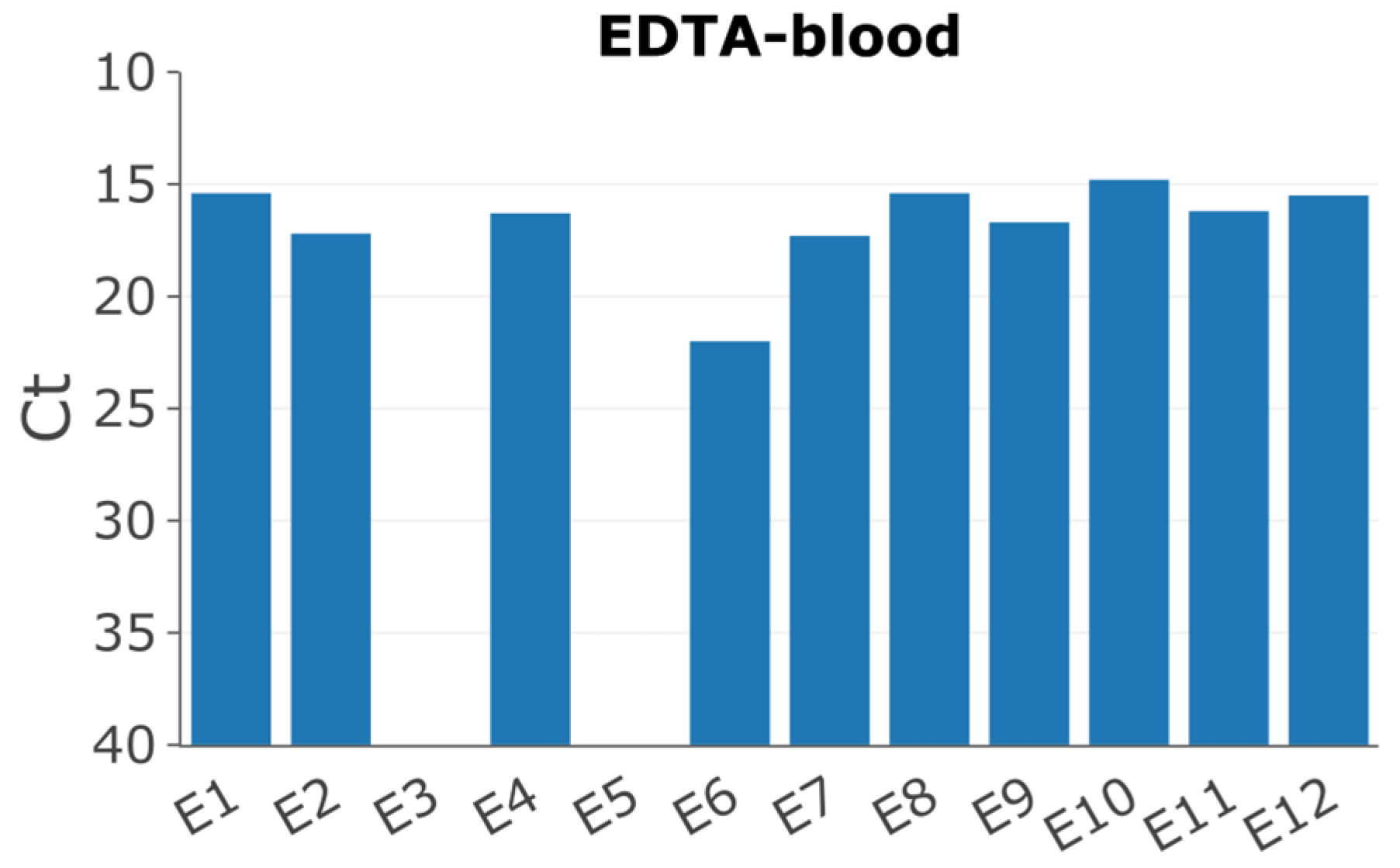
Figure 2.
PCR schematic of the overlapping long PCRs.
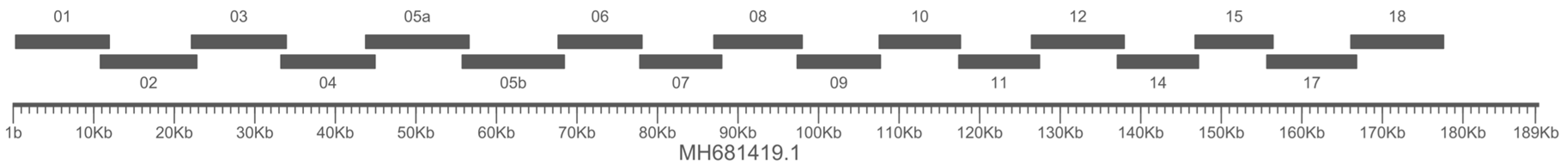
Figure 3.
