Submitted:
09 August 2023
Posted:
10 August 2023
You are already at the latest version
Abstract
Mallard (Anas platyrhynchos) are currently one of the most popular species in rare bird breeding in several southern provinces of China, but there are very few studies on the gut microbial communities of domestic and wild mallards. In this study, 16S rRNA gene high-throughput sequencing technology was used to compare the composition and diversity of gut microbial communities in domestic and wild mallards. Alpha diversity analysis revealed significant differences in the gut microbial communities of domestic and wild mallards. Beta diversity analysis showed that the two groups of stool samples were mostly separated on the principal coordinate analysis (PCOA) plot. In domestic mallards, Firmicutes (67.97%) was the most abundant bacterial phylum, followed by Proteobacteria (24.48%), Bacteroidetes (3.13%), Fusobacteria (2.21%), and Actinobacteria (1.10%). The dominant bacterial phyla in wild mallards were Firmicutes (79.02%), Proteobacteria (12.85%), Fusobacteria (3.37%), and Bacteroidetes (2.79%). At the genus level, a total of 10 dominant genera (Streptococcus, Enterococcus, Clostridium, Lactobacillus, Soilbacillus, Bacillus, Acinetobacter, Comamonas, Shigella, and Cetobacterium) with an average relative abundance greater than 1% were detected in the fecal samples of both groups. The average relative abundance of five potential pathogens (Streptococcus, Enterococcus, Acinetobacter, Comamonas, and Shigella) was higher in domestic mallards than in wild mallards. The enrichment of pathogenic bacteria in the intestinal tract of domestic mallards should be of sufficient concern.
Keywords:
mallard (Anas platyrhynchos)
; gut microbiome
; composition and diversity
; 16S rRNA gene high-throughput sequencing
; potential pathogens
1. Introduction
The collection of all microbial species and their associated genetic material in the host gut is called the gut microbiome [1]. The gut microbiota is considered the "second genome" of the animal and is intricately linked to the host. An increasing number of studies have shown that gut microbes play important physiological functions, such as nutrient absorption [2], energy supply and storage [3], development and maintenance of the intestinal mucosal barrier [4], establishment of the immune system, and resistance to pathogens [5]. In addition, gut microbes have been associated with the development of host obesity [6], cancer [7], diabetes [8], and other neurological diseases [9]. Therefore, the study of gut microbes is of physiological importance.
Birds are an important member of the biodiversity family and an important indicator species in ecosystems. More than 10,000 species of birds have been identified worldwide [10], more than twice as many as mammals. Research on wild birds' gut microbes lags far behind that of mammals because of factors such as the difficulty of sampling wild birds under natural conditions and the difficulty of preserving samples. Birds have complex and unique foraging strategies, physiological characteristics, and phylogenetic relationships [11], and their gut microbes may be influenced by both intrinsic and extrinsic factors [12]. Among these, environment and food are considered to be the main factors shaping the diversity of gut microbial communities in birds, and they strongly influence the composition of the gut microbiota. For example, gut microbial composition and diversity tend to be more similar between the same species in the same habitat than between species that are distantly related [13,14]. Significant differences in gut microbial communities may also exist between the same species in different geographical locations [15].
With the rapid development of high-throughput sequencing technologies, coupled with efficient bioinformatics analysis tools, it has been possible to conduct in-depth research on the vertebrate microbiome. An earlier report on the application of high-throughput sequencing technology to the intestinal flora of birds was seen in 2013 in a study on the diversity of the cecum flora of emus (Dromaius novaehollandiae) [16]. Current research on gut microbes in captive and wild birds is focused on a few species of economic and conservation value. For example, Wang et al. showed significant differences in microbial communities between wild and farmed swan geese (Anser cygnoides) in a study of their gut microbes [17]. In addition, Wang et al. also compared the intestinal microbial communities of bar-headed goose (Anser indicus) in three different rearing modes: artificial breeding, semi-artificial breeding, and wild, and found that the highest diversity and richness of gut microbial communities were found in the semi-artificial breeding group, followed by the wild group, and finally the artificial breeding group [18]. Jiang et al. studied the gut microbial composition of wild and captive Chinese monals (Lophophorus ihuysii), showing that the alpha diversity of the gut microbial community was significantly higher in the wild group than in the captive group, and that the core bacterial groups of the two groups differed significantly at the level of phylum, class, order, and family [19]. Xie et al. conducted research on three groups of red-crowned cranes (Grus japonensis) and showed that captive cranes had the greatest gut microbial alpha diversity, while wild cranes had the least gut microbial alpha diversity [20]. In summary, we can make the hypothesis that there may be significant differences in the intestinal microbiota of the same species (mallards) in different breeding environments (domestic and wild).
The wild mallard is one of the ancestors of domestic ducks in China [21], has a long history of domestication and breeding, and is one of the most popular species in rare wildfowl farming in China. Domestic mallards have the advantages of strong disease resistance, wide adaptability, diversified diet, high feed remuneration, and a short feeding cycle. Its meat is delicious, nutritious, low in fat, and high in protein content, so it is very popular in domestic and foreign markets. In this paper, 16S rRNA gene high-throughput sequencing technology was used to compare the composition and diversity of the gut microbiota of domestic and wild mallards. The study of microbial communities in the intestinal tract of wild and domestic mallards is not only beneficial to the expansion of wild populations and artificial domestication but also provides a basic basis for the health and scientific breeding of these ducks, which has important ecological and economic values.
2. Materials and Methods
2.1. Wild Mallards Fecal Sample Collection
Eight wild mallard fecal samples were collected from Caohai National Nature Reserve (26.84921854° N, 104.28497294° E), Guizhou Province, China, in March 2022. First, use binoculars to observe the range of the mallards and wait for the ducks to fly away to collect fresh droppings immediately. To avoid collecting samples from the same mallards, samples were taken at a minimum distance of 5 m apart, and only the middle part of the feces was collected. The stools were collected in sterile 15 mL centrifuge tubes filled with ethanol [22] and transported to the laboratory for freezing and storage at -80 °C.
2.2. Domestic Mallards Fecal Sample Collection
Twelve fecal samples of domesticated mallards were collected from June-August 2022 at Guangxi University. Sampling and preservation methods were as above.
2.3. Fecal DNA Extraction and PCR Amplification
Genomic DNA was extracted by CTAB, and the purity and concentration of DNA were checked by 1% agarose gel electrophoresis, take an appropriate amount of sample in a centrifuge tube and dilute the sample to 1 ng/ µL with sterile water. PCR amplification of the V3-V4 variable region was performed using 341F (5'-CCTAYGGGRBGCASCAG-3') and 806R (5'-GGACTACNNGGGTATCTAAT-3') primers. PCR reaction system (30µL): Phusion Master Mix (2×) 15 µL; Forward Primer (1 µM/µL) 1 µL; Reverse Primer (1 µM/µL) 1µL; gDNA (1 ng/µL) 10 µL; H2O 2 µL. Reaction procedure: 98 ℃ pre-denaturation for 1min; 30 cycles including (98 ℃, 10 s; 50 ℃, 30 s; 72 ℃, 30 s); 72℃, 5 min.
2.4. Mixing and Purification of PCR Products
The PCR products were purified by agarose gel electrophoresis using 1 x TAE 2% concentration, and the target bands were recovered using the Universal DNA (TianGen, China) purification recovery kit.
2.5. Libraries Generated and IIIumina NovaSeq Sequencing
Sequencing libraries were generated using the NEB Next® Ultra DNA Library Prep Kit (Illumina, USA) following the manufacturer's recommendations, and index codes were added. The library quality was assessed on the Agilent 5400 (Agilent Technologies Co., Ltd., USA). At last, the library was sequenced on an Illumina NovaSeq platform, and 250 bp paired-end reads were generated.
2.6. Data Analysis
The analysis was conducted by following the "Atacama soil microbiome tutorial" of Qiime2docs along with customized program scripts (https://docs.qiime2.org/2019.1/). Briefly, raw data FASTQ files were imported into the format that could be operated by the QIIME2 system using QIIME Tools import program. Demultiplexed sequences from each sample were quality filtered and trimmed, de-noised, and merged, and then the chimeric sequences were identified and removed using the QIIME2 dada2 plugin to obtain the feature table of an amplicon sequence variant (ASV) (Callahan et al. 2016). The QIIME2 feature-classifier plugin was then used to align ASV sequences to a pre-trained GREENGENES 13_8 99% database (trimmed to the V3V4 region bound by the 338F/806R primer pair) to generate the taxonomy table (Bokulich et al. 2018). Any contaminating mitochondrial and chloroplast sequences were filtered using the QIIME2 feature-table plugin. Characteristic sequence level Alpha diversity indices, including Chao1, and Shannon index. Beta diversity indices, including unweighted UniFrac and weighted UniFrac indices, were used to assess the structural variability of microbial communities between samples and were subsequently presented using PCoA plots. In addition, we also applied the PICRUSt software to predict the possible functional composition of the microbial community. Unless otherwise noted, the parameters applied in the above analysis are the default settings.
