
Submitted:
07 August 2023
Posted:
10 August 2023
You are already at the latest version
Abstract
Adipose tissue has acquired considerable importance due to its role in maintaining body homeostasis. Beyond storing energy, adipose tissue is essential in numerous processes of energy metabolism, food intake, endocrine and immunological functions as well as neromodulatory functions by secreting hormones that participate in the regulation of homeostasis and being responsive to soluble proteins secreted by other tissues. An imbalance of these functions will generate structural and functional changes in the adipose tissue, favouring the secretion of deleterious adipocytokines that benefit a pro-inflammatory state, allowing the development of metabolic and cardiovascular diseases. Obesity is a disease that has increased rapidly in recent decades and is caused by environmental, hormonal, and genetic factors, which probably work in combination. Environmental factors that contribute to the increase in obesity include decreased physical activity, associated with increased screen time, increased accessibility and consumption of foods rich in fats and sugars. Weight loss should be recommended for all obese patients and overweight patients with comorbidities. For many patients who need to lose weight for medical reasons, the initial goal is to lose 5-10% of their body weight in the first 6 months. A common theme worldwide has been the development of professional guidelines for the control and treatment of obesity and in Chile, the Chilean Society of Metabolic and Bariatric Surgery led the process of adopting the adult obesity clinical practice guideline, which establishes changes in the approach to managing obesity as a chronic disease, with an emphasis on multifactorial lifestyle interventions that include dietary changes, increased physical activity, and behavioural modifications. Pharmacotherapy, medical devices, and bariatric surgery are other options for patients who need additional interventions, so the objective of this review is to know the pathophysiological mechanisms of obesity from its origin to the current treatments available for treatment.
Keywords:
adipose tissue
; adipocytokines
; overweight
; obesity
; molecular mechanism
; treatments
; myocyotokines
Adipose Tissue
In recent years, adipose tissue has acquired considerable importance for its role in maintaining body homeostasis. Beyond storing energy, adipose tissue is fundamental in numerous processes, such as control of energy metabolism, food intake regulation, and the immune system [1]. Adipose tissue is considered an endocrine tissue since it produces and secrets peptides that can exert their action in surrounding or distant tissues [2]. Hormones and adipocytokines produced by adipocytes can exert their action in the central nervous systems, skeletal muscle, liver, bone, or other tissues, which have been extensively studied in the last two decades, establishing that the factors secreted by the adipose tissue play a preponderant role in the homeostasis of body glucose, through endocrine, autocrine and paracrine processes. Adipocytokines are essential for the balance between appetite and satiety, body fat reserve and energy expenditure, glucose tolerance, release, insulin sensitivity, cell growth, inflammation, angiogenesis, and reproduction [3].
Adipose tissue is composed of adipocytes, pre-adipocytes, endothelial cells, fibroblasts, and some immune cells such as macrophages, dendritic cells and T cells. These cells contribute to the release of metabolites, lipids, cytokines, and adipocytokines. In physiological conditions, adipose tissue plays a role in the homeostasis of the entire body due to lipid storage that is used as a source of energy during fasting, thus preserving proteins, regulating metabolism and food intake, reproduction, and enhancing the immune response in pathogenic invasion [1].
Adipose or adipocyte cells that have a large drop of lipid are called unilocular adipose cells and are part of the white adipose tissue, and cells with multiple small drops of lipids, are called multilocular adipose cells and constitute the adipose brown tissue [4]. White adipose tissue is the most abundant and is distributed throughout the body, mainly as, perivascular, and visceral fat. Its functions include mechanical protection and support, lipid storage, regulation of carbohydrate, lipids, and proteins metabolism, as well as regulation of appetite. It also produces and secrets adipocytokines, glucocorticoids, and sex hormones [5]. Brown adipose tissue is considered thermogenic, and its colour reflects the cytochromes present in its numerous mitochondria. UCP1 decoupling protein is responsible for modifying oxidative phosphorylation in mitochondria, causing a decrease in ATP production, and increasing heat production by this tissue [6]. Brown adipose tissue has a regulatory function in body temperature via adaptive thermogenesis, regulating the concentration of circulating triglycerides, storing glucose, and secreting prostaglandins, nitric oxide, adipsin, and other adipocytokines [7].
The beige adipose tissue has an intermediate diameter between white and brown adipose tissue. Originally it was observed by increasing in response to cold, however, factors such as diet, physical, pre and probiotic activity, and drugs, among others, can induce transdifferentiation (begging or browning) of white adipose tissue [8]. This tissue has the function of storing or eliminating energy according to physiological needs [6].
The excessive adiposity that occurs in obesity (excess of white adipose tissue) makes the metabolic balance of adipose tissue to be lost, producing a negative effect on health. Therefore, obesity is the causality of a group of chronic and complex diseases such as cardiovascular diseases, metabolic syndrome, type 2 diabetes [9], and cancer [10].
Adipose Tissue as an Endocrine Organ
Adipose tissue, besides being an energy reservoir, is an important endocrine organ, since it produces several hormones that participate in the regulation of homeostasis. A positive imbalance of adipose tissue (accumulation of fatty acids over BMI 24.9 kg/m2) generates structural and functional changes in this tissue, favouring the secretion of deleterious adipocytokines related to insulin signaling and those that benefit a pro-inflammatory state, which could promote the development of metabolic and cardiovascular diseases (see Figure 1) [11]. One of the first molecules described, secreted by adipocytes with hormonal activity was leptin [12], which suppresses food intake by inducing satiety at the level of the hypothalamus, along with increasing energy expenditure. Leptin levels are positively correlated with the amount of adipose tissue in an organism, being secreted mainly by visceral white adipose tissue [13,14]. Adiponectin is another hormone secreted by subcutaneous white adipose tissue. This hormone has anti-inflammatory and insulin-sensitizing functions [15]. In overweight or obese people with insulin resistance, plasma adiponectin levels are low [16]. Resistin is another hormone secreted by adipose tissue and has a close relationship between obesity and diabetes, notably contributing to insulin resistance and vascular inflammation [17]. Fibroblast Growth Factor 21 (FGF21) is a protein produced by adipose tissue and other tissues, with thermogenic effects that promote transdifferentiation from beige to brown adipose tissue. Like adiponectin, it has insulin-sensitizing effects, and in overweight or obese patients it is found in high plasmatic concentrations, suggesting resistance to FGF21 in these people [11]. Vaspina is a protein that is increased in obese patients, promoting insulin resistance and decreasing glucose tolerance [18]. Visfatin is a hormone involved in glucose homeostasis and, like acylation-stimulating protein (ASP), it is preferentially involved in fat storage [19].
Adipose tissue secretes inflammatory cytokines such as interleukin-6 (IL-6), IL-8, interferon-γ (INFγ), plasminogen activator inhibitor-1 (PAI-1) as well as anti-inflammatory cytokines, such as adiponectin [20]. Many other molecules are secreted by adipose tissue, such as retinol-binding protein 4 (RBP4), omentin, angiotensinogen, macrophage migration inhibitory factor (MIF), lipoprotein lipase (LPL), prostaglandins, estrogen, glucocorticoid, etc [12]. All these molecules influence homeostatic processes, affecting health positively or negatively. Adipose tissue can be involved in the development of many diseases, such as type 2 diabetes mellitus (T2DM), metabolic syndrome, several types of cancer (breast, cervical, endometrial, renal and gastrointestinal). Even more, adipose tissue dysfunction can lead to psychiatric disorders, such as depression, dementia, insomnia and many other [1].
Not just the adipokines secreted by the adipose tissue and its functions in other organs or systems, but the myokines secreted by muscle and the hepatokines by the liver, are relevant and associated with all the alterations caused by obesity in all the tissues and systems [21].
Definition and Incidence of Overweight and Obesity
The Body Mass Index [BMI (kg/m2, weight of the person divided by the square of their height)], is used to define and diagnose obesity according to the clinical guidelines of the World Health Organization (WHO) [22]. In adults, the WHO defines overweight as a BMI of 25.0 to 29.9 kg/m2 and obese as a BMI > 30.0 kg/m2. In addition, obesity is classified into three levels of severity: class I (BMI 30.0 - 34.9 kg/m2), class II (BMI 35.0 - 39.9 kg/m2) and class III (BMI > 40 .0kg/m2) [23]. For every 5-unit increase in BMI above 25.0 kg/m2, overall mortality increases by 29%, vascular mortality by 41%, and diabetes-related mortality by 210% [24].
Over the past 50 years, the global obesity rate has increased significantly [25] and above all, in the last 30 years there has been an exponential growth in the prevalence of global obesity, doubling the rate in adults and children (6-11 years) and tripling in the adolescent population (12-19 years) [26]. The BMI reflects body mass and in many, but not all cases, correlates with the degree of obesity and is a significant predictor of overall mortality with a reduction in median survival of approximately 2 to 4 years in persons with a BMI from 30 to 35 kg/m2 and from 8 to 10 years with a BMI of 40 to 45 kg/m2 [27]. Obesity is often stigmatized and carries with it a false perception that it is primarily caused by a lack of willpower, leading to inappropriate dietary choices and physical inactivity. However, there is rich literature-based evidence presenting obesity as a complicated chronic medical condition, caused by multiple genetic, environmental, metabolic, and behavioural factors [28].
Obesity increases the probability of contracting several diseases and conditions that are linked to increased mortality, such as type 2 diabetes mellitus (T2DM), cardiovascular disease (CVD), metabolic syndrome (MetS), chronic kidney disease (CKD), hyperlipidaemia, hypertension, non-alcoholic fatty liver disease (NAFLD), certain cancers, obstructive sleep apnea, osteoarthritis, and depression [29].
Obesity is a public health problem with epidemic characteristics in the world, both, in developed and developing countries, affecting low- and middle-income populations [30]. According to the WHO, in 2016 more than 1.9 billion adults in the world were overweight and 650 million were obese [31]. In Latin America and the Caribbean, more than half of the adult population is considered overweight or obese. In particular, a report in 2016 showed that in Chile, 63% of the population presented malnutrition due to excess, where 33.7% corresponded to women and 28.6% to men [32]. In the same country, the National Board of School Aid and Scholarships (JUNAEB) established that in 2019 52% of schoolchildren were overweight or obese and the prevalence increased to 60% for those who were in the 5th grade [33].
The pathogenesis of obesity is complex, with environmental, socio-cultural, physiological, medical, behavioural, genetic, epigenetic, and numerous other factors contributing to the cause and persistence of this condition [34].
Causes or Mechanisms of Obesity
Obesity is a disease that has increased rapidly in recent decades and is caused by environmental, hormonal, and genetic factors, which probably work in combination. Environmental factors that contribute to the increase in obesity include, among others, decreased physical activity, increased hours of television viewing, and a sedentary lifestyle [35], increased consumption of foods, particularly rich in energy, calories and tasty served in larger portions [36], as well as the use of medications with weight gain as a side effect [37].
Genetic Factors
Data shows that around 40 to 70% of the variations in obesity in humans are the result of genetic factors [38]. While environmental changes have increased the rate of obesity, genetic factors play a key role in the development of this condition, identifying nearly 100 genes related to obesity and fat distribution [28,39]. It is clear that in the same environment, some people become obese, and others do not. Many factors are involved in this differential response and some of them will be mentioned below.
Genetic causes of obesity can be classified as A) monogenic causes that result in a single mutation, located mainly in the Leptin-Melanocortin pathway. Many of the genes, such as AgRP (Agouti-related peptide), PYY (peptide tyrosine tyrosine, orexigenic), or MC4R (melanocortin-4 receptor), were identified as causing monogenic obesity, deregulating the appetite and body weight control system, where hormonal signalling (ghrelin, leptin, insulin) are sensed by receptors located in the hypothalamus (arcuate nucleus) [40]. B) Syndromic obesity is the result of severe obesity due to neurodevelopmental abnormalities and other organ/system malformations. This can be caused by mutations in a single gene or a chromosomal region that spans multiple genes [41]. C) Polygenic obesity is caused by the cumulative contribution of several genetic alterations. The presence of these types of alterations can cause an increase in caloric intake, an increase in hunger levels, a reduced control of overeating, a reduction in satiety, a greater tendency to store body fat and a greater tendency to a sedentary lifestyle [42].
