
Submitted:
26 September 2023
Posted:
27 September 2023
You are already at the latest version
Abstract
BACKGROUND AND OBJECTIVES. Rod-Cone Dystrophies (RCDs) are genetically determined retinal dystrophies, resulting in nyctalopia, visual field progressive reduction and visual acuity decay in the late stages. Retinitis Pigmentosa (RP) is the most described type. All modes of transmission can be identified in RP, with the autosomal dominant form (ADRP) accounting for about 20% of cases. To date, over 30 genes have been related to ADRP, with RP1 gene estimated to be involved in 5-10% of cases. In a cohort of patients affected by RCD and sharing the same geographic origin from Palermo province in Western Sicily, we identified a prevalent causative variant in the RP1 gene (NM_006269.2; c.2219C>G, p.Ser740*). Objective of our study was to analyse clinical and molecular data of this population and define the entity and the meaning of this finding.
MATERIALS AND METHODS. Eighty-four patients with a diagnosis of RCD or RP underwent deep phenotyping, pedigree definition and molecular characterization, mostly by Next Generation Sequencing (NGS). All potentially pathogenic variants have been confirmed through direct sequencing and familial segregation. Statistical evaluation of resulting data has been performed.
RESULTS. In 28 probands we identified a pathogenic variant (p.Ser740*) in the RP1 gene that resulted significantly prevalent. Some patients, after careful interviewing, were found to share the same pedigree; we ultimately defined 20 family groups with no traceable consanguinity.
CONCLUSIONS. The high prevalence of the p.Ser740* variant in RP patients from Western Sicily unveils the presence of a founder effect, which has implications for the molecular diagnosis of RCD in patients coming from this Italian region. This variant can be primarily searched in RP affected subjects displaying compatible mode of transmission and phenotype, with an advantage in terms of costs and analysis time. Moreover, given its high prevalence, RP1 p.Ser740* variant could represent a potential candidate for the development of therapeutic strategies based on gene editing or translational read-through therapy for suppression of nonsense variants.
Keywords:
rod-cone dystrophy
; retinitis pigmentosa
; RP1 gene
; founder effect
; inherited retinal dystrophies
; founder mutation
1. Introduction
Inherited Retinal Dystrophies (IRDs) include a spectrum of retinal diseases, characterised by genetically determined dysfunctions and progressive degeneration of the retina. Retinitis Pigmentosa (RP) is the traditional definition of a subgroup of IRDs in which rods are primarily affected before cones (rod-cone dystrophies, RCDs), with a prevalence estimated as about 1/3000-1/5000 [1,2]. RCD affected patients experience a visual impairment characterised by nyctalopia, progressive concentric reduction of the visual field and visual acuity decay in the advanced stages. Classic signs include optic disc pallor, attenuated retinal vessels, and diffuse pigmentary changes of the retina. An abnormal or unrecordable electroretinogram (ERG), with the scotopic component being altered more severely and earlier than the photopic one, is the hallmark of this type of retinal dystrophy. Age at onset can be very variable, usually being within the first two decades, but not uncommonly later [3]. In RP all modes of transmission can be recognised, with different percentages: autosomal dominant (ADRP) 15-25%, autosomal recessive (ARRP) 5-20%, X-linked (XLRP) 5-15%, and rarely mitochondrial or digenic. Forty to fifty percent of patients are sporadic (simplex RP), with unremarkable family history for the condition [4,5,6].
Non-syndromic RP is genetically highly heterogeneous. To date, about 31 genes have been associated with ADRP, 66 with ARRP and 3 genes with the XLRP form, in addition to several more loci [7,8]. However, in a consistent number of patients, the causative molecular defect cannot be identified, leading to the conclusion that other genes have to be be characterised yet, or that different mechanisms can play a pathogenic role.
The RP1 gene (OMIM # 603937) has been identified in 1999 as underlying the RP9 locus-linked form of RP. It is located on chromosome 8q12.1 and encompasses 4 exons of which 3 are coding [10]. It encodes an oxygen-regulated photoreceptor protein that is localized in the connecting cilia of both cones and rods photoreceptors and has a role in the transport of proteins between the inner and outer segments of photoreceptors, cilial structure maintenance, and stabilization of disc membranes in the outer segments [11].
Incomplete penetrance and variable expressivity are described for RP1-related RP [10].
Most pathogenic variants have a dominant effect, but a recessive mode of transmission is described as well [12,13,14].
Several dominant pathogenic variants are located in exon 4 of RP1 gene and generate stop codons that cause the formation of truncated proteins. The c.2029C>T (p.Arg677*) nonsense change is the most reported pathogenic variant in RP1 [15,16].
Mutation of the RP1 gene is considered to underlie 5-10% of ADRP cases, but its prevalence varies between studies and populations [5,17].
Individuals affected by dominantly inherited RP1 variants usually display a classic phenotype of RP and suffer from nyctalopia and visual field concentric progressive reduction. Most patients preserve a relatively acceptable visual acuity and reduced - but still present - residual central visual field for many years. As in other forms of RP, early cataract, cystoid macular oedema (CME) and pre-retinal fibrosis can complicate the visual outcome in some patients.
We performed molecular genetic characterization of a cohort of patients affected by RCD/RP, sharing the same geographic origin from the Palermo province in Western Sicily, Italy (an area of about 5000 sqKm and about 1275000 inhabitants) and we identified a recurrent nonsense variant in the RP1 gene (NM_006269.2; c.2219C>G, p.Ser740*).
Aim of this study was to assess the prevalence of this variant in this population, and to investigate the possible presence of a founder effect mechanism. Such finding can have a considerable impact on the strategy for molecular testing in patients having compatible geographic origin, transmission mode and phenotype, where this variant could be primarily searched. Furthermore, it could potentially direct towards the identification of a considerably large number of patients who could then be the target for the development of new molecular therapies.
2. Materials and Methods
Eighty-four knowingly unrelated probands (50% males, 50% females) that independently accessed our medical retina clinics and had a clinical and instrumental diagnosis of RCD or RP, underwent genetic testing.
All patients or their guardians have given written informed consent. This study was conducted adhering to the tenets of the Declaration of Helsinki.
- Clinical and instrumental evaluation
Patients were diagnosed in different centres with variable equipment. In most cases we could obtain comprehensive ophthalmic examinations including best-corrected visual acuity (BCVA), intraocular pressure (IOP) measurement (noncontact tonometer), slit-lamp anterior and posterior segment biomicroscopy, colour fundus photography, autofluorescence, OCT and OCT angiography (the latter in a limited number of patients), stationary perimetry tests, and electrodiagnostic tests (EDTs) performed according to ISCEV standards. When not performed personally by any of the authors of this study, exhibited documentation has been evaluated to establish or confirm a diagnosis. Clinical data are summarized in Table 1. Multimodal imaging of patients nos. 8, 9, 12, 13, 28 is shown in Figure 1, Figure 2, Figure 3, Figure 4 and Figure 5.
- Transmission mode definition
Genealogies were established by interviews to patients and their family members. Particularly surnames of the maternal branches have been recorded, in order to unmask less obvious eventual common origins. When no positive family history was reported, patients have been defined as sporadic. A consistent number of family members of the probands were self-reporting as unaffected but found to carry the pathogenic variant; in these cases, clinical and instrumental testing has been carried out, and the pathologic phenotype definition has followed the molecular diagnosis. Pedigrees are shown in Figure 6, Figure 7, Figure 8 and Figure 9.
- Statistical evaluation
Descriptive statistics were presented as median (interquartile range [IQR]), and percentages. Differences regarding CRD/RP modes of transmission between our cohort of patients and the general population, as well as regarding the causative genes for the AD form, have been evaluated by two-sample test of proportions (based on a z-statistic).
- Genetic analytic strategy
Patients of the selected cohort were analysed in different centres and with different molecular methodologies depending on the year of examination.
In two probands (# 10 and # 26) the RP1 p.Ser740* variant has been identified through Sanger sequencing of candidate genes for ADRP at CEINGE (Naples, Italy) in 2005. All other patients underwent NGS analysis. Patients # 2, 4, 6, 7, 11, 14, 15, 16, 20, 27, 28 were tested at AOOR Villa Sofia-Cervello (Palermo, Italy) on a panel of 70 RP-related genes (Supplementary Material S1), while patients # 1, 3, 5, 8, 9, 12, 13, 17, 18, 19, 21, 22, 23, 24, 25, were tested at MAGI’S in Bolzano (Italy) on a panel of 73 genes (Supplementary Material S2). Sanger direct sequencing was used to validate variants that were considered potentially pathogenic.
All variants were sought in the Human Gene Mutation Database, professional version 2023.1 (https://my.qiagendigitalinsights.com/bbp/view/hgmd/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), LOVD, Leiden Open Variation Database (https://www.lovd.nl/) and gnomAD (https://gnomad.broadinstitute.org/) databases. The pathogenic role of the identified variants was predicted by using the bioinformatic prediction tool Varsome, which classifies variants according to the American College of Medical Genetics and Genomics standards and guidelines [18,19].
Segregation analysis was also performed for informative and available family members, often following specific request for pre-symptomatic status definition. Only variants confirmed as pathogenic or likely pathogenic, through in silico tools and familial segregation, are reported.
3. Results
In the selected cohort of 84 probands with a clinical diagnosis of RP or RCD, a molecular genetic analysis has been carried out with the following results.
