
Submitted:
28 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
Recent advancements in the study of the protein complex photosystem II have clarified the sequence of events leading to the formation of oxygen during the S3->S4->S0 tran-sition, wherein the inorganic Mn4Ca(µ-O)6(OHx)4 cluster finishes photo-catalyzing the water splitting reaction (Greife et al, Nature 2023, 617, 623-628; Bhowmick et al, Nature 2023, 617, 629-636) . During this final step a tyrosine radical (TyrZ), stable for a couple of milliseconds, oxidizes a cluster bound oxygen while the hydrogen bonding patterns of nearby waters shift a proton away. A treatment of this redox reaction within the context of accepted transition state theories predicts rate constants that are significantly higher than experimentally recovered values (10^12s^-1 versus 10^3s^-1). In an effort to understand this disparity, temperature dependent experiments have revealed large entropic con-tributions to the rates with only a moderate energy of activation. We suggest that the entropic source may be related to the observed proton rearrangements, and further possible near isoenergetic variations in the nearby extended h-bonding network de-laying the realization of an ‘ideal’ transition state. In the following we explore this re-lation in the context of Eyring’s transition state theory and Marcus’ electron transfer theory, and evaluate their compatibility with the experimental evidence.
Keywords:
Photosystem II
; Oxygen Evolution Reaction
; Proton Coupled Electron Transfer
; Transition State Theory
1. Introduction
In oxygenic photosynthesis, water is oxidized at a Mn4Ca(µ-O)6(OHx)4 complex (denoted here as the Mn4Ca-cluster) bound to the proteins of photosystem II (PSII)[1,2]. Light drives water oxidation at the Mn4Ca-cluster indirectly[3,4], with a primary charge separation occurring at the electron donor side of PSII resulting in the formation of a delocalized chlorophyll cation radical (P680+). This is followed by the oxidation of a specific tyrosine residue, denoted as TyrZ or YZ . The subsequent electron transfer (ET) steps from the Mn4Ca cluster to TyrZox are closely related to the water oxidation chemistry coupled with proton movements over a large area on various length scales, see Figure 1. Herein we will discuss, in a broad sense, whether the Mn4Ca cluster – TyrZox ET can be described favorably by transition state (TS) theory in the framework of the Eyring-Polanyi equation[5,6,7] or whether elements of ET theories, e.g. the well-known semi-classical Marcus theory[8,9], are applicable or even essential for a correct description. We note that the following Results and Discussion section corresponds in parts to Sections SII.8 and SII.9 of the Supplementary Material of Greife et al.[10]. Our discussion addresses specifically the water oxidation reaction (or oxygen evolution reaction) in PSII, but it may, in parts, also be of relevance for redox reactions of other proteins, molecules, and redox-active electrodes in electrocatalysis.
2. Results and Discussion
2.1. TS theory applied to oxygen-evolution step of PSII
In Eyring’s TS theory, the reaction rate (kTST) is governed by the Eyring-Polanyi equation[5,6] involving the entropy (ΔS‡) and enthalpy (ΔH‡) of activation:
with kB being the Boltzmann constant, h the Planck constant, and T the absolute temperature in Kelvin. The κ parameter describes the transmission probability, i.e., the probability that the reaction will occur once the TS is reached; typically κ-values of 0.5 or 1.0 are assumed to be reasonable[5,6]. In Equ. 1, the activation enthalpy, ΔH‡, describes the temperature dependence of the rate constant and is closely related the activation energy, Ea, as determined from a conventional Arrhenius analysis (ΔH‡ ≈ Ea - kBT0, the difference ΔH‡ and Ea stems from the weak temperature dependence of in Equ. 1, see ref.[12]). The activation entropy, ΔS‡, is usually a negative number that corresponds to a temperature-independent reduction of the kTST.
In research on PSII water oxidation, Equ. 1 has been applied to the rate constant of the oxygen-evolution transition[10,13,14], which is initiated by the third flash of saturating light applied to dark-adapted PSII[15]. The oxygen evolution transition is referred to as the S3->S0 transition and involves an ET from the Mn4Ca-cluster to TyrZox. This ET step precedes O-O bond formation and O2-release. It is generally believed that the rate-determining step of the overall reaction is the Mn4Ca-cluster—TyrZox ET step, which we have suggested to be coupled to the movement of four protons[10] see Figure 2.
Based on experimental determination of the rate constants for the O2-formation at temperatures ranging from -5°C to 40°C step, we have calculated (by application of Equation 1) the enthalpy and entropy of activation of the rate-determining (proton-coupled) ET step for PSII of the thermophilic cyanobacterium Thermosynechococcus elongatus (T. elongatus), see Extended Data Figure 5 and elsewhere in Greife et al. We obtained values for the activation enthalpy of 310 meV (7.15 kcal/mol) and activation entropy of 284 meV (6.55 kcal/mol, value of -T0ΔS‡ for T0 = 293 K), for κ = 1 [10]. A variation of the κ-value from 1.0 to 0.5 would leave the activation enthalpy unchanged and affects the numerical value of the activation entropy only marginally.
For the PSII of T. elongatus and a closely related thermophilic cyanobacterium, Thermosynechococcus vulcanus (T. vulcanus), detailed crystallographic data is available, not only for the dark-stable state, but also for intermediates of the reaction cycle[11,16,17,18]. Due to the availability of atomic-resolution structures, computational studies on the mechanism of PSII water oxidation mostly have been based on coordinates obtained for PSII of T. elongatus or T. vulcanus[19,20,21,22,23,24,25]. Therefore, herein we focus on the enthalpy and entropy of activation experimentally determined for the O2-formation step of T. elongatus.
