Submitted:
14 November 2023
Posted:
15 November 2023
You are already at the latest version
Abstract
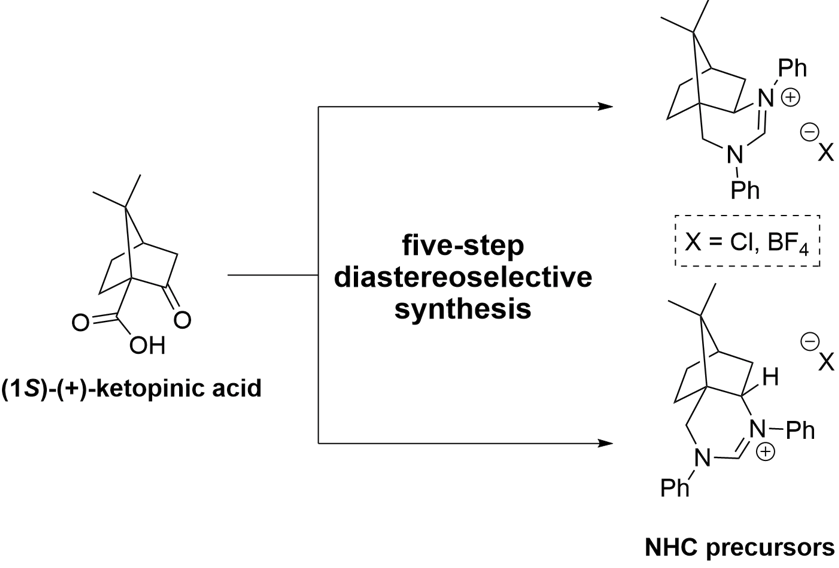
The endo- and exo-N-heterocyclic carbene precursors based on camphor were prepared diastereoselectively in five synthetic steps starting from (1S)-(+)-ketopinic acid. The obtained N-heterocyclic carbene precursors were investigated in an asymmetric benzoin reaction. All new compounds were fully characterized and the absolute configurations were determined by X-ray diffraction and NOESY measurements.
Keywords:
ketopinic acid
; amidation
; reduction
; diamines
; N-heterocyclic carbene precursors
; asymmetric catalysis
; amidinium salts
; hydrolysis
1. Introduction
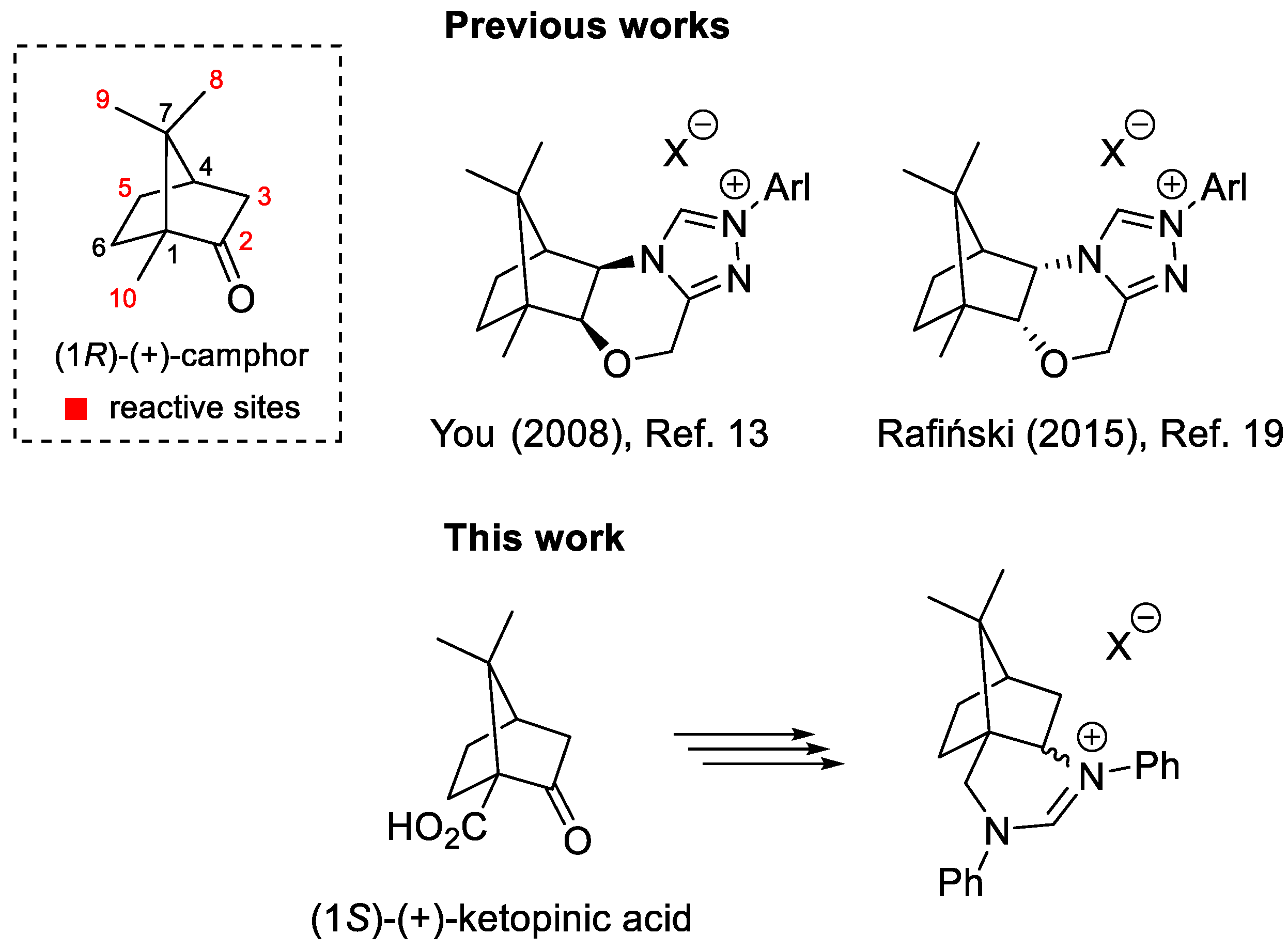
Camphor is a privileged chiral pool building block available in both enantiomeric forms, which undergo a wide range of different chemical transformations. These include fragmentation reactions and rearrangements, such as the Wagner-Meerwein rearrangement, which functionalize at first glance inactivated positions (Figure 1) and enable the synthesis of structurally and functionally very different products [1,2].
Numerous camphor derivatives have found their application in the field of asymmetric synthesis and catalysis. For example, camphorsultam has been widely used as an efficient chiral auxiliary [3,4], while the α-aminoisoborneol derivatives DAIB [5] and MIB [6] have been used as efficient ligands for the enantioselective addition of organozinc reagents to aldehydes. In the field of asymmetric organocatalysis [7,8], the first efficient camphor-derived organocatalyst was published in 2005 [9]. Both, covalent and non-covalent organocatalysts based on camphor backbone were developed [10]. While the first efficient N-heterocyclic carbene (NHC) organocatalyst was introduced by Enders and Kallfass in 2002 [11], efficient camphor-based NHC analogues appeared from 2008 [12,13,14,15,16,17,18,19,20]. The NHC precursors developed by You [13] and Rafiński [19] are particularly efficient in enantioselective catalysis (Figure 1).
Recently, we prepared camphor-derived 1,2-, 1,3-, and 1,4-diamines as potential building blocks for bifunctional organocatalysts with camphor as the exclusive chiral scaffold [21]. These camphor-derived diamines were used for the preparation of bifunctional non-covalent thiourea and squaramide organocatalysts [21,22] and bifunctional quaternary ammonium salt phase transfer organocatalysts [23]. The camphor-1,3-diamine-derived squaramide organocatalyst exhibited excellent catalytic activity in enantioselective conjugative additions of 1,3-dicarbonyls and α-amino acid-derived pyrrolin-4-ones to trans-β-nitrostyrenes [22,24]. In extension, we reasoned that camphor-based 1,3-diamines [21,22] could be transformed into cyclic amidinium salts as interesting non-racemic precursors of NHC. In this article, we report the synthesis of a novel type of six-membered NHC precursors in five steps from commercially available (1S)-(+)-ketopinic acid (Figure 1).
Figure 1.
