Submitted:
12 December 2023
Posted:
14 December 2023
You are already at the latest version
Abstract
Background: Mounting evidence supports the role of the endocannabinoid system in neurophysiology, includ-ing blood-brain barrier (BBB) function. Recent work has demonstrated that activation of endocannabinoid re-ceptors can mitigate insults to the BBB during neurological disorders, like traumatic brain injury, cortical spreading depression and stroke. As alterations to the BBB are associated with worsening clinical outcomes in these conditions, studies herein sought to examine the impact of endocannabinoid depletion on BBB integrity. Methods: Barrier integrity was investigated in vitro via bEnd.3 cell monolayers to assess endocannabinoid syn-thesis, barrier function, calcium influx, junctional protein expression, and proteome-wide changes. Results: In-hibition of 2-AG synthesis using DAGLα inhibition and siRNA inhibition of DAGLα led to loss of barrier in-tegrity via altered expression of VE-cadherin, which could be partially rescued by exogenous application of 2-AG. Moreover, the deleterious effects of DAGLα inhibition on BBB integrity showed both calcium and PKC (protein kinase C)-dependency. Conclusions: These data indicate that disruption of 2-AG homeostasis in brain endothelial cells, in the absence of insult, is sufficient to disrupt BBB integrity thus supporting the role of the endocannabinoid system in neurovascular disorders.
Keywords:
endocannabinoid
; blood-brain barrier
; 2-AG
; DAGLα
; VE-cadherin
1. Introduction
Alterations to the integrity of the blood-brain barrier (BBB) are well documented within neurodegenerative and neuroinflammatory disorders, such as Alzheimer’s disease and migraine headache [1,2]. BBB integrity is maintained by the neurovascular unit (NVU), which comprises endothelial cell tight junctions, astrocyte foot processes, and pericytes [3,4]. Loss of integrity of the barrier may stem from alterations to any of these elements of NVU [5,6,7]. Increased BBB permeability has been linked to accumulation of toxic metabolites, alterations to blood flow, cerebral edema, and other adverse outcomes; however, the processes that drive these changes are not fully understood [2,8,9].
One mechanism implicated in BBB integrity is the endocannabinoid system (ECBS). Multiple GPCRs (e.g., CB1R and CB2R), lipid metabolizing enzymes (e.g., monoacylglycerol lipase (MAGL); α/β serine hydrolase 6, (ABHD6)), and two main lipid mediators, AEA and 2-AG compose the ECBS [10,11,12]. 2-AG and AEA are produced on demand to serve as retrograde neuromodulators which act at CB1R and CB2R to negatively regulate cell-cell communication; 2-AG binds to and activates both receptors [10,12]. Synthesis of 2-AG requires activation of PLC to produce inositol triphosphate (IP3) and diacylglycerol (DAG) from PIP2 [10,12], which is followed by activation of DAGLα and β that converts DAG into 2-AG and a free fatty acid; DAGLα is the dominant isoform in the CNS [12]. In brain homogenates, 2-AG hydrolysis is mediated by MAGL (80%) and ABDH6 (6-8%) [10], which generate arachidonic acid, a precursor for inflammatory eicosanoids (e.g., PGE2). The ECBS serves several physiologic functions, including neuroprotection and restoration of tissue homeostasis following stress, inflammation, and tissue injury [13,14,15,16,17]. At the BBB, however, the role of the ECBS remains elusive. Activation of the cannabinoid receptors by exogenous ligands and by inhibition of 2-AG synthesis has been shown to mitigate BBB impairment in models of TBI and stroke, indicating a homeostatic role of 2-AG in BBB function [18,19,20,21]. Despite these advances in knowledge, the mechanisms by which the ECBS affects BBB structural integrity have yet to be elucidated.
To fill this gap in knowledge we utilized an in vitro model of bEnd.3 cell monolayers to examine the effects of 2-AG depletion, via the DAGLα inhibitor LEI-106 and siRNA inhibition of DAGLα, on synthesis of endocannabinoid lipids, monolayer functional integrity, cell viability and morphology, junctional protein expression, and changes in the phosphoproteome. We hypothesized that endothelial derived 2-AG would maintain BBB homeostasis and that DAGLα inhibition would impair BBB integrity. Our data supports a role for endothelial derived 2-AG in BBB endothelial cell homeostasis through regulation of cell structure and VE-cadherin function. The impact of 2-AG depletion on kinase activity along with cytoskeletal organization was also supported by phospho-proteomics data.
2. Results
2.1. Blockade of DAGLα decreases 2-AG levels in Endothelial Cells without altering AEA levels
The first experiment was designed to determine whether cultured brain endothelial cells (bEnd.3) synthesized endocannabinoid lipids, 2-AG and AEA using LC-MS. Basal levels of 2-AG and AEA synthesized by bEnd.3 cells were determined to be 25.27 ± 2.89 pmol/g and 0.324 ± .02516 pmol/g, respectively. We next examined whether blockade of DAGLα in the brain endothelial cells (bEnd.3) altered endocannabinoid lipid levels. Cells were treated with LEI-106 (1.3mM equimolar to the 40 mg/kg dose in vivo) for 15 min followed by a wash step before harvest by cell scraping (Figure 1A). The cell pellet was lysed and subjected to LC-MS to measure 2-AG and AEA levels. The treatment of bEnD.3 cells with LEI-106 significantly reduced the level of 2-AG without changing to levels of AEA as compared to vehicle (Figure 1B, C; LEI-106 vs. vehicle, 2-AG: p=0.001, as assessed by unpaired t-test, n=4-10 in each group; AEA: p=0.1984, as assessed by unpaired t-test, n=4-10 in each group), suggesting on-target actions of LEI-106 at DAGLα. It is noteworthy that 2-AG and AEA were detected in the pmol and fmole ranges, respectively, mirroring tissue observations of a roughly 1000x fold difference in concentration.
To determine if LEI-106 induced depletion in 2-AG was coupled to changes in DAG levels, quantitative ELISA was performed using the same experimental setting. No significant difference was observed between LEI-106 and vehicle-treated cells (Figure 1D) (DAG level in LEI-106 (650 µM) group: 4.713±0.2946 ng/mL, DAG level in LEI-106 (1.3 mM) group: 4.701±0.327 ng/mL, DAG level in vehicle group: 4.259±0.485 ng/mL, LEI-106 (650 µM) vs. vehicle: p=0.4364; LEI-106 (1.3 mM) vs. vehicle: p=0.4626, as assessed by unpaired t-test, n=8/group).
The next investigation employed genetic manipulation of DAGLα to assess endocannabinoid levels in bEnd.3 cells to complement pharmacology with LEI-106. bEnD.3 cells were transiently transfected with siRNA targeting DAGLα or a scrambled, non-targeting control. Cells were lysed 72 h post-transfection and lysates subjected to Western blot to detect DAGLα expression. The transfection of DAGLα -targeting siRNA significantly decreased the detection of DAGLα protein as compared to the non-targeting control (Figure 1E) (DAGLα siRNA vs. non-targeting control siRNA: p<0.0001, as assessed by unpaired t-test, n=8/group). siRNA transfected cells were assessed as well for levels of 2-AG (Figure 1F) and AEA (Figure 1G) via LC-MS. siRNA knock down of DAGLα in bEnD.3 cells caused significant reduction of 2-AG levels as compared to non-targeting control (Figure 1F) (DAGLα siRNA vs. non-targeting control siRNA: p=0.0266, as assessed by unpaired t-test, n=4/group). However, AEA level in bEnd.3 cells was significantly increased after DAGLα siRNA transfection compared to non-targeting control, which suggests that the cells can compensate the loss of 2-AG by increasing AEA level (Figure 1G) (DAGLα siRNA vs. non-targeting control siRNA: p=0.031, as assessed by unpaired t-test, n=4/group). Overall, these data support that brain endothelial cells have DAGLα, make detectable 2-AG under physiological conditions, and are sensitive to treatment with LEI-106 in vitro.
Figure 1.
The pharmacological or genetic manipulation of DAGLα decrease the level of 2-AG in bEnD.3 endothelial cells.
Figure 1.
The pharmacological or genetic manipulation of DAGLα decrease the level of 2-AG in bEnD.3 endothelial cells.
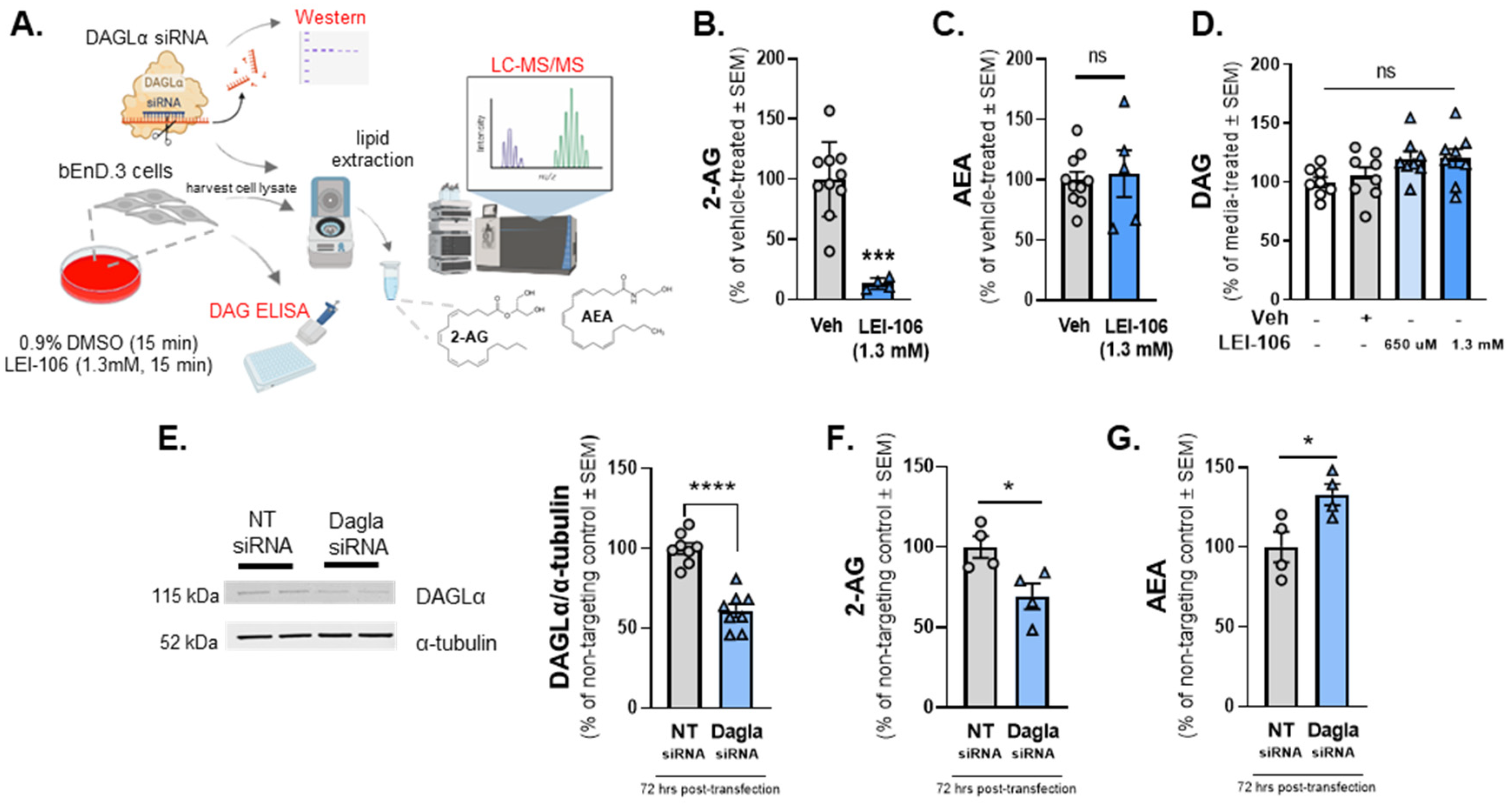
bEnD.3 endothelial cells were treated with LEI-106 (1.3 mM) or vehicle (0.9% DMSO in media) for 15 min, then subjected to LC-MS to measure 2-AG and AEA levels. In a separate set, level of DAG was quantified by ELISA. In the siRNA transfection experiments, bEnD.3 cells were transiently transfected with siRNA targeting DAGLα or a scrambled, non-targeting control, then harvested 72 hs post-transfection followed by a Western-blot to detect DAGLα expression. In a separate set of experiment, the cell pellet was lysed and subjected to LC-MS to measure 2-AG and AEA levels. (A) The schema of experimental setting. (B) The pharmacological blockade of DAGLα with LEI-106 significantly reduced the 2-AG level in endothelial cells (LEI-106 vs. vehicle, p=0.001, t(12)=5.447 as assessed by unpaired t-test). Data are shown as % of vehicle-treated ± SEM (n=4-10 in each group). *** denotes significantly different (p<0.001). (C) After LEI-106 treatment, the level of AEA was unchanged (LEI-106 vs. vehicle, p=0.7578, t(13)=0.3149 as assessed by unpaired t-test). Data are shown as % of vehicle-treated ± SEM (n=5-10 in each group). ns=non-significant. (D) The LEI-106 treatment did not significantly change the level of DAG as compared to vehicle control (LEI-106-650 µM vs. vehicle: p=0.4364, t(14)=0.8011; LEI-106-1.3 mM vs. vehicle: p=0.4626, t(14)=0.7553 as assessed by unpaired t-test). Data are presented as % of media-treated ± SEM (n=8/group). ns=non-significant. (E) Representative image showing the expression of DAGLα with α-tubulin as loading control in bEnd.3 cell lysate harvested72 h after siRNA transfection. The transfection of DAGLα -targeting siRNA significantly reduced detection of DAGLα protein as compared to the non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p<0.0001, t(14)=7.227, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n=8/group). **** denotes significantly different (p<0.0001). (F) siRNA knock down of DAGLα in bEnD.3 cells significantly reduced 2-AG levels as compared to non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p=0.0266, t(6)=2.919, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n=4/group). * denotes significantly different (p<0.05). (G) AEA level of bEnd.3 cells was significantly increased after DAGLα siRNA transfection compared to non-targeting control, suggesting the presence of a compensatory mechanism after the loss of 2-AG (DAGLα siRNA vs. non-targeting control siRNA: p=0.031, t(6)=2.804, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n=4/group). * denotes significantly different (p<0.05).