There are also several genetic, neuroendocrine and chromosomal syndromes that can cause obesity, such as Prader-Willi syndrome (PWS), which is a neurodevelopmental disorder with hypothalamic dysfunction with the impaired secretion of various hormones [43], polycystic ovary syndrome, which is an endocrine disorder that can lead to increased body fat mass [44] and deletions such as 16p11.2, 2q37 (brachydactyly mental retardation syndrome), 1p36 (monosomy 1p36 syndrome), 9q34 (Kleefstra syndrome), 6q16 (PWS-like syndrome), 17p11.2 (Smith Magenis syndrome) and 11p13 (WAGR syndrome) [45]. All of these conditions show an energy imbalance between calories consumed and spent, as the main cause of obesity [25].
Fat Cells
The excess calories from food results in an accumulation of fat in adipocytes [28]. This enlargement and/or increase in the number of fat cells to adapt to excessive fat storage constitutes the initial pathological lesion in obesity. The accumulation of ectopic fat, such as visceral, cardiac and muscle fat, is associated with several factors, among others when adipocytes have already reached their maximum storage capacity [46]. Here it is relevant to note that the laboratory of Philipp Scherer showed that the complete abolition of adipose tissue associated inflammation was associated with several adverse metabolic readouts, in other words, inflammation in a healthy dose is important during healthy adipose expansion [47]. The increase in the size of the adipocyte can eventually generate an inflammatory microenvironment due to an alteration of the homeostasis between the adipocyte and the cells of its microenvironment, particularly the macrophages [3]. The alteration of a healthy expansion in the size of the adipocyte produces an increase in the secretion of several inflammatory adipocytokines (some seen in the section Adipose tissue as an endocrine organ), such as Leptin, IL-6, TNF-α, angiotensinogen, adipsin, free fatty acids and lactate, while the concentration of anti-inflammatory molecules secreted by the adipocyte, such as adiponectin, decreases [48]. These metabolic changes lead to a variety of other pathological processes, such as dyslipidemia, physical stress, arterial hypertension, hyperinsulinemia, atherosclerosis, diabetes, heart attack, and cancer, among others [26].
Dysregulation of Energy Balance
Genes and the environment interact in a complex way that regulates energy balance, linked to physiological processes as well as body weight control [49]. Two groups of neurons located in arcuate nucleus of the hypothalamus are inhibited or stimulated by ghrelin and leptin, hormones that control energy balance by regulating food intake and energy expenditure: AgRP and POMC neuron. Brain regions external to the hypothalamus also contribute to the regulation of energy balance through sensory signals, cognitive processes, the hedonic effect of food consumption, memory, and attention [50].
Reducing food intake or increasing physical activity will generate a negative energy balance, activating adaptive compensatory mechanisms that preserve vital functions [51], and conversely, in a state of rest, there is a relative reduction in energy expenditure, concern for food and other metabolic processes that depend on the magnitude and duration of caloric restriction [52]. An increase in the stimulation of the orexigenic centre can explain a subtle and often inappropriate increase in appetite and food intake, limiting weight loss associated with interventions such as physical exercise programs. It is important to always consider obesity as a chronic disease, which requires long-term monitoring and weight control since there is a high relapse rate in those people who have managed to lose weight [34].
Metabolic and Physiological Effects
Adipocytes synthesize signalling molecules (adipocytokines) and hormones, whose secretion and effects are influenced by the distribution and amount of adipose tissue present in a person [53]. Excessive secretion of proinflammatory adipocytokines by adipocytes and macrophages within adipose tissue leads to a low-grade systemic inflammation in people with obesity [34].
Triglyceride breakdown within adipocytes releases free fatty acids, which are transported in the plasma to sites where they can be metabolized. In overweight people and to a greater extent in those with obesity, free fatty acid levels are often elevated, reflecting the increased mass of adipose tissue [53].
Lipids are not only stored in adipose tissue, we can also find them in various types of cells in organelles called liposomes located near mitochondria [54]. When overweight or obese, liposomes in hepatocytes increase in size, forming large vacuoles that are accompanied by a series of pathological states such as non-alcoholic fatty liver disease, steatohepatitis, and cirrhosis [55], which can generate cell dysfunction and apoptosis [34].
Elevated levels of free fatty acids, proinflammatory cytokines, and intermediary lipids, such as ceramides, in non-adipose tissues, contribute to impaired insulin signalling and a state of insulin resistance [56]. All these metabolic and anatomical findings are some of the pathophysiological mechanisms caused by dyslipidaemia in obesity, type 2 diabetes, obesity-related liver disease and osteoarthritis, as well as implicated in the development of some cancers, likely by the association with elevated levels of tumour-promoting molecules [10].
Finally, sympathetic nervous system hyperactivity in some overweight or obese people can produce multiple pathophysiological processes (see Figure 2) such as arterial hypertension, heart disease, heart attacks and chronic kidney disease, all associated with insulin resistance, dyslipidaemia and type 2 diabetes [57].
Complications and Comorbidities Associated with Obesity
Insulin Resistance (IR)
Obesity is associated with increased mortality. For every 5 kg/m2 increase in BMI above 25 kg/m2, overall mortality increases by approximately 30%, vascular mortality by 40%, and diabetic, renal, and hepatic mortality by 60% to 120%. With a BMI of 30 to 35 kg/m2, median survival is reduced by 2 to 4 years, and a BMI of 40 to 45 kg/m2 by 8 to 10 years [27]. The main cause of death in obesity includes cardiac ischemia, heart attack and complications associated with diabetes [58], incorporating components of insulin resistance (IR) as well as Metabolic Syndrome (hypertension, hyperglycaemia, dyslipidaemia, among others) [26].
Obesity is associated with an increased risk of IR. The HOMA-IR (HOmeostatic Model Assessment – IR) relationship is strongly correlated with visceral fat mass, total fat, and waist circumference [59]. Adipose tissue controls metabolism by regulating the levels of non-esterified fatty acids (NEFAs), glycerol, proinflammatory cytokines, immune system cells (lymphocytes, macrophages), and hormones [60]. Many of these molecules are increased in obesity, affecting insulin sensitivity through various mechanisms. First, the increased transport of NEFAs and consequently high intracellular levels compete with glucose for the oxidation of substrates, resulting in the inhibition of important enzymes involved in the glycolytic pathway [61]. In addition, metabolites from NEFAs (ceramides, diacylglycerol, acyl-coenzyme A, among others) are increased, affecting the insulin receptor signalling pathway by phosphorylation of the insulin receptor substrate type 1 and 2 (IRS-1 and - 2), which reduces the activity of phosphatidyl-3-kinase (PI3K) [62]. Second, an increase in proinflammatory cytokines, as well as immune cells, overstimulates inflammatory processes, causing tissue dysfunction, hypoxia and injury. Obesity causes chronic low-grade inflammation, increasing the secretion by this tissue (mainly visceral adipose tissue) of TNF-α, IL-6 and MCP-1 [63]. Proinflammatory signalling pathways involve JNK activation and IKK inhibition, again leading to phosphorylation of IRS-1 and IRS-2, as well as increased transcription of proinflammatory genes [64]. And third, increased levels of proteins such as RBP-4 (retinol-binding protein -4) and Leptin, and reduced levels of adiponectin affect insulin sensitivity by altering the PI3K signalling pathway in muscle, inducing the expression of gluconeogenic enzymes and the oxidation of fatty acids in the liver, giving as a final result, in obese people, liver and muscle IR, causing abnormal glucose production from the liver [65] and reduced glucose uptake by the muscle [66]. See Figure 3.
Type 2 Diabetes
Obesity is strongly associated with the development of type 2 diabetes (T2DM). IR demands in the liver, muscle, and adipose tissue an increase in insulin delivery by pancreatic β cells to maintain euglycemia [67]. β cells can improve their functionality and mass to meet increased demand for a limited time [26]. Muscular IR affects the metabolism of the whole organism, generating hepatic steatosis and an increase in adipose tissue [60]. The liver finely regulates postprandial glucose levels by suppressing hepatic glucose production and stimulating glucose storage as glycogen, being the primary source of glucose production during fasting [68]. In patients with T2DM, insulin cannot regulate glycogen synthesis or glucose production, so increased hepatic gluconeogenesis is the leading cause of hyperglycaemia in T2DM [65]. The combination of hyperglycaemia and dyslipidaemia (glucolipotoxicity) accelerates β-cell death, reducing insulin secretion, and further aggravating hyperglycaemia [69]. Finally, the relative risk of incidence of diabetes is approximately 1.87 per standard deviation of the body mass index or the waist-hip ratio [70].
Figure 3.
Insulin–resistance systemic level model. FFA, Free Fatty Acids; ER, Endoplasmic Reticulum; ROS, Reactive Oxygen Species; GluT4, Glucose Transporter 4; IRS-1, Insulin Receptor Substrate type 1, MAPK, Mitogen-Activated Protein Kinase; PI3K, Phosphatidyl Inositol 3 Kinase; PBK, Protein Kinase B or Akt; JNK, c-Jun N-terminal kinase.
Figure 3.
Insulin–resistance systemic level model. FFA, Free Fatty Acids; ER, Endoplasmic Reticulum; ROS, Reactive Oxygen Species; GluT4, Glucose Transporter 4; IRS-1, Insulin Receptor Substrate type 1, MAPK, Mitogen-Activated Protein Kinase; PI3K, Phosphatidyl Inositol 3 Kinase; PBK, Protein Kinase B or Akt; JNK, c-Jun N-terminal kinase.
