
Submitted:
04 August 2023
Posted:
07 August 2023
You are already at the latest version
Abstract
Interleukin (IL)-23 is a central proinflammatory cytokine with a broad range of effects on immune responses. The pathological consequences of excessive IL-23 signaling have been linked to its ability to promote the production of inflammatory mediators such as IL-17, IL-22 by stimulating the differentiation and proliferation of T helper type 17 (Th17 cells). Emerging evidence suggests a potential pro-fibrotic role for IL-23 in the development of chronic inflammatory autoimmune diseases characterized by intense fibrosis. In this review, we summarized the biological features of IL-23 and gathered recent research on the role of IL-23 in fibrotic autoimmune conditions, which could provide a theoretical basis for clinical targeting and drug development.
Keywords:
IL-23
; autoimmunity
; inflammation
; fibrosis
1. Introduction
Multiple autoimmune diseases are a group of clinically heterogeneous conditions that show common inflammatory signaling pathways arising from aberrant immune responses [1]. Some of these disorders are characterized by intense and severe fibrotic processes as the result of a complex interplay between different immune cell types following persistent inflammatory activity [2,3]. Several cytokines were well studied for their ability to generate inflammatory loops through positive feedback mechanisms [4]. Breaking these loops through cytokine neutralization proved that inflammation attenuates and ameliorates the disease [5]. One of these cytokines is interleukin (IL)-23, a multifunctional proinflammatory cytokine that is involved in a variety of biological processes [6]. Although IL-23 assumes a central role in the protective immune response against several bacterial and viral infections [7], its deregulation has been demonstrated to aggravate chronic inflammatory status, contributing to the development of autoimmune diseases [8,9]. As shown in an increasing number of experimental studies, IL-23 is involved in the pathogenesis of several autoimmune diseases [10,11,12,13]. The importance of IL-23 implicated in the evolution of autoimmune pathologies was demonstrated by analyzing the susceptibility of IL-12 or IL-23-deficient mice [10,11]. Indeed, mice that have a deletion of IL-23 were protected from disease in several experimental models of autoimmunity. Importantly, treatment of mice with anti-IL-23 prevents the development of autoimmune conditions [12].
In this review, we provide an overview of the most recent studies focusing on the role of IL-23 in fibrotic pathways and on its role in the pathogenesis of inflammatory autoimmune diseases characterized by fibrotic evolution.
2. Structure of IL-23 and Its Receptor
IL-23 is a heterodimeric pro-inflammatory cytokine that belongs to the special IL-12 family, which is composed of two different subunits: p19 and p40 [14]. The p40 subunit, which is also part of IL-12, is a glycosylated type I soluble protein with a molecular weight of 34.7 kDa, positioned on the 11q1.3 chromosome [15,16]. The unique p19 subunit is a non-glycosylated protein with a molecular weight of 18.7 kDa located on chromosome 12q13.2 [8]. Both subunits are linked by a disulfide bond, and they are attached only if they are synthesized in the same cell [8,14]. Specifically, IL-23 is expressed and secreted by activated macrophages and dendritic cells located in several tissues, such as the skin, intestinal mucosa, joints, and lungs (Figure 1).
Interestingly, it is also secreted by non-immune cells such as keratinocytes, synoviocytes, and salivary gland epithelial cells [17,18,19]. The IL-23 signaling pathway occurs through a link with its receptor. IL-23 receptor (IL-23R) is a heterodimeric structure composed of a heterodimer with the IL-12Rβ1 subunit and its own unique IL-23R subunit, located on human chromosome 19 encoding the gene that forms the IL-12Rβ1 subunit and on human chromosome 1 encoding the gene that forms the IL-23R subunit [20]. IL-12Rβ1 subunit is mainly expressed on T cells, monocytes/macrophages, natural killer T cells, and dendritic cells [7,21] with minor expression on B cells and lymphoid cells [22] (Figure 2).
3. Regulation of IL-23 Signaling
Since its discovery, IL-23 has received widespread attention, and although it has a similar structure to IL-12, its role is totally different. Indeed, despite the protective role played by IL-23 against bacterial, fungal, and viral infections, extensive knowledge supports the contribution of its dysregulation in triggering chronic inflammation and autoimmunity, providing a solid substrate for the development of several autoimmune diseases like psoriasis, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjogren syndrome (SS), and multiple sclerosis (MS) [8,11,23,24].
Currently, the IL-23 signalling pathway remains largely uncharacterized. IL-23 involves the activation of the members of the Janus family of tyrosine kinases (JAKs), their downstream factors, and the signal transducers and activators of transcription (STATs) family [25]. In particular, Il-23, through binding to its IL-23 receptor, provokes phosphorylation and activation of JAK/STAT signaling molecules (Jak2, Tyk2), promoting STAT3 and STAT4 phosphorylation and activation [25]. Subsequently, active STAT3 up-regulates the expression of the transcription factor RORγt, which is critical for IL-17 production [26]. Indeed, STAT3 plus RORγt cooperate to facilitate a positive loop that increases IL23R, IL-17, and IL-22 expression and stabilizes the Th17 phenotype. Thus, STAT4 phosphorylation and activation promote upregulation of IL23R, IL-17, and IL-22 expression, and the increase of Th17 [26]. However, other IL-23-regulated mechanisms are involved in the evolution of disease. For example, STAT3 activation is not restricted to IL-23 because other cytokines such as, IL-6, IL-21, IL-10, or IL-27 induce STAT3 activation without triggering adverse effects, rather, in some cases, exerting anti-inflammatory events [25] (Figure 2).
To date, multiple cytokines play complex roles in IL-23 regulation. IL-23 was shown to be upregulated in fibroblast-like synoviocytes in response to IL-1β, and TNF-α can increase IL-23 expression in fibroblast-like synoviocytes [27,28], while TNF-α receptor 1 can decrease IL-23 expression by downregulating subunit p40 [29]. Likewise, the cytokine IL-10, which has an anti-inflammatory effect, can also decrease IL-23 expression [30].
A mounting number of studies has evidenced [23,31] that the main role of IL-23 is to induce the differentiation of T CD4+ naive cells into Th17 cells [32,33], which leads to enhanced IL-17 production, considered a pivotal player in the pathogenesis of inflammatory and autoimmune diseases [34,35]. Thus, well documented experimental data have highlighted that IL-23/IL-17 axis activation contributes to the development of several inflammatory autoimmune diseases. Direct evidence obtained from experimental mouse models confirms the critical role of the IL-23/IL-17 axis in the pathogenesis of various autoimmune conditions, such as arthritis [36]. Consequently, suppressing the trigger of the IL-23/IL-17 axis improves the inflammatory condition, and is considered a promising therapeutic approach in patients with these disorders [34].
Recent advances have reported that IL-23 induction can also occur through Toll-like receptor signalling. It has been demonstrated that Theiler's murine encephalomyelitis virus (TMEV), which leads to infection of central nervous system microglia and macrophages in mice, provoking a disorder similar to MS in humans, stimulates the expression of IL-23 via binding to TLR3 and TLR7, contributing to the development of experimental autoimmune MS [37]. Additionally, other research findings have reported that IL-23 induction can also occur through TLR9 or cooperatively with other TLRs [37,38].
More recently, IL-23 has been demonstrated to be regulated during tumor promoting development and to have protumor immunity [39]. An interesting study has demonstrated that Stat3 induces expression of IL-23, which is mainly secreted by macrophages in tumor microenvironment, via transcriptional activation of the IL-23/p19 gene and through NF-κB/p65 activation promoting the tumor development. In contrast, Stat3 also inhibits NF-kappaB/c-Rel-dependent IL-12/p35 gene expression in cancer-linked dendritic cells. Furthermore, tumor-associated regulatory T cells (Tregs) express the IL-23 receptor, which stimulates the expression of Stat3 in dendritic cells, leading to upregulation of the Treg-specific transcription factor Foxp3 and the immunosuppressive cytokine IL-10. These results demonstrate that Stat3 induces IL-23-mediated tumorigenes [40]. However, findings of IL-23’s antitumorigenic and antimetastatic characteristics demonstrated that IL-23 induced long-term regression of tumors similar to that of IL-12-transduced cancers. Other studies have also shown that CD40 ligand expression on lung tumor cells activates the immune response, determining an increased transcription of p19 and p40 subunits and influencing the regression of the tumors [41,42].