Deletion screening on samples collected on day of euthanasia. (A) PCR with primers covering nt 6188-17145. L1: Genomic Ladder, L2: GeneRuler 1kb plus, Inoc: Inoculum, E1: Pig 1 EDTA-blood, E2: Pig 2 EDTA-blood, E4: Pig 4 EDTA-blood, E7: Pig 7 EDTA-blood, E8: Pig 8 EDTA-blood, E9: Pig 9 EDTA-blood, E10: Pig 10 EDTA-blood, E11: Pig 11 EDTA-blood, E12: Pig 12 EDTA-blood. (B) PCR with primers covering nt 6188-17145. L1: Genomic Ladder, L2: GeneRuler 1kb plus, E8: Pig 8 EDTA-blood, B8: Pig 8 bone-marrow, SP8: Pig 8 spleen, S8: Pig 8 serum. (C) PCR with primers covering nt 6708-7668. L1: D5000 Ladder, L2: GeneRuler 1kb plus, Inoc: Inoculum, E1: Pig 1 EDTA-blood, E2: Pig 2 EDTA-blood, E4: Pig 4 EDTA-blood, E7: Pig 7 EDTA-blood, E8: Pig 8 EDTA-blood, E9: Pig 9 EDTA-blood, E10: Pig 10 EDTA-blood, E11: Pig 11 EDTA-blood, E12: Pig 12 EDTA-blood. (D) PCR with primers covering nt 6708-7668. L1:D5000 Ladder, L2: GeneRuler 1kb plus, E8: Pig 8 EDTA-blood, B8: Pig 8 bone-marrow, SP8: Pig 8 spleen, S8: Pig 8 serum.
Figure 3.
Deletion screening on samples collected on day of euthanasia. (A) PCR with primers covering nt 6188-17145. L1: Genomic Ladder, L2: GeneRuler 1kb plus, Inoc: Inoculum, E1: Pig 1 EDTA-blood, E2: Pig 2 EDTA-blood, E4: Pig 4 EDTA-blood, E7: Pig 7 EDTA-blood, E8: Pig 8 EDTA-blood, E9: Pig 9 EDTA-blood, E10: Pig 10 EDTA-blood, E11: Pig 11 EDTA-blood, E12: Pig 12 EDTA-blood. (B) PCR with primers covering nt 6188-17145. L1: Genomic Ladder, L2: GeneRuler 1kb plus, E8: Pig 8 EDTA-blood, B8: Pig 8 bone-marrow, SP8: Pig 8 spleen, S8: Pig 8 serum. (C) PCR with primers covering nt 6708-7668. L1: D5000 Ladder, L2: GeneRuler 1kb plus, Inoc: Inoculum, E1: Pig 1 EDTA-blood, E2: Pig 2 EDTA-blood, E4: Pig 4 EDTA-blood, E7: Pig 7 EDTA-blood, E8: Pig 8 EDTA-blood, E9: Pig 9 EDTA-blood, E10: Pig 10 EDTA-blood, E11: Pig 11 EDTA-blood, E12: Pig 12 EDTA-blood. (D) PCR with primers covering nt 6708-7668. L1:D5000 Ladder, L2: GeneRuler 1kb plus, E8: Pig 8 EDTA-blood, B8: Pig 8 bone-marrow, SP8: Pig 8 spleen, S8: Pig 8 serum.