Efficient camphor-derived N-heterocyclic carbene precursors (top); six-membered N-heterocyclic carbene precursors (bottom).
Figure 1.
Efficient camphor-derived N-heterocyclic carbene precursors (top); six-membered N-heterocyclic carbene precursors (bottom).
2. Results and Discussion
2.1. Synthesis
The starting point for the synthesis was commercially available (1S)-(+)-ketopinic acid (1) (Scheme 1), which can alternatively be prepared from the much cheaper (1S)-(+)-10-camphorsulfonic acid according to procedures described in the literature [25,26,27]. First, (1S)-(+)-ketopinic acid (1) was treated with thionyl chloride. After removal of volatiles, crude acid chloride 2 was reacted with aniline in the presence of excess triethylamine in anhydrous toluene to give amide 3 [28] in 94% yield. Treatment of ketone 3 with excess aniline in the presence of catalytic amounts of para-toluenesulfonic acid with azeotropic removal of water using 4 Å molecular sieves gave imine 4 in 49% yield. Attempts to reduce both amide and imine functionality in one step to obtain diamines 6a/6b with excess LiAlH4 or BH3•THF resulted in complex product mixtures. Therefore, sequential reduction was performed. Diastereoselective reduction of imine 4 with NaCNBH3 [29] in methanol in the presence of acetic acid afforded exo-aminoamide 5a in 91% yield and high diastereoselectivity (dr 93:7). Reduction of imine 4 with sodium [21,22] in n-propanol at 95°C gave a mixture of products containing endo-epimer 5b in an estimated 94% combined yield. The diastereoselectivity of the reduction could not be determined. Reduction of imine 4 with Zn in the presence of KOH and catalytic hydrogenation with Pd-C in methanol failed. Reduction of epimeric amides 5a and 5b with excess LiAlH4 gave diamines 6a and 6b in 64% and 61% yields, respectively. Diamines 6a and 6b were isolated with a diastereoselectivity of 99:1 and 90:10, respectively. Finally, the cyclic amidinium salts 7a–c were isolated in 65–72% yield by treating diamines 6a and 6b in triethyl orthoformate in the presence of ammonium tetrafluoroborate or ammonium chloride at elevated temperature [30,31]. The exo-amidinium slats 7a and 7b were isolated in 99:1 dr, while the endo-salt 7c was isolated in 91:9 dr (Scheme 1). Recrystallization of 7c from i-PrOH did not improve the diastereomeric ratio.
2.2. Structure determination
The new compounds were characterized by spectroscopic methods (1H and 13C NMR, 2D NMR, HRMS, and IR). Compound 5b could not be isolated in pure form and was used in further transformation without additional purification.
The configuration of the newly formed stereocenter at C-2 for endo-isomers 6b and 7c was determined by NOESY spectroscopy. The NOEs between the exo-H(2) and the 8-Me group agreed with the (2S) configuration (Figure 2). Similarly, the (2R) configuration at the chiral C-2 center of compound 5a was in line with the cross peak between the 8-Me group and the exo-H-N proton observed in the NOESY spectra [32]. The endo-stereochemistry of isomers 6b and 7c was additionally confirmed based on the chemical shift and multiplicity correlations of the endo-H(3) proton at position 3, which appears as a doublet of doublet at 0.93 and 0.97 ppm, respectively (Figure 2) [21,23,32].
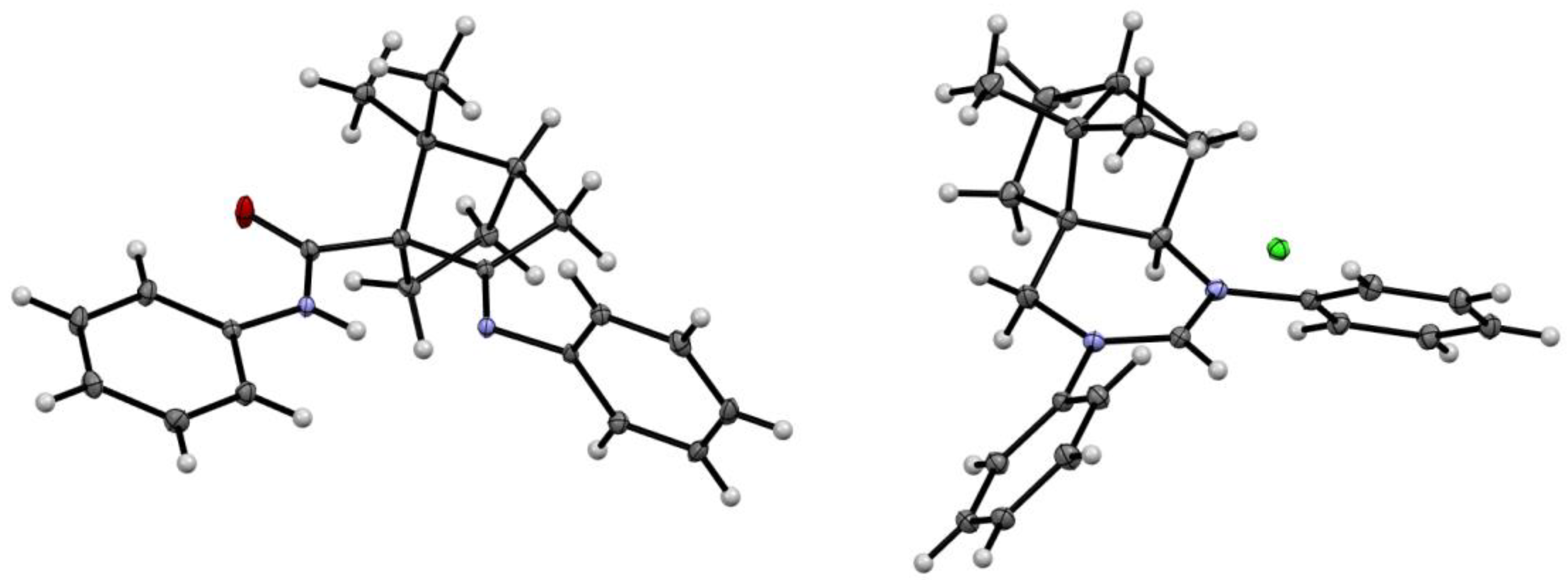
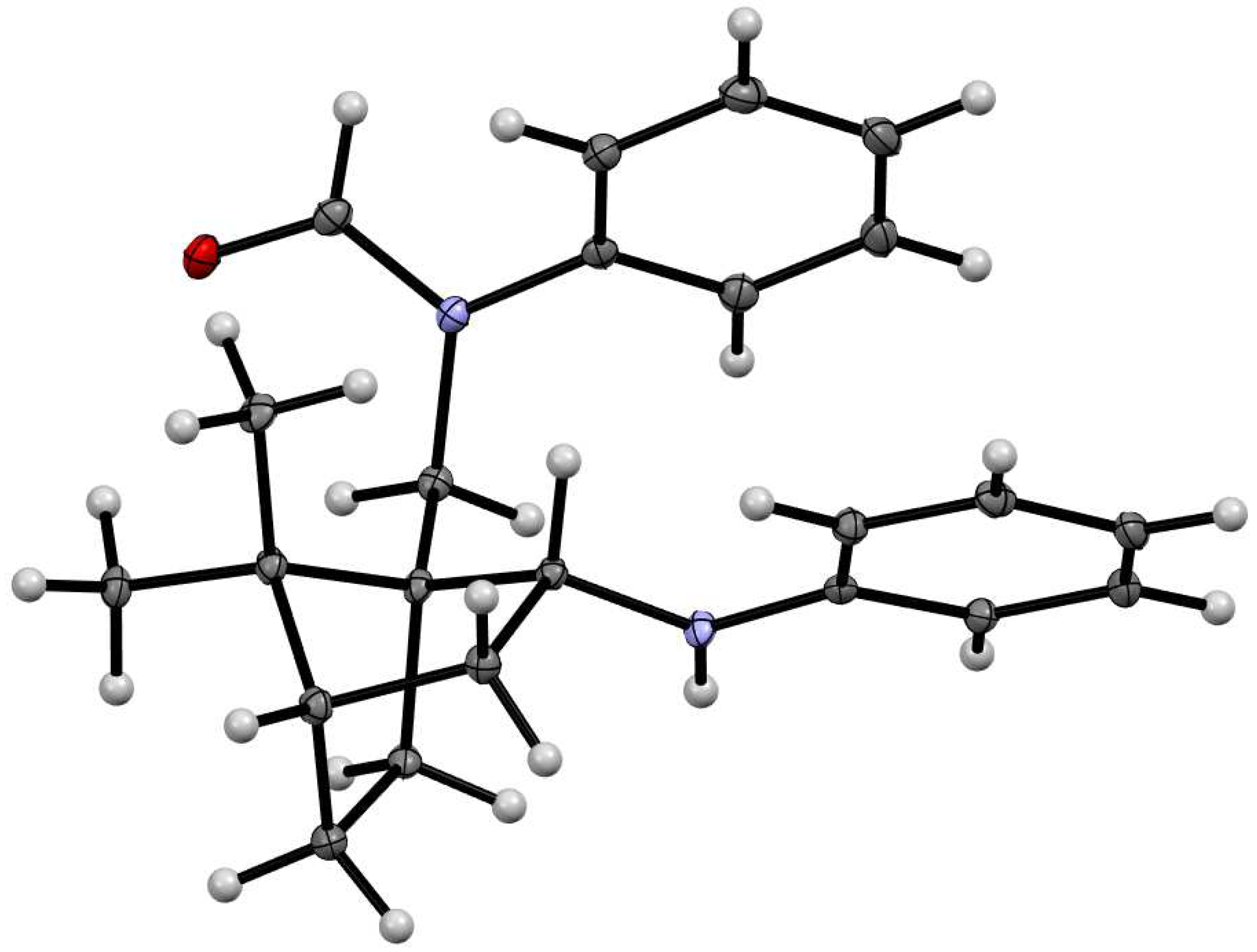
The structures of compounds 4 and 7a were also determined by single crystal X-ray analysis (Figure 3) [32].