It has been suggested, based largely on in vitro observations, that IL-23 stimulation increases the number of already-differentiated Th-17 cells, maintains IL-17 production from Th17 cells. For example, the addition of IL-23 during the culture of activated or memory T cells results in an increase in their proliferation and the frequency of IL-17+ T cells produced [43]; IL-23 is also required during restimulation of Th17 cells (i.e., cells previously stimulated with TGF-β and IL-6, to maintain IL-17 production from the Th-17 cells) [44]. Similarly, it has been suggested that IL-23 may stabilize the phenotype of Th17 cells through mechanisms dependent on the transcription factor STAT3 [45,46]. Two other cytokines thought to be involved in Th17 differentiation, IL-6 and IL-21, also share the STAT3-dependent signaling pathway with IL-23.
Emerging evidence has highlighted that IL-23 can be considered a survival factor for Th-17 cells [35]. All these data are confirmed by the observation of reduced frequencies of Th-17 cells in mice lacking the IL-23 gene [43].
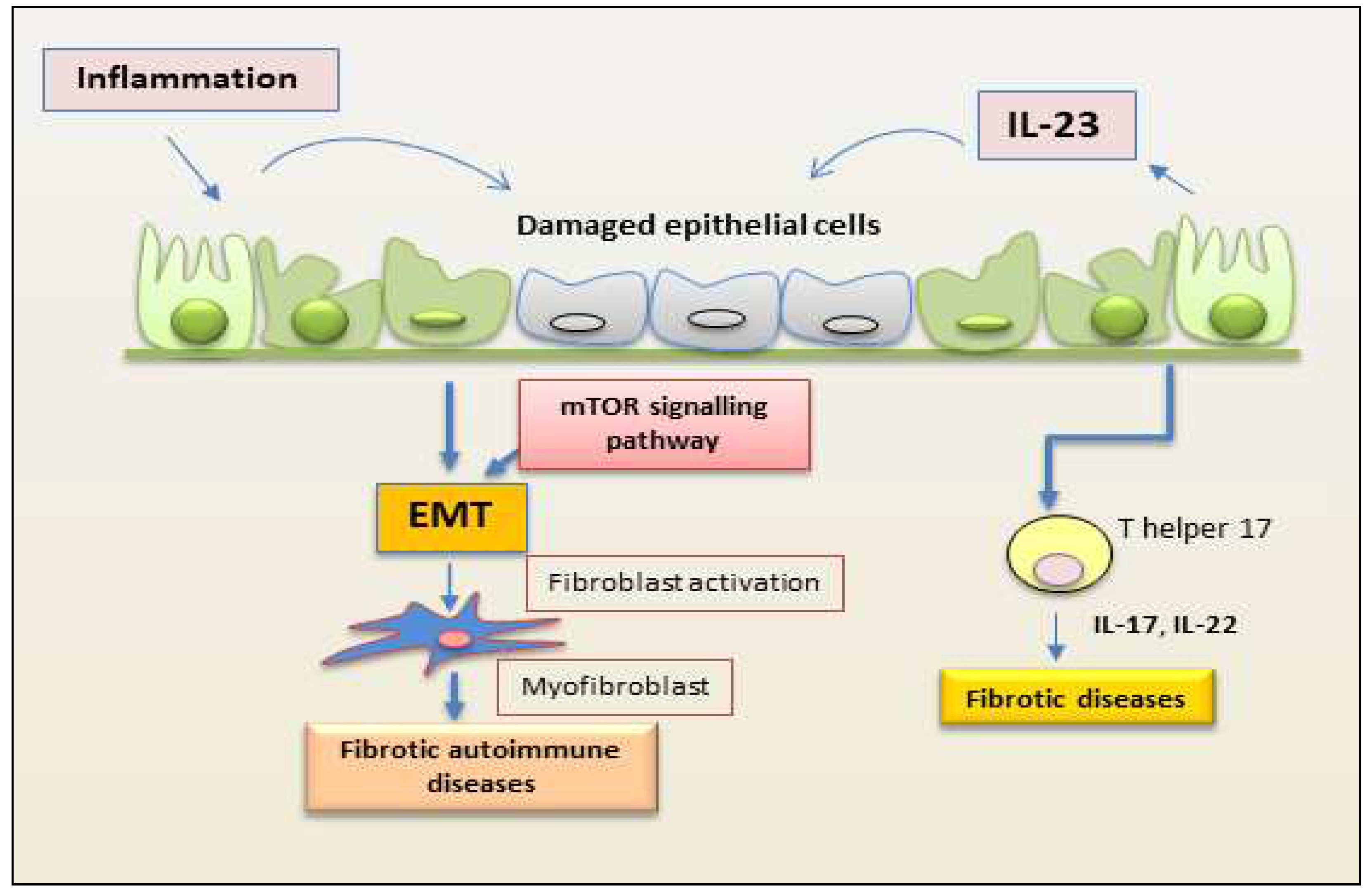
Finally, several findings highlighted the profibrogenic role of IL-23 as a promoter of the epithelial mesenchymal transition (EMT) process, an aberrant profibrotic response to repetitive injury of epithelia, and the acquisition of a mesenchymal phenotype. High levels of IL-23 have been found in some chronic inflammatory autoimmune disorders characterized by fibrosis [13,14,28].
4. Role of IL-23 in the Fibrotic Process
IL-23 is known to mediate inflammatory conditions through the induction of Th-17 cells which produce the pro-inflammatory IL-17 [Bianchi E, 2021 Sep 23;12:770275]. Since the prevailing hypothesis is that the fibrotic evolution of diseases is preceded by chronic inflammation, therapeutic strategies blocking IL-23 were suggested as a promising approach, though the specific role of IL-23 in fibrosis needs to be clarified. This section summarizes the most recent findings regarding the role of IL-23 in fibrotic diseases.
4.1. Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is characterized by chronic progressive fibrosis of the lung [47]. In pulmonary fibrosis, in vitro experiments demonstrated that treatment with an anti-interleukin-23 specific antibody attenuated airway inflammation and reduced fibrosis by blocking interleukin-17A and -22 release. The role of IL-23 in the pathophysiology of respiratory diseases is attracting increasing attention from researchers. An important role of IL-23 in the pathological features of pulmonary emphysema was demonstrated [48], and the involvement of IL-23 in the pathogenesis of a Lipopolysaccharide-induced animal model of acute respiratory distress syndrome was also demonstrated [49]. Also, in the overall accepted bleomycin or IL-1β-induced pulmonary fibrosis, both IL-23 and IL-17A play a significant role in this context. All the data collected suggest that IL-23 and IL-17A are essential for fibrosis pathway activation [50]. Moreover, the data seem to point towards a priority role of IL-23, which would also be responsible for the consequent secretion of IL-17 and acute exacerbation of pulmonary fibrosis; in fact, by blocking the secretion of IL-23, the release of IL-17 is also decreased [50]. In addition, IL-22, IL-23, as well as IL-17 were significantly increased in the serum of lung cancer patients associated with IPF; the levels of these cytokines found in the serum show such significant values that they represent a parameter used to discriminate between the lung cancer patients and the lung cancer-associated IPF group; finally, the expression of IL-22, IL-23, and IL-17 was positively correlated with the degree of differentiation and tumor metastasis [51].
