Submitted:
02 December 2023
Posted:
04 December 2023
You are already at the latest version
Abstract
We used ab initio molecular dynamics simulations to compare the activation of 2-propanol on the low index Co3O4 (111), (110) and (001) surfaces in dry conditions. The thermal and surface assisted decomposition of a film of 2-propanol to 2-propoxide on the B-termination of each surface was monitored and analyzed. The investigations suggest an activity order of Co3O4 (111) > (110) > (001). On the B-terminated (111) surface, full dissociation of all 2-propanol molecules at the interface is observed, accompanied by a Mars-van Krevelen-type mechanism upon hydroxylation of the surface. On the (110) surface, 2-propanol dissociation is driven by temperature, which activates the two-fold coordinatively unsaturated surface oxygens. The (001) surface on which almost no dissociation occurs is the least active. No formation of acetone is observed in the simulations conditions.
Keywords:
activation
; active sites
; oxidation
; surfaces
; temperature
; structure to activity
1. Introduction
Selective oxidation of primary alcohols plays a crucial role in environmentally friendly synthesis of organic oxygenated compounds. While gas phase oxidation processes have been studied to a respectable level, the understanding of these processes in liquid phase, on which rely most industrial processes, is still at its enfancy. Addressing the complexity of liquid phase catalytic processes in operando conditions requires the development of robust and systematic approaches in order to better manipulate the different factors governing the catalysts efficiency1. These include the nature of reaction medium, structure of the catalysts, oxidant selection, temperature or light.
Transition metal oxides (TMOs) are promising candidates for heterogeneous catalysis due to their abundance, low cost and good catalytic activity. Among the TMOs, Co3O4 catalysts have demonstrated potential application in energy production and environmentally friendly technological processes. For example, they have shown remarkable performance in methane combustion2, water oxidation3, CO oxidation4, or the selective oxidation of hydrocarbons5. Partial oxidation of propanol for the production of acetone has recently captured particular attention and has triggered studies on the selective oxidation of 2-propanol on Co3O4 spinel catalysts as they show beneficial electronic, magnetic and redox properties.6,7
Anke et al.8 observed high activity and selectivity on bulk Co3O4 catalysts during gas phase selective oxidation of 2-propanol. The highest conversion rates of 2-propanol to acetone were observed in between 373 K and 573 K, with a maximum conversion rate close to 100% achieved at 430 K. A similar finding was reported in Ref.9, where complete conversion and 100% selectivity for 2-propanol oxidation to acetone was observed at 430 K on Co3O4 nanospheres exposing the (110) surface orientation. In another study10, the impact of Co concentration on 2-propanol oxidation on Co1+xFe2-xO4 spinel oxides was investigated, both in gas and liquid phases. The study revealed that iron-free Co3O4 samples have the highest activity as the catalytic activity increased with with Co content. Moreover, XPS analysis showed that Co3+ sites were more active than Co2+ sites as also found by complementary density functional theory (DFT) calculations performed on the (100) and (101) surfaces8,9.
For a better understanding of the factors governing the catalytic performance of Co3O4 spinel nanocatalysts with respect to 2-propanol oxidation to acetone, more theoretical insights need to be provided. We recently used ab initio molecular dynamics (AIMD) simulations to study the role of temperature, surface structure and electrochemical environment on the oxidation process11. In a following study, we combined static DFT+U calculations and theoretical XANES to investigate the electronic properties of the active sites during 2-propanol oxidation to acetone on the B-termination of the Co3O4 (001) surface in dry and humid conditions12. Recently, we studied the interaction of 2-propanol with Co3O4 (001) surface by vibrational sum frequency spectroscopy and AIMD simulations, both near ambient conditions13. This allowed insight at the molecular level, into the pathways of catalytic oxidation. Particularly the multifaceted role of water at the interface for the electrochemical oxidation of 2-propanol in aqueous solution was elucidated to a good extend.
The above-mentioned studies shed light on key catalytic properties such as the local environment of active sites, the binding mechanisms, coordination environment, spin, and oxidation states of active sites , the influence of aqueous solvation and the elemental composition of the surface as well as the vapour pressure of alcohol on the reactivity. However, these studies were restricted to the (001) surfaces. For a complete picture of the interplay between the parameters that governs the catalytic performance of Co3O4 spinel nanocatalysts, a comprehensive study of the (111) surfaces, highly exposed on naturally grown nanoparticles as well as the (101) exposed on nanospheres and found to be reactive to 2-propanol oxidation needs to be performed.
In this study, we use AIMD simulations to investigate the interaction of a film of 2-propanol with the low index Co3O4 (111) and (110) surfaces. As a first step towards the partial oxidation and formation of acetone, we investigate the activation of 2-propanol and decomposition to 2-propoxide in dry conditions. We monitor the surfaces reactivity towards 2-propanol adsorption at catalytically relevant temperatures and compare with the previously studied (001) surface. To shed some light on the structure-activity relationship, we analyze the structural response of the surfaces to adsorption and provide fundamental insight into the underlying surface assisted mechanisms that promote 2-propanol activation.
2. Computational details
We considered orthorombic simulation cells, with (2x2) periodicity in the lateral directions (x,y) corresponding to 19.81 Å × 11.44 Å and 22.88 Å × 16.18 Å, in which Co3O4 (111) and (101) surfaces interact with a film of eight 2-propanol molecules, respectively. The catalytically active B-terminations14,15,16 of the surfaces were considered. The respective surfaces were modelled by slabs consisting of 13 and 8 atomic layers and the bottom 5 and 3 layers of the slab were fixed at their bulk positions, respectively. Because of the one-sided adsorption of the film of molecules and the asymmetry of the slab, a dipole correction was introduced in the z-direction of the slab. A vacuum region of 30 Å was also introduced in the same direction to decouple the system from its periodic images.
Spin-polarized Born-Oppenheimer molecular dynamics simulations were conducted using the CP2K/Quickstep package17. The Generalized Gradient Approximation (GGA) within the PBE formulation18 was used to treat the exchange and correlation effects, plus a U correction of the Hubbard type19 to accurately describe the Co 3d states. The value of U = 2 eV for both Co2+ and Co3+ was used as in revious studies.20,21,22,23 The Grimme D3 correction24 was considered to account for dispersion interactions.
Core electrons were described using Goedecker-Teter-Hutter (GTH) pseudopotentials. The O 2s and 2p electrons and Co 3s, 3p, 3d, and 4s electrons were considered as valence electrons. A mixed representation of plane wave functions with an energy cutoff of 500 Ry and a double-ζ quality local basis set with a single set of polarization functions (DZVP) were used as basis sets. The same type of pseudopotentials (GTH) and polarization functions (DZVP) are also usede for carbon and hydrogen atoms. All simulations were conducted at the Γ point.
NVT conditions were imposed on the simulated systems using a Nosé-Hoover thermostat with a time constant of 1 ps and target temperatures of 300 K and 450 K. A simulation time step of 0.5 fs was used to propagate the atomic positions. Following an equilibration period of at least 1 ps for all systems under study, a production phase of approximately 20 ps was considered to compute and anayze the properties of interest.
