
Submitted:
23 November 2023
Posted:
23 November 2023
You are already at the latest version
Abstract
Benign recurrent intrahepatic cholestasis (BRIC) is a rare genetic cause of cholestasis. It is considered as part of inherited intrahepatic cholestasis syndromes, such as progressive familial intrahepatic cholestasis (PFIC), and intrahepatic cholestasis of pregnancy. BRIC is presented in infancy or in early adulthood. It is characterized by exacerbations and remissions of jaundice with accompanying intense itching, lasting from weeks to years throughout lifetime. Normal gamma-glutamyl transferase (GGT) is a characteristic laboratory finding. Contrary to PFIC, which may progress to cirrhosis, BRIC does not progress to chronic liver disease or cirrhosis. However, incessant episodes of cholestasis result in marked reduction in quality of life and distinct mutations increase the risk of hepatobiliary malignancy. In intervals between the exacerbations, the histological findings of centrilobular cholestasis together with the abnormal laboratory parameters return to normal. In this context, liver biopsy might be avoided. In this review, we will focus on the genetic aspects of BRIC, its pathophysiology, clinical presentation, and prognosis of this autosomal recessive genetically determined cholestatic disorder. Moreover, triggering factors as well as treatment options will be further elucidated.
Keywords:
Benign recurrent intrahepatic cholestasis
; mutations
; elevated conjugated bilirubin
; normal gamma-glutamyl transferase
1. Introduction
Benign recurrent intrahepatic cholestasis (BRIC) is a genetically determined, autosomal recessive disorder [1-2]. It was first described by Summerskill and Walshe in 1959 as a cause of jaundice [3]. Characterized by recurrent episodes of jaundice, pruritus, anorexia, nausea, vomiting, and steatorrhea, BRIC manifests symptoms that can mimic a malignant condition, leading to weight loss [1-9]. However, BRIC is a benign clinical entity, as between the episodes, patients experience good clinical condition with normal laboratory values and histologic findings in liver histology. This return to normality is typical of BRIC. The attacks can last for weeks to years, presenting with cholestatic laboratory profiles and abnormal histopathology [10]. Despite its characterization as a benign syndrome, BRIC carries the risk of developing hepatobiliary malignancy, estimated at around 15% over a lifetime [10].
During attacks, liver biopsy specimens reveal intrahepatic non-inflammatory cholestasis and hyperplasia of Kupffer cells [11-12]. Elevated levels of serum conjugated bilirubin and alkaline phosphatase (ALP) are observed, while serum gamma-glutamyl transferase (GGT) levels and transaminases remain normal or only slightly increased [11-12]. It is noteworthy that the normality in serum GGT values, contrasting with the abnormal biochemical liver profile, should raise the clinical suspicion of BRIC. This review aims to explore the genetic aspects of BRIC, its triggering factors, pathophysiology, clinical presentation, diagnosis, prognosis, and treatment options.
2. Literature Search
On September 18, 2023, we conducted a literature search using the term “benign recurrent intrahepatic cholestasis” in the PubMed database. The search yielded 24 case reports, 38 systematic/narrative reviews, and 2 guidelines published in the English language between 2013 and 2023.
3. History and Epidemiology of Cholestatic Syndromes: BRIC and PFIC
BRIC, formerly known as Summerskill-Walshe-Tygstrup syndrome [3], made its debut in medical literature during the latter half of the 20th century, with awareness limited to a minority of clinicians. In 1959, Summerskill and Walshe initially reported two unrelated patients from the United Kingdom with recurrent intrahepatic cholestasis [3]. Subsequently, in 1960, Tygstrup detailed a similar condition in two distantly related 15-year-old boys in a Faroe Islands village, with disease onset occurring within the first two years of life, marked by cholestasis confirmed through liver biopsy and direct cholangiography [4]. By 1969, Tygstrup and Jensen added five new cases of intermittent intrahepatic cholestasis in young males from the Faroe Islands to the literature. Exacerbations were characterized by intense pruritus and jaundice, with disease-free intervals showing no clinical or biochemical abnormalities. Remission episodes lasted for months or even years [5]. A 1999 follow-up by Tygstrup et al. revealed that none of the patients had progressed to chronic liver disease, and with increasing age, episodes of cholestasis tended to diminish. One patient, aged 25 with 16 episodes lasting about 6 months each, underwent a liver transplant, after which no further episodes were recorded one-year post-transplantation [6].
In the contemporary era of remarkable medical advances, particularly in the fields of genetics and molecular medicine, the incidence of BRIC is on the rise. The global incidence is estimated at approximately 1 in 50,000 to 100,000 people [7]. While the exact prevalence of BRIC remains unknown, it is less common than the related cholestatic disorder progressive familial intrahepatic cholestasis (PFIC) [7]. Intrahepatic cholestasis symptoms may manifest in infancy and early adulthood, with the mean age at diagnosis typically ≤ 20 years old in reported cases. Notably, 10-15% of cases with marked cholestasis in the pediatric population are attributed to PFIC, constituting 10% of liver transplant candidates in infancy [8]. Sex differences are not significant, as males and females are affected at the same ratio [7].
BRIC, along with PFIC types 1 and 2, has been reported in diverse racial groups, including Northern and Mediterranean Europe, Africa, North and South America, and Japan. PFIC3 has been documented in Western European, White, and North African Arabic populations. A family history of cholestatic disease is present in 50% of patients suffering from cholestasis, although sporadic cases have also been described [9].
4. Genetic Aspects of BRIC and Progressive Familial Intrahepatic Cholestasis (PFIC)
BRIC, PFIC, and Intrahepatic Cholestasis of Pregnancy (ICP) are disorders affecting similar genes and associated with intrahepatic cholestasis, collectively considered as part of intrahepatic cholestasis syndromes. However, they exhibit differences in prognosis and outcomes [1-3, 10].
BRIC is further categorized into two subtypes: BRIC-1 and BRIC-2, resulting from mutations in ATP8B1 and ABCB11 genes, respectively [7, 13]. Mutations in SLC51A have also been reported in BRIC patients [14]. All affected genes lead to decreased bile salt (BS) secretion.
Progressive Familial Intrahepatic Cholestasis (PFIC) is a distinct family of inherited cholestatic syndromes of autosomal recessive origin, categorized into PFIC1, PFIC2, and PFIC3. PFIC1 and PFIC2 result from mutations in ATP8B1 (chromosome 18q21-22), encoding a defective Familial Intrahepatic Cholestasis 1 protein (FIC1), and mutations in ABCB11 (FIC2) (chromosome 2q24), encoding a defective bile salt export pump protein (BSEP), respectively [14].