3. Results
3.1. 16S rRNA Gene Data
Using the DADA2 plug-in of Qiime2 to quality control, denoise, merged, and de-chimerism all raw sequences of all samples, we obtained 1024791 high-quality sequences (Supplementary Table S1), with an average of 51239 ± 12487 sequences per sample (mean ± SD). A total of 7555 ASVs were identified based on the 99% sequence similarity threshold, and these ASVs were classified into 38 phyla, 101 classes, 168 orders, 236 families, and 409 genera (Supplementary Table S2). The rarefaction curve indicated that new species could not be detected by continuing to increase the sequencing depth (Figure 1A). As shown in Figure 1B, the Shannon diversity curve gradually reached a plateau as the sequencing depth increased, which indicates that the sequencing depth is sufficient to reflect the diversity of the samples and the data are suitable for subsequent analysis.
3.2. Alpha Diversity and Beta Diversity Analyses
We analyzed the diversity and richness of microbial communities in our samples by Shannon index (Figure 2A) and Chao1 index (Figure 2B). The results showed that there were significant differences between the diversity and richness of the gut microbial communities of the two groups of mallards (Shannon index: p=0.02; Chao1 index: p=0.03), and the diversity and richness of the gut microbial communities of the WM group were significantly higher than those of the DM group (p<0.05).
The composition of microbial communities was compared between samples by principal coordinate analysis (PCOA). According to the weighted (Figure 3A) and unweighted UniFrac (Figure 3B) distances, the two groups of samples were mostly separated. This suggests that gut microbial communities differ between the same species.
3.3. Comparison of The Intestinal Microflora of Two Groups of Mallards at The Phylum and Genus Levels
A total of 38 bacterial phyla were identified in the 20 stool samples, and Figure 4A shows the average relative abundance of bacterial phyla in the top 10 in both groups. In the wild mallard (WM) group, the total sequences were identified as four major phyla, Firmicutes were absolutely dominant with an average relative abundance of 79.02%, followed by Proteobacteria (12.85%), Fusobacteria (3.37 %), Bacteroidetes (2.79%). In contrast, in the domestic mallard (DM) group, Firmicutes was also the most abundant, with an average relative abundance of 67.97%, followed by Proteobacteria (24.48%), Bacteroidetes (3.13%), Fusobacteria (2.21%), and Actinobacteria (1.10%). Table 1 lists the bacterial phyla with an average relative abundance greater than 1% in both groups. The average relative abundance of the Firmicutes and Fusobacteria in the WM group was higher than that in the DM group, but the average relative abundance of the Proteobacteria, Bacteroidetes, and Actinobacteria in the WM group was lower than that in the WM group.
At the genus level, a total of 409 bacterial genera were identified from the 20 sample sequences. The top 10 bacterial genera in terms of average relative abundance are listed in Figure 4B, and sequences that could not be classified as known genera are indicated as "unclassified". The dominant bacterial genera with an average relative abundance greater than 1% are listed in Table 2. In the DM group, the average relative abundance of Streptococcus, Enterococcus, Acinetobacter, Comamonas, and Shigella is higher than that in the WM group. In the WM group, the average relative abundance of Clostridium, Lactobacillus, Cetobacterium, Solibacillus, and Bacillus is higher than that in the DM group.
LEfse analyzed those strains that contributed more to the differences. At the phylum level (Figure 5), the biomarkers with significant differences in the wild group were the Deferribacteres (p < 0.05), the Tenericutes (p < 0.001), and the Fusobacteria (p < 0.01). At the genus level (Figure 5), the biomarkers with significant differences in the wild group were Lactobacillus (p < 0.001), Bacillus (p < 0.05), Cetobacterium (p < 0.001), Clostridium (p < 0.05), Solibacillus (p < 0.001), and Clostridium (p < 0.001). There was no significant difference in biomarkers at the phylum and genus levels for the domestic group.
3.4. Prediction of Gut Microbiome Function
We predicted gut microbial function in domestic and wild mallards by PICRUSt2 analysis and annotated 47 functional pathways at KEGG level 2. Among the gut microorganisms of domestic mallards, carbohydrate metabolism (11.54%), amino acid metabolism (10.02%), metabolism of cofactors and vitamins (9.10%), metabolism of other amino acids (7.97%), global and overview maps (5.56%), replication and repair (5.38%), lipid metabolism (5.28%), and biosynthesis of other secondary metabolites (5.11%) of the KEGG pathway were very abundant. Similarly, the gut microorganisms of wild mallards had high abundances in Amino acid metabolism (10.52%), Carbohydrate metabolism (10.29%), Metabolism of cofactors and vitamins (9.76%), Metabolism of other amino acids (7.97%), Lipid metabolism (6.22%), Global and overview maps (5.54%), and Replication and repair (5.20%), with a high abundance of KEGG pathways. Figure 6 Shows the top 10 level 2 pathways in the DM and WM groups in terms of predicted abundance of gut microbial function.
Among 47 level 2 functional pathways, the two groups of mallards differed significantly in Cancer: overview, Digestive system, Glycan biosynthesis and metabolism, and Lipid metabolism (Figure 7). The abundance of these four different functional pathways was higher in wild mallards than in domestic mallards.
4. Discussion
The composition and diversity of the bacterial community are considered important parts of the gut microbiome, and gut microbes, as a dynamic ecosystem, are easily influenced by multiple factors. In most of the available studies, food is considered to be the underlying cause of the differences in the gut microbial community. Birds in different locations feed on different foods, and microorganisms ingested with the food may be one of the main pathways for microbial colonization of the gastrointestinal tract of birds [24]. Environmental spatial heterogeneity plays a dominant role in the formation of the gut microbial community in birds, sometimes even exceeding genetic factors [25]. Birds are exposed to different microorganisms through the environmental conditions of their habitat (including food, water, soil, nesting, and social activities), which are potential sources of microorganisms in the gastrointestinal tract of birds.
In this paper, we studied the gut microbial communities of domestic and wild mallards based on 16S rRNA gene high-throughput sequencing technology. Alpha diversity analysis confirmed significant differences in diversity and richness of the gut microbiota between domestic and wild mallards. This difference may be related to the fact that domestic and wild mallards live in different habitats and feed on different diets. This result is consistent with the study by Jiang et al. [19] on the gut microbiota of wild and captive Chinese monals. PCOA revealed that the two groups of mallards gut microbial communities were mostly separated. The higher specificity of gut microbes in the two groups of mallards indicated that there was no correlation between close evolutionary relationships and the composition of gut microbial communities. From this, we hypothesize that food and environmental factors have a greater influence on the gut microbial community of mallards than genetic factors.
The gut microbial community composition of the two groups of mallards showed that Firmicutes, Proteobacteria, Bacteroidetes, Fusobacteria, and Actinobacteria were the dominant phyla, with an average relative abundance of more than 1%. These dominant phyla are similar to studies of gut microbiota in other wild birds, such as the bar-headed Goose [26], whooper Swan (Cygnus cygnus) [27], black-necked Crane (Grus nigricollis) [28], hoatzin (Opisthocomus hoazin) [29], and turkey (Meleagris gallopavo) [30]. Firmicutes are the most prevalent and common bacterial phylum among all vertebrates. In mice and humans, Firmicutes have been shown to positively correlate with the ability to obtain energy and nutrient absorption from food [31,32]. Currently, we have not found any research on the function of Firmicutes in wild birds, but studies in domestic chickens have found a positive correlation between the abundance of Firmicutes and body weight gain and immune function. We speculate that Firmicutes may have similar roles in mammals and birds [33,34].
Proteobacteria were the second-most abundant bacterial phylum in both stool samples. The function of Proteobacteria in the gut of wild birds is unknown, and further studies are needed to confirm it. According to previous studies, Proteobacteria have multiple physiological functions, are able to utilize a large amount of carbon sources and play an important role in the energy accumulation of the host [35,36,37]. The phylum Proteobacteria include five major groups (α, β, γ, δ, ε) that vary greatly in their occurrence and function inside and outside the gastrointestinal tract, with proteobacteria being involved in the degradation of acidic herbicides [38], suggesting a possible detoxification role in the gastrointestinal tract of mallards.
