
Submitted:
03 August 2023
Posted:
04 August 2023
You are already at the latest version
Abstract
Gene or genome editing (GE) revises, removes, or replaces a mutated gene at the DNA level; it is a tool. Gene therapy (GT) offsets mutations by introducing a "normal" version of the gene into the body while the diseased gene remains in the genome; it is a medicine. So far, no in vivo GE product has been approved, as opposed to 22 GT products approved by the FDA. However, several are under development, and the FDA anticipates their fast entry, as evidenced by the FDA's actions of issuing GE-specific guidelines. The potential of GE in treating diseases far supersedes any other modality due to the recognized role of genes in most human diseases. While GT products add missing genes, GE tools can overcome many aberrations. However, GE presents many safety challenges, including off-target impact, delivery consistency, and long-term effects of gene-fixing. However, most of these concerns will be resolved once the FDA begins approving them, leading to hundreds of products capable of treating untreatable diseases.
Keywords:
gene editing 1
; CRISPR 2
; gRNA 3
; autoimmune disorders 4
; nuclease 5
; hereditary disorders 6
Introduction
The first reported instance of gene editing using “recombinant DNA” technology occurred in the early 1970s.[1] In 1972, Paul Berg, a biochemist at Stanford University, conducted an experiment where he successfully spliced together DNA from two different viruses. This experiment marked the first time scientists could manipulate and edit genes in a controlled laboratory. While this experiment was not directly editing genes in the modern sense, it marked a critical milestone in genetic engineering. It set the stage for future advancements in gene editing technologies. Berg’s pioneering work in gene editing was significant because it laid the foundation for developing modern gene-editing techniques. The term “gene editing,” as we commonly understand it today, is more closely associated with technologies like CRISPR-Cas9, which emerged much later in the 2010s. Since then, gene-editing technologies have advanced considerably, and in recent years, the CRISPR-Cas9 system has become the most widely used and versatile tool for gene editing.
GE has found many applications in agriculture, biomaterials, biodiversity, biosensors, climate change, and even backtracking evolutionary cycles. (Table 1). The first reported gene editing in plants dates to the early 1980s, specifically in 1983, when researchers were able to introduce new genes into plant cells using recombinant DNA technology.[2] This study reported on successfully introducing chimeric genes into tobacco plant cells. The researchers inserted genes from the bacterium Agrobacterium tumefaciens into the tobacco plant cells, resulting in genetically modified tobacco plants.
The Science of Gene Editing
The impact of GE is still not fully predictable but realizing that inheritance patterns (recessive or dominant) and genetic etiology, many single-gene disorders that have been identified, lead to hundreds of incurable diseases, bring great promise to the utility of GE. Figure 1 shows the chromosomes related to disorders that have been confirmed, making these disorders an excellent target for gene editing. The single-gene diseases like sickle cell anemia, Tay-Sachs, Tay-Sachs, and hemochromatosis can be treated, as well as all cancers. Many GE products are under development[19] [20,21,22,23] as ex and in vivo products.[24] In vivo GE is the focus of GE tools, delivering the gene editing machinery into the body; the ex vivo editing is considered at GT.
Ex vivo editing involves modifying cells outside the body and reintroducing them into the patient.[25] An example of the latter type is using CRISPR/Cas9 to modify T cells ex vivo to express a novel T-cell receptor (TCR) that targets a cancer-specific antigen.[26] These modified T cells are then re-infused into patients with advanced refractory cancers. Ex vivo approaches are currently more commonly employed in GT, particularly immuno-oncology. Chimeric antigen receptor (CAR) T cell therapies have shown significant promise, which involves modifying T cells to recognize and attack cancer cells. Two such treatments, Kymriah and Yescarta, have already received FDA approval, although they do not involve gene editing.[27]
The ex vivo GE target cells include stem cells and immune cells that can be cultured and transplanted into the patient, such as SCID, ß-thalassemia, sickle cell anemia, and CAR-T therapy, including hematopoietic stem cells, T cells, and NK cells. These products fall under the category of GT, wherein modified cells are introduced, leaving the mutated genes in place.[28]
The greater utility of GE comes from in vivo strategy that eliminates cell collection, isolation, expansion, editing, selection, and transplantation, making it more accessible and effective at targeting a single organ than the entire organism.[29] The GE is also ideally suited for precision or personalized therapies,[30,31,32] primarily cancer treatment. In 2021, the approval of 17 personalized (individualized, precision) medicines represented approximately 35 percent of all newly approved therapeutic molecular entities.[33] The next-generation sequencing (NGS) technologies to discover new uncommon genetic illnesses have made individualized therapies more practical.