3.1. Molecular data analysis
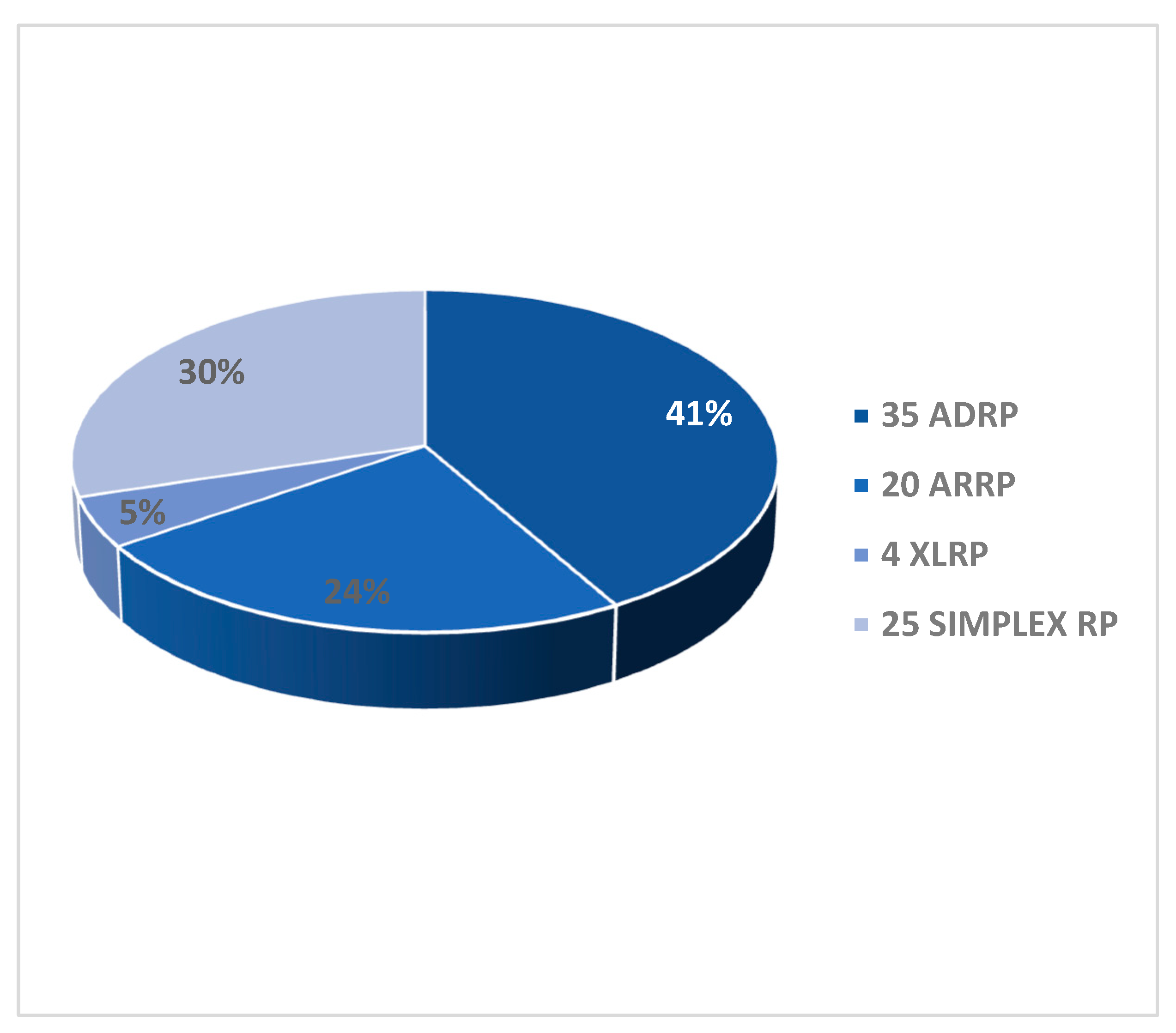
Pedigrees of the 84 RCD/RP probands revealed different patterns of Mendelian inheritance. We identified 35 AD (41%), 20 AR (24%), 4 XL (5%) putative transmission modes. Twenty-five probands (30%) did not report any family history and were therefore defined as sporadic (or simplex) (Graph 1).
Graph 1.
Transmission mode of RCD/RP in the 84 patients cohort. On the right the number of patients with the different modes of transmission by pedigree analysis. In the graph the relative percentages.
Graph 1.
Transmission mode of RCD/RP in the 84 patients cohort. On the right the number of patients with the different modes of transmission by pedigree analysis. In the graph the relative percentages.
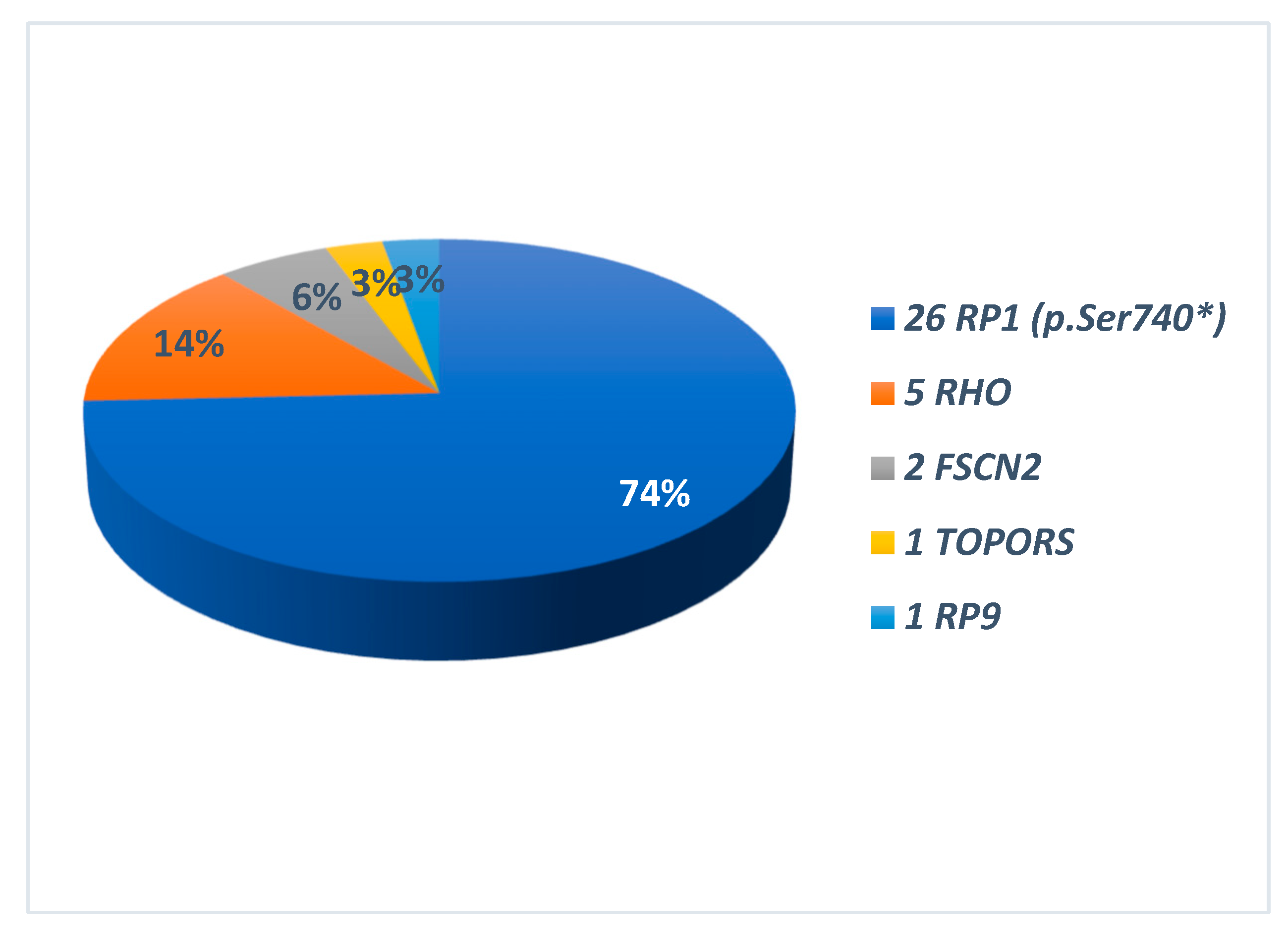
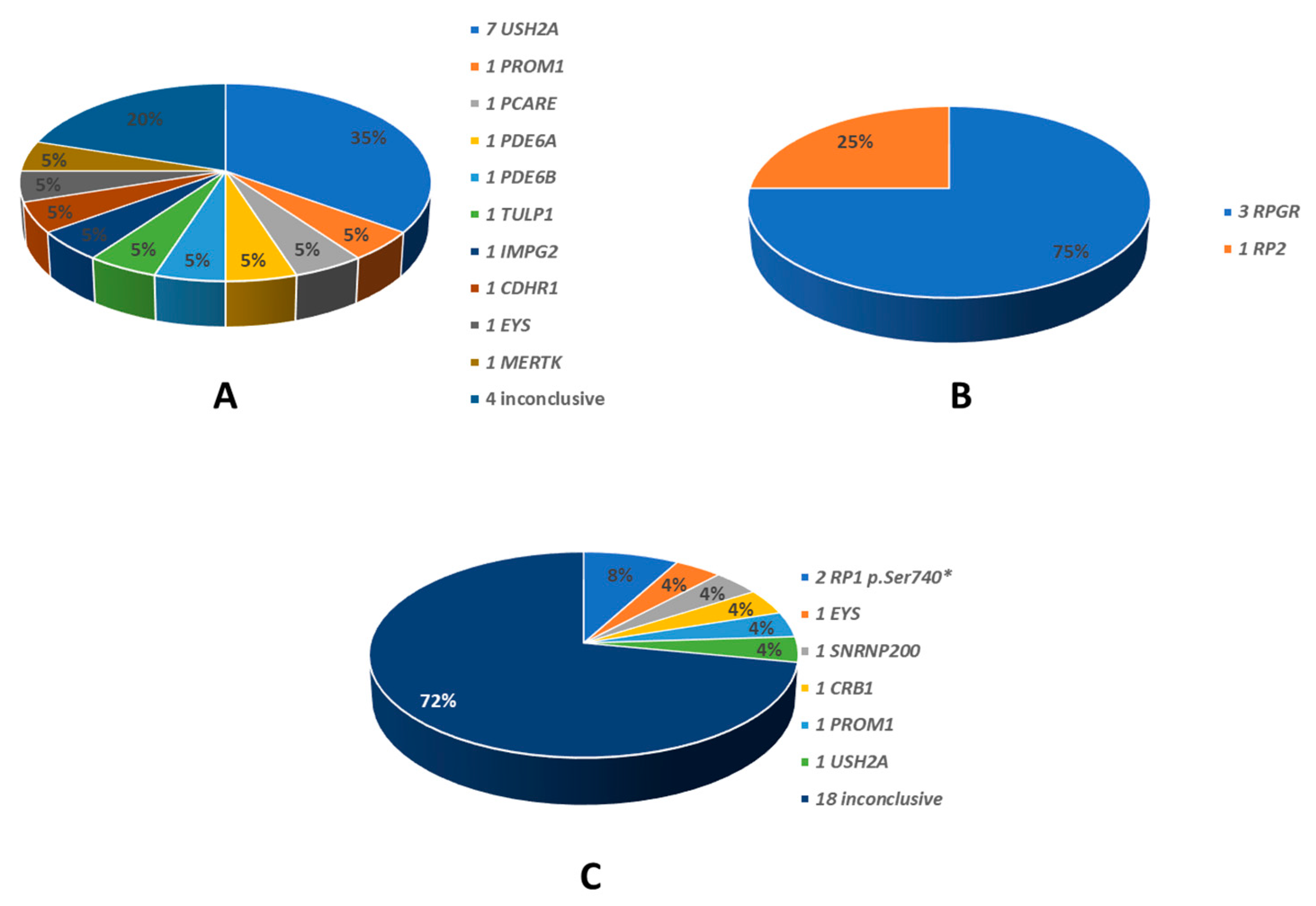
The identified disease-causing variants, which were consistent with the different patterns of transmission, are also summarised in Graphs 2 and 3. All the genes found to be causative, are described in the literature as RP-related [7,8]. Among the 35 ADRP patients, 26 had heterozygous variants in RP1 (p.Ser740* being the only variant identified in all of them), 5 in RHO, 2 in FSCN2, 1 for each in TOPORS and RP9 (Graph 2). The 20 ARRP patients included 7 probands affected by biallelic variants in USH2A, 1 patient each with mutation of PROM1, PCARE, PDE6B, CDHR1, IMPG2, MERTK, TULP1, EYS, PDE6A; in 4 patients the molecular analysis was inconclusive (Graph 3-A). Three patients with XLRP were carrying RPGR variants, and 1 patient was hemizygous for RP2 mutation (Graph 3-B). Molecular analysis performed for the 25 sporadic patients was inconclusive in 18 cases; 2 patients resulted positive for the RP1 p.Ser740* variant, while the remaining patients were carrying pathogenic variants in SNRNP200, CRB, PROM1, EYS and USH2A (Graph 3-C). Clinical and molecular details of patients carrying variants in genes other than RP1 in this cohort are the subject of a further work still in progress. Results of genetic analysis were considered conclusive only when unambiguously verified by in silico tools, coupled with consistent familial segregation.