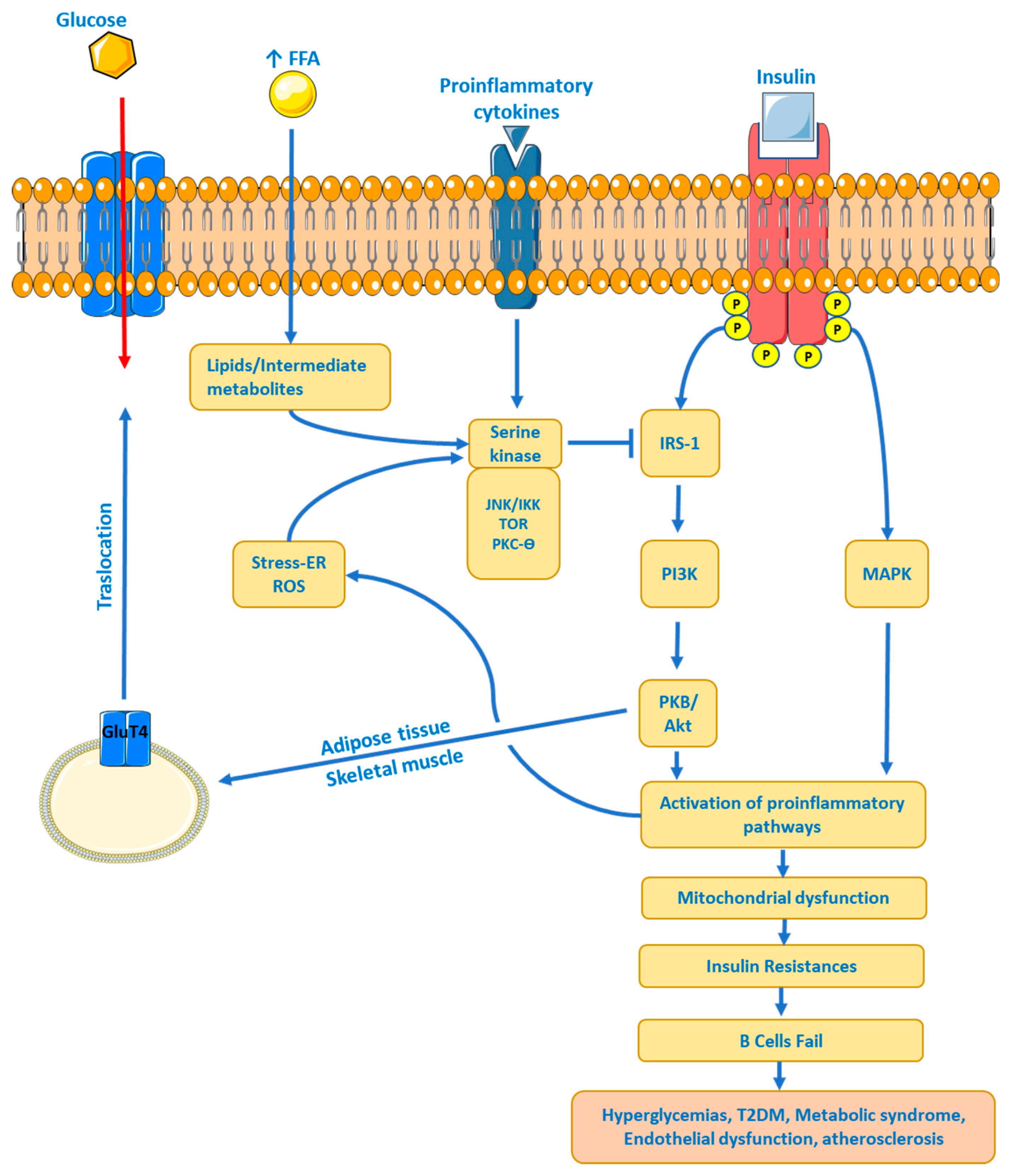
Dyslipidaemia
Obesity is associated with dyslipidaemia, which is characterized by increased plasma total cholesterol, triglycerides, VLDL-apoB, postprandial hyperlipidaemia, low HDL cholesterol (HDL-c) levels, and a predominance of small dense LDL cholesterol particles [26,71]. The accumulation of lipolytically active visceral fat in combination with IR leads to a marked increase in the flow of fatty acids in the portal vein and subsequently, an increase in hepatic synthesis of triglycerides, observing a higher risk of coronary disease in those patients with hypertriglyceridemia [72]. IR and low HDL-c levels present a common mediator, TNF. TNF is involved in insulin resistance in obese patients and is recognized for lowering HDL-c levels [73]. Several enzymes involved in HDL-c metabolism are altered in IR [74], which further increases the atherogenic risk.
Hypertension
Several mechanisms have been implicated in obesity-associated hypertension. First, obesity is characterized by hemodynamic changes due to volume overload, causing increased cardiac output, peripheral resistance, and increased blood pressure [75]. Second, high salt intake due to high food intake alters sodium homeostasis, promoting hypertension [76]. High sodium reabsorption combined with high renal blood flow plus hyperfiltration leads to changes in renal structure and dysfunction, contributing to further elevation of blood pressure [77]. In addition, hormonal changes such as hyperaldosteronism, hyperinsulinemia, and hyperleptinemia result in activation of the renin-angiotensin-aldosterone system (RAAs), the sympathetic autonomic nervous system, and decreased parasympathetic activity [78]. And third, endothelial dysfunction in combination with vascular stiffness increases the production of reactive oxygen species, reducing the bioavailability of nitric oxide [79], leading to decreased acetylcholine-dependent endothelial relaxation [26]. All these hormonal and vascular changes, plus the low-grade chronic inflammation that patients with obesity present increase blood pressure, which leads them to suffer from hypertension.
The high risk of cardiovascular events such as heart attack, heart failure or cardiac arrest, would be partly explained by IR, dyslipidaemia, hypertension, hyperglycaemia and diabetes that obese patients can present, mediating 44% of the risk of coronary disease and in 69% the risk of cardiac arrest [80].
Other Complications and Comorbidities
Other complications and comorbidities associated with obesity are a) Polycystic Ovarian Syndrome. The worldwide increase in the prevalence of obesity has led to an increase in comorbidities such as polycystic ovary syndrome (PCOS). In a genetic susceptibility setting, PCOS often manifests after weight gain, commonly in adolescence. PCOS is a common endocrinopathy that affects between 6 and 10% of women of childbearing age and presents characteristics such as hyperandrogenism, metabolic and reproductive dysfunction, as well as IR, independent of obesity, although it is amplified when it is present [81]. Although the mechanism of IR in PCOS is not fully elucidated, the reported defects would be within the insulin receptor signaling pathway and low-grade inflammation that occurs in PCOS [82,83]. b) Obstructive Sleep Apnea. Obstructive sleep apnea (OSA) occurs in high prevalence in patients with obesity and coincides with several comorbidities such as hypertension, type 2 diabetes, dyslipidemia, non-alcoholic fatty liver disease, heart failure, and atrial fibrillation [84]. It has been estimated that 58% of moderate to severe OSA is due to obesity [85], and OSA is an independent riek factor for stroke (Jehan S, Farag M, Zizi F, Pandi-Perumal SR, Chung A, Truong A, Jean-Louis G, Tello D, McFarlane SI. Obstructive sleep apnea and stroke. Sleep Med Disord. 2018;2(5):120-125. Epub 2018 Nov 30. PMID: 30680373; PMCID: PMC6340906.). However, one should be aware of other risk factors, such as advanced age, male gender, peri- or postmenopausal states in the female gender, and craniofacial abnormalities [86]. c) Cancer. Obesity is strongly related to increased susceptibility to various diseases, including different types of cancers, such as thyroid, uterine, liver, pancreatic, colorectal, breast, esophageal, and kidney cancers [10,26]. It is estimated that one in five cancers is related to obesity [87]. Several mechanisms linking cancer to obesity have been proposed, such as IR, high levels of IGF-1, chronic low-grade inflammation in obese patients, deregulation of factors/hormones secreted by adipose tissue, and alterations in sex hormones [88], as well as changes in the population of immune cells [10]. See Figure 4.
Management of the Overweight-Obesity Patient
Weight loss should be recommended for all obese patients and also for overweight patients with comorbidities such as insulin resistance, diabetes, hypertension, and dyslipidemia [89]. For many patients who need to lose weight for medical reasons, the initial goal is to lose 5-10% of their body weight in the first 6 months [90]. A common theme worldwide has been the development of professional guidelines and in the case of Chile, the Chilean Society of Metabolic and Bariatric Surgery led the process of adapting the adult obesity clinical practice guideline [91], which establishes changes in the approach to managing obesity as a chronic disease, with emphasis on multifactorial lifestyle interventions that include dietary changes, increased physical activity, and behaviour modifications. Pharmacotherapy, medical devices and bariatric surgery are other options for patients who need additional interventions [92].
Lifestyle Modification
To successfully achieve significant 5-10% weight loss requires comprehensive and intensive patient intervention within the first 6 months [90]. Effective weight loss interventions include dietary modifications via prescription of low-calorie diets, increased physical activity or exercise, and behavioural strategies to encourage adherence to dietary and physical activity recommendations [93]. Common strategies include self-monitoring of diet and physical activity, daily or regular weighing, goal setting, and stimulus control, such as limiting eating places [94]. Significant weight loss in a short time can be achieved by controlled consumption of small portions of food [95]. The long-term weight control can be achieved via high levels of physical activity and continuous doctor-patient contact. In many cases, lifestyle modification results in a dramatic loss of body weight, leading to a significant reduction in cardiovascular risk [96].
Pharmacotherapy: Who Are Candidates to Receive Anti-Obesity Drugs?
Drugs approved for the management or control of body weight should be considered for patients with a BMI ≥30 kg/m2 and those with a BMI of at least 27 kg/m2 in the presence of weight-related comorbidities [90]. Pharmacotherapy may be considered for patients with excess body weight who (i) achieve modest benefit with lifestyle intervention and require further reduction in body weight; (ii) who has lost some weight with the lifestyle intervention, but is having difficulty maintaining that loss; (iii) you have made numerous unsuccessful attempts to lose weight through diet and exercise; and (iv) are unable to comply with lifestyle change recommendations due to serious chronic medical conditions [89].
Orlistat
Orlistat is a pancreatic lipase inhibitor that reduces intestinal fat absorption [89], resulting in approximately 30% of ingested triglycerides being eliminated via faecal. To date, it is the only drug available for obesity that does not target satiety or appetite mechanisms [97]. Orlistat 120 mg three times a day for one year, achieves a reduction in body weight of approximately 3% [98] and is approved for patients with BMI ≥ 27 kg/m2 in the presence of comorbidities such as hypertension, type 2 diabetes, dyslipidaemia, among others [97]. Orlistat in low dose (60 mg three times a day), managed to reduce body weight between 1.6% to 2.4% in 6 months of treatment [89].
Treatment with orlistat generates gastrointestinal adverse effects, such as oily staining and oils in stools, farting with discharge, faecal urgency, and increased defecation [99]. These side effects may cause patients to discontinue the treatment.
Liraglutide
Liraglutide is a subcutaneous injectable Glucagon-Like Peptide-1 (GLP-1) agonist approved in 2010 for the treatment of T2DM at a dose of 1.8 mg per day. The data show that liraglutide acts on POMC/CART (pro-opiomelanocortin/The cocaine-and amphetamine-regulated transcript) neurons, decreasing appetite and increasing levels of satiety, in addition to generating a transient effect of slowing gastric emptying [100,101]. Another effect of liraglutide is to increase insulin release and suppress glucagon secretion when elevated blood glucose occurs [102].Nausea, vomiting, heartburn, constipation and diarrhoea are the most common adverse effects of liraglutide, especially nausea, which has an incidence of at least 40%, which is due to a transient decrease in gastric emptying [89,103].
Phentermine
Phentermine, chemically similar to amphetamine, is a sympathomimetic amine with an anorexigenic effect, which increases the secretion of norepinephrine and dopamine in the central nervous system. The action of phentermine is due to the inhibition of neuropeptide Y/agouti-related peptide (NPY/AGRP) secreting neurons and an increase in energy expenditure [104].
Phentermine is approved for use for 3 months. Formulations of 18.75 and 37.5 mg are found, although a 30 mg prolonged-release formulation was recently incorporated [97].Headache, insomnia, increased blood pressure, tachycardia and palpitations, rhabdomyolysis, and intracranial bleeding/stroke are the most common adverse effects of phentermine use [105,106].
Naltrexone/Bupropion
Bupropion, an approved smoking-cessation antidepressant agent, has been shown to promote significant weight loss in obese patients ,inducing satiety by enhancing the production and secretion of α-MSH (α-melanocyte-stimulating hormone) and β-endorphins from pro-opiomelanocortin cells in the arcuate nucleus of the hypothalamus [107]. Naltrexone, an antagonist of µ-opioid receptor, used for the treatment of alcohol and opiate dependence, can decrease food cravings and intake, causing weight loss in subjects receiving this treatment [108].The most common adverse effects of the naltrexone/bupropion combination are nausea, constipation, headache, vomiting, insomnia, dry mouth, dizziness, and diarrhoea [91].
Phentermine/Topiramate
As described above, phentermine has sympathomimetic actions, a noradrenergic agonist, which increases the secretion of noradrenaline, dopamine and serotonin [109]. Topiramate is currently used in the treatment of epilepsy and migraines, since it is an α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid/kainate (AMPA/KA) glutamate receptor antagonist. Topiramate has a GABAergic mechanism of action, but it is also an anorexigenic and is currently used for the treatment of obesity [110].
The most common adverse effects are paraesthesia, dry mouth, constipation, insomnia, dizziness, and dysgeusia [111].
Diethylpropion
Diethylpropion is an anorexigenic agent with a mechanism of action is similar to the antidepressant agent bupropion. The drugs used for the treatment of obesity are commonly called anorexigenics and their primary action is the suppression of appetite, either at the central or metabolic level. Diethylpropion acts at the level of the central nervous and cardiovascular systems, raising blood pressure, by inhibiting the reuptake of dopamine and norepinephrine [112].The most common side effects are dry mouth, insomnia, and stimulation of the nervous system [112], Therefore, extreme precautions should be taken when using this drug , since it may generate dependence [113].