3. Results and discussions
3.1. Co3O4 (111) surface
We performed AIMD simulations of eight 2-propanol molecules interacting with the B-terminated (111) surface of a pristine Co3O4 at 300 K , for 20 ps of total simulation time. Figure 1 shows snapshots of the initial and final equilibrium trajectories. All eight 2-propanol molecules dissociate on the Co3+ sites on which they sit all along the simulation. The perspective views in Figure 1 provide a clear evidence of the surface hydroxylation as a result of eight proton transfers to the surface lattice oxygens. No further deshydrogenation of 2-propanol leading to the formation of acetone was observed during the whole simulation period.
We monitored the changes into the 2-propanol molecules intramolecular bond lengths, O–H, C–H, C–O, and the Co3+–O bond length for a dissociatively adsorbed 2-propanol molecule during the 20 ps of simulation as displayed in Figure 2. it is seen that the O–H bond cleavage happens after approximately 2.5 ps of simulation and leads to a shortening of the Co3+–O bond length. Additionally, a relatively small shortening of the C–O bond is observed, while the C–H bond remains relatively unchanged throughout the simulation. This supports that no dehydrogenation (C–H bond cleavage) has occurred.
The B-terminated (111) surface exposes Co3+–O2-propoxide–Co2+ bridges between the adsorption sites and the adsorbates as illustrated by the by black lines in Figure 3. The formation of these bridges originates from the unique characteristics of the surface’s tetrahedrally coordinated Co2+ ions. In fact, unlike their counterparts that are coordinatively saturated on the B-terminated (001) surface,13 these ions are undercoordinated. Their three-fold coordinatively unsaturated nature dispose them to participate to 2-propanol adsorption and decomposition.
On the B-terminated (001) surface, only the topmost layer and, to a lesser extent, the first inner layer were involved into 2-propanol activation11,12. In the case of B-terminated (111) surface, 2-propanol activation is assisted by inner surface layers as illustrated by the charge density difference (CDD) calculated for the final equilibrium trajectory (Figure 4). The Co2+ ion involved in the Co3+–O2-propoxide–Co2+ bridge (Figure 3) and encircled with a dashed black lign in Figure 4 contributes significantly to 2-propanol decomposition as it losses electron density upon 2-propanol adsorption. Moreover, Co3+ ions located in the third inner layer of the slab(orange rectangle), is also electronically involved, as it can be seen by the charge reorganization in the vicinity of these sites.
However, in the adsorbate film, the interaction between the H in CH3 groups and the surface oxygens is less pronounced than on the B-terminated (001) surface. No electron density depletion or accumulation is observed in the hydrogens of the CH3 groups (black rectangles). This stems from the fact that proton transfer to the surface leads to the hydroxylation of all coordinatively unsaturated surface oxygens (Figure 1), while on the B-terminated (001) surface most of such surface oxygens remain intact.13
Interestingly, for a surface pre-exposed to water, with subsequent formation of surface OH groups, a Mars-van Krevelen mechanism happens leading to the formation of water from lattice oxygen. This is illustrated in Figure 5. A lattice OH group receives an hydrogen from 2-propanol, resulting into the formation of a water molecule, 2-propoxide and an oxygen vacancy ( black circle).
3.2. Co3O4 (110) surface
For the B-terminated pristine Co3O4(110), the adsorption and decomposition of eight 2-propanol molecules were addressed at 300 K and 450 K during a simulation time of 20 ps. Snapshots of the initial (left) and final (right)of equilibrium trajectories at 300 K are displayed in Figure 6 . Six out of the eight 2-propanol molecules adsorb molecularly ontop Co3+ sites and the two remaining molecules dissociate. The single deprotonation of these two molecules leads the formation of two 2-propoxide molecules. Like on (001) and (111) surfaces, the Co3+ adsorption sites remain unchanged throughout the simulation, as it can be seen from the top views in Figure 6. Also, no partial oxidation of 2-propanol and subsequent formation of acetone is observed.
Figure 7 shows the impact of temperature on the reactivity of 2-propanol the (110) surface. 2-propanol activation increases considerably when the temperature is raised to 450 K. Six 2-propanol molecules dissociate on the surface, resulting in the formation of six surface OH groups. For deeper insights into this temperature driven reactivity, the nature of the lattice oxygen species has been analyzed. They have a lower coordination number and are therefore highly reactive compared to the surface lattice oxygens on the (111) and (001) and A- and B-terminations. The former are 2-fold coordinated while the latter are either three-fold coordinatively unsaturated or four-fold coordinatively saturated. This is supported by Figure 7 which shows that all protons are transferred to the “two-fold” coordinatively unsaturated oxygens (in violet). This temperature driven reactivity (110) surface has been also confirmed experimentally9.
3.3. Structure to activity relationship
Table 1 summarizes the activity, with respect to 2-propanol convertion to 2-propoxide, of all three surfaces at two different temperatures, 300 K and 450 K. The (111) surface has the highest activity at 300 K or 450K. The lowest activity is observed on the (001) surface at 450 K where no molecule dissociate13. The activity of the (110) surface lies in between that of the (111) and the (001) surfaces, both at 300 K and 450 K. Temperature has the strongest impact on the (110) surface by increasing the activity by overall 60 percent. No formation of acetone is observed on any of the surfaces.
Figure 8 shows the structural response of surface atoms during the 20 ps of simulation of the interaction of the B-terminated (001), (111) and (110) surfaces with 2-propanol at 300 K. On the left, the starting surface atom configurations (t = 0) are shown and their trajectories traces are displayed on the right. The (001) surface is found to be the least dynamic compared to the other two surfaces, whereas the (111) surface is the most dynamic as supported by the highest mobility of the Co3+ ions. This is in line with the pronounced surface relaxations and the formation of Co3+–O2-propoxide–Co2+ bridges as mentioned above. The most dynamic atoms of the (110) surface are the two-fold unsaturated surface oxygen atoms identified as those activating the 2-propanol dissociation on the surface. The magnitude of relaxation of atoms on this surface lies in between that of the atoms on the (001) and (111) surfaces.
Recently, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)24 was used to obtain the Vibrational Density of States (power spectra) of 2-propanol adsorbed on various transition metal oxides including Co3O4. However some peaks in the spectra could not be ascribed to specific bands. To shed light on this, we calculated power spectra of adsorbed 2-propanol on B-terminated (001), (111), and (110) surfaces (Figure 9). The main difference between the three spectra is the region between 2800-3100 cm-1. The peaks around 3050 cm-1 and 3100 cm-1 can be assigned to the symmetric and asymmetric stretching modes of CH3, respectively, and the small band at 2850 cm-1 to the C–H bonds. Additionally, the difference in the 1100-1300 cm-1 region can be attributed to the O–H bending mode. There is no C–O bending mode, contributing to the bands around 1700 cm-1 and indicative of acetone formation. Similarly the O–H stretching mode (peak around 3600-3700 cm-1) does not appear, which supports the involvement of the respective H into H-bonding or transfer to the surface.
4. Conclusion
Our investigations showed that the (111) surface exhibits higher surface reactivity towards 2-propanol activation compared to the (110) and (001) surfaces. Complete dissociation of 2-propanol to 2-propoxide is observed on the (111) surface at 300 K and 450 K. The Co3+ are the adosrption sites. However, the neighbouring coordinatively unsaturated three-fold Co2+ also participate to the reaction via the formation of Co3+–O2-propoxide–Co2+ bridges. Upon hydroxylation of the surface at room temperature, 2-propanol activation is accompagnied by a Mars-van Krevelen mechanism which yields the formation of 2-propoxide and molecular water. On the (101) surface, the temperature activates the surface two-fold coordinatively unsaturated oxygens as illustrated by the improvement in the activity by 60% when temperature is increased to 450 K. The (001) surface studied in previous works is the least active and the activity order of magnitude reads (111) > (110) > (001).