Graph 2.
Causing genes in 35 patients with autosomal dominant transmission (ADRP). On the right the different genes names. In the graph the relative percentages. In all RP1-related ADRP p.Ser740* was the only identified variant.
Graph 2.
Causing genes in 35 patients with autosomal dominant transmission (ADRP). On the right the different genes names. In the graph the relative percentages. In all RP1-related ADRP p.Ser740* was the only identified variant.
Graph 3.
Genes identified in patients with other than autosomal dominant transmission modes. A: 20 autosomal recessive; B: 4 X-linked; C: 25 sporadic. On the right the different gene names. In the graph the relative percentages.
Graph 3.
Genes identified in patients with other than autosomal dominant transmission modes. A: 20 autosomal recessive; B: 4 X-linked; C: 25 sporadic. On the right the different gene names. In the graph the relative percentages.
Notably, the pathogenic RP1 nonsense variant c.2219C>G (p.Ser740*) was identified at the heterozygous state in 28 patients with RP1-related RCD, 26 with an AD pedigree and 2 sporadic, representing the 33.3% of all the examined probands.
This prevalence results in a statistically significant change in the percentages of modes of transmission in this cohort of RP/RCD patients, compared to the general population (see introduction), displaying an unbalance towards the autosomal dominant mode, with the following p-values: 35 ADRP patients (p=0.0181*), 20 ARRP patients (p=0.1284), 4 XLRP patients (p=0.1465), 25 Simplex (sporadic) patients (p=0.0257*).
According to the classification proposed by Chen et al. (2010) [12], this sequence variant would fall under the class II, which includes nonsense-mediated-decay (NMD)-insensitive truncations with a dominant negative effect leading to RP.
The 28 probands found to carry this variant, independently accessed the molecular genetic testing. After careful interviewing, we identified a shared pedigree for a proportion of them, thereby recognizing a total of 20 independent family groups with no traceable consanguinity. Examination of accessory variants identified in other genes by multigene NGS analysis did not reveal any other shared variant, with the exception of a benign CA4 variant (c.700G>A, p.Val234Ile) that was present in both families #1 and #18 (Table 1); the two families could not be otherwise conducted to a common ancestor through interviewing. Among the 20 pedigrees, 4 were then including more than one of our probands with a total of 12 patients (Figure 6 and Figure 7), while for the remaining, we could not trace any consanguinity by focused interviewing (Figure 8 and Figure 9).
Inheritance was consistent with autosomal dominant transmission mode in all the pedigrees, except 2 sporadic cases (patients #21, #24), not reporting any affected ascendant or sibling, and not providing availability for family members phenotyping and/or genotyping. In one exceptionally large pedigree (fig.6, pedigree CLB), we also identified an elevated degree of consanguinity, but none of the patients, either probands or tested affected family members, was homozygous for the variant, nor particularly severe cases were reported from family history.
In many cases, due to the relatively mild form of RP caused by the described variant, family members reported as non-affected, were then found as displaying RP signs at a careful clinical and instrumental evaluation. Due to this, and to the late onset of symptoms and signs, the definition of the relatives’ status was considered only partially reliable when based exclusively on the patients’ interview.
3.2. Phenotype-genotype correlation of c.2219C>G variant
The phenotype related with RP1 p.Ser740* appeared very consistent with the diagnosis of RCD or RP in all patients. Indeed, all patients experienced nyctalopia as the first sign, with a reported median age at onset of 35 years (IQR 26.7 - 41.3) (Table 1). Visual field concentric reduction, resulting in the typical "tunnel vision", and nyctalopia, were present in all patients. Unsurprisingly, when a positive family history was present, symptoms appeared to be recognised earlier by the patients.
At fundus examination, the classic signs of retinitis pigmentosa (pale optic disc, vessel attenuation, bone-spicule shaped pigmented deposits) were present and imaging was consistent with the diagnosis in all patients (Fig. 1 to 5). In some patients early cataract, pre-retinal fibrosis and cystoid macular oedema (CME) were reported complications (Table 1), consistent with the condition. In particular, CME was present in 4 patients who showed good response to topic carbonic anhydrase inhibitors. Most patients underwent electrodiagnostic testing, displaying severely reduced or unrecordable responses under scotopic conditions and reduced or absent response under photopic conditions in the more advanced stages.
The phenotype appears relatively benign in most affected patients, with preservation of central vision until later decades of age. Exceptions were two patients who underwent unconventional invasive treatments in the past, with complications such as endophthalmitis (patient # 26 OS) and laser-induced damage (patient # 2 OU). In several family members, clinical diagnosis followed the molecular diagnosis, even in older patients. This is expected, given the relatively mild phenotype associated with the p.Ser740* variant.
To explain the more severe presentation in some patients (excluding the aforementioned # 2 and # 26), we examined available molecular data regarding potential additional effect of accessory variants in the same or in other genes (Table 1). All additional variants were evaluated by in silico tools, with results reported in the table. Significantly, patient # 24 who showed the most severe phenotype especially in terms of age at onset, resulted compound heterozygous for a very rare intronic variant (c.788-12A>G) in RP1, which was predicted as variant of uncertain significance (VUS).
4. Discussion
In a cohort of patients affected by RP/RCD originating from Western Sicily, we found a statistically significant (P-value= 0.0073) high prevalence of the RP1 c.2219C>G (p.Ser740*) pathogenic variant, proven to be causative in 33.3% of the 84 examined probands and 74% of the 25 clearly AD pedigrees.
Interestingly, the high prevalence of this variant in Sicilian population has been recently mentioned by Karali et al [20], with the report of 13 cases from 9 apparently unrelated Sicilian families, but just as a general observation. The p.Ser740* variant in RP1 was first identified in a male patient belonging to a large cohort recruited in London at the Moorfields Eye Hospital [21], but with no specification about his geographic origin. We know that an affected son of patient #10 is based in London but couldn’t reconduct him to the described patient, due to lack of communication with his family. Again, the variant has been reported in a study conducted on 74 families from the United Arab Emirates [22], where it is generically mentioned that one of the probands was of Italian origin, but without a specific correlation between the identified variants and ethnic origin of single patients. We can speculate on different hypothesis, for instance that this latter patient was of Sicilian origin, or vice versa that the founder variant has an Arab origin, considering the very consistent presence of the Arab population in Western Sicily through the past centuries. It is also possible that in these sporadically described cases, the variant occurred spontaneously, without any link to the Sicilian population.
The statistically significant prevalence of a genetic variant can either be the result of the presence of a founder effect (a variant originating from a single individual being diffused through many descendants across generations in a specific population) or reflect the existence of a hotspot for mutation within a gene (an unstable DNA base pair, prone to mutation). We would exclude this second possibility, as the higher prevalence wouldn’t be confined to such a specific geographic area. Considering this, we didn’t perform a haplotype analysis showing co-segregation of specific markers with the variant, as the striking prevalence in this localised population makes the founder effect evident.
At the same time, we would exclude “bottleneck effects” of the specific geographic localization of this dominant variant as the result of other events, such as confinement, as Western Sicily has a wide territory and is a well-connected area, and/or a specific selection related to other genetically related conditions, as the RP1 protein is essentially expressed in the retina.
The wide spreading of this variant is probably related to the relatively benign phenotype and to the age of onset that, being after the primary reproductive age, does not alert patients about the transmission risk and the possible prevention by prenatal diagnosis [23].
Inherited retinal dystrophies still represent an important cause of visual impairment and blindness, with a great effort of the scientific community towards the characterization of pathogenic mechanisms and ultimately therapeutic strategies. Given the high genetic and phenotypic heterogeneity, molecular diagnosis is crucial for the correct definition of each patient condition, for familial genetic counselling, for better understanding pathogenic mechanisms and for creating datasets of patients, based on the phenotype-related genetic defect, rather than exclusively on the phenotype itself, especially in the perspective of developing potential innovative therapies [24].
In the described form of autosomal dominant rod-cone dystrophy, or retinitis pigmentosa, the phenotype is relatively mild with a generally late onset, leading to a widespread diffusion of the condition in the studied population, where the RP1 p.Ser740* pathogenic variant appears as the result of a founder effect. The described variant is significantly prevalent in the Palermo province in Western Sicily, thereby giving the possibility to modify the diagnostic strategy in this population. Indeed, whenever a compatible phenotype and mode of transmission is identified, the screening for this specific variant should precede the NGS testing, with great advantage in terms of resources employment and time of reporting for a significant proportion of RP patients of Western Sicily ancestry.
Finally, we identified a genetically homogeneous population affected by a condition that is caused by a nonsense variant.
Nonsense variants determine the generation of a stop codon (premature termination codon, PTC) that, in the translation process, results in the generation of a shorter and often non-funcional protein. Nonsense-mediated decay mechanism avoids the accumulation of misfolded proteins.
Translational read-through therapy is a recently studied approach aiming at the specific targeting of nonsense mutations. It is essentially based on the effect of specific molecules, known as translational read-through-inducing drugs (TRIDs), that through a ribosome-binding mechanism, can have the effect of suppressing a nonsense variant, therefore allowing the syntesis of a full length protein [25,26].
5. Conclusions
We identified a significantly prevalent pathogenic variant in the RP1 gene, causing RP/RCD in a particular area in Western Sicily, as the result of a founder effect. This finding might represent an advantage for genetic diagnostic strategies.