2.2. 2-AG Depletion Decreases Trans-Endothelial Electrical Resistance
Having established that bEnd.3 cells are responsive to LEI-106, in vitro analysis of barrier integrity was performed via testing of TEER (Figure 2) and 14C-sucrose transport (Figure 3). For TEER experiments, bEnd.3 cells were grown to confluency on trans-well membranes, and then treated with LEI-106 (15min, 650µM, 1.3mM, luminal side) or KCl as positive control (100mM, 5min, abluminal side) (Figure 2A). TEER values in monolayers treated with 0.9% DMSO vehicle were significantly decreased when media was changed at t=0 min; no further changes in TEER were observed across the experimental duration. As previously documented, KCl application significantly decreased TEER [22] (Figure 2B, C). TEER values were also significantly reduced from baseline and as compared to vehicle treatment at the end of 15 min incubation period (0 min time-point) of either concentration of LEI-106 (Figure 2B) (LEI-106-650 uM vs. vehicle: p=0.0017; LEI-106-1.3 mM vs. vehicle: p<0.0001, as assessed by two-way ANOVA). At additional time-points, TEER was normalized, however additional decrease of TEER values was detected at 2 and 3 h (Figure 2B) (2 h time-point: LEI-106-1.3 uM vs. vehicle: p=0.0075; 3 h: LEI-106-650 µM vs. vehicle: p=0.0254, LEI-106-1.3 mM vs vehicle: p<.0.022, as assessed by two-way ANOVA). The effect of LEI-106 on endothelial cell monolayer integrity over time was confirmed by AUC analysis (Figure 2C) (LEI-106-650 µM vs. vehicle: p=0.009; LEI-106-1.3 mM vs. vehicle: p<0.0001; LEI-106-650 µM vs. LEI-106-1.3 mM: p=0.01, as assessed by one-way ANOVA with Tukey post-test).
In the siRNA transfection experiments, bEnd.3 cells were seeded on trans-well inserts 24 h post-transfection, then TEER was performed 72 h after the transfection. Silencing DAGLα by siRNA transfection significantly reduced TEER, compared to non-targeting control (Figure 2D) (DAGLα siRNA vs. non-targeting siRNA: p=0.0003, as assessed by unpaired t-test, n=5). Together, these TEER data provide evidence that the integrity of the endothelial monolayer is disrupted following pharmacological or genetic blockade of DAGLα.
In a separate experiment designed to establish whether the decreases in TEER induced by LEI-106 were a result of reduced 2-AG, cells received treatment with LEI-106 followed by media spiked with 2-AG (15 min, 600pmol/well). Vehicle and LEI-106 were applied to the luminal side of the trans-well, to simulate blood stream delivery of drug to the BBB; 2-AG was added to the abluminal side of the trans-well insert to mimic CNS production of 2-AG (Figure 2A). 2-AG treatment (600pmol/well) in the abluminal chamber significantly increased TEER at individual time-points and as confirmed by AUC analysis (Figure 2E, F; 0 min time-point: LEI-106-650 µM vs. LEI-106-650 µM+2-AG: p=0.0002; LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by two-way ANOVA), (AUC: LEI-106-650 µM vs. LEI-106-650 µM+2-AG: p=0.0001; LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by one-way ANOVA). All measurements were repeated in triplicate (technical replicate) over three individual experiments (biological replicate). These data suggest that increasing 2-AG levels after DAGLα inhibitor restores endothelial monolayer electrical resistance.
Figure 2.
The depletion of 2-AG decreased Trans-Endothelial Electrical Resistance (TEER) of bEnD.3 cells, indicating the loss of barrier integrity.
Figure 2.
The depletion of 2-AG decreased Trans-Endothelial Electrical Resistance (TEER) of bEnD.3 cells, indicating the loss of barrier integrity.
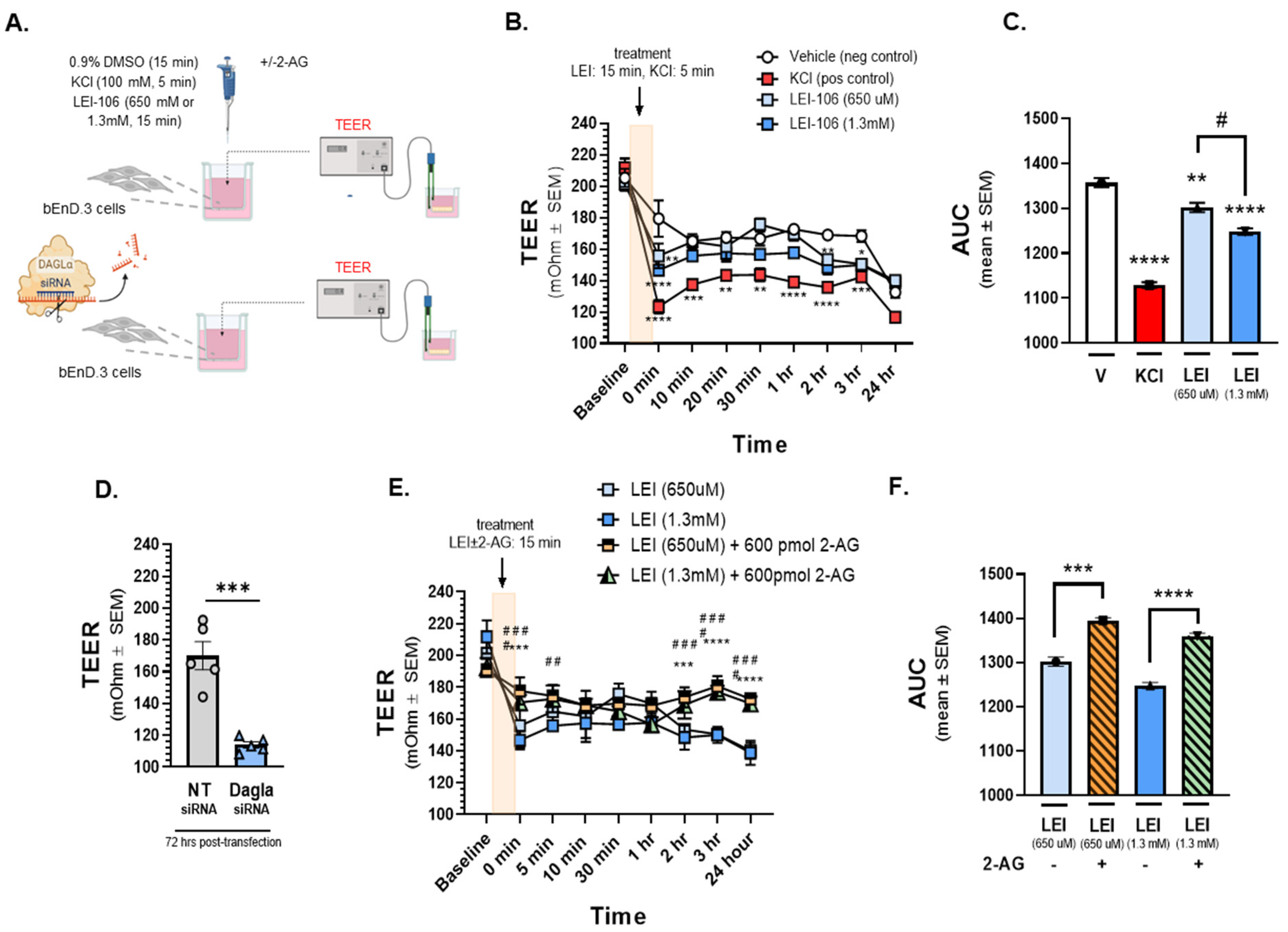
The bEnD.3 endothelial cells were cultured on trans-well insert, then treated with LEI-106 (650 uM or 1.3 mM) or vehicle (0.9% DMSO in media) with or without 2-AG (600 pmol) for 15 min. KCl pulse (100 mM, 5 min) was used as positive control. TEER was measured at baseline (before any treatment), right after the treatment (0 min), then 10, 20, 30 min, 1, 2, 3, and 24 h after post-treatment. In the transfection experiments, TEER was measured 72 h post-transfection. (A) This panel shows the schematic experimental setting. (B) The application of LEI-106 in either dose significantly decreased the TEER value, compared to vehicle control at 0 min time-point, suggesting the loss of barrier integrity (LEI-106-650 uM vs. vehicle: p=0.0017; LEI-106-1.3 mM vs. vehicle: p<0.0001, as assessed by two-way ANOVA, F(24,72)=3.723). At later time-points, the TEER was then normalized, followed by an additional significant decrease at 2 and 3 h (2 hr time-point: LEI-106-1.3 uM vs. vehicle: p=0.0075; 3 hr time-point: LEI-106-650 uM vs. vehicle: p=0.0254 LEI-106-1.3 mM vs vehicle: p<0.022, as assessed by two-way ANOVA, F(24,72)=3.723). All data represent the mean ± SEM of three independent experiment performed in triplicate. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 as compared to vehicle control. (C) The area under the curve (AUC) of the corresponding panel B. The AUC analysis further confirmed the significant reduction of TEER induced by LEI-106 treatment (LEI-106-650 uM vs. vehicle: p=0.009; LEI-106-1.3 mM vs. vehicle: p<.0.0001; LEI-106-650 uM vs.LEI-106-1.3 mM: p=0.01, as assessed by one-way ANOVA with Tukey post-test, F(3,8)=125.2). Data are shown mean ± SEM. **p<0.01, ****p<0.0001 as compared to vehicle control. #p<0.05: LEI-106-650 uM vs. LEI-106-1.3 mM. (D) bEnD.3 cells were transfected either with non-targeting control siRNA or siRNA targeting DAGLα. TEER was assessed 72 h post-transfection. Significant decrease of TEER was observed in DAGLα siRNA-transfected cells, compared to non-targeting control, indicating disruption of the barrier integrity after silencing of DAGLα (DAGLα siRNA vs. non-targeting siRNA: p=0.0003, t(8)= 6.203 as assessed by unpaired t-test, n=5). Data are shown mean ± SEM. ***p<0.001, as compared to the non-targeting control. (E) The application of 2-AG diminished the effect of LEI-106 on BBB integrity. Significant differences were observed between LEI-106 vs. LEI-106+2-AG groups at 0 min time-point (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.0002; hjnnnoLEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by two-way ANOVA, F(24,90)=7.318). All data represent the mean ± SEM of three independent experiment performed in triplicate. LEI-106-650 uM vs. LEI-106-650 uM+2-AG: ***p<0.001, ****p<0.0001. LEI-106-1.3 mM vs. LEI-106 1.3+2-AG: ##p<0.01, ###p<0.001, ####p<0.0001. (F) The area under the curve (AUC) for panel E. The AUC analysis also showed that 2-AG can mitigate the loss of barrier integrity caused by the inhibition of DAGLα (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.0001; LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by one-way ANOVA, F(3,8)=67.39). Data are shown mean ± SEM. ***p<0.001, ****p<0.0001 as compared to the corresponding 2-AG treatment.
2.3. 2-AG Depletion Increases in vitro 14C-Sucrose Transport
To determine if the observed TEER changes reflected functional changes in paracellular permeability, the next study examined 14C-sucrose transport across the bEnd.3 monolayer (Figure 3A). Cells were treated with 2-AG at concentrations aligning with what has been recorded in vivo from the parenchyma (300pmol/well-10nmol/well; 600µL/well) to determine basal responses to 2-AG on monolayer functionality (Figure 3B, C). Thirty min after treatment, 2-AG (300pmol/well) increased 14C-sucrose transport across the monolayer (2-AG-300 pmol vs. vehicle: p=0.0172, 2-AG at other doses vs. vehicle: p>0.05, F(5,20)=3.879, as assessed by one-way ANOVA with Bartlett`s test, n=3-10/group); no other significant effects of 2-AG alone on bEnd.3 monolayer integrity were observed. Separate sets of cells were incubated in LEI-106 (1.3mM), KCl (100mM), or vehicle (0.9% DMSO) with or without 2-AG application (600 pmol/insert). LEI-106 (1.3 mM) significantly increased the 14C-sucrose detection in the abluminal chamber at 5- and 30-min time-points as compared to vehicle control, indicating the presence of paracellular leak (Figure 3D, E) (5 min: LEI-106 vs. vehicle: p=0.0033, 30 min: LEI-106 vs. vehicle: p=0.0092, as assessed by unpaired t-test). We next investigated whether these paracellular breaches could be corrected with exogenously applied 2-AG. The application of 2-AG did not significantly influence the elevated sucrose uptake induced by LEI-106 at 5 min (Figure 3D) (LEI-106 vs. LEI-106+2-AG: p=0.0797, as assessed by unpaired t-test). However, 2-AG treatment significantly mitigated the increase of sucrose uptake caused by DAGLα inhibition at 30 min (Figure 3E) (LEI-106 vs. LEI-106+2-AG: p=0.05, as assessed by unpaired t-test). Data were obtained from three-four independent experiments using 4 trans-well inserts/group.
In the siRNA transfection experiments, bEnd.3 cells were seeded on trans-well-inserts 24 h post-transfection, then the 14C-sucrose assay was performed 72 h after the transfection. Transfection of DAGLα siRNA in bEnD.3 cells significantly increased the detection of 14C-sucrose in the abluminal chamber at 5 min, suggesting paracellular leak after DAGLα silencing. However no significant change between DAGLα siRNA and control was observed at 30 min time-point (Figure 3F, G) (5 min: DAGLα siRNA vs. non-targeting siRNA: p=0.0022, as assessed by unpaired t-test, n=5; 30 min: DAGLα siRNA vs. non-targeting siRNA: p=0.8204, as assessed by unpaired t-test, n=5). Together, these data suggest that 2-AG signaling in endothelial cells functionally maintains barrier integrity and that loss of endogenous 2-AG tone promotes paracellular leak.
Figure 3.
14C-Sucrose Transport through the bEnd.3 monolayer was increased after the blockade of DAGLα.
Figure 3.
14C-Sucrose Transport through the bEnd.3 monolayer was increased after the blockade of DAGLα.