Interestingly, similar genes are implicated in BRIC 1 and PFIC1, as well as among BRIC2 and PFIC2 patients [1-2]. Despite both BRIC1 and PFIC1 stemming from mutations in ATP8B1, there is substantial differentiation in gene expression, resulting in major clinical and prognostic differences between the two entities. BRIC1 is characterized by recurrent episodes of intrahepatic cholestasis, while PFIC1 progresses to a chronic state of cholestasis. This chronic cholestasis may lead to chronic lesions observed in liver biopsy, progressing from lobular fibrosis to cirrhosis and end-stage liver disease. Consequently, patients with PFIC1 or PFIC2 may ultimately require liver transplantation, whereas those with BRIC1 or BRIC2 experience a benign course [1-2, 10]. Similar distinctions exist between BRIC2 and PFIC2, as PFIC2 carries a 15% risk of cirrhosis progressing to hepatocellular carcinoma or cholangiocarcinoma [1]. PFIC3, characterized by mutations in ABCB4, which encodes the multi-drug resistant 3 protein (MDR3). MDR3 acts as a flippase, thus, catalyzing the movement of phospholipids from the inner to the outer leaflet of the cell's membrane. Unlike BRIC1, BRIC2, PFIC1 and PFIC2, in PFIC3, serum gamma-glutamyl-transferase levels are elevated [10]. Other genes, including SLC51A, TJP2, ATP8B1, NR1H4, and MYO5B have also been reported to be affected in PFIC [1, 10] (Figure 1).
The Bile Salt Export Pump (BSEP), a member of the adenosino-triphosphate (ATP)-Binding Cassette (ABC) transporter family, utilizes ATP to transport substrates, such as taurocholate and other BS [15-17]. Genetic variability is present, evidenced by various mutations in in aforementioned genes [15-21]. New mutations in ATP8B1, ABCB11, and SLC51A genes have recently been reported [13, 15-20]. Notably, over 100 mutations in ABCB11 have been described, with more than 50% being non-sense mutations [22-26].
In summary, BRIC, PFIC, and ICP share mutations in similar genes contributing to intrahepatic cholestasis. However, the severity of the diseases varies, with some mutations resulting in milder conditions like BRIC1 and BRIC 2, while others, as seen in PFIC1 and PFIC2, lead to more severe clinical entities that may necessitate liver transplantation or even result in death.
5. Pathophysiology of BRIC
Bile formation and outflow play a pivotal role in maintaining health. The hepatocytes produce bile acids, primarily cholic acid and chenodeoxycholic acid, which are then conjugated with taurine or glycine to form more soluble bile salts. These bile salts are subsequently secreted into the duodenum, where some are re-absorbed into the portal circulation, establishing an enterohepatic circulation. The liver produces approximately 200 mg to 600 mg of bile acids daily [1, 17].
BSEP, located on hepatocytes, is essential for the secretion of bile salts into the bile canaliculi, the interspace between hepatocytes. Disruption in this secretion results in the accumulation of bile salts in the canaliculi, leading to impaired bile outflow and intrahepatic cholestasis. In BRIC, the hallmark of pathophysiology is the accumulation of bile salts, particularly in the bile canaliculi. Although the exact mechanisms of intrahepatic cholestasis remain largely unknown, several hypotheses have been proposed.
In BRIC-1, it is suggested that reduced levels or impaired function of FIC1 lead to decreased plasma membrane stability. FIC1, a flippase, is involved in the translocation of various phospholipids across the membrane, contributing to membrane asymmetry and stability. Flippases, as type 4 P-ATPase efflux enzymes, facilitate the movement of phospholipids from the outer to the inner leaflet of the cell membrane, maintaining membrane stability. Conversely, floppases move phosphatidylcholine in the opposite direction, from the inner to the outer leaflet of the cell membranes. In other words, flippases and floppases are enzymes that participate in the movement of phospholipids in the inner and outer leaflets contributing to the charge gradient of cell membranes [27].
In BRIC-2, the impaired function of BSEP results in decreased elimination of bile salts, leading to their accumulation in the bile canaliculi [17, 27-28]. BSEP is the primary transporter of bile salts in the bile canaliculi, and its defect results in intrahepatic cholestasis [17, 27-28]. The rigid and detergent-stable membrane of the canaliculi may be compromised by the impaired FIC1 in BRIC-1, contributing to intrahepatic cholestasis. Overall, these disruptions in bile salt transport and membrane stability collectively contribute to the pathophysiology of BRIC [27].
6. Triggering Factors for BRIC
Various factors have been identified as potential triggers for exacerbations of BRIC, including hormonal influences, infections, and drugs (Table 1). Hormonal changes, such as in pregnancy and oral contraceptive drugs administration, have been linked to BRIC, particularly during the initial two trimesters [27, 29-36]. Pregnancy, in particular, may act as a triggering factor, especially during the first two trimesters. This can pose a diagnostic challenge when BRIC presents for the first time during pregnancy, requiring differential diagnosis from Intrahepatic Cholestasis of Pregnancy (ICP), viral hepatitis, acute fatty liver of pregnancy, drugs, pre-eclampsia with Hemolysis, Elevated Liver Enzymes, and Low Platelets (HELLP) syndrome, and neoplasms [29-36]. However, most of hepatic diseases related to pregnancy typically emerge in the third trimester or the later part of the second trimester [27]. Laboratory findings specific to viral hepatitis, pre-eclampsia, and HELLP syndrome aid in their exclusion. Imaging studies of the liver can reveal acute fatty liver of pregnancy and neoplasms. The association between BRIC and pregnancy or oral contraceptive drugs may involve hormonal factors, possibly implicating estrogen levels [27]. It is suggested that estrogen levels could modulate the function of bile salt transporters, such as BSEP. Estrogens may impact BSEP's function by inhibiting the Farsenoid X Receptor (FXR), a transcription factor via which BSEP is postulated to be involved in the excretion of bile salts [27].
Beyond pregnancy and oral contraceptive drug use, thyroid dysfunction has been linked to triggering BRIC episodes, with recent reports associating hyperthyroidism with BRIC [37]. Additionally, hypothyroidism has been proposed as an extra-hepatic manifestation of BRIC rather than a direct triggering factor [27].
Infections, particularly those involving gram-negative bacteria and their lipopolysaccharides (LPS), are considered potential triggers for BRIC. LPS can induce pro-inflammatory cytokine production from Kupffer cells, leading to the downregulation of canalicular bile salt transporters and predisposing individuals to intrahepatic cholestasis [27]. Viral diseases, including influenza and SARS-CoV-2 infections, have also been associated with BRIC attacks [27, 38].
While the association of BRIC with drug administration, especially antibiotics, has been rarely reported, it is important to note that antibiotics are often prescribed in the context of infection, making it challenging to establish a definite causal relationship between antibiotics and BRIC episodes. Furthermore, antibiotics appear to contribute to only a minority of the infrequent cases where exacerbation of BRIC is attributed to drugs [27].
