Submitted:
05 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
The feasibility of using zeolites, synthesized from components found in municipal solid waste fly ash (MSW-FA), as sorbents for the recovery of nutrients (nitrate and phosphate) and heavy metals is critically assessed in this review. The inherent drawbacks of utilising a highly contaminated, variable, and relatively Al- and Si-dilute source such as MSW-FA to synthesize zeolites are discussed, and different methods to extract and decontaminate zeolite precursor materials from MSW-FA are considered. Ways to synthesize tailored zeolites and how their properties as well as the operational conditions impact the adsorption of cations such as ammonium and heavy metals are summarized. The use of surface-modified zeolites to adsorb nitrate and phosphate is also reviewed. And subsequently, approaches to utilise directly or recover for reuse the adsorbed compounds are considered, discussing potential challenges and mitigating measures related to leaching of unwanted compounds from the zeolites. Moreover, the possibility to regenerate the adsorption capacity of the zeolites for multiple adsorption cycles is considered. In the final chapter of the review a more general discussion of the main challenges and existing research gaps is provided, giving directions for recommended studies.
Keywords:
waste-as-resource
; zeolite-precursor
; sorption
; surface-modified zeolites
; ammonium
; phosphate
; reuse
; regeneration
1. Introduction
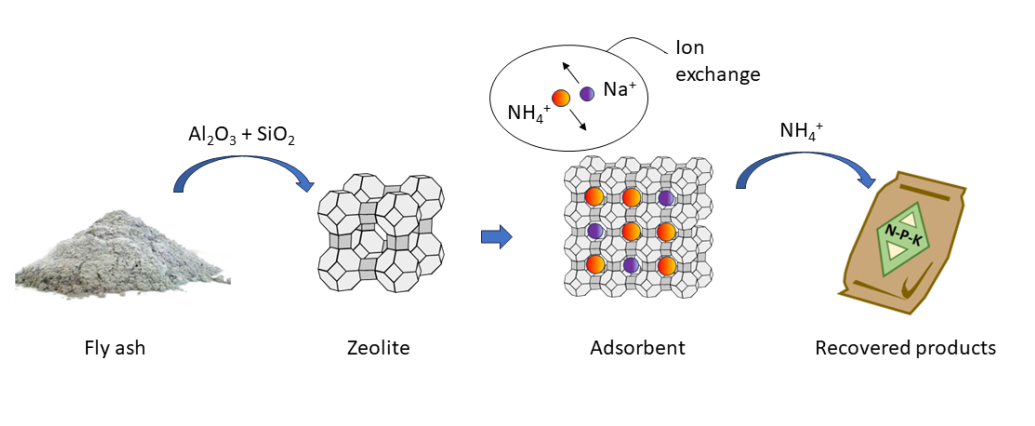
There is an increasing interest in using incineration to dispose of municipal solid waste (MSW) due to large reduction in weight (up 70%) and volume (up to 90%), a concomitant recovery of energy and heat, and destruction of most of the hazardous organic contaminants and pathogens. The globally generated amount of MSW increased significantly from 1.3 billion tons per annum in 2012 to 2.1 billion tons in 2018 [1], which is expected to increase further to 3.40 billion tons per year by 2050 [2]. Upon incineration, three main residues are produced: bottom ash, fly ash (FA) and air pollution control (APC) residues. Approximately 250-300 kg bottom ash and 25-30 kg fly ash and APC residues are generated for each ton of MSW incinerated [3]. The bottom ash residue is utilized in many applications, particularly in building and construction works. The FA residue, however, is generally regarded as toxic waste due to its high content of heavy metals, salts and organic micropollutants such as dioxins and furans [4] owing to the volatilization and condensation of different elements during the incineration. Therefore, nearly all MSW incineration FA (MSW-FA) are stabilized/solidified and deposited as toxic waste in landfills, and due to their enormous volumes, taking up valuable space. However, a shift in focus from environmental impacts to resource recovery has identified the potential value of embedded salts and metals in the MSW-FA, and some progress has been made in recovering these resources [5,6,7].
The principal components of FA are CaO, SiO2, Al2O3, and Fe2O3, resembling the main components of volcanic material, a precursor of natural zeolites. Zeolites are crystalline micro- and mesoporous materials widely used as catalysts and sorbents [8]. However, most of the zeolites that are used today are synthesized since natural zeolites vary too much in mineralogical and chemical composition, crystal structure and pore sizes, both between and within the mineral deposits they are extracted from to be used for applications that require zeolites with specific and predictable properties [9]. Zeolites are primarily synthesized from pure silica (SiO2) and alumina (Al2O3), though the search and use of alternative economical and abundant sources has increased in the last decades [10]. Due to its low cost, availability, abundance and resemblance with volcanic material, it is relatively common to synthesis zeolite from coal-derived FA [11]. MSW-FA usually contains much less silica and alumina than coal-derived FA [10,11]. Moreover, the often-high levels of leachable toxic compounds in this type of FA, need to be reduced [12].
Zeolites are commonly used in separation processes in e.g. the petrochemical and pharmaceutical industries as they can be made fairly specific to the target molecules, and since the main mechanisms behind the separation process (molecular sieving, electrostatic interaction and polarization) are always reversible, zeolites are believed (under ideal situations) to be able to undergo a virtually unlimited number of adsorption–desorption cycles [8]. This is important from a cost-efficiency perspective too, as the high initial costs can be compensated by the longer life, assured by excellent stability and ease to regenerate.
Zeolites, depending on their chemical composition and pore structures as well as the operating conditions, can be used as sorbents for the removal of mono-, di- and trivalent cations such as ammonium (NH4+) and heavy metals (Pb2+, Cu2+, Cd2+, Zn2+, Cr3+ and Ni2+) from industrial and municipal wastewaters [13]. And, by subsequent desorption and refinement, if needed, these adsorbates can be recovered for further downstream applications [11,14]. Furthermore, by certain surface modifications of the zeolites they can also be used as sorbents to remove anions such as phosphate (PO43-), nitrate (NO3-) and arsenate (H2AsO4-, HAsO42-, AsO43-). However, the recovery of the adsorbates and the regeneration of the SMZ may be low.
The need to drastically improve the recovery of phosphorous from available sources due to the forthcoming peak in readily available non-renewable phosphate rock is well known. Crop residues, food waste, manure and human excreta are such a source, and multiple methods and strategies are currently under development to facilitate cost-effective means to harvest phosphorous from these, including from domestic wastewater [15], primarily for use in agriculture [16]. Nitrogen, on the other hand, is not a scarce resource, but the industrial production of mineral fertilizers consumes extensive amounts of energy and natural gas as feedstock for the hydrogen gas in the production of ammonia. [17] estimated that the fertilizer production accounted for approximately 1.2% of the global energy consumption, of which about 93% was consumed by nitrogen-based fertilizers. Moreover, extensive nitrogen removal up to 85% at wastewater treatment plants (WWTP) as proposed by the European Commission in the revised version of the Urban Waste Water Treatment Directive [18] imply huge increase in investments, operating costs and energy consumption to comply with the more stringent discharge requirements for nitrogen, while losing approximately 60% of the nitrogen as nitrogen gas.
In this review we are focusing on the feasibility of extracting Al and Si from MSW-FA and use these as precursors in the synthesis of zeolites to be used as sorbents for the recovery of nutrients and heavy metals from sources rich in these adsorbates. Potential sources could for instance be liquid manure fractions, rejection water after anaerobic digestion of food or sewage sludge, urine, concentrated black water after fermentation, industrial waste streams or leachates from landfills or mines/mining areas [19,20,21,22]. Many of these sources are rich in constituents that may be transferred to the zeolite precursors if appropriate measures are not in place, which may then also end up as problematic contaminants in the recovered nutrients or heavy metals.
Though zeolites are frequently used as adsorbents in different applications, the appropriateness of using Al and Si extracted from MSW-FA as precursors to synthesize zeolites and using such zeolites to harvest and recover nutrients and heavy metals is much less studied. The final section of this review therefore discusses the possibilities and limitations in a somewhat broader perspective and pinpoints which topics that need to be addressed in the future to assess the feasibility of the implied strategy.
3. Zeolites
3.1. The crystalline structure of zeolites
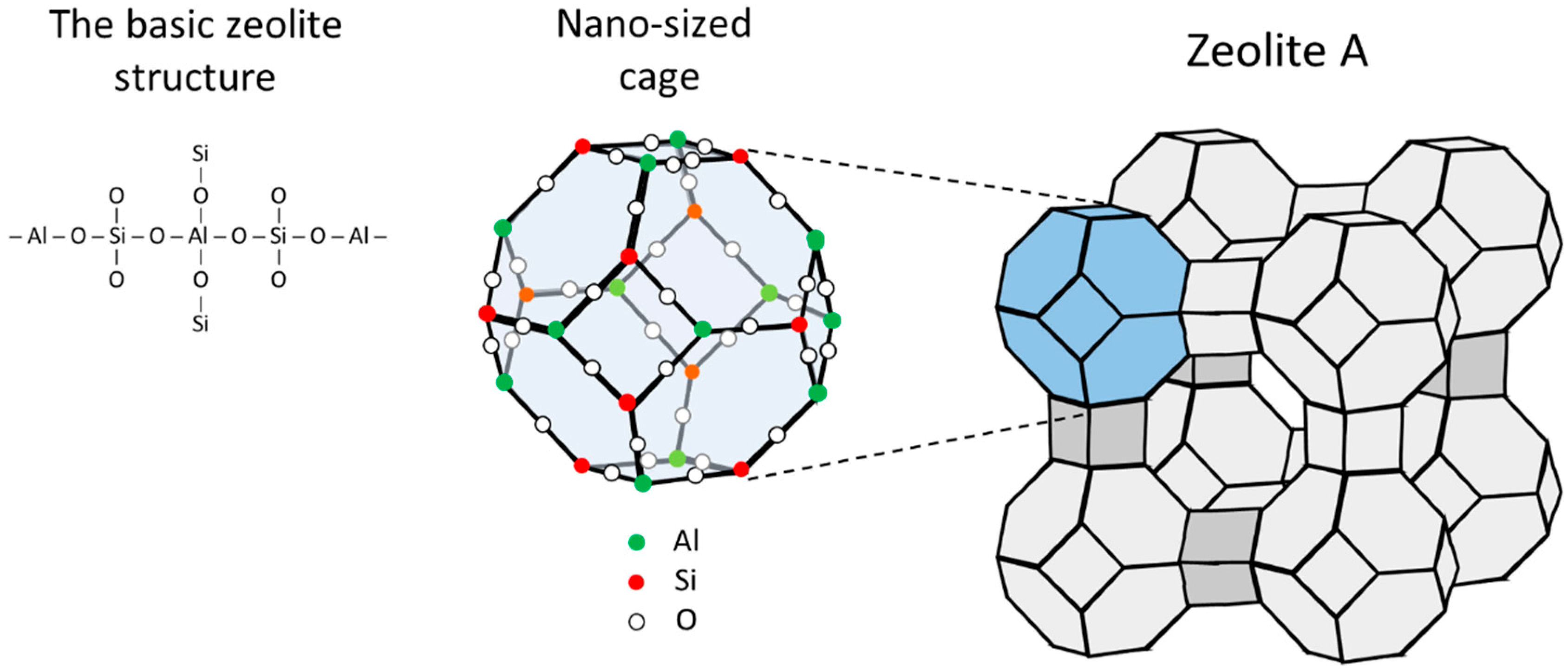
A zeolite is a crystalline substance with a structure characterized by a framework of linked tetrahedra, each consisting of four oxygen atoms surrounding a cation (typically AlO45- and/or SiO44-). The tetrahedra are linked in rings, normally between 4 and 12 atoms, that are units in a more complex three-dimensional framework containing open cavities in the form of channels and cages, as illustrated in Figure 1. The difference in valence between silicon (+4) and aluminium (+3) leads to an excess of negative charge in the crystalline structure, which is neutralized by the presence of compensation cations (Na+, K+, Li+, Ca2+, Mg2+ etc.) that do not occupy fixed positions but are free to move in the channels of the lattice framework [23]. Thus, the negative charge of zeolites is not localized but is more or less uniformly distributed in the framework, also called “framework charge” [24]. The compensation cations can be replaced by other cations (i.e. they are exchangeable). The channels or pores are typically smaller than 20 Å, hence zeolites are classified as microporous materials [8], yet the pores are large enough to allow the passage of small compounds [25]. Therefore, they are also called “molecular sieves”. Note that only the trivalent aluminium brings any negative charge into the frame, hence the lower the Si:Al ratio the more negatively charged and hydrophilic is the zeolite framework, and conversely, the higher the Si:Al ratio the more hydrophobic is the zeolite framework.
Figure 1.
Illustration of zeolite A from the basic structure via the nano-sized cage with a Si/Al ratio of 1 to the full characteristic zeolite A crystalline structure (LTA).
Figure 1.
Illustration of zeolite A from the basic structure via the nano-sized cage with a Si/Al ratio of 1 to the full characteristic zeolite A crystalline structure (LTA).
3.2. Naturally and synthesized zeolites
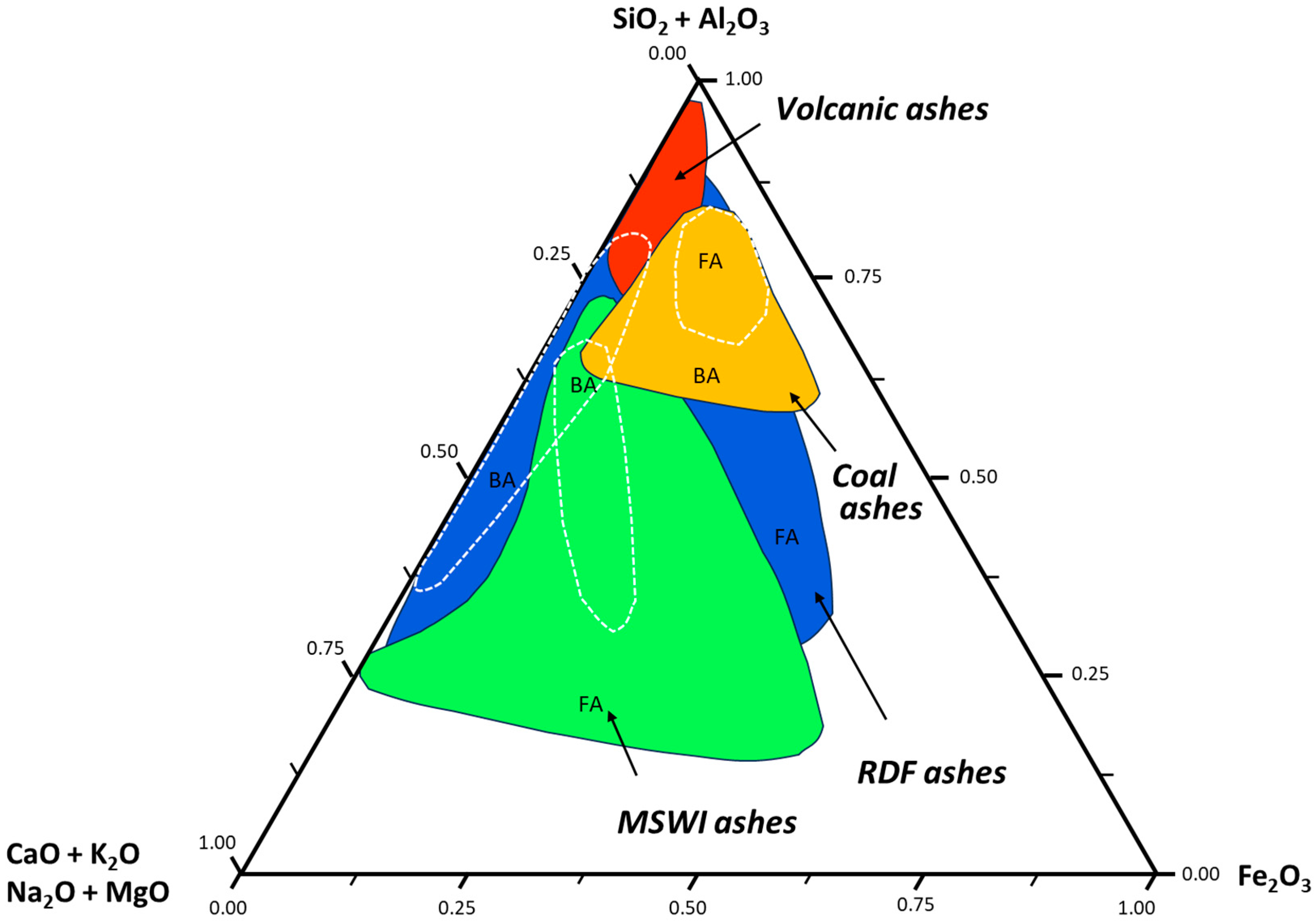
About 60 varieties of natural zeolites have been identified, distinguished by their framework structure and crystalline system [8,26,27]. Large deposits of sedimentary zeolite have been found, generally in volcanoclastic rocks, though only rocks with zeolite content >50% may be of economic interest [8]. As can be seen from the ternary diagram in Figure 2, the volcanic ashes that these natural zeolites are developed from are usually very rich in silica (SiO2) and/or alumna (Al2O3). However, as already mentioned, the mineralogical and chemical composition and the crystal structure and pore sizes are typically varying much in naturally occurring zeolites, both between and within the mineral deposit they are extracted [9]. Due to the possibility of synthesizing zeolites with a higher degree of ordering in the crystalline structure and with standardized pore sizes resulting in more uniform and predictable properties, a lot of research have put into developing zeolites that facilitate targeted applications in the process industry as heterogeneous catalysts, sorbents and ion exchange resins [8].
Figure 2.
Ternary diagram showing relative proportions of SiO2, Al2O3 and CaO, K2O, Na2O, MgO and Fe2O3 in various fly ashes (FA) and bottom ashes (BA). Refuse-derived fuel (RDF) are produced from various types of waste such as MSW, industrial waste or commercial waste. Reproduced from [28] with permission from the Royal Society of Chemistry.
Figure 2.
Ternary diagram showing relative proportions of SiO2, Al2O3 and CaO, K2O, Na2O, MgO and Fe2O3 in various fly ashes (FA) and bottom ashes (BA). Refuse-derived fuel (RDF) are produced from various types of waste such as MSW, industrial waste or commercial waste. Reproduced from [28] with permission from the Royal Society of Chemistry.
3.3. Zeolite synthesis
Historically, zeolites have mostly been prepared by hydrothermal synthesis at moderate temperatures (353-523 K) under autogenous pressure [8,29]. Until the end of the 1950s, only purely inorganic systems had been used. This imposed a major constraint on the Si/Al ratio of the framework, which was always very low, thereby generating very hydrophilic zeolites with great cation exchange properties. However, by introducing different organic additives to the reaction mixtures, the Si/Al ratio could be drastically raised; the first high-silica zeolite (beta) having a Si/Al ratio from 5 to 100, and then the first zeolite having a pure silica end-member (ZSM-5; Silicalite-1). This was possible since the organic molecules could act as void fillers (probably the role of small neutral organic molecules such as e.g. 1-propanol, 1-propanamine and 1,6-hexanediol in the synthesis of ZSM-5) or as structure directing agent (SDA) promoting the crystallization of different zeolite phases [8]. The relatively large organic molecules compensate a lower number of negative framework charges than the small inorganic cations (e.g. Na+, K+) used in the pure inorganic system, hence a lower number of trivalent metal ions (Al3+) are incorporated in the framework. By varying the size of the organic molecule, the maximum trivalent ion concentration can be modulated [8]. Hence, it has been possible to synthesize zeolites with larger pore volumes than those present in natural zeolites [30]. However, other parameters also influence the final structure of the synthesized zeolites; the composition of the reaction mixture, crystallization time, temperature and the SiO2/ Al2O3 molar ratio in the reaction mixture. Today several different methods (e.g. hydrothermal, solvothermal, ionothermal and solven-free methods) and strategies are applied to synthesize zeolites to improve specific qualities of the end product, to increase reaction rates and save energy [11,31]. Due to the obvious benefits of increased access to the internal structure while still having a high specific surface area, significant attention has been given to the possibility of producing zeolites with mesoporous characteristics (20 to 500 Å). According to [8], the most versatile and promising route to the preparation of micromesoporous hierarchical systems is probably the recrystallization of preformed zeolites in the presence of a surfactant [32,33]; the zeolite crystals are partially destroyed with an alkali solution and the zeolite fragments extracted from the crystals generate mesoporosity and are reassembled in a mesostructured phase with the help of the surfactant. The treatment conditions (OH-/zeolite ratio, temperature, time) determine the degree of dissolution of the zeolite crystals and, ultimately, influence the characteristics of the final products. Low dissolution degrees favour the formation of mesoporous crystals coated with a thin film of mesostructured phases, while composites of cocrystallized phases are obtained upon increasing the dissolution degree. Finally, when the dissolution of the zeolite crystals is complete, mesostructured materials with mesoporous walls formed by assembly of zeolite fragments are produced [8,34]. An alternative method to generate mesoporous structures is to synthesize nanosheets or nanosponges using a dual-porogenic surfactant as described by [34] for a ZSM-5-type zeolite, for a MFI-type zeolite as described by [35] or a *BEA-type zeolite as described by [36]. The dual-pyrogenic surfactant plays several roles: as a structuring agent of the zeolitic framework, as a crystal growth inhibitor along the b-axis (formation of the nanosheets) and as a mesopore generator [36].