References
- S. Najafishirtari, K. Friedel Ortega, M. Douthwaite, S. Pattisson, G. J. Hutchings, C. J. Bondue, K. Tschulik, D. Waffel, B. Peng, M. Deitermann, et al. A perspective on heterogeneous catalysts for the selective oxidation of alcohols. Chemistry–A European Journal, 27(68):16809–16833, 2021. [CrossRef]
- L. Hu, Q. Peng, and Y. Li. Selective synthesis of Co3 O4 nanocrystal with different shape and crystal plane effect on catalytic property for methane combustion. Journal of the American Chemical Society, 130(48):16136–16137, 2008. [CrossRef]
- F. Jiao and H. Frei. Nanostructured cobalt oxide clusters in mesoporous silica as efficient oxygen-evolving catalysts. Angewandte Chemie, 121(10):1873–1876, 2009. [CrossRef]
- X. Xie, Y. Li, Z.-Q. Liu, M. Haruta, and W. Shen. Low-temperature oxidation of CO catalysed by Co3 O4 nanorods. Nature, 458(7239):746–749, 2009. [CrossRef]
- F. Waidhas, S. Haschke, P. Khanipour, L. Fromm, A. Görling, J. Bachmann, I. Katsounaros, K. J. Mayrhofer, O. Brummel, and J. Libuda. Secondary alcohols as rechargeable electrofuels: Electrooxidation of isopropyl alcohol at pt electrodes. ACS Catalysis, 10(12):6831–6842, 2020. [CrossRef]
- Chen, J.; Wu, X.; Selloni, A. Electronic structure and bonding properties of cobalt oxide in the spinel structure. Phys. Rev. B 2011, 83, 245204. [CrossRef]
- Kenmoe, S.; Douma, D. H.; Raji, A. T.; M’Passi-Mabiala, B.; Gotsch, T.; Girgsdies, F.; Knop-Gericke, A.; Schlogl, R.; Spohr, E. X-ray absorption near-edge structure (xanes) at the o k-edge of bulk co3o4: Experimental and theoretical studies. Nanomaterials 2022, 12 (6), 921,. [CrossRef]
- Anke, S.; Bendt, G.; Sinev, I.; Hajiyani, H.; Antoni, H.; Zegkinoglou, I.; Jeon, H.; Pentcheva, R.; Roldan Cuenya, B.; Schulz, S.; Muhler, M. Selective 2-propanol oxidation over unsupported Co3O4 spinel nanoparticles: Mechanistic insights into aerobic oxidation of alcohols. ACS Catal. 2019, 9 (7), 5974– 5985. [CrossRef]
- Falk, T.; Anke, S.; Hajiyani, H.; Saddeler, S.; Schulz, S.; Pentcheva, R.; Peng, B.; Muhler, M. Influence of the particle size on selective 2-propanol gas-phase oxidation over co3o4 nanospheres. Catal. Sci. Technol. 2021, 11, 7552– 7562,. [CrossRef]
- T. Falk, E. Budiyanto, M. Dreyer, C. Pflieger, D. Waffel, J. Büker, C. Weidenthaler, K. F. Ortega, M. Behrens, H. Tüysüz, et al. Identification of active sites in the catalytic oxidation of 2-propanol over Co1+x Fe2−x O4 spinel oxides at solid/liquid and solid/gas interfaces. ChemCatChem, 13(12):2942–2951, 2021. [CrossRef]
- Omranpoor, T. Kox, E. Spohr, and S. Kenmoe. Influence of temperature, surface composition and electrochemical environment on 2-propanol decomposition at the Co3 O4 (001)/H2 O interface. Applied Surface Science Advances, 12:100319, 2022. [CrossRef]
- D. H. Douma, K. N. Nono, A. H. Omranpoor, A. Lamperti, A. Debernardi, and S. Kenmoe. Probing the local environment of active sites during 2-propanol oxidation to acetone on the Co3 O4 (001) surface: insights from first principles O K-edge XANES spectroscopy. The Journal of Physical Chemistry C, 2023. [CrossRef]
- H. Omranpoor, A. Bera, D. Bullert, M. Linke, S. Salamon, S. Webers, H. Wende, E. Hasselbrink, E. Spohr, and S. Kenmoe. 2-Propanol interacting with Co3 O4 (001): A combined vSFS and AIMD study. The Journal of Chemical Physics, 158(16), 04 2023. 164703. [CrossRef]
- Yan, G. And Sautet, P. Surface Structure of Co3O4 (111) under Reactive Gas-Phase Environments, ACS Catalysis 2019 9 (7), 6380-6392. [CrossRef]
- Selcuk, S and Selloni, A. DFT+U Study of the Surface Structure and Stability of Co3O4(110): Dependence on U. The Journal of Physical Chemistry C 2015, 119 (18), 9973-9979. [CrossRef]
- Creazzo, F., Ruth Galimberti, D., Pezzotti, S., and Gaigeot, M.-P.. (2019). DFT-MD of the (110)-Co3O4 cobalt oxide semiconductor in contact with liquid water, preliminary chemical and physical insights into the electrochemical environment. J. Chem. Phys. 150(4):041721. [CrossRef]
- CP2K Is Freely. T.C.d.g. 2016. Available online: http://www.cp2k.org/.
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [CrossRef]
- Hubbard, J.; Flowers, B.H. Electron correlations in narrow energy bands. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1963, 276, 238–257. [CrossRef]
- Kox, T.; Spohr, E.; Kenmoe, S. Impact of solvation on the structure and reactivity of the co3o4 (001)/h2o interface: Insights from molecular dynamics simulations. Frontiers in Energy Research 2020, 8, 312. [CrossRef]
- Kox, T; Omranpoor, A. H.;Kenmoe, S. Water adsorption on CoFe2O4 (001) surface: structure and reactivity from ab initio MD simulations, Physchem, 2022, 2, 321.
- Budiyanto, E.; Zerebecki, S.; Weidenthaler, C.; Kox, T.; Kenmoe, S.; Spohr, E.; DeBeer, S.; Rudiger, O.; Reichenberger, S.; Barcikowski, S.; Tuysuz, H. Impact of single-pulse, low-intensity laser post-processing on structure and activity of mesostructured cobalt oxide for the oxygen evolution reaction. ACS Appl. Mater. Interfaces 2021, 13 (44), 51962– 51973,. [CrossRef]
- Zerebecki, S.; Salamon, S.; Landers, J.; Yang, Y.; Tong, Y.; Budiyanto, E.; Waffel, D.; Dreyer, M.; Saddeler, S.; Kox, T.; Kenmoe, S.; Spohr, E.; Schulz, S.; Behrens, M.; Muhler, M.; Tuysuz, H.; Kramer Campen, R.; Wende, H.; Reichenberger, S.; Barcikowski, S. Engineering of cation occupancy of cofe2o4 oxidation catalysts by nanosecond, single-pulse laser excitation in water. ChemCatChem. 2022, 14, e202101785. [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [CrossRef]
- M. Dreyer, D. Cruz, U. Hagemann, P. Zeller, M. Heidelmann, S. Salamon, J. Landers, A. Rabe, K. F. Ortega, S. Najafishirtari, et al. The effect of water on the 2-propanol oxidation activity of Co-substituted LaFe1−x Cox O3 perovskites. Chemistry–A European Journal, 27(68):17127–17144, 2021. [CrossRef]
Figure 1.