The cohort of patients here studied is particularly numerous, and most have affected family members. In this scenario, we believe that the identification of such a large and genetically uniform population of patients affected by a dominant nonsense variant-related condition, can be of great interest for the scientific community for the development of potential diagnostic and/or therapeutic approaches.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, S1. Panel of tested genes at AOOR Villa Sofia-Cervello, Dipartimento di Genetica Oncoematologia e Malattie Rare, Palermo, Italy; S2. Panel of tested genes at MAGI Euregio, Bolzano, Italy.
Author Contributions
Conceptualization, Fabiana D’Esposito; Data curation, Fabiana D’Esposito, Viviana Randazzo, Marco Piergentili and Maria Francesca Cordeiro; Funding acquisition, Caterina Gagliano, Teresio Avitabile, Aurelio Maggio and Elena D'Alcamo; Investigation, Fabiana D’Esposito, Viviana Randazzo, Maria Igea Vega, Paolo Enrico Maltese, Salvatore Torregrossa, Paola Scibetta, Florinda Listì, Caterina Gagliano, Lucia Scalia, Antonino Pioppo, Antonio Marino, Emanuele Malvone, Tiziana Fioretti, Maria Piccione and Elena D'Alcamo; Methodology, Fabiana D’Esposito and Caterina Gagliano; Project administration, Matteo Bertelli, Aurelio Maggio and Elena D'Alcamo; Resources, Salvatore Torregrossa, Caterina Gagliano, Teresio Avitabile, Francesco Salvatore, Matteo Bertelli and Ciro Costagliola; Software, Maria Igea Vega, Paolo Enrico Maltese, Florinda Listì and Tiziana Fioretti; Supervision, Teresio Avitabile, Ciro Costagliola and Aurelio Maggio; Validation, Caterina Gagliano, Angela Vitrano and Elena D'Alcamo; Writing – original draft, Fabiana D’Esposito; Writing – review & editing, Fabiana D’Esposito, Paolo Enrico Maltese, Marco Piergentili, Francesco Salvatore and Maria Francesca Cordeiro.
Funding
This research was funded by Italian Multicentric Research Project: NET 2016-02363765 “Patients Phenotyping and genotyping and innovative treatments for retinitis pigmentosa”.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and under the ethic approvals of the different hospitals.
Informed Consent Statement
Informed consent to genetic analysis and anonymized data publication was obtained from all subjects involved in the study or their guardians.
Data Availability Statement
Data regarding the clinical and genetic results of our patients are available in the different centres involved in the study, although protected by privacy regulations.
Acknowledgments
The authors would like to thank: Foundation Franco and Piera Cutino based in Palermo for providing logistic support for the patients’ recruitment; Sicilia Regional Health Authority for cooperation at Villa Sofia-Cervello Hospital in Palermo, and patients, with their family members, for collaboration in the deep investigations finalized to the pedigrees completion.
Conflicts of Interest
The authors declare no conflict of interest. FDE is CEO of GENOFTA SRL (LTD), but with no competing interests in this research.
References
- Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;(233):1-34. [CrossRef]
- Hamel C. (https://www.orpha.net, updated 2014).
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. The Lancet 2006;368:1795-1809. [CrossRef]
- Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol 2007;125(2):151-8. [CrossRef]
- Colombo L, Maltese PE, Castori M, et al. Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Invest Ophthalmol Vis Sci. 2021;62(2):13. [CrossRef]
- Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. 2000 Aug 4 [Updated 2023 Apr 6]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. PMID: 20301590.
- RetNet (https://sph.uth.edu/retnet), accession 20th March 2023.
- OMIM (https://www.omim.org/).
- Pierce, EA, Quinn T, Meehan T, et al. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nature Genet. 1999;22: 248-254. [CrossRef]
- Audo I, Mohand-Saïd S, Dhaenens CM, et al. RP1 and autosomal dominant rod-cone dystrophy: novel mutations, a review of published variants, and genotype-phenotype correlation. Human Mutation 2012;33(1):73-80. [CrossRef]
- Liu Q, Zhou J, Daiger SP, et al. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43(1):22-32. PMID: 11773008; PMCID: PMC1963488.
- Chen LJ, Lai TY, Tam PO, et al. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 2010;51(4):2236-42. [CrossRef]
- Khaliq S, Abid A, Ismail M, et al. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J Med Genet 2005;42(5):436-8. [CrossRef]
- Wang J, Xiao X, Li S, et al. Dominant RP in the Middle While Recessive in Both the N- and C-Terminals Due to RP1 Truncations: Confirmation, Refinement, and Questions. Front Cell Dev Biol. 2021;9:634478. [CrossRef]
- Payne A, Vithana E, Khaliq S, et al. RP1 protein truncating mutations predominate at the RP1 adRP locus. Invest Ophthalmol Vis Sci. 2000;41(13):4069-73. PMID: 11095597.
- Schwartz SB, Aleman TS, Cideciyan AV, Swaroop A, Jacobson SG, Stone EM. De novo mutation in the RP1 gene (Arg677ter) associated with retinitis pigmentosa. Invest Ophthalmol Vis Sci 2003;44(8):3593-7. [CrossRef]
- Pontikos N, Arno G, Jurkute N, et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology. 2020;127(10):1384-1394. [CrossRef]
- Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics 2019;35(11):1978-1980. [CrossRef]
- Fioretti T, Auricchio L, Piccirillo A, Vitiello G, Ambrosio A, Cattaneo F, Ammendola R, Esposito G. Multi-Gene Next-Generation Sequencing for Molecular Diagnosis of Autosomal Recessive Congenital Ichthyosis: A Genotype-Phenotype Study of Four Italian Patients. Diagnostics (Basel). 2020 Nov 24;10(12):995. [CrossRef]
- Karali M, Testa F, Di Iorio V, et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci Rep. 2022;12(1):20815. [CrossRef]
- Carss KJ, Arno G, Erwood M, et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am J Hum Genet 2017;100(1):75-90. [CrossRef]
- Méjécase C, Kozak I, Moosajee M. The genetic landscape of inherited eye disorders in 74 consecutive families from the United Arab Emirates. Am J Med Genet C Semin Med Genet. 2020;184(3):762-772. [CrossRef]
- Esposito G, Ruggiero R, Savarese M, et al. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy, myotonic dystrophy type 1 and spinal muscular atrophy. Clin Chem Lab Med. 2013;51(12):2239-45. [CrossRef]
- Raimondi R, D'Esposito F, Sorrentino T, Tsoutsanis P, De Rosa FP, Stradiotto E, Barone G, Rizzato A, Allegrini D, Costagliola C, Romano MR. How to Set Up Genetic Counselling for Inherited Macular Dystrophies: Focus on Genetic Characterization. Int J Mol Sci. 2023 Jun 3;24(11):9722. [CrossRef]
- Vössing C, Owczarek-Lipska M, Nagel-Wolfrum K, Reiff C, Jüschke C, Neidhardt J. Translational Read-Through Therapy of RPGR Nonsense Mutations. Int J Mol Sci. 2020;21(22):8418. [CrossRef]
- Samanta A, Stingl K, Kohl S, Ries J, Linnert J, Nagel-Wolfrum K. Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int J Mol Sci. 2019 Dec 12;20(24):6274. [CrossRef]
Figure 1.
Multimodal imaging in patient # 8. Fundus colour pictures (A-B) showing pale optic disc, attenuated vessels and generalised thinning of Retinal Pigment Epithelium (RPE). Blue autofluorescence (C-D) showing diffused areas of hypo-autofluorescence with relative macular sparing. OCT (E-F) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere.
Figure 1.
Multimodal imaging in patient # 8. Fundus colour pictures (A-B) showing pale optic disc, attenuated vessels and generalised thinning of Retinal Pigment Epithelium (RPE). Blue autofluorescence (C-D) showing diffused areas of hypo-autofluorescence with relative macular sparing. OCT (E-F) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere.
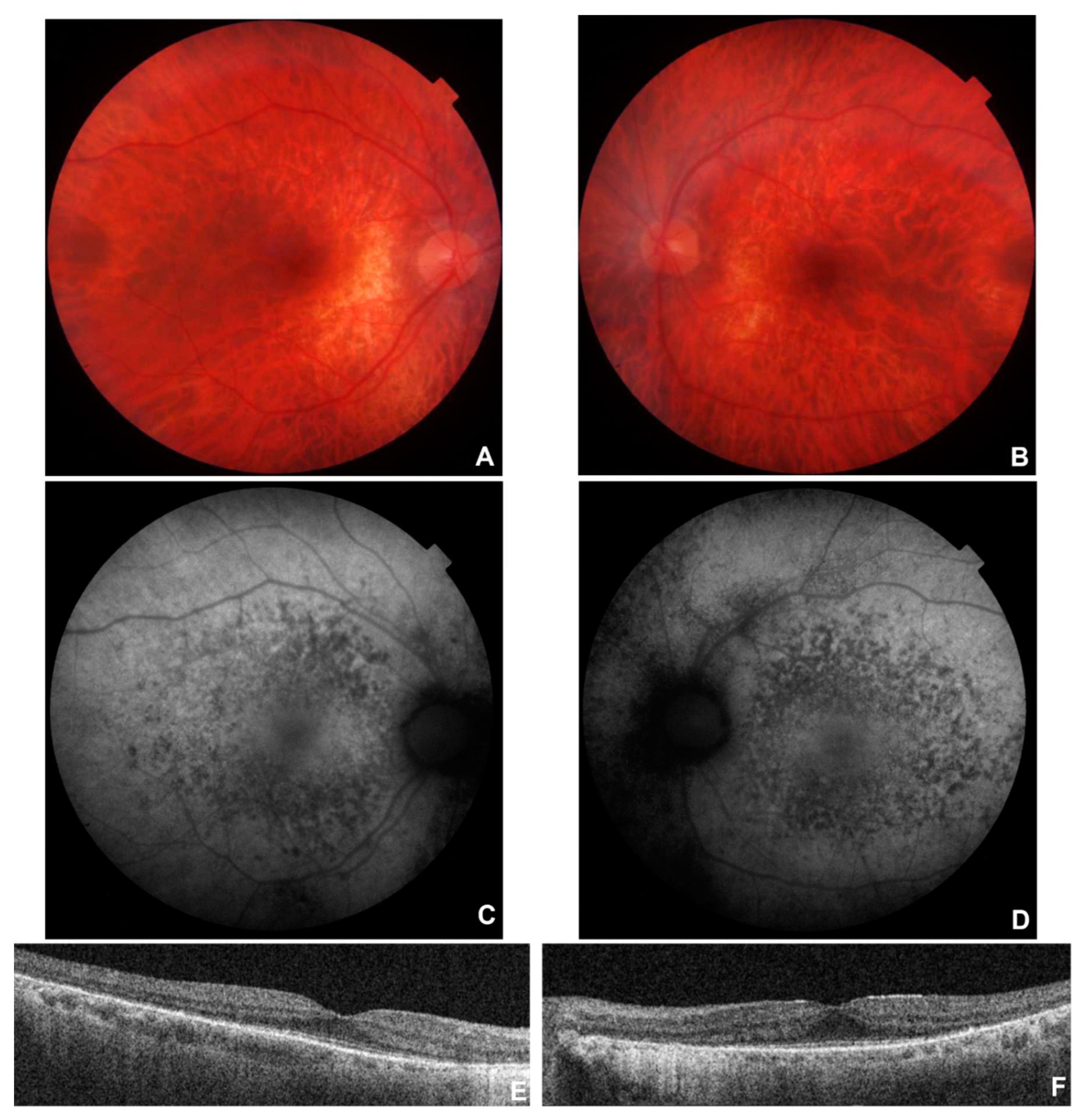
Figure 2.