Figure 2.
Determination of the absolute configuration at the C-2 based on the observed NOE correlation spectroscopy cross peaks (top) and chemical shift correlations in the series of C-2 endo-isomers (bottom).
Figure 2.
Determination of the absolute configuration at the C-2 based on the observed NOE correlation spectroscopy cross peaks (top) and chemical shift correlations in the series of C-2 endo-isomers (bottom).
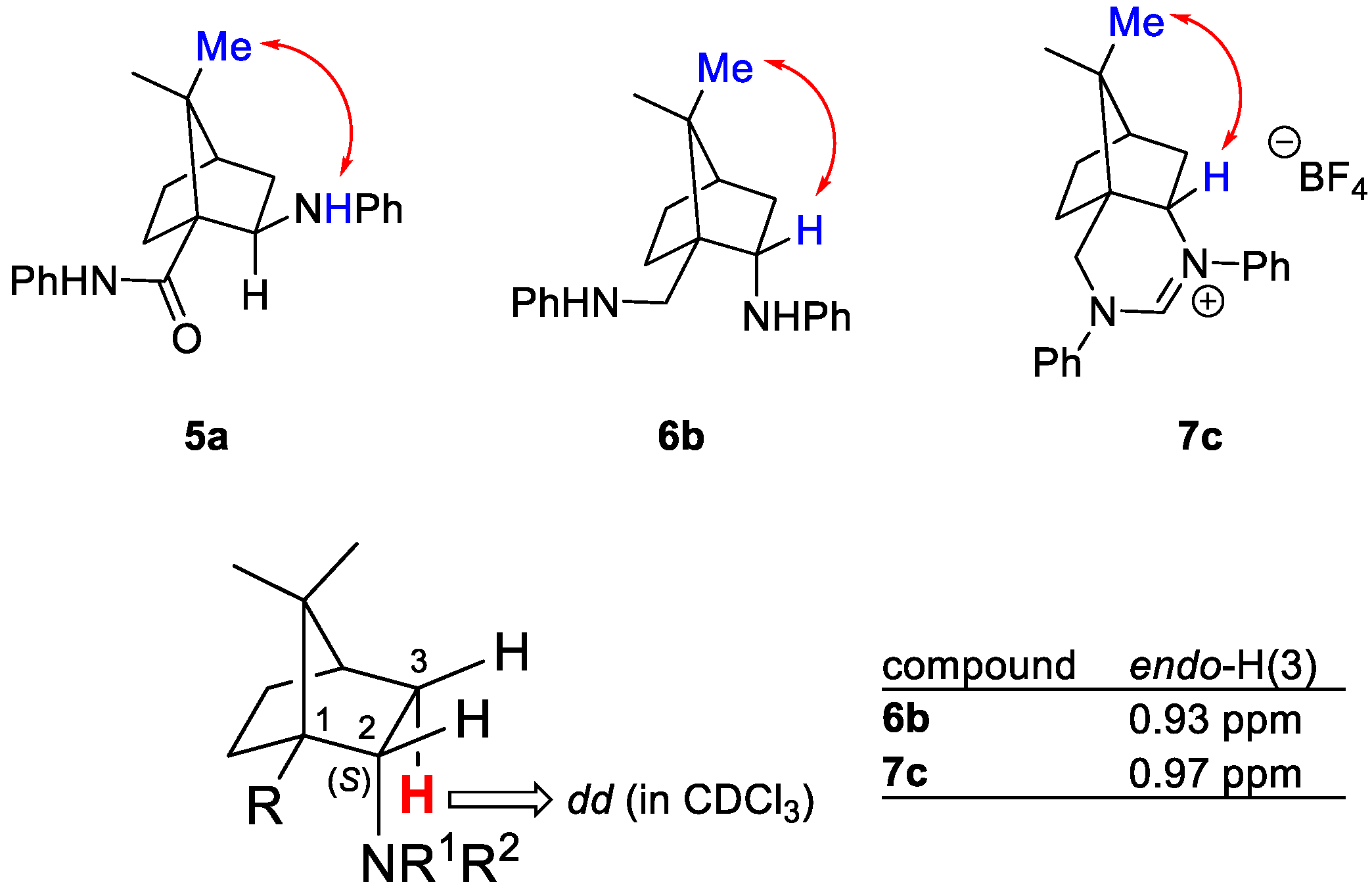
Figure 3.
Molecular structures of compound 4 (left) and 7a (right). Thermal ellipsoids are shown at 50% probability.
Figure 3.
Molecular structures of compound 4 (left) and 7a (right). Thermal ellipsoids are shown at 50% probability.
2.3. Performance of camphor-derived NHC precursors in benzoin reaction
The model reaction for evaluating the efficiency of the procatalyst 7a–c was the benzoin reaction with benzaldehyde, in which 10 mol% of the procatalyst was used (Scheme 2). Various bases were used for the in situ formation of the nucleophilic carbene catalyst. With aqueous Na2CO3 [33] as base, no reaction took place and the NHC precursors remained unchanged. Since the estimated pKa values of the amidinium salts 7a–c are in the range of 24 to 26 [34,35,36], stronger bases were used to obtain NHCs. Reactions in the presence of tBuONa, LiHDMS and LDA in anhydrous THF or 1,4-dioxane [37] did not give the benzoin product, while the amidinium salts 7a–c decomposed presumably due to traces of water present in the reaction mixture or during workup. The attempts to confirm the formation of the carbene catalyst in situ (in the NMR tube) were also unsuccessful, only the decomposition products were observed. To verify the hydrolysis of the amidinium salt 7 under basic conditions and to identify the decomposition product, the amidinium salt ent-7c (dr = 82:18; prepared from (1R)-(‒)-ketopinic acid) was hydrolyzed in a mixture of THF and water with two equivalents of NaOH. After 18 hours, the amidinium starting salt ent-7c was quantitatively hydrolyzed to an aminoamide mixture 8/8’ in the ratio 81:19. Crystallization of the crude product yielded single crystals of 8 (dr = 95:5) suitable for X-ray analysis, which confirmed the structure of the hydrolyzed product 8 (Figure 4). The absolute configuration at position 2 was additionally confirmed by NOESY measurement [21,23,32]. The amidinium salts ent-7c are hydrolytically unstable even in the presence of traces of water and are therefore not suitable as precursors of NHCs under applied reaction conditions.