The application of Eq. 1 to experimental data is straightforward and has resulted in values for enthalpy and entropy of activation for the rate-determining ET transfer step in the oxygen evolution transition. The question arises as to how the application of TS theory relates to ET theories.
2.2. Relation to ET theory
In the framework of the Eyring-Polanyi equation, essentially instantaneous ET would have to occur whenever the appropriate nuclear coordinates are reached, corresponding to a κ-value near unity in Equ. 1. In terms of non-adiabatic ET theory, however, the relocation of electron density (and thus spin density) does not always occur once a suitable nuclear geometry has been reached, but with a finite probability that is determined by the electron-tunneling distance. A tunneling probability well below the frequency factor in Equ. 1 might mimic an entropic contribution to the activation energy. Therefore, the possibility of whether non-adiabaticity of the ET step could contribute significantly to the experimentally determined entropy of activation is explored in the following.
Following Moser et al.[26], we estimate the tunneling distance as the molecular edge-to-edge distance between donor and acceptor moiety, where herein the acceptor moiety is the tyrosine radical and the donor being the Mn4Ca cluster. The ‘edges’ or rather the extension of the relevant orbital systems are not obvious and cannot reliably be identified without using computational approaches. We calculated several potentially relevant interatomic (internuclear) distances, see Figure 3 and Table 1. An internuclear distance close to 5.7 Å is obtained between TyrZ-O and O6. This distance could be especially relevant because in the rate-limiting ET step of the S3->S0 transition, formation of the O6• radical is facilitated by ET to the oxidized TyrZ, with high spin density expected on the phenolic oxygen. This corresponds to a Van-der-Waals tunneling distance of only about 2.5 Å. Other potentially relevant internuclear distances in Table 1 are even shorter, predicting a particularly high tunneling probability. According to Moser et al., the tunneling distance of 2.5 Å corresponds to a free-energy optimized rate constant for electron tunneling on the order of 1012 s-1, or a time constant of 1 ps. The frequency that describes nuclear movements in the TS regime is about 6 times greater (kBT/h in Equ. 1, about 6∙1012 s-1 at 20°C) suggesting that for oxygen-evolution steps in PSII a minor slow-down resulting from limiting ET probability cannot be excluded. Staying in the framework of TS theory, such a slowdown could be described by a reduced transmission probability (reduced κ-value), which would result in only a small reduction of the activation entropy that we calculated from experimental data using the Eyring-Polanyi equation (Equation 1).
The above considerations involve several approximations and estimates. Presumably the tunneling probability exceeds the above estimate of 1012 s-1 because the ‘medium’ between the donor and acceptor moieties comprises atom groups (specifically W3 and W4) that could lower the tunneling barrier significantly. In addition, multiple parallel tunneling paths could also increase the overall tunneling probability. Thus, it is well conceivable that the adiabatic limit, where electron tunneling probabilities become irrelevant, is reached.
In summary, uncertainties remain regarding the exact numerical value of the activation entropy, because of possible limitations due to electron tunnelling. However, it is likely that the limiting case of adiabatic ET is almost, or even fully reached. If the adiabatic ET limit were not reached, the activation entropy would be moderately lower than the value formally determined by application of Equ. 1 to experimental data. However, this does not affect the central conclusion that there is a pronounced entropic slowdown of the oxygen-evolution transition.
Based on minimum energy path (MEP) calculations, we have proposed that the ET from the Mn4Ca cluster to TyrZox is coupled to the movement of four specific protons[10]. However, the used computational approach cannot clarify whether concerted ET and proton transfer (PT) is involved, that is, the simultaneous tunneling of the electron and one or more protons at the TS. This question is of relevance when assessing the significance of activation entropy formally calculated by application of Equ. 1 to experimental data, because in the case of a truly concerted electron-proton transfer (CEPT), the proton tunneling probability could influence the reaction rate and mimic an entropic slowdown. However, the low value of the experimentally observed H/D kinetic isotope effect (about 1.2[27,28]) clearly disfavors a limitation of the reaction rate by the proton-tunneling probability.
Further evidence supporting the entropic slowdown hypothesis comes from experimental indications for entropy-enthalpy compensation in the total free energy of activation of the oxygen-evolution step. In PSII of higher plants (spinach) at room temperature, the rate constant is very similar to that of the thermophilic cyanobacterium (T. elongatus). However, the Arrhenius activation energy and consequently the activation enthalpy are significantly lower for the higher-plant PSII[14]. For the cyanobacterium Synechocystis sp. PCC 6803, again another combination of action enthalpy and entropy has been reported[13]. We propose that compensation of the decreased activation enthalpy by an increased activation entropy explains identical rate constant values at isokinetic temperatures in the physiological temperature range. This entropy-enthalpy compensation in the activation energy of the oxygen-evolution step is still insufficiently understood and requires further investigation (work in progress). In general, entropy-enthalpy compensation is well known for the free energy of activation relating to various catalytic processes in biological and non-biological systems [29,30,31,32]. Finding entropy-enthalpy compensation in PSII oxygen evolution supports the existence of a major entropic contribution to the total free energy of activation, and it disfavors alternative explanations like a dominating role of electron or proton tunneling probabilities.