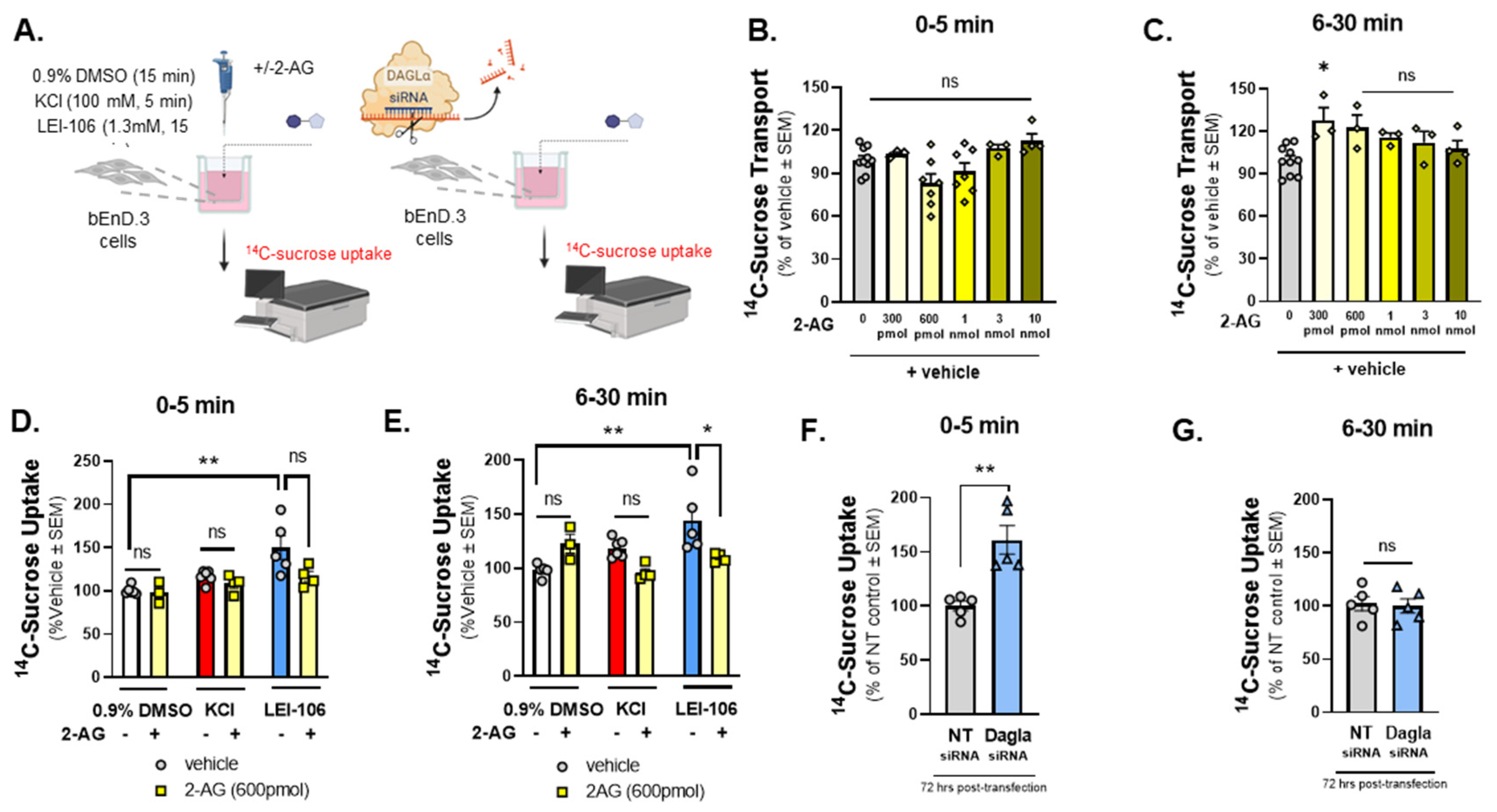
The bEnD.3 endothelial cells were cultured on trans-well insert, then treated with increasing amount of 2-AG (0-10 nmole). In a separate experiment, cells were treated with LEI-106 (1.3 mM) or vehicle (0.9% DMSO in media) with or without 2-AG (600 pmol) for 15 min. KCl pulse (100 mM, 5 min) was used as positive control. Following treatment, luminal media was replaced with fresh media containing 14C-sucrose (0.25uCi/mL). Abluminal media was collected at 5 min and 30 min post treatment and subjected to measure radioactivity with a liquid scintillation counter. 14C-sucrose uptake assay was performed 72 h after transfection of DAGLα siRNA or non-targeting control. 14C-sucrose (0.25uCi/mL) was added to the luminal side, abluminal media was collected at two time-points (5 min and 30 min), then radioactivity was measured. (A) Schema of experimental setting. (B) 2-AG treatment at any dose did not cause significant change in 14C-sucrose uptake at 5 min time-point as compared to vehicle control (2-AG at any dose vs. vehicle: p>0.05, F(5,28)=4.010, as assessed by one-way ANOVA with Bartlett`s test) All data represent the % of vehicle-treated ± SEM (n=3-10/group). ns=non-significant. (C) A significant increase of 14C-sucrose uptake after 300 pmol of 2-AG treatment was observed at 6-30 min time-point as compared to vehicle control (2-AG-300 pmol vs. vehicle: p=0.0172, 2-AG at other doses vs. vehicle: p>0.05, F(5,20)=3.879, as assessed by one-way ANOVA with Bartlett`s test) All data represent the % of vehicle-treated ± SEM (n=3-10/group). ns=non-significant, *p<0.05. (D) LEI-106 (1.3 mM) increased the 14C-sucrose uptake at 5 min time-point as compared to vehicle control, indicating the presence of paracellular leak (LEI-106 vs. vehicle: p=0.0033, t(9)=3.965 as assessed by unpaired t-test). The application of 2-AG did not significantly reduce the elevated sucrose uptake induced by LEI-106 (LEI-106 vs. LEI-106+2-AG: p=0.0797, t(7)=2.049 as assessed by unpaired t-test). All data represent the % of vehicle-treated ± SEM from three-four independent experiments using 4 trans-well inserts/group. **p<0.01 as compared to vehicle control. ns=non-significant. (E) Increased 14C-sucrose uptake was also observed at the 30 min time-point after LEI-106 treatment (LEI-106 vs. vehicle: p=0.0092, t(8)=3.415 as assessed by unpaired t-test). 2-AG treatment significantly mitigated the increase of sucrose uptake caused by DAGLα inhibition (LEI-106 vs. LEI-106+2-AG: p=0.05, t(7)=2.273 as assessed by unpaired t-test). All data represent the % of vehicle-treated ± SEM from three-four independent experiments using 4 trans-well inserts/group. *p<0.05, **p<0.01 as compared to corresponding controls. (F) Silencing of DAGLα significantly increased the sucrose uptake in the first 5 min, compared to non-targeting control, suggesting reduced barrier integrity caused by genetic inhibition of DAGLα (DAGLα siRNA vs. non-targeting siRNA: p=0.0022, t(8)=4.420 as assessed by unpaired t-test, n=5). Data are shown mean ± SEM. **p<0.01, as compared to the non-targeting control. (G) No significant difference between DAGLα siRNA and control was observed at the later time-point (6-30 min) (DAGLα siRNA vs. non-targeting siRNA: p=0.8204, t(8)=0.2347 as assessed by unpaired t-test, n=5). Data are shown mean ± SEM. ns=non-significant, as compared to the non-targeting control.
2.4. Immunocytochemistry of 2-AG Depleted Cells Reveals Morphological Changes
With confirmation that LEI-106 was reducing 2-AG within endothelial cells, and that this loss functionally reduced monolayer paracellular integrity, we investigated possible underlying mechanisms including cell viability, morphological changes, and junctional protein expression (Figure 4A). To test whether changes in barrier permeability reflect loss of cell viability, an XTT cell viability assay was employed. Cells were treated with titrated doses of LEI-106 (100nM, 10µM, 100µM, 325µM, 650µM, and 1.3mM). No significant differences in oxidative capacity were noted at all LEI-106 doses (Figure 4B) (LEI-106 at any doses vs. vehicle: p>0.05, as assessed by one-way ANOVA with Bartlett post-test, n=4/group) suggesting DAGLα inhibition and loss of 2-AG does not play a role in bEnd.3 cell viability.
ICC revealed significant changes in endothelial cell morphology following LEI-106 (650µM, 1.3mM; Figure 4C, D). Total cell area was significantly decreased with a significant increase in nuclear: cytoplasmic (N:C) ratio following LEI-106. 2-AG treatment following LEI-106 significantly reversed these effects (Figure 4D) (LEI-106-650 µM vs. vehicle: p<0.0001, LEI-106-1.3 mM vs. vehicle: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, n=9-12/conditions) (LEI-106-650 µM vs. LEI-106-650 µM+2-AG: p<0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, n=9-12/conditions) suggesting that endothelial derived 2-AG plays a role in maintenance of endothelial cell structural integrity.
Using the same cells as morphology experiments, immunoreactivity of VE-cadherin and claudin 5 were assessed to determine the impact of LEI-106 exposure on endothelial cell junctional proteins. LEI-106 (1.3 mM) significantly reduced VE-cadherin corrected total cell fluorescence (CTCF); no significant differences were detected at the lower dose (650 µM) as compared to vehicle control (Figure 4E; LEI-106-650 µM vs. vehicle: p=0.9052, LEI-106-1.3 mM vs. vehicle: p=0.0233, as assessed by one-way ANOVA with Bartlett post-test, n=9-12/condition). The administration of 2-AG increased the VE-cadherin CTCF, mitigating the loss of VE-cadherin expression after LEI-106 treatment (LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.005, as assessed by one-way ANOVA with Tukey post-test, n=9-12/conditions). The blockade of DAGLα with LEI-106 at either 650 µM or 1.3mM did not induce significant changes in the CTCF of claudin-5 as compared to the vehicle control (Figure 4F) (LEI-106-650 µM vs. vehicle: p=0.8982, LEI-106-1.3 mM vs. vehicle: p=0.6161, as assessed by one-way ANOVA with Tukey post-test, n=9-12/condition). These data indicate that the concentrations of LEI-106 utilized in this study do not reduce endothelial cell viability and that the alterations observed following LEI-106 treatment reflect changes to endothelial morphology and expression of the adherens junctional protein, VE-cadherin.
Figure 4.
The inhibition of DAGLα induced morphological changes in bEnD.3 cells accompanied with reduced expression of VE-cadherin.
Figure 4.
The inhibition of DAGLα induced morphological changes in bEnD.3 cells accompanied with reduced expression of VE-cadherin.
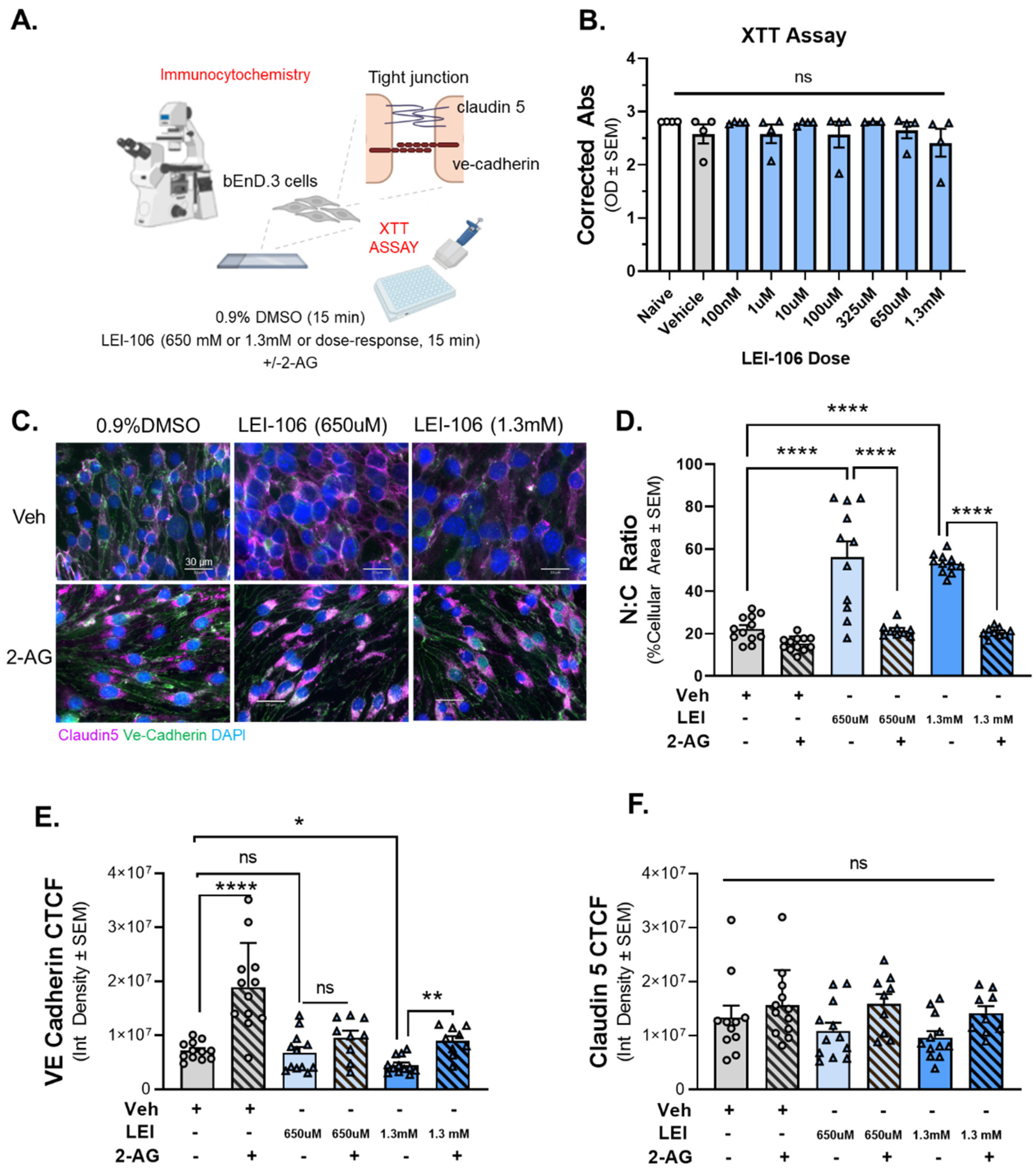
Cell viability assay on bEnD.3 cells was performed using increasing doses of LEI-106 (100 nM-1.3 mM). For ICC experiments, the bEnD.3 cells were treated with two doses of LEI-106 (650 uM and 1.3 mM) for 15 min, followed by 2-AG or vehicle application. ICC was performed to detect possible change in the expression of two tight junction protein, claudin 5 and VE-cadherin. (A) The panel represents the setting of experiments. (B) LEI-106 did not cause significant change in the cell viability at any doses applied in the study. (LEI-106 at any doses vs. vehicle: p>0.05, as assessed by one-way ANOVA with Bartlett post-test, F(8,26)=0.7738). All data are expressed as mean of optical density ± SEM (n=4 in each group). ns=non-significant. (C) Representative ICC images of bEnd.3 cells treated with LEI-106+/-2-AG application. (D) Significant changes in the nuclear: cytoplasmic ratio were observed following LEI-106 treatment, suggesting possible morphological alteration caused by 2-AG depletion (LEI-106-650 uM vs. vehicle: p<0.0001, LEI-106-1.3 mM vs. vehicle: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, F(8,85)=30.50). 2-AG treatment following LEI-106 application significantly reversed these effects (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p<0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, F(8,85)=30.50). All data are shown as the mean % of cellular area ± SEM (n=9-12/condition). ****p<0.0001, as compared to corresponding controls. (E) The higher dose of LEI-106 (1.3 mM) significantly reduced VE-cadherin CTCF, however no significant change was detected at the lower dose (650 uM) as compared to vehicle control (LEI-106-650 uM vs. vehicle: p=0.9052, LEI-106-1.3 mM vs. vehicle: p=0.0233, as assessed by one-way ANOVA with Bartlett post-test, F(2,33)=4.100). The administration of 2-AG increased the VE-cadherin CTCF, moderating the loss of VE-cadherin expression after LEI-106 treatment (LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.005, as assessed by one-way ANOVA with Tukey post-test, F(4,49)=5.568). All values are expressed as the mean of corrected total cell fluorescence (CTCF) ± SEM (n=9-12/condition). *p<0.05, **p<0.01, ****p<0.0001, compared to the corresponding controls. ns=non-significant. (F) Neither doses of LEI-106 treatment significantly influence the CTCF of claudin-5 as compared to vehicle control (LEI-106-650 uM vs. vehicle: p=0.8982, LEI-106-1.3 mM vs. vehicle: p=0.6161, as assessed by one-way ANOVA with Tukey post-test, F(5,59)=2.276). All values are expressed as the mean of corrected total cell fluorescence (CTCF) ± SEM (n=9-12/condition). ns=non-significant.
