
Submitted:
04 August 2023
Posted:
08 August 2023
You are already at the latest version
Abstract
Low nasal nitric oxide (nNO) is a typical feature in Primary Ciliary Dyskinesia (PCD). nNO is part of the PCD diagnostic algorithm due to its discriminative power against other lung diseases such cystic fibrosis (CF). However, the underlying pathomechanisms are elusive. To better understand NO dysregulation in PCD, the L-arginine/NO (Arg/NO) pathway in patients with PCD (pwPCD) and CF (pwCF) and in healthy control (HC) subjects was investigated. In a prospective, controlled study, we measured in 24 pwPCD, 25 age-matched pwCF, and 14 HC the concentrations of the NO precursors Arg and homoarginine (hArg), the arginase metabolite ornithine (Orn), the NO inhibitor asymmetric dimethylarginine (ADMA), and the major NO metabolites (nitrate, nitrite) in sputum, plasma and urine by validated methods. In comparison to HC, the sputum contents (in µmol/mg) of L-Arg (PCD 18.43 vs. CF 329.46 vs. HC 9.86, p < 0.001) and of ADMA (PCD 0.055 vs. CF 0.015 vs. HC 0.010, p < 0.001) were higher. In contrast, the sputum contents (in µmol/mg) of nitrate and nitrite were lower in PCD compared to HC (nitrite 4.54 vs. 9.26, p = 0.023; nitrate 12.86 vs. 40.33, p = 0.008), but higher in CF (nitrite 16.28, p <0.001; nitrate 56.83, p = 0.002). The metabolite concentrations in urine and plasma were similar in all groups. The results of our study indicate that PCD, unlike CF, is associated with impaired NO synthesis in the lung, presumably due to mechano-chemical uncoupling.
Keywords:
primary ciliary dyskinesia
; nitric oxide
; nitric oxide synthetase
; cystic fibrose
; L-arginine
1. Introduction
Primary ciliary dyskinesia (PCD) is a rare, autosomal recessively inherited disease mainly associated with respiratory symptoms. In PCD, various genetic defects of the motile respiratory cilia lead to an impairment of ciliary function which in turn compromises mucociliary clearance [1]. Sequelae caused by reduced mucociliary clearance include upper and lower airway infections eventually resulting in progressive bronchiectasis and impairment of lung function. The diagnostic algorithm includes high frequency video microscopy (HFVM), transmission electron microscopy (TEM), nasal nitric oxide (nNO) measurement and genetic analysis [2,3]. Low nNO levels are characteristic for PCD and of diagnostic value [4]. However, the pathogenesis of low nNO levels as a hallmark in PCD is still unclear.
Nitric oxide (NO), originally known as endothelium-derived relaxing factor (EDRF), is a small gaseous free radical with many physiological homeostatic and immunological functions [5]. The half-life of NO in blood is less than 0.1 second. NO is oxidized to nitrite and nitrate which are useful measures of NO synthesis. NO is produced by C- and N-oxidation of the guanidino group of L-arginine (Arg) catalyzed by the nitric oxide synthase (NOS) family, with L-citrulline (Cit) being the second product of this reaction. At least three NOS isoforms are known: the constitutive and Ca2+-dependent neuronal NOS (nNOS) and endothelial NOS (eNOS), and the inducible Ca2+-independent NOS (iNOS) [5,6]. iNOS is expressed in cells activated by cytokines and lipopolysaccharides (LPS) and acts via a high-affinity calmodulin-binding site [5,7]. iNOS is expressed across a wide range of tissues; co-expression of iNOS and nNOS with differential regulation has been demonstrated in lung epithelial cells [7]. nNOS is expressed in many different cell types such as skeletal, cardiac and smooth muscle cells, pancreatic islet cells, macula densa cells of the kidney as well as in alveolar and bronchial epithelial cells [7]. Recently, nNOS has been localized in murine ciliated tracheal epithelial cells in close proximity to the apical cortical actin grid, basal bodies and in the ciliary axonemes, and was linked to physiological ciliary function [8,9]. The activity of all NOS isoforms is mainly inhibited by asymmetric dimethylarginine (ADMA) [6]. ADMA is formed by post-translational modification, i.e., asymmetric dimethylation of Arg residues is proteins. ADMA is metabolized by dimethylarginine dimethylaminohydrolase (DDAH) to Cit and dimethylamine (DMA) [10]. High ADMA levels were found in many cardiovascular (e.g., heart failure and pulmonary hypertonia), in pulmonary diseases (e.g., bronchial asthma and cystic fibrosis) and in renal failure [10,11].
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene coding for the chloride channel situated in the apical membrane of epithelial cells of exocrine tissue. CFTR dysfunction results in viscous mucus impairing mucociliary clearance [12]. Patients with CF (pwCF) also show low nNO levels lying between those of patients with PCD (pwPCD) and healthy humans [13], as well as an upregulated Arg/NO pathway as evidenced by increased plasma NO metabolites [14]. Higher ADMA concentrations were found in sputum of pwCF compared to healthy humans [15]. ADMA concentrations decreased after treatment of pulmonary exacerbations, whereas sputum NO metabolites and fractionally exhaled NO (FENO) increased [16]. High levels of ADMA are suspected to lead to bronchial obstruction via NOS inhibition leading to lower NO production [15,16]. Higher Arg, ADMA, nitrite and nitrate concentrations in plasma and of nitrate in urine were measured in pediatric pwCF [14].
Compared to the considerable amount of data on the Arg/NO pathway in CF such data is lacking in PCD. Few hypotheses deal with the lower nNO levels in PCD. They include impaired NO biosynthesis, elevated NO oxidation to nitrite and nitrate, or NO trapping in obstructed paranasal sinuses [17]. A positive correlation between nNO levels and expression of the iNOS gene in pwPCD was reported [18]. A correlation between low nNO levels and increased quantity of immotile cilia has also been observed [18]. In vitro, the ciliary beat frequency (CBF) was elevated by treatment with Arg of human paranasal sinus mucosa, which expresses iNOS and eNOS, indirectly indicating an effect of NO synthesis on CBF [19,20]. However, a firm attribution of low nNO levels to impaired activity of a specific NOS isoform has not been proven yet. Recently the nNOS isoform has been linked to ciliary function [8].
The primary aim of this study was to analyze whether the L-Arg/NO pathway is different in PCD, CF and healthy controls (HC). We hypothesized that pwPCD and pwCF show higher sputum concentrations of Arg and ADMA in comparison to HC. Yet, we hypothesized that the Arg/NO pathway behaves differently in PCD and CF, and that these differences account for the PCD-immanent very low nNO levels.
2. Materials and Methods
2.1. Study design and population
We conducted a single-center, prospective, controlled, clinical trial to assess the L-Arg/NO pathway in pwPCD in comparison to pwCF and HC. PwPCD and pwCF were recruited within trimestral routine examination in the PCD special outpatient clinic or the CF Centre at University Children’s Hospital Bochum. Between 12/2017-05/2020, overall 34 of 63 pwPCD and 47 of 88 pwCF above the age of ten years, registered in the hospital, were asked about interest in study participation. The recruitment process is depicted in Figure 1. For an age-matched HC group, children and adults from outside the clinic were invited.