3.4. MSW-FA as source to silicate and alumina in zeolite synthesis
Using silicate and aluminate from pure chemicals makes the production of zeolites expensive, hence a lot of research has been carried out to find alternative sources to silicate and aluminate from cheaper and abundant sources during the last decades [10]. The prerequisites of such sources are that they must be cheap, readily available, generated in large quantities and rich in silica and/or alumna with low content of contaminating elements [11]. Fly ash from coal-burning is one such source that has been researched considerably. As can be seen from the ternary diagram in Figure 2, these fly ashes are very rich in silica and alumna, and more than the bottom ashes (BA) from coal-burning.
The fly ash fraction of the MSW incineration (MSWI) residues consist of greyish, fine, irregular-shaped dust-like particles [37]. The size of the particles are in the range of ca. 1-2000 µm, but the main weight fraction appears to be within a relatively narrow size range of 50-200 µm [38,39,40,41,42]. The average specific surface area of FA is typically in the range of 250-850 m2/kg [43,44], while the specific densities of the particles are typically 1.7-2.4 kg/m3 [12].
In terms of the silica and alumna content in ashes from the incineration of MSW, the situation is the opposite that of coal-burning ashes; the BA are much richer in silica and alumina than MSW-FA, and similar to BA from coal-burning. The reported concentrations of Al and Si in MSW-FA indicate that the relative contents of SiO2 and Al2O3 are in the range of 12-41 weight% and 5-24 weight%, respectively, with typical values of ca. 20 weight% SiO2 and ca. 10 weight% Al2O3. A summary of reported elemental composition of MSW-FA is shown in Table 1.
MSW-FA also contain significant amounts of Cl, Ca, Na and K, where most of the alkali metals (Na and K) are present as water soluble chlorides. Most of the Ca is present as calcium oxide (CaO), probably due to the decomposition of calcite (CaCO3) [45] and to the excessive use of lime in the scrubbing process [46]. The high levels of chloride probably originate from significant volumes of polyvinyl chloride (PVC) present in MSW [46]. The high content of certain heavy metals (Pb, Cd, Zn, Cd, etc.) in the MSW-FA (see Table 1) is also problematic. Moreover, the FA may contain significant concentrations of organic micropollutants such as polycyclic hydrocarbons (PAH; 18-5600 µg/kg), chlorobenzenes (CB; 0.03-890 µg/kg), polychlorinated biphenyls (PCB; <40 µg/kg), polychlorinated dibenzo-p-dioxins (PCDD; 0.7-1000 µg/kg) and furans (PCDF; 1.4-370 µg/Kg) [4]. Therefore, proper management of MSW-FA involves some sort of stabilization or solidification of the FA prior to landfilling or other confined placement to minimize potential leakage of salts, heavy metals and organic micropollutants [47].
Another important issue regarding MSW-FA is that their composition will vary much more than FA from incineration of fuel that is more homogeneous (e.g. coal-burning). The relative abundance of each component is influenced by 1) the composition of the incinerated waste and 2) the conditions during incineration. The former is usually affected by human habits, socioeconomic factors and the recycling system before incineration [48], while the latter is affected by the types of fuel, operating parameters, types of furnaces and the APC system design used [49].
Hence, both the inherent low and potentially highly variable concentrations of silica and alumina in MSW-FA and the potentially high contamination level of inorganic and organic micropollutants disqualifies it from being an interesting raw source for zeolite synthesis directly [50,51].

Table 1.
Chemical composition range of FA/APC residues from MSW incineration based on reported literature values [12,47,52,53,54,55,56,57,58,59,60,61,62].
| Element | Unit | Fly ash/APC residues | ||
|---|---|---|---|---|
| Min | Max | Median | ||
| Main elements | ||||
| Si | g/kg | 36 | 190 | - |
| Al | g/kg | 6.4 | 93 | - |
| Fe | g/kg | 0.76 | 71 | - |
| Ca | g/kg | 46 | 361 | - |
| Mg | g/kg | 1.1 | 19 | - |
| K | g/kg | 17 | 109 | - |
| Na | g/kg | 6.2 | 84 | - |
| Ti | g/kg | 0.7 | 12 | - |
| S | g/kg | 1.4 | 32 | - |
| Cl | g/kg | 45 | 380 | - |
| P | g/kg | 1.7 | 9.6 | - |
| Mn | g/kg | 0.2 | 1.7 | - |
| TOC | g/kg | 4.9 | 17 | - |
| LOI | g/kg | 11 | 120 | - |
| SiO2 | % | 11,5 | 41,4 | 19,1 |
| Al2O3 | % | 4,7 | 24,3 | 10,9 |
| CaO | % | 17 | 31,5 | 22,0 |
| SO3 | % | 3 | 10,2 | 6,4 |
| Na2O | % | 3,8 | 9,6 | 5,9 |
| K2O | % | 2 | 8,1 | 4,5 |
| Fe2O3 | % | 1,3 | 5,9 | 2,5 |
| MgO | % | 1,7 | 6,9 | 2,7 |
| Minor elements | ||||
| As | mg/kg | 18 | 960 | - |
| Cd | mg/kg | 16 | 1 660 | - |
| Cr | mg/kg | 72 | 570 | - |
| Cu | mg/kg | 16 | 2 220 | - |
| Hg | mg/kg | 0.1 | 51 | - |
| Ni | mg/kg | 19 | 710 | - |
| Pb | mg/kg | 254 | 27 000 | - |
| Zn | mg/kg | 4 308 | 41 000 | - |
3.5. Producing zeolite-like material from MSW fly ash
As described above, there are three main limitations using MSW-FA as source for producing zeolites; i) the Al and Si content is too low, ii) the elemental composition is variable and partially unpredictable, and iii) there is a high level of salts and contaminants that may leach out and have negative health and/or environmental implications depending on the applications of the zeolites or how any residues from the production process are managed.
Ideally, as much of the Al and Si should be separated from the MSW-FA, while generating two distinct pure compounds (e.g., Na2SiO3, Al(OH)3) or solutions (Na2O· SiO2, NaAlO2) e.g. to facilitate tailored synthetization of zeolites. These fractions would add to the salt and heavy metals (i.e., Zn and Cu) fractions that may have commercial interest.
3.5.1. Generating Al- and Si-containing zeolite precursors
The main property to play with for separating Al and Si from the other compounds in the MSW-FA are their pH-dependent solubilities. Most metal hydroxides, including aluminium hydroxides, are readily soluble at acidic conditions, while both crystalline and amorphous silicas are stable [63]. When clay minerals and materials based on silicates and aluminosilicates are treated with acids (typically hydrochloric or sulphuric acid) all the components of the clay minerals except SiO2 are substantially leached out by the acid, leaving internal cavities as pores in the final product, making acid leaching a simple and cost-effective method to prepare quite pure silica [64]. Since silicas are completely dissolved at basic conditions (pH>10), alkali leaching (usually with soda) at pH 11-12 can be used to produce sodium silicate (Na2O·SiO2) solutions either from the silica separated by acid leaching or directly from Si-rich waste [65]. At this pH also aluminium is highly soluble, while most transition metals and alkali earth metals remain largely insoluble [66]. Hence, aluminium metal can be leached with soda to generate sodium aluminate (NaAlO2) solution [67]. Iron should be oxidized to Fe3+ by air or oxidants prior to the alkali leaching to precipitate it as stable Fe(OH)3 at pH 11 [68]. Alkali fusion with soda to reduce the necessary temperature (from 1800 K to 1100 K) may also be used to produce sodium silicate directly from Si-rich waste [69]. The leachate from the acid leaching will contain the aluminium fraction together with the clay minerals. Aluminium can be precipitated as aluminium hydroxides by neutralisation [70]. The different options to extract and purify Si and Al to generate the Al- and Si-containing zeolite precursors from Si- and/or Al-rich wastes discussed above are briefly illustrated in Figure 3. Table 2 lists other Al-rich (>10% Al2O3) waste sources that may be used as secondary sources to generate zeolite precursors [31,66].
Figure 3.
Simplified experimental flowsheet for the extraction of Si and Al and subsequently the preparation of microporous materials from Si and Al wastes.
Figure 3.
Simplified experimental flowsheet for the extraction of Si and Al and subsequently the preparation of microporous materials from Si and Al wastes.
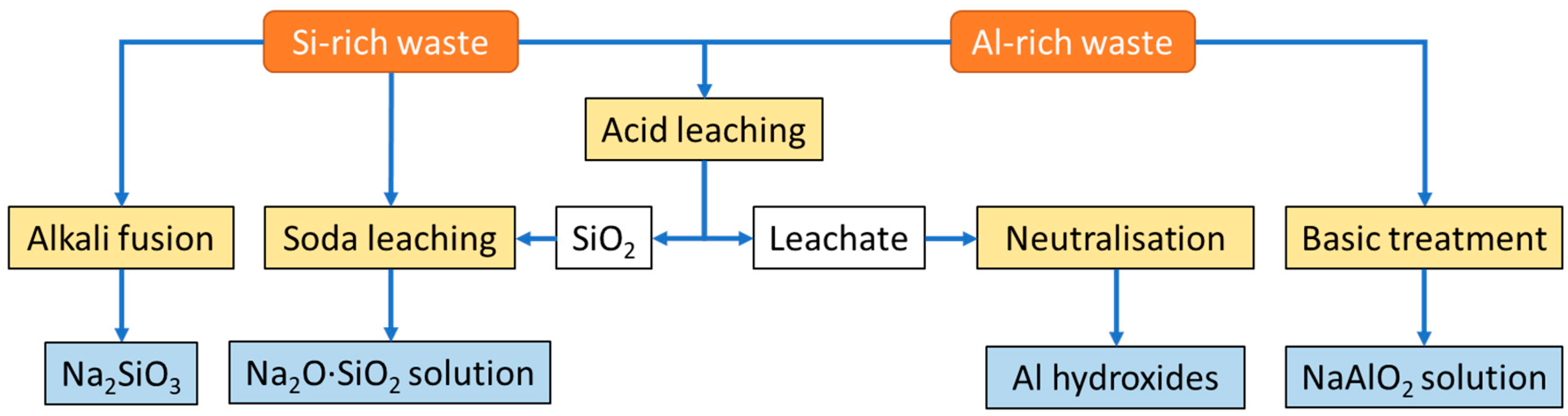
Table 2.
Al and Si sources from different waste products that contain a minimum of up to 10% Al. Sources: [66,71]; MSW-FA see Table 1.
| Waste material | Al2O3 | SiO2 | CaO |
|---|---|---|---|
| Aluminium scrap | Almet >90–99% | ||
| Aluminium dust | Altotal 25–40 Almet 15–25 |
6–11 | 1–4 |
| Black aluminium dross | 42–88 | 1.3–14 | 0.6–1 |
| White aluminium dross | 40–50 | ||
| Spent Fluid Catalytic Cracking catalysts | 40–50 | 40–50 | 0–1 |
| Coal combustion ashes | 15–40 | 40–60 | 3–15 |
| Aluminium salt slag | 20–30 Almet 5–10 |
2–10 | |
| Coal gasification ashes | 5–30 | 25–60 | 2–30 |
| Liquid Crystal Displays glass panel | 15–25 | 50–75 | 0–7 |
| MSW-FA | 5-24 | 12-41 | 15–50 |
| Electric furnace steel reduction slag | 15–20 | 15–20 | 50–60 |
| Lithium slag | 15–20 | 50–55 | 10–12 |
| Red mud from the Bayer process (dried) | 10–20 | 3–50 | 2–40 |
| Drilling and cutting muds (dried) | 5–20 | 30–70 | 2–30 |
| MSW-BA | 1–20 | 5–50 | 10–50 |
| Waste porcelain | 19 | 70 | 3 |
| Blast furnace iron slag | 10–15 | 30–40 | 40–50 |
| Wood ash | 0.5–15 | 10–70 | 10–70 |
| Waste foundry sand | 0–15 | 75–90 | 0–5 |
| Palm oil fuel ash (POFA) | 0.5–12 | 45–75 | 3–15 |
| Zinc slag | 7–10 | 15–20 | 15–20 |
| Electric furnace steel oxidation slag | 5–10 | 10–15 | 20–25 |
3.5.2. Specific leaching of salt and heavy metals
Washing the FA with clean water has been shown to efficiently leach out salts, primarily chlorides (e.g. NaCl, KCl, CaCl2 and CaCl2·Ca(OH)2·H2O) [46]. [72] found that a single wash cycle with water (L/S ratio of 2) for 60 min extracted more than 65% of Cl, greater than 50% of K and Na and about 20% of Ca. When the L/S ratio was increased to 20, ca. 95% of Cl and ca. 50% of Ca were washed out, but with no additional effect on K and Na. However, by increasing the L/S ratio, the leaching of certain heavy metals (Pb, Cu, Zn), and particularly Cr (8%), was increased. A limited loss of Al and Si was observed, but the total loss of mass was not provided. Though, [57] did a similar washing test with MSW-FA (L/S ratio of 2) but for a shorter time (15 min) and measured a total mass loss of ca. 12% due to the removal of chloride- and sulphate-based salts as well as elevated levels of Pb in the leachate (34 mg/L). [45] observed fairly similar levels of loss of Cl (73%), K (ca. 67%) and Na (58%) from MSW-FA as [72] when using treated process water (pH 7.9-9,5) from the incineration plant in the washing step with a L/S ratio of 10 for 1 hour with a final pH of 10.8. In contrast to [72], they observed no depletion of Cr, Pb, Cu or Zn. Pb-bearing sulphates were dissolved but was cemented as PbCu0 on the surface of Al0 particles. Aluminium was dissolved and recovered as precipitated aluminium hydroxide (Al(OH)3. However, significant amounts of Si (21%) were lost during the washing process. It must be noted that this MSW-FA had very low CaO content (<1%) compared to “normal” MSW-FA (17-31.5%; Table 1) as it contained no lime from the scrubbing treatment. [73] found that injecting CaO in the scrubber could make the fly ash alkaline and promote the formation of Ca3(AsO4)2, CaCrO4 and PbCl2 process, which significantly reduced the leaching rate of Pb and As from the FA. The cementation of soluble metal salts into less soluble silicates, hydroxides and carbonates was also observed by [74] for Hg, Mn, Pb and Zn, and notably also for Al and Si (leaching less than 3%), in compacted APC samples. In addition to the presence and nature of the cementitious material, the process is dependent on the amount of water that is added, the temperature and the time it is allowed to work; it may to take weeks or month to complete [74,75].
As already mentioned in Section 3.5.2, most heavy metals show increased leaching rates at low pH making acid leaching a commonly used method to facilitate their recovery from the MSW-FA [6,12,57]. Though, some of the most abundant heavy metals in MSW-FA (Pb, Zn, Cu and Cd) are amphoteric, meaning that they are characterized by a high leaching potential under both high- and low-pH conditions but not in between [52]. However, the same is the case for Al as discussed in Section 3.5.2 [76].
Leaching of heavy metals has been done in full scale in Switzerland since 1997 by the FLUWA process, currently treating >60% of all FA in Switzerland [77]. The process uses scrub water from wet flue gas cleaning to leach heavy metals from MSW-FA. [45] showed that the main parameters controlling the mobilization of the heavy metals were pH and redox conditions, the L/S ratio, the extraction time and temperature. The exothermic reaction between CaO in the FA and the scrub water brought the temperature in the process to up to ca. 60°C. The ideal leaching pH was determined to be ≤3.8, but the variable buffer capacity of the FA (due to carbonates) made it challenging to control the pH by the addition of HCl (32%), particularly in a continuous process [6]. They argued that the process must be individually adjusted to the composition of the FA being processed and the scrub water used in each specific FLUWA process. The acidic FA leaching resulted in depletion factors of 40% for Zn, 53% for Cd, 8% for Pb and 6% for Cu, with the extraction of Pb and Cu primarily limited by the cementation process and the formation of a PbCu0-alloy-phase [45]. The addition of hydrogen peroxide (H2O2; 40 L 35% per ton FA) during the acid FA leaching (termed optimized acidic leaching) and a high L/S ratio (3.5) prevented this reduction through oxidation of the redox-sensitive metals (mainly Pb, Cu and Cd) and thus significantly higher depletion factors for Pb (57%), Cu (30%) and Cd (92%) were achieved with an extraction time of 75 min [45]. Fe2+ was converted into Fe3+, which was precipitated as Fe-hydroxide and accumulated in the remaining filter cake. And moreover, Al0 was oxidized by the H2O2 and quickly precipitated as aluminium-hydroxide and thus no Al0 was available for contact-reduction of more noble dissolved metals [45]. By twerking the different parameters as much as 60–80% Zn, 80–95% Cd and 50–85% Pb and Cu is stated to be extracted by optimized acidic leaching [6,45,77].
The treated FA was dewatered on a vacuum belt filter with the heavy metals being accumulated in the leachate, which can later be used for heavy metal recovery. In 2012 the FLUREC process was implemented in full scale at the MSW incineration plant in Zuchwil, Switzerland to recover ca. 300 tons of high-purity Zn (>99.995%) from the heavy-metal-enriched filtrate from the FLUWA process [7].
The total mass loss during the optimized acid leaching was 32% but the concomitant losses of Al and Si were 14% and 15%, respectively, hence the overall enrichments of Al and Si were relatively marginal; from 31.8 g Al/kg to 35.9 g Al/kg (13% increase) and 69.5 g Si/L to 77.9 g/kg (12% increase). If calculated as Al2O3 and SiO2, these concentrations make up 6.4% and 15.6%, respectively. FA from coal-burning, which has been used as an Al- and Si-source for zeolite synthesis extensively, contains Al2O3 and SiO2 in the ranges of 12.5-35.6% and 28.5-59.7% with mean values of 24.3% and 46.6%, respectively [78]. However, as shown in Table 1, alumna and silica contents vary considerably in MSW-FA, and with median concentrations of ca. 10.9% and 19.1% for Al2O3-and SiO2, respectively, the potential concentrations after optimized acid leaching would be 12.3% Al2O3 and 21.4% SiO2. These values are still below the indicated range of their contents in FA from coal burning, indicating that additional Al and Si need to be supplemented to synthesize zeolites from MSW-FA.
4. Targeted sorption of cations
4.1. Zeolites as cation exchange resins
A wide range of zeolites, both natural and synthetic, have been used to capture different compounds in different types of matrices. Zeolites with a low Si:Al ratio will be negatively charged (and hydrophilic) and the dominating mechanism for capturing compounds will most probably be the exchange of the compensation cations (e.g., Na+, K+, Ca2+, etc.) with the target cations [13]. Hence, cations that fit well into the three-dimensional crystalline structure and show stronger affinity to the negatively charged framework are more likely to be adsorbed. A theoretical cation exchange capacity (CEC) of a zeolite can be calculated as the amount of exchangeable cations that can be accommodated by a weight unit of zeolite material using the idealized chemical formula of the zeolite and assuming all exchangeable cations are sodium. Table 3 shows the CEC range of natural and synthetic zeolites typically used in wastewater treatment together with key structural and chemical features. According to [13], CEC of natural zeolites depends on several factors including the structure and electrostatic field strength of the framework, its cation charge density, the composition and pH of the contacting solution, the raw mineral composition and work-up as well as the process apparatus and operating conditions (e.g., continuous stirred tank vs. fixed bed column, contact time and solid:liquid ratio) used for the measurements. Hence, comparison of reported CEC in the literature is extremely difficult and often contradictory, and the operating CEC is generally far below (till a half) theoretical one [13].