2.5. Endothelial Tight Junction Protein Detection is Modulated Following DAGLα Inhibition
The next series of experiments sought to validate the ICC obervations of VE-cadherin and claudin 5 immunofluorescence by performing Western blot analysis following LEI-106 treatment or gene editing (Figure 5A). Cell lysates harvested after LEI-106 (650µM, 1.3mM; 15min) were first probed for VE-cadherin. VE-cadherin was detected at 130 kDa in all treatment conditions. In LEI-106-treated cell lysates, a notable fragment at approximately 100kDa was also detected, indicating that VE-cadherin had undergone a fragmenting event following DAGLα inhibition. The treatment of LEI-106 at 650 uM dose did not cause significant change in the detection of VE-cadherin main 130kDa band as compared to vehicle control (Figure 5B) (vehicle vs. LEI-106-650 uM: p=0.3565 as assessed by one-way ANOVA with Bartlett`s test, n=8-12/group), but it significantly increased the detection of 100kDa fragment (vehicle vs. LEI-106-650 uM: p=0.0014 as assessed by one-way ANOVA with Bartlett`s test, n=8-12/condition). The higher dose of LEI-106 (1.3 mM) significantly reduced the detection of VE-cadherin main band and increased the detection of the fragmented one as compared to vehicle control (Figure 5B) (main band: vehicle vs. LEI-106-1.3 mM: p<0.0001 as assessed by one-way ANOVA with Bartlett`s test; fragment: vehicle vs. LEI-106-1.3 mM: p<0.0001 as assessed by one-way ANOVA with Bartlett`s test, n=8-12/group). Interestingly, the application of 2-AG further decreased the detection of main VE-cadherin band, but it did not significantly influence the fragmentation as compared to corresponding controls (Figure 5B) (main band: LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.0092, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.011 as assessed by one-way ANOVA with Bartlett`s test; fragment: LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.9665, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.0769 as assessed by one-way ANOVA with Bartlett`s test, n=8-12/condition). The expression of VE-cadherin was also assessed at 72 h after siRNA transfection in bEnD.3 cells. The transfection of DAGLα -specific siRNA significantly decreased the detection of VE-cadherin as compared to the non-targeting control (Figure 5C) (DAGLα siRNA vs. non-targeting control siRNA: p<0.0001, as assessed by unpaired t-test, n=8/group); this strategy did not lead to detection of the VE-cadherin fragment. These data, taken with the ICC data suggest regulation of endothelial derived 2-AG regulates the detection of VE-cadherin in bEnd.3 cells
Claudin 5 was detected in all cell lysates at 18kDa (Figure 5D). WB analysis of claudin-5 signal was significantly decreased after LEI-106 treatment at the higher dose (vehicle vs. LEI-106-650 uM: p=0.0759; vehicle vs. LEI-106-1.3 mM: p=0.0076 as assessed by one-way ANOVA with Tukey`s test, n=8-10/group). The application of 2-AG did not cause significant changes as compared to the corresponding controls (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.9870, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.6535 as assessed by one-way ANOVA with Tukey`s test, n=8-10/condition). No significant difference in the detection of claudin-5 was observed after the transfection of DAGLα -specific siRNA either (Figure 5E) (DAGLα siRNA vs. non-targeting control siRNA: p=0.9666, as assessed by unpaired t-test, n=6/group). Together with the ICC results, these data indicate that the pharmacological inhibition of DAGLα has a limited impact on detection of claudin 5 in bEnd.3 cells.
Figure 5.
The detection of VE-cadherin was altered after DAGLα blockade in bEnd.3 cells.
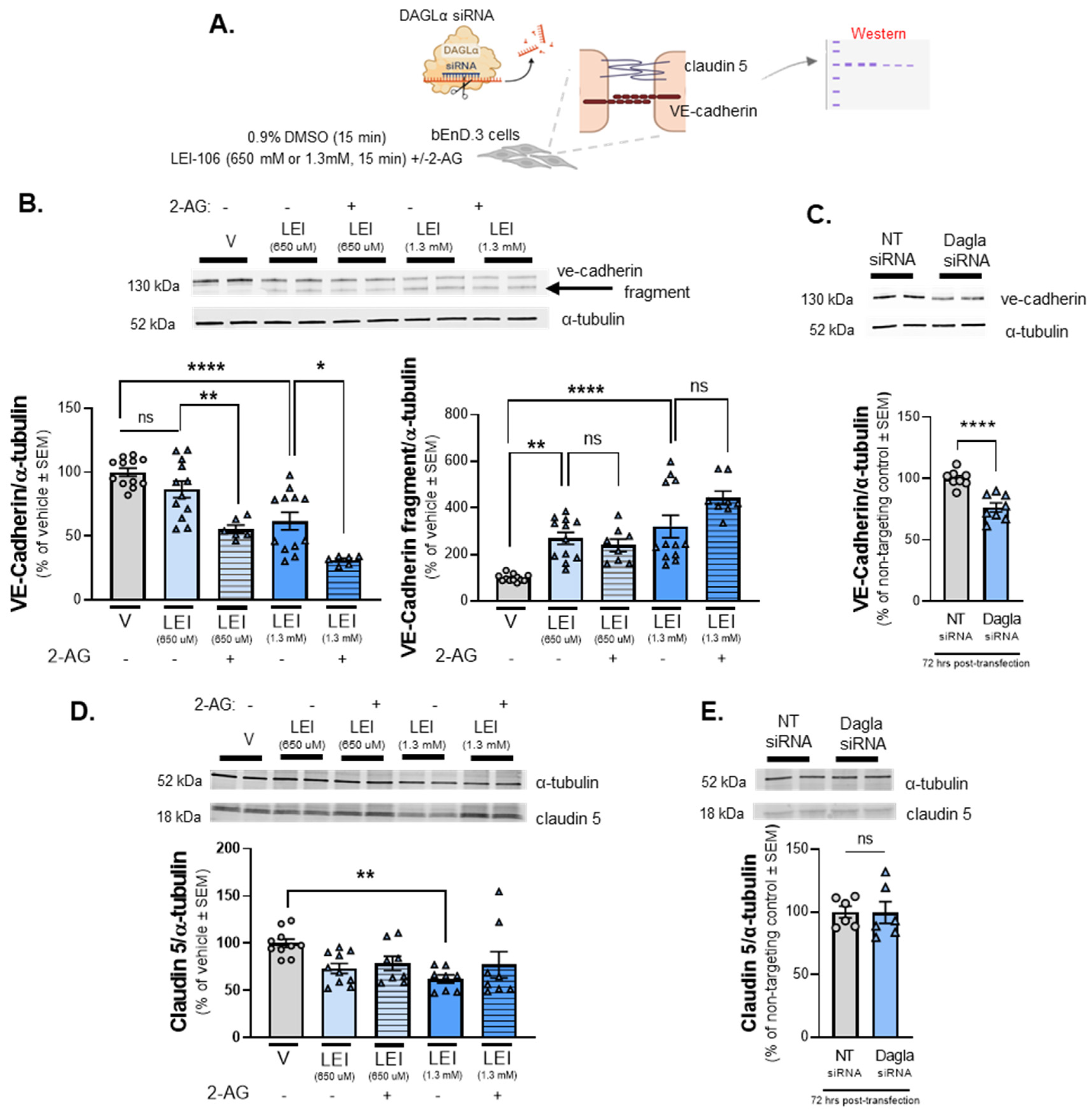
The bEnD.3 cells were treated with LEI-106 (650 uM or 1.3 mM) for 15 min with or without 2-AG (600 pmol) application. The cell lysate was subjected to Western to detect the expression of VE-cadherin and claudin 5. In the siRNA transfection experiment, samples were harvested 72 h post-transfection and subjected to Western to detect the expression of VE-cadherin and claudin 5. (A) Scheme of experimental outline. (B) Representative image showing the expression of VE-cadherin with α-tubulin used as loading control. The pharmacological blockade of DAGLα induced fragmentation of VE-cadherin. The treatment of LEI-106 at 650 uM dose did not cause significant change in the detection of VE-cadherin main band as compared to vehicle control (vehicle vs. LEI-106-650 uM: p=0.3565 as assessed by one-way ANOVA with Bartlett`s test, F(4,43)=19.62), but it significantly increased the detection of VE-cadherin fragment (vehicle vs. LEI-106-650 uM: p=0.0014 as assessed by one-way ANOVA with Bartlett`s test, F(4,47)=15.19). The higher dose of LEI-106 (1.3 mM) significantly reduced the detection of VE-cadherin main band and increased the detection of the fragmented one as compared to vehicle control (main band: vehicle vs. LEI-106-1.3 mM: p<0.0001 as assessed by one-way ANOVA with Bartlett`s test, F(4,43)=19.62; fragment: vehicle vs. LEI-106-1.3 mM: p<0.0001 as assessed by one-way ANOVA with Bartlett`s test, F(4,47)=15.19). The application of 2-AG further decreased the detection of main VE-cadherin band, but it did not significantly influence the fragmentation as compared to corresponding controls (main band: LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.0092, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.011 as assessed by one-way ANOVA with Bartlett`s test, F(4,43)=19.62; fragment: LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.9665, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.0769 as assessed by one-way ANOVA with Bartlett`s test, F(4,47)=15.19). All data presented as % of vehicle-treated ± SEM (n=8-12/condition). *p<0.05, **p<0.01, ****p<0.0001, compared to the corresponding controls. ns=non-significant. (C) Representative image showing the expression of VE-cadherin with α-tubulin as loading control in transfected cell samples. The transfection of DAGLα -specific siRNA significantly reduced the detection of VE-cadherin as compared to the non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p<0.0001, t(14)=5.376, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n=8/group). **** denotes significantly different (p<0.0001). (D) Representative image displaying the expression level of claudin 5. α-tubulin was used as loading control. The lower dose of LEI-106 (650 uM) did not significantly influence the detection of claudin 5 as compared to vehicle control (vehicle vs. LEI-106-650 uM: p=0.0759 as assessed by one-way ANOVA with Tukey`s test, F(4,39)=3.559). The treatment of LEI-106 at 1.3 mM dose significantly reduced the detection of of claudin 5 as compared to vehicle control (vehicle vs. LEI-106-1.3 mM: p=0.0076 as assessed by one-way ANOVA with Tukey`s test, F(4,39)=3.559). The application of 2-AG did not cause significant changes as compared to the corresponding controls (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p=0.9870, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p=0.6535 as assessed by one-way ANOVA with Tukey`s test, F(4,39)=3.559). All data presented as % of vehicle-treated ± SEM (n=8-10/condition). **p<0.01, compared to vehicle control. (E) Representative image of the expression of claudin 5 along with α-tubulin as loading control in transfected cell samples. No significant difference in the detection of claudin 5 was observed after the transfection of DAGLα -specific siRNA (DAGLα siRNA vs. non-targeting control siRNA: p=0.9666, t(10)=0.04292, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n=6/group). ns =non-significant.
2.6. DAGLα Inhibition Increases Cellular Calcium Detection with Engagement of Protein Kinase C (PKC) and Rho-Kinase Pathways
VE-cadherin is a calcium-dependent junctional protein, and prior reports indicate that VE-cadherin is fragmented by a calcium-dependent kinase, calpain [23]. The next experiments sought to determine whteher the fragment of VE-cadherin detected after LEI-106 treatment resulted from increasing intracellular calcium within bEnd.3 cells and calpain engagement. The first study used calcium imaging of bEnd.3 cells with the dye Fura-2, AM. Test cells underwent a 2-minute baseline observation in buffer, followed by 2-minute treatment with LEI-106 (650µM, 1.3mM), vehicle (0.9% DMSO), and a subsequent 2-minute washout phase. In a separate experiment, cells received buffer spiked with 2-AG (600pmol/coverslip) during the washout phase (Figure 6A). bEnd.3 cells treated with LEI-106 at both doses displayed significant increases in intracellular calcium as compared to vehicle control (Figure 6B) (LEI-106-650 uM vs. vehicle: p<0.0001, LEI-106-1.3 mM vs. vehicle: p<0.0001, as assessed by one-way ANOVA with Tukey post-test). Treatment with 2-AG spiked buffer following LEI-106 significantly decreased detection of intracellular calcium as compared to buffer alone (Figure 6C) (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p<0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by one-way ANOVA with Tukey post-test). To determine if this partial 2-AG effect was mediated by cannabinoid receptors (CB1R and CB2R), we repeated the experiment with the non-selective synthetic agonist, WIN-55,212. Application of WIN-55,212 after LEI-106 reduced the calcium signal as compared to LEI-106 alone (LEI-106-1.3 mM vs. LEI-106-1.3 mM+ WIN-55,212: p<0.0001, t(14.30)=856, as assessed by unpaired t-test). Together, these data indicate that LEI-106 treatment increases intracellular calcium concentration in a dose-dependent manner over time, which can be partially corrected with exogenous 2-AG supplementation and the non-selective CB1R/CB2R agonist WIN-55,212.
Having established that intracellular calcium levels are increased after treatement with LEI-106, we tested if the calcium-dependent kinase, calpain, played a role in the cleavage of VE-cadherin caused by DAGLα blockade. bEnD.3 cells were treated with LEI-106 in the combination with calpain inhibitor calpeptin (10µM, 30 min pretreatment). The calpain inhibitor did not significantly mitigate the fragmentation of VE-cadherin caused by LEI-106 treatment (Figure 6D) (LEI-106-650 uM vs. LEI-106-650 uM+calpeptin: p>0.9999, as assessed by one-way ANOVA with Tukey post-test, n=5-6/condition) suggesting that calpain did not play a role in the cleavage of VE-cadherin caused by DAGLα inhibition.
Previous work has shown as well that Ca-signalling along with activation of protein kinase C (PKC) has a critical role in mediating the disruption of VE-cadherin [24]. Therefore, the activity of PKC was determined in LEI-106 treated bEnD.3 cells. PKC activity, as assessed by ELISA, showed that LEI-106 treatment at both doses (15 min) significantly increased the PKC activity as compared to vehicle control (Figure 6E) (LEI-106-650 uM vs. vehicle: p=0.0289, t(8)=2.658; LEI-106-1.3 mM vs. vehicle: p=0.0099, as assesed by unpaired t-test, n=5/group). In order to test how PKC inhibitor can affect the fragmentation of VE-cadherin caused by LEI-106, bEnD.3 cells were pretreated with PKC inhibtor, G06938 (100 nM) 30 min before DAGLα inhibition. The PKC inhibitor did not cause significant changes in the fragmentation of VE-cadherin (Suppl Figure S1) (LEI-106-650 uM vs. LEI-106-650 uM+ G06938: p=0.9999, LEI-106-1.3 mM vs. LEI-106-1.3 mM+ G06938: p=0.9548, as assessed by one-way ANOVA with Tukey post-test, F(5,18)=21.88, n=4/condition).