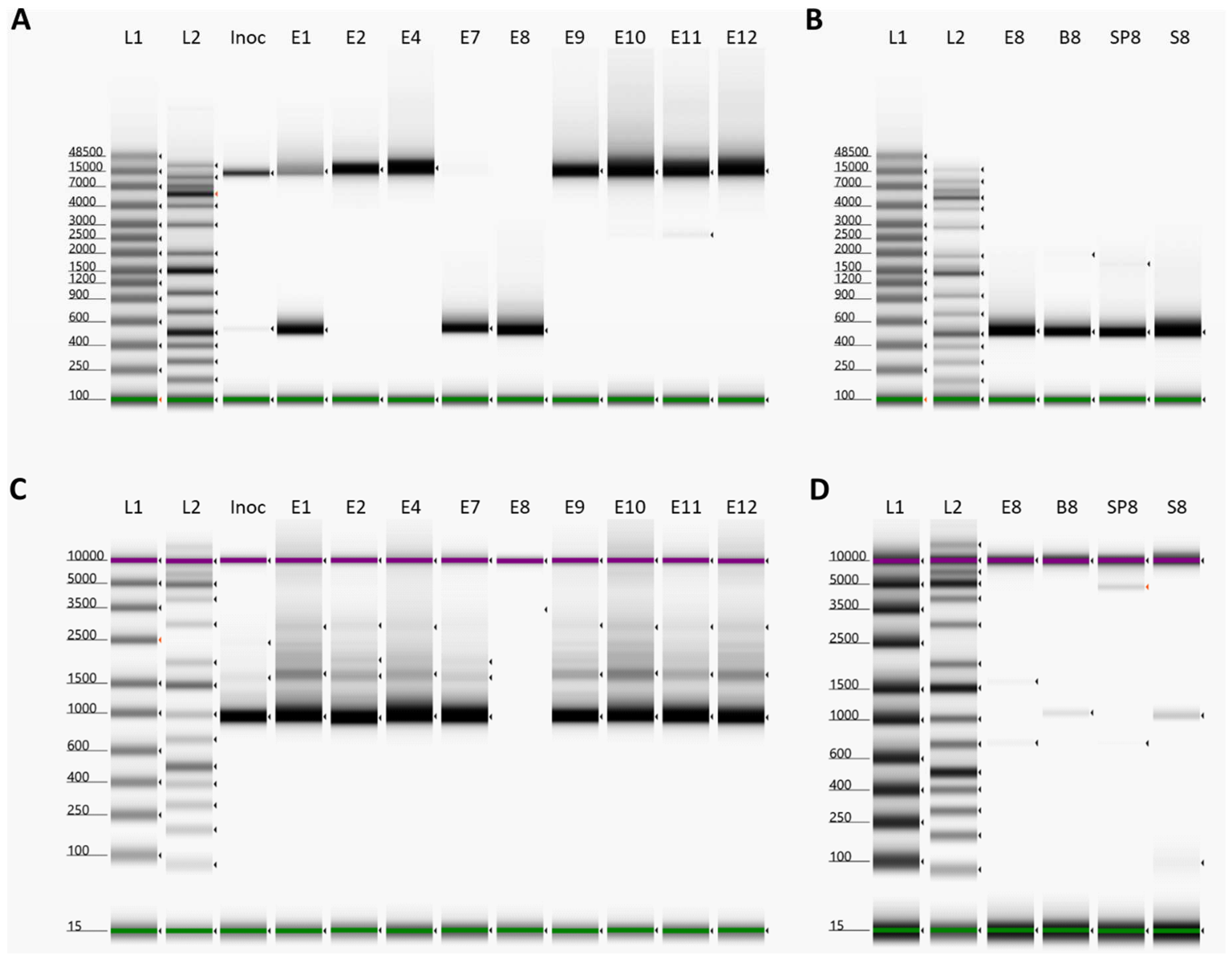
Figure 4.
SNP frequency plots of silent (green) and missense (red) mutations. Light grey background indicates areas with no PCR coverage, and dark grey background indicates areas with failed PCRs (due to the deletion). Genes on the forward strand are indicated in blue, and genes on the reverse strand are indicated in gold, with affected genes in dark grey. (A) SNP frequencies in Inoculum. (B) SNP frequencies in sample E1. (C) SNP frequencies in sample E2. (D) SNP frequencies in sample E4. (E) SNP frequencies in sample E7. (F) SNP frequencies in sample E8. (G) SNP frequencies in sample E9. (H) SNP frequencies in sample E10. (I) SNP frequencies in sample E11. (J) SNP frequencies in sample E12.
Figure 4.
SNP frequency plots of silent (green) and missense (red) mutations. Light grey background indicates areas with no PCR coverage, and dark grey background indicates areas with failed PCRs (due to the deletion). Genes on the forward strand are indicated in blue, and genes on the reverse strand are indicated in gold, with affected genes in dark grey. (A) SNP frequencies in Inoculum. (B) SNP frequencies in sample E1. (C) SNP frequencies in sample E2. (D) SNP frequencies in sample E4. (E) SNP frequencies in sample E7. (F) SNP frequencies in sample E8. (G) SNP frequencies in sample E9. (H) SNP frequencies in sample E10. (I) SNP frequencies in sample E11. (J) SNP frequencies in sample E12.