4.2. Inflammatory Bowel Diseases
Idiopathic inflammatory bowel diseases (IBD) represent chronic inflammatory diseases of the gastro-intestinal tract characterized by a strong inflammatory component, often on an autoimmune basis, against the intestinal microbiome; the main diseases that fall into this group are Crohn's disease and ulcerative colitis [52,53]. Intestinal fibrosis is an important complication of IBD. With the establishment of a fibrotic evolution, the intestinal wall undergoes substantial structural modifications that lead to stiffness and a reduction in caliber. These phenomena, in the long run, seriously compromise the quality of life of patients. Chronic inflammation is certainly a factor that predisposes to fibrotic evolution. Furthermore, currently, there are no drugs that have shown efficacy in blocking intestinal fibrosis or improving the pathological condition. For these reasons, surgical treatment remains the only intervention strategy in cases of intestinal fibrosis and stenosis. Interleukin (IL)-12 and IL-23, with their structural similarities, are important cytokines in the pathogenesis of IBD. Recent data reported the efficacy of p40 peptide-based vaccines on intestinal inflammation, in a colitis model made in a laboratory [54]. The results demonstrate that the administration of the vaccine reduces the clinical symptoms, slows down the fibrotic process with a significant attenuation of the inflammatory parameters, and reduces the release of pro-inflammatory cytokines, leading to an improvement in intestinal conditions in the mouse model used. In addition, in the intestinal lamina propria and in the local lymph nodes, a high ratio of Treg/Th1 and Treg/Th17 cells was detected [54]; furthermore, in CD11c+ dendritic cells, the vaccine stimulates the release of IL-10, which is critical to controlling small intestinal immune homeostasis by limiting the reactivation of local memory T cells and so attenuating excessive immune responses [54]. All these data confirm a key role for IL-23 in the modulation of the activation of Th17 cells involved in fibrosis pathway activation.
4.3. Liver Fibrosis
Non-alcoholic fatty liver disease (NAFLD) is a condition in which the liver has an excess of fat deposits [55]. Two types of NAFLD are non-alcoholic fatty liver (NAFL) and non-alcoholic steatohepatitis (NASH). People generally develop one form of NAFLD, but a good percentage appear to be predisposed to developing the second form in the long run as well. NAFL and NASH are constantly increasing around the world [56]. NASH is currently considered the advanced stage of NAFLD. It is characterized by liver steatosis, inflammation, and fibrosis with varying severity [57]. A predominant role in the inflammatory condition characterizing NASH is played by Th17 cells, which express high levels of IL-17 in response to IL-23. A vital transcription factor for Th17 is the retinoic acid receptor-related orphan receptor γt (RORγt), which enhanced expression and nuclear translocation is correlated with IL-23-dependent phosphorylation and dimerization of the signal transducer and activator of transcription 3 (STAT3) [58]. Since no targeted therapies have been identified for NAFLD/NASH, any factor that attenuates the inflammatory process and the related fibrotic process is evaluated experimentally in order to test its efficacy in these complex diseases [59]. In this respect, it is useful for scientists to hold in high regard the fact that hepatic IL-17 producing cells promote liver inflammation and dysfunction [60]. However, the genetic analysis carried out in subjects at risk of developing the NASH disease but who do not yet show clinical signs, has not yet given certain data regarding a possible alteration of the gene that synthesizes IL-23, and the candidacy of the IL-17/IL-23 axis in NASH is yet to be fully established. Experimental data obtained using a mouse model with a double mutation in the IL-23 gene have however shown that protection against chronic inflammation and fibrotic development is moderate but not total, and this seems to suggest that although the IL-17/IL-23 axis is involved, probably it is related to the activation of other inflammatory pathways, and targeting IL-23 signalling activation may not be the only therapeutic approach for NASH [61].
5. Novel role of IL-23 in Autoimmune Fibrotic Diseases
IL-23 is a factor involved in the development of autoimmune diseases such as multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus; it carries out its activity by stimulating and activating the pathogenic Th17 cells. Therefore, IL-23 is a potential target for modulating autoimmune responses and pathogenic Th17 cell effects. Recently developed clinical trials have shown the beneficial effects of blocking the IL-23/Th17 pathway in chronic inflammatory autoimmune diseases characterized by fibrotic damage to the organs. Here we report the most recent and pioneering discoveries in this field.
5.1. Rheumatoid Arthritis
The role of IL-23 in RA has been extensively studied in a co-morbidity that affects approximately 15% of RA patients and is termed RA interstitial lung disease (RA-ILD) [62]. How much an excessive or altered immune response mediated by Th17 activation is involved in this pathology remains to be demonstrated. Recently, insight on the direct role of IL-23 in lung fibrosis were obtained, experimentally investigating the responsiveness of lung fibroblasts to IL-23 stimulation. The induction of CCR2 expression that regulates monocyte chemotaxis, and the increased fibroblast migration suggest a direct role for IL-23 in fibrotic lung disease associated with a Th17 biased-immune response [63]. In this context, a process strictly correlated with the fibrotic evolution [2,3] plays a role, represented by EMT which seems to be activated in the lung by chronic inflammatory stimuli and tissue damage. The EMT process creates an environment that facilitates fibrosis when alveolar epithelial cells were injured. In this scenario, IL-23 exerts its pro-fibrogenic role on somatic alveolar type I (ATI) epithelial cells [63]. Primary ATI cells, after prolonged culture on rigid culture dishes, clearly show signs of a gradual transformation towards a mesenchymal phenotype characterized by the loss of epithelial proteins such as caveolin-1, and by a reorganization of the F-actin cytoskeleton, indicating the initiation of the EMT process. IL-23 appears to be actively involved in this process because the mesenchymal transformation process is accelerated by in vitro stimulation with this cytokine which results in loss of epithelial marker caveolin-1 and increased expression of mesenchymal markers such as α-smooth muscle actin (α-SMA) and collagen I/III protein. Furthermore, IL-23 significantly promotes cell migration, and regulates apoptotic resistance on Il-23-transitioning-treated ATI cells [63]. IL-23-induced EMT seems to be activated and regulated by the Target of Rapamycin (mTOR)/S6 signalling pathway, which has already been demonstrated to be the pathway involved in the pro-fibrotic activity of IL-23 [64]. The hypothesis of an involvement of IL-23 in the pathogenesis of RA-ILD exerted by promoting mTOR/S6 signalling-dependent EMT in alveolar epithelial cells was supported by transcriptional sequencing analysis of human lung fibrosis biopsy tissue [63], detecting a significant increase in IL-23 mRNA expression in RA-ILD lung sections positively correlated with transitioning ATI epithelial cell number [63] (Figure 3).
5.2. Crohn's Disease
Intestinal fibrosis is an important complication of Crohn’s disease (CD) characterized by exaggerated proliferation of myofibroblasts and increased deposition of collagen in response to prolonged injury or chronic inflammation typical of IBD [65,66]. The mechanism underlying this hyperproliferation of myofibroblasts in CD seems to be linked, as in other pathological conditions mentioned above, to an involvement of mTOR. Experimental data report that the inhibition of mTOR determines a decreased production of IL-23, which, in turn, negatively regulates IL-22 expression, and determining an improvement in the general conditions of the mouse model and a slowdown in the fibrotic evolution [66]. This inhibition of IL-23 expression is associated with elevated autophagy activity in intestinal Cx3cr1+ mononuclear phagocytes. This result paves the way to identify a new molecular pathway that can explain the intestinal fibrotic progression in CD. The autophagy gene Atg7 knockdown determines, in fact, increased IL-23 expression and, consequently, induces the release of IL-22, triggering the fibrotic molecular events’ activation [65]. These evidences suggest, once again, a correlation between the production of IL-23 and IL-22 and identify a new activation pathway of the fibrotic process that originates in Cx3cr1+ mononuclear phagocytes, in which the mTOR/autophagy pathway regulates the IL-23/IL-22 axis-dependent fibrosis. The synergistic action performed by IL-23 and IL-22 is elucidated and confirmed by the fact that the double inhibition of the release of both cytokines determines a decrease of all the parameters characterizing fibrosis [65] (Figure 3).