Figure 5.
Pharmacological and non-pharmacological management of the overweight-obesity patient.
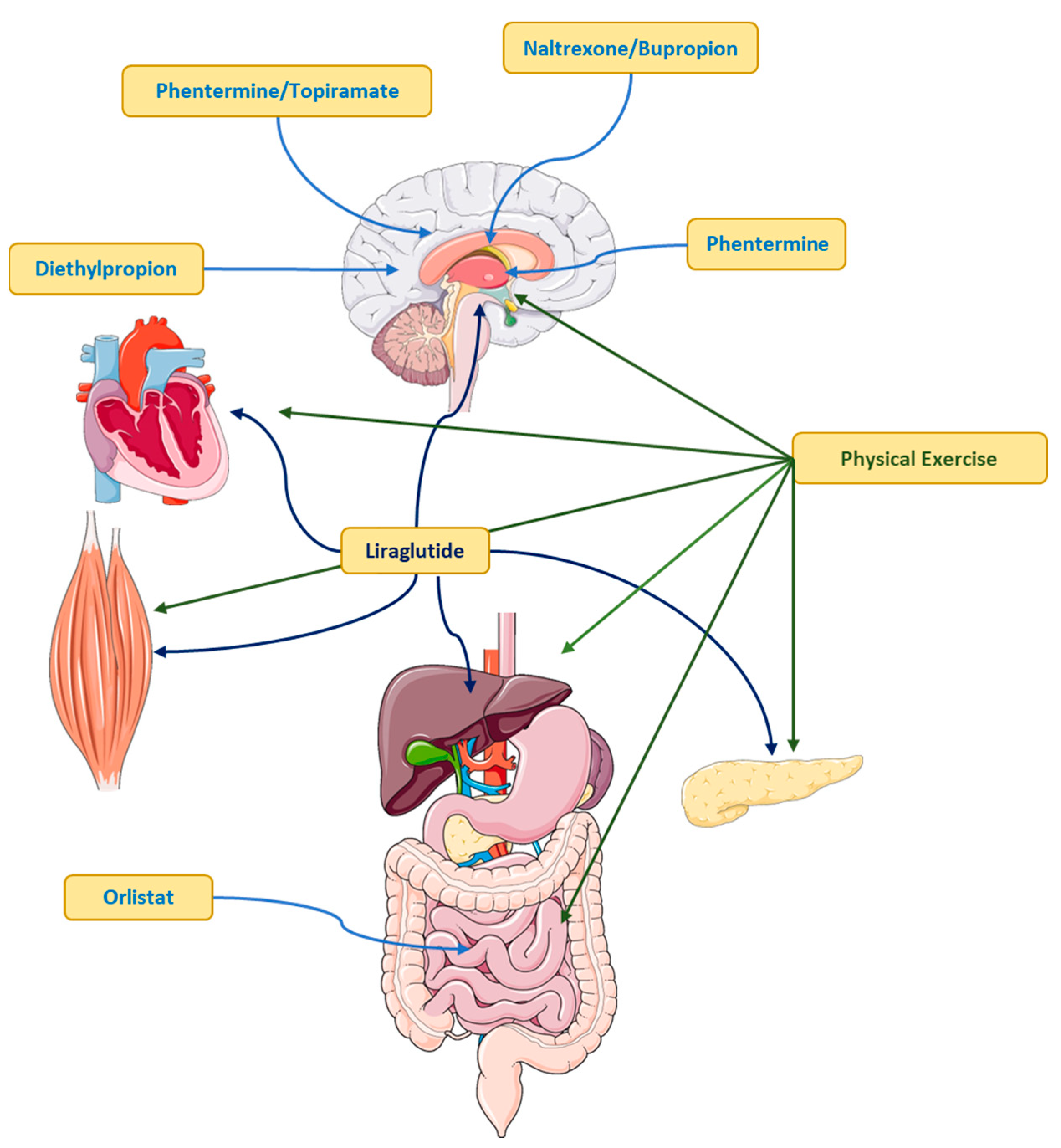
Physical Exercise as Treatment
The benefits of physical exercise have been shown to prevent all causes of mortality, including cardiovascular diseases, metabolic diseases and cancer [114]. Exercise reduces the risk of death by preventing the development of metabolic pathologies and protecting against chronic diseases. Organ-organ interaction involves muscle contraction at the molecular level as an emerging exercise-related field (see Figure 6) [115]. In addition, adipocytokines mediate the relationship between adipose tissue and other tissues, such as the brain, as well as metabolic functions during tissue activation [116]. The pro-inflammatory role of adipocytokines has been well identified (see above) and their secretion is increased in obesity, causing metabolic and cardiovascular diseases, among others [114].
The benefits induced by physical exercise are well known to prevent the harmful effects of proinflammatory adipocytokines through proteins secreted by the muscle (myocytokines or myokines) [115]. In recent years, several studies have shown that acute aerobic exercise is an important mediator of the systemic anti-inflammatory response [117]. Muscle contraction induced by physical exercise, regardless of intensity, volume, or density, produces an increase in gene expression and secretion of IL-6 into plasma [118]. After intense physical exercise, IL-1 receptor antagonist (IL1-ra) and soluble tumour necrosis factor I and II receptors (TNF I and II) are increased in plasma. This set of changes is the so-called “anti-inflammatory effect” [119].
Given this condition, the proposed hypothesis is that regular physical exercise (chronic effect) exerts an anti-inflammatory effect leading to protection against chronic inflammation produced by proinflammatory cytokines and C-reactive protein [118]. However, most adults, who have chronic non-communicable diseases, do not reach exercise levels according to guidelines [91], in addition to a reduction in physical activity levels and an increase in sedentary behaviour [120], therefore, overweight or obese adults should consider increasing physical activity as part of comprehensive obesity treatment. Physical exercise offers a wide range of health benefits and with just 150 minutes of physical activity a week (walking, cycling or resistance exercises) the expected benefits can be obtained.
Conclusions
Obesity epidemic is perhaps the single most extensive, expensive and penetrant condition or disease in the present time, caused by a multitude of factors, from genetics to environmental, with shared responsibilities through all aspects of human life. Far behind are the days when the pathophysiology of obesity was discussed and understood in the context of the of excessive caloric intake and its accumulation within the adipose tissue; now it is evident that the interplay of all systems needs to be considered to understand and approach its solution. An understanding that considers the contributions of secreted molecules, such as proteins, hormones, nucleic acids and metabolites by different tissues, that affects the functions and communication of the adipose tissue with the rest of the human body will be central to understand their particular role in sustaining a healthy metabolic homeostasis or not.
Improving our current understanding of the several dimensions of obesity, including propensity to regain weight, individual differences in pathogenesis and response to treatments, is necessary for designing effective interventions, especially considering how to address the additional complications associated to obesity such as diabetes, hypertension, and kidney failure, among others.
Small changes induced by physical exercise can produce significant benefits in patients. It is well known that physical activity or regular exercise can prevent the development of chronic diseases. Muscle contractions associated with physical activity or exercise increases the secretion of myocytokines, which could be potential candidates to provide beneficial effects by stimulating metabolic pathways, improving glucose uptake and fatty acid oxidation, as well as muscular growth and regeneration. The synergy between exercise, mental and physical health cannot be underscored.
What is not clear, are the precise mechanisms, the individual and specific differences, and how they could be tailor targeted, perhaps not individually as of yet, but narrowing down it terms of gender, ethnicity or age groups. The advent and eventual synergy of all the omics, artificial intelligence and other technological innovations will aid in this quest. Therapeutic strategies aimed at preserving or restoring adipose tissue functionality hold numerous unknown opportunities to improve human wellbeing. Nonetheless, significant efforts should be globally agreed in terms of education and lifestyles changes towards having more harmonious, sustainable and fulfilled lifestyles, where preventions efforts should diminish the need of reactive interventions.
References
- Booth, A. Magnuson, J. Fouts, M.T. Foster, Adipose tissue: an endocrine organ playing a role in metabolic regulation. Horm Mol Biol Clin Investig 2016, 26, 25–42. [CrossRef] [PubMed]
- D. García-Torres, Castellanos-González, M., Cedeño-Morales, R., Benet-Rodríguez, M., Ramírez-Arteaga, I., Adipose Tissue as an Endocrine Gland. Pathophysiological Implications, Revista Finlay [revista en Internet]. 2011 [citado 2017 Ene 5]; 1(2): [aprox. 20p.]. Disponible en: http://www.revfinlay.sld.cu/index.php/finlay/article/view/39, 2017.
- A.E. Ringel, J.M. Drijvers, G.J. Baker, A. Catozzi, J.C. García-Cañaveras, B.M. Gassaway, B.C. Miller, V.R. Juneja, T.H. Nguyen, S. Joshi, C.H. Yao, H. Yoon, P.T. Sage, M.W. LaFleur, J.D. Trombley, C.A. Jacobson, Z. Maliga, S.P. Gygi, P.K. Sorger, J.D. Rabinowitz, A.H. Sharpe, M.C. Haigis, Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.
- L. Gartner, Hiatt, J., Color Textbook of Histology, McGraw-Hill Interamericana Editores, S.A. de C.V., 2007.
- F. Wang, Vihma, V., Soronen, J., Turpeinen, U., Hämäläinen, E., Savolainen-Peltonen, H., et al., 17β estradiol and estradiol fatty acyl esters and estrogen-converting enzyme expression in adipose tissue in obese men and women, J Clin Endochrinol Metab, 2013, pp. 4923–4931.
- G. Vega-Robledo, Rico-Rosillo, MG., Adipose tissue: immune function and alterations caused by obesity, Rev Alerg Mex, 2016, pp. 340–353.
- K. Svensson, Long, JZ., Jedrychowski, MP., Cohen, P., Lo, JC., Serag, S., et al., A secreted Slit2 fragment regulates adipose tissue thermogenesis and metabolic function, Cell Metab, 2016, pp. 454–466.
- Chernukha, Fedulova, LV., Kotenkova, EA., White, beige and brown adipose tissue: structure, function, specific features and possibility formation and divergence in pigs, Foods and Raw Materials, 2022, pp. 10–18.
- S. O'Neill, L. O'Driscoll, Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev 2015, 16, 1–12. [CrossRef] [PubMed]
- J.C. Rathmell, Obesity, Immunity, and Cancer. N Engl J Med 2021, 384, 1160–1162. [CrossRef]
- M. Fasshauer, M. Blüher, Adipokines in health and disease. Trends Pharmacol Sci 2015, 36, 461–70. [CrossRef]
- L. Szablewski, Introductory Chapter: Adipose Tissue, IntechOpen, 2019.
- G. Atzmon, X.M. Yang, R. Muzumdar, X.H. Ma, I. Gabriely, N. Barzilai, Differential gene expression between visceral and subcutaneous fat depots, Horm Metab Res 34(11-12) (2002) 622-8.
- J.P. Bastard, M. Maachi, C. Lagathu, M.J. Kim, M. Caron, H. Vidal, J. Capeau, B. Feve, Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 2006, 17, 4–12.
- J.N. Fain, A.K. Madan, M.L. Hiler, P. Cheema, S.W. Bahouth, Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–82. [CrossRef]
- H.M. Choi, H.M. Doss, K.S. Kim, Multifaceted Physiological Roles of Adiponectin in Inflammation and Diseases. Int J Mol Sci 2020, 21.
- A.-R. Aida-Souki, N.J., Prieto-Fuenmayor, C., Cano-Ponce, C., Aspectos básicos en obesidad, Ediciones Universidad Simón Bolívar, Barranquilla, Colombia:, 2018, pp. 1–44.