Actinobacteria are the dominant bacterial phylum of the domestic mallard group. Turnbaugh et al. [39] showed that increased abundance of Actinobacteria was strongly associated with obesity and that 75% of obesity-enriched genes (involved in carbohydrate, lipid, and amino acid metabolism) were from Actinobacteria. In addition, the abundance of actinomycetes in stool samples from young children was reported to be positively correlated with the intake of barley dietary fiber [40]. Therefore, the physiological functions of actinomycetes in the gastrointestinal tract of mallards may be similar. However, more in-depth studies are needed to elucidate the role of specific members of the Actinobacteria phylum in the nutrition and health of mallards.
In this study, there were 10 bacterial genera with an average relative abundance greater than 1% (Table 2), which were distributed in three bacterial phyla. Among them, there are six genera (Streptococcus, Enterococcus, Clostridium, Lactobacillus, Solibacillus, and Bacillus) belongin to Firmicutes, Proteobacteria has three genera (Acinetobacter, Comamonas, and Shigella), and only one genus (Cetobacterium) belongs to Fusobacteria. Five potential pathogens (Streptococcus, Enterococcus, Acinetobacter, Comamonas, and Shigella) were identified in the DM group with a higher mean relative abundance than in the WM group. Streptococcosis is a general term for a variety of animal and human infections caused primarily by β-hemolytic streptococci, which can cause many serious diseases such as meningitis and toxic shock in humans [41]. Streptococcus is highly susceptible, and a variety of poultry (chickens, ducks, geese, and chickens) can be infected; the main clinical manifestation is acute septicemia, and some are chronic infections. Enterococcus are opportunistic pathogens prevalent in the gastrointestinal tract of humans and a variety of animals (mammals, reptiles, birds, and some invertebrates) [42] and can cause serious infections such as endocarditis, septicemia, and urinary tract infections [43]. They have become the third most prevalent nosocomial pathogen in the world [44]. Acinetobacter is receiving increasing attention because of its strong resistance to antibacterial drugs. The environment, soil, and animals are the natural habitats of Acinetobacter, which infects humans by contaminating food and water. Acinetobacter has been isolated from various animal sources, including birds [45], poultry (chicken, turkey), cattle, pigs [46], fish [47], etc. Acinetobacter is associated with diseases such as septicemia, pulmonary infections, meningitis, and diarrhea in humans and animals, with a mortality rate of about 20-60% [48]. Comamonas is a gram-negative pathogenic bacterium that causes steroid hormone degradation [49,50], is primarily associated with bacteremia [51], and occasionally causes low-virulence disease in humans and animals [52]. Shigella is a common and potentially pathogenic enteric pathogen that can cause bacterial food poisoning, typhoid fever, and uremia but is also present in small amounts in the feces of healthy individuals [53]. Comamonas, Acinetobacter, and Shigella all belong to Proteobacteria, and it has been shown that an increase in the relative abundance of Proteobacteria can lead to the development of intestinal diseases and reduced production performance in chickens [54,55]. Based on previous studies on the pathogenicity of these five conditional pathogens in humans and other animals, we cannot yet infer whether they are also pathogenic to domestic mallards; however, we can confirm that these pathogenic genera detected are conditional pathogenic microorganisms in the gut of domestic mallards.
Lactobacillus and Bacillus are two potential probiotics in the wild mallards, and Lactobacillus can enhance host digestion and inhibit the development of certain diseases [56]. Antibiotics produced by Bacillus have a broad antibacterial spectrum, can bind lipopolysaccharides, and neutralize endotoxin. Probiotics prepared with Bacillus play an important role in the treatment of intestinal flora disorders, candida infections, and wound infections. It is well known that wild populations face extreme survival pressure under natural conditions and are more resistant to disease than domestic populations. The enrichment of domestic mallards with pathogenic intestinal bacteria indicates that their health status is of concern and that some measures should be taken to prevent the spread of zoonotic infectious diseases.
5. Conclusions
It is important to study wild populations of species associated with poultry livestock to assess the potential health risks associated with these wild populations and to complete our ecological understanding of some pathogenic microorganisms. In this study, we analyzed the basic conditions of the intestinal microbiota of domestic and wild domestics using high-throughput sequencing of 16S rRNA genes. Significant differences were found in the diversity and richness of the gut microbial communities of the two groups of mallards, which may be closely related to the different food intake and different living environments of domestic and wild mallards. PCOA showed that gut microbial communities differed between the same species. From this, we hypothesize that differences in food and habitat environments may be potential drivers of significant differences in the gut microbial communities of domestic and wild mallards. This finding confirms our previous hypothesis that there are significant differences in the gut microbiota in different living environments for the same species. In addition, the enrichment of pathogenic bacteria in the intestinal tract of domestic mallards should attract enough attention. This study provides basic information for the conservation of wild populations and the prevention and control of diseases in domesticated mallards.
Supplementary Materials
The following supporting information can be downloaded at: https://doi.org/10.5281/zenodo.8218881, Table S1: Statistical table of sequencing quantity per sample; Table S2: ASV classification table for two groups of mallards.
Author Contributions
Conceptualization, Y.H. and L.Y.; Sample collection, C.D.; Statistical analysis, Y.H.; Methodology, Y.H.; Software, Y.H.; Investigation, M.Z.; Resources, M.Z. and C.D.; Data curation, Y.H., M.Z. and L.Y.; Writing—original draft, Y.H.; Project administration, L.Y.; Funding acquisition, C.D.; Supervision, L.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant No. 32060310).
Institutional Review Board Statement
Ethical review and approval were waived for this study, since only fecal samples were collected and analyzed.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data have been submitted to the NCBI Sequence Read Archive (BioProject ID: PRJNA995419).
Acknowledgments
We would like to thank the Caohai National Nature Reserve in Guizhou Province for supporting our work in the wild.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grond, Kirsten.; Sandercock, Brett K.; Jumpponen, Ari.; Zeglin, Lydia H. The avian gut microbiota: community, physiology and function in wild birds. Journal of Avian Biology. 2018. 49(11). [CrossRef]
- Wu, Gary D.; Chen, Jun.; Hoffmann, Christian.; Bittinger, Kyle.; Chen, Ying-Yu.; Keilbaugh, Sue A.; Bewtra, Meenakshi.; Knights, Dan.; Walters, William A.; Knight, Rob.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science. 2011. 334(6052): p. 105-108.
- 3. Turnbaugh, Peter J.; Gordon, Jeffrey I. The core gut microbiome, energy balance and obesity. Journal of Physiology-London. 2009. 587(17): p. 4153-4158.
- Margolis, Kara Gross.; Gershon, Michael David.; Bogunovic, Milena. Cellular Organization of Neuroimmune Interactions in the Gastrointestinal Tract. Trends in Immunology. 2016. 37(7): p. 487-501. [CrossRef]
- Ahern, Philip P.; Faith, Jeremiah J.; Gordon, Jeffrey I. Mining the Human Gut Microbiota for Effector Strains that Shape the Immune System. Immunity. 2014. 40(6): p. 815-823. [CrossRef]
- Sanmiguel, Claudia.; Gupta, Arpana.; Mayer, Emeran A. Gut Microbiome and Obesity: A Plausible Explanation for Obesity. Current obesity reports. 2015. 4(2): p. 250-61. [CrossRef]
- Pevsner-Fischer, Meirav.; Tuganbaev, Timur.; Meijer, Mariska.; Zhang, Sheng-Hong.; Zeng, Zhi-Rong.; Chen, Min-Hu.; Elinav, Eran. Role of the microbiome in non-gastrointestinal cancers. World journal of clinical oncology. 2016. 7(2): p. 200-13. [CrossRef]
- Forslund, Kristoffer.; Hildebrand, Falk.; Nielsen, Trine.; Falony, Gwen.; Le Chatelier, Emmanuelle.; Sunagawa, Shinichi.; Prifti, Edi.; Vieira-Silva, Sara.; Gudmundsdottir, Valborg.; Pedersen, Helle Krogh.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015. 528(7581): p. 262-+.