Table 3.
Selected natural and synthetic hydrophilic zeolites applied in wastewater effluent treatment. Framework type code (FTC); window size refers to the largest channel; Si/Al ratio range; exchangeable cations more frequently found; theoretical cation exchange capacity (CEC) range as calculated from the idealised chemical formula assuming sodium as only exchangeable cation. Sources: [79,80,81,82,83].
Table 3.
Selected natural and synthetic hydrophilic zeolites applied in wastewater effluent treatment. Framework type code (FTC); window size refers to the largest channel; Si/Al ratio range; exchangeable cations more frequently found; theoretical cation exchange capacity (CEC) range as calculated from the idealised chemical formula assuming sodium as only exchangeable cation. Sources: [79,80,81,82,83].
| Structure | Chemistry | ||||
|---|---|---|---|---|---|
| Zeolite | FTC | Window | Si/Al | Cation | CEC |
| Å | mol/mol | - | meq/g | ||
| Natural zeolites | |||||
| Clinoptilolite | HEU | 3.1x7.5 | 4.0-5.7 | Na, K, Ca | 2.0-2.6 |
| Chabazite | CHA | 3.8 | 1.4-4.0 | Na, K, Ca | 2.5-4.7 |
| Phillipsite | PHI | 3.8 | 1.1-3.3 | Na, K, Ca | 2.9-5.6 |
| Analcime | ANA | 1.6x4.2 | 1.5-2.8 | Na | 3.6-5.3 |
| Erionite | ERI | 3.6x5.1 | 2.6-3.8 | Na, K, Ca | 2.7-3.4 |
| Faujasite | FAU | 7.4 | 2.1-2.8 | Na, K, Mg | 3.0-3.4 |
| Ferrierite | FER | 4.2x5.4 | 4.9-5.7 | Ca | 2.1-2.3 |
| Heulandite | HEU | 3.1x7.5 | 4.0-6.2 | Na, K, Ca, Sr | 2.2-2.5 |
| Laumontite | LAU | 6.5x7.0 | 1.9-2.4 | Na, K, Mg | 3.8-4.3 |
| Synthetic zeolites | |||||
| X | FAU | 7.4 | 1.0-1.5 | - | 2.7-6.0 |
| Y | FAU | 7.4 | <3 | - | 3.9 |
| Mordenite 1 | MOR | 6.5x7.0 | 4.0-5.7 | Na, K, Ca | 2.0-2.4 |
| A | LTA | 4.1x4.5 | 1.0-3.2 | - | 3.9-5.3 |
| NaP1 | GIS | 2.9 | 1.7-3.9 | - | 2.0 |
1 Mordenite is a natural zeolite but the synthetic version of the zeolite is more in use.
A summary of the factors that determine the cation selectivity of the zeolites and their adsorption rates and capacities is provided below.
4.2. Sorption mechanisms
Adsorption is a surface phenomenon in which the adsorbate (i.e., cation) transfer onto the adsorbent (i.e., zeolite). The main adsorption mechanisms are chemisorption (involving strong covalent bonding), physisorption (involving weak van der Waals forces) and electrostatic interaction (involving attraction between ions or molecules with full permanent charges of opposite signs) [84]. For the latter to occur, an ion exchange with the already present compensation ions (or counterions) need to occur. Both physisorption and electrostatic interactions may govern the adsorption of cationic materials to zeolites, but the latter is generally believed to be the most important [85].
For a cation to be adsorbed to a particular site on a zeolite it first needs to travel by diffusion from the bulk liquid phase through the stagnant liquid film layer outside the zeolite surface. The concentration gradient over the stagnant film governs the driving force; higher bulk concentrations give faster diffusion. As most of the surface of the zeolite is within its porous structure, the cation needs to fit into these pores and diffuse through the interior voids of the crystalline structure of the zeolite. For the electrostatic interaction to be active, the cation needs to exchange place with the existing compensation ion; hence the electrostatic force between the cation and the anionic charge of the zeolite surface needs to be stronger than that between the surface and the compensation ion. In many cases, particularly for heavy metals, the cations are divalent (e.g. Pb2+, Cu2+, Zn2+, Cd2+, Ni2+) while the compensation ions are monovalent (e.g. K+, Na+) and therefore two compensation ions need to be exchanged for the cation to take their place [80,84]. Though, the opposite can also be the case; [86] measured the adsorption rate of ammonium (NH4+) in swine manure by a calcium-rich chabazite and argued that the diffusion rate of Ca2+ for the exchange with NH4+ was the rate limiting step in the adsorption as compared to the exchange between Na+ and NH4+.
4.2.1. Adsorption of heavy metals
As summarized by [80], the reported sorption kinetics of heavy metals such as Pb2+, Cd2+, Cr3+, Ni2+, Zn2+, Cu2+, As5+ and Cr6+ by zeolites with low Si/Al ratios (i.e. strongly negatively charged) all seems to be satisfactorily fitted by a pseudo-second-order kinetic model given by the general model equation [87]:
, where k_2 is the pseudo-second-order rate constant of adsorption (g/mg·min); q_e and q_t are the amount of metal ions adsorbed (mg/g) at equilibrium and at time t, respectively; t is adsorption time (min). The initial sorption rate is given by: h= k_2 q_e^2.
t/qt = 1/k2qe2 + t/qe
However, [80] argued that the intraparticle diffusion model, which assumes that the diffusion of the adsorbate into the pores of the adsorbate is the rate-limiting step in the adsorption process, would make a more appropriate model:
, where k_i is the intraparticle diffusion constant (mg/g·h0.5) and t is the time in hours.
qt = kit1/2
Empirical models such as the pseudo-second-order kinetic model cannot explain the adsorption mechanism [80]. The adsorption isotherm models, however, can do that from the amount of adsorbate at the adsorbent surface and the concentration of the adsorbate in the liquid phase, at a constant temperature. They can also be used to estimate the maximum adsorption capacity (mg/g). Two of the most commonly adopted models to represent the adsorption isotherms are the Langmuir and Freundlich models [80]. The Langmuir model [88], which assume a uniform distribution in a monolayer on the adsorbent surface that is also structurally and energetically homogeneous, takes the following form [87]:
, where Ce is the equilibrium concentration of heavy metals in solution (mg/L), qe is the amount adsorbed at equilibrium (mg/g), KL is the Langmuir constant (L/g) and qmax is the maximum adsorption capacity (mg/g). The Freundlich model [89], that assumes multilayer adsorption on a heterogeneous surface of the adsorbent, can be expressed by [90]:
, where Kf is the empirical constant of the Freundlich isotherm and n is the empirical parameter related to the intensity of the adsorption that depends on the heterogeneity of the material. If 1/n is between 0.1 and 1, the adsorption process is favourable.
Ce/qe = 1/KLqmax + Ce/qmax
qe = KfCe1/n
According to the summary made by [80], for most of the reported zeolites (except scolecite), the adsorption of the above-mentioned heavy metals (Pb2+, Cd2+, Cr3+, Ni2+, Zn2+, Cu2+, As5+ and Cr6+) all seem to be well described by the Langmuir isotherm, hence indicating that a monolayer of heavy metal ions is adsorbed to the surface of the zeolites. However, [91] reported better fit with the Freundlich isotherm at high metal concentrations when adsorbing Pb2+, Cu2+, Ni2+, and Cd2+ on clinoptilolite, hence indicating more a heterogenic adsorption coverage.
4.2.2. Adsorption of ammonium
Most studies appear to report good agreement between adsorption rates of ammonium by most zeolites and pseudo-second-order kinetics [80,92,93,94,95] under appropriate pH conditions (see Section 4.3.7). Though some studies report good agreement with the Langmuir isotherm for the adsorption of ammonium by natural zeolites [86,93,96,97], most studies have found that the Freundlich isotherm gave the best fit, indicating that the adsorption process occurred on a heterogeneous surface [80,90,92,94,95,98]. [95] found that the Langmuir isotherm gave best fit for zeolites synthesized from low-calcium FA, while the Freundlich isotherm gave best fit for zeolites synthesized from high-calcium FA.
4.3. Factors affecting the sorption of cations
As indicated above, there are many factors that influence both the sorption rate of a cation and the sorption capacity of the zeolite. The most important once are discussed briefly in the following.
4.3.1. Framework type vs size of the cation
The framework type of each zeolite determines the dimensions of its internal pores and channels, as illustrated in Figure 1. Table 3 gives the size of the largest channel in some framework types occurring in zeolites used in effluent treatment. These vary from up to 7.5 Å, 7.0 Å and 7.0 Å in HEU, MOR and LAU, respectively, to 2.9 Å, 3.8 Å and 3.8 Å in GIS, CHA and PHI, respectively. The actual size of the cation is dependent on to which degree it is hydrated. Table 4 shows the radii of selected ions when they are unhydrated and when they are fully hydrated. Looking at the heavy metals, the sizes of the fully hydrated ions rank them from smallest to largest: Pb2+< Ni2+< Cu2+< Cd2+< Zn2+< Cr3+ with Pb2+ being the smallest with an assumed maximum diameter of 8.02 Å. This is larger than the largest size of the channels in the framework.
Table 4 also shows the free energy of hydration (ΔhydG) that quantifies the amount of energy that is released when an ion is hydrated. The ions with the highest ΔhydG should prefer to remain in the solution phase [99]. To be able to pass through the pores/channels of the zeolite the ions need to release some of the water molecules; hence the ranking of the cations related to how easy they will do this is: Pb2+< Cd2+< Zn2+< Ni2+< Cu2+< Cr3+.
Table 5 summarize the ranking results from several adsorption studies that have included multiple heavy metals in each test. As can be seen, Pb2+ rank on top in all studies. However, Cr3+ that has the largest hydration radius and is the ion that is most resistant to dehydration, ranked on top or high up in several of the studies. [99] postulated that this was due to precipitation of Cr3+ (and Cu2+) as metal hydroxides on the surface of the zeolite (4A) or inside the pore walls. A similar observation was reported by [100] on adsorption of Cu2+ and Cr3+ ions on NaP1.
Synthetic zeolites are usually made to have larger cavities (i.e. a lower framework density) to facilitate the transport into the zeolite [8]. Synthetic zeolites with larger pore openings such as zeolites X and A showed the highest removal capacity for NH4+.
Table 4.
Unhydrated and hydrated radius and free energy of hydration (ΔhydG) of selected ions [101,102].
| Ion | Unhydrated radius |
Hydrated radius |
ΔhydG | Ion | Unhydrated radius |
Hydrated radius |
ΔhydG |
|---|---|---|---|---|---|---|---|
| Å | Å | kJ/mol | Å | Å | kJ/mol | ||
| Li+ | 0.60 | 3.82 | -475 | Cu2+ | 0.72 | 4.19 | -2010 |
| Na+ | 0.95 | 3.58 | -365 | Zn2+ | 0.74 | 4.30 | -1955 |
| K+ | 1.33 | 3.31 | -295 | Cd2+ | 0.97 | 4.26 | -1755 |
| Ca2+ | 0.99 | 4.12 | -1505 | Pb2+ | 1.32 | 4.01 | -1425 |
| NH4+ | 1.48 | 3.31 | -285 | Cr3+ | 0.64 | 4.61 | -4010 |
| NO3- | 2.64 | 3.35 | -300 | Ni2+ | 0.70 | 4.04 | -1980 |
| H2PO4- | - | 2.6 | - | ||||
| PO43- | - | 7.91 | -2765 |
1 Stokes radius.
Table 5.
Selectivity in competitive adsorption of heavy metals by different zeolites.
| Zeolite | Origin | Si/Al | Selectivity | References |
|---|---|---|---|---|
| Synthetic zeolites | ||||
| FAU-type | Coal FA | 2.5 | Pb2+>Cu2+>Cd2+>Zn2+>Co2+ | [103] |
| NaP1 | Coal FA | 1.7 | Cr3+>Cu2+>Zn2+>Cd2+>Ni2+ | [100] |
| 4A | Coal FA | 1.32 | Cu2+>Cr3+>Zn2+>Co2+>Ni2+ | [99] |
| X | Egyptian kaolin and Na2Si2O5 | 1.15 | Pb2+>Cd2+>Cu2+>Zn2+>Ni2+ | [104] |
| A | Egyptian kaolin and Na2Si2O5 | 1.04 | Pb2+>Cd2+>Cu2+>Zn2+>Ni2+ | [104] |
| Natural zeolites | ||||
| Mordenite | Natural | 4.4-5.5 | Cu2+>Co2+≈Zn2+>Ni2+ | [105] |
| Clinoptilolite | Natural | 4.9 | Pb2+>Zn2+>Cu2+>Ni2+ | [106] |
| Clinoptilolite | Natural | 4.8 | Cu2+>Cr3+>Zn2+>Cd2+>Ni2+ | [100] |
| Clinoptilolite | Natural | 4.2 | Pb2+>Cd2+>Zn2+≈Cu2+ | [107] |
| Clinoptilolite | Natural | 2.7-5.3 | Pb2+>Ag+>Cd2+≈Zn2+>Cu2+ | [105] |
| Phillipsite | Natural | 2.4-2.7 | Pb2+>Cd2+>Zn2+>Co2+ | [107] |
| Chabazite | Natural | 2.2-2.6 | Pb2+>Cd2+>Cu2+>Zn2+>Co2+ | [107] |
| Scolecite | Natural | 1.56 | Cu2+>Zn2+>Pb2+>Ni2+>Co2+>Co2+ | [108] |
4.3.2. Cation concentration and competing ions
As already mentioned, higher bulk concentrations give faster diffusion as the gradient gets steeper. However, studies with natural zeolites from Australia and the USA have reported that also the adsorption capacity of ammonium could be increased by 35-60% (up to 10-13 g NH4+/kg zeolite) by increasing the bulk concentration from 250 mg/L to 1000 mg/L [90] or by 230% (up to 13 g NH4+/kg zeolite) by increasing the concentration from 50 mg/L to 800 mg/L [109], respectively. However, [110] reported equilibrium uptake of ammonium (ca. 40% adsorbed) by 20 g/L clinoptilolite (grain size of 71-125 µm) from a 1 M NH4Cl solution (18 g NH4+/L) after just a few minutes contact time indicating an extreme adsorption capacity of 360 g N/kg zeolite (our calculations). Under normal conditions, there will often be several ions that compete for the adsorption sites on and in the zeolite. Besides the size of the ions compared to the cavities within the three-dimensional crystalline structure explained above, the concentrations of the competing ions impact the competition. These competing ions may, at least when considering the adsorption of NH4+, also be typical compensation ions such as Na+, K+, Ca2+ and Mg2+, as well as anions such as NO3-, SO42-, and Cl- [98,111]. Furthermore, at low pH the competition from H+ increases significantly [112].
4.3.3. Purity of the zeolite
Natural zeolites will usually contain a variety of other minerals that may act as contaminants as they have no or low sorption capacity. Such minerals that are frequently found are quartz, albite, biotite, illite, montmorillonite, feldspar, calcite, halite and heulandite [100,113,114]. Synthetic zeolites, however, usually contain a single framework type giving them high uniformity.
4.3.4. Hydrophilicity/hydrophobicity
Zeolites with low Si/Al ratios (e.g. zeolites A, X, Y and natural zeolites in general) have a the strong electrostatic field within their cavities resulting in very strong interactions with polar molecules. By increasing the Si/Al ratio the zeolite can be tuned to be more hydrophobic. This is beneficial when adsorbing non-polar compounds [80].
4.3.5. Compensation cations
Natural zeolites will usually have more than one type of compensation cation in their crystalline structure. Modification the zeolite with a solution of inorganic salts such as NaCl, CaCl2, NH4Cl etc. will contribute to uniform the compensation cations and thereby also potentially to optimize the cation exchange capacity [115]. For instance, [116] found that the removal of NH4+ by zeolite Y was significantly better using Na+ as compensation ion (92%) than with Cs+ (24.8%), K+ (24.3%), Mg2+ (18.5%), and Ca2+ (18.5%). Moreover, the effective dimensions of the pores of a given zeolite structure can be modulated by the appropriate choice of the extra-framework cations, located within the pores to balance the negative framework charge (see also Figure 1) [117]. It has been shown that following dehydration, subsequent heat treatment of the zeolite, salt molecules with anions of up to two times the size of the O6-ring’s “free” opening of the sodalite-cage structural units are occluded in these cages [118].
4.3.6. Available adsorption surface and size of the zeolite particles
Adsorption efficiency of an adsorbent is proportional to its specific surface area, defined as the total surface area available for adsorption. This area is dependent on the sizes of the zeolite particles, the pore size distribution and surface roughness [119]; smaller particles have larger specific surface areas per volume than larger particles. Due to their higher degree of crystalline ordering, compared to natural zeolites, synthesized zeolites have a greater specific surface area, which increases their efficiency as adsorbents [80].
Even if the availability of active sites is almost constant for both large and small particles, the accessibility of metal ions to adsorption sites is higher when the adsorbent consists of smaller particles because of the shorter diffusion paths for ions [120]. Hence, a smaller sized particle is expected to enhance the adsorption. The internal surface area is more important than external surface area in porous materials. Hence, changes on the external surface associated with grain size has very little effect on the adsorption efficiency [121].
By using more zeolite, the overall surface area will, of course, also increase.
4.3.7. pH
The pH affects both the surface charge (i.e., hydrophilicity) of the zeolite and the cationic character of the adsorbates; hence it is important to control the pH both to optimize the adsorption rate of specific compounds and to maximize the adsorption capacity. In general, at higher pHs the zeolite gets more negatively charged, but the adsorbates may lose their cationic character and be more neutrally charged or anionic. Apparently, most zeolites have points of zero charge (PZC) at a pH near neutral [80], though all PZCs of zeolites and other aluminosilicates reported by [122,123] were typically <3. [123] argued also that often PZC was measured using methods that produced unreliable results (e.g. mass titration), but that this less of a problem for metal oxides. Anyway, for zeolites to be negatively charged, the pH should be kept above their PZC. Hence, their PZC need to be determined properly [123].