Multimodal imaging in patient # 9. Fundus colour pictures (A-B) showing pale optic disc, attenuated vessels and generalised retinal thinning, with relative macular sparing that is particularly evident at blue autofluorescence (BAF) (C-D). OCT (E-F) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere, and preretinal fibrosis with a tractional effect in OS (F).
Figure 2.
Multimodal imaging in patient # 9. Fundus colour pictures (A-B) showing pale optic disc, attenuated vessels and generalised retinal thinning, with relative macular sparing that is particularly evident at blue autofluorescence (BAF) (C-D). OCT (E-F) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere, and preretinal fibrosis with a tractional effect in OS (F).
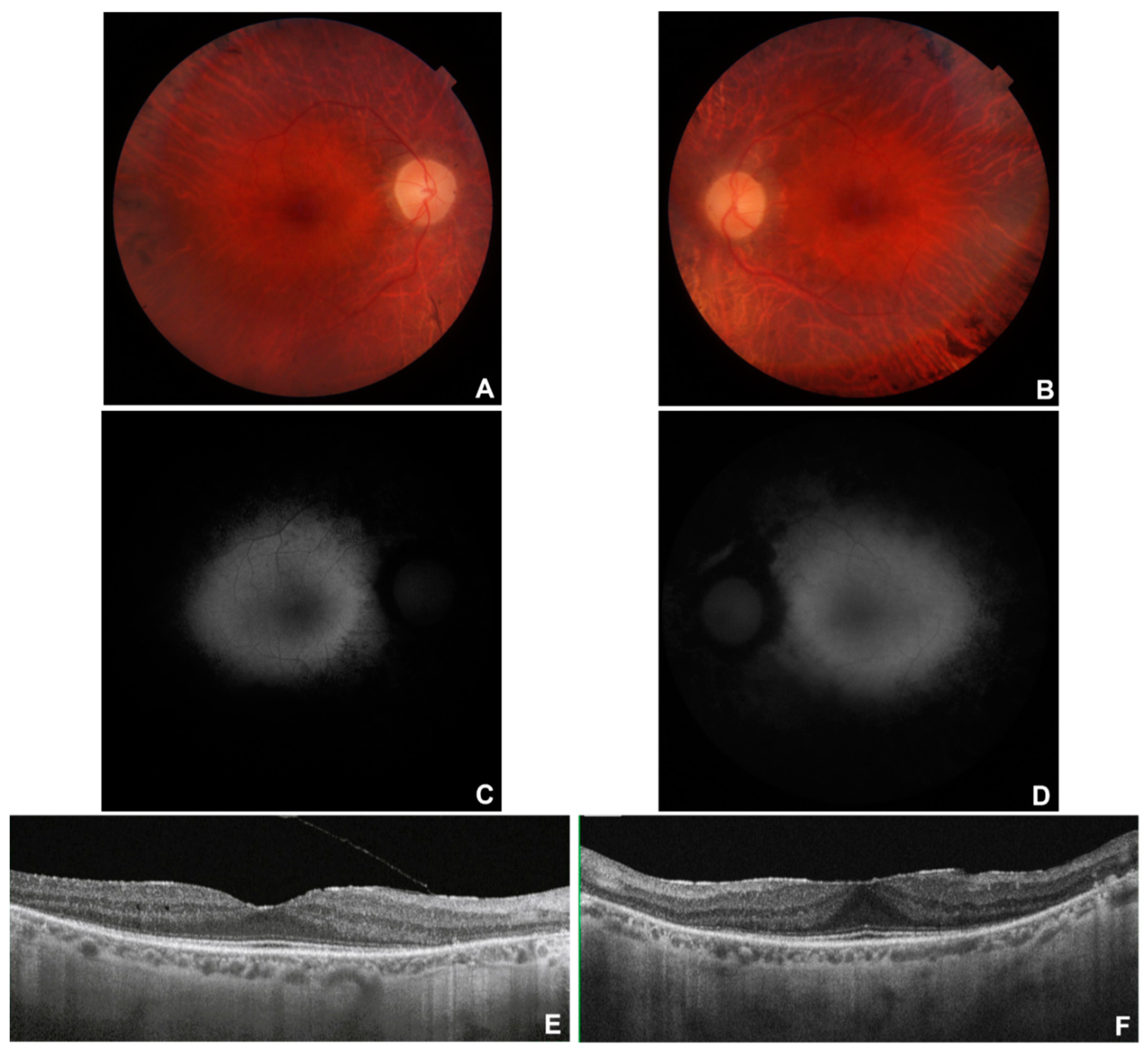
Figure 3.
Multimodal imaging in patient # 12. Posterior pole fundus colour pictures (A-B) showing pale optic disc and attenuated vessels. Posterior pole blue autofluorescence (BAF) showing the typical in RP perimacular hyper-autofluorescent ring (C-D) and hypo-autofluorescent peripheral areas corresponding to the bone-spicule-shaped pigment deposits (E-F). OCT (G-H) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere.
Figure 3.
Multimodal imaging in patient # 12. Posterior pole fundus colour pictures (A-B) showing pale optic disc and attenuated vessels. Posterior pole blue autofluorescence (BAF) showing the typical in RP perimacular hyper-autofluorescent ring (C-D) and hypo-autofluorescent peripheral areas corresponding to the bone-spicule-shaped pigment deposits (E-F). OCT (G-H) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere.
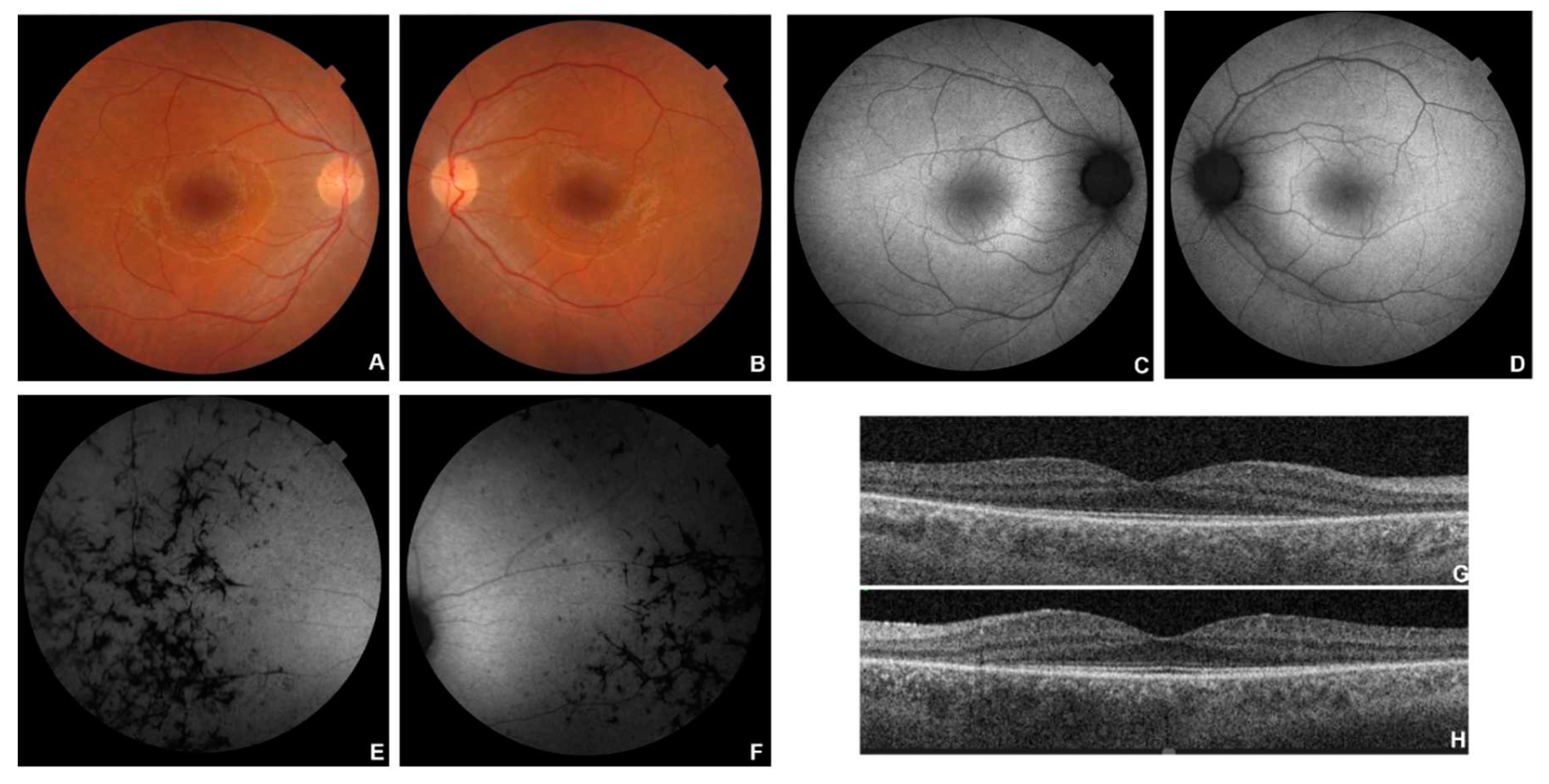
Figure 4.
Multimodal imaging in patient # 13. Posterior pole (A-B) and OD peripheral (C) fundus colour pictures showing attenuated vessels, diffuse atrophy and bone-spicule-shaped pigment deposits. Corresponding blue autofluorescence (BAF) (C-F) showing the typical in RP perimacular hyper-autofluorescent ring (D) and diffuse hypo-autofluorescence corresponding to the atrophic areas and to the bone-spicule-shaped pigment deposits. OCT (G-H) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere and preretinal fibrosis, evident in OS (H).
Figure 4.