- Montiel-Castro, Augusto J.; Gonzalez-Cervantes, Rina M.; Bravo-Ruiseco, Gabriela.; Pacheco-Lopez, Gustavo. The microbiota-gut-brain axis: neurobehavioral correlates, health and sociality. Frontiers in integrative neuroscience. 2013. 7: p. 70-70. [CrossRef]
- Prum, Richard O.; Berv, Jacob S.; Dornburg, Alex.; Field, Daniel J.; Townsend, Jeffrey P.; Lemmon, Emily Moriarty.; Lemmon, Alan R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature. 2015. 526(7574): p. 569-U247. [CrossRef]
- Alexandra Garcia-Amado, M.; Shin, Hakdong.; Sanz, Virginia.; Lentino, Miguel.; Margarita Martinez, L.; Contreras, Monica.; Michelangeli, Fabian.; Dominguez-Bello, Maria Gloria. Comparison of gizzard and intestinal microbiota of wild neotropical birds. Plos One. 2018. 13(3). [CrossRef]
- Ruiz-Rodriguez, Magdalena.; Martin-Vivaldi, Manuel.; Martinez-Bueno, Manuel.; Jose Soler, Juan. Gut Microbiota of Great Spotted Cuckoo Nestlings is a Mixture of Those of Their Foster Magpie Siblings and of Cuckoo Adults. Genes. 2018. 9(8).
- Eckburg, PB.; Bik, EM.; Bernstein, CN.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, SR.; Nelson, KE.; Relman, DA. Diversity of the human intestinal microbial flora. Science. 2005. 308(5728): p. 1635-1638. [CrossRef]
- Yang, Yuzhan.; Deng, Ye.; Cao, Lei. Characterising the interspecific variations and convergence of gut microbiota in Anseriformes herbivores at wintering areas. Scientific Reports. 2016. 6. [CrossRef]
- Zhang, Jiachao.; Guo, Zhuang.; Xue, Zhengsheng.; Sun, Zhihong.; Zhang, Menghui.; Wang, Lifeng.; Wang, Guoyang.; Wang, Fang.; Xu, Jie.; Cao, Hongfang.; et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. Isme Journal. 2015. 9(9): p. 1979-1990. [CrossRef]
- Bennett, Darin C.; Tun, Hein Min; Kim, Ji Eun; Leung, Frederick C.; Cheng, Kimberly M. Characterization of cecal microbiota of the emu (Dromaius novaehollandiae). Veterinary Microbiology. 2013. 166(1-2): p. 304-310. [CrossRef]
- Wang, Wen.; Zheng, Sisi.; Sharshov, Kirill.; Cao, Jian.; Sun, Hao.; Yang, Fang.; Wang, Xuelian.; Li, Laixing. Distinctive gut microbial community structure in both the wild and farmed Swan goose (Anser cygnoides). Journal of Basic Microbiology. 2016. 56(11): p. 1299-1307. [CrossRef]
- Wang, Wen.; Cao, Jian.; Li, Ji-Rong.; Yang, Fang.; Li, Zhuo.; Li, Lai-Xing. Comparative analysis of the gastrointestinal microbial communities of bar-headed goose (Anser indicus) in different breeding patterns by high-throughput sequencing. Microbiological Research. 2016. 182: p. 59-67. [CrossRef]
- Jiang, Dandan.; He, Xin.; Valitutto, Marc.; Chen, Li.; Xu, Qin.; Yao, Ying.; Hou, Rong. Wang, Hairui Gut microbiota composition and metabolomic profiles of wild and captive Chinese monals (Lophophorus lhuysii). Frontiers in Zoology. 2020. 17(1).
- Xie, Yuwei.; Xia, Pu.; Wang, Hui.; Yu, Hongxia.; Giesy, John P.; Zhang, Yimin.; Mora, Miguel A.; Zhang, Xiaowei. Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Scientific Reports. 2016. 6. [CrossRef]
- Li, Hui-Fang.; Zhu, Wen-Qi.; Song, Wei-Tao.; Shu, Jing-Ting.; Han, Wei.; Chen, Kuan-Wei. Origin and genetic diversity of Chinese domestic ducks. Molecular Phylogenetics and Evolution. 2010. 57(2): p. 634-640. [CrossRef]
- Hale, Vanessa L.; Tan, Chia L.; Knight, Rob.; Amato, Katherine R. Effect of preservation method on spider monkey (Ateles geoffroyi) fecal microbiota over 8 weeks. Journal of Microbiological Methods. 2015. 113: p. 16-26. [CrossRef]
- Caterino, MS.; Cho, S.; Sperling, FAH. The current state of insect molecular systematics: A thriving Tower of Babel. Annual Review of Entomology. 2000. 45: p. 1-54. [CrossRef]
- Grond, Kirsten.; Lanctot, Richard B.; Jumpponen, Ari.; Sandercock, Brett K. Recruitment and establishment of the gut microbiome in arctic shorebirds. Fems Microbiology Ecology. 2017. 93(12). [CrossRef]
- Hird, Sarah M.; Carstens, Bryan C.; Cardiff, StevenW.; Dittmann, Donna L.; Brumfield, Robb T. Sampling locality is more detectable than taxonomy or ecology in the gut microbiota of the brood-parasitic Brown-headed Cowbird (Molothrus ater). Peerj. 2014. 2. [CrossRef]
- Dong, Shixiong.; Xu, Shijun.; Zhang, Jian.; Hussain, Riaz.; Lu, Hong.; Ye, Yourong.; Mehmood, Khalid.; Zhang, Hui.; Shang, Peng. First Report of Fecal Microflora of Wild Bar-Headed Goose in Tibet Plateau. Frontiers in Veterinary Science. 2022. 8. [CrossRef]
- Wang, Wenxia.; Huang, Songlin.; Yang, Liangliang.; Zhang, Guogang. Comparative Analysis of the Fecal Bacterial Microbiota of Wintering Whooper Swans (Cygnus Cygnus). Frontiers in Veterinary Science. 2021. 8. [CrossRef]
- Wang, Wen.; Wang, Fang.; Li, Laixing.; Wang, Aizhen.; Sharshov, Kirill.; Druzyaka, Alexey.; Lancuo, Zhuoma.; Wang, Shuoying.; Shi, Yuetong. Characterization of the gut microbiome of black-necked cranes (Grus nigricollis) in six wintering areas in China. Archives of Microbiology, 2020. 202(5): p. 983-993. [CrossRef]
- Godoy-Vitorino, Filipa.; Ley, Ruth E.; Gao, Zhan.; Pei, Zhiheng.; Ortiz-Zuazaga, Humberto.; Pericchi, Luis R.; Garcia-Amado, Maria A.; Michelangeli, Fabian.; Blaser, Martin J.; Gordon, Jeffrey I.; et al. Bacterial community in the crop of the hoatzin, a neotropical folivorous flying bird. Applied and Environmental Microbiology. 2008. 74(19): p. 5905-5912. [CrossRef]
- D'Andreano, Sara.; Bonastre, Armand Sanchez.; Francino, Olga.; Marti, Anna Cusco.; Lecchi, Cristina.; Grilli, Guido.; Giovanardi, Davide.; Ceciliani, Fabrizio. GENETICS AND GENOMICS Gastrointestinal microbial population of turkey (Meleagris gallopavo) affected by hemorrhagic enteritis virus. Poultry Science. 2017. 96(10): p. 3550-3558.
- Jumpertz, Reiner.; Duc Son Le.; Turnbaugh, Peter J.; Trinidad, Cathy.; Bogardus, Clifton.; Gordon, Jeffrey I.; Krakoff, Jonathan. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. American Journal of Clinical Nutrition. 2011. 94(1): p. 58-65. [CrossRef]
- Turnbaugh, Peter J.; Baeckhed, Fredrik.; Fulton, Lucinda.; Gordon, Jeffrey I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host & Microbe. 2008. 3(4): p. 213-223. [CrossRef]
- Liao, X. D.; Ma, G.; Cai, J.; Fu, Y.; Yan, X. Y.; Wei, X. B.; Zhang, R. J. Effects of Clostridium butyricum on growth performance, antioxidation, and immune function of broilers. Poultry Science. 2015. 94(4): p. 662-667.
- Zhang, L.; Li, J.; Yun, T. T.; Qi, W. T.; Liang, X. X.; Wang, Y. W.; Li, A. K. Effects of pre-encapsulated and pro-encapsulated Enterococcus faecalis on growth performance, blood characteristics, and cecal microflora in broiler chickens. Poultry Science. 2015. 94(11): p. 2821-2830. [CrossRef]
- Lu, Hsiao-Pei.; Wang, Yu-bin.; Huang, Shiao-Wei.; Lin, Chung-Yen.; Wu, Martin.; Hsieh, Chih-hao.; Yu, Hon-Tsen. Metagenomic analysis reveals a functional signature for biomass degradation by cecal microbiota in the leaf-eating flying squirrel (Petaurista alborufus lena). Bmc Genomics. 2012. 13. [CrossRef]
- Samanta, Ashis K.; Torok, Valeria A.; Percy, Nigel J. Abimosleh, Suzanne M.; Howarth, Gordon S., Microbial Fingerprinting Detects Unique Bacterial Communities in the Faecal Microbiota of Rats with Experimentally-Induced Colitis. Journal of Microbiology. 2012. 50(2): p. 218-225. [CrossRef]
- Zhu, Lifeng.; Wu, Qi.; Dai, Jiayin.; Zhang, Shanning.; Wei, Fuwen. Evidence of cellulose metabolism by the giant panda gut microbiome. Proceedings of the National Academy of Sciences of the United States of America. 2011. 108(43): p. 17714-17719.