7. Clinical Presentation of BRIC
The defining clinical manifestations of BRIC encompass jaundice accompanied by intense pruritus. Additional symptoms, including anorexia, nausea, vomiting, and weight loss, may also manifest. BRIC-1 can present with pancreatitis, diarrhea, and hearing loss, attributed to the expression of mutated ATP8B1 in the pancreas, intestines, and cochlear cells, respectively. Cholelithiasis has been reported in BRIC-2 [27, 39-42].
The onset of the first BRIC attack typically occurs in childhood or early adolescence, although cases can present in infancy or adulthood. The disease follows a course marked by exacerbations lasting from weeks to months or even several years. During symptom-free intervals, which may extend for months to several years, patients experience good health. Table 1 provides details on BRIC cases and their characteristics over the past decade. Liver biopsy abnormalities are observed only during the attacks, with intrahepatic cholestasis resolving during asymptomatic intervals [27, 39-42]. A distinctive feature is the absence of progression to cirrhosis, setting BRIC apart from PFIC. While BRIC generally has a favorable prognosis, its repetitive cholestatic episodes can significantly impact the quality of life. It is important to note that any deficiency in BSEP heightens the risk of hepatobiliary malignancy, estimated at a 15% lifetime risk [10]. Despite not progressing to advanced liver disease, BRIC underscores the importance of regular monitoring and management to mitigate the impact on the patient's well-being.
8. Diagnosis of BRIC
In 1999, Luketic and Shiffman proposed six criteria for diagnosing BRIC, which included cholestasis with intense pruritus, at least two episodes of jaundice with asymptomatic intervals, laboratory findings of intrahepatic cholestasis, normal intra-hepatic and extra-hepatic bile ducts on imaging, a liver biopsy consistent with centrilobular cholestasis during attacks, and the absence of other known causes of cholestasis [9]. While these criteria have been helpful, they are somewhat dated. Advances in technology, particularly Next Generation Sequencing (NGS), now allow for genetic confirmation of BRIC [46-49]. Genetic information, obtained through NGS, has the potential to obviate the need for a liver biopsy in certain cases.
It is important to note that serum conjugated bilirubin and ALP levels are typically significantly elevated in BRIC, in contrast to serum transaminases levels and GGT, which remain normal or only mildly increased. The divergence between ALP and GGT levels can serve as a diagnostic clue for BRIC [28]. However, it's worth mentioning that PFIC and the recently described Ubiquitin-Specific Protease 53 (USP53) disease also exhibit normal or mildly elevated levels of serum GGT [45, 50-52]. USP53 disease, appearing in early infancy to adolescence, manifests as intrahepatic cholestasis and has been associated with hearing loss in animal models. While the cholestasis in USP53 disease may be relapsing, it can progress to chronic liver disease with fibrosis [45, 50-52]. Consequently, genetic testing becomes crucial in differentiating among BRIC, PFIC, and USP53 disease, enabling a more personalized evaluation of patients' prognosis.
Notably, genetic sequencing may identify mutations in ATP8B1 and ABCB11 genes, but it doesn't provide information about their functional impact. Although the discovery of mutations may support a diagnosis of BRIC or PFIC, understanding the activity of these mutated genes is paramount due to the significant differences in prognosis and monitoring approaches for BRIC and PFIC. Early detection is essential, especially since PFIC may necessitate liver transplantation. To address the limitations of genetic sequencing, alternative techniques have emerged to evaluate gene function, such as in vitro mutagenesis. However, in vitro mutagenesis is technically demanding and time-consuming [53-54].
Mizutani et al. proposed an alternative method to assess ATP8B1 deficiency in BRIC1 or PFIC1 using human peripheral blood monocyte-derived macrophages (HMDMs). They found that ATP8B1 is expressed in HMDMs and is associated with the polarization of HMDMs into M2 macrophages through treatment with interleukin-10 (IL-10). Flow cytometry analysis demonstrated a significant reduction in M2 macrophage markers CD14 and CD163 in PFIC1, but only a slight reduction in BRIC1 [54]. M2 macrophages, known for their anti-inflammatory effects, secrete cytokines like IL-10, moderating the host response to inflammatory stimuli. This reduction in host response protects against overactive reactions, contributing to homeostasis. Conversely, M1 macrophages, associated with inflammation, secrete pro-inflammatory cytokines, potentially contributing to cholestatic chronic liver injury. The diminished or dysfunctional M2 macrophages in PFIC1 may account for chronic inflammation, liver tissue damage, cirrhosis, and end-stage liver disease [54].
In summary, the evolution of sophisticated molecular techniques allows for easier differentiation between BRIC and PFIC, even in the early stages of these inherited cholestatic disorders.
9. Treatment Options and Future Challenges in BRIC
Several compounds have been utilized to alleviate symptoms in BRIC, with varying degrees of success. Anti-histamine drugs, though limited in efficacy for pruritus, do not address the underlying cholestasis-induced pruritus pathogenesis [52]. Opioids, believed to play a role in cholestasis-induced pruritus by acting on the mu opioid receptor, have led to the exploration of opioid antagonists, such as intravenous naloxone or oral naltrexone. While providing substantial relief, caution is warranted in severe liver disease, and the issue of opioid withdrawal syndrome restricts their use [55].
Cholestyramine and ursodeoxycholic acid (UDCA) are commonly employed in BRIC treatment. Cholestyramine, a non-absorbable polystyrene, binds bile acids (BAs) in the gut lumen, inhibiting their re-absorption by approximately 90%, thereby reducing BA levels [55-56]. Rifampin, acting on the Pregnane X Receptor (PXR), exhibits anti-pruritic effects in cholestasis but requires vigilant liver function monitoring due to the potential for drug-induced liver injury [17, 57, 58]. UDCA, a dihydroxy bile acid, alters the bile acids pool to a more hydrophilic mixture, and its favorable safety profile has contributed to its increased use, even during pregnancy [2, 16, 20, 21, 28, 31, 59].
Combination therapies, such as cholestyramine (4g daily) with UDCA (15-20 g daily) and or rimfampicin have been explored in non-responsive BRIC cases [13, 15, 18, 19, 38, 55]. Nasobiliary drainage [60, 61-63] and, rarely, hemodialysis with specific filters have been attempted with encouraging outcomes in certain cases [56, 64-65].
Fibrates, such as fenofibrate, inhibit bile acid synthesis and enhance bile excretion, potentially offering a management option for BRIC [52]. Obeticholic acid, an FXR agonist, which is FDA approved for the treatment of primary biliary cholangitis in patients non-responding to UDCA in doses 5-10 mg daily [52]. Even though obeticholic acid is a rather novel treatment option, it seems very promising with regards to cholestasis-induced pruritus. Nonetheless, due to its very recent development, it has not been widely used in inherited cholestasis disorders. It is noteworthy that, as shown in Table 1, obeticholic acid has not been administered in any BRIC case during the last 10 years.