2.3. Origin of activation entropy and its relation to Marcus theory
In PSII water oxidation, the environment of the two key protagonists, TyrZ and the Mn4Ca cluster, is characterized by an extended network of H-bonded water molecules and amino-acid residues. See Figure 4 for a two-dimensional mapping of the H-bond network (HBN) within 20 Å of the Mn4Ca cluster. There are over 40 resolved waters to be found, grouped into several different water channels. Some of these groups are likely involved in proton and water transport and have been shown to be variable during the S3->S0 transition[10,11]. We propose that multiple conformations, that is, multiple H-bond patterns with multiple orientations of water molecules and residues can explain the experimentally found activation entropy. The interplay between evolutionary optimized order (stabilization of favorable conformations) and thermally driven dynamics needs to be considered when discussing the functional relevance of the extended H-bonded protein-water network. Following Greife et al (Supplementary Information, section SII.9), we point out:
(1) It is expected and has been verified by extensive molecular dynamics simulations on PSII that the protein internal H-bond networks (HBN) are highly dynamic, especially regarding water positions and H-bonding directionality, with a multitude of roughly isoenergetic conformations reached within nanoseconds at room temperature (see, e.g., ref.[35]). Even though these HBN dynamics maybe in part conducive regarding PT reactions, they are largely ‘inevitable’ thermodynamic fluctuations.
(2) The arrangement of the water molecules in the extended H-bonded protein-water network surrounding the Mn4CaO5 cluster and the TyrZ is well resolved in the crystallographic structures determined at both cryogenic and room temperature. This implies that the HBN dynamics evolve around the mean positions of the individual nuclei that are detected by protein crystallography. The HBN and the related mean-value atom coordinates are likely evolutionarily optimized for efficient (fast) water oxidation.
In light of (1) and (2), the qualitative explanation for the energy of activation in Greife et al. appears plausible: ‘Although well-defined coordinates of individual oxygen atoms are resolved in crystal structures, the presence of an HBN that is at the same time and in every detail perfectly arranged for the here discussed proton-coupled ET, will still be a rare event. The limited probability to reach this perfect conformation of all atoms of the HBN explains the entropic contributions to the activation energy.’
We emphasize that even if the optimal HBN conformation perfectly matches the atomic coordinates resolved by protein crystallography, it will still be rare that all atoms are simultaneously at their evolutionarily optimized positions at physiological temperatures.
3. Conclusions
Marcus and related theories on ET or CEPT reactions are powerful tools because they can predict rate constants based on a small number of clearly defined parameters, which are often experimentally accessible. The extended H-bonded protein-water network in PSII might be seen as a special case of the solvent environment that in Marcus theory is described involving a quadratic energy dependence with the outer-sphere reorganization energy being the (only) decisive parameter [9]. However, such an approximative treatment does not consider individual solvent molecules. Instead, Marcus theory assumes a homogeneous solvent environment which responds to charges at the redox factors by a linear dielectric response; the total energy of charge solvation is decisive for the contribution of the outer-sphere reorganization to the activation energy.
In contrast to the consideration on the ET in PSII of the Results and Discussion section, in Marcus theory there is no special conformation of the solvent environment foreseen that uniquely supports the ET step; it is the total electrostatic solvation energy that matters, realized by a multitude of conformations of the solvent environment. Therefore, the underlying approximations of Marcus theory render its applicability to redox reactions in a protein environment with multiple conformations of a specific, evolutionary optimized protein-water network problematic, inter alia because it cannot easily capture the here discussed entropic contributions to the free energy of activation. Whether the predictive power of ET theory can be fruitfully used regarding the contribution of inner-sphere processes of contributing redox factors (in PSII and other redox proteins), remains to be answered. In conclusion, the common assumption (in ET theories) of nuclear movements in approximately quadratic potentials needs to be scrutinized for redox factors surrounded by a fluctuating protein-internal HBN.
In summary, our considerations support the appropriateness of discussing the rate-limiting proton-coupled ET step in photosynthetic water oxidation in terms of Eyring's TS theory. We point out the important but apparently poorly understood role of activation entropy and hope that future work can shed more light on the interplay between order and statistical dynamics in PSII water oxidation, biological redox processes in general, homogeneous and heterogeneous catalysis.
Author Contributions
The main text was written, edited, and reviewed by HD and PG, with initial conceptualization by HD. Data analysis and visualization was performed by PG. Supervision, project administration and funding acquisition was performed by HD.
Funding
The authors gratefully acknowledge the financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) provided to the collaborative research center on Protonation Dynamics and Protein Function (SFB 1078, project A4/Dau) under Germany’s Excellence Strategy EXC 2008/1-390540038-UniSysCat.