Inclusion criteria for pwPCD and pwCF were a confirmed diagnosis, age above ten years and a written consent to study participation. Diagnosis of PCD was verified by HFVM, TEM, IF and molecular genetics, according to ERS guidelines [3]. For details on the diagnostic parameters, see Table S1. CF diagnosis was verified by pathological sweat test and molecular genetics. Inclusion criteria for HC were absence of any acute infection or chronic lung disease, age above ten years and written consent to study participation. Study participants were excluded when canned fish was consumed the day before and in case a special diet was followed in the last two weeks before the examination date. We also excluded patients with colonization with resistant strains due to hygienic restrictions and with inability to produce sputum. As part of the routine examinations, body weight and length were measured. Oral and inhaled antibiotic therapy over three months was classified as long-term antibiotic therapy.
2.2. Biochemical analysis in sputum, plasma and urine
After a fasting period not less than four hours, 5 mL venous blood was sampled using Lithium-Heparin Monovettes (Kabe Labortechnik, Nümbrecht-Elsenroth, Germany), brought to the laboratory on ice and centrifuged immediately (2000×g, 10 min, 4°C) using a Megafuge 1.0R from Heraeus (Stadt, Germany). Urine was collected by spontaneous micturition. At least 1 mL aliquots of plasma and urine were stored at -80°C until further analysis. Sputum samples were immediately quick-frozen in liquid nitrogen and stored at -80°C until further analysis. Lung healthy individuals do not produce sputum, therefore by inhalation of hypertonic saline under constant control of the maximum flow rate using a peak flow meter according to ERS guideline [21] sputum was induced in HC. From pwPCD and pwCF additional blood samples were taken to measure C-reactive protein (CRP) and leukocytes, and sputum for culture-based microbiology.
Plasma, urine and sputum samples were analyzed by high performance liquid chromatography (HPLC) for ornithine (Orn) and citrulline (Cit) and by gas chromatography–mass spectrometry (GC-MS) for all other parameters as described previously [22,23,24]. The sputum parameters are reported as µmol analyte per mg sputum. Analytes concentrations in urine were corrected for urinary creatinine excretion and are reported as µmol analyte per mmol creatinine. All analyses for the Arg/NO pathway were performed at the same time in one laboratory.
2.3. Lung function
The values of nNO and FENO were assessed with EcoMedics CLD 88sp (Duernten, Schweiz) according to current recommendations [25]. Both values were measured in parts per billion (ppb) and converted into nanoliters per minute (nL/min). Spirometry was performed using the MasterScreen Body/Diff (Vyaire, Hoechberg, Germany) according to ATS/ERS guidelines [26]. Multiple breath washout was measured with the Exhalyzer D (EcoMedics AG, Duernten, Switzerland) according to current guidelines [27]. We reported the Lung Clearance Index (LCI) 2.5 as recommended [28]. All pulmonary function tests were done by specially trained staff.
2.4. Statistical Analyses and sample size
As there are no previous values for Arg/NO metabolism in pwPCD, we assumed that a difference of a factor of 2 in the normal values for arginase and ADMA in sputum is clinically relevant. Using 90% power (alpha=0.05), the sample size calculation resulted in a ratio of pwPCD and the HC of 24:12. Statistical analyses were performed by using the statistical software package IBM® SPSS® Statistics Version 29.01.0 for Mac OS (IBM Corp., Armonk, NY, USA). Equality of variances was tested by Levene test. When variance of mean values was equal, the significance (p <0.05) was tested by ANOVA. In case of unequal variance of mean values, the significance (p < 0.05) was tested by the Welch-test. Post-hoc analysis was performed by Bonferroni (equal variance of mean values) or Dunnett-T3 (unequal variance of mean values). Data are presented as mean ± standard deviation (SD) or median [25-75th interquartile range]. Missing data were considered in the analyses and are marked separately in the results.
3. Results
3.1. Characterization of participants, bacterial colonization and lung function
Twenty-four pwPCD (age, 9.7 – 31.6 years), 25 pwCF (age, 11.0 – 34.7 years) and 14 HC (age, 10.1 – 33.3 years) were enrolled. Half of the pwPCD had a situs inversus, consistent with PCD features. Pancreatic insufficiency was present in 92% of the pwCF. Three (12.5%) pwPCD and one (4%) pwCF were overweight (BMI z-Score >1.96). Nine (36%) pwCF received CFTR modulator treatment before enrolment into the study (see Table S2). Apart from the CFTR moderators, pwPCD and pwCF received similar supportive therapy (secretolysis, antibiotics) according to therapy standard. Demographic and functional data of patients and controls are shown in Table 1. Pathological airway colonization was more frequently detectable in pwCF, especially with those infected by Pseudomonas aeruginosa (Table S3). This may explain the higher number of long-term antibiotic treatments (Table 1). nNO levels were significantly lower in pwPCD compared to pwCF and HC (Table 1, Figure 2A). Lung function between patients of both lung diseases did not differ significantly (Table 1).
3.2. Characterization of the L-Arg/NO pathway in the sputum
The L-Arg/NO pathway was found to be altered in PCD and CF compared to HC (Table 2). In sputum, the concentrations of Arg, hArg and ADMA were significantly higher in the patient groups, being more pronounced in CF than in PCD. The sputum concentration of ADMA was 5.5 times higher in PCD and 15 times higher in CF than in HC. The Arg/ADMA ratio, a measure of NO biosynthesis capacity, was significantly lower in both diseases. The Orn/Cit ratio, a measure of arginase activity, was markedly higher in both lung diseases. This ratio was higher in CF (14.3-fold) than in PCD (6.4-fold). In the sputum, the Orn/Cit ratio correlated with the Arg concentration in all three groups. The highest Pearson correlation was observed in PCD (R 0.94; Figure S4).
3.3. Characterization of the L-Arg/NO pathway in plasma and urine
In plasma and urine (Table S4), less significant differences between these three groups were observed. In PCD and CF, slightly higher plasma concentrations of nitrite were measured. In pwPCD, plasma nitrate and also urinary nitrate excretion (p = 0.006) were lower compared to health, whereas in CF, urinary nitrate excretion was higher. The plasma Orn/Cit ratio was higher in CF, but comparable in PCD and HC. Only in CF disease a higher concentration of L-Arg (however not reaching significance) was measured in plasma and urine.
4. Discussion
To our knowledge, this is the first study to analyze the Arg/NO pathway in pwPCD. In comparison to HC and pwCF, considerable differences in the Arg/NO pathway were observed in the sputum of the pwPCD. Higher sputum levels of ADMA, Arg and the Orn/Cit ratio were found in both, PCD and CF. However, nasal NO levels were lower in PCD compared to CF and HC, and the concentrations of the NO metabolites nitrite and nitrate were lower in the pwPCD while being higher in the sputum of the pwCF, when compared to HC. These results point to different states of the Arg/NO pathway in health and disease, and clearly discriminate the Arg/NO pathway in the lung compartment in PCD from CF disease.