Recent discoveries of ileal bile acid transport (IBAT) inhibitors, like odevixibat and maralixibat, approved by the FDA for Alagille syndrome treatment, present additional options for BRIC patients [66-67].
In summary, gaining a deeper understanding of the molecular pathways involving FIC1 and BSEP in both healthy individuals and BRIC patients is crucial. Exploring the biosynthesis and various factors influencing FIC1 and BSEP at the transcriptional and translational levels may provide insights into this clinically benign yet troublesome entity. Continued research and the development of targeted therapies hold promise for improving the management of BRIC in the future.
10. Conclusions
In conclusion, BRIC is characterized by recurrent episodes of intrahepatic cholestasis, yet it does not progress to cirrhosis. Clinicians should remain vigilant regarding this inherited disorder, particularly as it can be triggered by factors such as viral infections and pregnancy. A deeper understanding of the molecular pathogenesis of BRIC holds the promise of identifying potential therapeutic interventions in the near future. Molecular genetic testing emerges as a valuable tool for diagnosing this clinically benign entity. The critical differentiation between BRIC and PFIC is crucial, especially in the early stages of these conditions. As highlighted earlier, PFIC may necessitate liver transplantation, which is not required for BRIC. Thus, beyond traditional sequencing methods, novel techniques for assessing the residual function of ATP8B1 and ABCB11 could elucidate the distinctions between BRIC and PFIC at an early stage, facilitating accurate and timely diagnoses. Continued research and innovative diagnostic approaches will contribute to refining our understanding and management of BRIC.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Henkel, S.A.; Squires, J.H.; Ayers, M.; Ganoza, A.; Mckiernan, P.; Squires, J.E. Expanding etiology of progressive familial intrahepatic cholestasis. World. J. Hepatol.2019, 11(5), 450-463. [CrossRef]
- Gupta, S.; Ali, I.A.; Abreo, E.; Gujju, V.; Hayat, M. The Mystery of Episodic Recurrent Jaundice in a Young Male: Cholestasis With a normal gamma-glutamyl Transferase. Cureus.2021, 13(3):e13834. [CrossRef]
- Summerskill, W.H.; Walshe, J.M. Benign recurrent intrahepatic “obstructive” jaundice. Lancet.1959, 2, 686-690. [CrossRef]
- TYGSTRUP N. Intermittent possibly familial intrahepatic cholestatic jaundice. Lancet. 1960;1(7135):1171-1172.
- Tygstrup N, Jensen B. Intermittent intrahepatic cholestasis of unknown etiology in five young males from the Faroe Islands. Acta Med Scand. 1969;185(6):523-530.
- Tygstrup N, Steig BA, Juijn JA, Bull LN, Houwen RH. Recurrent familial intrahepatic cholestasis in the Faeroe Islands. Phenotypic heterogeneity but genetic homogeneity. Hepatology. 1999;29(2):506-508.
- Srivastava, A. Progressive familial intrahepatic cholestasis. J. Clin. Exp. Hepatol. 2014, 4(1), 25-36. [CrossRef]
- Siddiqi I, Tadi P. Progressive Familial Intrahepatic Cholestasis. [Updated 2023 Jul 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-.
- V.A.; Shiffman, M.L. Bening Recurrent intrahepatic cholestasis. Clin. Liver. Dis. 1999, 3(3), 509-528, viii. [CrossRef]
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1. Published 2009 Jan 8. [CrossRef]
- Ganesh, R.; Suresh, N.; Sathiyasekeran, M.; Venkatakrishnan, L. Benign recurrent intrahepatic cholestasis Unravelling the paradox. Indian. Pediatr. 2021, 58(5), 486-487.
- García-Romero, R.; Morlan-Herrador, L.; Ros-Arnal, I.; Miramar, M.D.; Molera-Busons, C. Intrahepatic cholestasis, sometimes benign recurrent. Gastroenterol. Hepatol.2021, 44(10), 719-720. [CrossRef]
- Akbulut, U.E.; Randa, N.C.; Işık, İ.A.; Atalay, A. Beinign recurrent intrahepatic cholestasis type 2 in a child. A case report and novel mutation. Turk. Arch. Pediatr. 2021, 1, 56(1), 72-74. [CrossRef]
- Lu L; Chinese Society of Hepatology and Chinese Medical Association. Guidelines for the Management of Cholestatic Liver Diseases (2021). J Clin Transl Hepatol. 2022;10(4):757-769. [CrossRef]
- Miura, R.; Kawaoka, T.; Imamura, M.; Kosaka, M.; Johira, Y.; Shirane, Y.; Murakami, S.; Yano, S.; Amioka, K.; Naruto, K.; Ando, Y.; Kosaka, Y.; Kodama, K.; Uchikawa, S.; Fujino, H.; Ono, A.; Nakahara, T.; Murakami, E.; Yamauchi, M.; Hinoi, T.; Aikata, H. Benign Recurrent Intrahepatic Cholestasis Type 1 with Novel Nonsense Mutations in the ATP8B1 Gene. Case. Rep. Gastroenterol.2022, 16(1), 110-115. [CrossRef]
- Kalaranjini, K.V.; Glaxon, J.A.;, Vasudevan, S.; Arunkumar, M.L. Benign recurrent intrahepatic cholestasis 2 deficiency in a child. Report from a tertiary care center in South India. Indian. J. Pathol. Microbiol. 2021, 64(Supplement), S146-S148. [CrossRef]
- Sohn, M.J.; Woo, M.H.; Seong, M.W.; Park, S.S.; Kang, G.H.; Moon, J.S.; Ko, J.S. Benign Recurrent Intrahepatic Cholestasis Type 2 in Siblings with Novel ABCB11 mutations. Pediatr. Gastroenterol. Hepatol. Nutr. 2019, 22(2), 201-206. [CrossRef]
- Suzuki, H.; Arinaga-Hino, T.; Sano, T.; Mihara, Y.; Kusano, H.; Mizuochi, T.; Togawa, T.; Ito, S.; Ide, T.; Kuwahara, R.; Amano, K.; Kawaguchi, T.; Yano, H.; Kage, M.; Koga, H.; Torimura, T. Case Report: A Rare Case of Benign Recurrent Intrahepatic Cholestasis Type 1 with a Novel Heterozygous Pathogenic Variant of ATP8B1. Front. Med. (Lausanne). 2022, 9, 891659. [CrossRef]