- Liu, Ya-Jun.; Liu, Shuang-Jiang.; Drake, Harold L.; Horn, Marcus A. Alphaproteobacteria dominate active 2-methyl-4-chlorophenoxyacetic acid herbicide degraders in agricultural soil and drilosphere. Environmental Microbiology. 2011. 13(4): p. 991-1009. [CrossRef]
- Turnbaugh, Peter J.; Hamady, Micah.; Yatsunenko, Tanya.; Cantarel, Brandi L.; Duncan, Alexis.; Ley, Ruth E.; Sogin, Mitchell L.; Jones, William J.; Roe, Bruce A.; Affourtit, Jason P.; et al. A core gut microbiome in obese and lean twins. Nature. 2009. 457(7228): p. 480-U7.
- Dominianni, Christine.; Sinha, Rashmi.; Goedert, James J.; Pei, Zhiheng.; Yang, Liying.; Hayes, Richard B.; Ahn, Jiyoung.; Sex, Body Mass Index, and Dietary Fiber Intake Influence the Human Gut Microbiome. Plos One. 2015. 10(4). [CrossRef]
- Yu, Hongjie.; Jing, Huaiqi.; Chen, Zhihai.; Zheng, Han.; Zhu, Xiaoping.; Wang, Hua.; Wang, Shiwen.; Liu, Lunguang.; Zu, Rongqiang.; Luo, Longze.; et al., Human Streptococcus suis outbreak, Sichuan, China. Emerging Infectious Diseases. 2006. 12(6): p. 914-920.
- Torres, Carmen.; Alonso, Carla Andrea.; Ruiz-Ripa, Laura.; Leon-Sampedro, Ricardo.; Del Campo, Rosa.; Coque, Teresa M. Antimicrobial Resistance in Enterococcus spp. of animal origin. Microbiology spectrum. 2018. 6(4).
- Arias, Cesar A.; Murray, Barbara E. The rise of the Enterococcus: beyond vancomycin resistance. Nature Reviews Microbiology. 2012. 10(4): p. 266-278. [CrossRef]
- Fisher, Katie.; Phillips, Carol. The ecology, epidemiology and virulence of Enterococcus. Microbiology-Sgm. 2009. 155: p. 1749-1757. [CrossRef]
- Lopinska, Andzelina.; Indykiewicz, Piotr.; Skiebe, Evelyn.; Pfeifer, Yvonne.; Trcek, Janja.; Jerzak, Leszek.; Minias, Piotr.; Nowakowski, Jacek.; Ledwon, Mateusz.; Betleja, Jacek.; et al., Low Occurrence of Acinetobacter baumannii in Gulls and Songbirds. Polish Journal of Microbiology. 2020. 69(1): p. 85-90. [CrossRef]
- Francey, T.; Gaschen, F.; Nicolet, J.; Burnens, AP., et al., The role of Acinetobacter baumannii as a nosocomial pathogen for dogs and cats in an intensive care unit. Journal of Veterinary Internal Medicine. 2000. 14(2): p. 177-183. [CrossRef]
- Guardabassi, L.; Dalsgaard, A.; Olsen, JE. Phenotypic characterization and antibiotic resistance of Acinetobacter spp. isolated from aquatic sources. Journal of Applied Microbiology. 1999. 87(5): p. 659-667. [CrossRef]
- Doughari, Hamuel James.; Ndakidemi, Patrick Alois.; Human, Izanne Susan.; Benade, Spinney. The Ecology, Biology and Pathogenesis of Acinetobacter spp.: An Overview. Microbes and Environments. 2011. 26(2): p. 101-112. [CrossRef]
- Das, Taraprasad.; Jayasudha, Rajagopalaboopathi.; Chakravarthy, SamaKalyana.; Prashanthi, Gumpili Sai.; Bhargava, Archana.; Tyagi, Mudit.; Rani, Padmaja Kumari.; Pappuru, Rajeev Reddy.; Sharma, Savitri.; Shivaji, Sisinthy. Alterations in the gut bacterial microbiome in people with type 2 diabetes mellitus and diabetic retinopathy. Scientific Reports. 2021. 11(1). [CrossRef]
- Xu, Miao.; Han, Shining.; Lu, Ningning.; Zhang, Xin.; Liu, Junmei.; Liu, Dong.; Xiong, Guangming.; Guo, Liquan. Degradation of Oestrogen and an Oestrogen-like Compound in Chicken Faeces by Bacteria. Water Air and Soil Pollution. 2018. 229(10). [CrossRef]
- Opota, Onya.; Ney, Barbara.; Zanetti, Giorgio.; Jaton, Katia.; Greub, Gilbert.; Prod'hom, Guy. Bacteremia Caused by Comamonas kerstersii in a Patient with Diverticulosis. Journal of Clinical Microbiology. 2014. 52(3): p. 1009-1012.
- Wan, Yi.; Ma, Ruiyu.; Chai, Lilong.; Du, Qiang.; Yang, Rongbin.; Qi, Renrong.; Liu, Wei.; Li, Junying.; Li, Yan.; Zhan, Kai. Determination of bacterial abundance and communities in the nipple drinking system of cascading cage layer houses. Scientific Reports. 2021. 11(1). [CrossRef]
- Bin, Peng.; Tang, Zhiyi.; Liu, Shaojuan.; Chen, Shuai.; Xia, Yaoyao.; Liu, Jiaqi.; Wu, Hucong.; Zhu, Guoqiang. Intestinal microbiota mediates Enterotoxigenic Escherichia coli-induced diarrhea in piglets. Bmc Veterinary Research. 2018. 14. [CrossRef]
- Morales Fenero, Camila Ideli.; Colombo Flores, Alicia Angelina.; Camara, Niels Olsen Saraiva. Inflammatory diseases modelling in zebrafish. World journal of experimental medicine, 2016. 6(1): p. 9-20.
- Yu Xi.; Niu Shuling.; Tie Kunyuan.; Zhang Qiuyang.; Deng Hewen.; Gao ChenCheng.; Yu Tianhe.; Lei Liancheng.; Feng Xin. Characteristics of the intestinal flora of specific pathogen free chickens with age. Microbial Pathogenesis. 2019. 132: p. 325-334. [CrossRef]
- Rajoka, Muhammad Shahid Riaz.; Wu, Yiguang.; Mehwish, Hafiza Mahreen.; Bansal, Manisha.; Zhao, Liqing. Lactobacillus exopolysaccharides: New perspectives on engineering strategies, physiochemical functions, and immunomodulatory effects on host health. Trends in Food Science & Technology. 2020. 103: p. 36-48. [CrossRef]
Figure 1.
The rarefaction curves of observed (A) and Shannon index (B) for the 20 samples. D01-D12 represent the samples collected in domestic mallards, and W1-W8 represent the samples collected in wild mallards.
Figure 1.
The rarefaction curves of observed (A) and Shannon index (B) for the 20 samples. D01-D12 represent the samples collected in domestic mallards, and W1-W8 represent the samples collected in wild mallards.