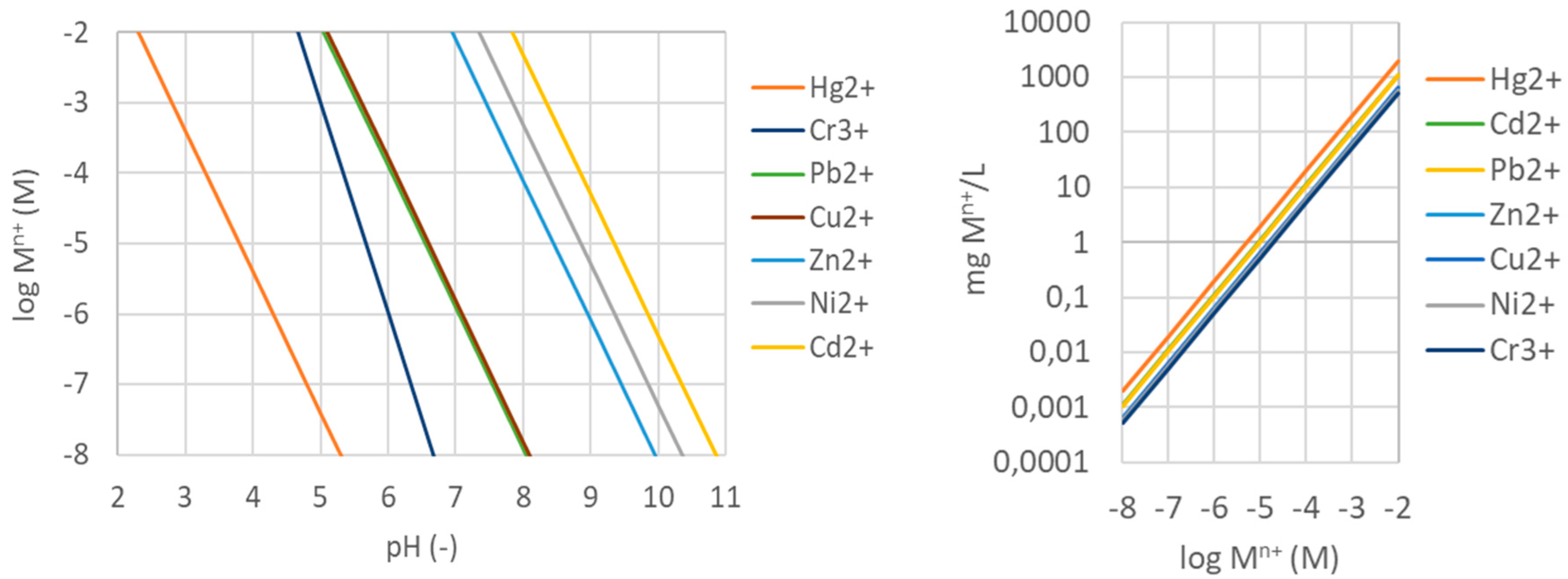
At increasing pH, heavy metals tend to precipitate as metal oxides and hydroxides depending on their solubilities, thereby limiting their possibility to be adsorbed. This occurs at highly different pHs for the different metals, as indicated in Figure 4 by the theoretical hydroxide precipitation diagram based on the solubility constants (KS0) of the different metal hydroxides at 25°C [124]:
, where M(n+) is the molar activity (or equilibrium concentration) of the metal ion with valence n, while Kw is the dissociation constant of water. The diagram says that above a given metal concentration and pH the metal hydroxide will precipitate out of solution. As this occurs at very different concentrations for different heavy metals, this have been used extensively in the separation of different metals in hydrometallurgical processes [124]. Note that at higher metal concentrations, the precipitation starts at a lower pH. However, the precipitation behaviour is also very dependent on the presence of other anions such as carbonate, sulphate and phosphate that can contribute to the precipitation of metal ions [124].
pH = (log KS0 – n log Kw – log [1])/n
Also, the adsorption of ammonium (NH4+) is strongly influenced by pH as at high pH NH4+ is converted into ammonia (NH3) (pKa of 9.24) that will not have any affinity for hydrophilic zeolites due to the lack of any electrostatic attraction [80].
Figure 4.
A: Theoretical hydroxide precipitation diagram of selected heavy metals at pH 1-10 based on their respective metal hydroxide solubilities at 25°C. To the left of each line the hydroxide is still in solution, while to the right they are expected to precipitate. Based on [124]. B: Calculated relationships between metal ion concentrations in molar (M) and mg/L within the concentration range used on the left side.
Figure 4.
A: Theoretical hydroxide precipitation diagram of selected heavy metals at pH 1-10 based on their respective metal hydroxide solubilities at 25°C. To the left of each line the hydroxide is still in solution, while to the right they are expected to precipitate. Based on [124]. B: Calculated relationships between metal ion concentrations in molar (M) and mg/L within the concentration range used on the left side.
4.3.8. Temperature
An increase in temperature leads to changes in both process kinetics and equilibrium including increased diffusion rate, increased kinetic energy resulting in greater access of metal ions to adsorption sites, increased activity on the surface of the adsorbent resulting in greater affinity and decrease in mass transfer resistance [120]. The temperature may also expand the crystalline structure itself, thereby increasing the accessibility for the adsorbate to the interior of the zeolite. Furthermore, an increase in temperature is associated with a decrease in thickness of the boundary layer surrounding the adsorbent (reduced hydration) such that the mass transfer resistance of the adsorbate decreases, which facilitates the diffusion of metals in the adsorbent. However, the electrostatic interactions are weaker at higher temperatures [120].
4.3.9. Contact time
The adsorption process is quicker when the adsorbate concentration is high and/or when there is more space available for sorption on the zeolite. Hence, an increase in contact time allows reaching closer to equilibrium conditions and hence full utilization of the adsorption capacity. However, an increased contact time reduces the overall process efficiency as the adsorption rate is drastically reduced with time requiring long retention/contact times. In a continuous filtration process the contact time is given by the flow rate and liquid bed volume, and when the necessary exposure time to get additional adsorption is exceeded, there will be a breakthrough of the filter.
5. Sorption of nitrate and phosphate using zeolites
Anions such as nitrate (NO3-) and phosphate (H2PO4-, HPO42- and PO43-) will not be efficiently captured by the hydrophilic zeolites described above as they will be repelled by the electrostatic forces otherwise attracting the cations, though zeolites derived from coal-FA has been shown to adsorb phosphate moderately well [125,126,127]. Moreover, increasing the Si/Al ratio making the zeolite more hydrophobic is not very useful either since zeolites need to be modified to be able to adsorb these anions. Two different strategies can be used to modify zeolites:
- Lowering the pH to make the zeolite cationic
- Modifying the surface of the zeolite by cationic metal-doping or using surfactants.
5.1. pH-derived cationic zeolites
The zeolite needs to be predominantly positively charged to be cationic. This may only occur at pHs below its PZC. As described above in section 4.3.7, the actual PZC may, however, be challenging to measure correctly.
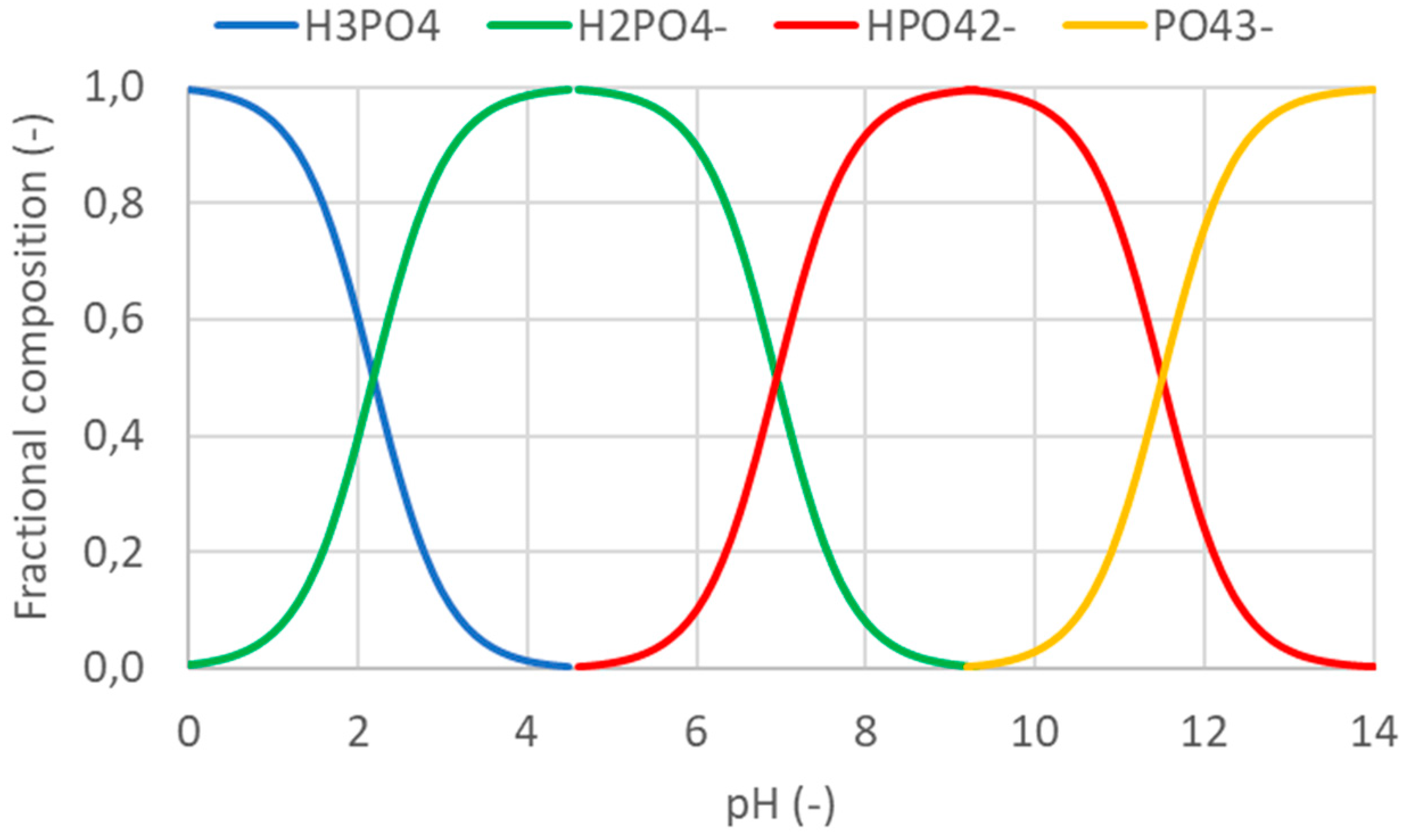
Phosphoric acid (H3PO4) is a triprotic acid with three dissociation constants: pKa,1 = 2.13 (to H2PO4-), pKa,2 = 7.20 (to HPO42-) and pKa,3 = 12.36 (to PO43-) [128]. The ratio of the four different phosphate compounds is determined by the pH of the solution, as depicted in Figure 5. As shown in Table 4, the stokes radius of PO43- (7.9 Å) is very much larger than the hydrated radius of H2PO4- (2.6 Å), hence H2PO4- is more likely to penetrate the micropores of the zeolites.
Relatively few studies have apparently been conducted that have tried to adsorb phosphate to pH-derived cationic zeolites (see Table 6). However, [129] reported an adsorption capacity of 52.9 mg P/g for a synthetic zeolite A from purified clay T just by lowering the pH to 4-5. The zeolite had a measured PZC of 5.5. [127] reported efficient removal of phosphate at pHs between 3.5 and 9 when testing 15 different coal ash-derived zeolites. The zeolites were not metal-doped (see section 5.2.1), but had variable levels of Ca2+, Mg2+, Al3+ and Fe2+ in their framework due to their different origins. The good removal at high pH (5.5-9) was attributed to precipitation of calcium phosphate due to the Ca2+-rich zeolite that was used. At acidic conditions (pH 3.5-5.5) the observed immobilization of phosphate was assumed due to adsorption to Fe2O3 through ligand exchange. [130] treated a coal ash-derived zeolite with a low concentrated (0.01 M) H2SO4 solution that lowered the equilibrium pH of the zeolite to <10.5. This resulted in efficient removal of both NH4+ and phosphate over the whole concentration range from 0.5 mg P/L to ca. 500 mg P/L with a S/L ratio of 10 g/L for 24 hours. The formation of calcium phosphate precipitates was assumed to be the predominant mechanism for phosphate removal.
By decreasing the pH to <2, the dominating phosphate fraction will be H3PO4, which is uncharged (see Figure 5). At this low pH, zeolites are very likely to be neutral or positively charged [123]. [131] demonstrated that it was possible to adsorb phosphate (as H3PO4 and/or H2PO4-) at pH <2. However, this requires considerable amounts of acid for pH adjustments and subsequently a potential equivalent amount of base to compensate the low pH prior to release.
In theory, it should be possible to adsorb nitrate to positively charged zeolites as has been documented for phosphorous (see above). Nitrate is 100% negatively charged at pH >1.0 (pKa = -1.37) and its hydrated radius is close to that of NH4+ and K+ (see Table 4). To the best of our knowledge, there has been no studies so far documenting the removal of nitrate by non-modified zeolites.
Figure 5.
Fractional composition of different phosphate compounds depending on the pH. Based on the dissociation constants of phosphoric acid [128].
Figure 5.
Fractional composition of different phosphate compounds depending on the pH. Based on the dissociation constants of phosphoric acid [128].
5.2. Modification of zeolites
The dominating method to increase the adsorption rate and capacity of anionic (and hydrophobic) compounds to zeolites has been to prepare modified zeolites. Two main methods have been used to modify the zeolites; metal doping and cationic surfactants as discussed below.
5.2.1. Metal-doped zeolites
Transition metal cations or oxides are doped into the zeolite framework typically using oxides of lanthanides, aluminium or zirconium to optimize the adsorption of anions [125,132,133]. The charge properties of the modified zeolites depend both on the kind of the modifier and conditions of preparation (temperature, amount of modifier) [133,134]. Calcination at rather high temperatures (200-800°C) is frequently applied [135].
5.2.2. Surfactant-modified zeolites (SMZs)
A cationic surfactant is used to create a hydrophobic monolayer (targeting hydrophobic compounds) or a cationic bilayer (targeting anions) on the external surface of the zeolite. Surfactants such as quaternary ammonium, sulphonium, arsonium, phosphonium or idonium have typically been used [136,137,138].
Hexadecyltrimethylammonium (HDTMA) is a long-chain quaternary ammonium surfactant that has been much used surfactant to generation SMZs. The large head group of the HDTMA molecule (three of the protons in NH4+ are replaced by three methyl groups, while the fourth is replaced by the long tail group) makes it too large to enter the zeolite channels for internal sorption sites, and the sorption of HDTMA is therefore limited to external cation-exchange sites on the zeolite surface [139]. The process of generating, first a monolayer, then a bilayer of the surfactant on the zeolite is illustrated in Figure 6 and a more detailed description is given in the figure text. It is important to note that a bilayer of the surfactant is needed for efficient adsorption of anions to the external surface of the SMZ by ion exchange, while hydrophobic compounds may still be adsorbed within the solvent-like hydrophobic bilayer [137,139,140]. According to [140] even cations still have access to the microporous structure of the zeolite despite the bilayer surrounding the zeolite. However, it has been challenging to find solid documentation that support this statement or documentation of how much they are affected by this bilayer; first by the electrostatic repulsion exserted by the cationic heads of the surfactant, then by the hydrophobic region within the bilayer. A study by [141] indicates that some reduced adsorption capacity and rate can be expected. Though, the adsorption of phosphate and nitrate to SMZs has been well documented as summarized below.
Figure 6.
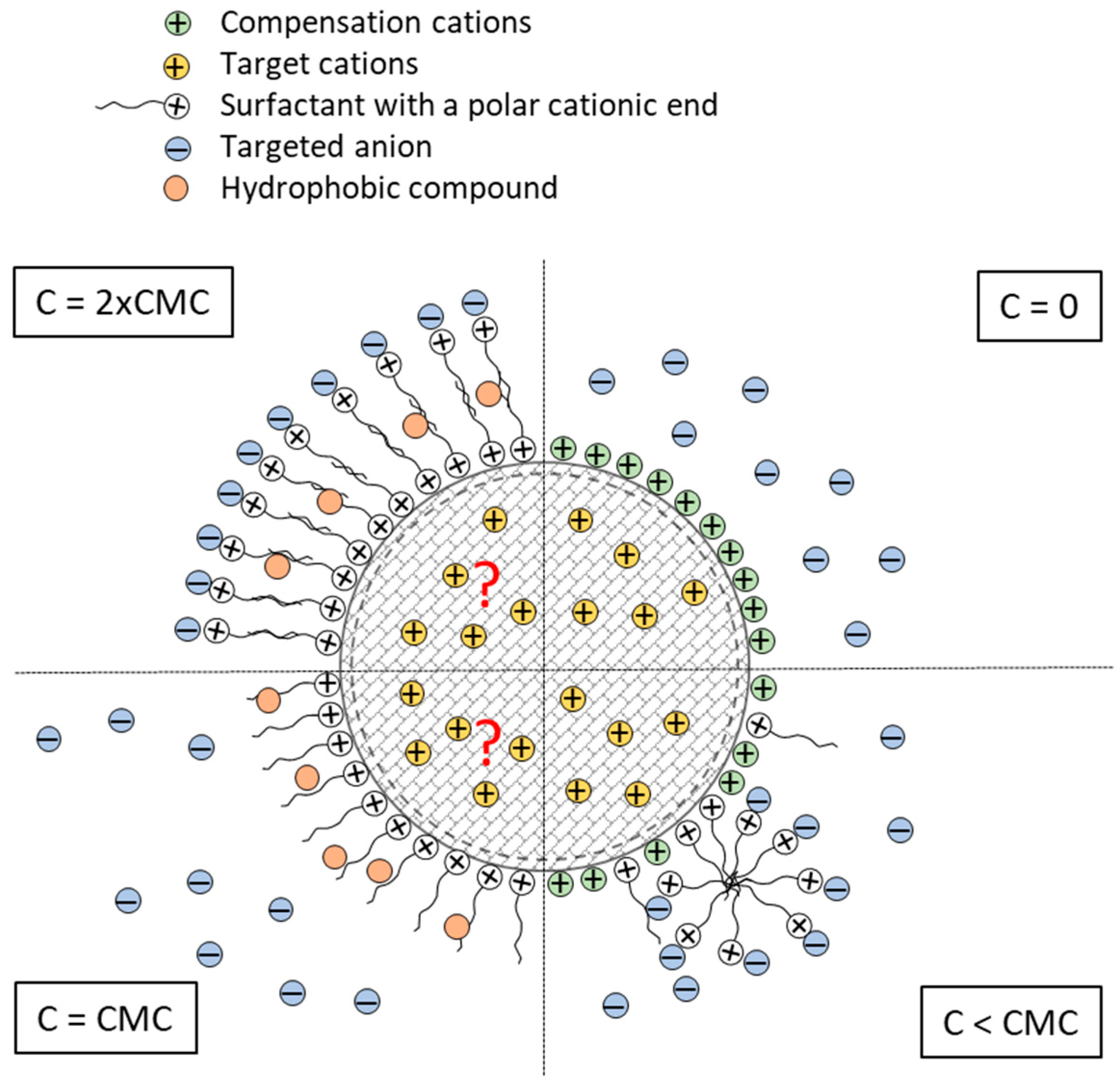
Illustration of the stepwise generation of a surface-modified zeolite using a surfactant: C = 0: the zeolite prior to any addition of surfactant attracting targeted cations that primarily adsorb to the micropores structure; C < CMC: the surfactant forms micelles that start adhering to the surface of the zeolite, replacing external compensation cations. When more surfactant molecules attach to the surface and the critical micelle concentration has been passed breaking up the micelle, the surfactants spreads out on the surface [139]; C = CMC: Just enough surfactant has been added to create a monolayer of surfactants on the surface of the zeolite, held in place by electrostatic interaction between the negative surface charge of the zeolite and the positively charged end of the surfactant [137]. The hydrophobic tail of the surfactant attracts hydrophobic compounds in the water; C > CMC: a bilayer of the surfactant is evolving driven by the hydrophobic affinity between surfactant tail groups, and eventually form a fully saturated bilayer. The original net negative charge of the zeolite has been reversed to a net positive charge attracting target anions in the water phase. Hydrophobic compounds are still attracted to the hydrophobic region within the bilayer [139,140], and according to [140] targeted cations still have access to the microporous structure, though a somewhat reduced adsorption capacity and adsorption rate can be expected (as indicated by red question marks) [141].
Figure 6.
Illustration of the stepwise generation of a surface-modified zeolite using a surfactant: C = 0: the zeolite prior to any addition of surfactant attracting targeted cations that primarily adsorb to the micropores structure; C < CMC: the surfactant forms micelles that start adhering to the surface of the zeolite, replacing external compensation cations. When more surfactant molecules attach to the surface and the critical micelle concentration has been passed breaking up the micelle, the surfactants spreads out on the surface [139]; C = CMC: Just enough surfactant has been added to create a monolayer of surfactants on the surface of the zeolite, held in place by electrostatic interaction between the negative surface charge of the zeolite and the positively charged end of the surfactant [137]. The hydrophobic tail of the surfactant attracts hydrophobic compounds in the water; C > CMC: a bilayer of the surfactant is evolving driven by the hydrophobic affinity between surfactant tail groups, and eventually form a fully saturated bilayer. The original net negative charge of the zeolite has been reversed to a net positive charge attracting target anions in the water phase. Hydrophobic compounds are still attracted to the hydrophobic region within the bilayer [139,140], and according to [140] targeted cations still have access to the microporous structure, though a somewhat reduced adsorption capacity and adsorption rate can be expected (as indicated by red question marks) [141].
5.2.3. Adsorption of phosphate by modified zeolites
The majority of studies have been assessing the effect of metal-doped zeolites on the adsorption of phosphate, very few using surfactant-modified zeolites. Some literature reported studies are shortly summarised in Table 6 and briefly described below.