- Piazzolla, M.; Castellaneta, N.; Novelli, A.; Agolini, E.; Cocciadiferro, D.; Resta, L.; Duda, L.; Barone, M.; Ierardi, E.; Di Leo, A. Nonsense variant of ATP8B1 gene in heterozygosis and benign recurrent intrahepatic cholestasis: a case report and review of the literature. World. J. Hepatol. 2020, 12(2), 64-71. [CrossRef]
- Kornitzer, G.A.; Alvarez, F. Case Report: A Novel Single Variant TJP2 Mutation in a Case of Benign Recurrent Intrahepatic Cholestasis. JPGN. Rep.2021, 2(3), e087. [CrossRef]
- Chen, H.; Wu, D.; Jiang, W.; Lei, T.; Lu, C.; Zhou, T. Case report: a novel homozygous variant identified in a Chinese patient with benign recurrent intrahepatic cholestasis type 1. Front. Med. (Lausanne). 2021, 8, 705489. [CrossRef]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Meier, Y.; Antoniou, A.; Stieger, B.; Arnell, H.; Ozçay, F.; Al-Hussaini, H.F.; Bassas, A.F.; Verkade, H.J.; Fischler, B.; Németh, A.; Kotalová, R.; Shneider, B.L.;, Cielecka-Kuszyk, J.; McClean, P.; Whitington, P.F.; Sokal, E.; Jirsa, M.; Wali, S.H.; Jankowska, I.; Pawłowska, J.; Mieli-Vergani, G.; Knisely, A.S.; Bull, L.N.; Thompson, R.J. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology2008, 134, 1203-1214. [CrossRef]
- Henkel, S.A.; Squires, J.H.; Ayers, M.; Ganoza, A.; Mckiernan, P.; Squires, J.E. Expanding etiology of progressive familial intrahepatic cholestasis. World. J. Hepatol. 2019, 11(5), 450-463. [CrossRef]
- Fotoulaki, M.; Giza, S.; Jirsa, M.; Grammatikopoulos, T.; Miquel, R.; Hytiroglou, P.; Tsitouridis, I.; Knisely, A.S. Beyond an Obvious Cause of Cholestasis in a Toddler: Compound Heterozygosity for ABCB11 Mutations. Pediatrics. 2019, 143(5), e20182146. [CrossRef]
- Corpechot, C.; Barbu, V.; Chazouillères, O.; Broué, P.; Girard, M.; Roquelaure, B.; Chrétien, Y.; Dong, C.; Lascols, O.; Housset, C.; Jéru, I. Genetic contribution of ABCC2 to Dubin Johnson syndrome and inherited cholestatic disorders. Liver. Int. 2020, 40(1), 163-174. [CrossRef]
- Schonfeld, E.A.; Brown, R.S.;Jr. Genetic causes of liver disease: when to suspect a genetic etiology, initial lab testing and the basics of management. Med. Clin. North. Am.2019, 103(6), 991-1003. [CrossRef]
- Halawi, A.; Ibrahim, N.; Bitar, R. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: a review of literature. Acta. Gastroenterol. Belg. 2021, 84(3),477-486. [CrossRef]
- Kumar, P.; Charaniya, R.; Ahuja, A.; Mittal, S.; Sahoo, R. Benign Recurrent Intrahepatic Cholestasis in a Young Adult. J. Clin. Diagn. Res. 2016, 10(6), OD01-2. [CrossRef]
- Sohail, M.I.; Dönmez-Cakil, Y.; Szöllősi, D.; Stockner, T.; Chiba, P. The Bile Salt Export Pump: Molecular Structure, Study Models and Small-Molecule Drugs for the Treatment of Inherited BSEP Deficiencies. Int. J. Mol. Sci.2021, 22(2), 784. [CrossRef]
- Corpechot, C.; Barbu, V.; Chazouillères, O.; Broué, P.; Girard, M.; Roquelaure, B.; Chrétien, Y.; Dong, C.; Lascols, O.; Housset, C.; Jéru, I. Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited cholestatic disorders. .Liver. Int. 2020, 40(1), 163-174. [CrossRef]
- Ayyash, M.; Smith, N.; Keerthy, M.; Singh, A.; Shaman, M. Bening recurrent intrahepatic cholestasis in pregnancy. Fetal death at 36 weeks of gestation. Case. Rep. Obstet. Gynecol.2021, 6, 2021, 5086846. [CrossRef]
- Nguyen, K.D.; Sundaram, V.; Ayoub, W.S. Atypical causes of cholestasis. World. J. Gastroenterol. 2014, 20(28), 9418-9426. [CrossRef]
- Sticova, E.; Jirsa, M.; Pawłowska, J. New insights in genetic cholestasis. From molecular mechanisms to clinical implications. Can. J. Gastroenterol. Hepatol. 2018, 2018, 2313675. [CrossRef]
- Nakanishi, Y.; Saxena, R. Pathophysiology and Diseases of the Proximal Pathways of the Biliary System. Arch. Pathol. Lab. Med. 2015, 139(7), 858-866. [CrossRef]
- Henkel, S.A.; Squires, J.H.; Ayers, M.; Ganoza, A.; Mckiernan, P.; Squires, J.E. Expanding etiology of progressive familial intrahepatic cholestasis. World. J. Hepatol. 2019, 11(5), 450-463. [CrossRef]
- Dietrich, C.G.; Geier, A. Effect of drug transporter pharmacogenetics on cholestasis. Expert. Opin. Drug. Metab. Toxicol. 2014, 10(11), 1533-1551. [CrossRef]
- Halawi, A.; Bitar, R.; Ibrahim, N. Hyperthyroidism as a potential trigger for benign recurrent intrahepatic cholestasis. ACG. Case. Rep. J. 2020, 7(7), e00423. [CrossRef]
- Calhan, T, Yivli, E. Coronavirus disease 2019 (COVID-19) as a potential trigger for benign recurrent intrahepatic cholestasis. Clin. Case. Rep. 2022, 10(3), e05557. [CrossRef]
- Soroka CJ, Boyer JL. Biosynthesis and trafficking of the bile salt export pump, BSEP: therapeutic implications of BSEP mutations. Mol. Aspects. Med. 2014, 37, 3-14. [CrossRef]
- Huynh, M.T.; Nguyen, T.T.; Grison, S.; Lascols, O.; Fernandez, E.; Barbu, V. Clinical characteristics and genetic profiles of young and adult patients with cholestatic liver disease. Rev. Esp. Enferm. Dig. 2019, 111(10), 775-788. [CrossRef]
- Provenzano, A.; Farina, A.; Seidenari, A.; Azzaroli, F.; Serra, C.; Della Gatta, A.; Zuffardi, O.; Giglio, S.R. Prenatal Noninvasive Trio-WES in a Case of Pregnancy Related Liver Disorder. Diagnostics. (Basel). 2021, 11(10), 1904. [CrossRef]