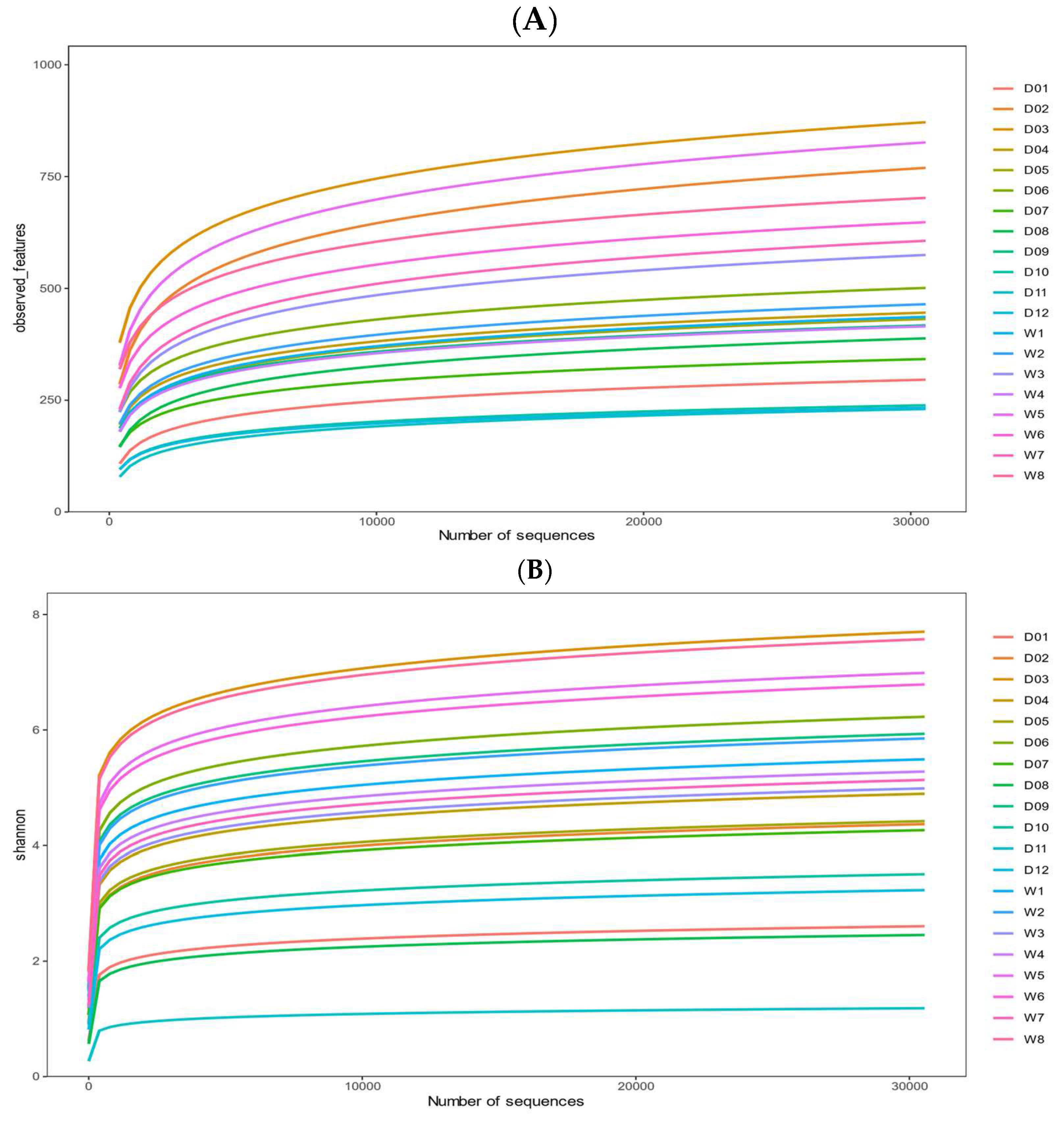
Figure 2.
Alpha diversity indices of gut microbial communities in domestic mallards (DM) and wild mallards (WM). Shannon index (A) and chao1 index (B) for DM and WM groups, * indicates p < 0.05.
Figure 2.
Alpha diversity indices of gut microbial communities in domestic mallards (DM) and wild mallards (WM). Shannon index (A) and chao1 index (B) for DM and WM groups, * indicates p < 0.05.
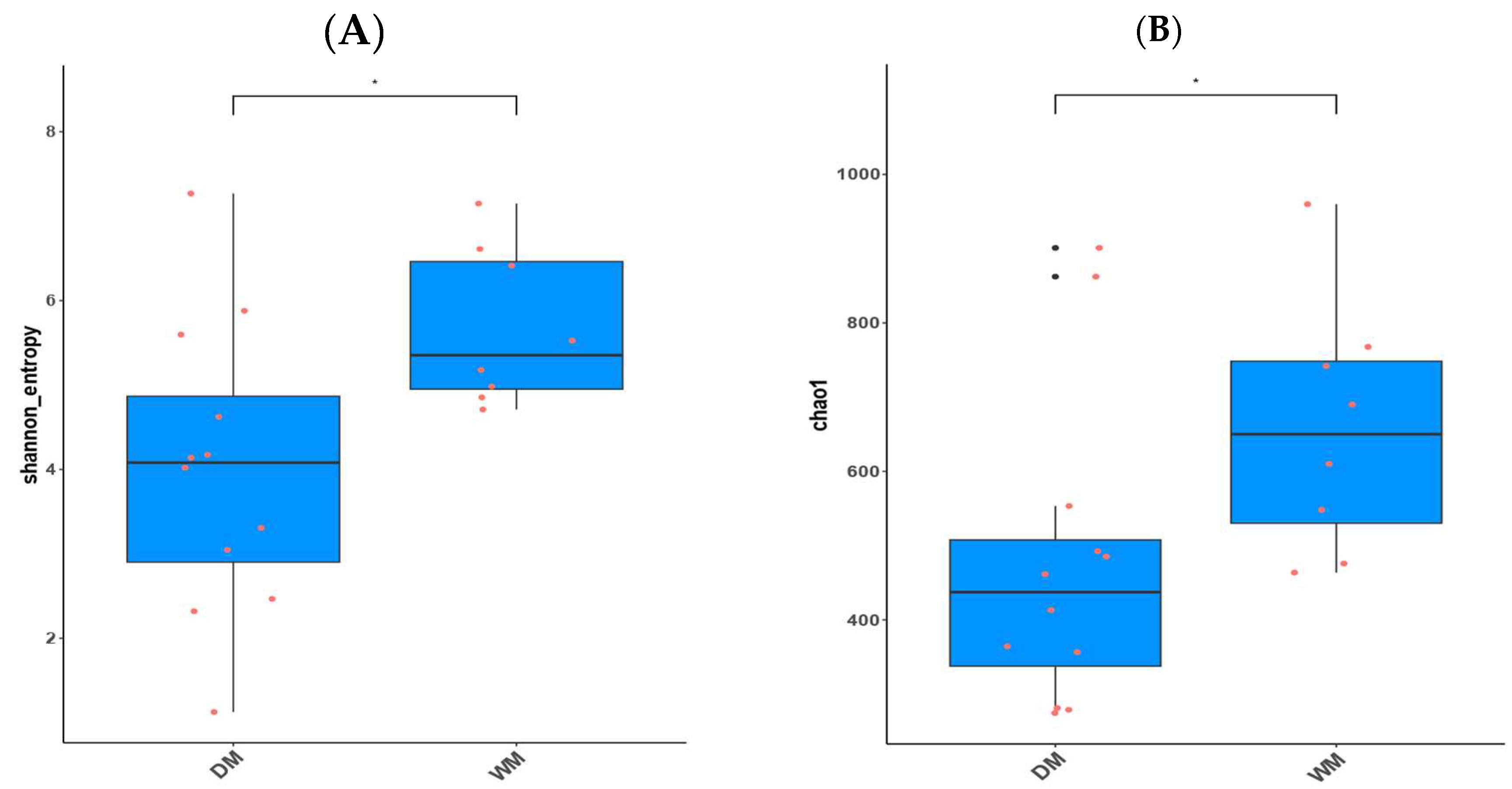
Figure 3.
PCoA plot of samples using the weighted (A) and unweighted (B) UniFrac distance metric.