Data Availability Statement
No new data were created in this study, structural data used publicly available int the protein data bank (PDB).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Junge, W. Oxygenic photosynthesis: History, status and perspective. Q. Rev. Biophys. 2019, 52, e1. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.; Pantazis, D.A.; Lubitz, W. Current Understanding of the Mechanism of Water Oxidation in Photosystem II and Its Relation to XFEL Data. Annu. Rev. Biochem. 2020, 89, 795–820. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Haumann, M. Eight steps preceding O-O bond formation in oxygenic photosynthesis--a basic reaction cycle of the Photosystem II manganese complex. Biochim. Biophys. Acta 2007, 1767, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Tran, R.; Kern, J.; Hattne, J.; Koroidov, S.; Hellmich, J.; Alonso-Mori, R.; Sauter, N.K.; Bergmann, U.; Messinger, J.; Zouni, A.; et al. The Mn4Ca photosynthetic water-oxidation catalyst studied by simultaneous X-ray spectroscopy and crystallography using an X-ray free-electron laser. Philos. Trans. R. Soc. B 2014, 369, 20130324. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. J. Chem. Soc. Faraday Trans. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Mortimer, R.G.; Eyring, H. Elementary transition state theory of the Soret and Dufour effects. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 1728–1731. [Google Scholar] [CrossRef]
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Greife, P.; Schönborn, M.; Capone, M.; Assunção, R.; Narzi, D.; Guidoni, L.; Dau, H. The electron–proton bottleneck of photosynthetic oxygen evolution. Nature 2023, 617, 623–628. [Google Scholar] [CrossRef]
- Bhowmick, A.; Hussein, R.; Bogacz, I.; Simon, P.S.; Ibrahim, M.; Chatterjee, R.; Doyle, M.D.; Cheah, M.H.; Fransson, T.; Chernev, P.; et al. Structural evidence for intermediates during O2 formation in photosystem II. Nature 2023, 617, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sekharan, S.; Liu, J.; Batista, V.S.; Tully, J.C.; Yan, E.C.Y. Unusual kinetics of thermal decay of dim-light photoreceptors in vertebrate vision. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10438–10443. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Burnap, R.L. Structural rearrangements preceding dioxygen formation by the water oxidation complex of photosystem II. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E6139–E6147. [Google Scholar] [CrossRef] [PubMed]
- Assunção, R.; Zaharieva, I.; Dau, H. Ammonia as a substrate-water analogue in photosynthetic water oxidation: Influence on activation barrier of the O2-formation step. Biochim. Biophys. Acta 2019, 1860, 533–540. [Google Scholar] [CrossRef]
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic O2 evolution - I. A linear four-step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok's photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Kato, K.; Miyazaki, N.; Hamaguchi, T.; Nakajima, Y.; Akita, F.; Yonekura, K.; Shen, J.R. High-resolution cryo-EM structure of photosystem II reveals damage from high-dose electron beams. Commun. Biol. 2021, 4, 382. [Google Scholar] [CrossRef]
- Hussein, R.; Ibrahim, M.; Bhowmick, A.; Simon, P.S.; Chatterjee, R.; Lassalle, L.; Doyle, M.; Bogacz, I.; Kim, I.S.; Cheah, M.H.; et al. Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat. Commun. 2021, 12, 6531. [Google Scholar] [CrossRef]
- Siegbahn, P.E. Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys. Chem. Chem. Phys. 2012, 14, 4849–4856. [Google Scholar] [CrossRef]
- Wang, J.; Askerka, M.; Brudvig, G.W.; Batista, V.S. Crystallographic Data Support the Carousel Mechanism of Water Supply to the Oxygen-Evolving Complex of Photosystem II. ACS Energy Lett. 2017, 2, 2299–2306. [Google Scholar] [CrossRef]
- Narzi, D.; Capone, M.; Bovi, D.; Guidoni, L. Evolution from S3 to S4 state of the oxygen evolving complex in Photosystem II monitored by QM/MM dynamics. Chem. Eur. J. 2018. [Google Scholar] [CrossRef]
- Shoji, M.; Isobe, H.; Shigeta, Y.; Nakajima, T.; Yamaguchi, K. Concerted Mechanism of Water Insertion and O2 Release during the S4 to S0 Transition of the Oxygen-Evolving Complex in Photosystem II. J. Phys. Chem. B 2018, 122, 6491–6502. [Google Scholar] [CrossRef] [PubMed]
- Capone, M.; Guidoni, L.; Narzi, D. Structural and dynamical characterization of the S-4 state of the Kok-Joliot's cycle by means of QM/MM Molecular Dynamics Simulations. Chem. Phys. Lett. 2020, 742, 137111. [Google Scholar] [CrossRef]
- Allgöwer, F.; Gamiz-Hernandez, A.P.; Rutherford, A.W.; Kaila, V.R.I. Molecular Principles of Redox-Coupled Protonation Dynamics in Photosystem II. J. Am. Chem. Soc. 2022, 144, 7171–7180. [Google Scholar] [CrossRef]
- Isobe, H.; Shoji, M.; Suzuki, T.; Shen, J.-R.; Yamaguchi, K. Exploring reaction pathways for the structural rearrangements of the Mn cluster induced by water binding in the S3 state of the oxygen evolving complex of photosystem II. J. Photochem. Photobiol. A: Chem. 2021, 405, 112905. [Google Scholar] [CrossRef]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef]
- Gerencser, L.; Dau, H. Water oxidation by Photosystem II: H2O−D2O Exchange and the influence of pH support formation of an intermediate by removal of a proton before dioxygen creation. Biochemistry 2010, 49, 10098–10106. [Google Scholar] [CrossRef]
- Klauss, A.; Haumann, M.; Dau, H. Alternating electron and proton transfer steps in photosynthetic water oxidation. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16035–16040. [Google Scholar] [CrossRef]
- Losi, A.; Wegener, A.A.; Engelhard, M.; Braslavsky, S.E. Enthalpy−Entropy Compensation in a Photocycle: The K-to-L Transition in Sensory Rhodopsin II from Natronobacterium pharaonis. J. Am. Chem. Soc. 2001, 123, 1766–1767. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Q.-X. Isokinetic Relationship, Isoequilibrium Relationship, and Enthalpy−Entropy Compensation. Chem. Rev. 2001, 101, 673–696. [Google Scholar] [CrossRef]
- Ferrante, A.; Gorski, J. Enthalpy-entropy compensation and cooperativity as thermodynamic epiphenomena of structural flexibility in ligand-receptor interactions. J. Mol. Biol. 2012, 417, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Mobley, D.L. Entropy-enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142. [Google Scholar] [CrossRef]
- Bondar, A.-N.; Dau, H. Extended protein/water H-bond networks in photosynthetic water oxidation. Biochim. Biophys. Acta 2012, 1817, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, N.; Ishikita, H.; Saito, K. Rigidly hydrogen-bonded water molecules facilitate proton transfer in photosystem II. Phys. Chem. Chem. Phys. 2020, 22, 15831–15841. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Siemers, M.; Mielack, C.; Bondar, A.N. Dynamics of Long-Distance Hydrogen-Bond Networks in Photosystem II. J. Phys. Chem. B 2018, 122, 4625–4641. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
H-bonded protein-water network surrounding the Mn4Ca cluster of PSII (S3-state structure, PDB 8EZ5 of T. elongatus [11]). The water molecules ligated to Mn1 and Ca (W1, W2, W3 and W4) are part of an extended H-bonding network reaching from the redox-active TyrZ towards the Glu65/Glu312 ‘proton gate’. H-bond distances of less than or equal to 3.4 Å are indicated by dashed yellow lines. The depicted amino-acid sidechain either are part of the PsbA (D1), PsbC (CP43) or PsbD (D2) subunits.