In plasma and urine differences in the Arg/NO pathway were discrete or not detectable in the three groups, suggesting no major differences in the systemic and whole body NO synthesis, respectively. Thus, the differences found in the sputum of the subjects of the groups are indicative of lung-specific alterations in PCD and CF.
4.1. Similarities between PCD and CF
The hypothesis of an altered L-Arg/NO pathway in the pulmonary compartment in PCD was motivated by the facts that PCD and CF diseases share a profound neutrophilic inflammatory airway pathology and that in CF L-Arg/NO pathway alterations linked to the CF airway inflammation have already been described: In CF low exhaled airway NO [13,30,31,32] goes along with increased sputum arginase activity [32], increased levels of the endogenous NOS inhibitor ADMA [15], reduced L-Arg/ADMA ratio [15] and increased polyamines [33]. In our study we found higher L-Arg, hArg and ADMA levels, a lower L-Arg/ADMA ratio and a higher Orn/Cit ratio in the lung compartment of our CF cohort, being in line with previous data and expanding them. For the first time we show these alterations also in PCD disease, paralleling those in CF.
The higher Orn/Cit ratio in sputum of both pwCF and pwPCD suggest a shift of the balance between arginase and NOS activity towards Orn production, possibly mediated or augmented through inhibition of the NOS by high ADMA levels, as ADMA is an endogenous NOS inhibitor. Hence, it can be speculated that a reduced bioavailability of the substrate L-Arg for the NOS through NOS inhibition by ADMA, and an increased activity of the competing arginase enzymes shown in CF [32,34] also may play a role in PCD due to similar airway inflammatory states. In our study differences in L-Arg and ADMA levels were significantly more pronounced in sputum from the pwCF than from the pwPCD, which could be attributed to a more pronounced airway inflammation in CF. Airway inflammation has not been measured in our study, but bacterial colonization was significantly higher in the CF cohort compared to the PCD cohort, and only a minority of pwCF were treated with CFTR modulators at the time of study conductance (Table S2).
All in all, elevated concentrations of ADMA, L-Arg, an increased Orn/Cit ratio and decreased L-Arg/ADMA ratio are present in both lung diseases PCD and CF, and thus are not disease-specific but might rather be a function of airway inflammation shared by both diseases.
4.2. PCD specific effects
Previous studies reported higher concentrations of the NO metabolites nitrite and nitrate in CF sputum compared to healthy controls [35] despite low exhaled NO in CF [13,26,28,32]. This is believed to be due to an increased NO metabolism in the CF sputum by NO retention in the airway surface liquid compartment and / or degradation by denitrifying bacteria [36] and phagocytes. In the present study, we found significantly higher nitrite and nitrate concentrations in CF sputum compared to sputum from HC, consistent with previous data [33]. Remarkably, in the present study in PCD sputum nitrite and nitrate levels were lower compared to HC and to CF, strongly discriminating PCD L-Arg/NO metabolism from that in CF. This marked reduction of NO downstream metabolites confined to the PCD cohort parallels their distinctly low nasal NO being even lower than in CF (Table 1), which is in line with nasal NO known to be specifically low in PCD [34] and thus being used in the PCD diagnostic algorithms [25,37]. Low NO downstream metabolites in PCD sputum but not in CF sputum suggest that the PCD-specifically low nasal NO levels result from a reduction in NO production, i.e., reduced NOS activity, due to mechanism(s) not shared between PCD and CF. Several hypotheses about NO dysregulation in PCD have been proposed in the literature, comprehensively discussed by Walker et al. [17]. However, the underlying mechanism still has not been fully elucidated by now. One hypothesis for decreased nasal NO in PCD is a local inhibition of NOS activity by ADMA. Although we found significantly increased ADMA levels in PCD sputum compared to HC, the amount of ADMA in CF sputum was even higher than in PCD sputum. Therefore, NOS inhibition by ADMA cannot account for the severely low nNO levels in PCD. Furthermore, a substrate deficiency for the NOS was discussed [17], but based on our data this can be excluded as a reason for decreased NO production in PCD, as L-Arg concentration in PCD sputum is significantly higher than in HC. Increased NO consumption by NO-consuming / denitrifying bacteria and / or by sputum phagocytes as the cause of the very low nasal NO in PCD seems also unlikely, as in our study pwCF showed significantly more bacterial colonization, higher nasal NO and higher sputum NO downstream metabolites than the pwPCD. The hypothesis that pathology of the PCD upper airways could result in low nNO [17] by gas trapping or reduced NO production capacity confined to the paranasal sinuses is not supported by our measurements: NO was low in both upper and lower airways in PCD and NO metabolites were low in sputum, hinting at a rather global mechanism not confined to the upper airways.
The results of the present study suggest that a specific pathomechanism is active in PCD, but not in CF, and accounts for the reduced airway NO production in PCD leading to the PCD-immanently low nasal NO. Our data preclude low NO production in PCD being attributable to NOS inhibition by ADMA or to bronchial inflammation, as these mechanisms would also work in pwCF.
As discussed elsewhere [17,38], low NOS activity in PCD could potentially result from mechanochemical uncoupling. This would imply that normal NOS activity requires ciliary function [20,39]. A “mechano-chemical decoupling” resulting in low NO production by a decreased NOS activity due to reduced NOS mechanical “loading” was discussed in children with Duchenne muscular dystrophy (DMD) [40]. In DMD a dystrophin deficiency in muscle cells due to mutations on the dystrophin gene, goes along with reduced intracellular NO production [41]. In the plasma of pediatric DMD patients, lower nitrate concentrations were measured compared to HC [42]. In muscle cells the neuronal NOS (nNOS) is anchored to sarcolemma by a dystrophin-glycoprotein complex. In DMD the missing of dystrophin results in an aberrant translocation and accumulation of nNOS in the cytosol leading to reduced nNOS enzyme activity [43]. However, a PCD-immanent candidate mechanism able to account for a reduced NOS activity in respiratory epithelial cells throughout PCD disease types would not be attributable to one gene. This is because PCD is a very heterogenous genetic disease [1] with PCD-causative mutations in at least 50 different genes. Thus, in PCD a mechanism depending on a single protein defect is not plausible. Already a decade ago it has been hypothesized [18] that low nasal NO in PCD might be due to an uncoupling of the contractile process of ciliary beating from the enzyme NOS, leading to failure in NO production. Interestingly, in that study low nasal NO correlated with reduced ciliary beat frequency and inversely with the amount of immotile cilia not only in PCD as a genetic disease but also in secondary (i.e., acquired) ciliary defects, linking mechanical ciliary malfunctioning to low NO irrespective of a genetic defect.