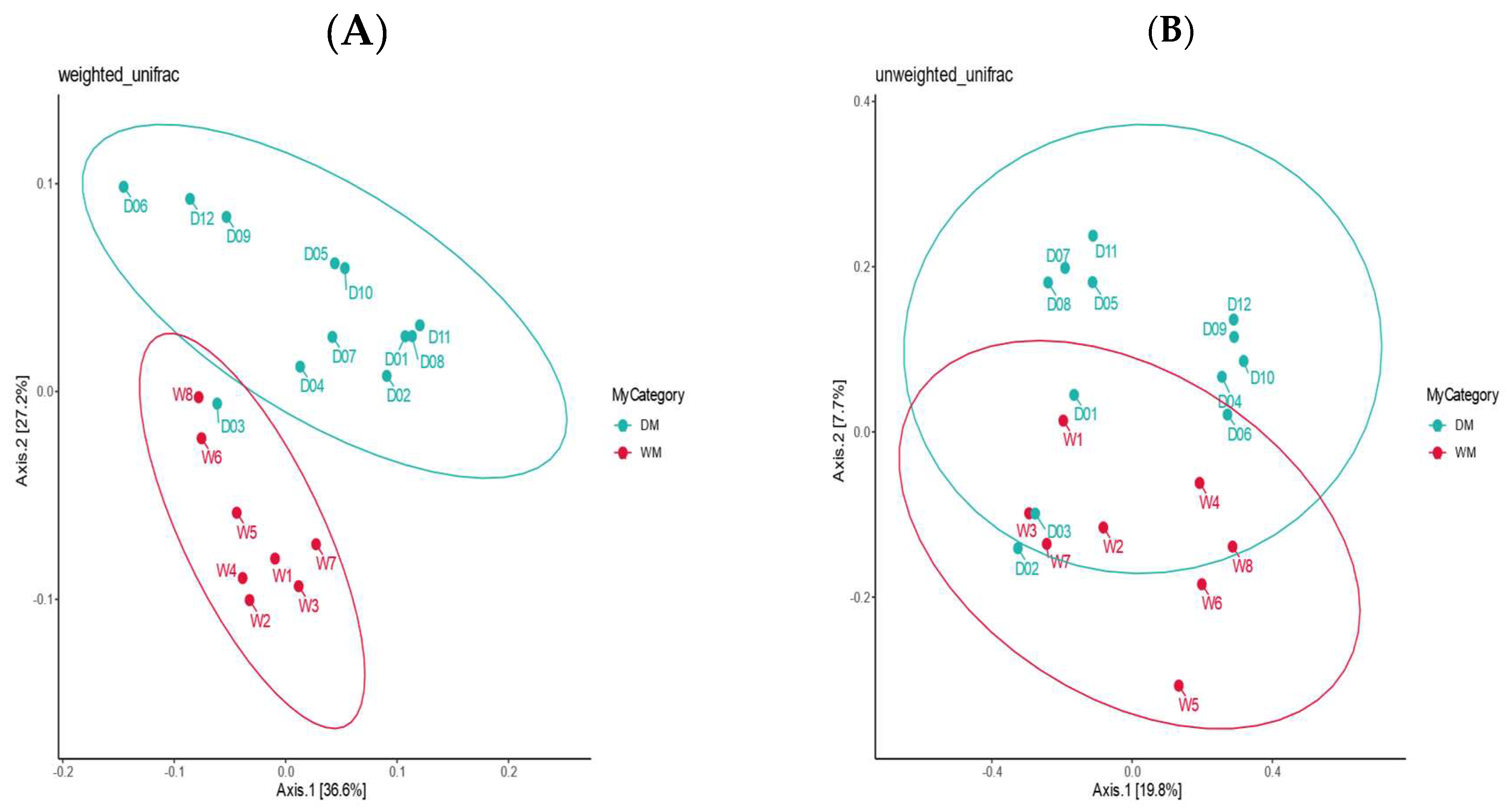
Figure 4.
Top 10 phyla (A) and genera (B) in terms of mean relative abundance in the two sample groups.
Figure 4.
Top 10 phyla (A) and genera (B) in terms of mean relative abundance in the two sample groups.
Figure 5.
LEfSe analysis of cladogram plots. The cladogram diagram corresponds to the different taxonomic levels of the kingdoms, phyla, orders, families, and genera from the inside to the outside, and the lines between the levels represent the affiliation. Each circle node represents a species, and a yellow node means that the difference between groups is not significant, while a non-yellow node means that the species is a characteristic microorganism of the corresponding color group (with significantly higher abundance in that group). The colored sectors mark the subordinate taxonomic intervals of the characteristic microorganisms.
Figure 5.
LEfSe analysis of cladogram plots. The cladogram diagram corresponds to the different taxonomic levels of the kingdoms, phyla, orders, families, and genera from the inside to the outside, and the lines between the levels represent the affiliation. Each circle node represents a species, and a yellow node means that the difference between groups is not significant, while a non-yellow node means that the species is a characteristic microorganism of the corresponding color group (with significantly higher abundance in that group). The colored sectors mark the subordinate taxonomic intervals of the characteristic microorganisms.
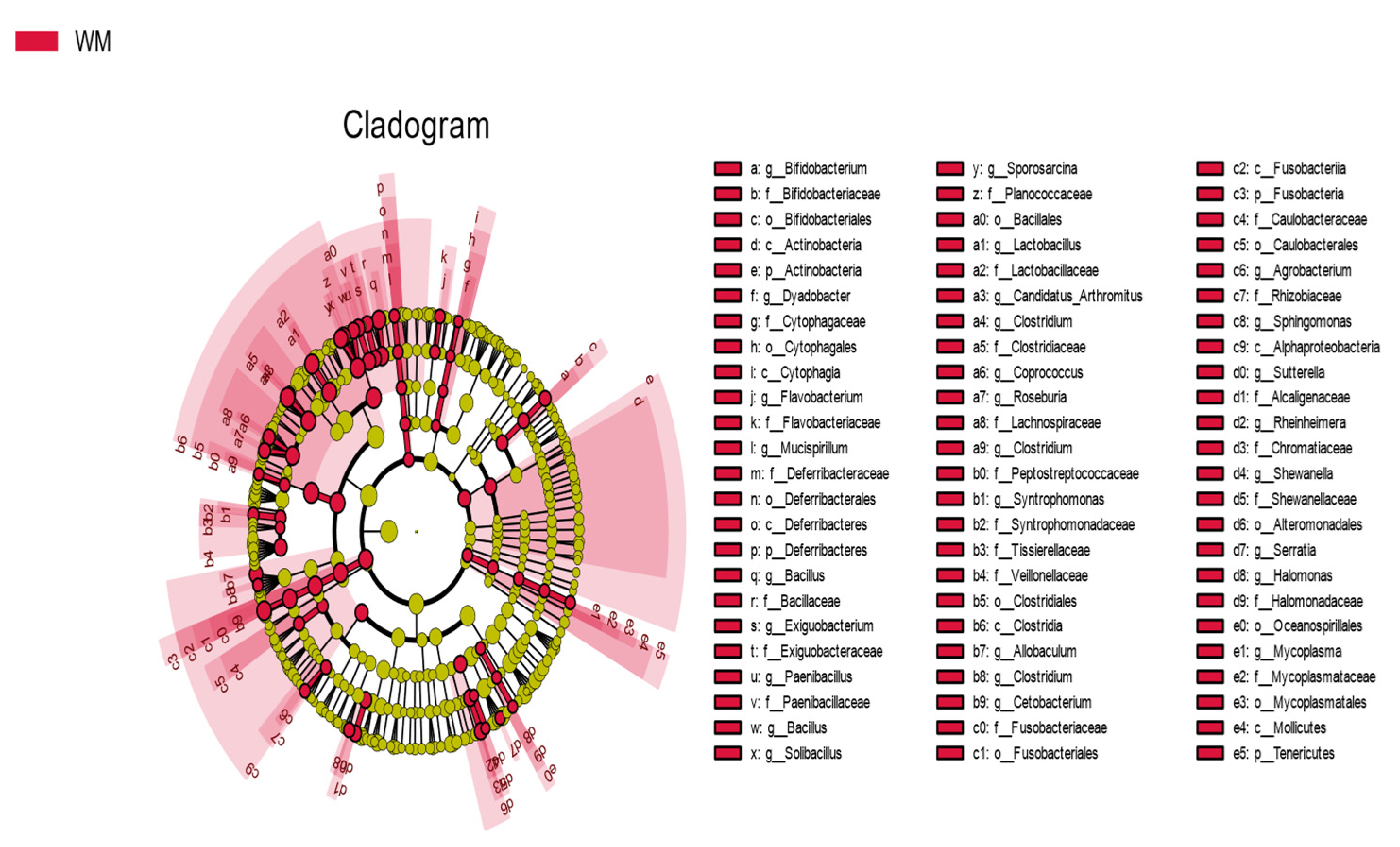
Figure 6.
Top 10 level 2 pathways with predicted abundances of gut microbial function in the DM and WM groups.
Figure 6.
Top 10 level 2 pathways with predicted abundances of gut microbial function in the DM and WM groups.