Multimodal imaging in patient # 13. Posterior pole (A-B) and OD peripheral (C) fundus colour pictures showing attenuated vessels, diffuse atrophy and bone-spicule-shaped pigment deposits. Corresponding blue autofluorescence (BAF) (C-F) showing the typical in RP perimacular hyper-autofluorescent ring (D) and diffuse hypo-autofluorescence corresponding to the atrophic areas and to the bone-spicule-shaped pigment deposits. OCT (G-H) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere and preretinal fibrosis, evident in OS (H).
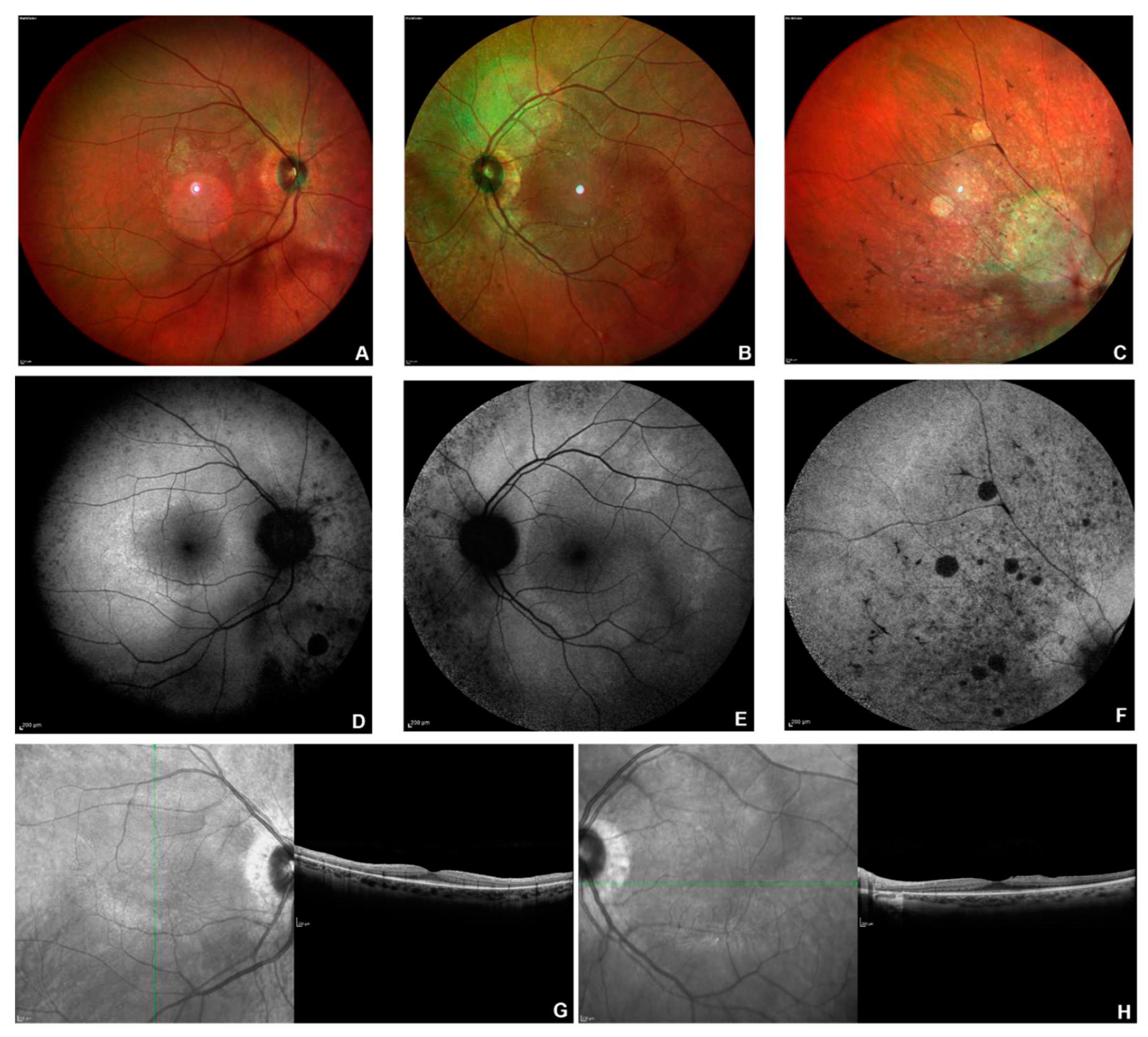
Figure 5.
Multimodal imaging in patient # 28. Posterior pole fundus colour pictures (A-B) showing pale optic disc, attenuated vessels and diffuse retinal thinning, relatively sparing the macular region. Posterior pole blue autofluorescence (BAF) showing the macular hyper-autofluorescent ring, typical in RP (C-D). OCT (E-F) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere and vitreomacular traction in OS (F).
Figure 5.
Multimodal imaging in patient # 28. Posterior pole fundus colour pictures (A-B) showing pale optic disc, attenuated vessels and diffuse retinal thinning, relatively sparing the macular region. Posterior pole blue autofluorescence (BAF) showing the macular hyper-autofluorescent ring, typical in RP (C-D). OCT (E-F) showing a portion of subfoveal preserved ellipsoid zone, with diffuse disruption elsewhere and vitreomacular traction in OS (F).
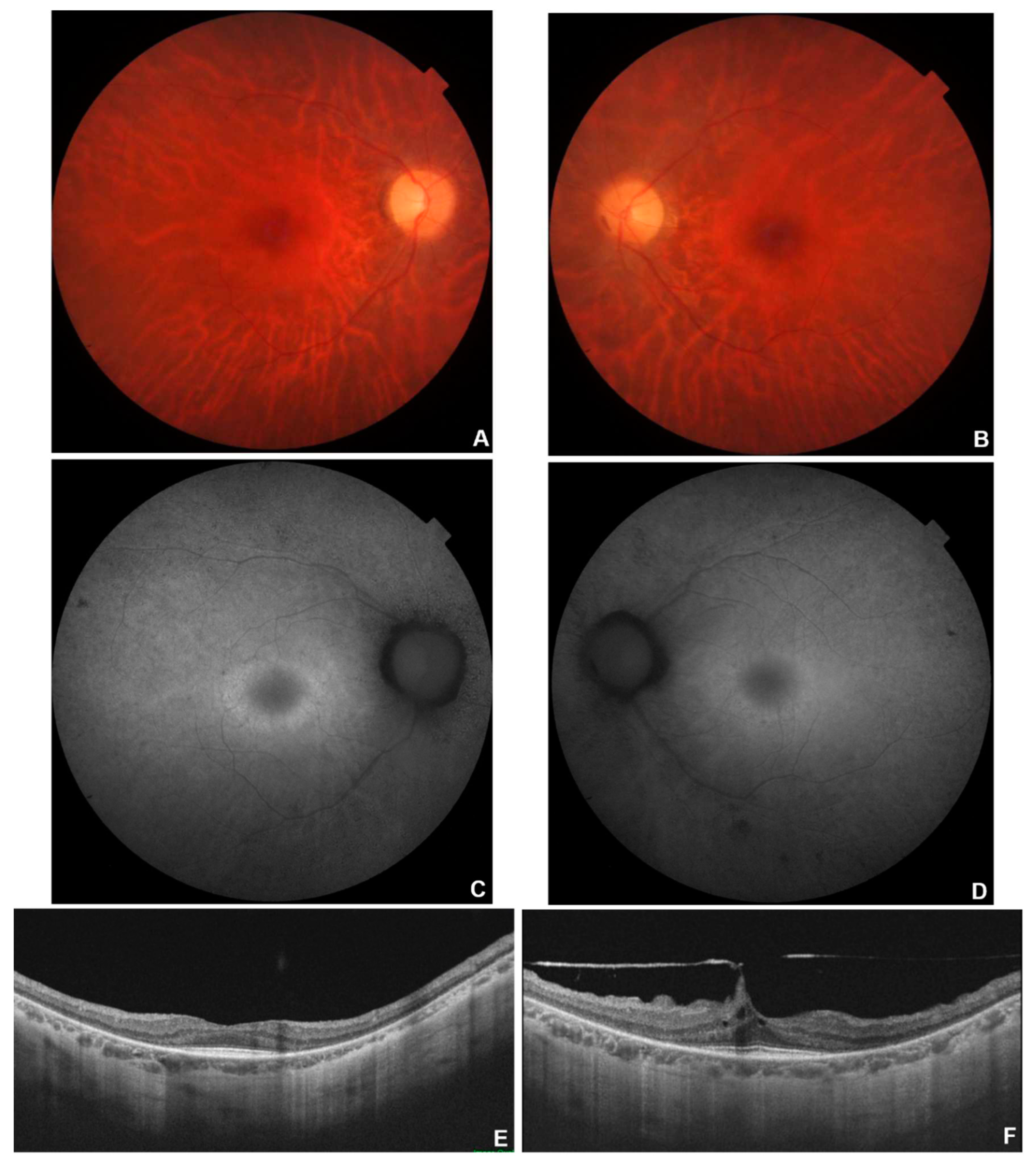
Figure 6.
Pedigrees of related families CLB and CR. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate the initial probands. Probands subsequently identified as in the same pedigree are indicated by the number assigned in this work. Double lines indicate consanguineous marriages. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing. Pedigree of family CLB: patients nos. 4, 6, 7, 9 are indicated. Pedigree of family CR: patients nos. 5 and 27 are indicated.
Figure 6.
Pedigrees of related families CLB and CR. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate the initial probands. Probands subsequently identified as in the same pedigree are indicated by the number assigned in this work. Double lines indicate consanguineous marriages. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing. Pedigree of family CLB: patients nos. 4, 6, 7, 9 are indicated. Pedigree of family CR: patients nos. 5 and 27 are indicated.