Results with metal-doped zeolites
[126] tested the ability of salt-modified (Ca2+, Mg2+, Al3+ and Fe3+) NaP1-zeolite derived from coal-FA to concomitantly adsorb ammonium and phosphate compared to non-modified NaP1. Only the Ca-modified zeolite improved the adsorption capacity for phosphate (from 34.7 mg/g to 49.5 mg/g), but Al- and Fe-modified zeolites improved the removal at relatively low phosphate concentrations (0.5-10 mg P/L) where the Ca-modified zeolite showed limited P removal. The mechanisms behind the phosphate removals were believed to be precipitation of calcium phosphate by the Ca-modified zeolite and ligand exchange for the Fe- and Al-zeolites. [142] observed efficient adsorption of phosphate using a coal FA-derived zeolite modified with lanthanum hydroxide (La(OH3)) at pH>2.5 with an adsorption capacity of 71.9 mg P/g. The phosphate precipitated as LaPO4 on the zeolite. The sorption appeared to be insensitive to pH in the 3-10 range for low (5 mg P/L) and medium (100 mg P/L) concentrations but increased in the whole pH range 1-11 (highest at pH 1) at the highest tested concentration (500 mg P/L). At pH 3 La started to desorb from the zeolite, and at pH 2.3 99.8% had desorbed. A similar study was conducted by [125] comparing the adsorption of phosphate by coal FA-derived zeolites (NaP1 and A) with that of a natural zeolite (clinoptilolite), all modified with lanthanum. The observed adsorption capacities were significantly higher with the coal FA-derived zeolites (44.0-58.2 mg P/g) than that with the natural zeolite (24.6 mg P/g), all with an apparent pH optimum at pH 5.3. The adsorption capacities appeared, however, to be only moderately affected by the pH in the range 2-7. The experimental data fitted very well with the Langmuir isotherm model (much better than the Freundlich isotherm), hence that only a monolayer of phosphate was adsorbed to the surfactant-modified zeolite surface, and the thermodynamics showed that the phosphate adsorption was a spontaneous and endothermic process. In a recent study by [143] they used zirconium oxide-modified merlinoite obtained from coal fly to adsorb phosphate. The adsorption capacity was determined to be 67.7 mg P/g at pH <5 (PZC = 4.6). Electrostatic interaction was found to be the predominant mechanism behind the observed adsorption. However, a part of the phosphate ions was adsorbed to the zeolite surface via strong bonding, presumably by coordination bonding, resulting in reduced desorption of the phosphate even if the pH was turned to strong alkaline conditions. The Freundlich isotherm gave an adequate fit to the data, indicating that the phosphate had adsorbed as a multilayer coverage over the heterogeneous distribution of active sites on the metal-doped zeolite.
Results with surfactant-modified zeolites
[144] tested the adsorption capacity of phosphate by HDTMA bromide-modified clinoptilolite and hexadecylpridinium (HDP) bromide clinoptilolite and found the optima at pH 12 at 675 mmol/kg (20.9 mg/g) and 376 mmol/kg (11.6 mg/g), respectively. Unmodified clinoptilolite had the optimum adsorption capacity at pH 3 with 25 mmol/kg (0.8 mg/kg). They explained the increased adsorption at high pH with a stronger attraction between the cationic head groups of the surfactants and the PO43- form than any of the other phosphate forms. The Freundlich isotherm gave the best fit to the adsorption data, though also the Langmuir isotherm gave acceptable correlation, suggesting that in addition to the monolayer of phosphate that was provided by the head ammonium group of the surfactant, some phosphate species may have been trapped between the organic chains within the bilayer [144].
Table 6.
Reported apparent sorption capacity of phosphate for non-modified and modified zeolites with test conditions (phosphate concentration range, S/L ratio, contact time and temperature) and pH at optimum adsorption.
Table 6.
Reported apparent sorption capacity of phosphate for non-modified and modified zeolites with test conditions (phosphate concentration range, S/L ratio, contact time and temperature) and pH at optimum adsorption.
| Zeolite | App. sorption capacity |
Conc. range |
S/L ratio | Contact time |
Temp. | pH | Ref. |
|---|---|---|---|---|---|---|---|
| mg/g | mg P/L | g/L | h | oC | - | ||
| Non-modified zeolites | |||||||
| NaP1 | 11.4 | 12.5-200 | 1 | 24 | 25 | 5.3 | [125] |
| NaA | 15.7 | ||||||
| Clinoptilolite | 20.2 | ||||||
| A | 52.9 | 50-1000 | 6.6 | 4 | 70 | 5.5 | [129] |
| Clinoptilolite | 1.3 | 10-100 | 48 | 2 | 25 | 2 | [132] |
| Zeolite from coal-FA | 11.7-42.4 | 1000 | 10 | 24 | room | 3.5-9 | [127] |
| Clinoptilolite | 0.77 | 0.03-3.1 | 8 | 24 | room | 3.0 | [144] |
| NaP1-zeolite from coal-FA | 34.7 | 0.5-1000 | 10 | 24 | 18-22 | - | [126] |
| Salt-modified zeolites | |||||||
| LaP1 | 58.2 | 12.5-200 | 1 | 24 | 25 | 5.3 | [125] |
| LaA | 48.9 | ||||||
| La-clinoptilolite | 25.5 | ||||||
| TiO2-modified clinoptilolite | 34.2 | 10-100 | 20 | 2 | 25 | 2 | [132] |
| Ca-bearing K-zeolite | 142-250 | 100-16000 | 16.7 | 0.8-2.2 | 22 | 6-9 | [145] |
| Zr oxide merlinoite | 67.7 | 5-200 | 0.2-2 | 4 | 40 | <5 | [133] |
| CaP1-zeolite from coal-FA | 49.5 | 0.5-1000 | 10 | 24 | 18-22 | - | [126] |
| MgP1-zeolite from coal-FA | 31.3 | ||||||
| AlP1-zeolite from coal-FA | 29.9 | ||||||
| FeP1-zeolite from coal-FA | 30.9 | ||||||
| Cu-zeolite X | 87.7 | 10-200 | 1 | 24 | 25 | 5.0 | [146] |
| Surfactant-modified zeolites | |||||||
| HDTMA-Br clinoptilolite | 20.9 | 0.03-3.1 | 8 | 24 | room | 12.0 | [144] |
| HDP-Br clinoptilolite | 11.6 | ||||||
5.2.4. Adsorption of nitrate by surfactant-modified zeolites
As far as the authors have found, all available studies on the adsorption of nitrate on modified zeolites have been done on zeolites modified with different types of surfactants, primarily HDTMA. Table 7 summarizes the literature findings which are also briefly presented below.
[147] found that a clinoptilolite-rich turf modified with HDTMA-Br was able to sorb 5.0 mg NO3/g. In a later study with clinoptilolite from the same area, [148] reported that the HDTMA-Cl was better at exploiting its anion exchange capacity compared to the same zeolite modified with HDTMA-Br. [149] ran small-scale filtration tests on a polydopamine-coated clinoptilolite in fixed-bed columns for the removal of nitrate from deionized water with and without other competing anions. 2.47 mg NO3/g was adsorbed to the zeolite. Lower flow rate, pH (3-9) and temperature (10-35°C), but higher influent concentration and bed height, increased the adsorption of nitrate. [150] used natural zeolite modified with HDTMA-Br in ballasted flocculation to improve the settling properties of activated sludge and at the same time remove nitrate and phosphate. When dosing 0.91 g modified zeolite per L sludge slurry with 4 g suspended solids/L and 5 mg NO3/L, the nitrate removal was ca. 44% (2.42 mg NO3/g) (our calculations). The adsorption of nitrate was only marginally impacted by competing anions when these were increased from 0 mg/L to 5 mg/L: 2.6% reduction by Cl- and 3.2% reduction by SO42-. When modifying natural zeolite with cetylpyridinium bromide (CPB), [151] determined the maximum adsorption capacity to be 9.7 mg NO3/g from the Langmuir isotherm model, and the uptake of nitrate was only slightly impacted by competing anions (Cl-, HCO3- and SO42-) in the concentration range 0-200 mg/L. Interestingly, the adsorption was reported to be decreasing with temperature in the temperature range 15-35°C. They also found that the adsorption of nitrate onto the zeolite followed the pseudo-second-order kinetic model for the entire adsorption process, and that both the Langmuir and the Freundlich isotherm models gave good fits with the adsorption data. [152] measured the adsorption of nitrate by a HDTMA-Br-modified clinoptilolite in batch and found that the maximum capacity was 6.1 mg NO3/g from the Langmuir isotherm model. The equilibrium time for uptake of nitrate was only 0.5-1 hour, and the competition with other anions (Cl-, HCO3- and SO42-) only reduced the kinetics (uptake rate) not the absolute removal capacity. However, in a later study with the same HDTMA-Br-modified clinoptilolite tested in a filter column, [153] reported that nitrate was significantly reduced in the presence of SO42- with SO42- and NO3- being similarly adsorbed to the zeolite. The same research group has also tested the adsorption of nitrate by differently nanostructured MFI-, ZSM-5- and *BEA-type zeolites modified with HDTMA-Br [34,35,36]. First, [35] obtained an adsorption of 37.2 mg NO3/g with HDTMA-Br-modified MFI-zeolite nanosheets. Then [36], testing a HDTMA-Br-modified *BEA-type zeolite nanosponge, reached a maximum adsorption capacity of 83 mg NO3/g. This was considerably better than the same HDTMA-Br-modified *BEA-zeolite nanocrystals (19 mg NO3/g). Finally, [34] tested different HDTMA-Br-modified nanostructured zeolites (nanosheets, nanocrystals and nanosponges). Again, the nanosponge performed best with 132 mg NO3/g compared to 120 mg NO3/g with nanosheets and 50 mg NO3/g with nanocrystals. The adsorption occurred very quickly; equilibrium within about 2 min fitting well with the pseudo-second order model. A good fit with the Langmuir and the Dubinin-Radushkevich isotherm models confirmed that the adsorption mechanism was by ion exchange and that the nitrate molecules formed a saturated monolayer on the surfactant at equilibrium [34,36].
Table 7.
Reported apparent sorption capacity of nitrate for modified zeolites with test conditions (nitrate concentration range, S/L ratio, contact time and temperature) and pH at optimum adsorption.
Table 7.
Reported apparent sorption capacity of nitrate for modified zeolites with test conditions (nitrate concentration range, S/L ratio, contact time and temperature) and pH at optimum adsorption.
| Zeolite | Surfactant | Amount adsorbed |
Conc. Range |
S/L ratio |
Contact time |
Temp. | pH | Ref |
|---|---|---|---|---|---|---|---|---|
| mg NO3/g | mg NO3/L | g/L | h | °C | - | |||
| Clinoptilolite | polydopamine | 2,47 | 150 | - | 0,30 | 10 | 3 | [149] |
| ZSM-5 nanocrystals | HDTMA-Br | 50 | 50-2500 | 0,5 | 24 | room | 6 | [34] |
| ZSM-5 nanosheets | HDTMA-Br | 120 | ||||||
| ZSM-5 nanosponges | HDTMA-Br | 132 | ||||||
| clinoptilolite-rich turf | HDTMA-Br | 4,96 | 124-1240 | 100 | 24 | room | - | [147] |
| Natural zeolite | HDTMA-Br | 2,42 | 5 | 0,91 | 2 | room | 7 | [150] |
| *BEA-type zeolite nanosponge | HDTMA-Br | 83 | 50-1500 | 2 | 2 min | room | 5,5 | [36] |
| *BEA-type zeolite nanocrystals | HDTMA-Br | 19 | 25 | 5 min | ||||
| Clinoptilolite-rich tuf | HDTMA-Br | 6,07 | 1-113 | 20-200 | 24 | room | 5-6 | [152] |
| Natural zeolite | CPB | 9,68 | 89 | 2 | 0,5 | 15 | 6 | [151] |
5.2.5. Leaching of surfactants – a potential setback
An inherent challenge with using surfactant that are only adsorbed to the surface of the zeolites by ion exchange or electrostatic forces is that they may leach from the zeolite during actual use out in the field. Not surprisingly, the majority of available studies on surfactant-leaching from zeolites have been done on HDTMA [147,154,155,156,157,158].
A fully saturated bilayer of HDTMA on a clinoptilolite contains typically approximately 200 mmol HDTMA /kg (57 g/kg) [139]. [147] studied the loss of HDTMA from clinoptilolite placed in a filter column continuously operated for 130-150 days. In one of the tests only the medium sized fraction (14-40-mesh) of the zeolite was used, resulting in a relatively high hydraulic conductivity of 0.01 cm/s, and the zeolite was only partially covered with a HDTMA-Br bilayer (150 mmol/kg). In this test, at the end of 150 days and 100 pore volumes (PV), 17% of the HDTMA had been lost. The loss followed a first-order model, but during the initial 70 PV, the loss was markedly faster (k = 0.003) than the later 30 PV (k = 0.0003) with a distinct transition between the two, corresponding to a quicker loss of the bilayer than the monolayer. In a second test, only the smallest size fraction (<40 mesh) of the zeolite was used (hydraulic conductivity of 9·10-6 cm/s), and this time it was fully covered with a HDTMA-Br bilayer (230 mmol/kg). Due to the smaller pore volume in the column, the linear velocity through the column was about 10 times faster and at the end of the 130 days test period 3000 PV had passed through and 60% of the HDTMA was lost from the zeolite. In this case there were two distinct transitions between an initial very rapid loss period (0-300 PV; k = 0.007), a rapid loss period (300-1500 PV; k = 0.0003) and a slow loss period (1500-3000 PV; k = 0.00008), with the second transition corresponding to the external cation exchange capacity of the clinoptilolite. It was not suggested by [147], but the first initial very rapid loss may potentially have been excess HDTMA not fully bound within the bilayer by hydrophobic forces. In summary, many factors may influence the loss of HDTMA from the modified zeolites, including the HDTMA coverage, flow rate and conductivity (low conductivity increases the loss; [154]).
HDTMA has been shown to be toxic to soil bacteria in the dissolved state, but not when bound to the cation exchange sites present in soil [160]. It is also not biodegraded when bound to zeolites [154]. [160] estimated the 50% growth inhibition concentration of HDTMA-Br (EC50) towards Acinetobacter junii, a phosphate-accumulating bacteria important for biological phosphorous removal in wastewater treatment plants, to be 3.27 ± 1.12·10-7 mol/L (120 µg/L). [137] pointed to the lack of data connected to the toxicity of HDTMA-Br that is needed to determine the suitability of surfactant-modified zeolites in water remediation applications. [153] suggested that the residual amounts of surfactants leaching out could be lowered below the maximum residual concentration limit of ca. 10-7 mol/L by filtration through a second column filled with activated carbon.
6. Reuse of adsorbed compounds
There are two main strategies for reusing the adsorbed compounds:
- Use them as they are, embedded in zeolite, typically as slow-release compounds, for instance in fertilizers.
- Recover them from the zeolite by controlled release.
There are at least two strengths of the first strategy: 1) There are no further treatments needed, implying limited added costs, and 2) depending on how it is applied, when the slow-released compounds in the zeolite is exhausted, potentially no residual waste-zeolite needs to be managed.
The second strategy has potentially several strengths: 1) The compounds can be recovered as a rich concentrate 2) that, depending on its purity and legal restrictions, can be utilized as a secondary raw material in commercial products, 3) which could drastically broaden the range of reuse applications, 4) hence also strengthening its potential role in the circular economy. 5) This also potentially enables multiple reuse cycles of the zeolite as an adsorbent, 6) thereby reducing the upstream costs per cycle, and 7) generate less residual zeolite waste that needs to be managed.
Both strategies require that it is possible to release the compounds of interest under conditions that can be controlled or naturally occur, and that the release of unwanted compounds (e.g., toxic substances or compounds that limit further application) is within acceptable limits. These two topics are discussed in somewhat more detail in the following.
6.1. Slow release of compounds from the zeolite during application
Zeolites have been shown to be valuable in agriculture as soil conditioners due to their ability to improve soil chemical and physical properties such as the saturated hydraulic conductivity, the infiltration rate, its cation exchange capacity and great water-holding capacity [161]. They may also help the farmer with soil or water pollution, contamination by heavy metals, loss of nutrients and loss of water-use efficiency of drylands [161]. Moreover, they have a large carrying capacity of slow-release macro- and micronutrients [20,161,162]. Slow release of ammonium from zeolites have been studied for decades [162]. Slow release from surfactant-modified zeolites loaded with nitrate have also been investigated [163]. In the case of phosphate, the ability to produce slow-dissolving calcium phosphate compounds such as brushite (CaHPO4·2H2O (s)) during sorption in the pH range 6-9 is important, avoiding the formation of relatively insoluble Ca-phosphate minerals such as hydroxyapatite [145].
6.2. Controlled release of compounds of interest
As discussed earlier, the main mechanism governing the adsorption of heavy metals and ammonium (Section 4.2) and phosphate and nitrate (sections 5.1 and 5.2) to any zeolite, natural or synthetic or modified by metal-doping or by surfactants appeared to be electrostatic interactions. Since electrostatic interactions by nature are reversible, it may be suggested that zeolites, under ideal conditions, could undergo a virtually unlimited number of adsorption–desorption cycles [8]. However, the real situation is never ideal, and especially not when the raw materials that the zeolites are synthesized from are far from homogeneous.
6.2.1. Methods used to release the compounds from zeolites
Table 8 summarises results from studies that have included the release of the compounds that have already been adsorbed to the zeolite, most often focusing on the possibility to regenerate the zeolite for additional sorption cycles to reduce the operational process costs. In general, to optimise the desorption of the compounds, the conditions that led to their optimum adsorption need to be reversed. There are typically two factors that are easy to manipulate that have been used extensively to desorb compounds from sorbents: the pH and the concentration of competing ions.
As mentioned in Section 4.3.7, at pHs below their PZC, zeolites become neutral or positively charged, hence at these pHs the electrostatic forces diminish or gets reversed. Since heavy metals tend to precipitate as metal oxides and hydroxides at high pHs (see Figure 4), they are typically desorbed using strong acids (Table 8).
Ammonium can also be desorbed by pH adjustment, either by increasing the pH to drive the equilibrium between ammonium and ammonia towards ammonia and releasing the compound as partially dissolved and gaseous NH3 [164] or by decreasing the pH to reverse the electrostatic forces. However, the desorption process is typically further accelerated by adding competing ions (i.e., salts) as this will also contribute to the regeneration of the zeolite (ref).
Adsorption of phosphate is usually done using metal-doped zeolites at slightly acidic conditions (Table 6). [142] recovered the phosphate by hydrothermal treatment at 250°C in 3 M NaOH as this also regenerated the La-doped zeolite.
Nitrate is typically captured using surfactant-modified zeolites (Table 7), and since the inner monolayer of surfactant is also kept in place by electrostatic forces [137], it is important to selectively target the nitrate during desorption. [149] did this by adding a weak basic solution (0.01-0.05 M NaOH), thereby reducing the positive charge of the polydopamine-coated clinoptilolite and increasing the concentration of OH- as competing ion. However, the fraction of recovered nitrate was rather low (59-71%), and the loss of polydopamine during desorption was not reported, but the 80-87% of the adsorption capacity of the polydopamine-coated clinoptilolite was recovered after three adsorption-desorption cycles. [153] recovered ca. 100% of nitrate from a HDTMA-modified clinoptilolite using 1 M NaBr. The loss of HDTMA from the surface of the clinoptilolite during desorption was apparently not substantial but the slow leaching of HDTMA both during application (see Section 5.2.5) and desorption is a challenge both for repeated use of the adsorbent, for the environmental application and potentially for the application of the recovered compounds.
Table 8.
Reported desorption of selected compounds from different types of zeolites under given conditions.
Table 8.