Of note, there is a spatial proximity of the NOS, actin and microtubules in respiratory epithelial cells [8] as well as in muscle cells [43], and the contractile mechanisms of structures at the ciliary base in respiratory epithelial cells and of contractile structures in muscle cells are similar. In an in vitro animal model mechanically induced muscular action increased neuronal NOS (nNOS) activity, going along with NO formation in muscle cells a phenomenon called "mechanical loading" [44]. Hence, we postulate that in PCD the mechanical ciliary malfunction itself, caused by various genetic PCD defects, might be the origin of the markedly low nasal NO via reduced “mechanical loading” of the nNOS ("mechano-chemical decoupling"). This hypothesis is strengthened by the observation of higher nasal NO in pwPCD with residual ciliary function [45], and by very recent data linking nNO production rates to specific PCD genotypes: pwPCD with normal ciliary ultrastructure compared with abnormal ultrastructure showed higher ciliary motility and higher nNO production rates [46], suggesting that higher NO production and higher residual ciliary motility are connected.
A recent work [8] studied the spacial and functional connection of the neuronal NOS to ciliary structures and the impact of NO donors in a knock-out mice model in ciliary beating: In murine respiratory epithelial cells the nNOS is localized around the docking points of the ciliary bases at the apical cytoskeleton forming a tight network around the ciliary bases. In nNOS knock-out mice, a lack of neuronal NOS and thus of NO production in the respiratory epithelium was linked to a loss of ciliary beating activity, with cilia losing their synchronized beating direction and epithelial cells losing polarity in the absence of NO [8]. After treatment with NO donors synchronized ciliary function was restored. In summary, this study demonstrated a link between reduced NO production in ciliated epithelial cells and impaired cilia beating function. Thus, impaired ciliary function might not only compromise NOS activity by mechano-chemical uncoupling but might itself be a secondary consequence of reduced NO production. Similarly, Pifferi et al. [18] speculated that reduced nasal NO levels, as well as being a marker for PCD, may contribute to its pathophysiology. Very recently, in a large PCD cohort lower nNO observed in patients with more severe genetic and / or ultrastructural PCD types was shown to correlate with greater lung function decline over time, and higher nNO with a reduced likelihood of bacterial infection [47]. Taken together, NO production and motile ciliary defects seem to be interrelated in a complex way forming a vicious circle. Up to now it can only be speculated whether treatment strategies aiming at increasing NO production (such as pharmacological NO donors) might be able to improve the disease course or prognosis in PCD.
4.3. Strength and Limitations
A strength of the study is the use of reliable GC-MS methods for the quantitative measurement of numerous parameters of the Arg/NO pathway in sputum, plasma and urine samples of patients and HC. Although groups of patients and HC are relatively small, group effects can clearly be detected. Patients and HC were similar in age distribution. However, due to the limited availability of pwPCD and pwCF, it was not possible to achieve a completely balanced gender distribution. Yet, gender-specific differences in the Arg/NO pathway have not been described so far. The recruitment period of our study was relatively long, but the biosamples were frozen at -80 °C immediately. All samples were transported to the laboratory together at frozen state (dry ice) and analyzed at one single time period. A subgroup analysis to correlate between differential genetic or ultrastructural defects and the Arg/NO pathway results was not performed due to small numbers of various genetic backgrounds: High proportion of pwPCD has genetic defects known to cause low nNO (i.e., dynein arm defects), only two patients have ciliary defects known to cause either low or normal nNO [45]. However, while this circumstance precludes insights into Arg/NO pathway diversity due to genetic differences it has been an advantage in terms of recognizing the Arg/NO effects typical for most PCD genotypes.
5. Conclusions
In the present study, the Arg/NO pathway was comprehensively studied for the first time in pwPCD and was compared to pwCF and HC. In sputum, we found marked differences between pwPCD and pwCF and compared to HC. In PCD lower nitrate and nitrite concentrations in the sputum and lower nasal NO levels suggest a decreased NOS activity in the respiratory epithelium in PCD compared to CF and HC. Driven by the presented data a mechano-chemical uncoupling could be assumed as causative for low nasal NO in PCD, because mechanical impairment of motile ciliary function is the hallmark of PCD pathology across many various genetic defects. Dependence of NO production on mechanical loading may have therapeutical implications: In PCD, physical exercise may improve endogenous NO synthesis in the lung by positively affecting mechanical loading of the NOS in the respiratory epithelium. Pharmacological treatment with NO donors may also be an option. Further studies are needed to confirm the proposed mechano-chemical decoupling of the NOS in PCD and to address its therapeutic augmentation in the clinical course of this disease.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Diagnostic parameters of PCD patients; Table S2: Data of patients with cystic fibrosis on treatment with CFTR modulators; Table S3: Pulmonary bacterial colonization in patients with primary ciliary dyskinesia (PCD) and cystic fibrosis (CF); Table S4: Contents in plasma and urine of metabolites of the L-Arg/NO pathway in patients with PCD or CF and in healthy controls (HC); Figure S5: Correlation between L-Arginine and Ornithine/Citrulline ratio in sputum of patients with primary ciliary dyskinesia (PCD), cystic fibrosis (CF) and healthy control (HC).
Author Contributions
Conceptualization, C.K-R., T.L.; methodology, L.E., A.S.; validation, C.K-R., D.T.; formal analysis, L.E., C.M.; investigation, D.T., B.B., A.S., L.E., C.K.-R.; data curation, L.E., C.M.; writing—original draft preparation, L.E., C.K-R.; A.S. writing—review and editing, T.L., C.M., F.B.; visualization, L.E.; supervision, C.K-R., T.L., D.T.; project administration, T.L.;. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The ethics committee of Ruhr-University Bochum, Bochum, Germany, approved the study in 2017 (Ethics Committee No. 17-6079 dated Sept 3, 2017).