To further investigate the possible signaling mechanism underlying how DAGLα inhibition resulted in VE-cadherin fragmentation, the pan-Rho-kinase inhibitor, SR3677 was tested in a combination with LEI-106. bEnD.3 cells were treated with SR3677 (100 nM) 30 min prior LEI-106 (650 uM), then subjected to Western-immunoblotting to probe for VE-cadherin. Rho-kinase inhibitor significantly mitigated the fragmentation of VE-cadherin caused by DAGLα inhibitor (Figure 6F) (LEI-106-650 uM vs. LEI-106-650 uM+SR3677: p<0.0001, as assessed by one-way ANOVA, n=8-10/condition). Together, these data suggest a role for increased intracellular Ca-signal, elevated PKC activity, and the engagement of Rho-kinase pathway in the effect of DAGLα inhibition on VE-cadherin.
Figure 6.
Increased intracellular calcium level and elevated PKC activity after DAGLα inhibition in bEnD.3 cells.
Figure 6.
Increased intracellular calcium level and elevated PKC activity after DAGLα inhibition in bEnD.3 cells.
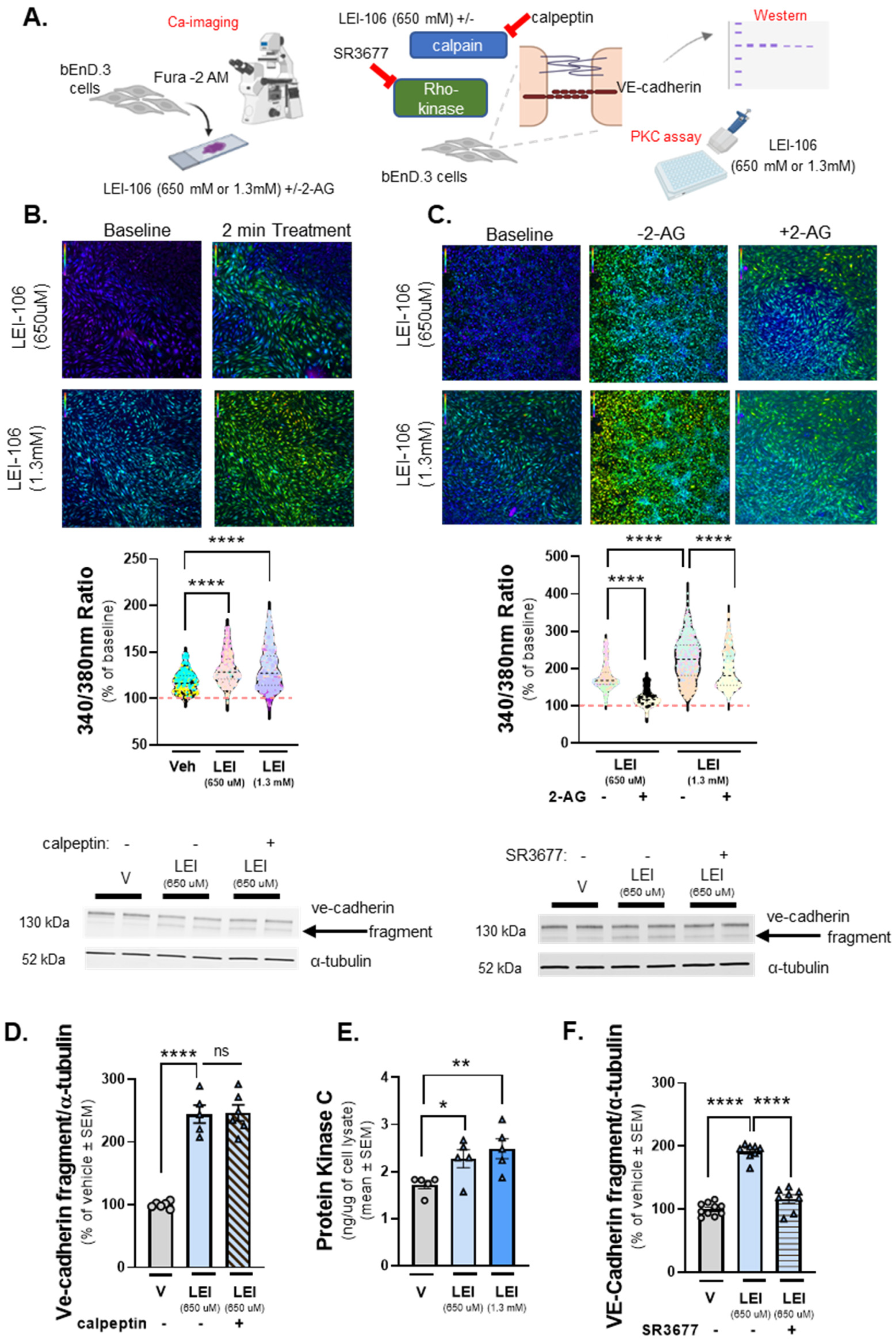
The bEnd.3 cells plated on collagen coated coverslips were subjected to calcium imaging with the dye Fura-2. After 2-minute baseline observation in buffer, cells were treated with LEI-106 (650µM or 1.3mM) or vehicle (0.9% DMSO) for 2 minutes, followed by a subsequent 2-minute washout phase. In a separate experiment, cells received buffer spiked with 2-AG (600pmol/coverslip) during the washout phase. Images used for analysis were captured at the beginning of the treatment application and after removal of the treatment, approximately 2 minutes apart. In a separate experiment, protein kinase C (PKC) activity was measured by ELISA in bEnD.3 cells after LEI-106 treatment. bEnD.3 cells were treated with calpain inhibitor, calpeptin (10µM) or Rho-kinase inhibitor, SR3677 (100 nM) 30 min prior the application of LEI-106 (650 µM), then subjected to Western blot to detect changes in the fragmentation of VE-cadherin caused by DAGLα inhibition. (A) Schema of experimental setting. (B) Representative calcium images of bEnD.3 cells at baseline, then treated with two different doses of LEI-106 (650 uM, 1.3 mM). LEI-106 at both doses significantly increased the intracellular calcium levels as compared to vehicle control (LEI-106-650 uM vs. vehicle: p<0.0001, LEI-106-1.3 mM vs. vehicle: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, F(3,3295)=482.1). All values are % of baseline obtained from three independent experiment using three coverslips in each. In each coverslip, 100 cells were analyzed by Image J software. ****p<0.0001 as compared to vehicle control. Red dashed line represents the baseline. (C) Representative images showing intracellular calcium levels in bEnD.3 cells treated with LEI-106 (650 uM or 1.3 mM) with or without 2-AG. The application of 2-AG significantly reduced the elevation of intracellular calcium caused by LEI-106 treatment (LEI-106-650 uM vs. LEI-106-650 uM+2-AG: p<0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM+2-AG: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, F(3,2700)=889.9). All data are shown as % of baseline obtained from three independent experiment using three coverslips in each. In each coverslip, 100 cells were analyzed by Image J software. ****p<0.0001 as compared to vehicle control. Red dashed line represents the baseline. (D) Representative image of Western blot showing VE-cadherin expression in bEnD.3 cells treated with LEI-106 with or without calpain inhibitor, calpeptin (10µM). α-tubulin was used as loading control. The calpain inhibitor did not significantly change the fragmentation of VE-cadherin caused by LEI-106 treatment (LEI-106-650 uM vs. LEI-106-650 uM+calpeptin: p>0.9999, as assessed by one-way ANOVA with Tukey post-test, F(4,20)=84.40). All data are shown as % of vehicle-treated ± SEM (n=5-6/condition). ****p<0.0001, ns=non-significant. (E) The assessment of PKC activity by ELISA showed that LEI-106 treatment (15 min) significantly increased the activity level of PKC as compared to vehicle control (LEI-106-650 uM vs. vehicle: p=0.0289, t(8)=2.658; LEI-106-1.3 mM vs. vehicle: p=0.0099, t(8)=3.362 as assessed by unpaired t-test). Data are presented as mean of PKC in ng/µg of cell lysate ± SEM (n=5/group). (F) Representative image of immunoblot targeting VE-cadherin and α-tubulin as loading control in bEnD.3 cells treated with LEI-106 (650 uM) with or without Rho-kinase inhibitor, SR3677 (100 nM). The pretreatment (30 min) with Rho-kinase inhibitor significantly mitigated the fragmentation of VE-cadherin caused by LEI-106 (LEI-106-650 uM vs. LEI-106-650 uM+SR3677: p<0.0001, as assessed by one-way ANOVA with Tukey post-test, F(2,23)=110.3). All data are shown as % of vehicle-treated ± SEM (n=8-10/condition). ****p<0.0001, compared to the LEI-106 (650 uM) treatment.
To further elucidate the effect of DAGLα inhibition in endothelial cells, an unbiased phosphoproteomics screen was performed on bEnd.3 cells treated with either vehicle or the DAGLα inhibitor LEI-106 (Figure 7A). Mass spectrometry analysis of the phosphopeptides purified from the bEnd.3 lysates detected a total of 24,505 phosphopeptide ions (Supplemental Tables S1–S5), of which 8,166 posessed significantly different abundances between the two groups (Figure 7B - VOLCANO). Unbiased principal component analysis (PCA) of the significantly affected phosphopeptide ions clustered the samples according to treatment, supporting the strength of the effect of LEI-106 inhibitor on changes in phosphorylation (Figure 7C - PCA). While within-group variance along component 2 accounted for only 7.7% of the variance, the LEI-106-treated samples did exhibit more pronounced phosphopeptide ion abundance heterogeneity as compared to the vehicle controls. Hierarchical protein clustering, heatmap visualization and protein cluster profiles of the 8,166 significantly affected phosphopeptide ions confirmed that samples within groups cluster together, supporting reproducibility of the findings (Figure 7D – HEAT MAP). Of the 8,166 significantly affected phosphopeptide ions, 5,589 (68.4%) exhibited a greater abundance after the LEI-106 treatment (Supplemental Table S3) while 2,577 phosphopeptide ions underwent a decrease as compared to vehicle treatment (Figure 7D – HEAT MAP). Of these, and related to the pharmacological experiment in Figure 6, ROCK 2 had sites (1121-SQLQALHIGMDSSSIGSGPGDAEPDDGFPESR-1152 that were significantly decreased by LEI106 (p<0.001, Supplemental Table S1). Gene Ontology enrichment analysis of the proteins displaying significant changes in phosphopeptide ion abundance after LEI-106 treatment was performed for biological processes, cell compartment, and molecular function; Reactome pathways were also assessed (Figure 7 E-H). Proteins displaying a significant loss in phosphopeptide ion abundance after LEI-106 treatment revealed gene ontology enrichment for cytoskeletal organization, activation of GTPase activity including RHO isoform cycling, actin binding, focal adhesions, and VEGFA-VEGFR2 pathways. LEI-106 significantly increased phosphorylation of proteins involved in mRNA processing and binding as well as pathways regulating mRNA splicing, RUNX1 mediated gene regulation, and estrogen dependent gene expression. Proteomics data also suggets changes of ZO-1 after DAGLα inhibition. In order to confirm that finding, Western experiment was peformed to detect ZO-1 expression after LEI-106 treatment. The blockade of DAGLα significantly reduced the detection of ZO1 as compared to vehicle control (Suppl Figure S2) (vehicle vs. LEI-106-650 uM: p=0.034, vehicle vs. LEI-106-1.3 mM: p=0.0004, as assessed by one-way ANOVA with Tukey post-test, n=3-4/condition). Together, these data suggest that disruption of 2-AG homeostasis by DAGLα inhibition significantly alters the phospho-proteome of bEnd.3 cells.
3. Discussion
While it is accepted that (endo)cannabinoids can enhance the integrity of the blood-brain barrier (BBB) in pathologic states, the exact mechanisms by which this occurs are not yet fully understood [21]. The above studies aimed to gain insight into the role of DAGLα, the 2-AG synthetic enzyme, and endothelial-derived 2-AG in BBB function using a bEnd.3cell monolayer model. Using both pharmacological inhibition and siRNA manipulation to decrease DAGLα function, endothelial-derived 2-AG was decreased. This loss of 2-AG compromised bEnd.3 monolayer paracellular integrity by increasing intracellular calcium signaling, activity of protein kinase C, and fragmenting VE-cadherin. The reduced functional expression of VE-cadherin was dependent on Rho-kinase but not calpain. Thus, constitutive production of 2-AG in brain endothelium maintains BBB homeostasis by regulating junctional integrity through phosphorylation and not protein internalization. Figure 8 summarizes all our findings.
The inhibition of DAGLα either with LEI-106 or siRNA lead to loss of 2-AG and induced paracellular leak manifesting by increased C14-surose transport through the monolayer of bEnd.3 cells. Disruption of 2-AG homeostasis was connected to increased intracellular calcium signaling, enhanced activity of protein kinase C (PKC), and fragmentation of tight junction protein, VE-cadherin. Inhibition of Rho-kinase mitigated the fragmentation of VE-cadherin induced by the blockade of DAGLα.
Evidence has increased in the last decade of decreased endocannabinoid tone in neuropathologies including dementias, traumatic brain injury, and migraine [15,25,26,27]. These neuropathologies also disrupt BBB integrity and methods to increase endocannabinoid tone have shown promise as therapeutic interventions, suggesting a role for the endocannabinoid system in BBB homeostasis [2,15,18,19,20,26,27,28]. The current data using selective DAGLα inhibition with LEI-106 and siRNA targeting of DAGLα showed that significant decreases in endothelial derived 2-AG levels, but not AEA or DAG, in endothelial cell lysates impaired endothelial cell monolayer integrity as indicated by reduced TEER and increased uptake of 14C sucrose across the monolayer. These data are consistent with prior in vivo evidence of 2-AG mitigating BBB impairment, further supporting the role of 2-AG tone in endothelial cells in BBB homeostasis [18,19,20].
Breaches of BBB integrity during CNS disorders and diseases have been attributed to endothelial cell death [29,30,31], changes in morphology (e.g., stress fiber formation) and junctional protein instability [32,33]. Following LEI-106, cell viability was not changed, suggesting that acute depletion of 2-AG did not facilitate cell death. However, ICC showed cell morphology shifted away from a confluent monolayer and towards isolated cell clusters 15 min after LEI-106. Co-treatment of LEI-106 with exogenous 2-AG prevented these morphological shifts suggesting that endothelial cells require tonic 2-AG tone to maintain shape and functional integrity.