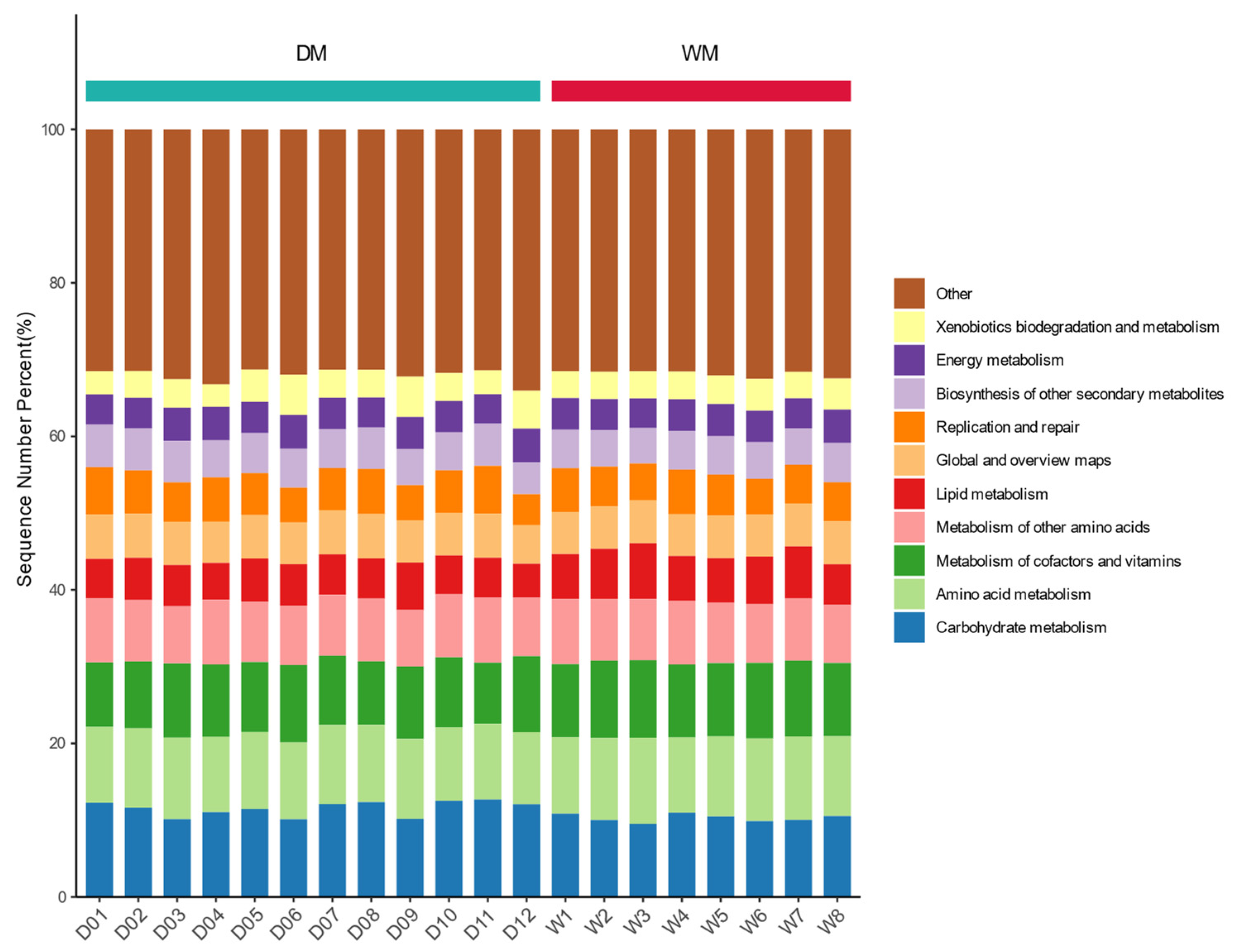
Figure 7.
ANOVA and Duncan's test were obtained for all significant differences in MetaCyc pathways. The horizontal coordinate is the name of the pathway; for each pathway, different colors are used to indicate different subgroups, and if there are the same letters above two subgroups, it means the difference is not significant; otherwise, the difference is significant.
Figure 7.
ANOVA and Duncan's test were obtained for all significant differences in MetaCyc pathways. The horizontal coordinate is the name of the pathway; for each pathway, different colors are used to indicate different subgroups, and if there are the same letters above two subgroups, it means the difference is not significant; otherwise, the difference is significant.
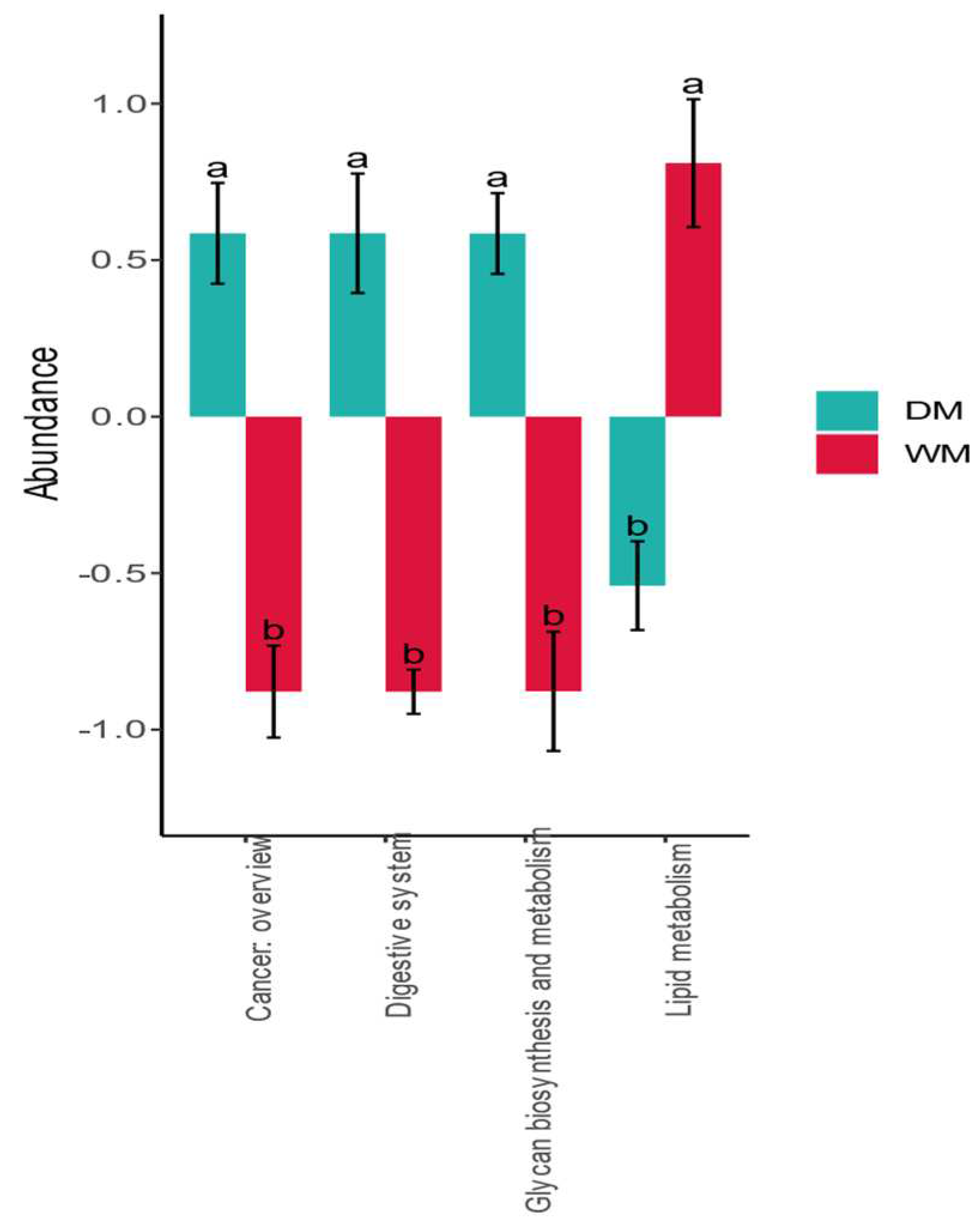

Table 1.
Mean relative abundance >1% of phyla in the two sample groups.
| Category | Firmicutes | Proteobacteria | Bacteroidetes | Fusobacteria | Actinobacteria |
|---|---|---|---|---|---|
| WM | 79.02% | 12.85% | 2.79% | 3.37% | — |
| DM | 67.97% | 24.48% | 3.13% | 2.21% | 1.10% |
Table 2.
Genera with a mean relative abundance >1% in each phylum.
| Phylum | Genus | DM | WM |
|---|---|---|---|
| Firmicutes | Streptococcus | 35.07% | 7.62% |
| Enterococcus | 13.67% | 3.80% | |
| Clostridium | 1.53% | 2.96% | |
| Lactobacillus | — | 2.83% | |
| Solibacillus | — | 1.87% | |
| Bacillus | — | 1.28% | |
| Proteobacteria | Acinetobacter | 5.09% | 1.16% |
| Comamonas | 2.37% | — | |
| Shigella | 2.50% | — | |
| Fusobacteria | Cetobacterium | — | 2.22% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