Informed Consent Statement
Informed consent was obtained from all subjects and / or their parents involved in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zariwala, M.A.; Omran, H.; Ferkol, T.W. The Emerging Genetics of Primary Ciliary Dyskinesia. Proc. Am. Thorac. Soc. 2011, 8, 430–433. [Google Scholar] [CrossRef]
- Lucas, J.S.; Burgess, A.; Mitchison, H.M.; Moya, E.; Williamson, M.; Hogg, C. Diagnosis and Management of Primary Ciliary Dyskinesia. Arch. Dis. Child. 2014, 99, 850–856. [Google Scholar] [CrossRef]
- Lucas, J.; Barbato, A.; Collins, S.; Goutaki, M.; Behan, L.; Caudri, D.; Eber, E.; Escudier, E.; Hirst, R.; Hogg, C.; et al. ERS Task Force Guideline for the Diagnosis of Primary Ciliary Dyskinesia. Eur. Respir. J. 2016, 49, 1–54. [Google Scholar] [CrossRef]
- Leigh, M.W.; Hazucha, M.J.; Chawla, K.K.; Baker, B.R.; Shapiro, A.J.; Brown, D.E.; Lavange, L.M.; Horton, B.J.; Qaqish, B.; Carson, J.L.; et al. Standardizing Nasal Nitric Oxide Measurement as a Test for Primary Ciliary Dyskinesia. Ann. Am. Thorac. Soc. 2013, 10, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P. Nitric Oxide and Immune Response. Indian J. Biochem. Biophys. 2007, 44, 310–319. [Google Scholar] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric Oxide Synthases in Mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Chee, C.B.E.; Gaston, B.; Lilly, C.M.; Gerard, C.; Drazen, J.M.; Stamler, J.S. Constitutive and Inducible Nitric Oxide Synthase Gene Expression, Regulation, and Activity in Human Lung Epithelial Cells. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 10089–10093. [Google Scholar] [CrossRef]
- Mikhailik, A.; Michurina, T. V.; Dikranian, K.; Hearn, S.; Maxakov, V.I.; Siller, S.S.; Takemaru, K.I.; Enikolopov, G.; Peunova, N. NNOS Regulates Ciliated Cell Polarity, Ciliary Beat Frequency, and Directional Flow in Mouse Trachea. Life Sci. Alliance 2021, 4, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Degano, B.; Valmary, S.; Serrano, E.; Brousset, P.; Arnal, J.F. Expression of Nitric Oxide Synthases in Primary Ciliary Dyskinesia. Hum. Pathol. 2011, 42, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leiper, J. Cardiovascular Biology of the Asymmetric Dimethylarginine:Dimethylarginine Dimethylaminohydrolase Pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef]
- Scott, J.A.; North, M.L.; Rafii, M.; Huang, H.; Pencharz, P.; Subbarao, P.; Belik, J.; Grasemann, H. Asymmetric Dimethylarginine Is Increased in Asthma. Am. J. Respir. Crit. Care Med. 2011, 184, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Naehrig, S.; Chao, C.M.; Naehrlich, L. Cystic Fibrosis - Diagnosis and Treatment. Dtsch. Arztebl. Int. 2017, 114, 564–573. [Google Scholar] [CrossRef]
- Grasemann, H.; Michler, E.; Wallot, M.; Ratjen, F. Decreased Concentration of Exhaled Nitric Oxide (NO) in Patients with Cystic Fibrosis. Pediatr. Pulmonol. 1997, 24, 173–177. [Google Scholar] [CrossRef]
- Brinkmann, F.; Hanusch, B.; Ballmann, M.; Mayorandan, S.; Bollenbach, A.; Chobanyan-Jürgens, K.; Jansen, K.; Schmidt-Choudhury, A.; Derichs, N.; Tsikas, D.; et al. Activated L-Arginine/Nitric Oxide Pathway in Pediatric Cystic Fibrosis and Its Association with Pancreatic Insufficiency, Liver Involvement and Nourishment: An Overview and New Results. J. Clin. Med. 2020, 9, 1–21. [Google Scholar] [CrossRef]
- Grasemann, H.; Al-Saleh, S.; Scott, J.A.; Shehnaz, D.; Mehl, A.; Amin, R.; Rafii, M.; Pencharz, P.; Belik, J.; Ratjen, F. Asymmetric Dimethylarginine Contributes to Airway Nitric Oxide Deficiency in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Hanusch, B.; Brinkmann, F.; Mayorandan, S.; Chobanyan-Jürgens, K.; Wiemers, A.; Jansen, K.; Ballmann, M.; Schmidt-Choudhury, A.; Bollenbach, A.; Derichs, N.; et al. Local and Systemic Alterations of the L-Arginine/Nitric Oxide Pathway in Sputum, Blood, and Urine of Pediatric Cystic Fibrosis Patients and Effects of Antibiotic Treatment. J. Clin. Med. 2020, 9, 1–19. [Google Scholar] [CrossRef]
- Walker, W.T.; Jackson, C.L.; Lackie, P.M.; Hogg, C.; Lucas, J.S. Nitric Oxide in Primary Ciliary Dyskinesia. Eur. Respir. J. 2012, 40, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, M.; Bush, A.; Maggi, F.; Michelucci, A.; Ricci, V.; Conidi, M.E.; Cangiotti, A.M.; Bodini, A.; Simi, P.; Macchia, P.; et al. Nasal Nitric Oxide and Nitric Oxide Synthase Expression in Primary Ciliary Dyskinesia. Eur. Respir. J. 2011, 37, 572–577. [Google Scholar] [CrossRef]
- Kim, J.W.; Min, Y.G.; Rhee, C.S.; Lee, C.H.; Koh, Y.Y.; Rhyoo, C.; Kwon, T.Y.; Park, S.W. Regulation of Mucociliary Motility by Nitric Oxide and Expression of Nitric Oxide Synthase in the Human Sinus Epithelial Cells. Laryngoscope 2001, 111, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shirakami, G.; Zhan, X.; Johns, R.A. Regulation of Ciliary Beat Frequency by the Nitric Oxide-Cyclic Guanosine Monophosphate Signaling Pathway in Rat Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Weiszhar, Z.; Horvath, I. Induced Sputum Analysis: Step by Step. Breathe 2013, 9, 301–306. [Google Scholar] [CrossRef]
- Tsikas, D. Simultaneous Derivatization and Quantification of the Nitric Oxide Metabolites Nitrite and Nitrate in Biological Fluids by Gas Chromatography/Mass Spectrometry. Anal. Chem. 2000, 72, 4064–4072. [Google Scholar] [CrossRef] [PubMed]
- Hanff, E.; Lützow, M.; Kayacelebi, A.A.; Finkel, A.; Maassen, M.; Yanchev, G.R.; Haghikia, A.; Bavendiek, U.; Buck, A.; Lücke, T.; et al. Simultaneous GC-ECNICI-MS Measurement of Nitrite, Nitrate and Creatinine in Human Urine and Plasma in Clinical Settings. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1047, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Hanff, E.; Ruben, S.; Kreuzer, M.; Bollenbach, A.; Kayacelebi, A.A.; Das, A.M.; von Versen-Höynck, F.; von Kaisenberg, C.; Haffner, D.; Ückert, S.; et al. Development and Validation of GC–MS Methods for the Comprehensive Analysis of Amino Acids in Plasma and Urine and Applications to the HELLP Syndrome and Pediatric Kidney Transplantation: Evidence of Altered Methylation, Transamidination, and Arginase Acti. Amino Acids 2019, 51, 529–547. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society ATS/ERS Recommendations for Standardized Procedures for the Online and Offline Measurement of Exhaled Lower Respiratory Nitric Oxide and Nasal Nitric Oxide, 2005. Am. J. Respir. Crit. Care Med. 2005, 171, 912–930. [CrossRef]