In addition to cytoskeletal remodeling, instability of junctional proteins is attributed to multiple mechanisms including protein cleavage and internalization as well as post-translational modification [34,35,36]. Of note, both cytoskeletal remodeling and protein cleavage have calcium-dependent and -independent mechanisms. Our data support a role for endothelial derived 2-AG in regulating intracellular Ca2+ levels that could be reversed by 2-AG application. In both ICC and Western blot analysis of bEnd.3 cell lysates, detection of the TJ protein, VE-cadherin was decreased with increased fragmentation after pharmacological blockade of DAGLα. Reduced detection of claudin 5 was observed after LEI-106 treatment in Western experiments. Detection of VE-cadherin was significantly decreased in cells transfected with DAGLα-targeting siRNA, however neither the fragmentation of VE-cadherin, nor the reduced expression of claudin-5 were observed after genetic inhibition of DAGLα, suggestive of differing regulation of junctional proteins with loss of protein vs pharmacological inhibition. The time-line difference between pharmacological (15min) and siRNA manipulation (72h) can also provide a possible rationale, since the pharmacological blockade can induce rapid changes, whereas the mRNA silencing does not immediately impact protein synthesis; the detection difference in VE-cadherin main band and fragment after LEI-106 but not DAGLα silencing exemplifies this idea. With respect to claudin-5, the effect of LEI-106 might reflect an off-target effect since no changes were observes when the siRNA was introduced. The observed reduction in the detection of ZO-1 after LEI-106 can also contribute to the permeability change showed in functional assays, but further experiments need to be conducted to answer how the changes in ZO-1 and VE-cadherin after DAGLα inhibition are related to each other. Interestingly, exogenous 2-AG application was not able to diminish the effect of DAGLα inhibition on junctional protein detection, suggesting these changes in protein function were calcium independent. Thus, endothelial derived 2-AG plays a role in BBB tone in both Ca2+-dependent and -independent manners.
VE-Cadherin plays a critical role in maintaining cell-cell adhesions at the BBB, and complete loss of functional VE-cadherin is lethal [37]. In pathological states, multiple mechanisms are linked to regulation of VE-cadherin functional expression including fragmentation and internalization via calpain. Our results indicate increased Ca2+ influx in bEnD.3 cells after pharmacological inhibition of DAGLα, but not the involvement of calpain in the effect of LEI-106 on VE-cadherin. A recent review paper discusses the role of calcium in barrier integrity, highlighting that calcium influx triggers adherens junction disassembly and facilitates endothelial permeability [38]. However, this review paper focuses on the changes of barrier integrity during inflammation, another review article written by Brown and Davis also describes that calcium can regulate functional expression of the tight junction proteins on cerebral microvessels during stroke [39]. Based on our data, the loss of 2-AG via DAGLα inhibition did facilitate calcium influx in bEnD.3 cells, indicating a homeostatic role of 2-AG in these processes.
A study published by Sandoval et al showed that calcium signaling along with PKC activity has a critical role in mediating the disruption of VE-cadherin in human umbilical vein endothelial cells [24]. Therefore, to further explore the possible mechanism underlying the effect of DAGLα, we tested the involvement of protein kinase C (PKC)-related pathways. The activity of PKC in bEnD.3 cells was significantly increased after LEI-106 treatment, however the pretreatment with PKC inhibitor, G06938 did not prevent the fragmentation of VE-cadherin after DAGLα blockade. In contrast, the application of ROCK inhibitor, SR3677 mitigated the fragmentation of VE-cadherin caused by LEI-106. This data aligns well with former work published by Li et al [40]. Their results showed that ROCK pathway inhibitor, Y27632 increased VE-cadherin expression in human umbilical vein endothelial cells. The proteomics data also suggests the alteration of Rho-pathways, including Rho GTPase-activating proteins (Rho-GAPs) and Rho guanine nucleotide exchange factors (Rho-GEFs). GAPs and GEFs are highly expressed in endothelium, and they play a role in endothelial cell adhesion and vascular homeostasis [41]. Complete understanding of the connection between 2-AG signaling and Rho-pathways in endothelial cells requires future experiments.
Limitations: Multiple limitations are present within this study. While it was demonstrated that the in vitro dosing of LEI-106 did not decrease cell viability, the doses used in this study, chosen from Levine et al., 2021 [25], were intended to represent maximal bioavailability and likely do not reflect the true delivery of LEI-106 to the endothelial cells of the NVU. Future work is necessary to examine lower doses of LEI-106 on cell cultures. The mechanism by which LEI-106 increases intracellular calcium remains elusive as well. The reliance of Western blot and ICC on antibody binding limits the results of these studies as well. Finally, the in vitro culture of endothelial cells is not able to completely mimic the complexity of BBB in vivo; therefore it is necessary to evaluate the molecular observations presented in this study, including levels of calcium, VE-cadherin, and claudin 5 following in vivo administration of LEI-106.
4. Materials and Methods
Drugs
LEI-106, WIN-55,212, calpeptin, G06983, SR3677, 2-AG, 2-AG-d5, and AEA-d4 were purchased from Cayman Chemicals (Ann Arbor, MI). Cells received LEI-106 (650µM, 1.3mM) dissolved in 0.9% DMSO in cell media 15 minutes prior to analysis. The doses of LEI-106 used for cell assays were derived as equimolar doses to previous in vivo assays assuming 50% and 100% bioavailability. Titrated doses of LEI-106 in 0.9% DMSO (100nM, 10µM, 100µM, 325µM, 650µM, 1.3mM, 10mM) were utilized to assess for cell viability. 2-AG was dissolved in 0.9% DMSO in cell media and was administered to cells in 600 pmol dose. Calpeptin (10µM), G06983 (100nM), and SR3677 (100nM) were applied 30 min before LEI-106 treatment.
Cell Culture
Murine brain endothelial cells, bEnd.3 (CRL-2299, ATCC) were cultured in DMEM (Gibco, 11995–065), supplemented with 2 mM L-glutamine (ThermoScientific, 25030081), 10% fetal bovine serum (Gibco, 10082139), and penicillin (100 UI/mL)-streptomycin (100 μg/mL) (Invitrogen, 15140122). Cells were cultured at 37°C in a humidified 5% CO2/95% air atmosphere and were grown to 80% confluence in all experiments. 24 h before treatment, the media of bEnd.3 cells were changed to astrocyte-conditioned media (ACM) harvested from confluent C8-D1A flasks. The C8-D1A cells (CRL-2541, ATCC) were cultured in DMEM (Gibco, 11995–065), supplemented with 10% fetal bovine serum, and penicillin-streptomycin. To mimic CSD event in vitro, bEnd.3 cells were treated with 100 mM KCl for 5 minutes, which is a typical condition to evoke potassium-triggered spreading depolarization in live brain slices [42].
Liquid Chromatography-Mass Spectrometry (LC-MS)
Cellular samples for LC-MS were purified by organic solvent extraction with a protocol modified from Wilkerson et al [43]. 2x106 cells were seeded into 10cm2 culture dishes and allowed to grow for 7 days. On the day of processing, cells were treated and then collected via cell scraping and placed into microcentrifuge tubes. Samples were then centrifuged (1000 x g, for 10 min at 4°C) and the supernatant was removed. The resulting pellet was then weighed and homogenized in 1 ml of chloroform/methanol (2:1 v/v) supplemented with phenylmethylsulfonyl fluoride (PMSF) at 1 mM final concentration to inhibit the degradation by endogenous enzymes. Homogenates were then mixed with 0.3 ml of 0.7% w/v NaCl, vortexed, and then centrifuged for 10 min at 3,200 × g at 4°C. The aqueous phase plus debris were collected and extracted two more times with 0.8 ml of chloroform. The organic phases from the three extractions were pooled and internal standard was added to each sample. Mixed internal standard solutions were prepared by serial dilution of AEA-d4 and 2-AG-d5 in 80% acetonitrile. The organic solvents were evaporated under nitrogen gas. 6 uL of 30% glycerol in methanol per sample was added before evaporation. Dried samples were reconstituted with 0.2 ml of chloroform and mixed with 1 ml of ice-cold acetone to precipitate proteins. The mixtures were then centrifuged for 5 min at 1,800 × g at 4°C. The organic layer of each sample was collected and evaporated under nitrogen.
Analysis of 2-AG and AEA was performed on an Ultivo triple quadrupole mass spectrometer combined with a 1290 Infinity II UPLC system (Agilent, Palo Alto, CA). The instrument was operated in electrospray positive mode with a gas temperature of 150°C at a flow of 5 L/min, nebulizer at 15 psi, capillary voltage of 4,500 V, sheath gas at 400°C with a flow of 12 L/min and nozzle voltage of 300 V. Transitions monitored were 348.3 → 287.3 and 62, 352.3 → 287.4 and 65.9, 379.3 → 287.2 and 269.2, and 384.3 → 287.2 and 296.1 for AEA, AEA-d4, 2-AG, and 2-AG-d5. The first fragment listed was used for quantification and the second fragment was used for confirmation. The first 3 min of analysis time was diverted to waste. Chromatographic separation was achieved using an isocratic system of 21% 1 mM ammonium fluoride and 79% methanol on an Acquity UPLC BEH C-18 1.7u 2.1 × 100 mm column (Waters, Milford, MA) maintained at 60°C. After each injection the column was washed with 90% methanol for 1 min then re-equilibrated for 5 min prior to the next injection. Samples were maintained at 4°C. Mixed calibration solutions were prepared by serial dilution of AEA and 2-AG stock solutions in 80% acetonitrile. Calibration curves were prepared for each analysis by adding 10 µL internal standard solution to 20 µL standard solution. Prior to sample analysis, 200 µL of 80:20 acetonitrile:water was added to dried samples which were then vortexed and sonicated. The samples were centrifuged at 15,800 × g at 4°C for 5 min. Supernatant was transferred to autosampler vials and 5 µL was injected for analysis.
siRNA transfection of bEnD.3 cells to silence DAGLα gene
bEnd.3 cells were seeded onto 6-well plates at 200,000 cells/well density and cultured in regular media for 24 h. On the day of experiment, the cells were incubated in FBS and antibiotic free DMEM for 2h before the transfection. Double-stranded, siRNA targeting DAGLα was purchased from ThermoScientific (Silencer® Select Pre-designed siRNA, #s114215). The sequences targeting DAGLα are the following: CCAAGUACCUCGACCUCAAtt (sense) and UUGAGGUCGAGGUACUUGGtg (antisense). Non-targeting control siRNA (ThermoScientific, #4390843) was applied as negative control. Lipofectamine RNAiMAX (ThermoScientific, #13778) was used as transfection reagent. siRNAs were diluted in Opti-MEM for 50 pmol/well as final concentration. The cells were incubated with siRNA-Lipofectamine mixture in Opti-MEM for 4h, then media was changed to the regular one. Subsequent experiments, including Western blotting, TEER, sucrose assays, and LC-MS were performed 72 h post-transfection. For TEER and sucrose assays, the transfected cells were cultured on trans-well inserts from 24 h post-transfection. The transfection efficacy was validated by Western blotting probing for DAGLα (antibody: Cell Signaling #13626S) and α-tubulin as loading control. In a separate experiment, the linear relationship between different amount of loaded protein and signal of DAGLα antibody was measured using serial dilution of naïve bEnD.3 cell lysate (data not shown).
Trans-Endothelial Electrical Resistance (TEER)
To capture possible breaches in the active and passive transport, TEER experiments were conducted as described in Liktor-Busa et al. [44]. Briefly, bEnd.3 cells were seeded on collagen coated trans-well inserts (Corning) at 6.0 × 104 cells/cm2 density. The regular bEnd.3 media was used on the luminal side, and astro-conditioned media (ACM) was added to the abluminal side to facilitate barrier functions. ACM was collected as described above. LEI-106 (650µM or 1.3mM, 0.9% DMSO in Media) with or without 2-AG (600 pmol) was applied to the luminal side of the trans-well insert. KCl pulse (100 mM KCl in media) was added to the abluminal side. Baseline measurements were taken before any treatment, then right after treatment (0 time-point), then 10, 20, 30 min, 1, 2, 3, and 24 h post- treatment via 2-electrode, chopstick method (EVOM2). All measurements were repeated in triplicate over three individual experiments at temperature-controlled environment (at 37°C).
In Vitro 14C-Sucrose Transport
bEnd.3 cells (CRL-2299, ATCC) were cultured on collagen coated trans-well membranes as described in the former section and in our former paper [44]. LEI-106 (1.3mM, 0.9% DMSO in Media) was added to the luminal side of the membrane 15 minutes prior to analysis. Following pretreatment, luminal media was replaced with fresh bEnd.3 media containing 14C-Sucrose (0.25uCi/mL). Abluminal media was collected at 5 min and 30 min post treatment and analyzed for disintegrations per minute (dpm) using a model 1450 liquid scintillation counter (PerkinElmer). In a separate experiment, following pretreatment, 2-AG (600pmol) was supplemented into the abluminal media to simulate increased eCB signaling. Experiments were carried out in triplicate, with 4 trans-well inserts/group.
Immunocytochemistry (ICC)
bEnd.3 cells were grown on collagenated glass coverslips to confluence and treated as described above in “Materials and Methods: Drugs”. Cells were then washed with phosphate buffer saline (PBS) twice before fixing them with 1% Paraformaldehyde (PFA) for 15-minutes followed by a 1 h incubation in 10% BSA as a blocking agent. Following PBS washes, the cells were incubated in primary antibodies (Rabbit anti-VE-Cadherin, Cell Signaling 2500S; Mouse anti-Claudin 5, Invitrogen 35-2500) overnight in a 4°C room. After washing with PBS, the cells were incubated in secondary antibodies (Goat anti-Rabbit AlexaFluor 488, Invitrogen A-11008; Donkey anti-Mouse AlexaFluor 647, Invitrogen A-31571) at a 1:500 dilution in blocking buffer for 1 hour at room temperature. The coverslips were mounted after additional PBS washes using Prolong Gold Antifade with DAPI (ThermoScientific, P36941) mounting media and were then dried and sealed. The images of the slides were captured with an ECHO Revolve Microscope (ECHO, San Diego, CA) at 40X magnification. 4 slides were generated per condition with 3 images obtained for each slide. The corrected total cell fluorescence (CTCF) was calculated as the Integrated Density – (Area of selected cell X Mean fluorescence of background readings) for 5 cell ROIs/image and 5 background readings/image. All values determined in FIJI ImageJ as previously described [45].
Western Immunoblotting
bEnd.3 cells were cultured in 6-well plates in regular media. 24 h before experiment, media was changed to ACM, as described above. On the day of experiment, cells were treated with LEI-106 (650µM or 1.3 mM) for 15 min, then washed with ice-cold PBS. In a separate experiment, cells underwent DAGLα blockade via siRNA transfection, described above. The cells were lysed in ice cold lysis buffer (20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 2 mM MgCl2 hexahydrate, 1% (vol/vol) NP40, 0.5% (w/vol) sodium deoxycholate, 0.1% (w/vol) SDS supplemented with protease and phosphatase inhibitor cocktail (Halt Protease and Phosphatase inhibitor cocktail, ThermoScientific, #78441). The lysate was centrifuged at 12,000 x g for 10 min at 4°C, then the supernatant was collected. The protein content of supernatant was determined by BCA assay. 25 ug of total protein was loaded into TGX precast gels (4-20% CriterionTM, BioRad) and transferred to nitrocellulose membrane (AmershamTM ProtranTM, GE Healthcare). After transfer, the membrane was blocked at room temperature for 30 min in blocking buffer (5% dry milk in Tris-buffered saline (TBS)). The following primary antibodies were used: NPAS4 (ThermoScientific, MA5-27592, 1:500), Ve-cadherin (ThermoScientific, 36-1900, 1:500), claudin-5 (ThermoScientific, 35-2500, 1:500), and α-tubulin (Cell Signaling, 3873S, 1:10,000). The primary antibodies were diluted in 5% BSA in TBST (Tris-buffered saline with Tween 20). The membrane was incubated in diluted primary antibodies for 48 hours at 4˚C. The membrane was then washed three times in TBST for 5 minutes each followed by incubation with IRDye® 800CW Donkey anti-Rabbit IgG Secondary Antibody (Li-Cor, 926-32213) and IRDye® 680RD Donkey anti-Mouse IgG Secondary Antibody (Li-Cor, 926-68072) in 5% milk in TBST for 1 h rocking at room temperature. The membrane was washed again two times for five minutes each in TBST. TBS buffer was used for the last washing step. The membrane was imaged with an Azure Sapphire laser imager (Azure Biosystems). Un-Scan-It 6.1 software (Silk Scientific Inc.) was used for quantification.