3. Conclusions
Starting from (1S)-(+)-ketopinic acid, three NHC precursors based on camphor were prepared in a diastereoselective five-step synthesis. Both the exo- and endo-diastereomer of the NHC precursors were also analyzed in an asymmetric benzoin reaction. The desired benzoin product was not observed under any reaction conditions. On the other hand, we have shown that the amidinium salt procatalysts 7 decompose under basic conditions. All new compounds were fully characterized, including determination of the absolute configuration by X-ray diffraction, NOESY measurements and NMR data correlation.
4. Materials and Methods
4.1. Materials and General Methods
Solvents for extractions and chromatography were of technical grade and were distilled prior to use. Extracts were dried over technical grade anhydrous Na2SO4. Melting points were determined on a Kofler micro hot stage and on SRS OptiMelt MPA100 – Automated Melting Point System (Stanford Research Systems, Sunnyvale, California, United States). The NMR spectra were obtained on a Bruker UltraShield 500 plus (Bruker, Billerica, Massachusetts, United States) at 500 MHz for 1H and 126 MHz for 13C nucleus, using CDCl3 with TMS as the internal standard, as solvents. Mass spectra were recorded on an Agilent 6224 Accurate Mass TOF LC/MS (Agilent Technologies, Santa Clara, California, United States), IR spectra on a Perkin-Elmer Spectrum BX FTIR spectrophotometer (PerkinElmer, Waltham, Massachusetts, United States). Column chromatography (CC) was performed on silica gel (Silica gel 60, particle size: 0.035-0.070 mm (Sigma-Aldrich, St. Louis, Missouri, United States)). All the commercially available chemicals used were purchased from Sigma-Aldrich (St. Louis, Missouri, United States).
4.1.1. Synthesis of (1R,4R)-7,7-dimethyl-2-oxo-N-phenylbicyclo[2.2.1]heptane-1-carboxamide (3) [28]
SOCl2 (8 mL) was added to the flask containing (1S)-(+)-ketopinic acid (1) (20 mmol, 3.644 g) under argon. The reaction mixture was stirred for 2 h at room temperature and then for 1 h under reflux. Excess SOCl2 was evaporated in vacuo. The crude acid chloride 2 was immediately reacted further.
To a solution of acid chloride 2 (20 mmol) in anhydrous toluene at 0°C (20 mL) were added dropwise aniline (20 mmol, 1.823 mL) and Et3N (7 mL). The reaction mixture was stirred at room temperature for 20 hours. Ethyl acetate (20 mL) was added to the reaction mixture, followed by extraction with NaCl (aq. sat., 2 × 10 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The crude amide 3 was further reacted without additional purification. The crude amide 3 can, if needed, be purified by recrystallization from EtOH. Yield: 3.838 g (18.8 mmol, 94%) of white solid. 1H-NMR (500 MHz, CDCl3) δ (ppm): 1.04 (s, 3H) 1.34 (s, 3H), 1.45 – 1.52 (m, 1H), 1.68 – 1.75 (m, 1H), 2.05 (d, J=18.8 Hz, 1H), 2.12 (t, J=4.5 Hz, 1H), 2.16 – 2.25 (m, 1H), 2.55 – 2.65 (m, 2H), 7.07 – 7.12 (m, 1H), 7.29 – 7.35 (m, 2H), 7.59 – 7.64 (m, 2H), 9.70 (br s, 1H).
4.1.2. Synthesis of (1R,4R,E)-7,7-dimethyl-N-phenyl-2-(phenylimino)bicyclo[2.2.1]heptane-1-carboxamide (4)
To a solution of amide 3 (10 mmol, 2.573 g) in anhydrous toluene (40 mL) under argon were added aniline (50 mmol, 4.556 mL) and para-toluenesulfonic acid monohydrate (2 mmol, 380 mg). The flask was fitted with a Dean–Stark trap filled with activated 4 Å molecular sieves and a reflux condenser. The reaction mixture was refluxed for 20 h. To a cooled reaction mixture were added EtOAc (30 mL) and H2O (30 mL) and the phases were separated. The aqueous phase was extracted with EtOAc (2 × 20 mL). The combined organic phase was washed with brine (10 mL), dried under anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The crude product was purified by CC (Silica gel 60, EtOAc/petroleum ether = 1:5). The fractions containing pure product 4 were combined and the volatiles evaporated in vacuo. Yield: 1.629 g (4.9 mmol, 49%) of orange solid; mp = 138–140°C. [α]Dr.t. = +76.9 (0.13, CHCl3). EI-HRMS: m/z = 333.1956 (MH+); C22H25N2O requires: m/z = 333.1961 (MH+); νmax 3238, 3177, 3116, 3022, 3000, 2970, 2954, 1680, 1594, 1547, 1487, 1455, 1443, 1391, 1375, 1322, 1297, 1253, 1229, 1206, 1196, 1154, 1102, 1073, 1041, 1026, 966, 905, 885, 870, 833, 816, 790, 760, 712, 695, 662 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 1.05 (s, 3H), 1.37 (s, 3H), 1.34 – 1.41 (m, 1H), 1.83 (ddd, J=4.8, 9.4, 13.7 Hz, 1H), 1.91 – 1.99 (m, 2H), 2.10 – 2.19 (m, 1H), 2.41 (dt, J=4.1, 18.1, 1H), 2.67 – 2.76 (m, 1H), 6.81 – 6.91 (m, 2H), 7.03 – 7.09 (m, 1H), 7.13 – 7.19 (m, 1H), 7.26 – 7.34 (m, 2H), 7.35 – 7.42 (m, 2H), 7.58 – 7.67 (m, 2H), 11.49 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 19.64, 20.51, 27.58, 30.56, 35.99, 43.28, 50.25, 60.70, 118.93, 119.26, 119.35, 123.05, 123.82, 128.10, 128.13, 128.64, 128.67, 128.70, 137.88, 148.56, 168.45, 182.04.
4.1.3. Synthesis of (1R,2R,4R)-7,7-dimethyl-N-phenyl-2-(phenylamino)bicyclo[2.2.1]heptane-1-carboxamide (5a)
NaCNBH3 (12 mmol, 794 mg, ω = 0.95) was added to a solution of imine 4 (332 mg, 1 mmol) in anhydrous MeOH (15 mL) under argon. Then a catalytic amount of anhydrous acetic acid (0.2 mL) was added and the reaction mixture was stirred at room temperature for 5 h. The reaction was stopped by adding a saturated solution of NaHCO3 (5 mL) and EtOAc (10 mL), and the phases were separated. The aqueous phase was extracted with EtOAc (10 mL) and the combined organic phase was washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. Yield: 304 mg (0.91 mmol, 91%, dr 93:7) of dirty white solid; mp = 174–175°C. [α]Dr.t. = –10.6 (0.12, CHCl3). EI-HRMS: m/z = 335.2112 (MH+); C22H27N2O requires: m/z = 335.2118 (MH+); νmax 3333, 2954, 1652, 1601, 1519, 1498, 1439, 1388, 1308, 1245, 1180, 1104, 1072, 869, 746, 690 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 1.10 (s, 3H), 1.22 – 1.29 (m, 1H), 1.31 (s, 3H), 1.46 – 1.53 (m, 1H), 1.81 – 1.87 (m, 1H), 1.89 (t, J=4.3 Hz, 1H), 1.91 – 1.98 (m, 1H), 2.15 (dd, J=8.6, 12.8 Hz, 1H), 2.58 (td, J=4.6, 12.4 Hz, 1H), 3.58 (dt, J=5.0, 9.3 Hz, 1H), 4.09 (d, J=4.9 Hz, 1H), 6.62 – 6.68 (m, 2H), 6.74 – 6.79 (m, 1H), 6.97 – 7.02 (m, 1H), 7.13 – 7.23 (m, 6H), 8.92 (br s, 1H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 21.48, 21.50, 26.91, 31.65, 42.35, 45.79, 50.79, 58.43, 63.09, 114.36, 119.09, 120.52, 123.98, 128.71, 129.52, 138.04, 147.47, 171.17.