- Graham, B.L.; Steenbruggen, I.; Barjaktarevic, I.Z.; Cooper, B.G.; Hall, G.L.; Hallstrand, T.S.; Kaminsky, D.A.; McCarthy, K.; McCormack, M.C.; Miller, M.R.; et al. Standardization of Spirometry 2019 Update an Official American Thoracic Society and European Respiratory Society Technical Statement. Am. J. Respir. Crit. Care Med. 2019, 200, E70–E88. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.D.; Stocks, J.; Aurora, P.; Lum, S. Abbreviated Multi-Breath Washout for Calculation of Lung Clearance Index. Pediatr. Pulmonol. 2013, 48, 336–343. [Google Scholar] [CrossRef]
- Robinson, P.D.; Latzin, P.; Verbanck, S.; Hall, G.L.; Horsley, A.; Gappa, M.; Thamrin, C.; Arets, H.G.M.; Aurora, P.; Fuchs, S.I.; et al. Consensus Statement for Inert Gas Washout Measurement Using Multiple- and Singlebreath Tests. Eur. Respir. J. 2013, 41, 507–522. [Google Scholar] [CrossRef]
- Grasemann, H.; Pencharz, P.B. Arginine Metabolism in Patients with Cystic Fibrosis. J. Pediatr. 2013, 163, 317–319. [Google Scholar] [CrossRef]
- Ho, L.P.; Innes, J.A.; Greening, A.R. Exhaled Nitric Oxide Is Not Elevated in the Inflammatory Airways Diseases of Cystic Fibrosis and Bronchiectasis. Eur. Respir. J. 1998, 12, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Balfour-Lynn, I.M.; Laverty, A.; Dinwiddie, R. Reduced Upper Airway Nitric Oxide in Cystic Fibrosis. Arch. Dis. Child. 1996, 75, 319–322. [Google Scholar] [CrossRef]
- Grasemann, H.; Schwiertz, R.; Matthiesen, S.; Racké, K.; Ratjen, F. Increased Arginase Activity in Cystic Fibrosis Airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Shehnaz, D.; Enomoto, M.; Leadley, M.; Belik, J.; Ratjen, F. L-Ornithine Derived Polyamines in Cystic Fibrosis Airways. PLoS One 2012, 7, e46618. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Schwiertz, R.; Grasemann, C.; Vester, U.; Racké, K.; Ratjen, F. Decreased Systemic Bioavailability of L-Arginine in Patients with Cystic Fibrosis. Respir. Res. 2006, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Ioannidis, I.; Tomkiewicz, R.P.; De Groot, H.; Rubin, B.K.; Ratjen, F. Nitric Oxide Metabolites in Cystic Fibrosis Lung Disease. Arch. Dis. Child. 1998, 78, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Stouthamer AH Metabolic Regulation Including Anaerobic Metabolism in Paracoccus Denitrificans. J Bioenerg Biomembr. 1991, 23, 163–185. [CrossRef] [PubMed]
- Kuehni, C.E.; Lucas, J.S. Diagnosis of Primary Ciliary Dyskinesia: Summary of the ERS Task Force Report. Breathe 2017, 13, 166–178. [Google Scholar] [CrossRef]
- Lundberg, J.O.N.; Weitzberg, E.; Nordvall, S.L.; Kuylenstierna, R.; Lundberg, J.M.; Alving, K. Primarily Nasal Origin of Exhaled Nitric Oxide and Absence in Kartagener’s Syndrome. Eur. Respir. J. 1994, 7, 1501–1504. [Google Scholar] [CrossRef]
- Jain B, Rubinstein I, Robbins RA, Leise KL, S. J. Modulation of Airway Epithelial Cell Ciliary Beat Frequency by Nitric Oxide. Biiochemical Biophys. Res. Commun. 1993, 191, 83–88. [Google Scholar] [CrossRef]
- Narang, I.; Ersu, R.; Wilson, N.M.; Bush, A. Nitric Oxide in Chronic Airway Inflammation in Children: Diagnostic Use and Pathophysiological Significance. Thorax 2002, 57, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Gücüyener, K.; Ergenekon, E.; Erbas, D.; Pinarli, G. üçlü; Serdaroglu, A. The Serum Nitric Oxide Levels in Patients with Duchenne Muscular Dystrophy. Brain Dev. 2000, 22, 181–183. [Google Scholar] [CrossRef]
- Hörster, I.; Weigt-Usinger, K.; Carmann, C.; Chobanyan-Jürgens, K.; Köhler, C.; Schara, U.; Kayacelebi, A.A.; Beckmann, B.; Tsikas, D.; Lücke, T. The L-Arginine/NO Pathway and Homoarginine Are Altered in Duchenne Muscular Dystrophy and Improved by Glucocorticoids. Amino Acids 2015, 47, 1853–1863. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric Oxide Synthase Complexed with Dystrophin and Absent from Skeletal Muscle Sarcolemma in Duchenne Muscular Dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Tidball, J.G.; Lavergne, E.; Lau, K.S.; Spencer, M.J.; Stull, J.T.; Wehling, M. Mechanical Loading Regulates NOS Expression and Activity in Developing and Adult Skeletal Muscle. Am. J. Physiol. - Cell Physiol. 1998, 275, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.K.; Ferkol, T.W.; Davis, S.D. Emerging Genotype-Phenotype Relationships in Primary Ciliary Dyskinesia. Int. J. Mol. Sci. 2021, 22, 8272. [Google Scholar] [CrossRef] [PubMed]
- Biebach, L.; Cindrić, S.; Koenig, J.; Aprea, I.; Dougherty, G.; Raidt, J.; Bracht, D.; Ruppel, R.; Schreiber, J.; Hjeij, R.; et al. Recessive Mutations in CFAP74 Cause Primary Ciliary Dyskinesia with Normal Ciliary Ultrastructure. Am. J. Respir. Cell Mol. Biol. 2022, 67, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, M.; Boner, A.L.; Gracci, S.; Fonnesu, R.; Maj, D.; Donzelli, G.; Michelucci, A.; Cangiotti, A.; Bertini, V.; Valetto, A.; et al. Longitudinal Nitric Oxide Levels and Infections by Ultrastructure and Genotype in Primary Ciliary Dyskinesia. Chest 2022, 126, 1265–1276. [Google Scholar] [CrossRef]
Figure 1.
Flow chart of the recruitment process of the study. * Age of one patient below 10 (9.7 years) due to falsely age information in time of recruitment.
Figure 1.
Flow chart of the recruitment process of the study. * Age of one patient below 10 (9.7 years) due to falsely age information in time of recruitment.
Figure 2.
Concentrations of (A) nasal nitric oxide (nNO, nl/min) and of its metabolites nitrite (B) and nitrate (A) in sputum (µmol/mg sputum) of patients with PCD (light grey) or CF (dark grey) and of healthy controls (HC, white). * = Significance PCD vs. CF, # = Significance PCD vs. HC, + = Significance CF vs. HC.
Figure 2.
Concentrations of (A) nasal nitric oxide (nNO, nl/min) and of its metabolites nitrite (B) and nitrate (A) in sputum (µmol/mg sputum) of patients with PCD (light grey) or CF (dark grey) and of healthy controls (HC, white). * = Significance PCD vs. CF, # = Significance PCD vs. HC, + = Significance CF vs. HC.