Reported desorption of selected compounds from different types of zeolites under given conditions.
| Compound | Zeolite | Release conditions | Important factors | Released compound | Desorption efficiency | Ref. |
|---|---|---|---|---|---|---|
| Cu2+ | Synthetic from FA | 0.1-0.8 M H2SO4 | High conc H2SO4 | CuSO4 | 96-102% (four cycles) | [165] |
| Ni2+ | Synthetic from FA | 0.1-0.8 M H2SO4 | High conc H2SO4 | NiSO4 | 84-98% (four cycles) | [165] |
| Cd2+ | Natural zeolites | 0.1 M HCl (54-80 bed volumes) | - | CdCl2 | 90% first cycle | [166] |
| Zn2+ | Natural zeolites | 0.1 M HCl (6-30 bed volumes) | - | ZnCl2 | 90% first cycle | [166] |
| Cr6+ | HDTMA-modified clinoptilolite-rich tuff | 0.28 M Na2CO3 and 0.5 M NaOH (L/S: 3 mL/g); regeneration with 3x 0.1 M HCl (L/S: 3 mL/g) | - | - | 90% first cycle (100% regeneration) | [158] |
| NH4+ | Alkali-treated clinoptilolite | 0.5 M HCl | - | NH4Cl | Adsorption unaffected after 12 cycles | [167] |
| NH4+ | Zeolite from FA | 1 M NaCl (3x25 mL/2 g zeolite) at 25°C for 1.25 h | - | NH4Cl | Ca. 10% loss in adsorbent capacity after one cycle | [168] |
| NH4+ | Clinoptilolite | 20 g NaCl/L for 15 h | High NaCl conc | NH4Cl | 100% (five cycles). Adsorption capacity increased from 9.2 mg/g to 10.9 mg/g (over first three cycles) | [169] |
| NH4+ | Clinoptilolite | 30 g NaCl/L (123-134 BV) | Low flow rate to get high conc | NH4Cl | 88-95% | [170] |
| NH4+ | Synthetic NaA | 30 g NaCl/L (43-46 BV) | 92-95% | |||
| NH4+ | Clinoptilolite | 10% NaCl and 0.6% NaOH | Increased desorption: 10-15% NaCl and 0-0.6% NaOH | NH3 | 100% | [164] |
| PO43- | La-doped zeolite from FA | 3 M NaOH (L/S ratio 80:1) at 250°C for 5 h | High conc NaOH (<4 M NaOH), high L/S ratio, high temp | Na3PO4 | 95% (five cycles) | [142] |
| NO3- | Polydopamin-coated clinoptilolite | 0.01 M and 0.05 M NaOH | - | NaNO3 | 59-71% (three cycles) | [149] |
| NO3- | HDTMA-modified clinoptilolite | 1 M NaBr (L/S: 5 mL/g) for 6 h | - | NaNO3 | Ca. 100% first cycle | [153] |
6.2.2. Downstream concentration and refinement
Ideally, the solution with the recovered compounds should be as concentrated and pure as possible, potentially just containing other compounds that would be beneficial for any downstream application. However, this is seldom the case. The concentrations are often low and variable, and the level of contaminants may be significantly higher than the concentrations of the compounds of interest. Hence, downstream processing to concentrate and refine the valuable compounds will normally be necessary. It is not within the scope of this review to summarise all these possibilities, but some general comments are discussed below.
Concentrating ammonia by stripping and condensation
If ammonium is desorbed as ammonia by adding NaOH and NaCl, the partially dissolved and gaseous NH3 can be stripped off directly using air or hot steam in a stripping column and recovered as ammonium sulphate or ammonium nitrate by subsequent acid-washing with H2SO4 or HNO3, respectively, in a condenser column [19,171]. The stripping process is highly dependent on temperature and pH since both influence the equilibrium between ammonium and ammonia (both with positive correlations). Hence, the process is more efficient at elevated temperatures due to higher concentrations of ammonia and reduced solubility of gaseous ammonia at high temperature as well as reduced water viscosity at higher temperatures [172]. At ambient temperatures, the typical operating pH is >11, while it may be <10 if the operating temperature is increased to 60°C [173]. To avoid clogging due to scaling in the stripping column, it is advisable to avoid using calcium and magnesium salts for pH adjustments. However, problem with clogging due to scaling is avoided using jet loop reactors [174].
Concentrating by precipitation
Precipitation as struvite (a slow-release fertilizer) is a common method to separate ammonium and phosphate from other dissolved compounds in a solution by adding a magnesium salt [171]. This may also be done to selectively separate different heavy metals in a stepwise precipitation process taking into account their pH-dependent solubilities in the presence of anions such as carbonate, sulphate and phosphate [124].
6.2.3. Regeneration of the zeolite’s adsorption capacity
To be able to reuse the zeolite as an adsorbent multiple times it needs to be regenerated. The methods mentioned in Section 6.2.1 for the desorption of the compounds of interest may fully or partially regenerate the zeolite, however particularly organic compounds may be difficult to detach from the zeolite. Then more harsh conditions must be applied in terms of forced oxidation and/or high temperature: e.g., high temperature calcination, washing with a mixed solution of NaClO and NaCl and Fenton oxidation [175,176,177]. In general, short contact times (<30 min) are needed to destroy organic contaminants [178], but thermal regeneration requires several hours regeneration [175]. One of the advantages of zeolites is that they are stable in oxidative conditions, but when exposing them to high temperatures it is important to take into account their temperature stability to not destroy their structure [179].
7. Discussion and need for further studies
7.1. MSW-FA as a source for synthetic zeolites
As outlined in Section 3.4, MSW-FA is far from being the best source for synthesizing zeolites meant for capturing cations. Its content of SiO2 and Al2O3, the primary constituent in zeolites, is both relatively low and variable (12-41 wt% and 5-24 wt%, respectively), and it usually has a relatively high level of contaminants that will hamper its appropriateness for any use. Hence, significant amendments are needed for it to be utilized as a source for zeolites, which may bring significant production costs.
On the other hand, the increasing awareness of the need to reduce waste, and particularly toxic waste, in combination with the large fractions of salts and certain heavy metals (e.g., zinc) in MSW-FA that could potentially have some commercial value, may assist additional options of utilizing these resources that are currently wasted. The processes needed to extract salts and heavy metals from MSW-FA are beneficial to the concomitant extraction and purification of zeolite precursor materials (e.g. Na2SiO3, Al(OH)3, Na2O· SiO2 and NaAlO2). A remaining important uncertainty is the level of impurities of residual heavy metals and organic micropollutants (e.g., dioxins and furans) in these precursor materials. The level of contamination will strongly restrict their range of applicability. Though additional downstream purification processes may drastically reduce the contamination level, this would also increase the size and probably also the complexity of the production plant and increase the consumption of chemicals and energy and, inevitably, add extra investment and production costs. However, the size of the hazardous waste fraction may be reduced, though its specific toxicity may increase.
An obvious drawback considering using MSW-FA as a source for Al- and Si-precursors for producing synthetic zeolites in the current situation is the almost complete lack of examples that it will be possible, not the least, feasible. Hence, more studies focusing on optimising the extraction and necessary purification steps to produce precursors with appropriate qualities are needed. Also, the impact of the composition of the initial MSW should be considered.
7.2. Capturing efficiency
Since cation exchange will, in most cases, be the dominating mechanism for capturing the targeted compound (e.g., ammonium, nitrate, phosphate or selected heavy metals), competition for available adsorption sites on the zeolite is crucial. Hence, tailoring the zeolite (i.e., framework type and necessary surface modifications) to the targeted compound will be important as well as optimizing the operating conditions (see Section 4.3). However, based on the rather large variability in the reported adsorption capacities and selectivity of the different types of zeolites, specific tailoring is challenging and probably need to be adapted to the conditions in each case. Furthermore, this is made further difficult by the fact that most studies are done under relative ideal conditions and not representative when applying zeolites as adsorbents in the “field”. From a recovery point of view, it is appropriate to harvest the targeted compounds from rich sources such as liquid manure fractions, rejection water after anaerobic digestion of food or sewage sludge, urine, concentrated black water after fermentation, industrial waste streams or leachates from landfills or mines/mining areas. At high compound concentrations, and low concentrations of competing ions, the need for tailored zeolites may be reduced [170]. In that regard it is also interesting to note the reported increased adsorption capacity of zeolites at elevated bulk concentrations, particularly for ammonium (see Section 4.3.2), which would be beneficial when harvesting from high-strength sources.
There is also a balance between utilizing the whole adsorption capacity of the zeolite and the reduced adsorption kinetics when the capacity is close to being exhausted. This implies a need for increased exposure times or increased risk of breakthrough (in flow-through applications).
7.3. Acceptance and need of recovered end-products
The above sections have described and discussed the feasibility of extracting Al- and Si-containing zeolite precursors from MSW-FA and using these precursors to synthesize tailored zeolites that could be used to harvest and recover nutrients and heavy metals as secondary raw materials. Hence, the intention is to turn a hazardous waste (MSW-FA) into valuable products (zeolites) and secondary raw materials (zeolite precursors and recovered compounds) in a stepwise approach. However, within the European Union and the European Economic Area waste management is strictly regulated through the Waste Framework Directive 2000/98 (WFD). As a general measure to minimize waste generation Article 6 (1) and (2) in WFD has defined common end-of-waste criteria that specify when a certain waste ceases to be waste and becomes a product, or a secondary raw material. End-of-waste may help to alleviate any user prejudice, to increase the confidence of the users on quality standards and to encourage the use of secondary materials [180].
The two first criteria are that the substance (or object) is commonly used for specific purposes, and that there is an existing market or demand for the substance (or object). Hence, end-of-waste criteria will not be defined for materials for which there are no demand or for which a market has not yet been developed. The global market for zeolites for all types of applications was ca. 6 million tons at a value of 13.2 billion US$ in 2022 and is expected to increase by 4% annually to ca. 8.5 million tons at a value of 21.7 billion US$ in 2030 [181]. The market for synthetic zeolites is still somewhat smaller than for natural zeolites, which are also expected to be dominating due to a growing demand in various applications such as constructing and building materials, soil remediation and wastewater treatment. The largest bulk market for synthetic zeolites is as water softeners in detergents. However, the fastest-growing market is for synthetic zeolites as adsorbents/desiccants [182]. About 60 million tons MSW-FA are generated globally each year. However, the typical contents of SiO2 and Al2O3 are only in the range of 20% and 10%, respectively. This would anyway more or less cover the current global need for zeolite precursors. Nevertheless, the local availabilities of MSW-FA and demands for zeolite precursors are, of course, crucial to minimize necessary transport from both cost and environmental perspectives. These need to be considered in each case.
The global market for recycled nutrients (i.e., N, P, K) from waste streams such as wastewater, agricultural residues and food waste is also relatively substantial at a value of 5.0 billion US$ in 2023 and is expected to reach 7.4 billion US$ in 2030 [183]. However, as for zeolite precursors only the local or reginal demands for recycled nutrients is relevant in this context, and these depend on local or regional traditional practices and are prone to seasonal changes. Moreover, this market is possibly more sensitive to transport costs due to smaller buyers. Hence, also here the market potentials (as a part of a more thorough market analysis) need to be considered in each local case.
The third end-of-waste criteria is that the use is lawful, i.e., that the substance (or object) fulfils the technical requirements for the specific purposes and meets the existing legislation and standards applicable to products. In zeolite synthesis both liquid and solid (crystalline or amorphous) Al- and Si precursors are utilized. The variation in precursors appears to be relatively broad and include those precursors referred to in Section 3.5.1 [66]. The MSW-FA management plant(s) that will extract and purify the zeolite precursors will most probably receive MSW-FA from a large range of incineration facility implying a huge variation in FA composition between a waste fraction that is inherently heterogeneous. Hence, it may be challenging to determine optimum operational conditions for the extraction and purification processes, which may impact the composition and quality of the final precursors. It will be important to assess how easy it will be to control and predict the final quality and identify any need for initial source separation to exclude problematic MSW-FA fractions [180] and make sure that local legal requirements are fulfilled.
The fourth end-of-waste criteria is the most critical in this regard and states that the use should not lead to overall adverse environmental or human health impacts. As discussed in detail in Section 3.5, proper strategies that prevent the heavy metals and organic micropollutants embedded in the MSW-FA from being transferred to the zeolite precursors and to the recovered secondary raw materials need to be in place. Also in this regard, it must be determined if any hazard(s) associated with a particular MSW-FA source can be adequately controlled in some way during processing or whether they need to be excluded at source to provide the requisite product quality [180]. The level of complexity needed in these assessments is difficult to infer at the current state of development and lack of results from properly designed studies. Anyway, the environmental risk assessment should include both chemical analyses, toxicity testing and leaching rates of all relevant toxic compounds at different stages along the process train until the final products or secondary raw material.
It should be noted that if a material does not meet the end-of-waste requirements, it does not imply that the material cannot be recycled and needs to be disposed. Materials not fulfilling the end-of-waste requirements can be recycled and reused under the waste regime [180].
8. Concluding remarks
MSW-FA is far from being the first option to use as a source for zeolite-precursor, but if its other contents such as salts and selected heavy metals are extracted, it should be possible to isolate Al and Si using a customized extraction process adapted to the quality of the initial MSW-FA, the processes used to extract the other compounds and the desired types and necessary qualities (including level of contaminants) of the zeolite-precursors. If it will be feasible economically and legal for the intended application need to be carefully considered, possibly in each case. The implications of the associated reduced residual hazardous waste volume, but with potentially increased toxicity, need to be taken into account.
It is well established that zeolites may be used to adsorb compounds such as ammonium and heavy metals, and with necessary modifications of their surface, they may also be used to adsorb nutrients such as nitrate and phosphate. However, the large variability in reported adsorption capacities and specificities of the different types of zeolites, as well as the inherent heterogenic nature and variable composition and concentrations of most nutrient- or heavy metals-rich waste streams (e.g., liquid manure fractions, reject water, urine, industrial waste streams or landfill leachate), makes it challenging to tailor the zeolite properties and specific adaptation to the conditions in each case may be needed. Therefore, recovery would be benefitted by high concentrations of the target compounds and low concentrations of competing ions. Though, potential sorption of polluting compounds (e.g., unwanted heavy metals and organic micropollutants), and loss of surfactants from SMZ during adsorption and/or desorption of nitrate and/or phosphate, need anyway to be considered.
The main purpose here is to turn a hazardous waste (MSW-FA) into valuable products (zeolites) and secondary raw materials (zeolite precursors and recovered compounds). The third and fourth end-of-waste criteria of EU’s WFD, relating to the lawful use of extracts from MSW-FA and potential environmental or human health impacts from their applications, respectively, are probably the most critical issues to consider in this process. Although fulfilling these criteria are currently far from being realized, and not absolutely required to recycle extracted fractions from MSW-FA, it is recommended to work towards such a goal since it would contribute to alleviate any user prejudice, increase the confidence of the users on quality standards, encourage the use of secondary materials, and support the EU common accepted end-of-waste strategy.
Author Contributions
Conceptualization, C.V.; investigation, C.V. and M.U.; writing—original draft preparation, C.V. and M.U.; writing—review and editing, C.V. and M.U.; visualization, C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Norwegian Institute for Water Research internal basic grant provided by the NORWEGIAN RESEARCH COUNCIL.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hoornweg, D.; Bhada-Tata, P. What a waste: a global review of solid waste management. 2012, The World Bank: Washington.
- Kaza, S.; Yao, L.; Bhada-Tata, P.; Van Woerden, F. What a Waste 2.0: A Global Snapshot of Solid Waste Management to 2050. Urban Development, 2018, Washington, DC.
- Chen, D.; Zhang, Y.; Xu, Y.; Nie, Q.; Yang, Z.; Shenga, W.; Qian, G. Municipal solid waste incineration residues recycled for typical construction materials—a review. RSC Adv 2022, 12, 6279. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, D.; Molin, C.; Hupa, M. Thermal treatment of solid residues from WtE units: A review. Waste Management 2015, 37, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Quina, M.J.; Bontempi, E.; Bogush, A.; Schlumberger, S.; Weibel, G.; Braga, R.; Funari, V.; Hyks, J.; Rasumssen, E.; Lederer, J. Technologies for the management of MSW incineration ashes from gas cleaning: New perspectives on recovery of secondary raw materials and circular economy. Sci Total Env 2018, 635, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Weibel, G.; Zappatini, A.; Wolffers, M.; Ringmann, S. Optimization of metal recovery from MSWI fly ash by acid leaching: findings from laboratory- and industrial-scale experiments. Processes 2021, 9, 352. [Google Scholar] [CrossRef]
- Kanhar, A.H.; Chen, S.; Wang, F. Incineration fly ash and its treatment to possible utilization: A Review. Energies 2020, 13, 6681. [Google Scholar] [CrossRef]
- Millini, R.; Bellussi, G. Zeolite science and perspectives. In Zeolites in Catalysis: Properties and Applications, Cejka, J., Morris, R.E., Nachtigall, P. The Royal Society of Chemistry 2017; RSC Catalysis Series No. 28.
- Ray, R.L.; Sheppard, R.A. Occurrence of zeolites in sedimentary rocks: an overview, in Natural Zeolites: Occurrence, Properties, Applications. Rev. Miner. Geochem 2001, 45, 17–34. [Google Scholar]
- MgBemere, H.E.; Ekpe, I.C.; Lawal, G.I. Zeolite synthesis, characterizations, and application areas - a review. Int. Res. J. Environ. Sci 2017, 6, 45–59. [Google Scholar]
- Khaleque, A.; Alam, M.M.; Hoque, M.; Mondal, S.; Haider, J.B.; Xuc, B.; Johir, M.A.H.; Karmakar, A.K.; Zhoud, J.L.; Ahmedb, M.B.; Moni, M.A. Zeolite synthesis from low-cost materials and environmental applications: a review. Environ. Adv 2020, 2, 100019. [Google Scholar] [CrossRef]
- Cho, B.H.; Nam, B.H.; An, J.; Youn, H. Municipal Solid Waste Incineration (MSWI) Ashes as Construction Materials-A Review. Materials 2020, 13(14), 3143. [Google Scholar] [CrossRef]
- Perego, C.; Bagatin, R.; Tagliabue, M.; Vignola, R. Zeolites and related mesoporous materials for multi-talented environmental solutions. Microp. Mesop. Mater 2013, 166, 37–49. [Google Scholar] [CrossRef]
- Karlfeldt Fedje, K.; Rauch, S.; Cho, P.; Steenari, B.M. ; Element associations in ash from waste combustion in fluidized bed. Waste Manag 2010, 30(7), 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Witek-Krowiak, A.; Gorazda, K.; Szopa, D.; Trzaska, K.; Moustakas, K.; Chojnacka, K. Phosphorus recovery from wastewater and bio-based waste: an overview. Bioeng 2022, 13(5), 13474–13506. [Google Scholar] [CrossRef] [PubMed]
- Cordell, D. Peak phosphorous and the role of P recovery in achieving food security. In: T.A. Larsen, K.M. Udert and J. Lienert (Eds.) Source Separation and Decentralization for Wastewater Management IWA Publishing, London, 2013, 29-44.
- Jenssen, T.K.; Kongshaug, G. Energy Consumption and Greenhouse Gas Emissions in Fertiliser Production. Proceeding No. 509, The International Fertilizer Society, 2003.
- European Commission Proposal for a DIRECTIVE OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL concerning urban wastewater treatment (recast) 541, 2022.
- van Eekert, M.; Weijma, J.; Verdoes, N.; de Buisonjé, F.; Reitsma, B.; van den Bulk, J.; van Gastel, J. Explorative research on innovative nitrogen recovery. STOWA Report 51 2012, Stichting Toegepast Onderzoek Waterbeheer, Amersfoort, The Netherlands.
- Ganrot Z.; Use of zeolites for improved nutrient recovery from decentralized domestic wastewater. Chapter 17 in Vassilis J.; Antonis, A.Z.; (Eds) Handbook of Natural Zeolites 2012, 410-435.
- Bandala, E.R.; Liu, A.; Wijesiri, B.; Zeidman, A.B.; Goonetilleke, A. Emerging materials and technologies for landfill leachate treatment: A critical review. Environ. Poll. 2021, 291, 118133. [Google Scholar] [CrossRef]
- Ferrel-Luna, R.; García-Arreola, M.E.; González-Rodríguez, L.M.; oredo-Cancino, M.; Escárcega-González, C.E.; de Haro-Del Río, D.A. Reducing toxic element leaching in mine tailings with natural zeolite clinoptilolite. Environ. Sci. Poll. Res 2023. [Google Scholar] [CrossRef] [PubMed]
- Helfferich, F. Ion Exchange. Dover Publishing, 1995, New York.