Figure 1.
H-bonded protein-water network surrounding the Mn4Ca cluster of PSII (S3-state structure, PDB 8EZ5 of T. elongatus [11]). The water molecules ligated to Mn1 and Ca (W1, W2, W3 and W4) are part of an extended H-bonding network reaching from the redox-active TyrZ towards the Glu65/Glu312 ‘proton gate’. H-bond distances of less than or equal to 3.4 Å are indicated by dashed yellow lines. The depicted amino-acid sidechain either are part of the PsbA (D1), PsbC (CP43) or PsbD (D2) subunits.
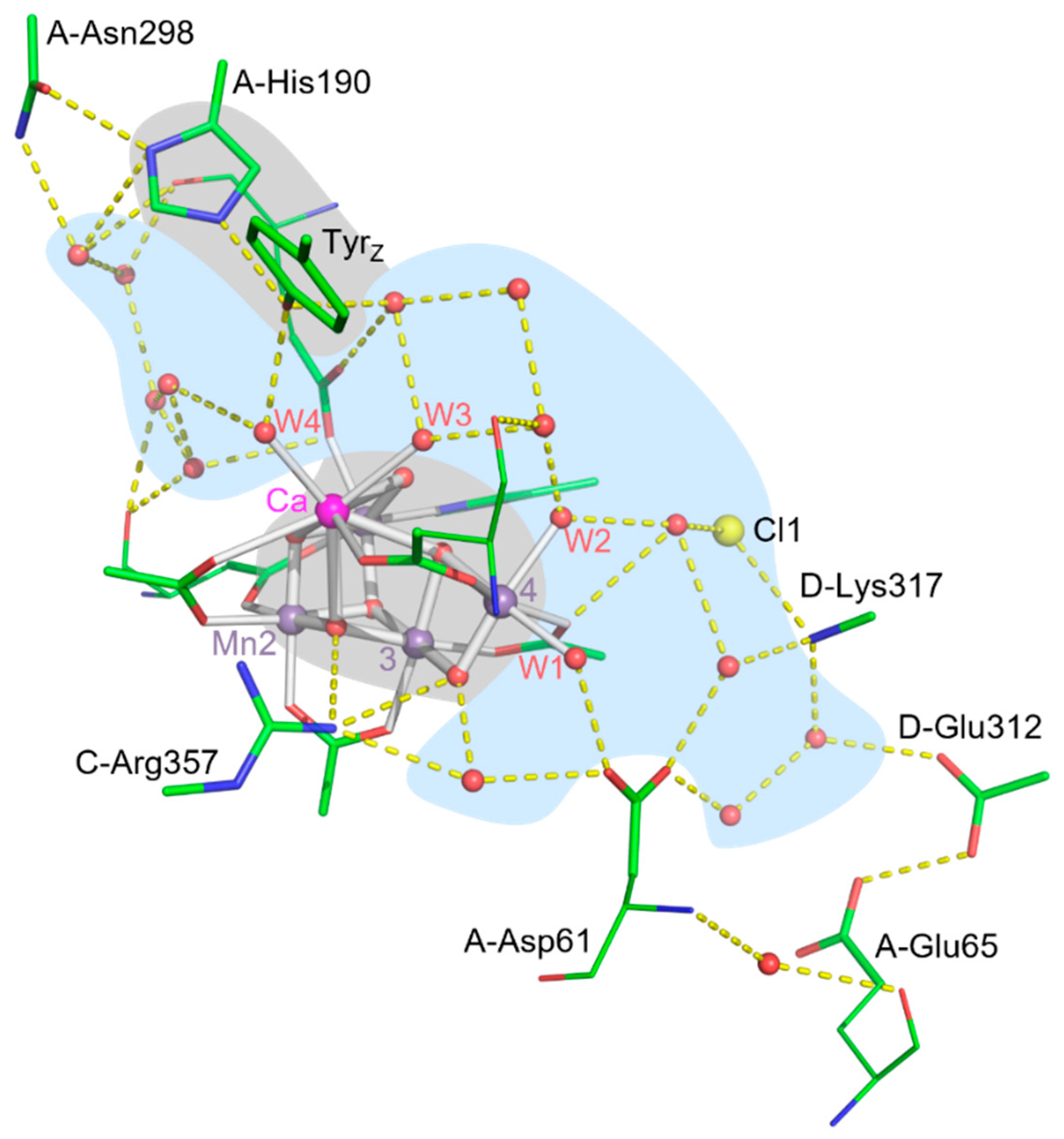
Figure 2.