Snapshots of the initial and final configurations of the AIMD simulation (top and perspective views) for 2-propanol adsorption on the B-terminated pristine at 300 K. Co3O4 (111) surface at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propanol (red), C (gray) and H (white).
Figure 1.
Snapshots of the initial and final configurations of the AIMD simulation (top and perspective views) for 2-propanol adsorption on the B-terminated pristine at 300 K. Co3O4 (111) surface at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propanol (red), C (gray) and H (white).
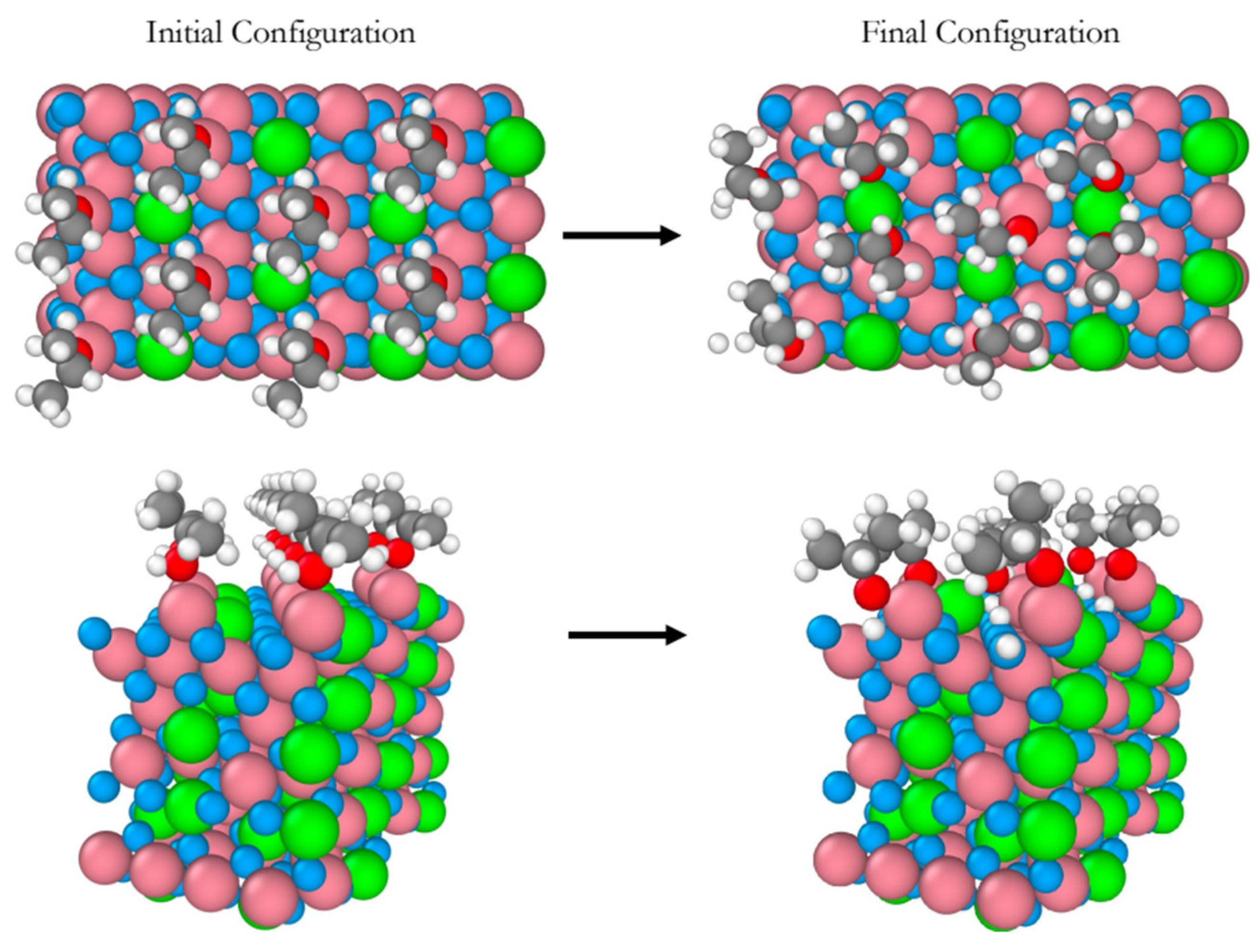
Figure 2.
Time dependence of O–H, C–H, C–O and Co3+–O bond lengths for a dissociatively adsorbed 2-propanol molecule.
Figure 2.
Time dependence of O–H, C–H, C–O and Co3+–O bond lengths for a dissociatively adsorbed 2-propanol molecule.
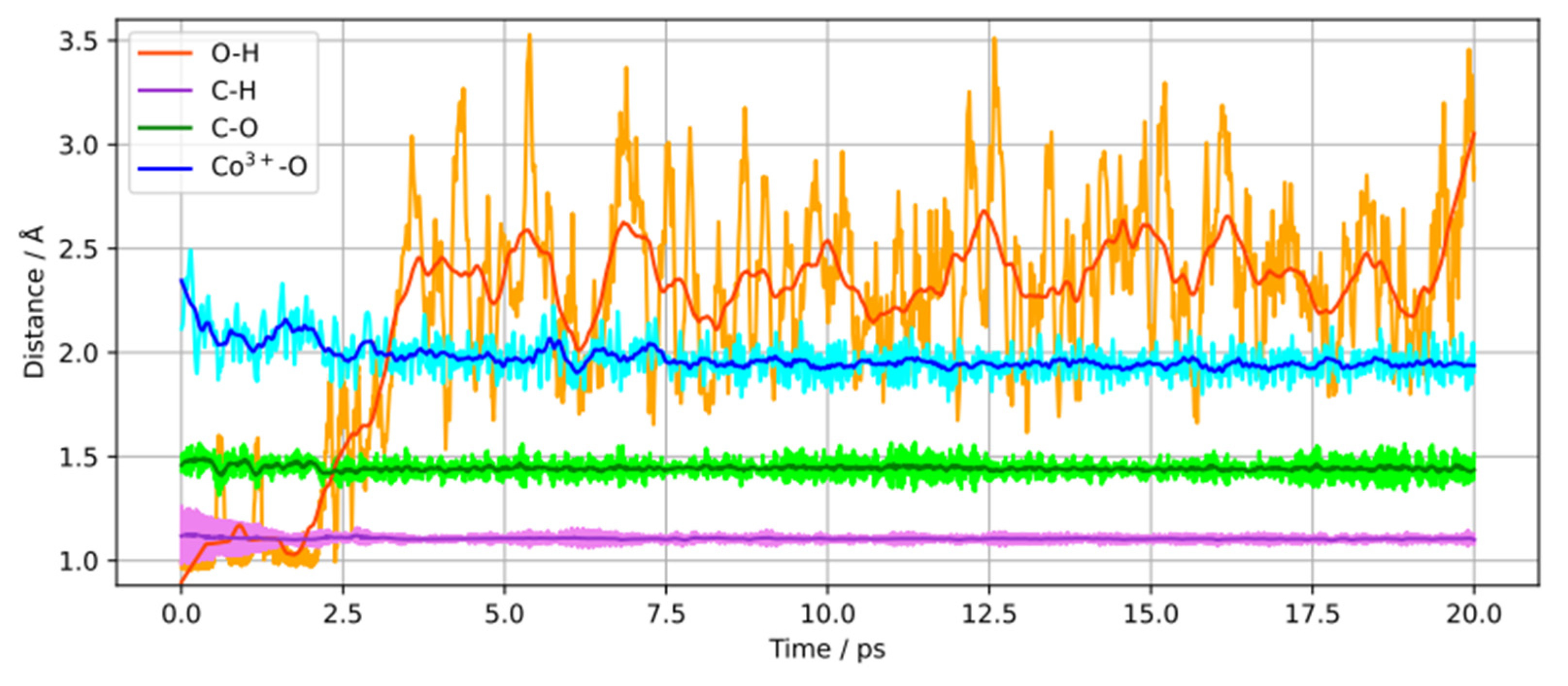
Figure 3.