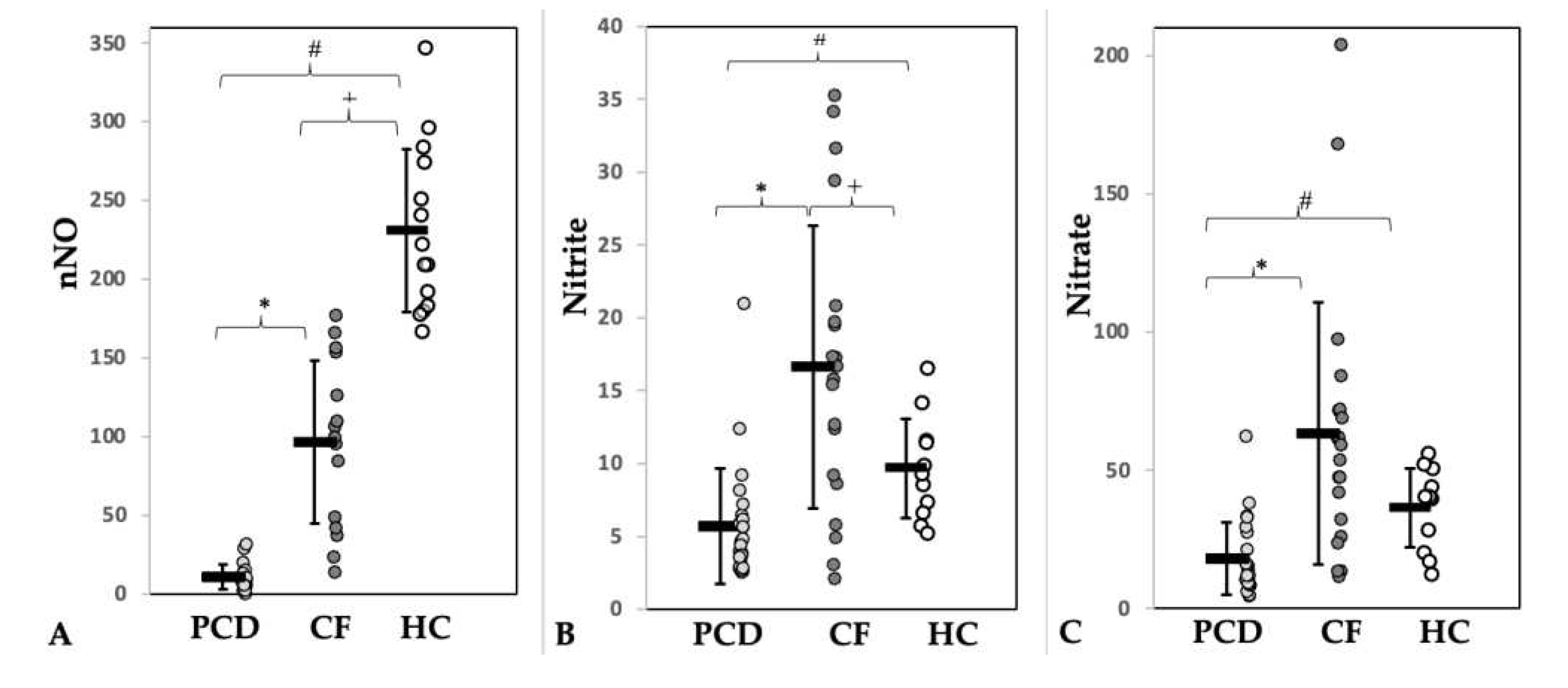
Figure 3.
Simplified schematic showing differences in L-Arg/NO pathway in the sputum in PCD (light grey) or CF (dark grey) in comparison to a healthy state. Modified according to Grasemann et al. [29]. Arrows indicate higher or lower. * = Significance PCD vs. CF, # = Significance PCD vs. HC, + = Significance CF vs. HC. Abbreviations. ADMA, asymmetric dimethylarginine; ADC, arginine decarboxylase; AGMAT, agmatinase; BH4, tetrahydrobiopterin; DDAH, dimethylarginine dimethylaminohydrolase; NO, nitric oxide; NOS, nitric oxide synthetase; OAT, ornithine aminotransferase; ODC1, ornithine decarboxylase; OTC, ornithine carbamoyltransferase; PRMTs, protein arginine N-methyltransferases; RARS, arginyl-tRNA-synthetase.
Figure 3.
Simplified schematic showing differences in L-Arg/NO pathway in the sputum in PCD (light grey) or CF (dark grey) in comparison to a healthy state. Modified according to Grasemann et al. [29]. Arrows indicate higher or lower. * = Significance PCD vs. CF, # = Significance PCD vs. HC, + = Significance CF vs. HC. Abbreviations. ADMA, asymmetric dimethylarginine; ADC, arginine decarboxylase; AGMAT, agmatinase; BH4, tetrahydrobiopterin; DDAH, dimethylarginine dimethylaminohydrolase; NO, nitric oxide; NOS, nitric oxide synthetase; OAT, ornithine aminotransferase; ODC1, ornithine decarboxylase; OTC, ornithine carbamoyltransferase; PRMTs, protein arginine N-methyltransferases; RARS, arginyl-tRNA-synthetase.
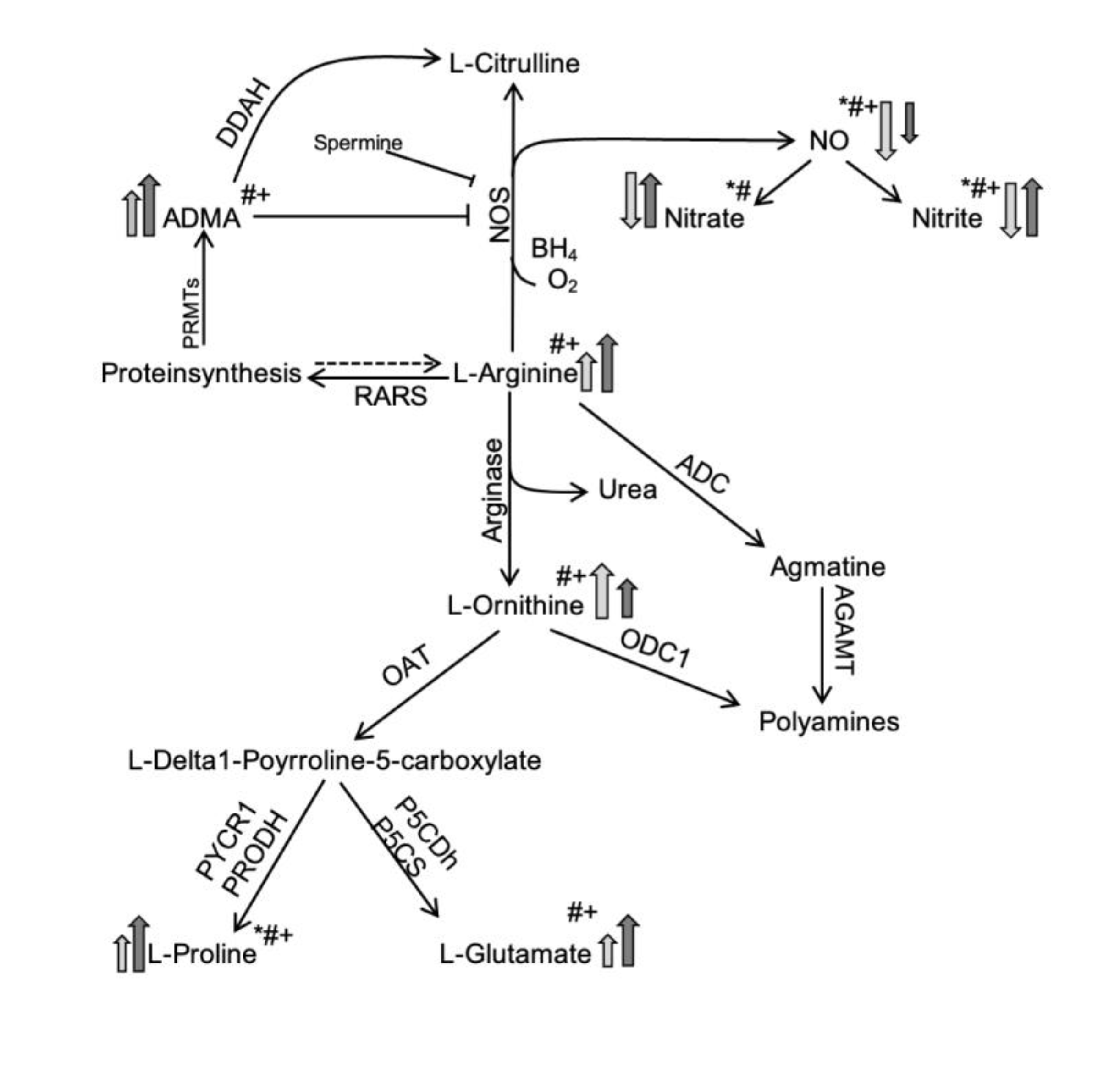

Table 1.