- Giovannoni, I.; Callea, F.; Bellacchio, E.; Torre, G.; De Ville De Goyet, J.; Francalanci, P. Genetics and Molecular Modeling of New Mutations of Familial Intrahepatic Cholestasis in a Single Italian Center. PloS. One. 2015, 10(12), e0145021. [CrossRef]
- Reichert, M.C.; Hall, R.A.; Krawczyk, M.; Lammert, F. Genetic determinants of cholangiopathies: Molecular and systems genetics. Biochim. Biophys. Acta. Mol. Basis. Dis.2018, 1864(4 Pt B), 1484-1490. [CrossRef]
- Togawa, T.; Sugiura, T.; Ito, K.; Endo, T.; Aoyama, K.; Ohashi, K.; Negishi, Y.; Kudo, T.; Ito, R.; Kikuchi, A.; Arai-Ichinoi, N.; Kure, S.; Saitoh, S. Molecular Genetic Dissection and Neonatal/Infantile Intrahepatic Cholestasis Using Targeted Next Generation Sequencing. J. Pediatr.2016, 171, 171-7.e1-4. [CrossRef]
- Bull LN, Ellmers R, Foskett P, Strautnieks S, Sambrotta M, Czubkowski P, Jankowska I, Wagner B, Deheragoda M, Thompson RJ. Cholestasis due to USP53 Deficiency. J. Pediatr. Gastroenterol. Nutr. 2021, 1, 72(5), 667-673. [CrossRef]
- Schonfeld EA, Brown RS Jr. Genetic Testing in Liver Disease: What to order, in Whom, and When. Clin. Liver. Dis. 2017, 21(4), 673-686. [CrossRef]
- Lee, S.J.; Kim, J.E.; Choe, B.H.; Seo, A.N.; Bae, H.I.; Hwang, S.K. Early Diagnosis of ABCB11 Spectrum Liver Disorders by Next Generation Sequencing. Pediatr. Gastroenterol. Hepatol. Nutr. 2017, 20(2), 114-123. [CrossRef]
- Naik, J.; de Waart, D.R.; Utsunomiya, K.; Duijst, S.; Mok, K.H.; Oude Elferink, R.P.; Bosma, P.J.; Paulusma, C.C. ATP8B1 and ATP11C: Two Lipid Flippases Important for Hepatocyte Function. Dig. Dis. 2015, 33(3), 314-318. [CrossRef]
- Helgadottir, H.; Folvik, G.; Vesterhus, M. Improvement of cholestatic episodes in patients with benign recurrent intrahepatic cholestasis (BRIC) treated with rifampicin. A long-term follow-up. Scand. J. Gastroenterol. 2023, 58(5), 512-520. [CrossRef]
- Zheng, Y.; Guo, H.; Chen, L.; Cheng, W.; Yan, K.; Zhang, Z.; Li, M.; Jin, Y.; Hu, G.; Wang, C.; Zhou, C.; Zhou, W.; Jia, Z.; Zheng, B.; Liu, Z. Diagnostic yield and novel candidate genes by next generation sequencing in 166 children with intrahepatic cholestasis. Hepatol. Int. 2023, Jun 14. [CrossRef]
- Zhang, J.; Yang, Y.; Gong, J.Y.; Li, L.T.; Li, J.Q.; Zhang, M.H.; Lu, Y.; Xie, X.B.; Hong, Y.R.; Yu, Z.; Knisely, A.S.; Wang, J.S. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization. Liver Int. 2020, 40(5), 1142-1150. [CrossRef]
- Lu, L. Chinese Society of Hepatology and Chinese Medical Association. Guidelines for the Management of Cholestatic Liver Diseases (2021). J. Clin. Transl. Hepatol. 2022, 10(4), 757-769. [CrossRef]
- Folmer, D.E., van der Mark, V.A., Ho-Mok, K.S., Oude Elferink, R.P., Paulusma, C.C. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology. 2009, 50(5), 1597-605. [CrossRef]
- Mizutani, A., Sabu, Y., Naoi, S., Ito, S., Nakano, S., Minowa, K., Mizuochi, T., Ito, K., Abukawa, D., Kaji, S., Sasaki, M., Muroya, K., Azuma, Y., Watanabe, S., Oya, Y., Inomata, Y., Fukuda, A., Kasahara, M., Inui, A., Takikawa, H., Kusuhara, H., Bessho, K., Suzuki, M., Togawa, T., Hayashi, H. Assessment of Adenosine Triphosphatase Phospholipid Transporting 8B1 (ATP8B1) Function in Patients With Cholestasis With ATP8B1 Deficiency by Using Peripheral Blood Monocyte-Derived Macrophages. Hepatol Commun. 2020, 5(1), 52-62. [CrossRef]
- van der Woerd, W.L.; Houwen, R.H.; van de Graaf, S.F. Current and future therapies for inherited cholestatic liver diseases. World. J.Gastroenterol.2017, 23(5), 763-775. [CrossRef]
- Koukoulioti, E.; Ziagaki, A.; Weber, S.N.; Lammert, F.; Berg, T. Long-Term Colestyramine Treatment Prevents Cholestatic Attacks in Refractory Benign Recurrent Intrahepatic Cholestasis Type 1 Disease. Hepatology. 2021, 74(1), 522-524. [CrossRef]
- Alhebbi, H.; Peer-Zada, A.A.; Al-Hussaini, A.A.; Algubaisi, S.; Albassami, A.; AlMasri, N.; Alrusayni, Y.; Alruzug, I.M.; Alharby, E.; Samman, M.A.; Ayoub, S.Z.; Maddirevula, S.; Peake, R.W.A.; Alkuraya, F.S.; Wali, S.; Almontashiri, N.A.M. New paradigms of USP53 disease. Normal CG, cholestasis, BRIC, cholangiopathy and responsiveness to rifampicin. J. Hum. Genet. 2021, 66(2), 151-159. [CrossRef]
- Salyani A, Barasa L, Rajula A, Ali SK. Benign Recurrent Intrahepatic Cholestasis (BRIC): An African Case Report. Case. Rep. Gastrointest Med. 2020, 10, 020, 894293. [CrossRef]
- Kagawa, T.; Orii, R.; Hirose, S.; Arase, Y.; Shiraishi, K.; Mizutani, A.; Tsukamoto, H.; Mine, T. Ursodeoxycholic acid stabilizes the bile salt export pump in the apical membranein MDCK II cells. J. Gastroenterol. 2014, 49(5), 890-9. [CrossRef]
- Choudhury, A.; Kulkarni, AV.; Sahoo, B.; Bihari, C. Endoscopic nasobiliary drainage: an effective treatment option for benign recurrent intrahepatic cholestasis(BRIC). BMJ. Case. Rep. 2017, 2017, bcr2016218874. [CrossRef]
- Dold, L.; Tschada, A.; Strassburg, C.P.; Weismüller, T.J. Percutaneous transgastral biliodigestive diversion as treatment option for benign recurrent intrahepatic cholestasis. Liver. Int. 2019, 39(1), 222. [CrossRef]
- Yakar, T.; Demir, M.; Gokturk, H.S.; Unler Kanat, A.G.; Parlakgumus, A.; Ozer, B.; Serin, E. Nasobiliary Drainage for Benign Recurrent Intrahepatic Cholestasis in Patients Refractory to Sandard Therapy. Clin. Invest. Med. 2016, 39(6), 27522.