4.1.4. Synthesis of (1R,2S,4R)-7,7-dimethyl-N-phenyl-2-(phenylamino)bicyclo[2.2.1]heptane-1-carboxamide (5b)
Imine 4 (3 mmol, 997 mg) was dissolved in n-PrOH (100 mL) and the mixture was heated to 95°C. Then the first sodium piece was added to the reaction mixture, followed by another sodium piece after the first sodium piece had reacted, then the third, and so on. After 2 hours at 95°C, when the last sodium piece had reacted, H2O (100 mL) and Et2O (100 mL) were added to the cooled reaction mixture and the phases were separated. The aqueous phase was extracted with Et2O (2 × 100 mL) and the combined organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles evaporated in vacuo. The crude amine 5b was further reacted without additional purification. Yield: 943 mg (2.82 mmol, 94%,) of grey oil.
Synthesis of (1S,2R,4R)-7,7-dimethyl-N-phenyl-1-((phenylamino)methyl)bicyclo[2.2.1]-heptan-2-amine (6a)
To a solution of compound 5a (0.6 mmol, 201 mg) in anhydrous THF (2 mL) under argon at room temperature was added LiAlH4 (2.4 M in THF, 1.0 mL) dropwise. After addition, the reaction mixture was stirred for 20 h at 60°C. The reaction was cooled (0°C) and quenched by careful addition of a mixture of H2O and THF in a 1:5 ratio. The reaction mixture was filtered and the cake was washed with EtOAc (3 × 15 mL). The collected liquid was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The crude product was purified by CC (Silica gel 60, EtOAc/petroleum ether = 1:10). The fractions containing the pure product 6a were combined and the volatiles were evaporated in vacuo. Yield: 123 mg (0.384 mmol, 64%, dr 99:1) of dirty white semisolid. [α]Dr.t. = –123.7 (0.12, CHCl3). EI-HRMS: m/z = 321.2323 (MH+); C22H29N2 requires: m/z = 321.2325 (MH+); νmax 3412, 3050, 2951, 2876, 1600, 1499, 1429, 1387, 1369, 1302, 1251, 1180, 1152, 1096, 1073, 1028, 992, 866, 744, 689 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.99 (s, 3H), 1.17 (s, 3H), 1.21 – 1.28 (m, 1H), 1.46 – 1.53 (m, 1H), 1.70 – 1.87 (m, 4H), 1.91 – 1.99 (m, 1H), 3.20 (d, J=12.4 Hz, 1H), 3.29 (d, J=12.3 Hz, 1H), 3.50 – 3.56 (m, 1H), 3.69 (br s, 1H), 4.05 (br s, 1H), 6.55 – 6.62 (m, 4H), 6.68 (t, J=7.3 Hz, 2H), 7.08 – 7.19 (m, 4H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 21.12, 21.24, 27.41, 34.22, 40.72, 43.58, 46.13, 47.69, 51.93, 59.63, 113.42, 113.53, 117.47, 117.64, 129.27, 129.39, 147.58, 149.05.
4.1.5. Synthesis of (1S,2S,4R)-7,7-dimethyl-N-phenyl-1-((phenylamino)methyl)bicyclo[2.2.1]-heptan-2-amine (6b)
To a solution of compound 5b (0.5 mmol, 201 mg) in anhydrous THF (2 mL) under argon at room temperature was added LiAlH4 (2.4 M in THF, 1.0 mL) dropwise. After addition, the reaction mixture was stirred for 20 h at 60°C. The reaction was cooled (0°C) and quenched by careful addition of a mixture of H2O and THF in a 1:5 ratio. The reaction mixture was filtered and the cake was washed with EtOAc (3 × 15 mL). The collected liquid was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The crude product was purified by CC (Silica gel 60, EtOAc/petroleum ether = 1:10). The fractions containing the pure product 6b were combined and the volatiles were evaporated in vacuo. Yield: 98 mg (0.305 mmol, 61%, dr 90:10) of grey semisolid. [α]Dr.t. = +25.0 (0.15, CHCl3). EI-HRMS: m/z = 321.2323 (MH+); C22H29N2 requires: m/z = 321.2325 (MH+); νmax 3403, 3050, 3020, 2951, 2875, 1600, 1499, 1430, 1388, 1372, 1310, 1276, 1252, 1179, 1153, 1100, 1072, 1045, 1028, 992, 866, 744, 689, 617 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.93 (dd, J=3.8, 13.1 Hz, 1H), 1.03 (s, 3H), 1.08 (s, 3H), 1.29 – 1.36 (m, 1H), 1.61 – 1.69 (m, 1H), 1.73 (t, J=4.6 Hz, 1H), 1.84 – 1.92 (m, 1H), 2.00 – 2.07 (m, 1H), 2.44 – 2.52 (m, 1H), 3.18 (d, J=11.7 Hz, 1H), 3.24 (d, J=11.7 Hz, 1H), 3.88 – 3.94 (m, 1H), 4.01 (s, 1H), 4.37 (s, 1H), 6.48 – 6.54 (m, 2H), 6.62 – 6.68 (m, 3H), 6.70 – 6.75 (m, 1H), 7.08 – 7.19 (m, 4H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 19.53, 20.54, 25.30, 28.31, 39.32, 45.67, 46.91, 48.80, 51.27, 58.44, 113.07, 114.48, 117.28, 118.31, 129.23, 129.48, 148.13, 149.17.
4.1.6. Synthesis of (7R,8aR)-9,9-dimethyl-1,3-diphenyl-3,5,6,7,8,8a-hexahydro-4H-4a,7-methanoquinazolin-1-ium chloride (7a)
A mixture of diamine 6a (0.25 mmol, 80 mg, dr 99:1), triethyl orthoformate (1.5 mL), and NH4Cl (0.26 mmol, 13 mg) was stirred for 5 h at 120°C. The reaction mixture was cooled to room temperature and then Et2O (5 mL) was added. The resulting precipitate was filtered off, and the filter cake was washed thoroughly with CH2Cl2. The filtrate was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The product 6a was recrystallized from i-PrOH at room temperature by slow evaporation. Yield: 62 mg (0.170 mmol, 68%, dr 99:1) of white solid; mp = 143–146°C. [α]Dr.t. = +3.6 (0.14, CHCl3). EI-HRMS: m/z = 331.2166 (M+); C23H27N2 requires: m/z = 331.2169 (M+); νmax 3013, 2953, 2933, 2880, 1663, 1592, 1497, 1455, 1381, 1330, 1300, 1240, 1212, 1030, 934, 767, 728, 704, 638 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 1.03 (s, 3H), 1.15 (s, 3H), 1.30 – 1.38 (m, 1H), 1.66 (dd, J=8.6, 13.7 Hz, 1H), 1.70 – 1.76 (m, 1H), 1.80 – 1.91 (m, 4H), 3.61 (d, J=13.9 Hz, 1H), 4.83 (d, J=13.9 Hz, 1H), 4.93 (dd, J=4.8, 8.7 Hz, 1H), 7.39 – 7.44 (m, 2H), 7.45 – 7.51 (m, 4H), 7.57 – 7.62 (m, 2H), 7.64 – 7.69 (m, 2H), 8.06 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 20.12, 20.21, 26.58, 32.21, 34.18, 45.66, 46.16, 47.97, 48.61, 60.65, 124.17, 124.70, 129.32, 129.40, 130.20, 130.33, 138.82, 141.81, 152.01.
4.1.7. Synthesis of (7R,8aR)-9,9-dimethyl-1,3-diphenyl-3,5,6,7,8,8a-hexahydro-4H-4a,7-methanoquinazolin-1-ium tetrafluoroborate (7b)
A mixture of diamine 6a (0.25 mmol, 80 mg, dr 99:1), triethyl orthoformate (1.5 mL), and NH4BF4 (0.26 mmol, 27 mg) was stirred for 5 h at 120°C. The reaction mixture was cooled to room temperature and then Et2O (5 mL) was added. The resulting precipitate was filtered off, and the filter cake was washed thoroughly with CH2Cl2. The filtrate was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The product 6a was recrystallized from i-PrOH at room temperature by slow evaporation. Yield: 75 mg (0.180 mmol, 72%, dr 99:1) of grayish white solid; mp = 280–285°C. [α]Dr.t. = +30.8 (0.25, CHCl3). EI-HRMS: m/z = 331.2170 (M+); C23H27N2 requires: m/z = 331.2169 (M+); νmax 2956, 2921, 1663, 1591, 1494, 1380, 1319, 1297, 1229, 1049, 1030, 916, 766, 696 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 1.04 (s, 3H), 1.13 (s, 3H), 1.31 – 1.38 (m, 1H), 1.64 – 1.92 (m, 6H), 3.61 (d, J=14.3 Hz, 1H), 4.46 (d, J=14.3 Hz, 1H), 4.55 (dd, J=5.3, 8.1 Hz, 1H), 7.37 – 7.53 (m, 10H), 7.85 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 20.22, 20.25, 26.56, 32.24, 34.35, 45.65, 46.12, 47.98, 48.05, 60.03, 123.99, 124.42, 129.52, 129.67, 130.42, 130.55, 138.69, 141.72, 151.62.