- Inglezakis, V.J. The concept of “capacity” in zeolite ion-exchange systems. J. Colloid. Inter. Sci 2005, 281, 68–79. [Google Scholar] [CrossRef]
- Coombs D.S.; Alberti A.; Armbruster T.; Artioli G.; Colella C.; Galli E.; Grice J.D.; Liebau F.; Mandarino J.A.; Minato H., Nickel E.H.; Passaglia E.; Peacor D.R.; Quartieri S.; Rinaldi R.; Ross M.; Sheppard R.A., Tillmans E. and Vezzalini G., Recommended nomenclature for zeolite minerals: Report of the subcommittee on zeolites of the international mineralogical association, commission on new minerals and mineral names. Can. Mineral., 1997, 35, 1571–1606.
- Barelocher, C.; McCuster, L.B.; Olson, H.D. Atlas of zeolite framework types, Elsevier, Amsterdam, 6th Ed. 2007.
- Yu, J. Synthesis of zeolites,” in (Eds.) J. Čejka, B. Hv, A. Corma, and F. Schüth, Introduction to zeolite science and practice. Vol. 2007, 168, Elsevier, 3rd Ed.
- Brännvall, E.; Kumpiene, J. Fly ash in landfill top covers – a review. Environ. Sci. Proc. Imp 2016, 18, 11–21. [Google Scholar] [CrossRef]
- Cundy, C.S.; Cox, P.A. The hydrothermal synthesis of zeolites: Precursors, intermediates and reaction mechanism. Review. Microp. Mesop. Mater 2005, 82, 1–78. [Google Scholar] [CrossRef]
- Na, K.; Choi, M.; Ryoo, R. Recent advances in the synthesis of hierarchically nanoporous zeolites. Review. Microp. Mesop. Mater 2013, 166, 3–19. [Google Scholar] [CrossRef]
- Gao, M.; Ma Q.; Lin Q.; Chang J.; Bao W.; Ma M.; Combined modification of fly ash with Ca(OH)2/Na2FeO4 and its adsorption of Methyl orange. App. Surf. Sci. 2015, 359, 323–330. [CrossRef]
- Perez-Ramırez, J.; Verboekend, D.; Bonilla, A.; Abello, S. Zeolite catalysts with tunable hierarchy factor by pore-growth moderators. Adv. Funct. Mater 2009, 19, 3972. [Google Scholar] [CrossRef]
- Ivanova I.I.; Knyazeva E.E.; Micro–mesoporous materials obtained by zeolite recrystallization: synthesis, characterization and catalytic applications. Chem. Soc. Rev 2013, 42, 3671–3688. [CrossRef] [PubMed]
- Hanache, L.E.; Sundermann, L.; Lebeau, B.; Toufaily, J.; Hamieh, T.; Daou, T.J. Surfactant-modified MFI-type nanozeolites: Super-adsorbents for nitrate removal from contaminated water. Microp. Mesop. Mater 2019, 283, 1–13. [Google Scholar] [CrossRef]
- Schick, J.; Daou, T.J.; Caullet, P.; Paillaud, J.L.; Patarin, J.; Mangold-Callarec, C. Surfactant-modified MFI nanosheets: a high capacity anion-exchanger. Chem. Comm 2011, 47, 902–904. [Google Scholar] [CrossRef]
- Hanache, L.E.; Lebeau, B.; Nouali, H.; Toufaily, J.; Hamieh, T.; Daou, T.J. Performance of surfactant-modified *BEA-type zeolite nanosponges for the removal of nitrate in contaminated water: Effect of the external surface. J. Hazard. Mater. 2019, 364, 206–217. [Google Scholar] [CrossRef]
- Mao, Y.; Wu, H.; Wang W.; Jia M.; Che X.; Pretreatment of municipal solid waste incineration fly ash and preparation of solid waste source sulphoaluminate cementitious material. J. Hazard. Mater 2020, 385, 121580. [CrossRef]
- Li, Y., R. Cui, T. Yang, Z. Zhai, and R. Li, Distribution characteristics of heavy metals in different size fly ash from a sewage sludge circulating fluidized bed incinerator. Energy & Fuels 2017, 31(2), 2044–2051.
- Fabricius A-L.; Renner M.; Voss M.; Funk M.; Perfoll A.; Gehring F.; Graf R.; Fromm S.; Duester L. Municipal waste incineration fly ashes: from a multi-element approach to market potential evaluation. Environ. Sci. Eur 2020, 32(1), 88.
- Bodénan, F.; Deniard, P. Characterization of flue gas cleaning residues from European solid waste incinerators: assessment of various Ca-based sorbent processes. Chemosphere 2003, 51(5), 335–347. [Google Scholar] [CrossRef]
- Chiang, K.-Y.; Jih J.-C.; Chien M-D.; The acid extraction of metals from municipal solid waste incinerator products. Hydrometallurgy 2008, 93(1), 16–22. [CrossRef]
- Zhu, J.; Hao, Q.; Chen, J.; Hu, M.; Tu, T.; Jiang, C. Distribution characteristics and comparison of chemical stabilization ways of heavy metals from MSW incineration fly ashes. Waste Manage. 2020, 113, 488–496. [Google Scholar] [CrossRef]
- Saakshy, K.; Singh, A.B.; Gupta, Sharma, A.K. Fly ash as low cost adsorbent for treatment of effluent of handmade paper industry-Kinetic and modelling studies for direct black dye. J. Clean. Prod. 2016, 112, 1227–1240. [CrossRef]
- Wesche, K. Fly ash in concrete properties and performance. RILEM Report. Ed. Hall C.A. 1990, International Union of Testing and Research Laboratories, France: London.
- Weibel, G.; Eggenberger, U.; Schlumberger, S.; Mäder, U.K. Chemical associations and mobilization of heavy metals in fly ash from municipal solid waste incineration. Waste Manag. 2017, 62, 147–159. [Google Scholar] [CrossRef]
- Rani, D.A.; Boccaccini, A.R.; Deegan, D.; Cheeseman, C.R. Air pollution control residues from waste incineration: Current UK situation and assessment of alternative technologies. Waste Manag. 2008, 28, 2279–2292. [Google Scholar] [CrossRef] [PubMed]
- IAWG (International Ash Working Group), Chandler, A.J.; Eighmy, T.T.; Hartlén, J.; Hjelmar, O.; Kosson, D.S.; Sawell, S.E.; van der Sloot H.; Vehlow, J.; Municipal Solid Waste Incinerator Residues. Studies Environ. Sci 1997, 67, Elsevier.
- Zhao, Y.; Li, H. Understanding municipal solid waste production and diversion factors utilizing deep-learning methods. Utilities Policy 2023, 83, 101612. [Google Scholar] [CrossRef]
- Joseph, A.M.; Snellings, R.; van den Heede, P.; Matthys, S.; de Belie, N. The use of municipal solid waste incineration ash in various building materials: A Belgian Point of View. Materials 2018, 11, 141. [Google Scholar] [CrossRef]
- Deng L.; Xu Q.; Wu H.; Synthesis of zeolite-like material by hydrothermal and fusion methods using municipal solid waste fly ash. Proc. Environ. Sci 2016, 31, 662–667. [CrossRef]
- Fan, X.F.; Yuan, R.; Gan, M.; Ji, Z.; Sun, Z. Subcritical hydrothermal treatment of municipal solid waste incineration fly ash: A review. Sci. Total Environ. 2023, 865, 160745. [Google Scholar] [CrossRef]
- Eighmy, T.T.; Eusden, J.D.; Krzanowski, J.E.; Domiago, D.S.; Stampfli, D.; Martin, P.M.; Erickson, P.M. Comprehensive approach toward understanding element speciation and leaching behavior in municipal solid waste incineration electrostatic precipitator ash. Environ. Sci. Technol. 1995, 29, 629–646. [Google Scholar] [CrossRef]
- Hjelmar, O. Disposal strategies for municipal solid waste incineration residues. J. Hazard. Mater. 1996, 47, 345–368. [Google Scholar] [CrossRef]
- Forestier, L.L.; Libourel, G. Characterization of flue gas residues from municipal solid waste combustors. Environ. Sci. Technol. 1998, 32, 2250–2256. [Google Scholar] [CrossRef]
- Quina, M.J.; Bordado, J.C.; Quinta-Ferreira, R.M. Treatment and use of air pollution control residues from MSW incineration: An overview. Waste Manag. 2008, 28, 2097–2121. [Google Scholar] [CrossRef]
- Song, G.J.; Kim, K.; Seo, Y.; Kim, S. ; Characteristics of ashes from different locations at the MSW incinerator equipped with various air pollution control devices. Waste Manage. 2004, 24, 99–106. [Google Scholar] [CrossRef]
- Vavva, C.; Voutsas, E.; Magoulas, K. Process development for chemical stabilization of fly ash from municipal solid waste incineration. Chem. Eng. Res. Des 2017, 125, 57–71. [Google Scholar] [CrossRef]
- Li, X.; Chen, Q.; Zhou, Y.; Tyrer, M.; Yu, Y. Stabilization of heavy metals in MSWI fly ash using silica fume. Waste Manage. 2014, 34, 2494–2504. [Google Scholar] [CrossRef]
- Karlfeldt, K.; Steenari, B.-M. Assessment of metal mobility in MSW incineration ashes using water as the reagent. Fuel 2007, 86, 1983–1993. [Google Scholar] [CrossRef]
- Alba, N.; Gasso, S.; Lacorte, T.; Baldasano, J.M. Characterization of municipal solid waste incineration residues from facilities with different air pollution control systems. J. Air Waste Manag. Assoc. 1997, 47, 1170–1179. [Google Scholar] [CrossRef]
- Romero, M.; Rincon, J.M., Rawlings, R.D., Boccaccini, A.R. Use of vitrified urban incinerator waste as raw material for production of sintered glass–ceramics. Mat. Res. Bull. 2001, 36, 383–395. [CrossRef]
- Cheng, T.W.; Chen, Y.S.; Characterisation of glass–ceramics made from incinerator fly ash. Ceram. Intern 2004, 30, 343–349. [CrossRef]
- Faust, S.D.; Aly, O.M. Chemistry of water treatment. CRC press, 1988, p. 224.
- Okada, K.; Arimitsu, N.; Kameshima, Y.; Nakajima, A.; MacKenzie, K.J.D.; Preparation of porous silica from chlorite by selective acid leaching. Appl. Clay Sci 2005, 30, 116–124. [CrossRef]
- Shoppert, A.A.; Loginova, I.V.; Chaikin, L.I.; Rogozhnikov, D.A.; Alkali fusion leaching method for comprehensive processing of fly ash. KnE Mater. Sci 2017, 2, 89–96. [CrossRef]
- Sayehi, M.; Tounsi, H.; Garbarino, G.; Riani, P.; Busca, G. Reutilization of silicon- and aluminum- containing wastes in the perspective of the preparation of SiO2-Al2O3 based porous materials for adsorbents and catalysts. Waste Manag. 2020, 103, 146–158. [Google Scholar] [CrossRef]
- Boukerche, I.; Djerada, S.; Benmansoura, L.; Tifoutia, L.; Saleh, K. Degradability of aluminum in acidic and alkaline solutions. Corros. Sci 2014, 78, 343–352. [Google Scholar] [CrossRef]
- Lenntech. Iron Removal by physical-chemical ways. Accessed from: https://www.lenntech.com/processes/iron-manganese/iron/iron-removal-physicalchemical-way.htm.
- Zaitsev, A.I.; Shelkova, N.E.; Lyakishev, N.P.; Mogutnov, B.M. Thermodynamic properties and phase equilibria in the Na2O-SiO2 system. Phys. Chem. Chem. Phys 1999, 1, 1899–1907. [Google Scholar] [CrossRef]
- Adans, Y.F.; Martins, A.R.; Coelho, R.E.; das Virgens, C.F.; Ballarini, A.D.; Carvalho, L.S. A simple way to produce c-alumina from aluminum cans by precipitation reactions. Mat. Res. 2016, 19, 977–982. [Google Scholar] [CrossRef]
- Wajima T..; Ikegami Y. Synthesis of zeolitic materials from waste porcelain at low temperature via a two-step alkali conversion. Ceram. Int. 2007, 33(7), 1269–1274. [CrossRef]
- Wang, K.-S.; Chiang, K.-Y.; Lin, K.-L.; Sun, C.-J. Effects of a water-extraction process on heavy metal behavior in municipal solid waste incinerator fly ash. Hydrometall. 2001, 62, 73–81. [Google Scholar] [CrossRef]
- Chuai, X.; Yang, Q.; Zhang, T.; Zhao, Y.; Wang, J.; Zhao, G.; Cui, X.; Zhang, Y.; Zhang, T.; Xiong, Z.; Zhang, J. Speciation and leaching characteristics of heavy metals from municipal solid waste incineration fly ash. Fuel 2022, 328, 125338. [Google Scholar] [CrossRef]
- Todorovic, J.; Ecke, H.; Lagerkvist, A. Solidification with water as a treatment method for air pollution control residues. Waste Manage 2003, 23, 621–629. [Google Scholar] [CrossRef]
- Glasser, F.P. Fundamental aspects of cement solidification and stabilisation. Journal of Hazardous Materials 1997, 52(2–3), 151–170. [Google Scholar] [CrossRef]
- Weibel, G.; Eggenberger, U.; Dmitrii, A.K.; Hummel, W.; Schlumberger, S.; Klink, W.; Fisch, M.; Mäder, U.K. Extraction of heavy metals from MSWI fly ash using hydrochloric acid and sodium chloride solution. Waste Manage. 2018, 76, 457–471. [Google Scholar] [CrossRef] [PubMed]
- AWEL, Office for Waste Management, Environmental Protection Agency of Canton Zurich (AWEL Zurich), 2013. Stand der Technik für die Aufbereitung von Rauchgasreinigungsrückständen (RGRR) aus Kehrichtverbrennungsanlagen.
- Vassilev, S.V.; Vassileva, C.G. A new approach for the classification of coal fly ashes based on their origin, composition, properties, and behaviour. Fuel 2007, 86(10), 1490–1512. [Google Scholar] [CrossRef]
- Colella, C.; Wise, W.S. The IZA Handbook of Natural Zeolites: A tool of knowledge on the most important family of porous minerals. Microp. Mesop. Mater 2014, 189, 4–10. [Google Scholar] [CrossRef]
- de Magalhães, L.F.; da Silva G.R.; Peres A.C.E.; Zeolite application in wastewater treatmen. Adsorp. Sci Techn. 2022, 4544104.
- Moshoeshoe, M.; Nadiye-Tabbiruka, M.S.; Obuseng, V.A. Review of the chemistry, structure, properties and applications of zeolites. Amer. J. Mater. Sci 2017, 7(5), 196–221. [Google Scholar] [CrossRef]
- Mumpton, F.A. La roca magica: Uses of natural zeolites inagriculture and industry. Proc. Natl. Acad. Sci. USA 1999, 96, 3463–3470. [Google Scholar] [CrossRef]
- Baerlocher, C.; McCusker, L.B.; Olson, D.H. Atlas of zeolite framework types. 6th Ed. Elsevier, B.V. Amsterdam.
- Yue, B.; Liu, S.; Chai, Y.; Wu, G.; Guan, N.; Li, L. Zeolites for separation: Fundamental and application. J. Ener. Chem. 2022, 71, 288–303. [Google Scholar] [CrossRef]
- Pérez-Botella, E.; Palomino, M.; Valencia, S.; Rey, F. In: Nanoporous materials for gas storage, Springer-Verlag GmbH & Co. KG, Singapore, 2019, 173–208.
- Lin, H.; Wu, X.; Zhu, J. Kinetics, equilibrium, and thermodynamics of ammonium sorption from swine manure by natural chabazite. Sep. Sci. Technol. 2015, 51(2), 202–213. [Google Scholar] [CrossRef]
- Dong, L.D.; Zhu, Z.; Qiu, Y.; Zhao, J. Removal of lead from aqueous solution by hydroxyapatite/magnetite composite adsorbent. Chem. Eng. J. 2010, 165, 827–834. [Google Scholar] [CrossRef]
- Langmuir, I. The constitution and fundamental properties of solid sand liquids. Part-I. Solids. J. Am. Chem. Soc 1916, 38, 2221–2295. [Google Scholar] [CrossRef]
- Freundlich, H. Colloid and Capillary Chemistry, Metheum, London, 1926, 993.
- Millar, G.J.; Winnett, A.; Thompson, T.; Couperthwaite, S.J. Equilibrium studies of ammonium exchange with Australian natural zeolites. J. Water Proc. Eng. 2016, 9, 47–57. [Google Scholar] [CrossRef]
- Sprynskyy, M.; Buszewski, B.; Terzyk, A.P.; Namieśnik, J. Study of the selection mechanism of heavy metal (Pb2+, Cu2+, Ni2+, and Cd2+) adsorption on clinoptilolite. J. Colloid Interface Sci. 2006, 304(1), 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, M.; Karaoglu, M.H. Adsorption of ammonium from an aqueous solution by fly ash and sepiolite: Isotherm, kinetic and thermodynamic analysis. Microp. Mesop. Mater. 2011, 139, 173–178. [Google Scholar] [CrossRef]
- Ji, X.D.; Zhang, M.L.; Ke, Y.Y.; Song, Y.C. Simultaneous immobilization of ammonium and phosphate from aqueous solution using zeolites synthesized from fly ashes. Water Sci. Technol. 2013, 67, 6. [Google Scholar] [CrossRef]
- Makgabutlane, B.; Nthunya, L.N.; Musyoka, N.; Dladla, B.S.; Nxumalo, E.N.; Mhlanga, S.D. Microwave assisted synthesis of coal fly ash-based zeolites for removal of ammonium from urine. RSC Adv 2020, 10, 2416–2427. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, H.; Xu, D.; Han, L.; Niu, D.; Zhang, L.; Wu, W. Tianm B. Ammonium removal from aqueous solution by zeolites synthesized from low-calcium and high-calcium fly ashes. Desalination 2011, 277, 46–53. [Google Scholar] [CrossRef]
- Beler-Baykal, B.; Allar, A.D. Upgrading fertilizer production wastewater effluent quality for ammonium discharges through ion exchange with clinoptilolite. Environ. Technol. 2008, 29, 665–672. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, C.; Zhao, J.; Zhang, Z.; Wang, H.; Zhou, S. Synthesis of zeolite P1 from fly ash under solvent-free conditions for ammonium removal from water. J. Clean. Prod. 2018, 202, 11–22. [Google Scholar] [CrossRef]
- Fu, H.; Li, Y.; Yu, Z.; Shen, J.; Li, J.; Zhang, M.; Ding, T.; Xu, L.; Lee, S.S. Ammonium removal using a calcined natural zeolite modified with sodium nitrate. J. Hazard Mater 2020, 393, 122481. [Google Scholar] [CrossRef]
- Hui K.S.; Chao C.Y.; Kot S.; Removal of mixed heavy metal ions in wastewater by zeolite 4A and residual products from recycled coal fly ash. J. Hazard. Mater 2005, 127(1-3), 89–101. [CrossRef] [PubMed]
- Alvarez-Ayuso E.; Garcia-Sanchez A.; Querol X.; Purification of metal electroplating waste waters using zeolites. Water Res 2003, 37, 4855–4862. [CrossRef] [PubMed]
- Nightingale E.R.; Phenomenological theory of ion solvation. effective radii of hydrated ions 1959, 1381-1387.
- Marcus, Y. Thermodynamics of solvation of ions. Part 5.—Gibbs free energy of hydration at 298.15 K. Trans. 1991, 87(18), 2995–2999. [Google Scholar] [CrossRef]
- Joseph I., V.; Tosheva, L.; Doyle, A.M. Simultaneous removal of Cd(II), Co(II), Cu(II), Pb(II), and Zn(II) ions from aqueous solutions via adsorption on FAU-type zeolites prepared from coal fly ash. J. Environ. Chem. Eng. 2020, 8, 103895. [Google Scholar] [CrossRef]
- Ibrahim H.S.; Jamil, T.S.; Hegazy E.Z.; Application of zeolite prepared from Egyptian kaolin for the removal of heavy metals: II. Isotherm models. J. Hazard. Mater. 2010, 182(1-3), 842–847.