- D. Ulbricht, J. Pippel, S. Schultz, R. Meier, N. Sträter, J.T. Heiker, A unique serpin P1' glutamate and a conserved β-sheet C arginine are key residues for activity, protease recognition and stability of serpinA12 (vaspin). Biochem J 2015, 470, 357–67. [CrossRef]
- Fukuhara, M. Matsuda, M. Nishizawa, K. Segawa, M. Tanaka, K. Kishimoto, Y. Matsuki, M. Murakami, T. Ichisaka, H. Murakami, E. Watanabe, T. Takagi, M. Akiyoshi, T. Ohtsubo, S. Kihara, S. Yamashita, M. Makishima, T. Funahashi, S. Yamanaka, R. Hiramatsu, Y. Matsuzawa, I. Shimomura, Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–30.
- J. DeFuria, A.C. Belkina, M. Jagannathan-Bogdan, J. Snyder-Cappione, J.D. Carr, Y.R. Nersesova, D. Markham, K.J. Strissel, A.A. Watkins, M. Zhu, J. Allen, J. Bouchard, G. Toraldo, R. Jasuja, M.S. Obin, M.E. McDonnell, C. Apovian, G.V. Denis, B.S. Nikolajczyk, B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A 2013, 110, 5133–8.
- Y. Ren, H. Y. Ren, H. Zhao, C. Yin, X. Lan, L. Wu, X. Du, H.R. Griffiths, D. Gao, Adipokines, Hepatokines and Myokines: Focus on Their Role and Molecular Mechanisms in Adipose Tissue Inflammation, Front Endocrinol (Lausanne) 13 (2022) 873699.
- C.o.O.G. WHO, Switzerland) & World Health Organization, Obesity: Preventing and Managing the Global Epidemic. Report of a WHO Consultation. Report of a WHO Consultation., World Health Organ Tech Rep Ser, 2000, pp. 1–253.
- P. Poirier, T.D. Giles, G.A. Bray, Y. Hong, J.S. Stern, F.X. Pi-Sunyer, R.H. Eckel, Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol 2006, 26, 968–76. [CrossRef]
- C.M. Apovian, Obesity: definition, comorbidities, causes, and burden, Am J Manag Care 22(7 Suppl) (2016) s176-85.
- X. Lin, H. Li, Obesity: Epidemiology, Pathophysiology, and Therapeutics, Front Endocrinol (Lausanne) 12 (2021) 706978.
- J. Upadhyay, O. Farr, N. Perakakis, W. Ghaly, C. Mantzoros, Obesity as a Disease. Med Clin North Am 2018, 102, 13–33.
- G. Whitlock, S. Lewington, P. Sherliker, R. Clarke, J. Emberson, J. Halsey, N. Qizilbash, R. Collins, R. Peto, P.S. Collaboration, Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet 2009, 373, 1083–96. [CrossRef]
- G.A. Bray, K.K. Kim, J.P.H. Wilding, W.O. Federation, Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev 2017, 18, 715–723. [CrossRef] [PubMed]
- B.A. Swinburn, G. Sacks, K.D. Hall, K. McPherson, D.T. Finegood, M.L. Moodie, S.L. Gortmaker, The global obesity pandemic: shaped by global drivers and local environments. Lancet 2011, 378, 804–14. [CrossRef] [PubMed]
- K. Sahoo, B. Sahoo, A.K. Choudhury, N.Y. Sofi, R. Kumar, A.S. Bhadoria, Childhood obesity: causes and consequences. J Family Med Prim Care 2015, 4, 187–92.
- WHO, Obesidad y Sobrepeso, 2021.
- D. Gómez-Pérez, M. Cancino, P.I. Moreno, M.S. Ortiz, Weight Stigma, Chronic Stress, Unhealthy Diet, and Obesity in Chilean Adults. Int J Behav Med 2021, 28, 292–298. [CrossRef]
- Instituto de Nutrición y Tecnología de los Alimentos, ESPECIAL: Obesidad, la otra pandemia, INTA, 2021.
- S.B. Heymsfield, T.A. Wadden, Mechanisms, Pathophysiology, and Management of Obesity. N Engl J Med 2017, 376, 1492.
- T.S. Church, D.M. Thomas, C. Tudor-Locke, P.T. Katzmarzyk, C.P. Earnest, R.Q. Rodarte, C.K. Martin, S.N. Blair, C. Bouchard, Trends over 5 decades in U.S. occupation-related physical activity and their associations with obesity. PLoS One 2011, 6, e19657.
- V.Y. Njike, T.M. Smith, O. Shuval, K. Shuval, I. Edshteyn, V. Kalantari, A.L. Yaroch, Snack Food, Satiety, and Weight. Adv Nutr 2016, 7, 866–78.
- V. Medici, S.A. McClave, K.R. Miller, Common Medications Which Lead to Unintended Alterations in Weight Gain or Organ Lipotoxicity. Curr Gastroenterol Rep 2016, 18, 2. [CrossRef] [PubMed]
- Y. Wu, H. Duan, X. Tian, C. Xu, W. Wang, W. Jiang, Z. Pang, D. Zhang, Q. Tan, Genetics of Obesity Traits: A Bivariate Genome-Wide Association Analysis, Front Genet 9 (2018) 179.
- D. Shungin, T.W. Winkler, D.C. Croteau-Chonka, T. Ferreira, A.E. Locke, R. Mägi, R.J. Strawbridge, T.H. Pers, K. Fischer, A.E. Justice, T. Workalemahu, J.M.W. Wu, M.L. Buchkovich, N.L. Heard-Costa, T.S. Roman, A.W. Drong, C. Song, S. Gustafsson, F.R. Day, T. Esko, T. Fall, Z. Kutalik, J. Luan, J.C. Randall, A. Scherag, S. Vedantam, A.R. Wood, J. Chen, R. Fehrmann, J. Karjalainen, B. Kahali, C.T. Liu, E.M. Schmidt, D. Absher, N. Amin, D. Anderson, M. Beekman, J.L. Bragg-Gresham, S. Buyske, A. Demirkan, G.B. Ehret, M.F. Feitosa, A. Goel, A.U. Jackson, T. Johnson, M.E. Kleber, K. Kristiansson, M. Mangino, I.M. Leach, C. Medina-Gomez, C.D. Palmer, D. Pasko, S. Pechlivanis, M.J. Peters, I. Prokopenko, A. Stančáková, Y.J. Sung, T. Tanaka, A. Teumer, J.V. Van Vliet-Ostaptchouk, L. Yengo, W. Zhang, E. Albrecht, J. Ärnlöv, G.M. Arscott, S. Bandinelli, A. Barrett, C. Bellis, A.J. Bennett, C. Berne, M. Blüher, S. Böhringer, F. Bonnet, Y. Böttcher, M. Bruinenberg, D.B. Carba, I.H. Caspersen, R. Clarke, E.W. Daw, J. Deelen, E. Deelman, G. Delgado, A.S. Doney, N. Eklund, M.R. Erdos, K. Estrada, E. Eury, N. Friedrich, M.E. Garcia, V. Giedraitis, B. Gigante, A.S. Go, A. Golay, H. Grallert, T.B. Grammer, J. Gräßler, J. Grewal, C.J. Groves, T. Haller, G. Hallmans, C.A. Hartman, M. Hassinen, C. Hayward, K. Heikkilä, K.H. Herzig, Q. Helmer, H.L. Hillege, O. Holmen, S.C. Hunt, A. Isaacs, T. Ittermann, A.L. James, I. Johansson, T. Juliusdottir, I.P. Kalafati, L. Kinnunen, W. Koenig, I.K. Kooner, W. Kratzer, C. Lamina, K. Leander, N.R. Lee, P. Lichtner, L. Lind, J. Lindström, S. Lobbens, M. Lorentzon, F. Mach, P.K. Magnusson, A. Mahajan, W.L. McArdle, C. Menni, S. Merger, E. Mihailov, L. Milani, R. Mills, A. Moayyeri, K.L. Monda, S.P. Mooijaart, T.W. Mühleisen, A. Mulas, G. Müller, M. Müller-Nurasyid, R. Nagaraja, M.A. Nalls, N. Narisu, N. Glorioso, I.M. Nolte, M. Olden, N.W. Rayner, F. Renstrom, J.S. Ried, N.R. Robertson, L.M. Rose, S. Sanna, H. Scharnagl, S. Scholtens, B. Sennblad, T. Seufferlein, C.M. Sitlani, A.V. Smith, K. Stirrups, H.M. Stringham, J. Sundström, M.A. Swertz, A.J. Swift, A.C. Syvänen, B.O. Tayo, B. Thorand, G. Thorleifsson, A. Tomaschitz, C. Troffa, F.V. van Oort, N. Verweij, J.M. Vonk, L.L. Waite, R. Wennauer, T. Wilsgaard, M.K. Wojczynski, A. Wong, Q. Zhang, J.H. Zhao, E.P. Brennan, M. Choi, P. Eriksson, L. Folkersen, A. Franco-Cereceda, A.G. Gharavi, Å. Hedman, M.F. Hivert, J. Huang, S. Kanoni, F. Karpe, S. Keildson, K. Kiryluk, L. Liang, R.P. Lifton, B. Ma, A.J. McKnight, R. McPherson, A. Metspalu, J.L. Min, M.F. Moffatt, G.W. Montgomery, J.M. Murabito, G. Nicholson, D.R. Nyholt, C. Olsson, J.R. Perry, E. Reinmaa, R.M. Salem, N. Sandholm, E.E. Schadt, R.A. Scott, L. Stolk, E.E. Vallejo, H.J. Westra, K.T. Zondervan, P. Amouyel, D. Arveiler, S.J. Bakker, J. Beilby, R.N. Bergman, J. Blangero, M.J. Brown, M. Burnier, H. Campbell, A. Chakravarti, P.S. Chines, S. Claudi-Boehm, F.S. Collins, D.C. Crawford, J. Danesh, U. de Faire, E.J. de Geus, M. Dörr, R. Erbel, J.G. Eriksson, M. Farrall, E. Ferrannini, J. Ferrières, N.G. Forouhi, T. Forrester, O.H. Franco, R.T. Gansevoort, C. Gieger, V. Gudnason, C.A. Haiman, T.B. Harris, A.T. Hattersley, M. Heliövaara, A.A. Hicks, A.D. Hingorani, W. Hoffmann, A. Hofman, G. Homuth, S.E. Humphries, E. Hyppönen, T. Illig, M.R. Jarvelin, B. Johansen, P. Jousilahti, A.M. Jula, J. Kaprio, F. Kee, S.M. Keinanen-Kiukaanniemi, J.S. Kooner, C. Kooperberg, P. Kovacs, A.T. Kraja, M. Kumari, K. Kuulasmaa, J. Kuusisto, T.A. Lakka, C. Langenberg, L. Le Marchand, T. Lehtimäki, V. Lyssenko, S. Männistö, A. Marette, T.C. Matise, C.A. McKenzie, B. McKnight, A.W. Musk, S. Möhlenkamp, A.D. Morris, M. Nelis, C. Ohlsson, A.J. Oldehinkel, K.K. Ong, L.J. Palmer, B.W. Penninx, A. Peters, P.P. Pramstaller, O.T. Raitakari, T. Rankinen, D.C. Rao, T.K. Rice, P.M. Ridker, M.D. Ritchie, I. Rudan, V. Salomaa, N.J. Samani, J. Saramies, M.A. Sarzynski, P.E. Schwarz, A.R. Shuldiner, J.A. Staessen, V. Steinthorsdottir, R.P. Stolk, K. Strauch, A. Tönjes, A. Tremblay, E. Tremoli, M.C. Vohl, U. Völker, P. Vollenweider, J.F. Wilson, J.C. Witteman, L.S. Adair, M. Bochud, B.O. Boehm, S.R. Bornstein, C. Bouchard, S. Cauchi, M.J. Caulfield, J.C. Chambers, D.I. Chasman, R.S. Cooper, G. Dedoussis, L. Ferrucci, P. Froguel, H.J. Grabe, A. Hamsten, J. Hui, K. Hveem, K.H. Jöckel, M. Kivimaki, D. Kuh, M. Laakso, Y. Liu, W. März, P.B. Munroe, I. Njølstad, B.A. Oostra, C.N. Palmer, N.L. Pedersen, M. Perola, L. Pérusse, U. Peters, C. Power, T. Quertermous, R. Rauramaa, F. Rivadeneira, T.E. Saaristo, D. Saleheen, J. Sinisalo, P.E. Slagboom, H. Snieder, T.D. Spector, K. Stefansson, M. Stumvoll, J. Tuomilehto, A.G. Uitterlinden, M. Uusitupa, P. van der Harst, G. Veronesi, M. Walker, N.J. Wareham, H. Watkins, H.E. Wichmann, G.R. Abecasis, T.L. Assimes, S.I. Berndt, M. Boehnke, I.B. Borecki, P. Deloukas, L. Franke, T.M. Frayling, L.C. Groop, D.J. Hunter, R.C. Kaplan, J.R. O'Connell, L. Qi, D. Schlessinger, D.P. Strachan, U. Thorsteinsdottir, C.M. van Duijn, C.J. Willer, P.M. Visscher, J. Yang, J.N. Hirschhorn, M.C. Zillikens, M.I. McCarthy, E.K. Speliotes, K.E. North, C.S. Fox, I. Barroso, P.W. Franks, E. Ingelsson, I.M. Heid, R.J. Loos, L.A. Cupples, A.P. Morris, C.M. Lindgren, K.L. Mohlke, A. Consortium, C.D. Consortium, C. Consortium, G. Consortium, G. Consortium, GLGC, ICBP, I.E. Consortium, L.C. Study, M. Investigators, M. Consortium, P. Consortium, R. Consortium, New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015, 518, 187–196. [Google Scholar]
- V.V. Thaker, GENETIC AND EPIGENETIC CAUSES OF OBESITY. Adolesc Med State Art Rev 2017, 28, 379–405.