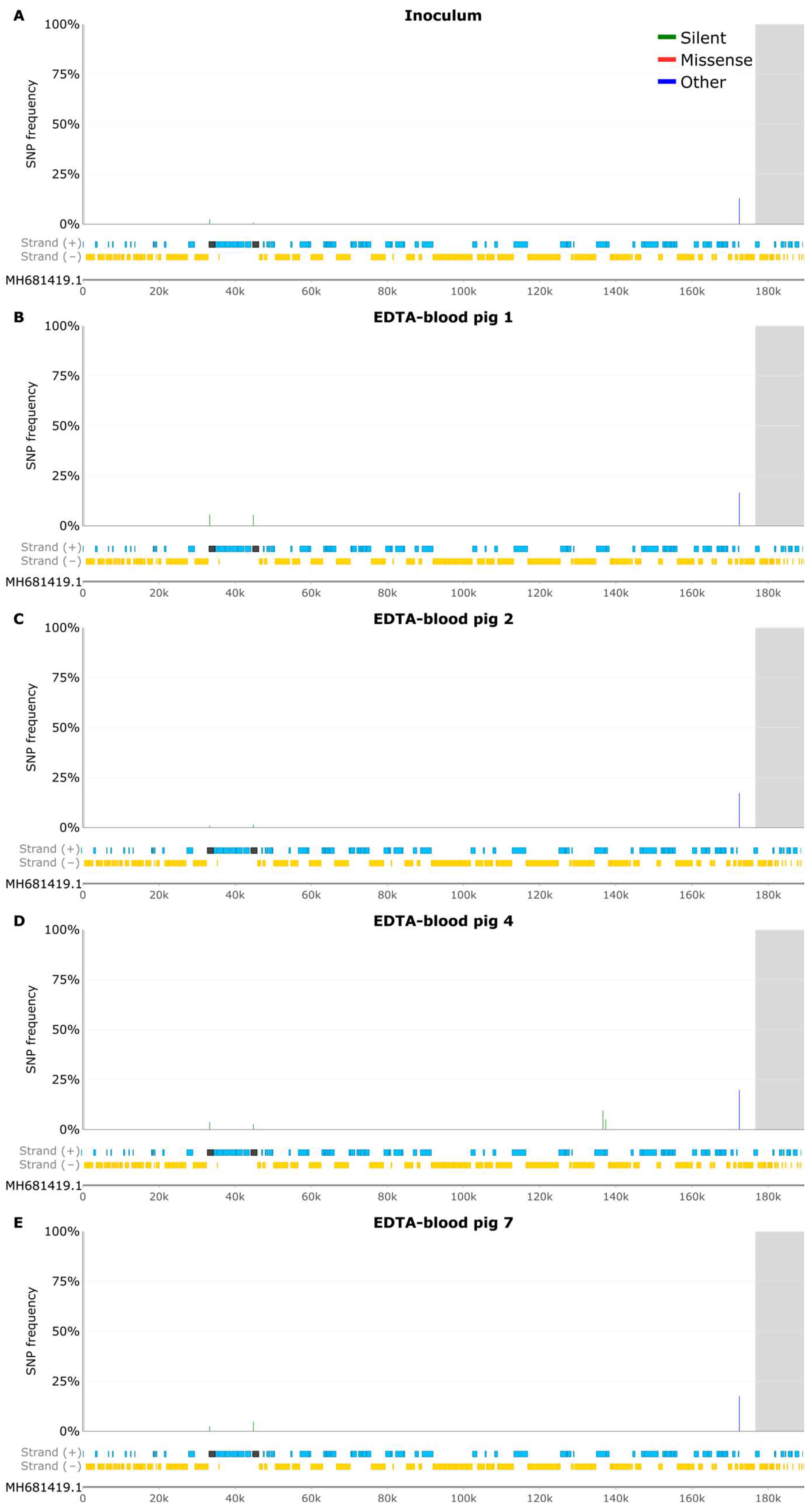
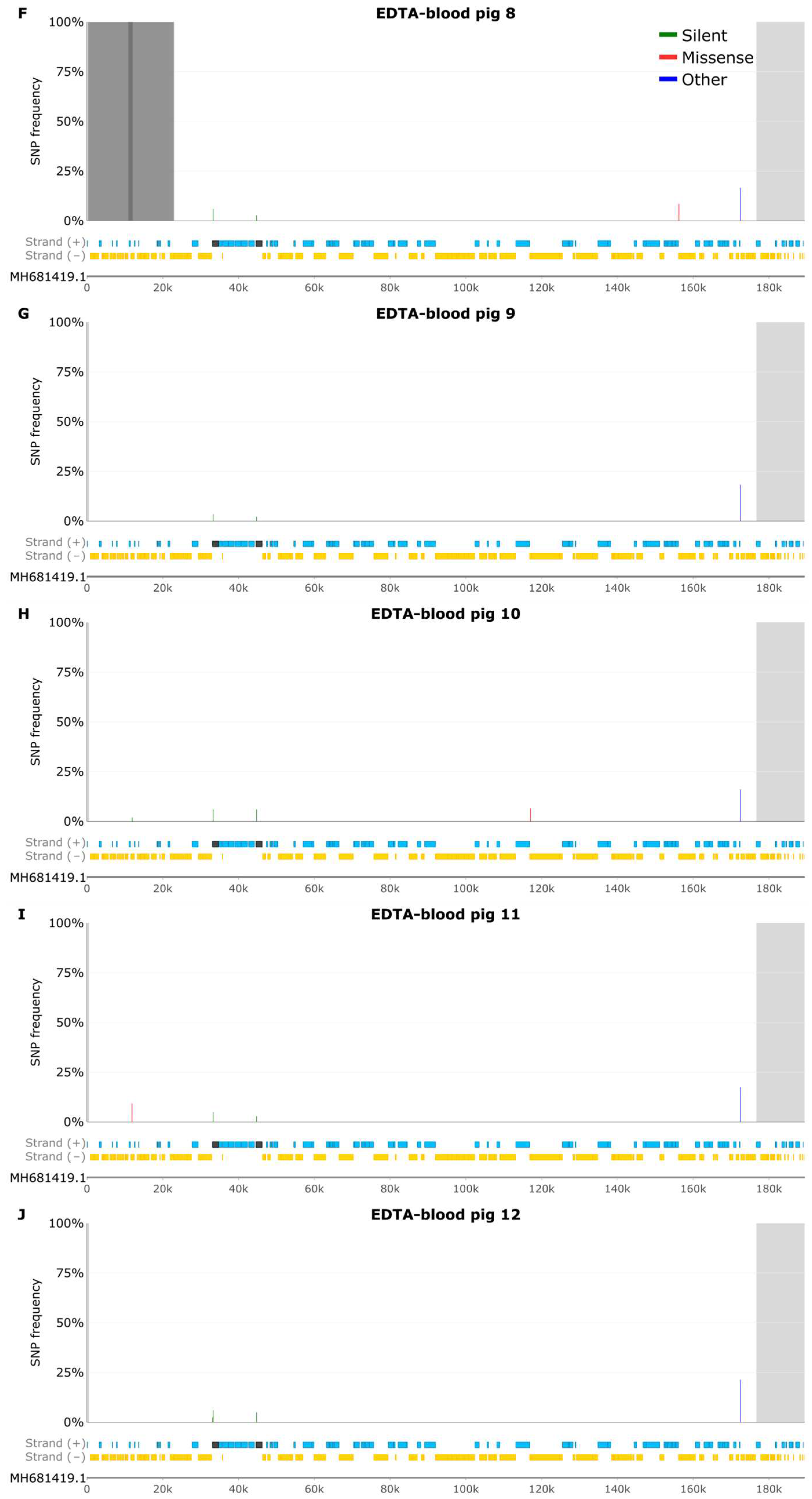
Figure 5.
SNP frequency plots of silent (green) and missense (red) mutations. Light grey background indicates areas with no PCR coverage. Genes on the forward strand are indicated in blue, and genes on the reverse strand are indicated in gold, with affected genes in dark grey. (A) SNP frequencies in Inoculum. (B) SNP frequencies in sample E2. (C) SNP frequencies in sample B2. (D) SNP frequencies in sample SP2. (E) SNP frequencies in sample S2.
Figure 5.
SNP frequency plots of silent (green) and missense (red) mutations. Light grey background indicates areas with no PCR coverage. Genes on the forward strand are indicated in blue, and genes on the reverse strand are indicated in gold, with affected genes in dark grey. (A) SNP frequencies in Inoculum. (B) SNP frequencies in sample E2. (C) SNP frequencies in sample B2. (D) SNP frequencies in sample SP2. (E) SNP frequencies in sample S2.