- Kesraoui-Ouki S.; Cheeseman C.R.; Perry R.; Natural zeolite utilization in pollution-control—a review of applications to metals effluents. J. Chem. Technol. Biotechnol 1994, 59, 121–126. [CrossRef]
- Oter, O.; Akcay, H. Use of natural clinoptilolite to improve, water quality: sorption and selectivity studies of lead(II), copper(II), zinc(II), and nickel(II). Water Environ. Res 2007, 79, 329–335. [Google Scholar] [CrossRef]
- Caputo, D.; Pepe, F. Experiments and data processing of ion exchange equilibria involving Italian natural zeolites: a review. Microp. Mesop. Mater 2007, 105(3), 222–231. [Google Scholar] [CrossRef]
- Bosso, S.T.; Enzweiler, J. Evaluation of heavy metal removal from aqueous solution onto scolecite. Water Res 2002, 36, 4795–4800. [Google Scholar] [CrossRef]
- Deng, Q.; Dhar, B.R.; Elbeshbishy, E.; Lee, H.S. Ammonium nitrogen removal from the permeates of anaerobic membrane bioreactors: economic regeneration of exhausted zeolite. Environ. Technol. 2014, 35, 2008–2017. [Google Scholar] [CrossRef]
- Lind, B.-B.; Ban, Z.; Byden, S. Nutrient recovery from human urine by struvite crystallization with ammonia adsorption on zeolite and wollastonite. Biores. Technol. 2000, 73, 169–174. [Google Scholar] [CrossRef]
- Zhao, Y.; Niu, Y.; Hu, X.; Xi, B.; Peng, X.; Liu, W.; Guan, W.; Wang, L. Removal of ammonium ions from aqueous solutions using zeolite synthesized from red mud. Desal. Water Treat 2016, 57(10), 4720–4731. [Google Scholar] [CrossRef]
- Zanin, E.; Scapinello, J.; de Oliveira, M.; Rambo, C.L.; Franscescon, F.; Freitas, L.; Maria, J.; de Mello, M.; Fiori, M.A.; Oliveira, J.V.; Dal Magro, J. Adsorption of heavy metals from wastewater graphic industry using clinoptilolite zeolite as adsorbent. Proc. Safe. Environ. Prot 2017, 105, 194–200. [Google Scholar] [CrossRef]
- Dal Bosco, S.M.; Jimenez, R.S.; Carvalho, W. Removal of toxic metals from wastewater by Brazilian natural scolecite. J. Colloid. Interface. Sci 2005, 281(2), 424–431. [Google Scholar] [CrossRef]
- Ćurković, L.; Cerjan-Stefanović, Š.; Filipan, T. Metal ion exchange by natural and modified zeolites. Water Res 1997, 31(6), 1379–1382. [Google Scholar] [CrossRef]
- Margeta, K.; Zabukovec, N.; Siljeg, M.; Farkas, A. Natural zeolites in water treatment–how effective is their use. Water Treat 2013, Chapter 5.
- Haji, S.; Al-Buqaishi, B.A.; Bucheeri, A.A.; Bu-Ali, Q.; Al-Aseeri, M.; Ahmed, S. The dynamics and equilibrium of ammonium removal from aqueous solution by Na-Y zeolite. Desal. Water Treat 2016, 57(40), 18992–19001. [Google Scholar] [CrossRef]
- Walton, K.S.; Abney, M.B.; LeVan, M.D. ; CO2 adsorption in Y and X zeolites modified by alkali metal cation exchange. Micropor Mesopor Mater 2006, 91, 78. [Google Scholar] [CrossRef]
- Rabo, J.A.; Schoonover, M.W. Early discoveries in zeolite chemistry and catalysis at Union Carbide, and follow-up in industrial catalysis. Appl. Catal. A 2001, 222, 261–275. [Google Scholar] [CrossRef]
- Bujnova, A.; Lesny, J. Sorption characteristics of zinc and cadmium by some natural, modified, and synthetic zeolites. Electron. J. Chem. Technol. Biotechnol 2004, 59, 121–126. [Google Scholar]
- Inglezakis, V.J.; Loizidou, M.M.; Grigoropoulou, H.P. Ion exchange studies on natural and modified zeolites and the concept of exchange site accessibility. J. Colloid Inter. Sci 2004, 275(2), 570–576. [Google Scholar] [CrossRef]
- Oren, A.H.; Kaya, A. Factors affecting adsorption characteristics of Zn2+ on two natural zeolites. J Hazard Mater 2006, 131(1-3), 59–65. [Google Scholar] [CrossRef]
- Kosmulski, M. The pH dependent surface charging and points of zero charge. VII. Update. Adv. Colloid Inter. Sci 2018, 251, 115–138. [Google Scholar] [CrossRef]
- Kosmulski, M. The pH dependent surface charging and points of zero charge. VIII. Update. Adv. Colloid Inter. Sci 2020, 275, 102064. [Google Scholar] [CrossRef]
- Monhemius, A.J. Precipitation diagrams for metal hydroxides, sulphides, arsenates and phosphates. Trans. Inst. Min. Metall 1977, 6, 203–206. [Google Scholar]
- Goscianska, J.; Ptaszkowska-Koniarz, M.; Frankowski, M.; Franus, M.; Panek, R.; Franus, W. Removal of phosphate from water by lanthanum-modified zeolites obtained from fly ash. J. Colloid Inter. Sci 2018, 513, 72–81. [Google Scholar] [CrossRef]
- Wu, D.Y.; Zhang, B.H.; Li, C.J.; Zhang, Z.J.; Kong, H.N. Simultaneous removal of ammonium and phosphate by zeolite synthesized from fly ash as influenced by salt treatment. J. Colloid Inter. Sci 2006, 304(2), 300–306. [Google Scholar] [CrossRef]
- Chen, J.G.; Kong, H.N.; Wu, D.Y.; Hu, Z.B.; Wang, Z.S.; Wang, Y.H. Removal of phosphate from aqueous solution by zeolite synthesized from fly ash. J. Colloid Interface Sci. 2006, 300(2), 491–497. [Google Scholar] [CrossRef]
- Aylward G.H.; Findlay T.J.V.; SI Chemical Data, 2nd Ed. 1974, John Wiley & Sons.
- Hamdi, N.; Srasra, E. Removal of phosphate ions from aqueous solution using Tunisian clays minerals and synthetic zeolite. J. Environ. Sci. 2012, 24(4), 617–623. [Google Scholar] [CrossRef]
- Zhang, B.H.; Wu, D.Y.; Wang, C.; He, S.B.; Zhang, Z.J.; Kong, H.N. Simultaneous removal of ammonium and phosphate by zeolite synthesized from coal fly ash as influenced by acid treatment. J. Environ. Sci. 2007, 19, 540–54. [Google Scholar] [CrossRef]
- Zhang, Y.; Kou, X.; Lu, H.; Lv, X. The feasibility of adopting zeolite in phosphorus removal from aqueous solutions. Desal. Water Treat. 2014, 52. [Google Scholar] [CrossRef]
- Alshameri A.; Yan C.; Lei X.; Enhancement of phosphate removal from water by TiO2/Yemeni natural zeolite: Preparation, characterization and thermodynamic. Micropor. Mesopor Mater 2014, 196, 145–157. [CrossRef]
- Ciesla, J.; Franus, W.; Franus, M.; Kedziora, K.; Gluszczyk, J.; Szerement, J.; Jozefaciuk, G. Environmental-friendly modifications of zeolite to increase its sorption and anion exchange properties. Physicoch. Stud. Modi. Mater 2019, 12, 3213. [Google Scholar] [CrossRef]
- Mahmoodi, N.; Saffar-Dastgerdi, M.H. Zeolite nanoparticle as a superior adsorbent with high capacity: Synthesis, surface modification and pollutant adsorption ability from wastewater. Microchem. J 2019, 145, 74–83. [Google Scholar] [CrossRef]
- Swiderska-Dabrowska R.; Schmidt R.; Sikora A. Effect of calcination temperature on chemical stability of zeolite modified by iron ions. In Polish Environ. Eng. 2012; Environmental Engineering Committee PAS, Ed.; Environmental Engineering Committee PAS: Lublin, Poland, 2012, 307–317.
- Glassman, H.N. ; Surface active agents and their application in bacteriology. Bacteriol. Rev. 1948, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Reeve, P.J.; Fallowfield, H.J. Natural and surfactant modified zeolites: A review of their applications for water remediation with a focus on surfactant desorption and toxicity towards microorganisms. J. Environ. Manag 2018, 205, 253–261. [Google Scholar] [CrossRef]
- Ivankovic, T.; Hrenovic, J. Surfactants in the environment. Arch. Ind. Hyg. Toxicol 2010, 61, 95–110. [Google Scholar] [CrossRef]
- Li, Z. Sorption kinetics of hexadecyltrimethylammonium on natural clinoptilolite. Langmuir 1999, 15, 6438–6445. [Google Scholar] [CrossRef]
- Bowman, R.S. Applications of surfactant-modified zeolites to environmental remediation. Micropor. Mesopor. Mater 2003, 61, 43–56. [Google Scholar] [CrossRef]
- Baez-Alvarado, M.D.; Olguín, M.T. Surfactant-modified clinoptilolite-rich tuff to remove barium (Ba2+) and fulvic acid from mono- and bi-component aqueous media. Micropor. Mesopor Mater 2011, 139, 81–86. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Z.; Fang, D.; Li, C.; Wu, D. Green synthesis of a novel hybrid sorbent of zeolite/lanthanum hydroxide and its application in the removal and recovery of phosphate from water. J. Colloid Inter. Sci. 2014, 423, 13–19. [Google Scholar] [CrossRef]
- Abdellaoui, Y.; Oualid, H.A.; Hsini, A.; Ibrahimi, B.E.; Laabd, M.; Ouardi, M.E.; Giácoman-Vallejos, G.; Gamero-Melo, P. Synthesis of zirconium-modified Merlinoite from fly ash for enhanced removal of phosphate in aqueous medium: Experimental studies supported by Monte Carlo/SA simulations. Chem. Eng. Journ 2021, 404, 126600. [Google Scholar] [CrossRef]
- Naghash, A.; Nezamzadeh-Ejhieh, A. Comparison of the efficiency of modified clinoptilolite with HDTMA and HDP surfactants for the removal of phosphate in aqueous solutions. J. Ind. Eng. Chem. 2015, 31, 185–191. [Google Scholar] [CrossRef]
- Hermassi, M.; Valderrama, C.; Font, O.; Moreno, N.; Querol, X.; Batis, N.; Cortina, J. Phosphate recovery fromaqueous solution by K-zeolite synthesized from fly ash for subsequent valorisation as slow release fertilizer. Sci. Total Environ 2020, 731, 139002. [Google Scholar] [CrossRef]
- Mokrzycki, J.; Fedyna, M.; Marzec, M.; Szerement, J.; Panek, R.; Klimek, A.; Bajda, T.; Mierzwa-Hersztek, M. Copper ion-exchanged zeolite X from fly ash as an efficient adsorbent of phosphate ions from aqueous solutions. J. Environ. Chem. Engin 2022, 10, 108567. [Google Scholar] [CrossRef]
- Li, Z.; Willims, C.; Roy, S.; Bowman, R.S. Desorption of hexadecyltrimethylammonium from charged mineral surfaces. Environ. Geosc 2017, 10(1), 37–45. [Google Scholar] [CrossRef]
- de Gennaro, B.; Catalanotti, L.; Bowman, R.S.; Mercurio, M. Anion exchange selectivity of surfactant modified clinoptilolite-rich tuff for environmental remediation. J. Colloid Inter. Sci 2014, 430, 178–183. [Google Scholar] [CrossRef]
- Gouran-Orimi, R.; Mirzayi, B.; Nematollahzadeh, Al.; Tardast, A. Competitive adsorption of nitrate in fixed-bed column packed with bio-inspired polydopamine coated zeolite. J. Environ. Chem. Eng 2018, 6, 2232–2240. [Google Scholar] [CrossRef]
- Lin, H.; Huang, X.; Chang, J.; Li, B.; Bai, Y.; Su, B.; Shi, L.; Dong, Y. Improving sludge settling performance of secondary settling tank and simultaneously adsorbing nitrate and phosphate with surfactant modified zeolite (SMZ) ballasted flocculation. J. Environ. Chem. Eng 2023, 11, 109650. [Google Scholar] [CrossRef]
- Zhan, Y.; Lin, J.; Zhu, Z. Removal of nitrate from aqueous solution using cetylpyridinium bromide (CPB) modified zeolite as adsorbent. J. Hazard. Mater. 2011, 186, 1972–1978. [Google Scholar] [CrossRef]
- Schick, J.; Caullet, P.; Paillaud, J.-L.; Patarin, J.; Mangold-Callarec, C. Batch-wise nitrate removal from water on a surfactant-modified zeolite. Micropor. Mesopor. Mater. 2010, 132, 395–400. [Google Scholar] [CrossRef]
- Schick, J.; Caullet, P.; Paillaud, J.-L.; Patarin, J.; Mangold-Callarec, C. Nitrate sorption from water on a Surfactant-Modified Zeolite. Fixed-bed column experiments. Micropor. Mesopor. Mater. 2011, 142, 549–556. [Google Scholar] [CrossRef]
- Li, Z.; Roy, S.J.; Zou, Y.; Bowman, R.S. Long-Term chemical and biological stability of surfactant-modified zeolite. Environ. Sci. Technol 1998, 32, 2628–2632. [Google Scholar] [CrossRef]
- Hrenovic, J.; Rozic, M.; Sekovanic, L.; Anic-Vucinic, A. Interaction of surfactant-modified zeolites and phosphate accumulating bacteria, J. Hazard. Mater. 2008, 156, 576–582. [Google Scholar] [CrossRef]
- Li, Z. Chromate transport through surfactant-modified zeolite columns. Ground Water Monit. Rem 2006, 26, 117–124. [Google Scholar] [CrossRef]
- Li, Z.; Hong, H. Retardation of chromate through packed columns of surfactant-modified zeolite. J. Hazard. Mater. 2009, 162, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bowman, R.S. Retention of inorganic oxyanions by organo-kaolinite. Wat. Res 2001, 35(16), 3771–3776. [Google Scholar] [CrossRef] [PubMed]
- Nye, J.V.; Guerin, W.F.; Boyd, S.A. Heterotrophic activity of microorganisms in soils treated with quaternary ammonium compounds. Environ. Sci. Technol 1994, 28, 944–951. [Google Scholar] [CrossRef]
- Hrenovic, J.; Ivankovic, T. Toxicity of anionic and cationic surfactant to Acinetobacter junii in pure culture Cent. Eur. J. Biol 2007, 2, 405–414. [Google Scholar]
- Cataldo, E.; Salvi, L.; Paoli, F.; Fucile, M.; Masciandaro, G.; Manzi, D.; Masini, C.M.; Mattii, G.B. Application of zeolites in agriculture and other potential uses: a review. Agronomy 2021, 11, 1547. [Google Scholar] [CrossRef]
- Pond, W.G.; Mumpton, F.A. Zeo-agriculture - use of natural zeolites in agriculture and aquaculture. Westview Press, 1984, Brockport, New York.
- Li, Z. Use of surfactant-modified zeolite as fertilizer carriers to control nitrate release. Micropor. Mesopor. Mater 2003, 61, 181–188. [Google Scholar] [CrossRef]
- Zhang, Y.; Prigent, B.; Geißen, S.-U. Adsorption and regenerative oxidation of trichlorophenol with synthetic zeolite: Ozone dosage and its influence on adsorption performance. Chemosphere 2016, 154, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Sireesha, S.; Agarwal, A.; Sopanrao, K.S.; Sreedhar, L.; Anitha, K.L. Modified coal fly ash as a low-cost, efficient, green, and stable adsorbent for heavy metal removal from aqueous solution. Biomass Conv Bioref 2022. [Google Scholar] [CrossRef]
- Batjargal, T.; Yang, J.-S.; Kim, D.-H.; Baek, K. Removal Characteristics of Cd(II), Cu(II), Pb(II), and Zn(II) by Natural Mongolian Zeolite through Batch and Column Experiments. Sep. Sci. Technol 2011, 46(8), 1313–1320. [Google Scholar] [CrossRef]
- Bolan N.S., Mowatt C., Adriano D.C. and Blennerhassett J.D. Removal of ammonium ions from fellmongery effluent by zeolite. Comm. Soil Sci. Plant Anal 2003, 34, 1861–1872. [CrossRef]
- Zhang, M.; Zhang, H.; Xu, D.; Han, L.; Niu, D.; Tian, B.; Zhang, J.; Zhang, L.; Wu, W. Removal of ammonium from aqueous solutions using zeolite synthesized from fly ash by a fusion method. Desalination 2011, 271, 111–121. [Google Scholar] [CrossRef]
- Karadag, D.; Tok, S.; Akgul, E.; Turan, M.; Ozturk, M.; Demir, A. Ammonium removal from sanitary landfill leachate using natural G¨ordes clinoptilolite. J. Hazard. Mater 2008, 153, 60–66. [Google Scholar] [CrossRef]
- Malovanyy, A.; Sakalova, H.; Yatchyshyn, Y.; Plaza, E.; Malovanyy, M. Concentration of ammonium from municipal wastewater using ion exchange process. Desalination 2013, 329, 93–102. [Google Scholar] [CrossRef]
- Lorick, D.; Ahlström, M.; Grimvall, A.; Harder, R. Effectiveness of struvite precipitation and ammonia stripping for recovery of phosphorus and nitrogen from anaerobic digestate: a systematic review. Environ Evid 2020, 9(27), 1–20. [Google Scholar] [CrossRef]
- Huang J.-C.; Shang C. Air stripping. In Handbook of Environmental Engineering. Vol. 4: Advanced Physicochemical Treatment Processes. Edited by: Wang, L.L.; Hung Y.-T.; Shammas N.K. The Humana Press Inc. 2006, Totowa, NJ.
- Katehis, D.; Diyamandoglu, V.; Fillos, J. Stripping and recovery of ammonia from centrate of anaerobically digested biosolids at elevated temperatures. Water Environ. Res. 1998, 70, 231–240. [Google Scholar] [CrossRef]
- Değermenci, N.; Yildiz, E. Ammonia stripping using a continuous flow jet loop reactor: mass transfer of ammonia and effect on stripping performance of influent ammonia concentration, hydraulic retention time, temperature, and air flow rate. Environ. Sci. Poll. Res 2021, 28, 31462–31469. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Xie, S.; Liu, S.; Xu, L. Physical and chemical regeneration of zeolitic adsorbents for dye removal in wastewater treatment. Chemosphere 2006, 65(1), 82–87. [Google Scholar] [CrossRef] [PubMed]
- Salvador, F.; Martin-Sanchez, N.; Sanchez-Hernandez, R.; Sanchez-Montero, M.J.; Izquierdo, C. Regeneration of carbonaceous adsorbents. Part I: Thermal Regeneration. Micropor. Mesopor. Mater 2015, 202, 259–276. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Shi, T.B.; Jia, C.Z.; Ji, W.J.; Chen, Y.; He, M.Y. Adsorptive removal of aromatic organosulfur compounds over the modified Na-Y zeolites. App. Catal. B: Environ 2008, 82(1), 1–10. [Google Scholar] [CrossRef]
- Fujita, H.; Izumi, J.; Sagehashi, M.; Fujii, T.; Sakoda, A. Decomposition of trichloroethene on ozone-adsorbed high silica zeolites. Water Res 2004, 38(1), 166–172. [Google Scholar] [CrossRef]
- Lee, D.-G.; Kim, J.-H.; Lee, C.-H. ; Adsorption and thermal regeneration of acetone and toluene vapors in dealuminated Y-zeolite bed. Sep. Pur. Technol. 2011, 77(3), 312–324. [Google Scholar] [CrossRef]
- Delgado, L.; Catarino, A.S.; Eder, P.; Litten, D.; Luo, Z.; Villanueva, A. End-of-Waste Criteria. EUR – Scientific and Technical Research series, 2009. [CrossRef]
- Grand View Research, Market Analysis Report: Zeolite Market Size, Share & Trends Analysis Report By Application (Catalyst, Adsorbent, Detergent Builder), By Product (Natural, Synthetic), By Region (North America, Europe, APAC, CSA, MEA), And Segment Forecasts, 2022 – 2030. Report 978-1-68038-601-1, pp. 114.
- Markets and Markets, Market Research report: Zeolites market by type (natural, synthetic), function (ion-exchange, catalyst, molecular sieve), synthetic zeolites application (detergent, catalyst), natural zeolites application, and regional-global forecast to 2026. Report CH 8006, 2021.
- Coherent Market Insights (2023) Nutrient Recycling Market. Report CM15972, 154.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