- H. Huvenne, B. Dubern, K. Clément, C. Poitou, Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes Facts 2016, 9, 158–73. [CrossRef] [PubMed]
- G. Koochakpour, Z. Esfandiar, F. Hosseini-Esfahani, P. Mirmiran, M.S. Daneshpour, B. Sedaghati-Khayat, F. Azizi, Evaluating the interaction of common FTO genetic variants, added sugar, and trans-fatty acid intakes in altering obesity phenotypes. Nutr Metab Cardiovasc Dis 2019, 29, 474–480. [CrossRef]
- N. Gupta, V. Jain, Prader Willi Syndrome - A Common Epigenetic Cause of Syndromic Obesity. Indian J Pediatr 2017, 84, 809–810. [CrossRef]
- H. Cena, L. Chiovato, R.E. Nappi, Obesity, Polycystic Ovary Syndrome, and Infertility: A New Avenue for GLP-1 Receptor Agonists. J Clin Endocrinol Metab 2020, 105.
- C.S. D'Angelo, C.P. Koiffmann, Copy number variants in obesity-related syndromes: review and perspectives on novel molecular approaches. J Obes 2012, 2012, 845480.
- M. Rosenbaum, R. Knight, R.L. Leibel, The gut microbiota in human energy homeostasis and obesity. Trends Endocrinol Metab 2015, 26, 493–501. [CrossRef]
- Wernstedt Asterholm, C. Tao, T.S. Morley, Q.A. Wang, F. Delgado-Lopez, Z.V. Wang, P.E. Scherer, Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 2014, 20, 103–18. [CrossRef] [PubMed]
- Y. Arita, S. Kihara, N. Ouchi, M. Takahashi, K. Maeda, J. Miyagawa, K. Hotta, I. Shimomura, T. Nakamura, K. Miyaoka, H. Kuriyama, M. Nishida, S. Yamashita, K. Okubo, K. Matsubara, M. Muraguchi, Y. Ohmoto, T. Funahashi, Y. Matsuzawa, Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999, 257, 79–83.
- M. Pigeyre, F.T. Yazdi, Y. Kaur, D. Meyre, Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin Sci (Lond) 2016, 130, 943–86. [CrossRef] [PubMed]
- A.A. van der Klaauw, I.S. Farooqi, The hunger genes: pathways to obesity. Cell 2015, 161, 119–132. [CrossRef]
- P.S. MacLean, J.A. Higgins, E.D. Giles, V.D. Sherk, M.R. Jackman, The role for adipose tissue in weight regain after weight loss, Obes Rev 16 Suppl 1 (2015) 45-54.
- C.N. Ochner, A.G. Tsai, R.F. Kushner, T.A. Wadden, Treating obesity seriously: when recommendations for lifestyle change confront biological adaptations. Lancet Diabetes Endocrinol 2015, 3, 232–4. [CrossRef]
- T. Tchkonia, T. Thomou, Y. Zhu, I. Karagiannides, C. Pothoulakis, M.D. Jensen, J.L. Kirkland, Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab 2013, 17, 644–656. [CrossRef]
- S.B. Heymsfield, H.H. Hu, W. Shen, O. Carmichael, Emerging Technologies and their Applications in Lipid Compartment Measurement. Trends Endocrinol Metab 2015, 26, 688–698. [CrossRef]
- A.J. McCullough, The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004, 8, 521–33. [CrossRef]
- J. Kaur, A comprehensive review on metabolic syndrome, Cardiol Res Pract 2014 (2014) 943162.
- J.E. Hall, A.A. da Silva, J.M. do Carmo, J. Dubinion, S. Hamza, S. Munusamy, G. Smith, D.E. Stec, Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 2010, 285, 17271–6. [CrossRef]
- M. Ortiz-Martínez, M. González-González, A.J. Martagón, V. Hlavinka, R.C. Willson, M. Rito-Palomares, Recent Developments in Biomarkers for Diagnosis and Screening of Type 2 Diabetes Mellitus. Curr Diab Rep 2022, 22, 95–115. [CrossRef]
- M. Zhang, T. Hu, S. Zhang, L. Zhou, Associations of Different Adipose Tissue Depots with Insulin Resistance: A Systematic Review and Meta-analysis of Observational Studies, Sci Rep 5 (2015) 18495.
- S.H. Lee, S.Y. Park, C.S. Choi, Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab J 2022, 46, 15–37. [CrossRef]
- Y.T. Wondmkun, Obesity, Insulin Resistance, and Type 2 Diabetes: Associations and Therapeutic Implications, Diabetes Metab Syndr Obes 13 (2020) 3611-3616.
- M.C. Petersen, G.I. Shulman, Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 2018, 98, 2133–2223. [CrossRef] [PubMed]
- T. Ota, Obesity-induced inflammation and insulin resistance, Front Endocrinol (Lausanne) 5 (2014) 204.
- U. Ozcan, E. Yilmaz, L. Ozcan, M. Furuhashi, E. Vaillancourt, R.O. Smith, C.Z. Görgün, G.S. Hotamisligil, Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–40. [CrossRef] [PubMed]
- M. Krssak, A. Brehm, E. Bernroider, C. Anderwald, P. Nowotny, C. Dalla Man, C. Cobelli, G.W. Cline, G.I. Shulman, W. Waldhäusl, M. Roden, Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 2004, 53, 3048–56.
- R.A. DeFronzo, D. Tripathy, Skeletal muscle insulin resistance is the primary defect in type 2 diabetes, Diabetes Care 32 Suppl 2(Suppl 2) (2009) S157-63.
- S.E. Kahn, R.L. Hull, K.M. Utzschneider, Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–6. [CrossRef] [PubMed]
- G.F. Lewis, A.C. Carpentier, S. Pereira, M. Hahn, A. Giacca, Direct and indirect control of hepatic glucose production by insulin. Cell Metab 2021, 33, 709–720. [CrossRef]
- A.E. Butler, J. Janson, S. Bonner-Weir, R. Ritzel, R.A. Rizza, P.C. Butler, Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–10. [CrossRef] [PubMed]
- G. Vazquez, S. Duval, D.R. Jacobs, K. Silventoinen, Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: a meta-analysis, Epidemiol Rev 29 (2007) 115-28.
- K.S.R. Thapa SD, Gautam S, Gyawali D., Dyslipidemia in Type 2 Diabetes mellitus, Journal of Pathology of Nepal, 2017, pp. 1149 - 1154.
- D.C. Chan, H.P. Barrett, G.F. Watts, Dyslipidemia in visceral obesity: mechanisms, implications, and therapy. Am J Cardiovasc Drugs 2004, 4, 227–46.
- Beers, M.J. Haas, N.C. Wong, A.D. Mooradian, Inhibition of apolipoprotein AI gene expression by tumor necrosis factor alpha: roles for MEK/ERK and JNK signaling. Biochemistry 2006, 45, 2408–13. [CrossRef] [PubMed]
- A.D. Mooradian, M.J. Haas, K.R. Wehmeier, N.C. Wong, Obesity-related changes in high-density lipoprotein metabolism. Obesity (Silver Spring) 2008, 16, 1152–60. [CrossRef]
- M.A. Alpert, J. Omran, B.P. Bostick, Effects of Obesity on Cardiovascular Hemodynamics, Cardiac Morphology, and Ventricular Function. Curr Obes Rep 2016, 5, 424–434. [CrossRef] [PubMed]
- Grillo, L. Salvi, P. Coruzzi, P. Salvi, G. Parati, Sodium Intake and Hypertension, Nutrients 2019, 11, 2019; 11.
- M.E. Hall, J.M. do Carmo, A.A. da Silva, L.A. Juncos, Z. Wang, J.E. Hall, Obesity, hypertension, and chronic kidney disease, Int J Nephrol Renovasc Dis 7 (2014) 75-88.
- D. Susic, J. Varagic, Obesity: A Perspective from Hypertension. Med Clin North Am 2017, 101, 139–157.
- Virdis, S. Taddei, Endothelial Dysfunction in Resistance Arteries of Hypertensive Humans: Old and New Conspirators. J Cardiovasc Pharmacol 2016, 67, 451–7. [Google Scholar] [CrossRef] [PubMed]
- Y. Lu, K. Hajifathalian, M. Ezzati, M. Woodward, E.B. Rimm, G. Danaei, G.B.o.M.R.F.f.C.D.C.B.M. Effects), Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1·8 million participants. Lancet 2014, 383, 970–83.
- T.M. Barber, S. Franks, Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf) 2021, 95, 531–541. [CrossRef]
- T.M. Barber, M.I. McCarthy, J.A. Wass, S. Franks, Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf) 2006, 65, 137–45.
- Purwar, S. Nagpure, Insulin Resistance in Polycystic Ovarian Syndrome. Cureus 2022, 14, e30351. [Google Scholar]
- W. Kositanurit, D. Muntham, S. Udomsawaengsup, N. Chirakalwasan, Prevalence and associated factors of obstructive sleep apnea in morbidly obese patients undergoing bariatric surgery. Sleep Breath 2018, 22, 251–256. [CrossRef]
- A.B. Newman, G. Foster, R. Givelber, F.J. Nieto, S. Redline, T. Young, Progression and regression of sleep-disordered breathing with changes in weight: the Sleep Heart Health Study. Arch Intern Med 2005, 165, 2408–13. [CrossRef]
- G.S. Hamilton, S.A. Joosten, Obstructive sleep apnoea and obesity. Aust Fam Physician 2017, 46, 460–463.
- E.E. Calle, C. Rodriguez, K. Walker-Thurmond, M.J. Thun, Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003, 348, 1625–38. [CrossRef] [PubMed]
- J. Park, D.M. Euhus, P.E. Scherer, Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev 2011, 32, 550–70. [CrossRef] [PubMed]
- K.M. Gadde, J.W. Apolzan, H.R. Berthoud, Pharmacotherapy for Patients with Obesity. Clin Chem 2018, 64, 118–129. [CrossRef] [PubMed]
- M.D. Jensen, D.H. Ryan, C.M. Apovian, J.D. Ard, A.G. Comuzzie, K.A. Donato, F.B. Hu, V.S. Hubbard, J.M. Jakicic, R.F. Kushner, C.M. Loria, B.E. Millen, C.A. Nonas, F.X. Pi-Sunyer, J. Stevens, V.J. Stevens, T.A. Wadden, B.M. Wolfe, S.Z. Yanovski, A.C.o.C.A.H.A.T.F.o.P. Guidelines, O. Society, 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society, J Am Coll Cardiol 63(25 Pt B) (2014) 2985-3023.