Two Co3+–O2-propoxide–Co2+ bridges on the B-terminated pristine Co3O4 (111) surface at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propoxide (red), C (gray) and H (white).
Figure 3.
Two Co3+–O2-propoxide–Co2+ bridges on the B-terminated pristine Co3O4 (111) surface at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propoxide (red), C (gray) and H (white).
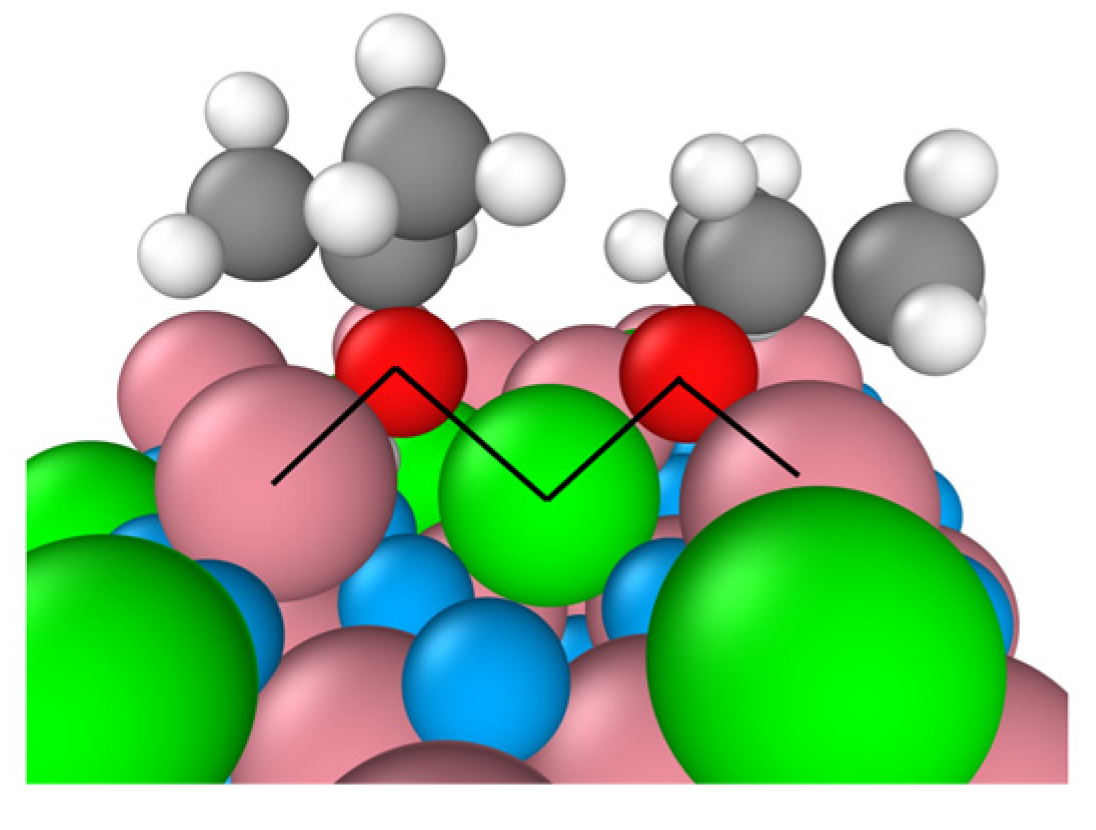
Figure 4.
Charge density difference plot for the adsorption of 2-propanol on Co3+ sites of the B-terminated Co3O4 (111) surface at 300 K. The iso-surfaces are drawn at density levels of – 0.003 (cyan) and + 0.003 (yellow) e Å-3 . The yellow regions indicate the accumulation of electron density, while the cyan regions indicate the depletion of electron density.
Figure 4.
Charge density difference plot for the adsorption of 2-propanol on Co3+ sites of the B-terminated Co3O4 (111) surface at 300 K. The iso-surfaces are drawn at density levels of – 0.003 (cyan) and + 0.003 (yellow) e Å-3 . The yellow regions indicate the accumulation of electron density, while the cyan regions indicate the depletion of electron density.
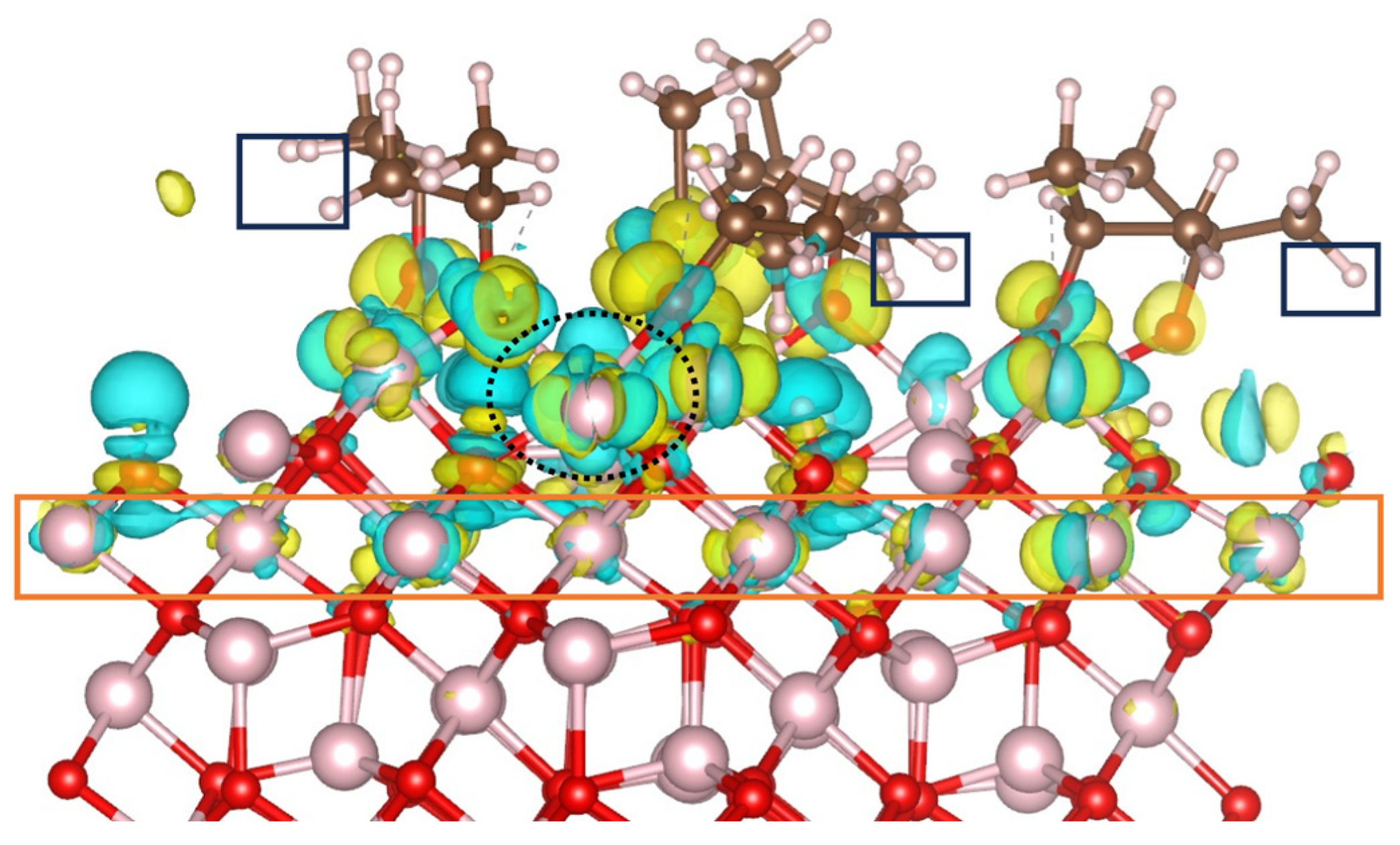
Figure 5.