Scheme of the electron transfer between O6 and TyrZ with accompanying proton rearrangements as reported in Greife et al. [10]. Alongside the ET, resulting in oxidation of O6, three protons move within an H-bond (from H-bond donor to H-bond acceptor) shifting the protonation by about 7 Å from O6 to Asp61. This proton-coupled electron transfer deprotonates and oxidizes O6, thereby allowing for the subsequent peroxide formation between O5 and O6. The ET step is coupled further to the movement of a fourth proton from His190 to TyrZ (not shown).
Figure 2.
Scheme of the electron transfer between O6 and TyrZ with accompanying proton rearrangements as reported in Greife et al. [10]. Alongside the ET, resulting in oxidation of O6, three protons move within an H-bond (from H-bond donor to H-bond acceptor) shifting the protonation by about 7 Å from O6 to Asp61. This proton-coupled electron transfer deprotonates and oxidizes O6, thereby allowing for the subsequent peroxide formation between O5 and O6. The ET step is coupled further to the movement of a fourth proton from His190 to TyrZ (not shown).
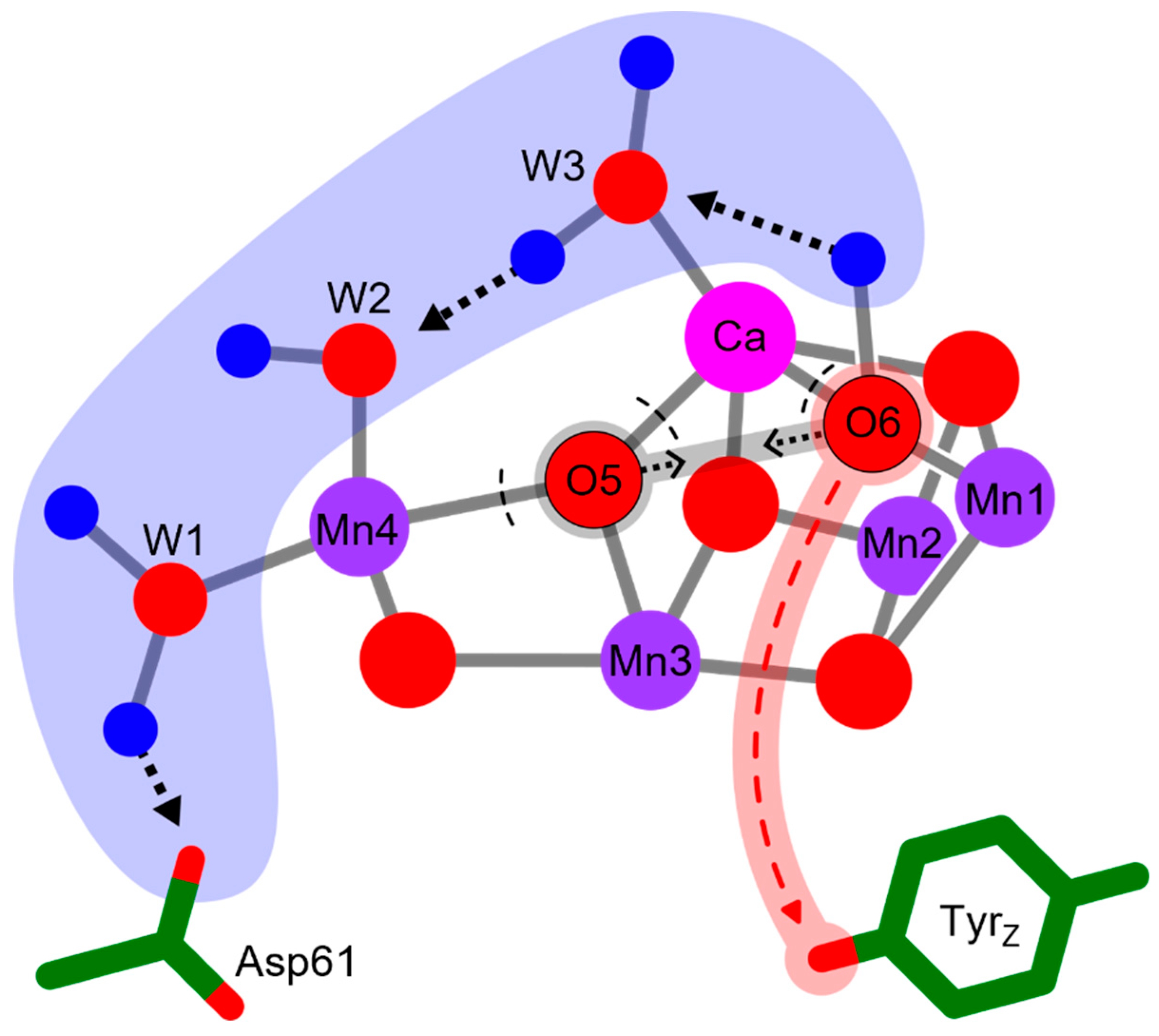
Figure 3.
Shortest distances between the sidechain of Tyr161 (TyrZ) and the first-sphere ligand atoms (all oxygen atoms) of the Mn and Ca atoms of the Mn4Ca cluster in the S3 state (PDB 8EZ5) [11]. For seven distances, connecting lines are indicated starting at four oxygen atoms, which are W4, W3, O6 an one carboxylate oxygen of the ligating Asp170. The corresponding seven distances and further distances are indicated in Table 1 with comparative distances for the structure PDB 6DHO (2nd flash, predominantly S3 state)[16].