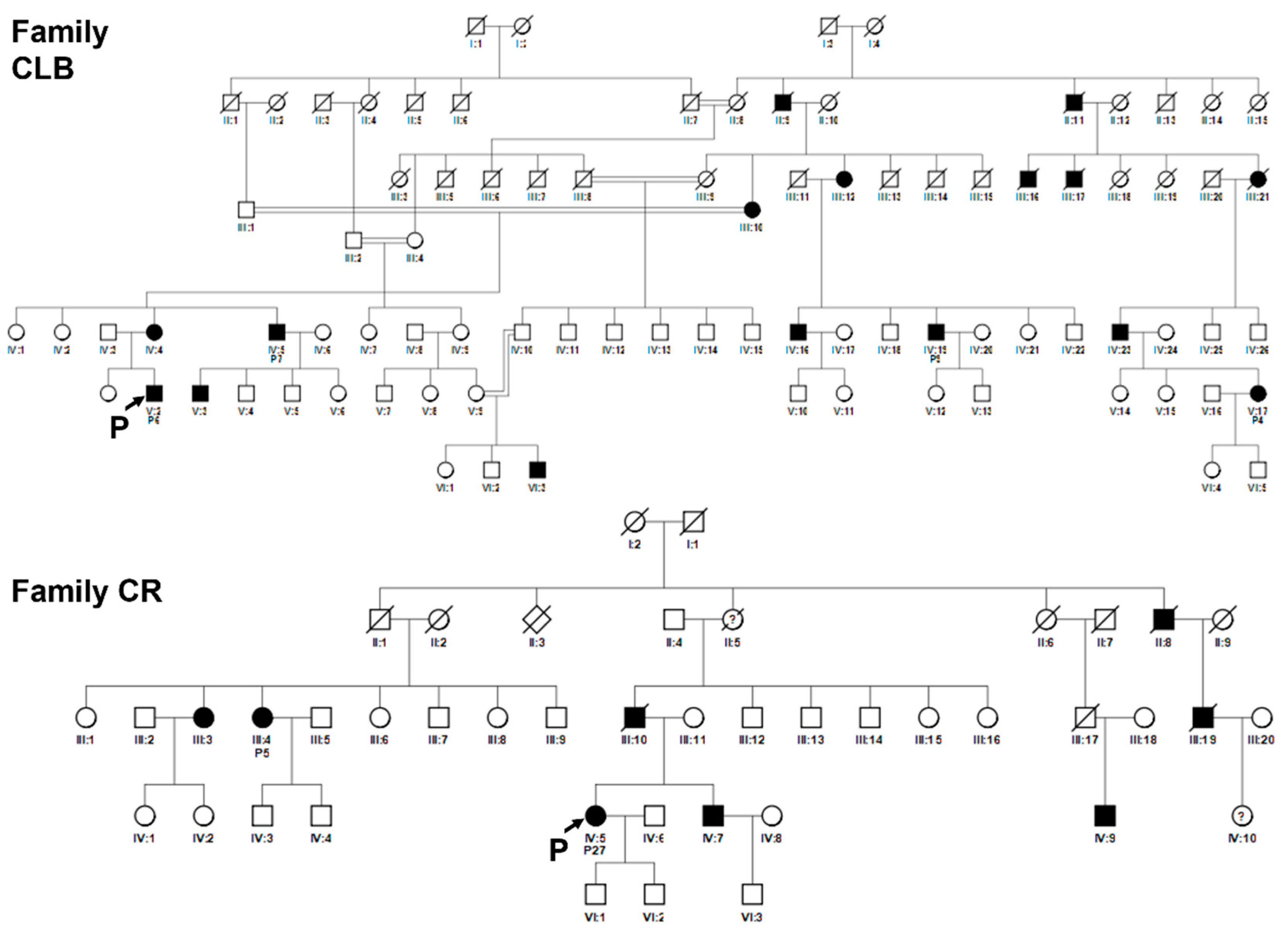
Figure 7.
Pedigrees of related families CPL and SMM. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate the initial probands. Probands subsequently identified as in the same pedigree are indicated by the number assigned in this work. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing. Pedigree of family CPL: patients nos. 8, 17, 25 are indicated. Pedigree of family SMM: patients nos. 19, 20, 22 are indicated.
Figure 7.
Pedigrees of related families CPL and SMM. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate the initial probands. Probands subsequently identified as in the same pedigree are indicated by the number assigned in this work. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing. Pedigree of family CPL: patients nos. 8, 17, 25 are indicated. Pedigree of family SMM: patients nos. 19, 20, 22 are indicated.
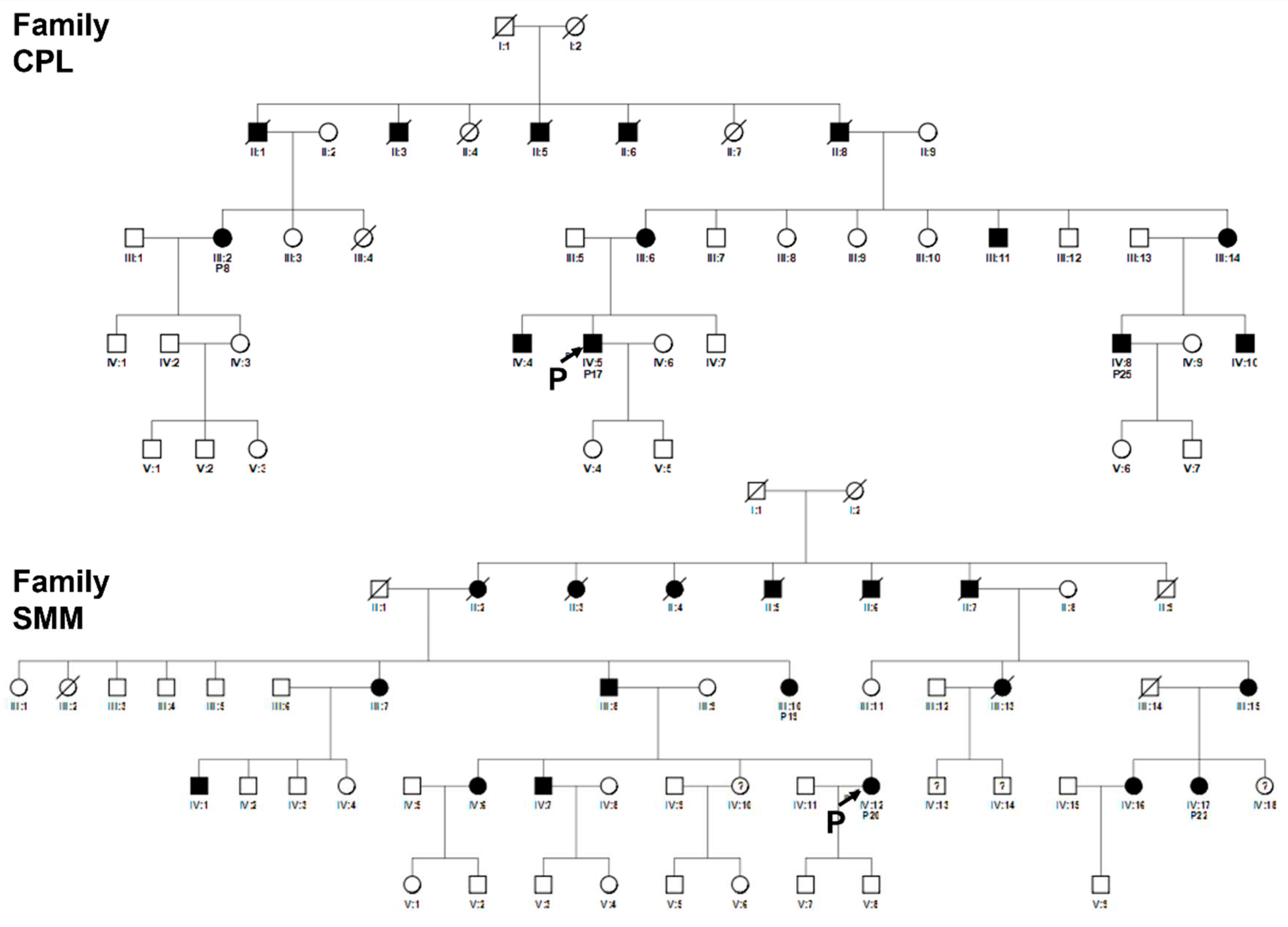
Figure 8.
Pedigrees of unrelated families 1, 2, 3, 10, 11 12, 13, 14, 15. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate probands. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing.
Figure 8.
Pedigrees of unrelated families 1, 2, 3, 10, 11 12, 13, 14, 15. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate probands. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing.
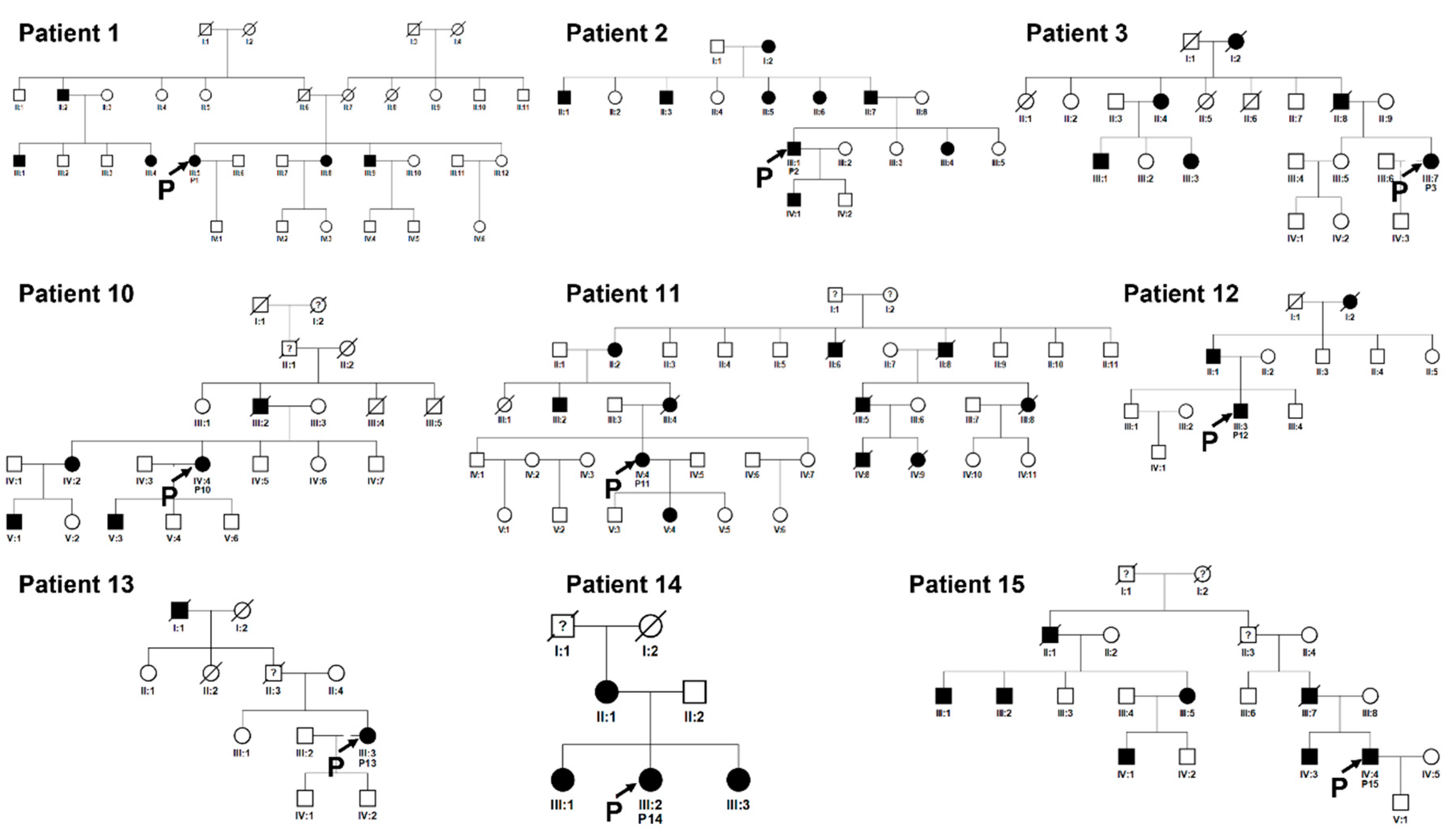
Figure 9.