5.3. Autoimmune Myocarditis
Recent observations have correlated IL-23 levels with the regulation of T cells function in autoimmune myocarditis, thus confirming the role of IL-23 in the regulation of inflammatory processes [67]. Using mice mutated for the IL-23 gene, unable to produce a functional interleukin, IL23a−/− mice, it was demonstrated that IL-23 is necessary for the induction of cardiac inflammation in experimentally- induced autoimmune myocarditis (EAM) [68]. EAM has been considered a disease characterized by altered function of CD4+ T cells [67]. In addition, transfection experiments demonstrated that IL-23 was able to restore pathogenicity to CD4+ T cells lacking the IL-23 gene. These results support the hypothesis of a direct involvement of IL-23 in the autoimmune reactions of the heart, which could occur either thanks to the action of IL-23 on the activity of T helper cells or by inducing the secretion by the T helpers of a combination of cytokines capable of triggering autoimmune responses [69]. Both hypotheses seem to be valid and have found scientific evidence; in fact, continuous stimulation through IL-23 is necessary to determine the production of IL-17A by T helper lymphocytes. On the other hand, even a temporary stimulation by IL-23 seems to be sufficient to determine a pathogenic activation of the T helper. Confirming this, the lack of IL-23 does not compromise the establishment of an EAM condition when the T helpers have become autoreactive [68]. Based on this evidence, since high levels of IL-23 were found in patients with autoimmune myocarditis, which proceeded to stimulate T helper cells determining an increased production and release of IL-17A, a therapy against IL-23 could be effective in blocking or delaying the fibrotic progression of the disease [68]. Moreover, elevated fibrosis and impaired heart function was detected in IL-23−/−mice, but not in mice lacking the IL-12 gene, at the chronic stage of the disease; this underlines the importance of IL-23-dependent T cell activation in the resolution phase of the acute stage of autoimmune myocarditis.
5.4. Sjӧgren’s Syndrome
Sjӧgren's syndrome (SS) is an autoimmune disease characterized by a chronic inflammatory response which causes a morphological and functional alteration of the exocrine glands, in particular the salivary and lacrimal glands and this seriously compromises the quality of life of these patients [2,3]. “Primary” SS is defined as a standalone entity occurring in the absence of another systemic autoimmune disease, whereas “secondary” disease is associated with the presence of others autoimmune conditions such as RA, SLE or systemic sclerosis (SSc). Currently, the data relating to an involvement of IL-23 in the pathogenesis of SS are still few, although very promising. Previous research revealed that IL-17/IL-23 expression was enhanced in mouse models with SS, indicating that Th17 participated in lymphocytic infiltration of salivary glands and contributed to lesion formation [70,71]. Another study showed that IL-22, IL-23, and IL-17 were increased in the peripheral blood of patients with SS, both at the protein and mRNA levels [72], assuming that the IL-23/IL-17/IL-22 axis could be one of the key mediators in the pathogenesis of primary SS [70,72,73,74]. Interestingly, in the absence of certain evidence of primary SS, the immunohistochemical detection of IL-17/IL-23 would classify these patients as involved in a Th17 reaction, and lead to the selection of patients to be referred for subsequent periodic diagnostic screening. Based on this assumption, the use the IL-17/ IL-23 immunohistochemical detection could be employed to improve the identification of SS patients with a possible diagnosis in all cases which do not fully meet the American-European criteria for pSS, in particular when the germinal center is not present at histopathological analysis and anti-SSA and anti-SSB antibody are undetectable in the serum [19]. Recently a role for the synergetic interaction between Il-23 and TLR was demonstrated in SS; TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in primary SS. Therefore, therapeutic strategies directed against TLR/IL-17 pathway might be valid candidates for treatment of SS [75]. In recent years, a thriving research sector has demonstrated that SS is often accompanied by fibrotic phenomena affecting the salivary glands mediated by the activation of the EMT program, triggered by the chronic inflammation that characterizes the disease [2,3]. Therefore, based on the attribution of an important role of IL-23 in the exacerbation of the disease, there are all the premises to identify a correlation between IL-23 expression, chronic inflammation and fibrosis in SS.
5.5. Systemic Sclerosis
Systemic sclerosis (SSc) is a heterogeneous chronic, autoimmune, multisystem connective tissue disorder characterized by vasculopathy, inflammation, and progressive fibrosis of the skin and internal organs [76]. Interstitial lung disease (ILD) is a major common complication, along with pulmonary arterial hypertension, which is the leading cause of morbidity and mortality in scleroderma patients [77]. The frequency of pulmonary fibrosis in patients with elevated IL-23 levels was significantly higher than in those with normal IL-23 levels [78]. IL-23 elevation was related to early-stage SSc and to the presence of pulmonary fibrosis but not to other clinical features of SSc [78]. IL-23 has been suggested to be involved in the induction of autoimmunity, including Experimental autoimmune encephalomyelitis (EAE), collagen-induced arthritis, and inflammatory bowel disease [79]. In SSc, chronic T cell activation characterized by increased T cell-associated cytokines is thought to contribute to tissue inflammation [76]. Since recent studies suggest that IL-23 can differentiate activated T cells into effector T cells [80], IL-23 may be associated with the process of T cell activation in SSc [78]. Th17 cells may cause vascular injury, and fibrosis as well as autoimmunity in SSc since the IL-17 receptor is expressed on fibroblasts and endothelial cells [81]. Consistently, IL-17 can cause fibroblast proliferation and upregulation of cell adhesion molecules on endothelial cells [81]. Thus, it is possible that IL-23-induced Th17 cells secrete IL-17, which causes autoimmunity, vascular injury and fibrosis in patients with SSc [78]. Since IL-17 is significantly related to the early stage of SSc but not to other clinical features of SSc, IL-17 plays an important role in the induction of SSc [81]. Recently, a strong correlation was demonstrated between the disease onset, and the triggering of pulmonary fibrosis, and the elevation of IL-23, although IL-23 was not associated with the extent of skin sclerosis. Overall, these pioneer data seem to suggest that Th17, under the stimulus of Il-23, is related to the induction of SSc, but not to progression or maintenance of the disease [78]. This hypothesis has also recently found radiological confirmation because, in SSc patients, there was a statistically significant difference as regards serum concentration of IL-23 in patients with pulmonary fibrosis by chest x-ray [82].
5.6. Multiple Sclerosis
MS is a chronic autoimmune disorder affecting an estimated two million people worldwide. The pathological hallmarks of MS include perivascular T-cell inflammation and disseminated demyelinating lesions [83]. Experimental autoimmune encephalomyelitis (EAE) is an inflammatory autoimmune pathology that can be induced in mice and, in addition, presents many similarities with human MS [79]. EAE is a complex disease in which the interaction between several immunopathological and neuropathological events determines an approximation of the pathological characteristics of MS manifested morphologically by inflammation, demyelination, axonal loss, and gliosis [79]. The main characteristic of the EAE condition is a fibrotic scar that determines an inhibitory environment hindering the remyelination; thus, anti-fibrotic drugs may serve as novel therapeutic targets for MS. As in MS, aberrant T lymphocytes traffic versus the brain and spinal cord, causing disruption of the myelin sheath integrity of the central nervous system (CNS), leading to paresthesia, paraparesis, neuritis, and ataxia [84]. Since EAE presents clinical features similar to human MS, it could be used as a model to identify the clinical efficacy of targeting the IL-23 immune pathway. Indeed, specific anti-IL-23p19 antibodies were produced to test whether blocking the functionality of IL-23 reduced the clinical symptoms of EAE and whether it could be used also in human disease [85]. The treatment of anti-IL-23p19 diminishes the serum level of IL-17 as well as the expression of IFN-γ, IP-10, IL-17, IL-6, and TNF in the CNS, inhibiting thus multiple inflammatory signalling pathways that drive CNS autoimmune inflammation. In addition, therapeutic efficacy of the anti-IL-23p19 was demonstrated to prevent disease relapsing [85]. Recently, the wealth of knowledge in this field has been enriched with new discoveries that have demonstrated IL-23 as a key factor driving inflammatory processes in the CNS [86]. In particular, it was used a transgenic mouse with astrocyte-specific expression of IL-23 that developed an ataxic phenotype and cerebellar infiltrates with high amounts of B lymphocytes. In these mice, in which was EAE was induced, it was demonstrated that the local IL-23 production in the CNS determines the aggravation of the disease course with severe paraparesis and an ataxic phenotype, leading to the enhancement of gliosis and neuroinflammation in the CNS [86,87]. Certainly, further studies will be necessary to identify the mechanisms that explain the key role of IL-23 in MS, but the premises are very interesting and the preliminary results very intriguing.