4.1.8. Synthesis of (4aS,7R)-9,9-dimethyl-1,3-diphenyl-3,5,6,7,8,8a-hexahydro-4H-4a,7-methanoquinazolin-1-ium tetrafluoroborate (7c)
A mixture of diamine 6a (0.25 mmol, 80 mg, dr 90:10), triethyl orthoformate (1.5 mL), and NH4BF4 (0.26 mmol, 27 mg) was stirred for 5 h at 120°C. The reaction mixture was cooled to room temperature and then Et2O (5 mL) was added. The resulting precipitate was filtered off, and the filter cake was washed thoroughly with CH2Cl2. The filtrate was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. The product 6a was recrystallized from i-PrOH at room temperature by slow evaporation. Yield: 68 mg (0.1625 mmol, 65%, dr 91:9) of grayish white solid; mp = 238–240°C. [α]Dr.t. = –25.5 (0.13, CHCl3). EI-HRMS: m/z = 331.2163 (M+); C23H27N2 requires: m/z = 331.2169 (M+); νmax 3082, 3064, 2952, 2881, 1645, 1591, 1494, 1457, 1419, 1396, 1368, 1330, 1285, 1232, 1162, 1050, 1026, 968, 829, 770, 753, 697, 656, 627 cm-1. 1H-NMR (500 MHz, CDCl3) δ (ppm): 0.97 (dd, J=5.9, 13.8 Hz, 1H), 1.08 (s, 3H), 1.20 (s, 3H), 1.23 – 1.29 (m, 1H), 1.69 – 1.78 (m, 1H), 1.87 – 2.00 (m, 3H), 2.19 – 2.28 (m, 1H), 3.49 (d, J=12.8 Hz, 1H), 4.44 (d, J=12.8 Hz, 1H), 4.91 (dd, J=6.0, 11.1 Hz, 1H), 7.36 – 7.54 (m, 10H), 7.83 (s, 1H). 13C-NMR (126 MHz, CDCl3) δ (ppm): 18.49, 20.07, 27.41, 28.05, 30.79, 45.49, 46.37, 47.61, 54.91, 59.40, 124.54, 124.78, 129.49, 129.70, 130.29, 130.41, 139.33, 141.95, 152.66.
4.1.9. Synthesis of N-(((1S,2S,4R)-7,7-dimethyl-2-(phenylamino)bicyclo[2.2.1]heptan-1-yl)methyl)-N-phenylformamide (8) and N-(((1S,2R,4R)-7,7-dimethyl-2-(phenylamino)bicyclo[2.2.1]heptan-1-yl)methyl)-N-phenylformamide (8’)
To a solution of ent-7c (0.0239 mmol, 10 mg, dr 82:18) in THF (1 mL) was added H2O (100 μL) and NaOH (0.0478 mmol, 1.9 mg). The resulting reaction mixture was stirred for 18 h at room temperature. The reaction mixture was diluted with Et2O (20 mL) and washed with H2O (1 mL) and brine (1 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and the volatiles were evaporated in vacuo. Yield: 6 mg (0.0172 mmol, 72%, dr 81:19) of colorless semisolid. 1H-NMR (500 MHz, CDCl3) for 8’: δ (ppm): 0.73 – 0.79 (m, 1H), 0.93 (s, 3H), 3.04 (dd, J=4.7, 7.3, 1H), 3.61 (d, J=11.1, 1H), 4.64 (d, J=14.9, 1H), 4.81 (s, 1H), 6.51 – 6.56 (m, 2H), 8.17 (s, 1H). 13C-NMR (126 MHz, CDCl3) for 8’: δ (ppm): 20.93, 21.01, 30.45, 35.33, 41.41, 43.11, 45.69, 48.85, 53.55, 59.23, 113.14, 116.55, 125.27, 127.41, 129.13, 129.64, 141.60, 147.75, 163.82.
The crude product was additionally crystallized from a mixture of CHCl3 and n-heptane by slow evaporation of chloroform at room temperature. Compound 8: Yield: 3 mg (0.0086 mmol, 36%, dr 95:5) of white solid; mp = 144–146°C. EI-HRMS: m/z = 349.2275 (M+); C23H29N2O requires: m/z = 349.2274 (M+); νmax 3383, 2965, 2943, 2863, 1660, 1592, 1517, 1495, 1432, 1390, 1358, 1313, 1257, 1215, 1180, 1128, 1069, 1022, 988, 917, 866, 826, 748, 691, 670 cm-1. 1H-NMR (500 MHz, CDCl3) for 8: δ (ppm): 0.89 (s, 3H), 0.87 – 0.93 (m, 1H), 1.03 (s, 3H), 1.14 – 1.21 (m, 1H), 1.44 – 1.52 (m, 1H), 1.61 – 1.77 (m, 3H), 2.29 – 2.38 (m, 1H), 3.63 (d, J=9.0, 1H), 3.87 (d, J=14.8, 1H), 4.07 (s, 1H), 4.14 (d, J=14.8, 1H), 6.43 – 6.48 (m, 2H), 6.66 (t, J=7.3, 1H), 7.09 – 7.16 (m, 4H), 7.17 – 7.21 (m, 1H), 7.29 – 7.36 (m, 2H), 8.31 (s, 1H). 13C-NMR (126 MHz, CDCl3) for 8: δ (ppm): 19.31, 20.14, 25.48, 27.95, 38.60, 44.43, 45.84, 48.67, 53.67, 56.60, 113.50, 117.18, 123.62, 126.57, 129.22, 129.71, 142.15, 148.49, 163.72.
4.2. General procedure for the catalytic asymmetric benzoin condensation reaction with benzaldehyde
To a solution/suspension of amidinium salt 7 (10 mol%) in anhydrous THF or 1,4-dioxane (in the case of Na2CO3, water was used as solvent), benzaldehyde (0.75 mmol) and then base (10 mol%; tBuONa, LiHDMS and LDA (1 M in THF/hexanes)) were added. The resulting reaction mixture was stirred for 24 h at room temperature. The reaction mixture was concentrated under reduced pressure. Part of the residue was used for 1H-NMR measurements, the rest was subjected to column chromatography.
4.3. X-ray Crystallography
Single-crystal X-ray diffraction data was collected on Agilent Technologies SuperNova Dual diffractometer with an Atlas detector using monochromated Cu-Kα radiation (λ = 1.54184 Å) at 150 K. The data was processed using CrysAlis PRO [38]. Using Olex2.1.2. [39], the structures were solved by direct methods implemented in SHELXS [40] or SHELXT [41] and refined by a full-matrix least-squares procedure based on F2 with SHELXT-2014/7 [42]. All nonhydrogen atoms were refined anisotropicallly. Hydrogen atoms were placed in geometrically calculated positions and were refined using a riding model. The drawings and the analysis of bond lengths, angles and intermolecular interactions were carried out using Mercury [43] and Platon [44]. Structural and other crystallographic details on data collection and refinement for compounds 4, 7a, and 8 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC Deposition Numbers 2302865, 2302861 and 2307379, respectively. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: deposit@ccdc.cam.ac.uk).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Copies of 1H- and 13C-NMR spectra; Copies of NOESY spectra; Copies of HRMS reports; Copies of IR spectra, Structure determination by X-ray diffraction analysis.
Author Contributions
Conceptualization, J.Š., L.C., U.G., J.S., and B.Š.; methodology, J.Š., L.C., and U.G.; software, J.Š., L.C., H.B., U.G., J.S., and B.Š.; validation, J.Š., L.C., H.B., U.G., J.S., F.P., and B.Š.; formal analysis, J.Š., U.G., H.B., and L.C.; investigation, J.Š., L.C., and U.G.; resources, L.C., U.G., and J.S.; data curation, J.Š., L.C., H.B., U.G., J.S., and B.Š.; writing—original draft preparation, U.G., J.S., and B.Š.; writing—review and editing, L.C., U.G., J.S., F.P., and B.Š.; visualization, L.C., H.B., U.G., B.Š., and J.S.; supervision, U.G.; project administration, U.G. and J.S.; funding acquisition, U.G. and J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovenian Research Agency through grant P1-0179.