Formation of water from surface oxygen through the Mars-van Krevelen mechanism. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propanol (orange), C (gray) and H (white). The lattice oxygen which participates to the Mars-van Krevelen mechanism is denoted in red. The created vacancy is illustrated with a black circle on the right side.
Figure 5.
Formation of water from surface oxygen through the Mars-van Krevelen mechanism. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propanol (orange), C (gray) and H (white). The lattice oxygen which participates to the Mars-van Krevelen mechanism is denoted in red. The created vacancy is illustrated with a black circle on the right side.
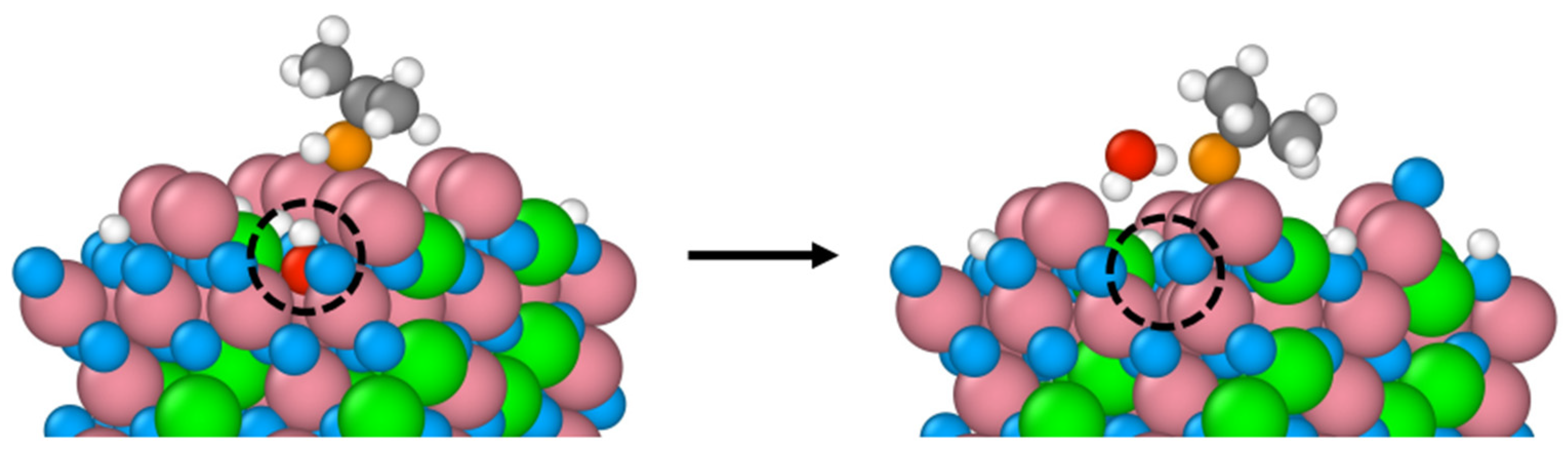
Figure 6.
Snapshots of the initial and final configurations of the AIMD simulation (top and perspective views) for 2-propanol adsorption on the B-terminated pristine Co3O4 (110) surface at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propanol (red), C (gray) and H (white).
Figure 6.
Snapshots of the initial and final configurations of the AIMD simulation (top and perspective views) for 2-propanol adsorption on the B-terminated pristine Co3O4 (110) surface at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4 (blue), O in 2-propanol (red), C (gray) and H (white).
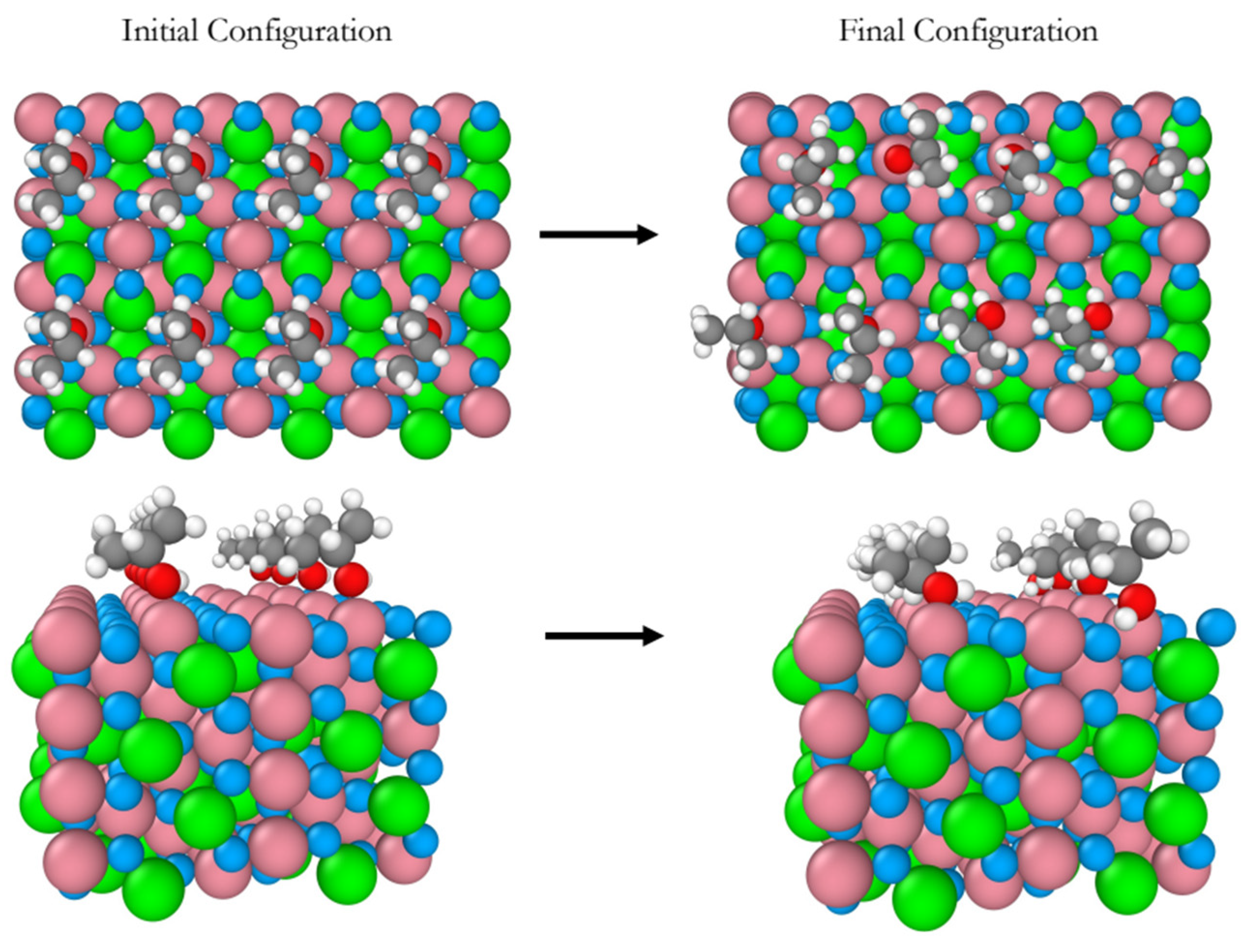
Figure 7.