6. Conclusions
Recent discoveries place IL-23 in a prominent position in the modulation of the immune response mediated by T helper lymphocytes. This has given rise to a series of investigations on the ability of IL-23 to modulate autoimmune processes, characterized by chronic inflammation, which, frequently, evolve towards fibrotic processes. Although the mechanisms underlying the pro-fibrotic activity of IL-23 are not clear, some underlying mechanisms common to several fibrotic and autoimmune diseases have been identified. This suggests that we are on the right track and that, soon, therapies that block the activity or the release of IL-23 could represent a valid therapeutic alternative in the course of autoimmune fibrotic diseases.
Author Contributions
all authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version for publication. M.S. and S.L. had full access to the data collected in the review, take responsibility for their integrity and performed a critical reading. All authors have read and agreed to the published version of the manuscript.
Funding
this research received no external funding.
Conflicts of Interest
the authors declare no conflict of interest.
References
- Pisetsky, D.S. Pathogenesis of autoimmune disease. Nat Rev Nephrol 2023, 19, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFβ-Dependent EMT. Biomolecules. 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S. Immune and Non-Immune Inflammatory Cells Involved in Autoimmune Fibrosis: New Discoveries. J. Clin. Med. 2023, 12, 3801. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From Clinical Significance to Quantification. Adv. Sci 2021, 8, e2004433. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. The 'cytokine storm': Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, E.; Semerano, L.; Assier, E.; Falgarone, G.; Boissier, M.C. Interleukin-23: A key cytokine in inflammatory diseases. Ann. Med. 2011, 43, 503–511. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Croxford, A.L.; Mair, F.; Becher, B. IL-23: One cytokine in control of autoimmunity. Eur. J. Immunol. 2012, 42, 2263–2273. [Google Scholar] [CrossRef]
- Xiong, D.K.; Shi, X.; Han, M.M.; Zhang, X.M.; Wu, N.N.; Sheng, X.Y.; Wang, J.N. The regulatory mechanism and potential application of IL-23 in autoimmune diseases. Front. Pharmacol. 2022, 13, 982238. [Google Scholar] [CrossRef]
- Abdo, A.I.K.; Tye, G.J. Interleukin 23 and autoimmune diseases: Current and possible future therapies. Inflamm Res. 2020, 69, 463–480. [Google Scholar] [CrossRef]
- Bianchi, E.; Vecellio, M.; Rogge, L. Editorial: Role of the IL-23/IL-17 Pathway in Chronic Immune-Mediated Inflammatory Diseases: Mechanisms and Targeted Therapies. Front. Immunol. 2021, 12, 770275. [Google Scholar] [CrossRef] [PubMed]
- Senoo, S.; Taniguchi, A.; Itano, J.; Oda, N.; Morichika, D.; Fujii, U.; Guo, L.; Sunami, R.; Kanehiro, A.; Tokioka, F.; et al. Essential role of IL-23 in the development of acute exacerbation of pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L925–L940. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology. 2012, 135, 112–124. [Google Scholar] [CrossRef]
- Gubler, U.; Chua, A.O.; Schoenhaut, D.S.; Dwyer, C.M.; McComas, W.; Motyka,R. ; Nabavi, N.; Wolitzky, A.G.; Quinn, P.M.; Familletti, P.C. Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc. Natl. Acad. Sci. USA 1991, 88, 4143–4147. [Google Scholar] [CrossRef]
- Shimozato, O.; Ugai, S.; Chiyo, M.; Takenobu, H.; Nagakawa, H.; Wada, A.; Kawamura, K.; Yamamoto, H.; Tagawa, M. The secreted form of the p40 subunit of interleukin (IL)-12 inhibits IL-23 functions and abrogates IL-23-mediated antitumour effects. Immunology. 2006, 117, 22–28. [Google Scholar] [CrossRef]
- McKenzie, B.S.; Kastelein, R.A.; Cua, D.J. Understanding the IL-23/IL-17 immune pathway. Trends Immunol. 2006, 27, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Brentano, F.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Abundant expression of the interleukin (IL)23 subunit p19, but low levels of bioactive IL23 in the rheumatoid synovium: Differential expression and Toll-like receptor-(TLR) dependent regulation of the IL23 subunits, p19 and p40, in rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 143–150. [Google Scholar] [CrossRef]
- Fusconi, M.; Musy, I.; Valente, D.; Maggi, E.; Priori, R.; Pecorella, I.; Mastromanno, L.; Di Cristofano, C.; Greco, A.; Armeli, F.; et al. Immunohistochemical detection of IL-17 and IL-23 improves the identification of patients with a possible diagnosis of Sjogren's syndrome. Pathol. Res. Pract. 2020, 216, 153137. [Google Scholar] [CrossRef]
- Zhu, C.; Nan, L.; Wu, K.; Zhai, J. Research progress on the role and therapeutic significance of IL-23/Th17 in the pathogenesis of inflammatory bowel disease. J. Liaoning Med. Coll. 2016, 37, 103–106. [Google Scholar]
- Pahan, K.; Jana, M. Induction of lymphotoxin-alpha by interleukin-12 p40 homodimer, the so-called biologically inactive molecule, but not IL-12 p70. Immunology 2009, 127, 312–325. [Google Scholar]
- Chognard, G.; Bellemare, L.; Pelletier, A.N.; Dominguez-Punaro, M.C.; Beauchamp, C.; Guyon, M.J.; Charron, G.; Morin, N.; Sivanesan, D.; Kuchroo, V.; et al. The dichotomous pattern of IL-12r and IL-23R expression elucidates the role of IL-12 and IL-23 in inflammation. PLoS ONE 2014, 9, 89092. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.M.; Way, S.S. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology. 2009, 126, 177–185. [Google Scholar] [CrossRef]
- Korta, A.; Kula, J.; Gomułka, K. The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 10172. [Google Scholar] [CrossRef]
- Salamero, A.C.; Castillo-González, R.; Pastor-Fernández, G.; Mariblanca, I.R.; Pino, J.; Cibrian, D.; Navarro, M.N. IL-23 signaling regulation of pro-inflammatory T-cell migration uncovered by phosphoproteomics. PLoS Biol. 2020, 18, e3000646. [Google Scholar]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.L.; Hughes, R.; Turner, M.; et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat Commun. 2019, 10, 9. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Synoviocytes and skin fibroblasts show opposite effects on IL-23 production and IL-23 receptor expression during cell interactions with immune cells. Arthritis Res. Ther. 2022, 24, 220. [Google Scholar] [CrossRef]