Acknowledgments
We thank the EN-FIST Centre of Excellence, Dunajska 156, 1000 Ljubljana, Slovenia, for the use of their BX FTIR spectrophotometer and Agilent 1260 Infinity LC for the HPLC analyses.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 7a–c are available from the authors.
References
- Money, T. Remote functionalization of camphor: application to natural product synthesis. Org. Synth.: Theory Appl. 1996, 3, 1–83. [Google Scholar]
- Money, T. Camphor: a chiral starting material in natural product synthesis. Nat. Prod. Rep. 1985, 2, 253–289. [Google Scholar] [CrossRef]
- Oppolzer, W. Camphor derivatives as chiral auxiliaries in asymmetric synthesis. Tetrahedron 1987, 43, 1969–2004. [Google Scholar] [CrossRef]
- Oppolzer, W. Camphor as a natural source of chirality in asymmetric synthesis. Pure Appl. Chem. 1990, 62, 1241–1250. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef]
- Nugent, W. A. MIB: an advantageous alternative to DAIB for the addition of organozinc reagents to aldehydes. Chem. Commun. 1999, 1369–1370. [Google Scholar] [CrossRef]
- Stereoselective Organocatalysis; Rios R., Ed.; John Wiley & Sons, Hoboken, United States, 2013.
- Jakab, G.; Schreiner, P. R. Brønsted Acids: Chiral (Thio)urea Derivatives In Comprehensive Enantioselective Organocatalysis; Dalko P. I., Ed.; Wiley-VCH, Weinheim, Germany, 2013; pp 315–342.
- Rajaram, S.; Sigman, M. S. Design of Hydrogen Bond Catalysts Based on a Modular Oxazoline Template: Application to an Enantioselective Hetero Diels−Alder Reaction. Org. Lett. 2005, 7, 5473–5475. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-Derivatives in Asymmetric Organocatalysis - Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Enders, D.; Kallfass, U. An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem., Int. Ed. 2002, 41, 1743–1745. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-derived heterocycles syntheses and potential applications. Targets Heterocycl. Syst. 2015, 19, 62–100. DOI: http://dx.medra.org/10.17374/targets.2016.19.62. [CrossRef]
- Li, Y.; Feng, Z.; You, S.-L. D-Camphor-derived triazolium salts for catalytic intramolecular crossed aldehyde–ketone benzoin reactions. Chem. Commun. 2008, 2263–2265. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.-Q.; Zheng, C.; You, S.-L. Highly enantioselective intramolecular Michael reactions by D-camphor-derived triazolium salts. Chem. Commun. 2009, 5823–5825. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.-Q.; Jia, M.-Q.; You, S.-L. Enantioselective N-Heterocyclic Carbene-Catalyzed Michael Addition to α,β-Unsaturated Aldehydes by Redox Oxidation. Org. Lett. 2011, 13, 4080–4083. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.-Q.; You, S.-L. N-Heterocyclic Carbene-Catalyzed Enantioselective Intramolecular N-Tethered Aldehyde–Ketone Benzoin Reactions. ACS Catal. 2013, 3, 622–624. [Google Scholar] [CrossRef]
- Jia, M.-Q.; You, S.-L. Desymmetrization of Cyclohexadienones via Intramolecular Stetter Reaction to Construct Tricyclic Carbocycles. Synlett 2013, 24, 1201–1204. [Google Scholar] [CrossRef]
- Rong, Z.-Q.; Li, Y.; Yang, G.-Q.; You, S.-L. D-Camphor-Derived Triazolium Salts for Enantioselective Intramolecular Stetter Reactions. Synlett 2011, 1033–1037. [Google Scholar] [CrossRef]
- Rafiński, Z.; Kozakiewicz, A. Enantioselective Synthesis of Chromanones Bearing Quaternary Substituted Stereocenters Catalyzed by (1R)-Camphor-Derived N-Heterocyclic Carbenes. J. Org. Chem. 2015, 80, 7468–7476. [Google Scholar] [CrossRef] [PubMed]
- Rafiński, Z.; Kozakiewicz, A.; Rafińska, K. Highly efficient synthesis of spirocyclic (1R)-camphor-derived triazolium salts: application in the catalytic asymmetric benzoin condensation, Tetrahedron 2014, 70, 5739–5745. [CrossRef]
- Ričko, S.; Požgan, F.; Štefane, B.; Svete, J.; Golobič, A.; Grošelj, U. Stereodivergent Synthesis of Camphor-Derived Diamines and Their Application as Thiourea Organocatalysts. Molecules 2020, 25, 2978. [Google Scholar] [CrossRef]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Waser, M.; Grošelj, U. Synthesis and Catalytic Activity of Bifunctional Phase-Transfer Organocatalysts Based on Camphor. Molecules 2023, 28, 1515. [Google Scholar] [CrossRef]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, A.; Štefane, B.; Svete, J.; Grošelj, U. Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [Google Scholar] [CrossRef]
- Bartlett, P. D.; Knox, L. H. DL-Ketopinic acid. Org. Synth. 1965, 45, 55–56. [Google Scholar]
- Bartlett, P. D.; Knox, L. H. D,L-10-Camphorsulfonyl chloride. Org. Synth. 1965, 45, 14–16. [Google Scholar]
- Huynh, U.; McDonald, S. L.; Lim, D.; Uddin, M. N.; Wengryniuk, S. E.; Dey, S.; Coltart, D. M. Formation, Alkylation, and Hydrolysis of Chiral Nonracemic N-Amino Cyclic Carbamate Hydrazones: An Approach to the Enantioselective α-Alkylation of Ketones. J. Org. Chem. 2018, 83, 12951–12964. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Bian, Y. Synthesis of novel chiral Schiff bases and their application in asymmetric transfer hydrogenation of prochiral ketones. Heteroat. Chem. 2008, 19, 682–687. [Google Scholar] [CrossRef]
- Grošelj, U.; Sevšek, A.; Ričko, S.; Golobič, A.; Svete, J.; Stanovnik, B. Synthesis and Structural Characterization of Novel Camphor-derived Amines. Chirality, 2012, 24, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qin, X.; Fu, J.; Wang, X.; Su, X.; Hu, F.; Jiao, J.; Shi, M. Fine-tunable 3, 4-dihydroquinazol-2-ylidene carbenes: Synthesis, rhodium (I) complexes, and reactivity. Organometallics 2012, 31, 8275–8282. [Google Scholar] [CrossRef]
- Mincheva, R.; Narayana Murthy Chilla, S.; Todd, R.; Guillerm, B.; De Winter, J.; Gerbaux, P.; Coulembier, O.; Dubois, P.; Raquez, J.-M. Reactive Extrusion and Magnesium (II) N-Heterocyclic Carbene Catalyst in Continuous PLA Production. Polymers 2019, 11, 1987. [Google Scholar] [CrossRef]
- For details see the Supporting Information.
- Yan, J; Sun, R; Shi, K; Li, K; Yang, L; Zhong, G. N-heterocyclic carbene-catalyzed asymmetric benzoin reaction in water. J. Org. Chem. 2018, 83, 7547–7552. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Xue, X.-S.; Ji, P. Acidity Scale of N-Heterocyclic Carbene Precursors: Can We Predict the Stability of NHC–CO2 Adducts? Org. Lett. 2018, 20, 6041–6045. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xue, X.-S.; Fu, Y.; Ji, P. Comprehensive Basicity Scales for N-Heterocyclic Carbenes in DMSO: Implications on the Stabilities of N-Heterocyclic Carbene and CO2 Adducts. Chem. Asian J. 2020, 15, 169–181. [Google Scholar] [CrossRef]
- Wang, N.; Xu, J.; Lee, J.K. The importance of N-heterocyclic carbene basicity in organocatalysis. Org. Biomol. Chem. 2018, 16, 8230–8244. [Google Scholar] [CrossRef]
- Soeta, T.; Mizuno, S.; Hatanaka, Y.; Ukaji, Y. Asymmetric cross-benzoin condensation promoted by a chiral triazolium precatalyst bearing a pyridine moiety. Tetrahedron 2017, 73, 3430–3437. [Google Scholar] [CrossRef]
- CrysAlis PRO; Agilent Technologies UK Ltd.: Yarnton, UK, 2011.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cristallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of NHC precursors 7a–c; a dr could not be determined.