- Y. Preiss Contreras, X. Ramos Salas, C. Ávila Oliver, M.A. Saquimux Contreras, R. Muñoz Claro, C. Canales Ferrada, C.C.p.e.E.d.l. Obesidad, Obesity in adults: Clinical practice guideline adapted for Chile. Medwave 2022, 22, e2649.
- K.M. Gadde, C.K. Martin, H.R. Berthoud, S.B. Heymsfield, Obesity: Pathophysiology and Management. J Am Coll Cardiol 2018, 71, 69–84.
- R.R. Wing, D.F. Tate, A.A. Gorin, H.A. Raynor, J.L. Fava, A self-regulation program for maintenance of weight loss. N Engl J Med 2006, 355, 1563–71. [CrossRef]
- C.L. Rock, S.W. Flatt, N.E. Sherwood, N. Karanja, B. Pakiz, C.A. Thomson, Effect of a free prepared meal and incentivized weight loss program on weight loss and weight loss maintenance in obese and overweight women: a randomized controlled trial. JAMA 2010, 304, 1803–10. [CrossRef]
- E.Y. Lee, K.H. Yoon, Epidemic obesity in children and adolescents: risk factors and prevention. Front Med 2018, 12, 658–666. [CrossRef]
- Nguyen, J. Clements, Obesity management among patients with type 2 diabetes and prediabetes: a focus on lifestyle modifications and evidence of antiobesity medications. Expert Rev Endocrinol Metab 2017, 12, 303–313. [Google Scholar] [CrossRef]
- M.P. Pedersen SD, Wharton S., Canadian Adult Obesity Clinical Practice Guidelines: Pharmacotherapy in Obesity Management, Obesity Canada, 2020.
- E.S. Leblanc, E. O'Connor, E.P. Whitlock, C.D. Patnode, T. Kapka, Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2011, 155, 434–47. [CrossRef]
- Rucker, R. Padwal, S.K. Li, C. Curioni, D.C. Lau, Long term pharmacotherapy for obesity and overweight: updated meta-analysis. BMJ 2007, 335, 1194–9. [CrossRef]
- Secher, J. Jelsing, A.F. Baquero, J. Hecksher-Sørensen, M.A. Cowley, L.S. Dalbøge, G. Hansen, K.L. Grove, C. Pyke, K. Raun, L. Schäffer, M. Tang-Christensen, S. Verma, B.M. Witgen, N. Vrang, L. Bjerre Knudsen, The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest 2014, 124, 4473–88. [Google Scholar]
- S. Sisley, R. Gutierrez-Aguilar, M. Scott, D.A. D'Alessio, D.A. Sandoval, R.J. Seeley, Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. J Clin Invest 2014, 124, 2456–63. [CrossRef] [PubMed]
- ISP, Ficha Registro ISP SAXENDA, 2022.
- N.N. Canada, SAXENDA® liraglutide, 2017.
- D.B. Allison, K.M. Gadde, W.T. Garvey, C.A. Peterson, M.L. Schwiers, T. Najarian, P.Y. Tam, B. Troupin, W.W. Day, Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring) 2012, 20, 330–42. [CrossRef]
- R.B. Rothman, E.J. Hendricks, Phentermine cardiovascular safety, Am J Emerg Med 2009, 27, 1010–3. [CrossRef]
- K.E. Steidl, W. Darko, L.A. Probst, J.A. Noviasky, S. Nasser, Rhabdomyolysis associated with phentermine. Am J Health Syst Pharm 2010, 67, 1929–32.
- K.M. Gadde, G.L. Xiong, Bupropion for weight reduction. Expert Rev Neurother 2007, 7, 17–24. [CrossRef] [PubMed]
- S.R. Smith, K. Fujioka, A.K. Gupta, S.K. Billes, C. Burns, D. Kim, E. Dunayevich, F.L. Greenway, Combination therapy with naltrexone and bupropion for obesity reduces total and visceral adiposity. Diabetes Obes Metab 2013, 15, 863–6. [CrossRef] [PubMed]
- R.B. Rothman, M.H. Baumann, Appetite suppressants, cardiac valve disease and combination pharmacotherapy. Am J Ther 2009, 16, 354–64. [CrossRef] [PubMed]
- M.A. Scudeler, S. Morreale, L. Doretto-Silva, G. Petri, J.F.R.D. Santos, C. Nassis, O.M.T. Correa, J.M. Veridiano, Effects of topiramate, bupropion and naltrexone isolated or combined on subcutaneous adipose tissue in obese rats, Einstein (Sao Paulo) 20 (2022) eAO5587.
- F. Food and Drug Administration, Advisory Committee Meeting for Phentermine/Topiramate (Qnexa), Division of Metabolism and Endocrinology Products (DMEP), Office of Drug Evaluation II, Center for Drug Evaluation and Research, Silver Spring, MD, 2010.
- H.R. Arias, A. Santamaría, S.F. Ali, Pharmacological and neurotoxicological actions mediated by bupropion and diethylpropion, Int Rev Neurobiol 88 (2009) 223-55.
- H.S. Jones, Diethylpropion dependence. Med J Aust 1968, 1, 267. [CrossRef]
- B. So, H.J. Kim, J. Kim, W. Song, Exercise-induced myokines in health and metabolic diseases. Integr Med Res 2014, 3, 172–179. [CrossRef] [PubMed]
- B.K. Pedersen, M.A. Febbraio, Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 2012, 8, 457–65. [CrossRef] [PubMed]
- P.E. Scherer, Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–45.
- B.K. Pedersen, Muscles and their myokines, J Exp Biol 214(Pt 2) (2011) 337-46.
- L.G. Leal, M.A. Lopes, M.L. Batista, Physical Exercise-Induced Myokines and Muscle-Adipose Tissue Crosstalk: A Review of Current Knowledge and the Implications for Health and Metabolic Diseases, Front Physiol 9 (2018) 1307.
- A.M. Petersen, B.K. Pedersen, The anti-inflammatory effect of exercise. J Appl Physiol (1985) 2005, 98, 1154–62. [CrossRef]
- S.W. Ng, B.M. Popkin, Time use and physical activity: a shift away from movement across the globe. Obes Rev 2012, 13, 659–80. [CrossRef]
Figure 1.
Adipocytokines and other molecules secreted by Adipose Tissue. (+) beneficial effect on energy homeostasis; (-) negative effect on energy homeostasis.
Figure 1.
Adipocytokines and other molecules secreted by Adipose Tissue. (+) beneficial effect on energy homeostasis; (-) negative effect on energy homeostasis.
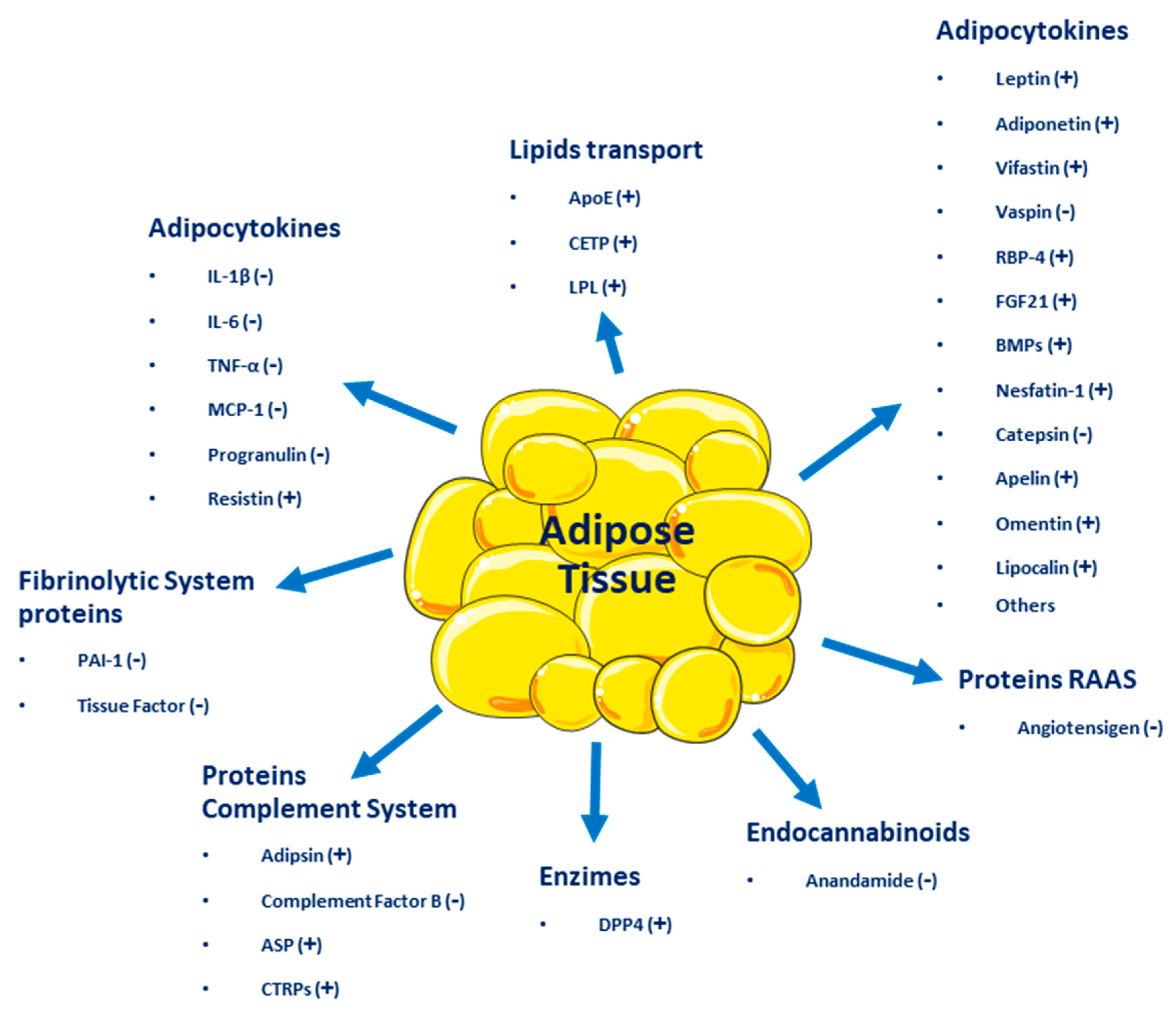
Figure 2.
Some pathways by which excess adipose tissue produces risk factors or chronic diseases. ANS, Autonomic Nervous System; RAAs, Renin-Angiotensin-Aldosterone System.
Figure 2.
Some pathways by which excess adipose tissue produces risk factors or chronic diseases. ANS, Autonomic Nervous System; RAAs, Renin-Angiotensin-Aldosterone System.