- Goldberg, M.; Nadiv, O.; Luknar-Gabor, N.; Agar, G.; Beer, Y.; Katz, Y. Synergism between tumor necrosis factor α and interleukin-17 to induce IL-23 p19 expression in fibroblast-like synoviocytes. Mol. Immunol. 2009, 46, 1854–1859. [Google Scholar] [CrossRef]
- Zakharova, M.; Ziegler, H.K. Paradoxical anti-inflammatory actions of TNF-alpha: Inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J. Immunol. 2005, 175, 5024–5033. [Google Scholar] [CrossRef]
- Mills, K.H.G. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol 2023, 23, 38–54. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ghilardi, N.; Xie, M.H.; de Sauvage, F.J.; Gurney, A.L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Blom, B.; Liu, Y.J.; de Waal Malefyt, R. From interleukin-23 to T-helper 17 cells: Human T-helper cell differentiation revisited. Immunol. Rev. 2008, 226, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Maddur, M.S.; Miossec, P.; Kaveri, S.V.; Bayry, J. Th17 cells: Biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am. J. Pathol. 2012, 181, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Tsukazaki, H.; Kaito, T. The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. Int. J. Mol. Sci. 2020, 21, 6401. [Google Scholar] [CrossRef]
- Al-Salleeh, F.; Petro, T.M. TLR3 and TLR7 are involved in expression of IL-23 subunits while TLR3 but not TLR7 is involved in expression of IFN-beta by Theiler's virus-infected RAW264.7 cells. Microbes Infect. 2007, 9, 1384–1392. [Google Scholar] [CrossRef]
- Bhan, U.; Ballinger, M.N.; Zeng, X.; Newstead, M.J.; Cornicelli, M.D.; Standiford, T.J. Cooperative interactions between TLR4 and TLR9 regulate interleukin 23 and 17 production in a murine model of gram negative bacterial pneumonia. PLoS ONE 2010, 5, e9896. [Google Scholar] [CrossRef]
- Yan, J.; Smyth, M.J.; Teng, M.W.L. Interleukin (IL)-12 and IL-23 and Their Conflicting Roles in Cancer. Cold Spring Harb Perspect Biol. 2018, 10, a028530. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Weiss, J.M.; Subleski, J.J.; Wigginton, J.M.; Wiltrout, R.H. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin. Biol. Ther. 2007, 7, 1705–1721. [Google Scholar] [CrossRef] [PubMed]
- Cheng, E.M.; Tsarovsky, N.W.; Sondel, P.M.; Rakhmilevich, A.L. Interleukin-12 as an in situ cancer vaccine component: A review. Cancer Immunol Immunother. 2022, 71, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; Mc Clanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGF beta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006, 24, 179–189. [Google Scholar] [CrossRef]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Morena, D.; Fernández, J.; Campos, C.; Castillo, M.; López, G.; Benavent, M.; Izquierdo, J.L. Clinical Profile of Patients with Idiopathic Pulmonary Fibrosis in Real Life. J. Clin. Med. 2023, 12, 1669. [Google Scholar] [CrossRef]
- Fujii, U.; Miyahara, N.; Taniguchi, A.; Waseda, K.; Morichika, D.; Kurimoto, E.; Koga, H.; Kataoka, M.; Gelfand, E.W.; Cua, D.J.; et al. IL-23 is essential for the development of elastase-induced pulmonary inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 2016, 55, 697–707. [Google Scholar] [CrossRef]
- Sakaguchi, R.; Chikuma, S.; Shichita, T.; Morita, R.; Sekiya, T.; Ouyang, W.; Ueda, T.; Seki, H.; Morisaki, H.; Yoshimura, A. Innate-like function of memory Th17 cells for enhancing endotoxin-induced acute lung inflammation through IL-22. Int. Immunol. 2016, 28, 233–243. [Google Scholar] [CrossRef]
- Gasse, P.; Riteau, N.; Vacher, R.; Michel, M.L.; Fautrel, A.; di Padova, F.; Fick, L.; Charron, S.; Lagente, V.; Eberl, G.; et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE 2011, 6, e23185. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, S.; Zhang, K.; Zhang, L. Serum cytokine levels and other associated factors as possible immunotherapeutic targets and prognostic indicators for lung cancer. Front. Oncol. 2023, 13, 1064616. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Kellerman, R. Gastrointestinal Conditions: Inflammatory Bowel Disease. FP Essent. 2022, 516, 23–30. [Google Scholar] [PubMed]
- Rodríguez-Lago, I.; Blackwell, J.; Mateos, B.; Marigorta, U.M. .; Barreiro-de Acosta, M.; Pollok, R. Recent Advances and Potential Multi-Omics Approaches in the Early Phases of Inflammatory Bowel Disease. J. Clin. Med. 2023, 12, 3418. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Weiss, C.R.; Wang, S.; Qing, G.; Yang, X.; Warrington, R.J.; Bernstein, C.N.; Peng, Z. Reversing Ongoing Chronic Intestinal Inflammation and Fibrosis by Sustained Block of IL-12 and IL-23 Using a Vaccine in Mice. Inflamm. Bowel Dis. 2018, 24, 1941–1952. [Google Scholar] [CrossRef]
- Kosmalski, M.; Frankowski, R.; Ziółkowska, S.; Różycka-Kosmalska, M.; Pietras, T. What's New in the Treatment of Non-Alcoholic Fatty Liver Disease (NAFLD). J. Clin. Med. 2023, 12, 1852. [Google Scholar] [CrossRef]
- Banini, B.A.; Sanyal, A.J. Nonalcoholic Fatty Liver Disease: Epidemiology, Pathogenesis, Natural History, Diagnosis, and Current Treatment Options. Clin. Med. Insights Ther. 2016, 8, 75–84. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Valencia-Rodríguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodríguez, G.; Zamora-Valdés, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Albhaisi, S.; Noureddin, M. Current and Potential Therapies Targeting Inflammation in NASH. Front. Endocrinol. 2021, 12, 767314. [Google Scholar] [CrossRef]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776. [Google Scholar] [CrossRef]
- Heredia, J.E.; Sorenson, C.; Flanagan, S.; Nunez, V.; Jones C; et al. IL-23 signaling is not an important driver of liver inflammation and fibrosis in murine non-alcoholic steatohepatitis models. PLoS ONE 2022, 17, e0274582. [Google Scholar] [CrossRef] [PubMed]