Calcium Imaging
Calcium imaging was performed as previously described [46]. bEnd.3 cells grown on coverslips were incubated at 37°C with 3 μM Fura-2AM (Cat#F-1221; Life technologies, stock solution prepared at 1 mM in DMSO, 0.02% pluronic acid, Cat#P-3000MP; Life technologies) for 30 minutes (Kd = 25 μM, λex 340, 380 nm/λemi 512 nm) to follow changes in intracellular calcium in DMEM (Gibco, 11995–065), supplemented with 2 mM L-glutamine (ThermoScientific, 25030081), 10% fetal bovine serum (Gibco, 10082139), and penicillin (100 UI/mL)-streptomycin (100 μg/mL) (Invitrogen, 15140122). All calcium imaging experiments were done at room temperature (∼23°C). Treatments were performed following 2 min baseline with 1X Phosphate Buffer Solution containing 2mM Calcium Chloride. Cells were treated with Vehicle (0.9% DMSO in 1XPBS), KCl (100mM), or LEI-106 (650µM, 1.3mM, 2min or 15 min) followed by 2 min washout with phosphate buffer with and without 2-AG (600pmol/cover slip). In a separate experiment, cells underwent DAGLα blockade via siRNA transfection, described below. Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse Ti-U (Nikon Instruments Inc, Melville, NY), using objective Nikon Super Fluor MTB FLUOR ×10 0.50 and a photometrics cooled CCD camera CoolSNAP ES2 (Roper Scientific, Planegg, Germany) controlled by NIS Elements software (version 4.20; Nikon instruments). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters (340 ± 5 and 380 ± 7 nm) were controlled by a Lambda 10 to 2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505-nm dichroic mirror at 535 ± 25 nm. To minimize photobleaching and phototoxicity, the images were taken every 10 seconds during the time-course of the experiment, using the minimal exposure time that provided acceptable image quality. The changes in calcium were monitored by following the ratio of F340/F380, calculated after subtracting the background from both channels. The region of interest (ROI) tool was used to define 150 cells per cover slip to measure calcium signaling in each individual cell, where one ROI covers the cell body of the whole cell. To verify that the cells did not shift during solution changes, the time lapse was checked to ensure that the correct cells were being measured. The time measurement software (NIS Software ND Acquisition) was utilized to collect time-lapse ratio measurements for each ROI in each image. These measurements were exported to Excel, in which baseline, treatment average, and percent change from baseline calculations were done. The baseline was taken as the average of the F340/F380 from zero to two minutes when the phosphate buffer was applied. The treatment average calculations were done as the average of the F340/F380 during each respective treatment application period. The percent response was calculated with the treatment average over the baseline multiplied by 100 to calculate the calcium response change for each treatment.
XTT Viability Assay
XTT Viability Assay was performed using CyQUANT™ XTT Cell Viability Assay kit (ThermoScientific, #X12223). Briefly, bEnD.3 cells were culture in 96-well plate, at 1x104 cells/well seeding density. On the day of experiment, cells were treated with different concentrations of LEI-106 (100nM, 1μM, 10μM, 100μM, 325μM, 625μM, and 1.3 mM) along with vehicle control for 15 min, followed by XTT assay performed according to manufacturer`s instructions.
DAG ELISA
DAG ELISA kit (Aviva System Biology, OKEH02607) was used according to manufacturer’s instruction. bEnD.3 cells were treated with LEI-106 (1.3 mM) or vehicle for 15 min followed by a wash step before harvest by cell scraping. The cell pellet was lysed, then centrifuged (5,000g, 10 min, 4°C). The supernatant was applied in the immunoassay.
PKC Kinase Activity Assay
PKC Kinase Activity Assay (ab139437) was used according to manufacturer`s instruction. bEnd.3 cells were treated with LEI-106 or vehicle for 15 min, as described above. The cells were lysed in lysis buffer, then centrifuged (13,000rpm, 15 min, 4°C). The supernatant was subjected to the assay.
Phosphoproteomics
Mouse bEnd.3 endothelial cells cultured on 150 mm plates were treated with vehicle (0.9% DMSO in media) or LEI-106 (650 μM) for 15 min, then washed with ice-cold PBS. The cells were lysed in ice cold lysis buffer (20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 2 mM MgCl2 hexahydrate, 1% (vol/vol) NP40, 0.5% (w/vol) sodium deoxycholate, 0.1% (w/vol) SDS) supplemented with protease and phosphatase inhibitor cocktail (Halt Protease and Phosphatase inhibitor cocktail, ThermoScientific, #78441). The lysate was incubated at 4°C with continuous rocking for 30 min. After centrifuged at 12,000 x g for 10 min at 4°C, the supernatant was collected. Protein concentration was determined using a BCA protein quantitation assay (ThermoScientific). To determine global differences in protein phosphorylation abundance between treatments, 5 mg of protein lysate per sample (n=4) was subjected to in-solution tryptic digestion and phosphopeptide enrichment using sequential enrichment from metal oxide affinity chromatography per manufacturer’s protocol (Thermo Scientific, San Jose, CA) similar to as previously described [47]. HPLC-ESI-MS/MS was performed in positive ion mode on a Thermo Scientific Orbitrap Fusion Lumos tribrid mass spectrometer fitted with an EASY-Spray Source (Thermo Scientific) as previously described [48].
Spectra were acquired using XCalibur, version 2.3 (ThermoFisher Scientific). Progenesis QI for proteomics software (version 2.4, Nonlinear Dynamics Ltd., Newcastle upon Tyne, UK) was used to perform ion-intensity based label-free quantification, as previously described [49,50]. In brief, .raw files were imported and converted into two-dimensional maps (y-axis = time, x-axis =m/z) followed by selection of a reference run for alignment purposes. An aggregate data set containing all peak information from all samples was created from the aligned runs, which was then further narrowed down by selecting only +2, +3, and +4 charged ions for further analysis. The samples were then grouped by treatment. A peak list of fragment ion spectra was exported in Mascot generic file (.mgf) format and searched against the mouse SwissProt database using Mascot (Matrix Science, London, UK; version 2.6). The search variables that were used were: 10 ppm mass tolerance for precursor ion masses and 0.5 Da for product ion masses; digestion with trypsin; a maximum of two missed tryptic cleavages; variable modifications of oxidation of methionine and phosphorylation of serine, threonine, and tyrosine; 13C=1. The resulting Mascot .xml file was then imported into Progenesis, allowing for peptide/protein assignment, while peptides with a Mascot Ion Score of <25 were not considered for further analysis. Precursor ion-abundance values for peptide ions were normalized to all proteins.
Statistical Analysis
GraphPad Prism 7.0 and 8.3.1 software (GraphPad Software) were used for statistical analysis. To determine numbers needed for each experiment G.Power3.1 was used for 80% power to detect a 20% difference when alpha = 0.05. The data were expressed as mean ± S.E.M, unless otherwise stated. Groups were compared by unpaired t-test or one-way ANOVA with Tukey’s post-test, as indicated. Differences were considered significant if p ≤ 0.05.
5. Conclusions
Clinically, increased permeability of the BBB is associated with worsened prognosis, increased risk of CNS infection, and advanced neuronal loss in neurodegenerative diseases [4]. It is therefore imperative to understand the mechanisms by which decreased endocannabinoid tone plays a role in compromising BBB integrity to facilitate novel therapeutic development Given the above findings, therapies targeting 2-AG hydrolysis or rho kinases have the potential to meet this need in addition to their ability to treat headache-like pain [15].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.A.L., E.L.B. and T.M.L.; methodology, A.A.L., E.L.B. and T.M.L..; formal analysis, A.A.L., E.L.B., S.B., P.R.L., and T.M.L.; data curation, A.A.L., E.L.B., S.M.P., S.B., A.M.B., S.A.C. and A.A.L; writing—original draft preparation, A.A.L. and E.L.B.; writing—review and editing, A.A.L., E.L.B., P.R.L. and T.M.L.; supervision, T.M.L.; project administration, E.L.B. and T.M.L; funding acquisition, T.M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (R01NS099292 and R01NS126752-01A1, TML) and Arizona Biomedical Research Commission (ABRC45952, TML), with monies from the Department of Pharmacology at the University of Arizona, the M.D.-Ph.D. Program at the University of Arizona, the University of Arizona Comprehensive Pain and Addiction Center, and the NIDA Center for Excellence in Addiction Studies (P30DA051355). Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA023074. Authors are solely responsible for the content which does not necessarily represent the official views of the National Institutes of Health, the State of Arizona, or the University of Arizona.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
University of Arizona Cancer Center Analytical Chemistry Shared Resources is acknowledged for the assistance of LC-MS service. The Ibrahim Lab at the University of Arizona is acknowledged for their assistance in Ca2+ Imaging.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Edvinsson, L.; Tfelt-Hansen, P. The blood-brain barrier in migraine treatment. Cephalalgia 2008, 28, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Reinhold, A.K.; Rittner, H.L. Barrier function in the peripheral and central nervous system-a review. Pflugers Arch 2017, 469, 123–34. [Google Scholar] [CrossRef]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front Neurosci 2020, 14, 334. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.L.; Brooks, T.A.; Davis, T.P. Chronic inflammatory pain and the neurovascular unit: a central role for glia in maintaining BBB integrity? Curr Pharm Des 2008, 14, 1625–1643. [Google Scholar] [CrossRef]
- Hamilton, N.B.; Attwell, D.; Hall, C. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics 2010, 2, 5. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Dos Santos, M.F.; Holanda-Afonso, R.C.; Lima, R.L.; DaSilva, A.F.; Moura-Neto, V. The role of the blood-brain barrier in the development and treatment of migraine and other pain disorders. Front Cell Neurosci 2014, 8, 302. [Google Scholar] [CrossRef]
- Ahn, K.; McKinney, M. K.; Cravatt, B. F. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev 2008, 108, 1687–1707. [Google Scholar] [CrossRef]
- Fezza, F.; Mastrangelo, N.; Maccarrone, M. Assay of NAPE-PLD activity. Methods Mol Biol 2016, 1412, 123–130. [Google Scholar] [CrossRef]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef]