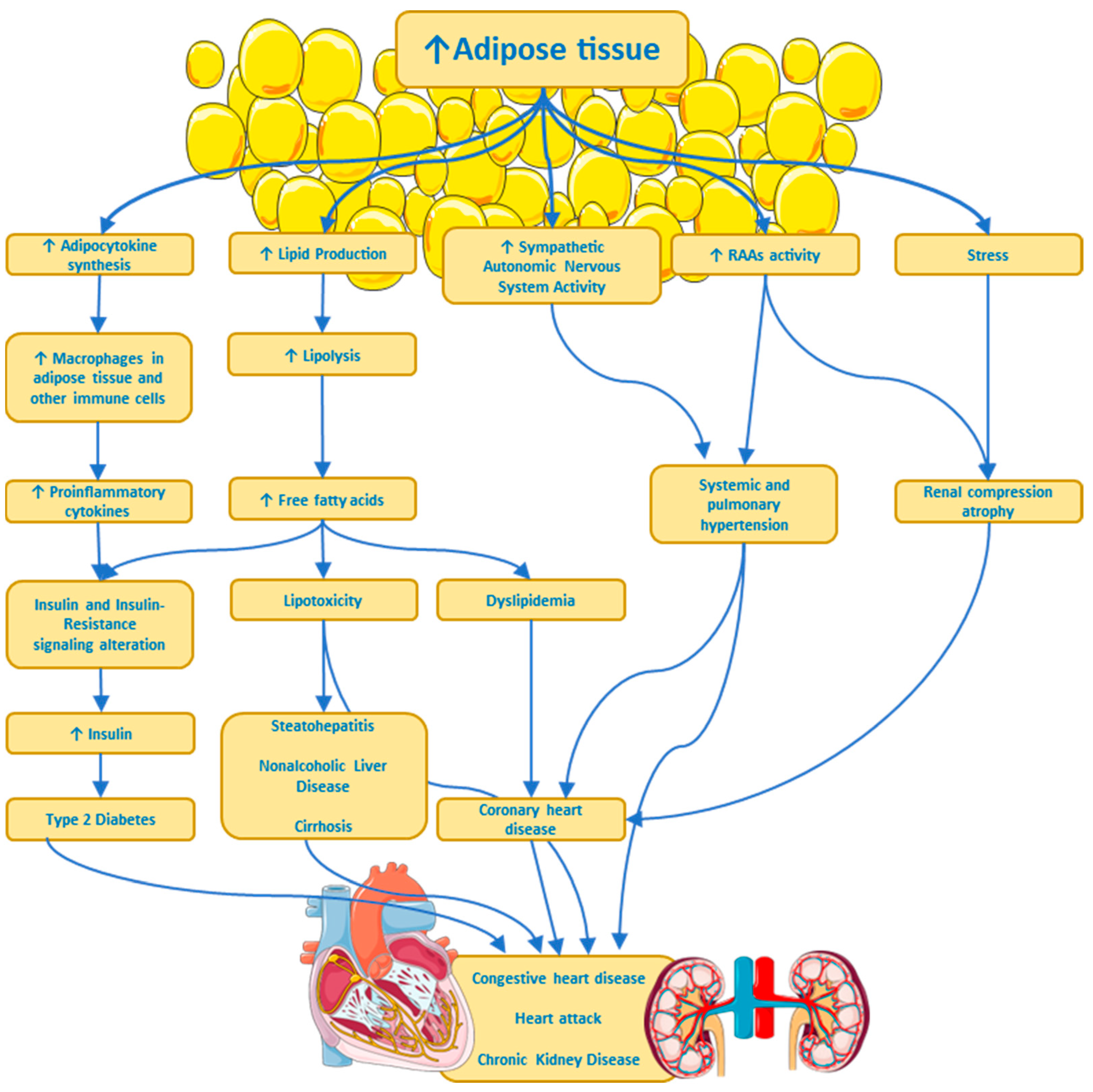
Figure 4.
Complications and Comorbidities associated with overweight - obesity.
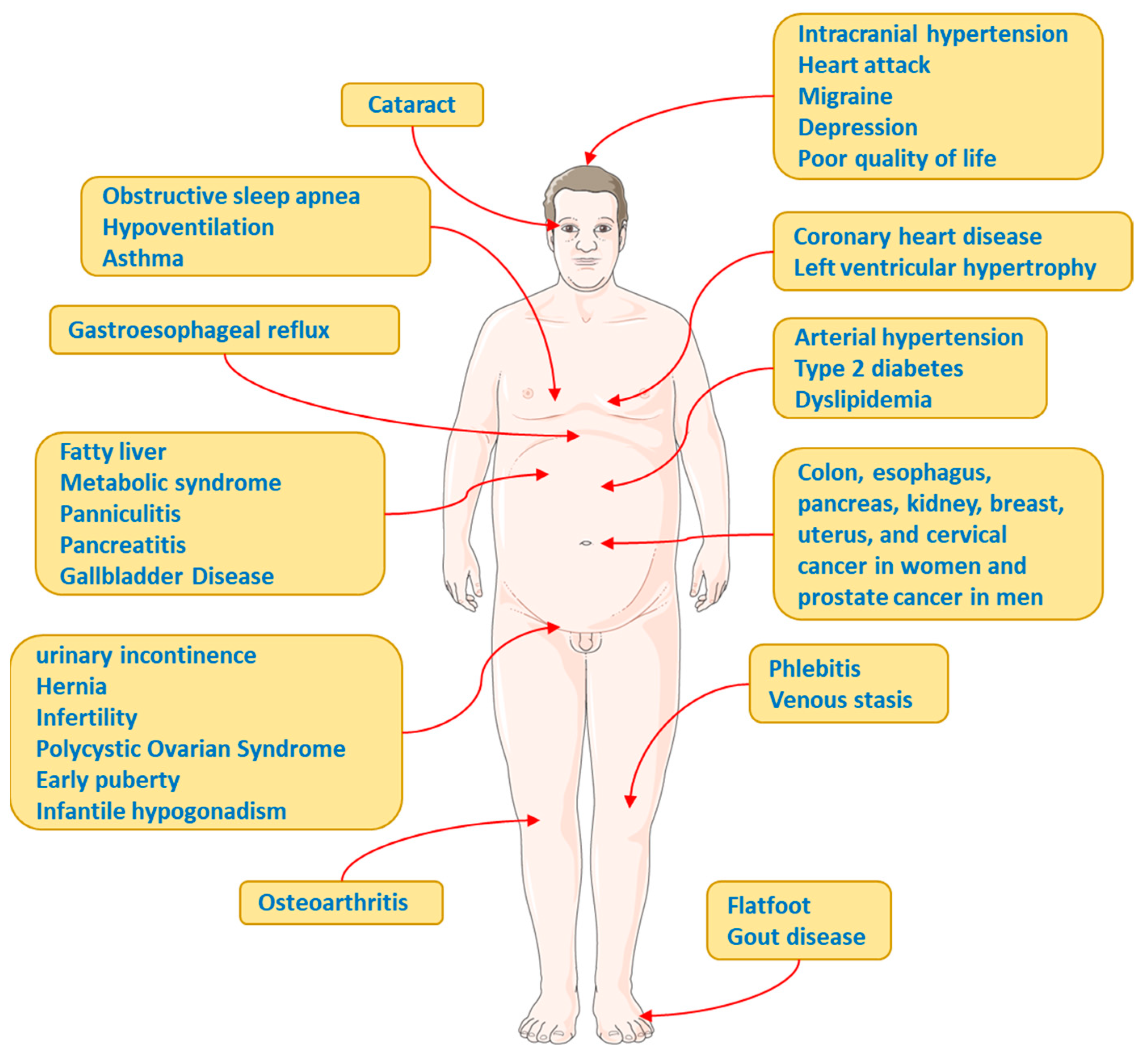
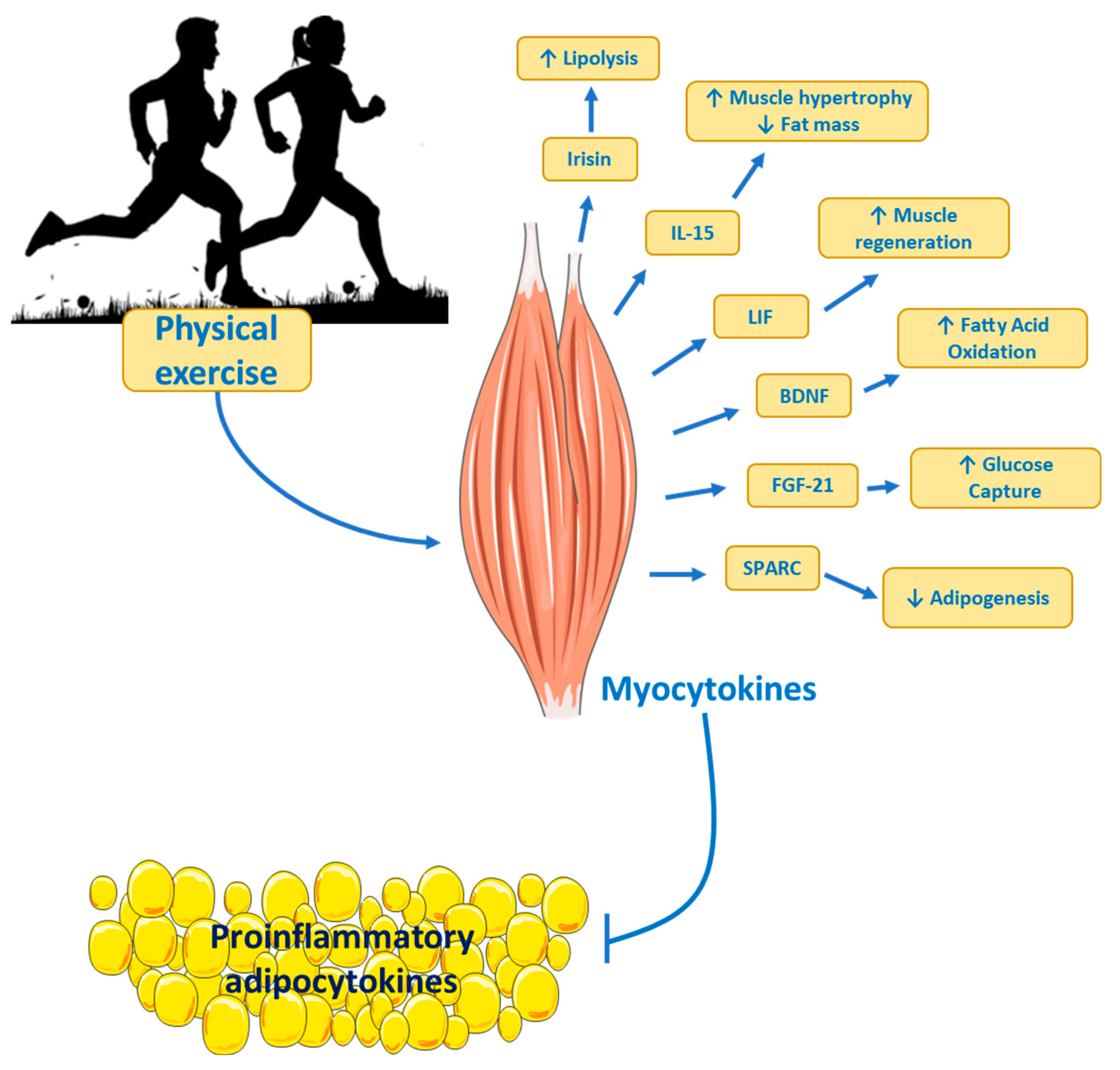
Figure 6.
Myocytokines and their potential role on metabolism. Skeletal muscle produces and secretes myocytokines into the circulation. Adipose tissue, under conditions of metabolic diseases, secretes proinflammatory adipocytokines that promote pathological processes such as insulin-resistance. However, physical exercise promotes the secretion of myocytokines that can counteract the effects of adipocytokines. IL, Interleukin; LIF, Leukaemia Inhibitory Factor; BDNF, Brain-derived Neurotrophic Factor; FGF-21, Fibroblast Growth Factor 21; SPARC, secreted protein rich in acid and cysteine.
Figure 6.
Myocytokines and their potential role on metabolism. Skeletal muscle produces and secretes myocytokines into the circulation. Adipose tissue, under conditions of metabolic diseases, secretes proinflammatory adipocytokines that promote pathological processes such as insulin-resistance. However, physical exercise promotes the secretion of myocytokines that can counteract the effects of adipocytokines. IL, Interleukin; LIF, Leukaemia Inhibitory Factor; BDNF, Brain-derived Neurotrophic Factor; FGF-21, Fibroblast Growth Factor 21; SPARC, secreted protein rich in acid and cysteine.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