- Laria, A.; Lurati, A.M.; Zizzo, G.; Zaccara, E.; Mazzocchi, D.; Re, K.A.; Marrazza, M.; Faggioli, P.; Mazzone, A. Interstitial Lung Disease in Rheumatoid Arthritis: A Practical Review. Front. Med. 2022, 9, 837133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.; Lau, J.; Roden, A.C.; Matteson, E.L.; Sun, J.; Luo, F.; Tschumperlin, D.J.; Vassallo, R. IL-23 amplifies the epithelial-mesenchymal transition of mechanically conditioned alveolar epithelial cells in rheumatoid arthritis-associated interstitial lung disease through mTOR/S6 signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L1006–L1022. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cao, A.; Yao, S.; Evans-Marin, H.L.; Liu, H.; Wu, W.; Carlsen, E.D.; Dann, S.M.; Soong, L.; Sun, J.; et al. mTOR Mediates IL-23 Induction of Neutrophil IL-17 and IL-22 Production. J. Immunol. 2016, 196, 4390–4399. [Google Scholar] [CrossRef]
- Mathur, R.; Alam, M.M.; Zhao, X.F.; Liao, Y.; Shen, J.; Morgan, S.; Huang, T.; Lee, H.; Lee, E.; Huang, Y.; et al. Induction of autophagy in Cx3cr1+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol. 2019, 12, 612–623. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, B.; Jin, T.; Ocansey, D.K.W.; Jiang, J.; Mao, F. Intestinal Fibrosis in Inflammatory Bowel Disease and the Prospects of Mesenchymal Stem Cell Therapy. Front. Immunol. 2022, 13, 835005. [Google Scholar] [CrossRef]
- Vdovenko, D.; Wijnen, W.J.; Zarak Crnkovic, M.; Blyszczuk, P.; Bachmann, M.; Costantino, S.; Paneni, F.; Camici, G.G.; Luescher, T.F.; Eriksson, U. IL-23 promotes T-cell mediated cardiac inflammation but protects the heart from fibrosis. Eur. Heart J. 2020, 41, ehaa946.3725. [Google Scholar] [CrossRef]
- Wu, L.; Diny, N.L.; Ong, S.; Barin, J.G.; Hou, X.; Rose, N.R.; Talor, M.V.; Čiháková, D. Pathogenic IL-23 signaling is required to initiate GM-CSF-driven autoimmune myocarditis in mice. Eur. J. Immunol. 2016, 46, 582–592. [Google Scholar] [CrossRef]
- Burkett, P.R.; Meyer, G.; Kuchroo, V.K. ; Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J. Clin. Invest. 2015, 125, 1–9. [Google Scholar] [CrossRef]
- Nguyen, C.Q.; Hu, M.H.; Li, Y.; Stewart, C.; Peck, A.B. Salivary gland tissue expression of interleukin-23 and interleukin-17 in sjogren's syndrome: Findings in humans and mice. Arthritis Rheum. 2008, 58, 734–743. [Google Scholar] [CrossRef]
- Luo, D.; Chen, Y.; Zhou, N.; Li, T.; Wang, H. Blockade of Th17 response by IL-38 in primary sjogren's syndrome. Mol. Immunol. 2020, 127, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Ferrante, A.; Raimondo, S.; Giardina, A.; Dieli, F. , Campisi, G.; Alessandro, R., Triolo, G. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren's syndrome. Ann. Rheum. Dis. 2012, 71, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Zhang, J.; Lin, Y.; Chen, W.; Fan, X.; Zhang, D. Pathogenesis and treatment of Sjogren's syndrome: Review and update. Front. Immunol. 2023, 14, 1127417. [Google Scholar] [CrossRef] [PubMed]
- Soypaçacı, Z.; Gümüş, Z.Z.; Çakaloğlu, F.; Özmen, M.; Solmaz, D.; Gücenmez, S.; Gercik, Ö.; Akar,S. Role of the mTOR pathway in minor salivary gland changes in Sjogren's syndrome and systemic sclerosis. Arthritis Res. Ther. 2018, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.K.; Cho, M.L.; Her, Y.M.; Oh, H.J.; Park, M.K.; Lee, S.Y.; Woo, Y.J.; Ju, J.H.; Park, K.S.; Kim, H.Y.; et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren's syndrome. Arthritis Res. Ther. 2012, 14, R64. [Google Scholar] [CrossRef]
- Truchetet, M.E.; Brembilla, N.C.; Chizzolini, C. Current Concepts on the Pathogenesis of Systemic Sclerosis. Clin. Rev. Allergy Immunol. 2023, 64, 262–283. [Google Scholar] [CrossRef]
- Mendoza, F.A.; Allawh, T.; Jimenez, S.A. Pharmacological treatment of systemic sclerosis-associated interstitial lung disease: An updated review and current approach to patient care. Clin. Exp. Rheumatol. 2023, 22. [Google Scholar] [CrossRef]
- Komura, K.; Fujimoto, M.; Hasegawa, M.; Ogawa, F.; Hara, T.; Muroi, E.; Takehara, K.; Sato, S. Increased serum interleukin 23 in patients with systemic sclerosis. J. Rheumatol. 2008, 35, 120–125. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O'Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and biology of IL-23 and IL-27: Related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef]
- Kurasawa, K.; Hirose, K.; Sano, H.; Endo, H.; Shinkai, H.; Nawata, Y.; Takabayashi, K.; Iwamoto, I. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum 2000, 43, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Hammad, G.A.; Eltanawy, R.M.; Fawzy, R.M.; et al. Serum interleukin 23 and its associations with interstitial lung disease and clinical manifestations of scleroderma. Egypt. J. Bronchol. 2018, 12, 69–75. [Google Scholar]
- Khan, Z.; Gupta, G.D.; Mehan, S. Cellular and Molecular Evidence of Multiple Sclerosis Diagnosis and Treatment Challenges. J. Clin. Med. 2023, 12, 4274. [Google Scholar] [CrossRef] [PubMed]
- El Behi, M.; Dubucquoi, S.; Lefranc, D.; Zéphir, H.; De Seze, J.; Vermersch, P.; Prin, L. New insights into cell responses involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Lett. 2005, 96, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Langrish, C.L.; McKenzie, B.; Joyce-Shaikh, B.; Stumhofer, J.S.; McClanahan, T.; Blumenschein, W.; Churakovsa, T.; Low, J.; Presta, L.; et al. . Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Invest. 2006, 116, 1317–1326. [Google Scholar] [CrossRef]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. . Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003, 421, 744–748. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef]
Figure 1.
IL-23 is expressed and secreted by activated macrophages and dendritic cells located in different tissues and by non-immune cells such as keratinocytes, synoviocytes, and salivary gland epithelial cells. IL-23, through several signalling pathways, provokes, on one side, the activation of the epithelial-mesenchymal transition (EMT) mechanism inducing fibrotic processes in autoimmune diseases, and, on the other side, chronic inflammatory that determines severe fibrotic evolution in several diseases.
Figure 1.
IL-23 is expressed and secreted by activated macrophages and dendritic cells located in different tissues and by non-immune cells such as keratinocytes, synoviocytes, and salivary gland epithelial cells. IL-23, through several signalling pathways, provokes, on one side, the activation of the epithelial-mesenchymal transition (EMT) mechanism inducing fibrotic processes in autoimmune diseases, and, on the other side, chronic inflammatory that determines severe fibrotic evolution in several diseases.
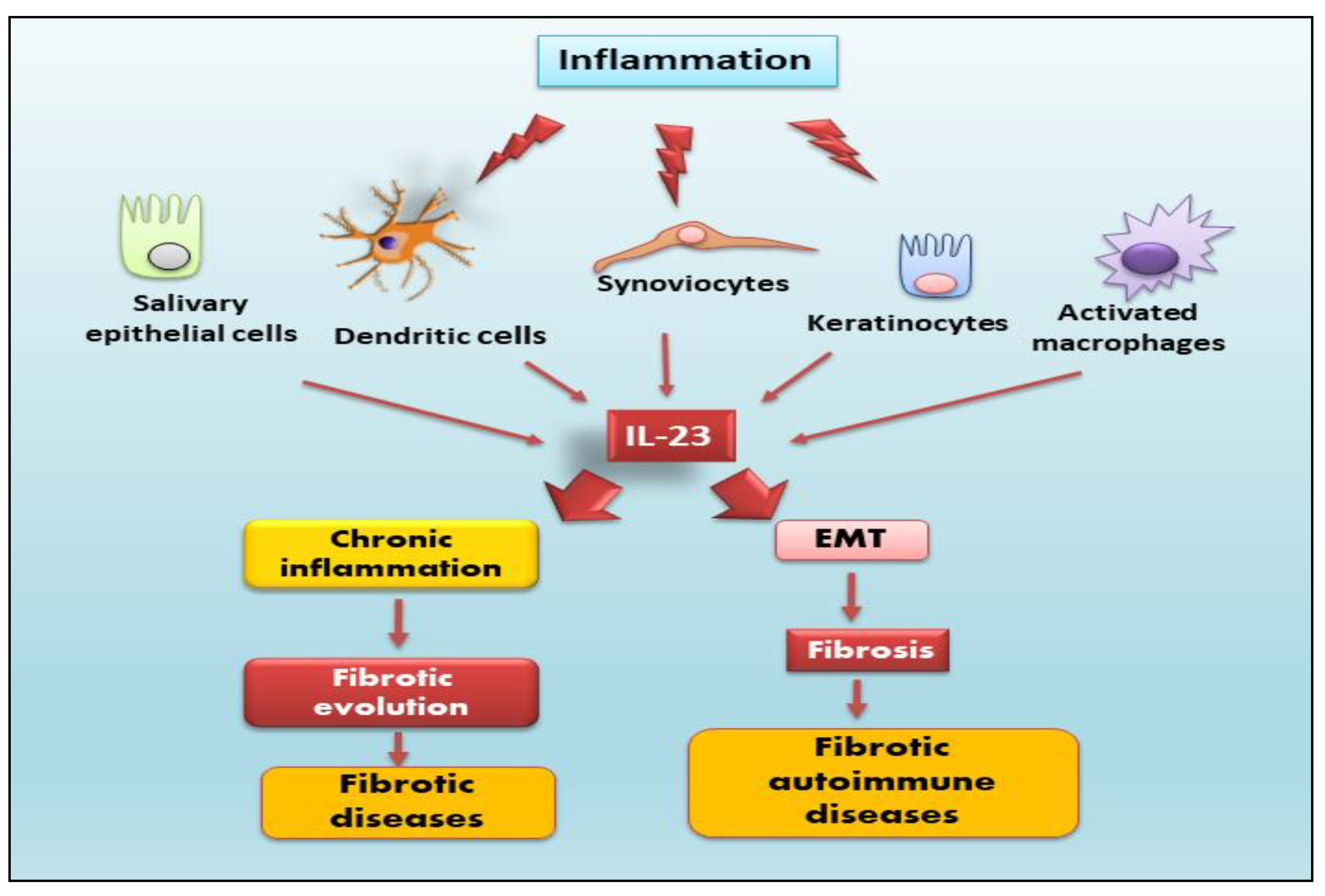
Figure 2.