- Russo, E.B. Clinical endocannabinoid deficiency reconsidered: current research supports the theory in migraine, fibromyalgia, irritable bowel, and other treatment-resistant syndromes. Cannabis Cannabinoid Res 2016, 1, 154–165. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Zanaboni, A. M.; Piomelli, D.; Tassorelli, C. Endocannabinoid system and migraine pain: an update. Front Neurosci 2018, 12, 172. [Google Scholar] [CrossRef] [PubMed]
- Liktor-Busa E; Levine A.A.; Palomino S.M.; et al. ABHD6 and MAGL control 2-AG levels in the PAG and allodynia in a CSD-induced periorbital model of headache. Front Pain Res. 2023, 4:1171188. [CrossRef]
- Moreno E.; Cavic M.; Canela EI. Functional Fine-Tuning of Metabolic Pathways by the Endocannabinoid System-Implications for Health and Disease. Int J Mol Sci. 2021, (7):3661. [CrossRef]
- Acharya N.; Penukonda S.; Shcheglova T.; et al. Endocannabinoid system acts as a regulator of immune homeostasis in the gut. Proc Natl Acad Sci USA. 2017, 114(19):5005-5010. [CrossRef]
- Panikashvili, D.; Shein, N.A.; Mechoulam, R.; Trembovler, V.; Kohen, R.; Alexandrovich, A.; Shohami, E. The endocannabinoid 2-AG protects the blood–brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis 2006, 22, 257–264. [Google Scholar] [CrossRef]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid Degradation Inhibition Improves Neurobehavioral Function, Blood–Brain Barrier Integrity, and Neuroinflammation following Mild Traumatic Brain Injury. J Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Xu J.; Zheng G.; Hu J.; Ge W.; Bradley J.L.; Ornato J.P.; Tang W. The monoacylglycerol lipase inhibitor, JZL184, has comparable effects to therapeutic hypothermia, attenuating global cerebral injury in a rat model of cardiac arrest. Biomed Pharmacother. 2022, 156:113847. [CrossRef]
- Hagan K.; Varelas P.; Zheng H. Endocannabinoid System of the Blood-Brain Barrier: Current Understandings and Therapeutic Potentials. Cannabis Cannabinoid Res. 2022, 7(5):561-568. [CrossRef]
- Cottier, K.E.; Galloway, E.A.; Calabrese, E.C.; Tome, M.E.; Liktor-Busa, E.; John Kim, J.; et al. Loss of Blood-Brain Barrier Integrity in a KCl-Induced Model of Episodic Headache Enhances CNS Drug Delivery. eNeuro 2018, 5, ENEURO.0116-18.2018. [Google Scholar] [CrossRef] [PubMed]
- Su W.; Kowalczyk A.P. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis. Mol Biol Cell. 2017, 28(1):76-84. [CrossRef]
- Sandoval R.; Malik A.B.; Minshall R.D.; Kouklis P.; Ellis C.A.; Tiruppathi C. Ca(2+) signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol. 2001 533(Pt 2):433-45. [CrossRef]
- Levine, A.; Liktor-Busa, E.; Karlage, K.L.; Giancotti, L.; Salvemini, D.; Vanderah, T.W.; Largent-Milnes, T.M. DAGLα Inhibition as a Non-invasive and Translational Model of Episodic Headache. Front Pharmacol 2021, 11, 615028. [Google Scholar] [CrossRef]
- Lins B.R.; Anyaegbu C.C.; Hellewell S.C.; Papini M.; et al. Cannabinoids in traumatic brain injury and related neuropathologies: preclinical and clinical research on endogenous, plant-derived, and synthetic compounds. J Neuroinflammation. 2023, 20(1):77. [CrossRef]
- Walther S.; Halpern M. Cannabinoids and Dementia: A Review of Clinical and Preclinical Data. Pharmaceuticals (Basel). 2010, 3(8):2689-2708. [CrossRef]
- Fried, N.T.; Maxwell, C.R.; Elliott, M.B.; Oshinsky, M.L. Region-specific disruption of the blood-brain barrier following repeated inflammatory dural stimulation in a rat model of chronic trigeminal allodynia. Cephalalgia 2018, 38, 674–689. [Google Scholar] [CrossRef]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis 2019, 10, 487. [Google Scholar] [CrossRef]
- Kim, K.A.; Kim, D.; Kim, J.H.; Shin, Y.J.; Kim, E.S.; Akram, M.; et al. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS 2020, 17, 21. [Google Scholar] [CrossRef]
- Fang, J.; Yuan, Q.; Du, Z.; Fei, M.; Zhang, Q.; Yang, L.; et al. Ferroptosis in brain microvascular endothelial cells mediates blood-brain barrier disruption after traumatic brain injury. Biochem Biophys Res Commun 2022, 619, 34–41. [Google Scholar] [CrossRef]
- Matsuoka, R.L.; Buck, L.D.; Vajrala, K.P.; Quick, R.E.; Card, O.A. Historical and current perspectives on blood endothelial cell heterogeneity in the brain. Cell Mol Life Sci 2022, 79, 372. [Google Scholar] [CrossRef]
- Manaenko, A.; Yang, P.; Nowrangi, D.; Budbazar, E.; Hartman, R.E.; Obenaus, A.; et al. Inhibition of stress fiber formation preserves blood-brain barrier after intracerebral hemorrhage in mice. J Cereb Blood Flow Metab 2018, 38, 87–102. [Google Scholar] [CrossRef]
- Huber, J.D.; Witt, K.A.; Hom, S.; Egleton, R.D.; Mark, K.S.; Davis, T.P. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol 2001, 280, H1241-8. [Google Scholar] [CrossRef]
- Witt, K.A.; Mark, K.S.; Hom, S.; Davis, T.P. Effects of hypoxia-reoxygenation on rat blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol 2003, 285, H2820-31. [Google Scholar] [CrossRef]
- Wacker, B.K.; Freie, A.B.; Perfater, J.L.; Gidday, J.M. Junctional protein regulation by sphingosine kinase 2 contributes to blood-brain barrier protection in hypoxic preconditioning-induced cerebral ischemic tolerance. J Cereb Blood Flow Metab 2012, 32, 1014–1023. [Google Scholar] [CrossRef]
- Li, W.; Chen, Z.; Chin, I.; Chen, Z.; Dai, H. The Role of VE-cadherin in Blood-brain Barrier Integrity Under Central Nervous System Pathological Conditions. Curr Neuropharmacol 2018, 16, 1375–1384. [Google Scholar] [CrossRef]
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial Cell Calcium Signaling during Barrier Function and Inflammation. Am J Pathol 2020, 190, 535–542. [Google Scholar] [CrossRef]
- Brown, R.C.; Davis, T.P. Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke 2002, 33, 1706–1711. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Sun, R.; Gao, M.; Wang, H. Cadmium exposure enhances VE-cadherin expression in endothelial cells via suppression of ROCK signaling. Exp Ther Med 2022, 23, 355. [Google Scholar] [CrossRef] [PubMed]
- van Buul, J.D.; Geerts, D.; Huveneers, S. Rho GAPs and GEFs: controling switches in endothelial cell adhesion. Cell Adh Migr 2014, 8, 108–24. [Google Scholar] [CrossRef]
- Andrew, R.D.; Hsieh, Y.T.; Brisson, C.D. Spreading depolarization triggered by elevated potassium is weak or absent in the rodent lower brain. J Cereb Blood Flow Metab 2017, 37, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, J.L.; Niphakis, M.J.; Grim, T.W.; Mustafa, M.A.; et al. The Selective Monoacylglycerol Lipase Inhibitor MJN110 Produces Opioid-Sparing Effects in a Mouse Neuropathic Pain Model. J Pharmacol Exp Ther. 2016, 357, 145–56. [Google Scholar] [CrossRef] [PubMed]
- Liktor-Busa, E.; Blawn, K.T.; Kellohen, K.L.; Wiese, B.M.; et al. Functional NHE1 expression is critical to blood brain barrier integrity and sumatriptan blood to brain uptake. PLoS One 2020, 15, e0227463. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.; Magalhães, P.; Ram, S. J. ; Image Processing with ImageJ. Biophotonics Int 2003, 11, 36–42. [Google Scholar]
- Moutal, A.; Chew, L.A.; Yang, X.; Wang, Y.; Yeon, S.K.; et al. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain 2016, 157, 1448–1463. [Google Scholar] [CrossRef] [PubMed]
- Keresztes, A.; Olson, K.; Nguyen, P.; Lopez-Pier, A.M.; Hecksel, R.; et al. Antagonism of the mu-delta opioid receptor heterodimer enhances opioid antinociception by activating Src and calcium/calmodulin-dependent protein kinase II signaling. Pain 2022, 163, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.S.; Krantz, J.; Kwak, E-A.; Barker, N.K.; Deer, C.G.; et al. Insulin Induces Microtubule Stabilization and Regulates the Microtubule Plus-end Tracking Protein Network in Adipocytes. Mol Cell Proteomics 2019, 18, 1363-1381. [CrossRef]
- Blawn, K.T.; Kellohen, K.L.; Galloway, E.A.; Wahl, J.; Vivek, A.; et al. Sex hormones regulate NHE1 functional expression and brain endothelial proteome to control paracellular integrity of the blood endothelial barrier. Brain Res 2021, 1763, 147448. [Google Scholar] [CrossRef]
- Levine, A.; Liktor-Busa, E.; Lipinski, A.A.; Couture, S.; Balasubramanian, S.; et al. Sex differences in the expression of the endocannabinoid system within V1M cortex and PAG of Sprague Dawley rats. Biol Sex Differ 2021, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol Biol 2018, 1711, 133–148. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009, 4, 44–57. [Google Scholar] [CrossRef]
Figure 7.
Quantitative Phospho-proteomics after DAGLα inhibition in bEnD.3 cells. bEnd.3 cells cultured on 150 mm plates were treated with vehicle (0.9% DMSO in media) or LEI-106 (650 μM) for 15 min, then subjected to quantitative phospho-proteomics. (A) Schema of the experiment. (B) Volcano plot of bEnd.3 of detected phospho-peptides showing a greater than 2-fold change after LEI-106 as compared to vehicle treatment. (C) Principle Component Analysis (PCA) of cells treated with vehicle or LEI106 (650 µM). (D) Heatmap of phospho-peptides increased by LEI-106 treatment as compared to vehicle or decreased as compared to vehicle. (E) GO Biological process functional analysis showing top 10 processes altered in each condition. (F) GO cellular compartment functional analysis showing top 10 compartments in which phospho-peptides were impacted each condition. (G) GO molecular function functional analysis showing top 10 in which phospho-peptides were impacted each condition. (H) Top 10 Reactome Pathways associated with changes in phospho-peptides in each outcome. n=4 biological replicates per condition.
Figure 7.
Quantitative Phospho-proteomics after DAGLα inhibition in bEnD.3 cells. bEnd.3 cells cultured on 150 mm plates were treated with vehicle (0.9% DMSO in media) or LEI-106 (650 μM) for 15 min, then subjected to quantitative phospho-proteomics. (A) Schema of the experiment. (B) Volcano plot of bEnd.3 of detected phospho-peptides showing a greater than 2-fold change after LEI-106 as compared to vehicle treatment. (C) Principle Component Analysis (PCA) of cells treated with vehicle or LEI106 (650 µM). (D) Heatmap of phospho-peptides increased by LEI-106 treatment as compared to vehicle or decreased as compared to vehicle. (E) GO Biological process functional analysis showing top 10 processes altered in each condition. (F) GO cellular compartment functional analysis showing top 10 compartments in which phospho-peptides were impacted each condition. (G) GO molecular function functional analysis showing top 10 in which phospho-peptides were impacted each condition. (H) Top 10 Reactome Pathways associated with changes in phospho-peptides in each outcome. n=4 biological replicates per condition.
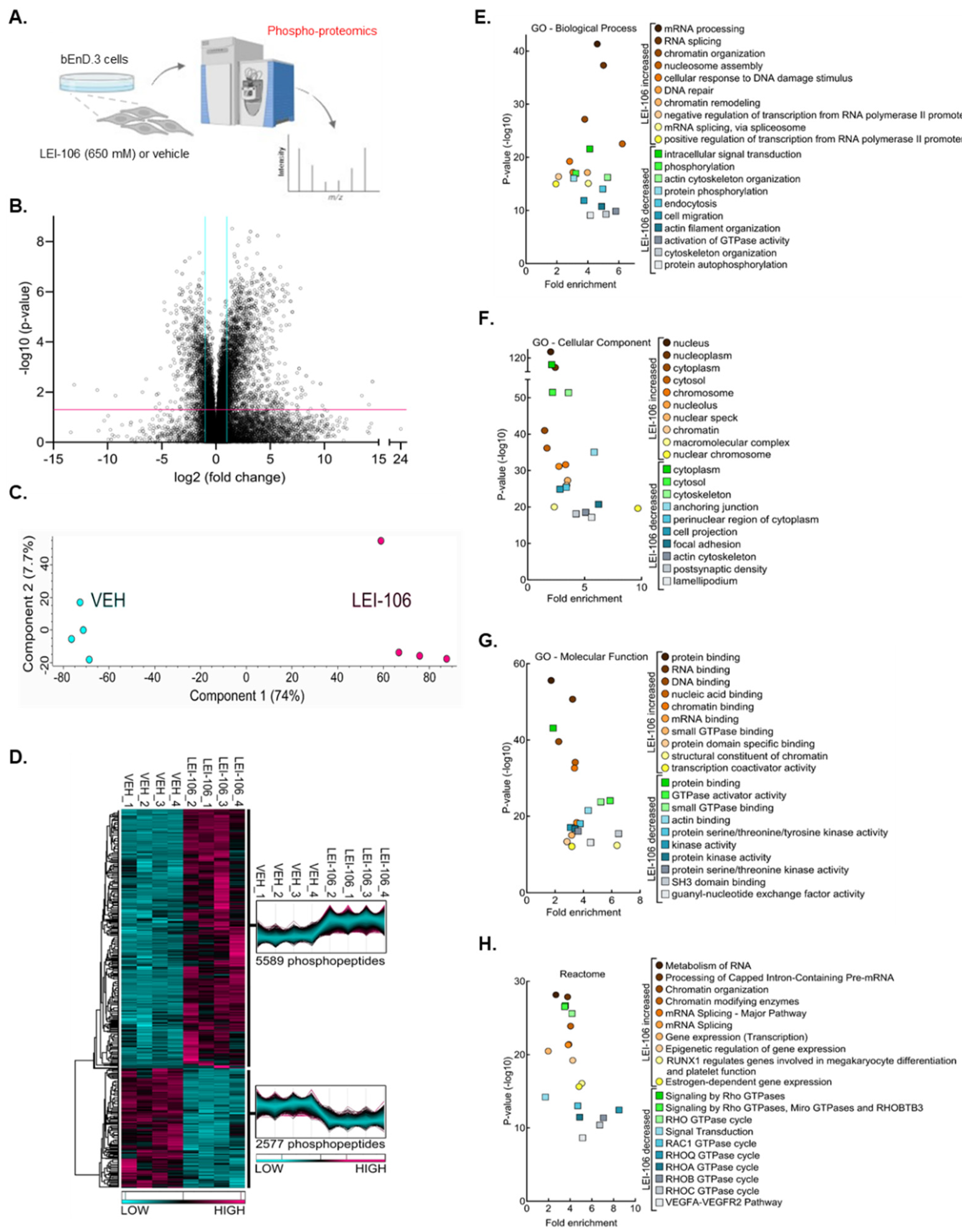
Figure 8.
2-AG depletion via inhibition of DAGLα disrupted integrity of bEnd.3 endothelial cells.
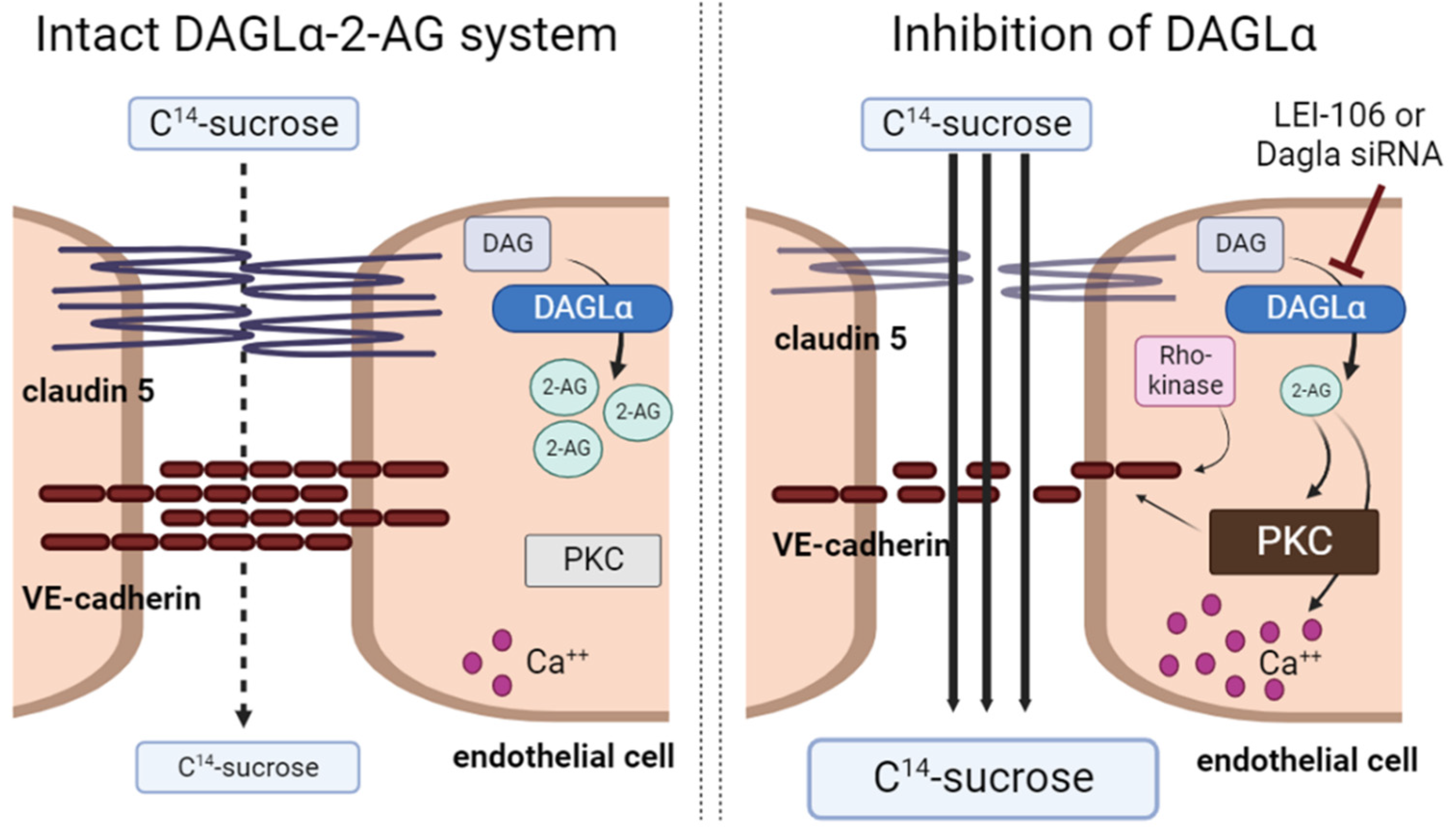
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