- Appleby, V.J.; Hutchinson, J.M.; Davies, M.H. Safety and efficacy of long term nasobiliary drainage to treat intractable pruritus in cholestatic liver disease. Frontline. Gastroenterol. 2015, 6(4), 252-254. [CrossRef]
- Schoeneich K, Frimmel S, Koball S. Successful treatment of a patient with benign recurrent intrahepatic cholestasis type 1 with albumin dialysis. Artif Organs. 2020, 44(3), 341-342. [CrossRef]
- Ołdakowska-Jedynak, U.; Jankowska, I.; Hartleb, M.; Jirsa, M.; Pawłowska, J.; CzubChoudhury, A.; Kulkarni, A.V.; Sahoo, B.; Bihari Ckowski, P.; Krawczyk, M. Treatment of pruritus with Prometheus dialysis and absorption system in a patient with benign recurrent intrahepatic cholestasis. Hepatol. Res.2014, 44(10), E304-E308. [CrossRef]
- Muntaha HST, Munir M, Sajid SH, et al. Ileal Bile Acid Transporter Blockers for Cholestatic Liver Disease in Pediatric Patients with Alagille Syndrome: A Systematic Review and Meta-Analysis. J Clin Med. 2022;11(24):7526. Published 2022 Dec 19.
- Kamath BM, Stein P, Houwen RHJ, Verkade HJ. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020;40(8):1812-1822.
- Moghadamrad, S.; Montani, M.; Weimann, R.; De Gottardi A. Cholestasis in a patient with gallstones and a normal gamma-glutamyl transferase. Hepatology. 2013, 57, 2539-2541. [CrossRef]
- Schreiner P, Stieger B, McLin V, Rougemont AL, Keitel V, Dröge C, Müllhaupt B. A rare cause of cholestatic jaundice in a North African teenager. Liver Int.2019, 39(11), 2036-2041. [CrossRef]
- Arthur Lorio, E.; Valadez, D.; Alkhouri, N.; Loo, N. Cholestasis in Benign Recurrent Intrahepatic Cholestasis 2. ACG. Case. Rep. J. 2020, 7(6), e00412. [CrossRef]
- Bing, H.; Li, Y.L.; Li, D.; Zhang, C.; Chang, B. Case Report: A Rare Heterozygous ATP8B1 Mutation in a BRIC1 Patient: Haploinsufficiency? Front. Med. (Lausanne). 2022, 16, 9, 897108. [CrossRef]
Figure 1.
Pathogenesis of BRIC 1 and 2, PFIC 1, 2 and 3 and ICP: Familial intrahepatic cholestasis protein 1 (FIC 1 protein) is a flippase that helps in the movement of aminophospholipids; phosphatidylserine and phosphatidylethanolamine from the outer to the inner space of the plasma membrane of hepatocyte; Dysfunction of FIC1 results in PFIC 1, BRIC 1 and ICP. Bile salt exporter pump (BSEP) exports bile acids – cholesterol’s catabolism products - from hepatocytes to bile canaliculus; Defective BSEP underlies the pathogenesis of PFIC 2, BRIC 2 and ICP. Multidrug resistance protein 3 is a flippase involved in transporting phosphatidylcholine into bile canaliculus. PFIC 3 and ICP result by a nonfunctional MDR3 protein. Genetic mutations in ATP8B1, ABCB11, and ABCB4 genes result in the impaired function of FIC1, BSEP and MDR3 respectively. ABCB4 gene: ATP binding cassette subfamily B member 4, ABCB11 gene: ATP binding cassette subfamily B member 11, AMP: Aminophospholipids, ATP8B1 gene: ATPase phospholipid transporting 8B1, BRIC: Benign Recurrent Intrahepatic Cholestasis, BS: Bile Salts, BSEP: Bile Salt Export Pump Protein, FIC: Familial Intrahepatic Cholestasis, ICP: Intrahepatic Cholestasis of Pregnancy, MDR3: Multi-Drug Resistant 3 Protein, PC: Phosphatidylcholine, PFIC: Progressive Familial Intrahepatic Cholestasis.
Figure 1.
Pathogenesis of BRIC 1 and 2, PFIC 1, 2 and 3 and ICP: Familial intrahepatic cholestasis protein 1 (FIC 1 protein) is a flippase that helps in the movement of aminophospholipids; phosphatidylserine and phosphatidylethanolamine from the outer to the inner space of the plasma membrane of hepatocyte; Dysfunction of FIC1 results in PFIC 1, BRIC 1 and ICP. Bile salt exporter pump (BSEP) exports bile acids – cholesterol’s catabolism products - from hepatocytes to bile canaliculus; Defective BSEP underlies the pathogenesis of PFIC 2, BRIC 2 and ICP. Multidrug resistance protein 3 is a flippase involved in transporting phosphatidylcholine into bile canaliculus. PFIC 3 and ICP result by a nonfunctional MDR3 protein. Genetic mutations in ATP8B1, ABCB11, and ABCB4 genes result in the impaired function of FIC1, BSEP and MDR3 respectively. ABCB4 gene: ATP binding cassette subfamily B member 4, ABCB11 gene: ATP binding cassette subfamily B member 11, AMP: Aminophospholipids, ATP8B1 gene: ATPase phospholipid transporting 8B1, BRIC: Benign Recurrent Intrahepatic Cholestasis, BS: Bile Salts, BSEP: Bile Salt Export Pump Protein, FIC: Familial Intrahepatic Cholestasis, ICP: Intrahepatic Cholestasis of Pregnancy, MDR3: Multi-Drug Resistant 3 Protein, PC: Phosphatidylcholine, PFIC: Progressive Familial Intrahepatic Cholestasis.