Figure 3.
Shortest distances between the sidechain of Tyr161 (TyrZ) and the first-sphere ligand atoms (all oxygen atoms) of the Mn and Ca atoms of the Mn4Ca cluster in the S3 state (PDB 8EZ5) [11]. For seven distances, connecting lines are indicated starting at four oxygen atoms, which are W4, W3, O6 an one carboxylate oxygen of the ligating Asp170. The corresponding seven distances and further distances are indicated in Table 1 with comparative distances for the structure PDB 6DHO (2nd flash, predominantly S3 state)[16].
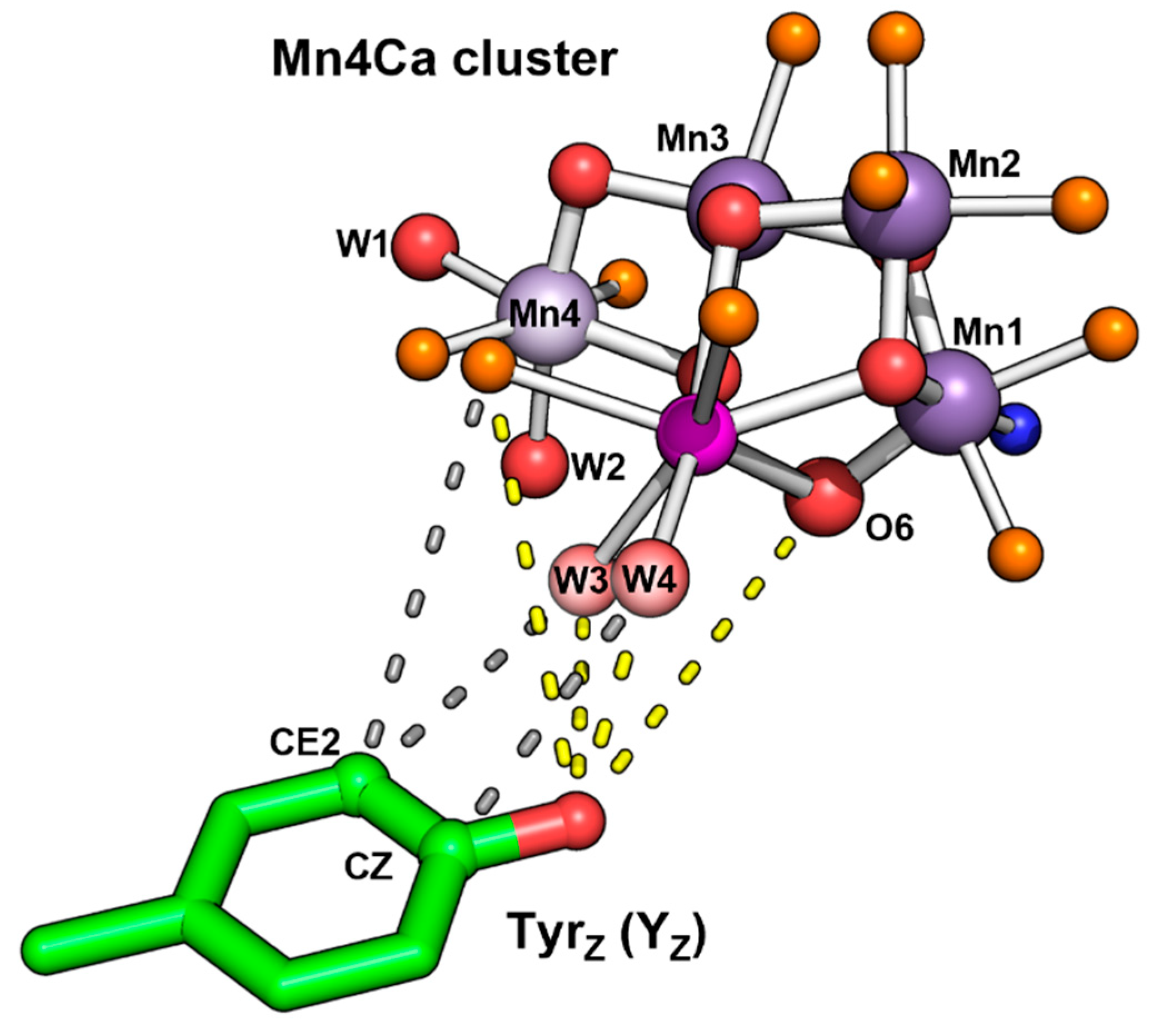
Figure 4.
Two-dimensional representation of the resolved extended H-bonded water (blue) and amino acid (red) network within 20 Å of the Mn4Ca cluster. Distances less than 2.7 Å are indicated with a solid line while distances less than 3.3 Å are dashed. A black line marks an association with a sidechain while a grey line indicates a backbone connection. Colored borders of water molecules represent participation in water channels possibly used for water and proton transport [18,33,34]. Only oxygen and nitrogen atoms were included in the analysis (H-bond distances measured between O/N donor and O/N acceptor). While the first-sphere ligands of the Mn4Ca cluster were included in the analysis, connections within are not shown for simplicity. Numbering is based on the structure PDB 8EZ5 and Supplementary table s1 of ref.[11]. Waters not present in the table are denoted by their chain and residue ID number.
Figure 4.