Snapshot of the final configuration of the B-terminated Co3O4 (110) surface at 300 K. The adsorbed 2-propoxide molecules are removed for visualization. The “two-fold” surface oxygens are denoted in violet. Co3+ (pink), Co2+ (green), two-fold O (violet), other O in Co3O4 (blue), and H (white).
Figure 7.
Snapshot of the final configuration of the B-terminated Co3O4 (110) surface at 300 K. The adsorbed 2-propoxide molecules are removed for visualization. The “two-fold” surface oxygens are denoted in violet. Co3+ (pink), Co2+ (green), two-fold O (violet), other O in Co3O4 (blue), and H (white).
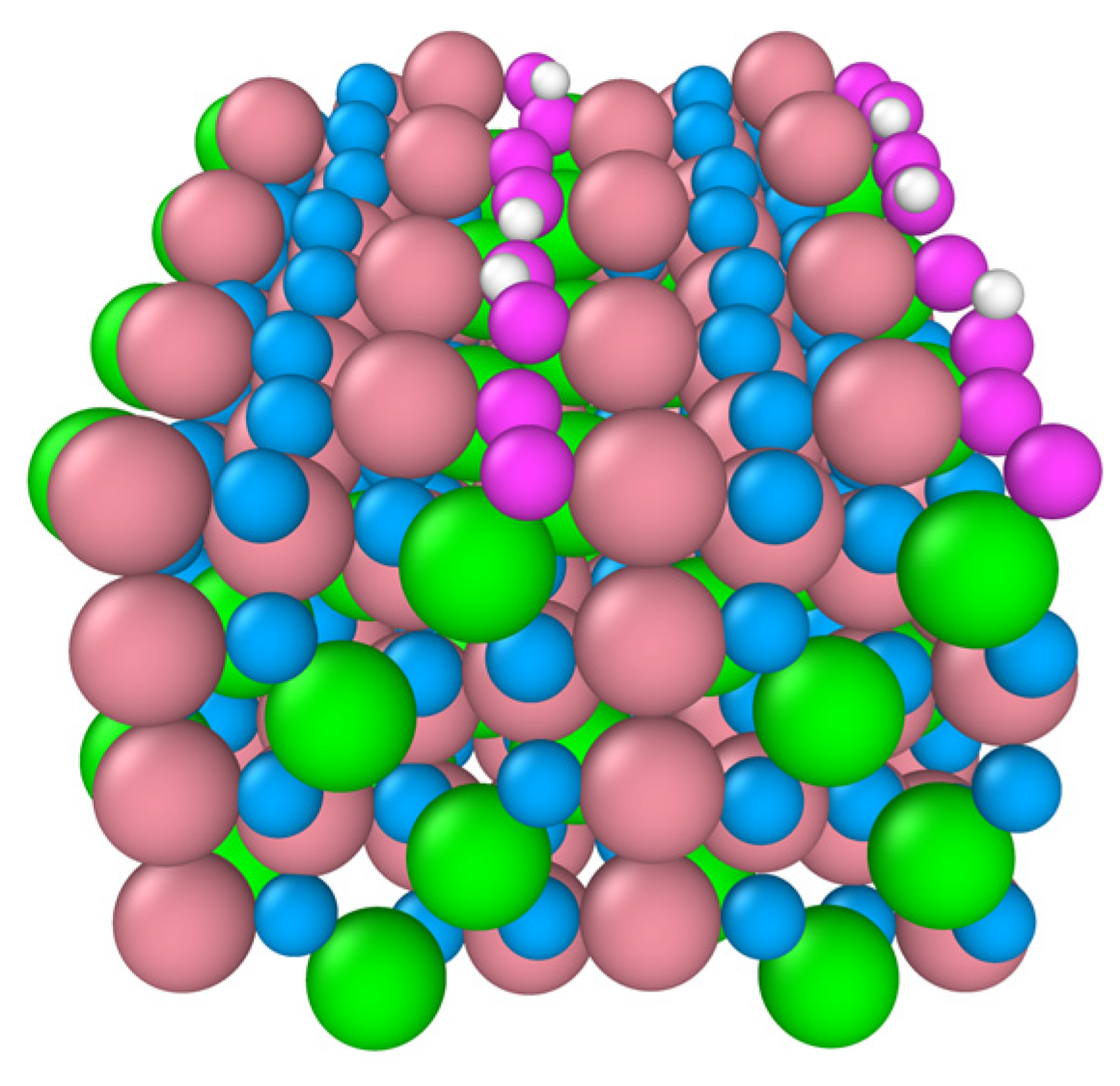
Figure 8.
Initial configuration (left) of AIMD simulation (top-view) and trajectory traces (right) of surface atoms for (001), (111), and (110) surfaces at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4, (blue)).
Figure 8.
Initial configuration (left) of AIMD simulation (top-view) and trajectory traces (right) of surface atoms for (001), (111), and (110) surfaces at 300 K. Co3+ (pink), Co2+ (green), O in Co3O4, (blue)).