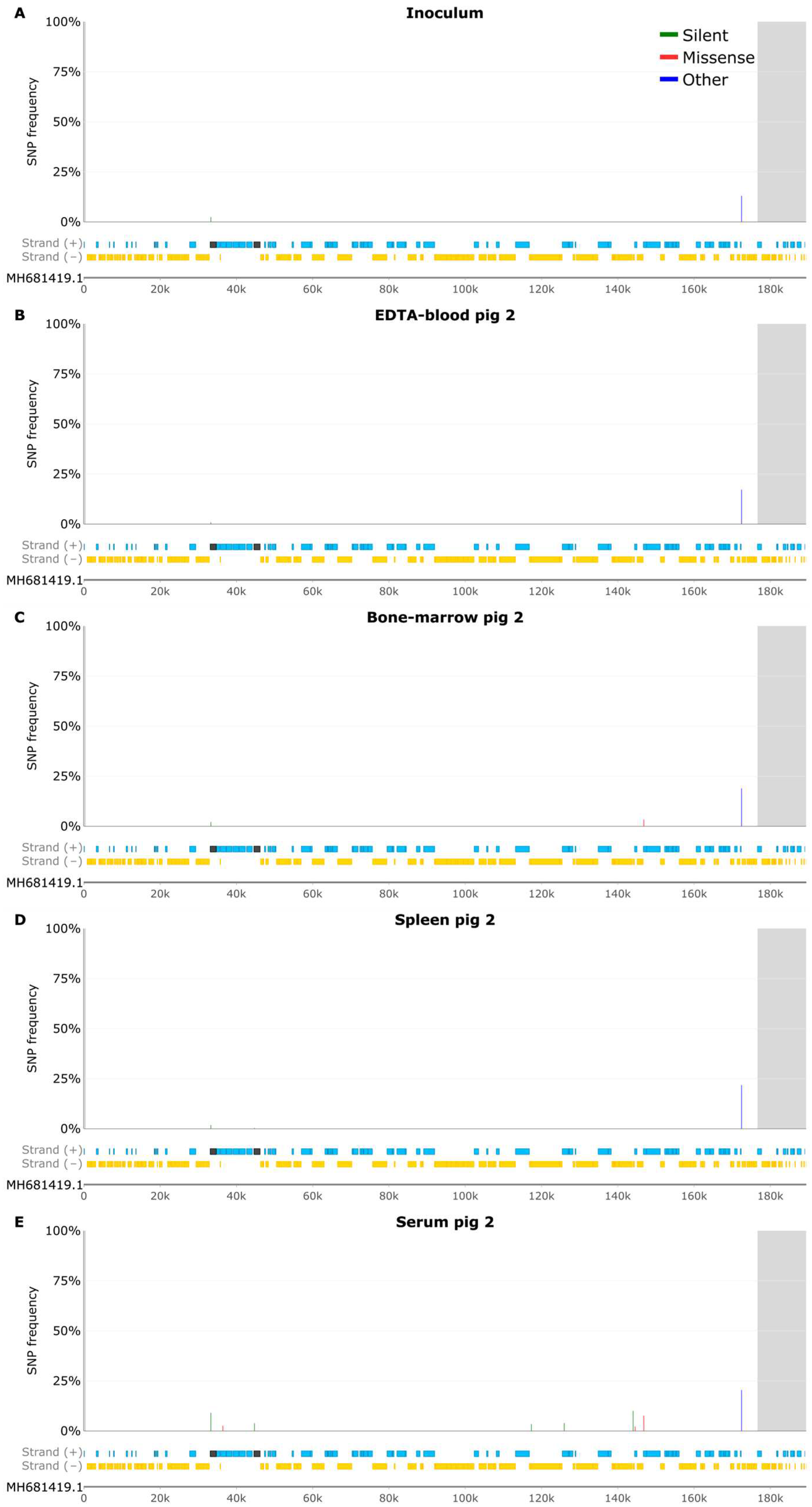
Table 3.
PCR matrix overview.
| Matrix | 1xα | 10xα | 100xα |
|---|---|---|---|
| EDTA-blood | +++ | ++ | + |
| Bone-marrow | - | ++ | + |
| Spleen | - | ++ | + |
| Serum | + | - | - |
αDilution of sample; - no band; + indicates strength of band
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