Two-dimensional representation of the resolved extended H-bonded water (blue) and amino acid (red) network within 20 Å of the Mn4Ca cluster. Distances less than 2.7 Å are indicated with a solid line while distances less than 3.3 Å are dashed. A black line marks an association with a sidechain while a grey line indicates a backbone connection. Colored borders of water molecules represent participation in water channels possibly used for water and proton transport [18,33,34]. Only oxygen and nitrogen atoms were included in the analysis (H-bond distances measured between O/N donor and O/N acceptor). While the first-sphere ligands of the Mn4Ca cluster were included in the analysis, connections within are not shown for simplicity. Numbering is based on the structure PDB 8EZ5 and Supplementary table s1 of ref.[11]. Waters not present in the table are denoted by their chain and residue ID number.
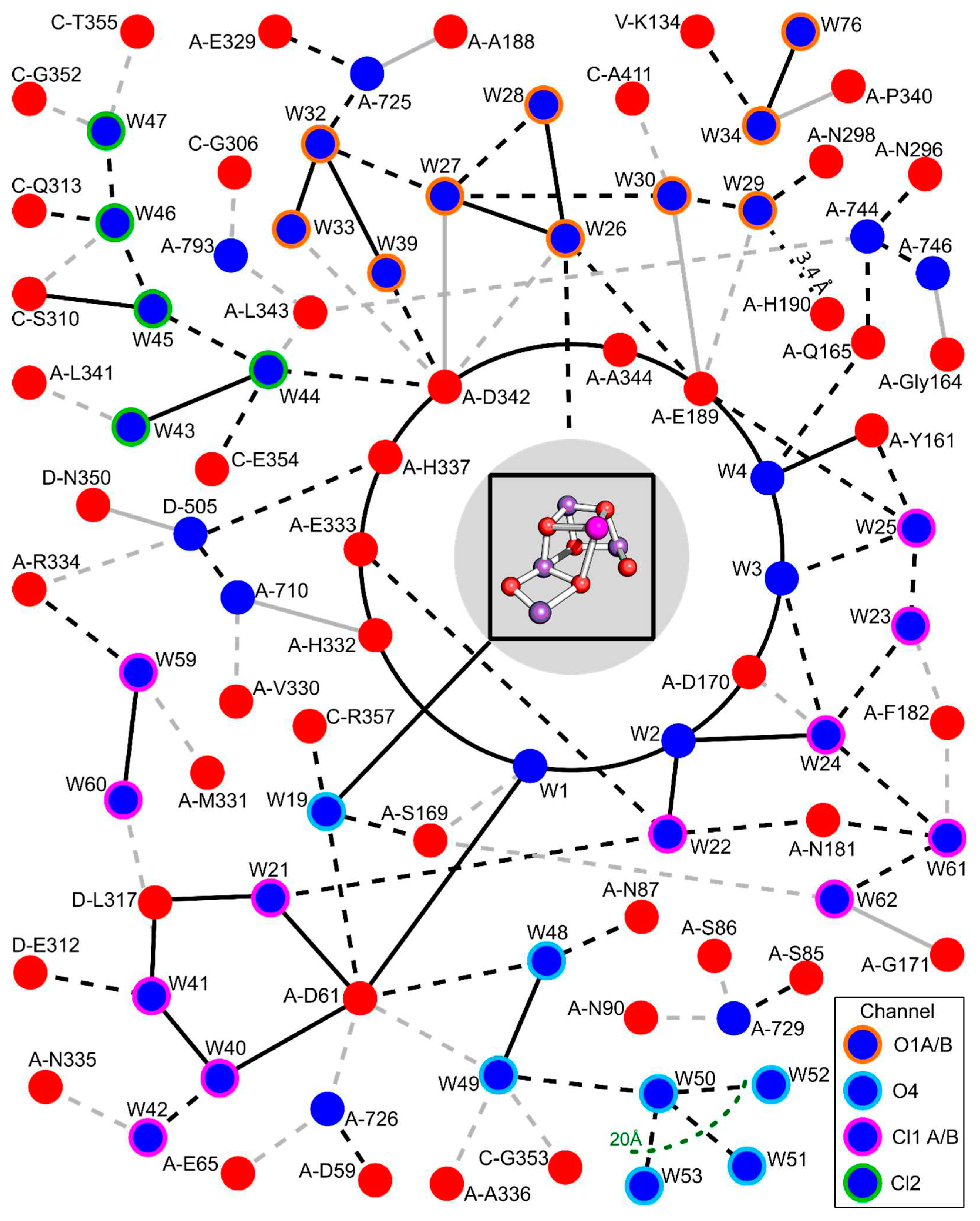

Table 1.
Distances between atoms of the Mn4Ca cluster and the nearest atoms of TyrZ for two S3-state coordinates sets (8EZ5 of ref.[11], 6DHO of ref.[16]).
| Atom | Distance to YZO (Å) | |
| 8EZ5 | 6DHO | |
| W4 | 2.7 | 2.9 |
| 3.6 (to CZ Ring) | 3.7 | |
| W3 | 3.9 | 4 |
| 4.1 (to CE2 Ring) | 4.2 | |
| Ca | 4.6 | 4.7 |
| (CA1-Asp170) OD2 | 4.9 (to CE2 Ring) | 5.1 |
| 5.3 | 5.4 | |
| (CA1-Ala344) O | 5.6 | 5.7 |
| O6 | 5.7 | 5.7 |
| (Mn4-Asp170) OD1 | 5.8 (to CE2 Ring) | 5.8 |
| O1 | 6.1 | 6.2 |
| (Mn1-Glu189) OE2 | 6.2 | 6.1 |
| Mn1 | 7.0 | 7.1 |
| Mn4 | 7.4 | 7.4 |
| Mn2 | 7.7 | 7.7 |
| Mn3 | 8.0 | 8.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