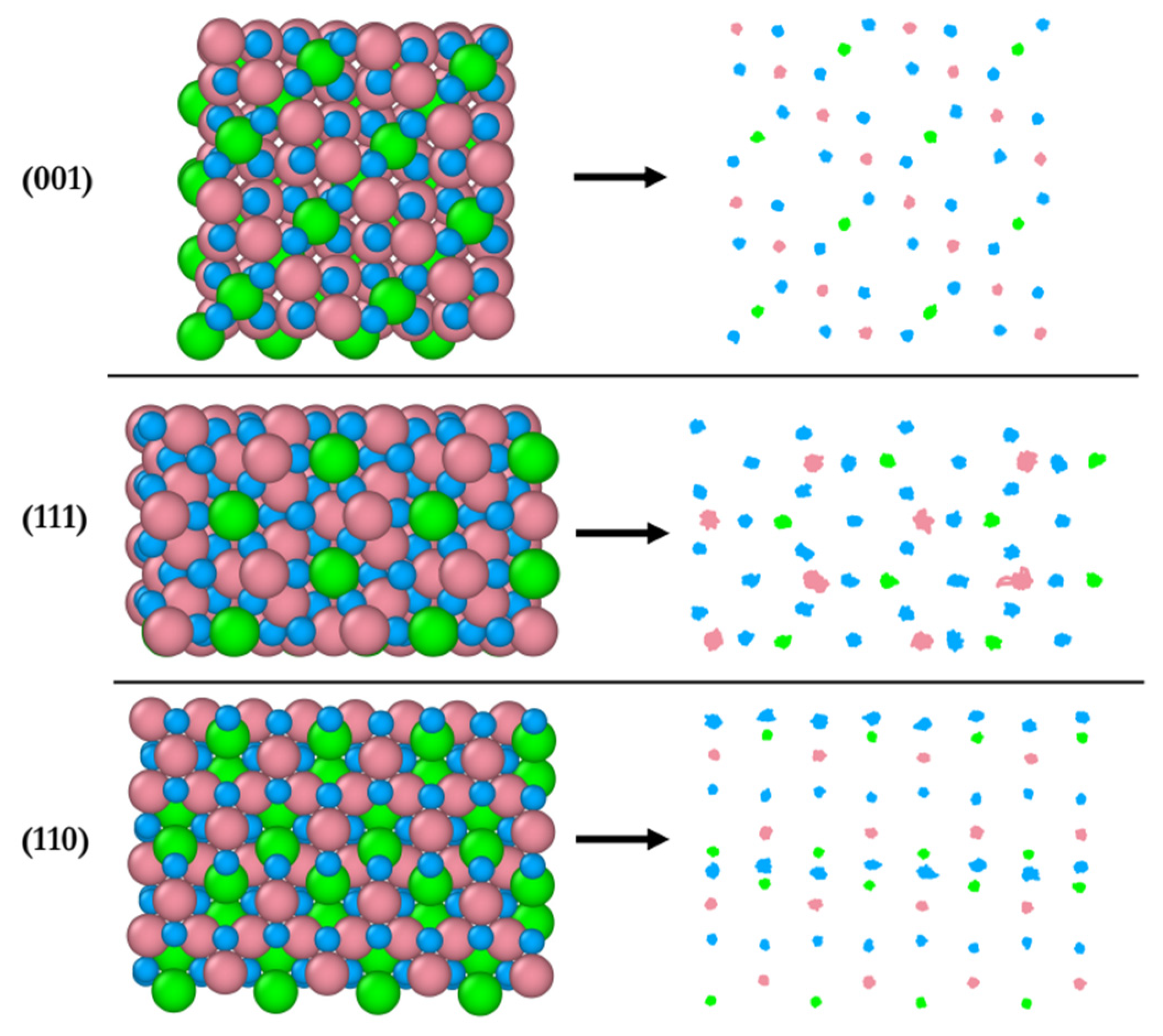
Figure 9.
Power spectra of a adsorbed 2-propanol molecule equilibrated at 300 K on B-terminated (001), (111) and (110) surfaces .
Figure 9.
Power spectra of a adsorbed 2-propanol molecule equilibrated at 300 K on B-terminated (001), (111) and (110) surfaces .
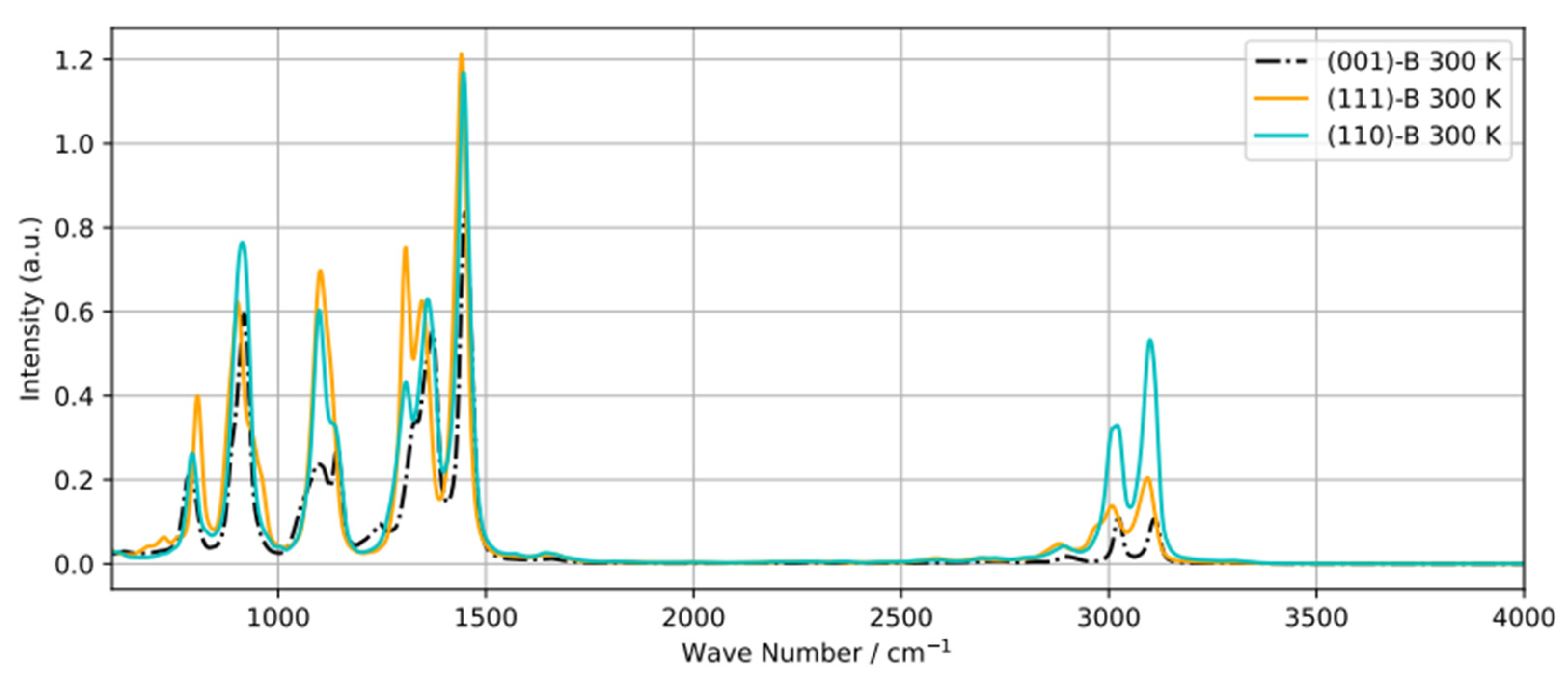

Table 1.
Number of initial and final 2-propanol, final 2-propoxide and acetone molecules on the (001), (111), and (110) surfaces at 300 K and 450 K .
Table 1.
Number of initial and final 2-propanol, final 2-propoxide and acetone molecules on the (001), (111), and (110) surfaces at 300 K and 450 K .
| (001) | (101) | (111) | ||||
| 300 K | 450 K | 300 K | 450 K | 300 K | 450 K | |
| Initial 2-propanol | 8 | 8 | 8 | 8 | 8 | 8 |
| Final 2-propanol | 7 | 8 | 6 | 2 | 0 | 0 |
| Final 2-propoxide | 1 | 0 | 2 | 6 | 8 | 8 |
| Final Acetone | 0 | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