Demographic and clinical data of patients with PCD or CF and of healthy controls (HC). Data are presented as mean ± standard deviation or median [25-75th interquartile range].
Table 1.
Demographic and clinical data of patients with PCD or CF and of healthy controls (HC). Data are presented as mean ± standard deviation or median [25-75th interquartile range].
| PCD | CF | HC | p value | |
|---|---|---|---|---|
| Subject (n) | 24 | 25 | 14 | |
| Female (n (%)) | 15 (62.5) | 8 (32.0) | 8 (57.1) | 0.083 |
| Age (years) | 16.6 [14.3 – 21.6] |
20.6 [13.7 – 23.9] |
23.7 [17.0 – 25.5] |
0.224 |
| BMI (kg/m2) | 21.77 ± 4.76 | 19.88 ± 3.38 | 21.04 ± 3.25 | 0.247 |
| BMI (z-Score) | -0.02 | -0.49 | -0.08 | 0.149 |
| [-0.56 – 0.99] | [-0.98 – 0.13] | [-1.66 – 1.57] | ||
| Subjects with: | ||||
| Short-term antibiotic therapy < 3 months (n (%)) | 4 (16.7) | 8 (32.0) | 0 (0) | |
| Long-term antibiotic therapy (n (%)) | 6 (25) | 10 (40) | 0 (0) | |
| CRP > 5mg/L (n (%)) | 6 (27.3) a | 8 (33.3) f | n.m. | 0.664 |
| Leucocytes >10.000/µL (n (%)) | 5 (23.8) b | 6 (24.0) | n.m. | 0.988 |
Bacterial colonization (n (%))
|
10 (43.5)c* 5 (21.7) 1 (4.4) 5 (21.7) |
20 (80) 12 (48) 7 (28) 0 (0) |
n.m. |
0.021 |
| FENO (nL/min) | 1.85 ± 1.14 d | 4.49 ± 4.24 g | 4.88 ± 4.31 | 0.051 |
| nNO (nL/min) | 10.67 ± 7.82c*# | 96.4 ± 53.73d+ | 230.98 ± 53.66 | < 0.001 |
| FEV1 (z-Score) | -1.88 [-2.59 – -1.0] |
-1.80 [-2.77 – -0.31] |
n.m. | 0.859 |
| LCI 2.5% (absolute value) | 11.36 ± 2.2 e | 10.45 ± 5.30 g | n.m. | 0.265 |
Incomplete data available for highlighted variables: a n=22, b n=21, c n=23, d n=15, e n=20, f n=24, g n=18. Statistics: Bold indicates statistical significance; * = Significance PCD vs. CF; # = Significance PCD vs. HC; + = Significance CF vs. HC. Abbreviations: Body Mass Index (BMI); C-reactive Protein (CRP); fractional exhaled nitric oxide (FENO); nasal nitric oxide (nNO); Forced expiratory volume in 1 second (FEV1); Lung Clearance Index (LCI); not measured (n.m.).
Table 2.
Contents in sputum (µmol/mg) of metabolites of the L-Arg/NO pathway in patients with PCD or CF and in healthy controls (HC). Data are presented as median [25-75th interquartile range].
Table 2.
Contents in sputum (µmol/mg) of metabolites of the L-Arg/NO pathway in patients with PCD or CF and in healthy controls (HC). Data are presented as median [25-75th interquartile range].
| Sputum | PCD | CF | HC | p value |
|---|---|---|---|---|
| Subjects (n) | 24 | 20 | 11 | |
| L-Arg | 18.43 # [9.50 – 44.63] |
29.46 + [18.48 – 47.22] |
9.86 [6.53 – 14.65] |
* 0.067 # 0.012 + <0.001 |
| h-Arg | 0.009 # [0.005 – 0.028] |
0.037 + [0.015 – 0.074] |
0.005 [0.003 – 0.007] |
* 0.067 # 0.019 + 0.011 |
| ADMA | 0.055 # [0.029 – 0.163] |
0.015 + [0.056 – 0.213] |
0.010 [0.008 – 0.018] |
* 0.687 # 0.002 + 0.003 |
| Nitrite | 4.54 *# [3.35 – 6.31] |
16.28 + [9.12 – 20.07] |
9.26 [6.98 – 11.53] |
* <0.001 # 0.023 + 0.026 |
| Nitrate | 12.86 *# [10.27 – 23.28] |
56.83 [31.12 – 72.06] |
40.33 [24.37 – 47.27] |
* 0.002 # 0.008 + 0.09 |
| Orn/Cit | 10.20 # [3.85 – 57.13] |
22.84 + [11.20 – 42.76] |
1.60 [0.86 – 3.58] |
* 0.884 # 0.002 + <0.001 |
| L-Arg/ADMA | 239.33 # [194.38 – 406.26] |
319.11 + [154.78 – 649.33] |
917.62 [693.49 – 1205.32] |
* 0.522 # <0.001 + <0.001 |
Statistics: Bold indicates statistical significance; * = Significance PCD vs. CF; # = Significance PCD vs. HC; + = Significance CF vs. HC. Abbreviations. L-Arg, L-Arginine; hArg, Homoarginine, ADMA, asymmetric dimethylarginine; Orn/Cit, Ornithine/Citrulline ratio.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