IL-23 is a heterodimeric cytokine composed of p40 and p19 subunits. It binds to its IL-23 receptor complex, composed of IL-12Rb1 and IL-23R subunits, which results linked to Jak2 and Tyk2 respectively. Active phosphorylated Jak2/Tyk2 leads to phosphorylation of STAT3 and STAT4. Phospho-STAT3 and phospho-STAT4 translocate into the nucleus inducing transcription of cytokines such as IL-17, IL-22 and differentiation of T helper 17. STAT3 plus RORγt cooperate to increase IL23R, IL-17 and IL-22 expression, and stabilize the Th17 phenotype. The binding of IL-23 to its receptor through NF-κB/p65 activation leads to tumor progression. Jak2, Janus kinase 2; RORγt, RAR-related orphan receptor gamma; Tyk2, tyrosine kinase 2.
Figure 2.
IL-23 is a heterodimeric cytokine composed of p40 and p19 subunits. It binds to its IL-23 receptor complex, composed of IL-12Rb1 and IL-23R subunits, which results linked to Jak2 and Tyk2 respectively. Active phosphorylated Jak2/Tyk2 leads to phosphorylation of STAT3 and STAT4. Phospho-STAT3 and phospho-STAT4 translocate into the nucleus inducing transcription of cytokines such as IL-17, IL-22 and differentiation of T helper 17. STAT3 plus RORγt cooperate to increase IL23R, IL-17 and IL-22 expression, and stabilize the Th17 phenotype. The binding of IL-23 to its receptor through NF-κB/p65 activation leads to tumor progression. Jak2, Janus kinase 2; RORγt, RAR-related orphan receptor gamma; Tyk2, tyrosine kinase 2.
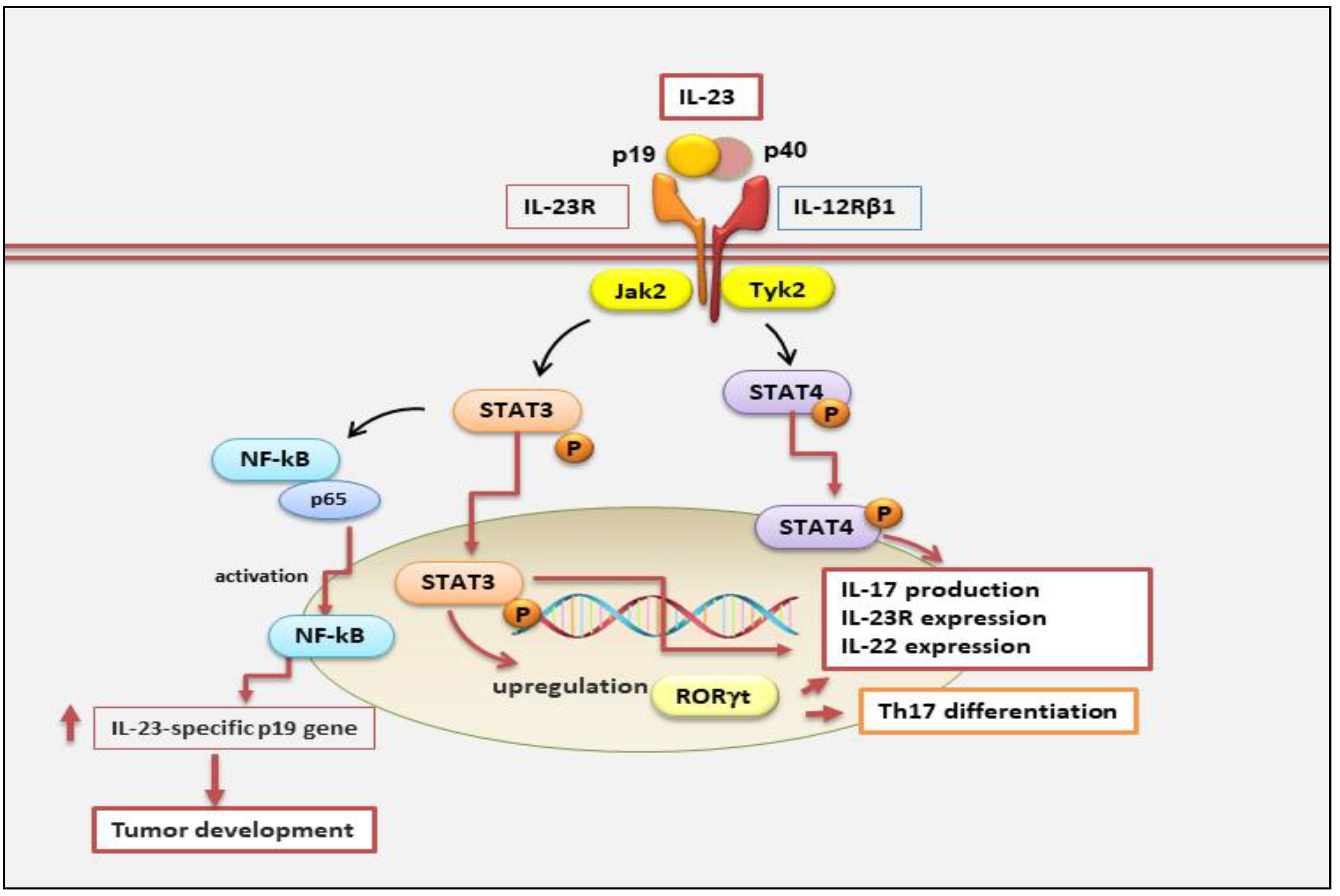
Figure 3.
Schematic representation of a proposed mechanism by which IL-23 mediates EMT in epithelial cells. Inflammation promotes repetitive injury to epithelial cells that leads to the secretion of IL-23, inducing the EMT process. Injured cells start transforming into mesenchymal cells, and IL-23 amplifies the EMT process. On binding to its receptor, IL-23 activates the kinase mTOR and promotes the expression of mesenchymal markers. This process induces fibrosis in autoimmune diseases. (EMT, epithelial-mesenchymal transition; mTOR, mammalian target of rapamycin).
Figure 3.
Schematic representation of a proposed mechanism by which IL-23 mediates EMT in epithelial cells. Inflammation promotes repetitive injury to epithelial cells that leads to the secretion of IL-23, inducing the EMT process. Injured cells start transforming into mesenchymal cells, and IL-23 amplifies the EMT process. On binding to its receptor, IL-23 activates the kinase mTOR and promotes the expression of mesenchymal markers. This process induces fibrosis in autoimmune diseases. (EMT, epithelial-mesenchymal transition; mTOR, mammalian target of rapamycin).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