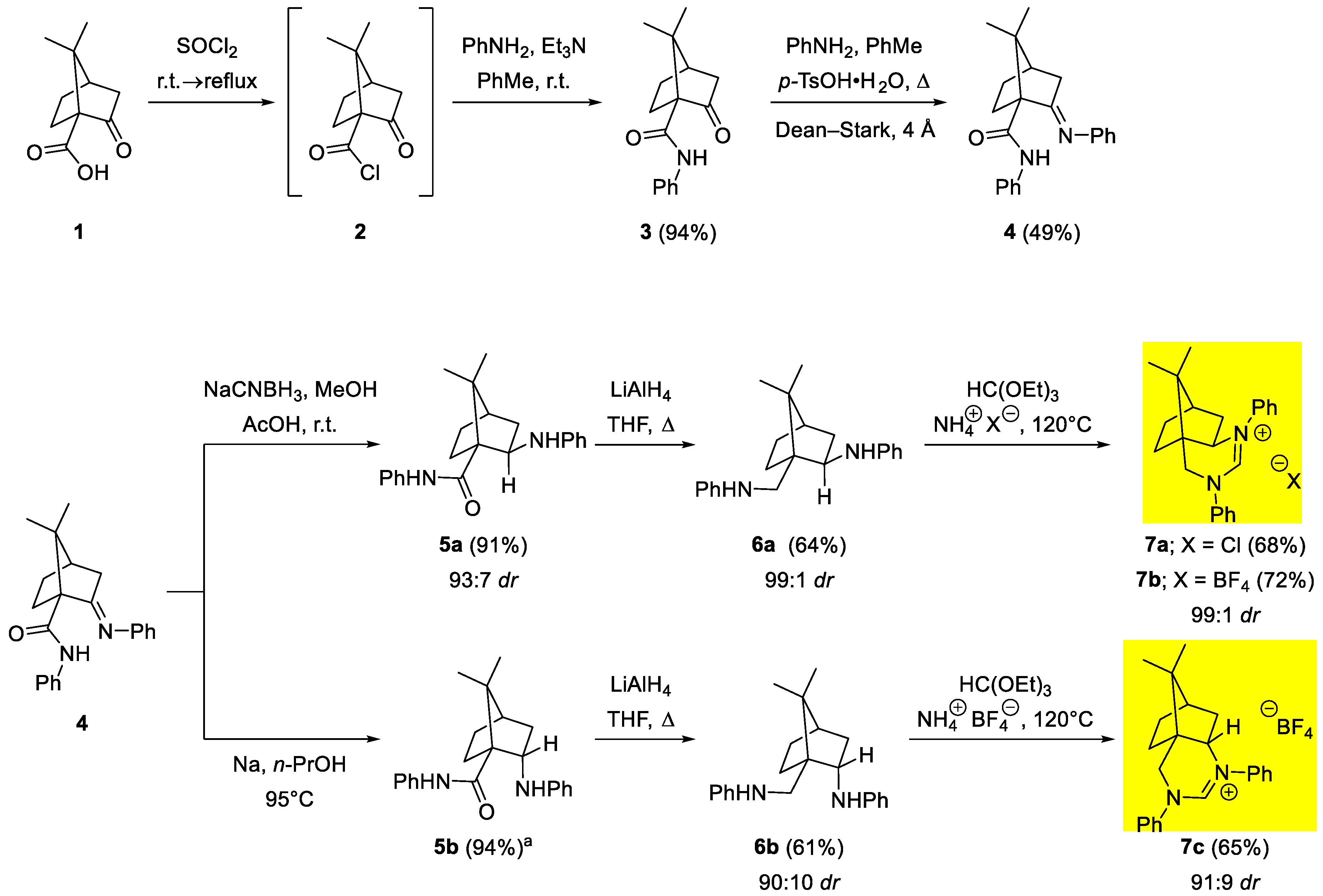
Scheme 2.
Benzoin reaction (top) and opening of the amidinium salt 7c (bottom).
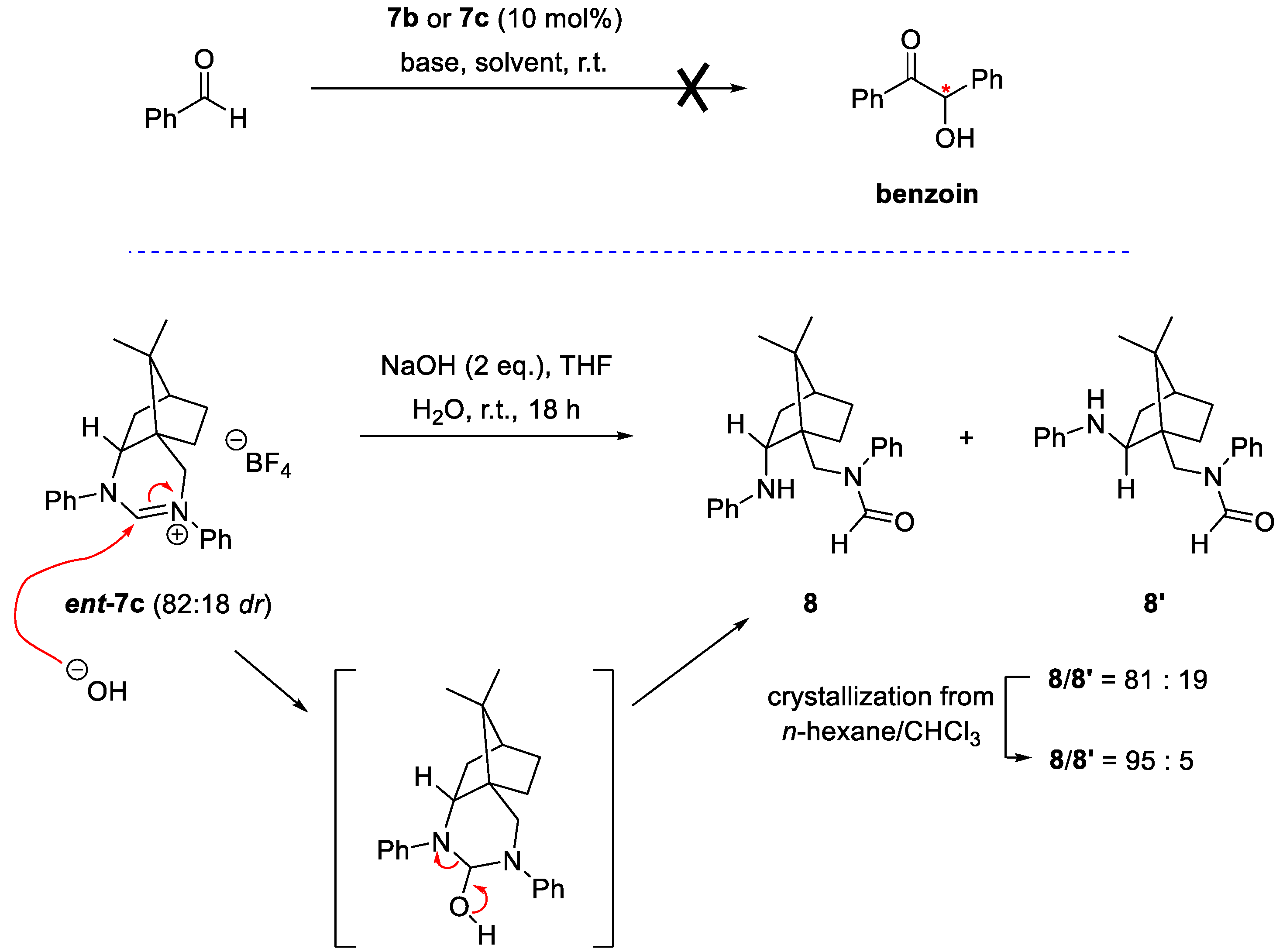
Figure 4.
Molecular structure of compound 8. Thermal ellipsoids are shown at 50% probability.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