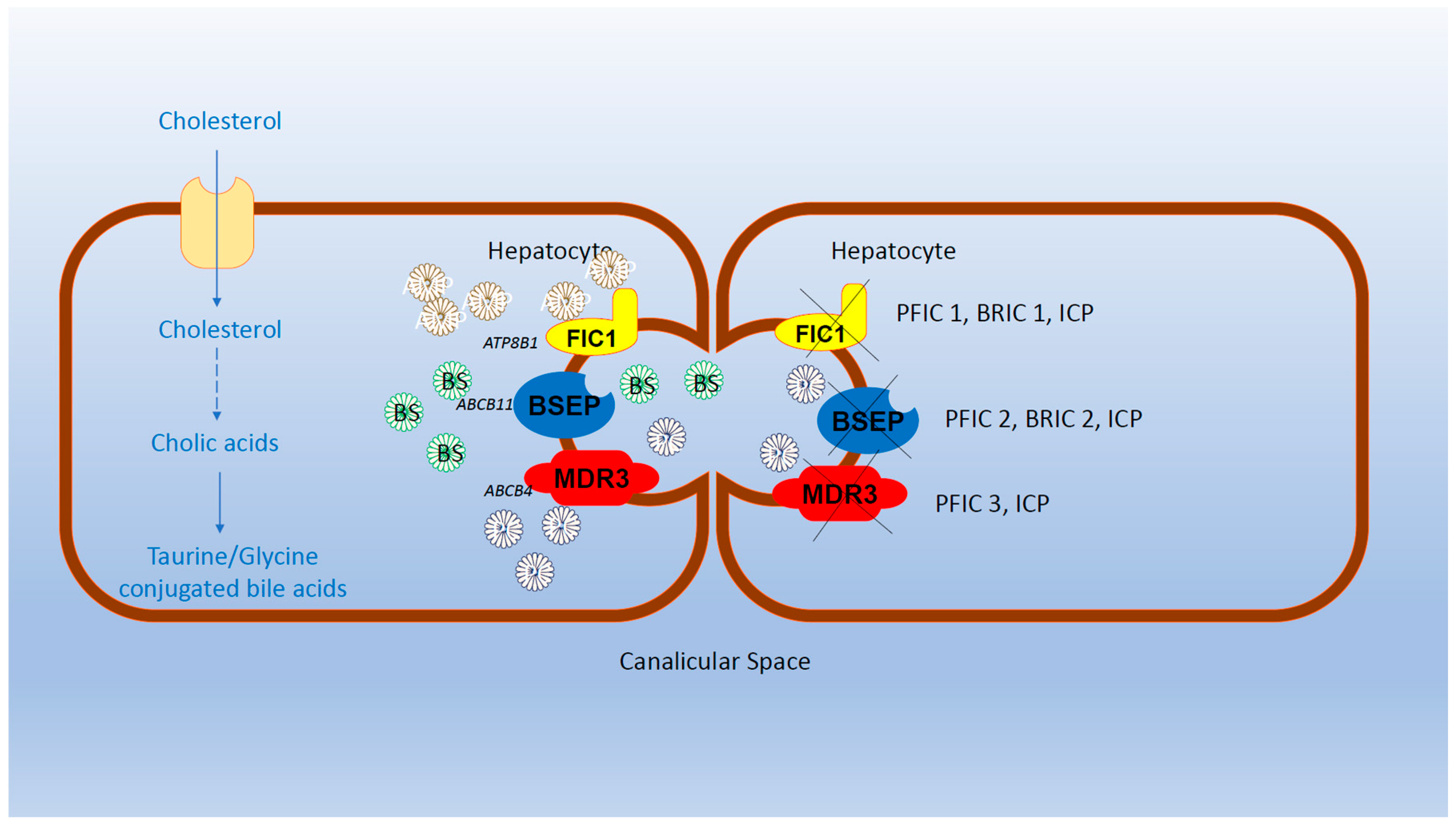

Table 1.
BRIC cases reported in the past 10 years and their characteristics.
| Author/Year | Age y.o. |
Number of attacks | Sex | BRIC-1/ BRIC-2 |
Triggering Factor | Treatment | Remarks |
|---|---|---|---|---|---|---|---|
| Moghadamrad et al, 2013 [68] | 44 | Unknown | F | BRIC-2 | Gastroenteritis | None | |
| Oldakowska Jedynak et al, 2014 [65] |
14 | Unknown | M | BRIC-1 | Skin infection or tetracycline | None | |
| Kumar et al., 2016 [28] | 27 | >10 | M | BRIC 1 | Unknown | UDCA | |
| Schreiner et al, 2019 [69] | 16 | Unknown | M | BRIC-2 | Pharyngitis | UDCA | |
| Dold et al, 2019 [61] | 51 | 1 | F | BRIC 2 | Unknown | Nasobillary Drainage+ Percutaneous Transgastral Biliodigestive Diversion |
|
| Sohn et al., 2019 [17] | 6 | 3 | F | BRIC 1 | Unknown | Phenylbutyrate and Rifampicin | |
| Fotoulaki et al., 2019 [24] | 27 months | 1 | F | BRIC 2+ PFIC 2 |
Cystitis | UDCA + Liver Transplantation after 5 years | |
| Halawi et al, 2020 [37] | 37 | 7 | F | BRIC-1 | Hyperthyroidism | UDCA + Thyroxine |
|
| Arthur et al, 2020 [70] | 27 | 2 | F | BRIC-2 | Pregnancy | None | |
| Piazzolla et al, 2020 [19] | 29 | Unknown | M | BRIC-1 | Unknown | UDCA 15mg/Kg + Cholestyramine 8 g/d | |
| Salyani et al, 2020 [58] | 21 | 4 | M | Unnknown | Skin abscess in 2 prior attacks | Rifampicin 150mg x 2/d | |
| Schoneich et al, 2020 [64] | 24 | Unknown | Unknown | BRIC-1 | No | Dialysis | Resistant to Cholestyramine+ Steroids +Rifampicin |
| Ayyash et al, 2021 [31] | 29 | 2 related to ICP |
F | Unknown | Pregnancy | UDCA 600mg x 2/d | Fetal Death at 36 ws of gestation |
| Akbulut et al, 2021 [13] | 16 | >10 | M | BRIC-2 | Unknown | UDCA 20mg /Kg/d + Cholestyramine 4g/d | |
| Provenzano et al., 2021 [41] | 35 | 1 | F | BRIC 1 | Pregnancy | Childbirth | |
| Kornitzer et al., 2021[20] | 15 | >10 | F | BRIC 1 | Unknown | UDCA | |
| Kalaranjini et al, 2021 [16] | 12 | >10 | M | BRIC-2 | Fever | UDCA | |
| Gupta et al, 2021 [2] | 26 | 3 | M | Unknown | Methamphetamine use | UDCA | |
| Chen et al, 2021 [21] | 34 | >10 | M | BRIC-1 | Pharyngitis | UDCA | |
| Koukoulioti et al, 2021 [56] | 47 | >10 | M | BRIC-1 | Unknown | Dialysis | Resistant to drugs on dialysis for many years |
| Bing et al, 2022 [71] | 17 | 1 | M | BRIC-1 | Unknown | UDCA Glycyrrhizin + glucocorticoids + Dialysis |
|
| Calhan et al, 2022 [38] | 59 | Unknown | M | Unknown | COVID-19 | UDCA + Cholestyramine + Rifampicin | |
| Miura et al, 2022 [15] | 16 | 1 | F | Unknown | Unknown | UDCA 300 mg/d + Cholestyramine 4 g/d | |
| Suzuki et al., 2022[18] | 17 | Unknown | M | BRIC 1 | Unknown | UDCA/ prednisolone/Rifampicin 450 mg/day |
Abbreviations: d, daily; F, Female; ICP, intrahepatic cholestasis of pregnancy; M, Male; Ws, Weeks; UDCA, Ursodeoxycholic acid; y.o., years old.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