Pedigrees of unrelated families 16, 18, 21, 23, 24, 26, 28. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate probands. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing.
Figure 9.
Pedigrees of unrelated families 16, 18, 21, 23, 24, 26, 28. Squared boxes indicate males, circles indicate females, closed symbols represent affected and open symbols represent unaffected persons. The arrows indicate probands. Question marks (?) indicate a possible but not proven affected status by relatives’ interviewing.
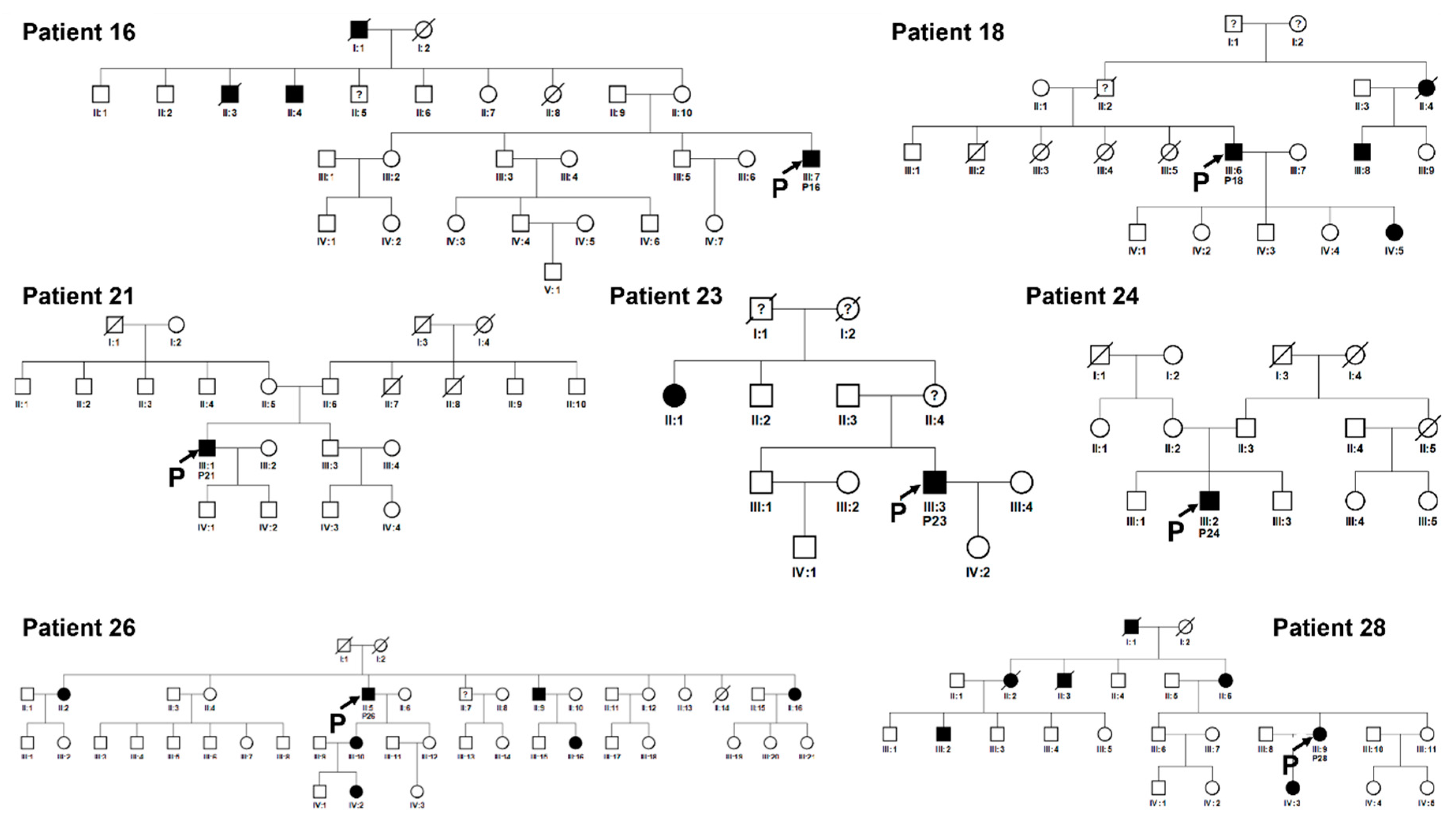

Table 1.
Patients’ clinical and genetic data. Caption: Pt: patient. OD: right eye. OS: left eye. OU: both eyes. HM: hand motion. CME: cystoid macular oedema. PRF: preretinal fibrosis. ND: not detected. VUS: variant of uncertain significance.
Table 1.
Patients’ clinical and genetic data. Caption: Pt: patient. OD: right eye. OS: left eye. OU: both eyes. HM: hand motion. CME: cystoid macular oedema. PRF: preretinal fibrosis. ND: not detected. VUS: variant of uncertain significance.
| Pt. no. | Birth date | Sex | Age at onset (years) |
Duration of symptoms at observation (years) | BCVA OD/OS Associated signs |
Family History |
Traceable consanguinity with other patients of this study |
Other possibly significant variants (and effect prediction) |
|---|---|---|---|---|---|---|---|---|
| # 1 | 1970 | F | 40 | 6 | OD 20/20 OS 20/20 CME |
+ | ND | CA4:c.700G>A:(p.Val234Ile) Benign |
| # 2 | 1958 | M | 30 | 35 | OD HM OS HM |
+ | ND |
USH2A:c.12112A>G:(p.Thr4038Ala) VUS |
| # 3 | 1953 | F | 26 | 39 | OD 20/40 OS 20/40 PSEUDOPHAKIC OU |
+ | ND | ND |
| # 4 | 1982 | F | 21 | 18 | OD 20/25 OS 20/25 PSEUDOPHAKIC OU |
+ | FAM CLB |
CNGB1:c.3122G>A:(p.Arg1041Gln) VUS |
| # 5 | 1949 | F | 45 | 20 | OD 20/25 OS 20/30 PSEUDOPHAKIC OU |
+ | FAM CR | ND |
| # 6 | 1989 | M | 27 | < 1 | OD 20/20 OS 20/20 REGRESSED CME OU |
+ | FAM CLB | ND (direct sequencing of RP1) |
| # 7 | 1948 | M | 50 | 25 | OD 20/100 OS 20/100 |
+ | FAM CLB | ND |
| # 8 | 1955 | F | 40 | 10 | OD 20/25 OS 20/30 PSEUDOPHAKIC OU |
+ | FAM CPL | ND |
| # 9 |
1960 | M | 40 | 63 | OD 20/25 OS 20/30 CATARACT OU PRF OU |
+ | FAM CLB | ND |
| # 10 | 1949 | F | 35 | 35 | OD 20/40 OS 20/100 PSEUDOPHAKIC, CORNEAL DYSTROPHY, CME OU |
+ | NO | ND (direct sequencing of RP1) |
| # 11 | 1961 | F | 55 | 1 | OD 20/30 OS 20/20 |
+ | NO | ND |
| # 12 | 1985 | M | 26 | 7 | OD 20/20 OS 20/20 |
+ | NO |
OFD1:c.815A>G:(p.His272Arg) VUS-Likely Benign |
| # 13 | 1975 | F | 45 | 1 | OD 20/25 OS 20/25 CATARACT OU PRF OS |
+ | NO | ND |
| # 14 | 1980 | F | 31 | 8 | OD 20/25 OS 20/30 |
+ | NO | ND |
| # 15 | 1985 | M | 26 | 11 | OD 20/100 OS 20/50 |
+ | NO | ABCA4:c.6089G>A:(p.Arg2030Gln) Pathogenic |
| # 16 | 1978 | M | 20 | 24 | OD 20/20 OS 20/30 SECONDARY IOL OPACITIES |
+ | NO | PROM1:c.652C>T:(p.Gln218Ter) Pathogenic |
| # 17 | 1976 | M | 35 | 5 | OD20/25 OS 20/30 CME OU |
+ | FAM CPL | ND |
| # 18 | 1944 | M | 50 | 23 | OD 20/70 OS 20/70 PSEUDOPHAKIC OU |
+ | ND |
IMPDH1:c.189A>G:(p.Ser63=) VUS-Likely Benign CA4:c.700G>A:(p.Val234Ile) Benign |
| # 19 | 1955 | F | 53 | 8 | OD 20/70 OS 20/200 PSEUDOPHAKIC OU |
+ | FAM SMM | ND |
| # 20 | 1990 | F | 31 | 1 | OD 20/20 OS 20/20 |
+ | FAM SMM | ND (direct sequencing of RP1) |
| # 21 | 1968 | M | 40 | 7 | OD 20/25 OS 20/40 CATARACT OU |
- | ND |
RP1:c.6166G>A:(p.Gly2056Ser) Likely Benign -VUS SNRNP200:c.1203+15G>A Likely Benign USH2A: c.12112A>G:(p.Thr4038Ala) VUS |
| # 22 | 1965 | F | 37 | 15 | OD 20/25 OS 20/25 PSEUDOPHAKIC OU |
+ | FAM SMM | ND |
| # 23 | 1966 | M | 48 | 4 | OD 20/20 OS 20/25 CATARACT OU |
+ | ND |
PROM1:c.1345G>A: (p.Val449Met) Likely Benign |
| # 24 | 1992 | M | 10 | 15 | OD 20/100 OS 20/100 |
- | ND |
RP1: c.788-12A>G VUS PRPF6:c.2638_2639delinsGC: (p.Phe880Ala) VUS FSCN2:c.619C>T:(p.Arg207Cys) Likely Benign |
| # 25 |
1979 | M | 30 | 6 | OD 20/25 OS 20/25 REGRESSED CME OU |
+ | FAM CPL | ND |
| # 26 | 1945 | M | 25 | 46 | OD 20/200 OS HM |
+ | ND | ND (direct sequencing of RP1) |
| # 27 | 1973 | F | 30 | 19 | OD 20/20 OS 20/20 |
+ | FAM CR | ND |
| # 28 | 1966 | F | 40 | 16 | OD 20/20 OS 20/20 PRF |
+ | ND | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