Gene editing treatments should target multiple, conserved, and functionally essential sites on genomes and use multiple gene editing events to prevent the development of resistance because gene editing encounters biological resistance immunologically or by selecting cells with a resistant target region. The modern understandings include the realization that while a single gene causes these disorders, different mutations can cause the same disease with a range of severity. In addition, several phenotypes can occur from the same mutation, brought on by variances in the patient’s surroundings and other genetic variations. For instance, additional genes have been shown to modify the cystic fibrosis phenotype in infants with the same mutation in the cystic fibrosis transmembrane conductance regulator (CFTR); for some diseases like galactosemia, mutations in various genes can produce similar manifestations.[34]
Table 3 lists the main identified gene editing applications that can significantly impact human health.
Challenges and Concerns
Applications for GE have sparked questions in science, safety, ethics, and legislation. GE in human embryos is not supported by NIH funding. The Dickey-Wicker Amendment[113] forbids the use of monies appropriated by Congress for research involving the creation of human embryos or the destruction of human embryos. The international community has called for a moratorium on heritable gene editing (HGE) since the birth of twins in China in 2019 that were genetically modified using CRISPR/Cas9. Several expert groups have been formed to create global governance frameworks.[114] The established[115] best practices require multi-stakeholder decision-making, including learned medical societies stating that “eugenic development of offspring or the genetic manipulation of non-disease traits may never be justifiable.”[116] Regulators should consider the need to support anti-editing tools like anti-CRISPR, which can prevent or undo unwanted gene editing, to counteract the misuse of gene editing tools.[117] However, there is a growing concern about these limitations that might slow the progress of GE technologies and the abuse of use technologies.[118]
A significant issue in GE pertains to biohacking, which presents serious health issues, left with little control, despite many incidences of self-medication and even producing gene-edited babies. Additionally, the widely available DIY kits pose a severe risk of off-target editing, unintended off-target editing consequences, and unknowable long-term off-target editing effects.[119]
Given these risks, the NIH guideline[120] to streamline gene therapy was established. However, in a recent revision of this guideline, the NIH dropped the requirement to register and report on human gene therapy protocols. As a result, only the FDA has regulatory oversight of all human GT and GE, treating them like any other biological product. The NIH now focuses on safety and ethical issues[121] [122] , yet much of the GE remains uncontrolled.
Current Status
Genetic mutations trigger human evolution and bring thousands of diseases. Until gene manipulation technologies arrived, treating the ailments of about a billion patients suffering from mutated, disease-causing genes was impossible. The GE technology is preceded by GT products that fall within the same definition, and understanding the nature of GT products is essential for GE development. The entry of GT products began in 2017 using an AAV-delivered ZFN.[123]
The FDA has approved 22 GT products[124] (Table 2), and the EMA[125] has approved 13 products (labeled as Advanced Therapy Medicinal Products) (ATMPs); neither agency has approved any GE product, though a large number are under development.[126] [127]
The first FDA-approved GT product Kymriah (tisagenlecleucel), an antigen receptor T cell that is chimeric (CAR-T),[128] costs about half a million dollars per dose; the most recent gene therapy product, Hemgenix, a hemophilia treatment, costs $3.5 million per dose.[129] This high cost comes from the amortization of development costs of about $1-5 billion per product[130] distributed over a smaller number of patients. When they arrive, the GE tools are also expected to have a high cost, opening the debates about the affordability of these products. The potential of gene editing as a therapeutic intervention for human disease extends across multiple diseases, such as cancer, blood disorders, genetic diseases, and viral diseases.[131] Gene editing technologies can correct disease-causing mutations in these diseases or equip cells with new functionalities to combat disease progression.[132] However, translating these technologies from bench to bedside has various hurdles, including ethical and regulatory challenges and scientific and technological limitations.[133] The advent of gene editing technologies raises critical societal questions, particularly related to the accessibility and equity of these therapies. The development and manufacturing of gene editing therapies are complex and costly, potentially limiting their accessibility to a small fraction of patients who can afford them.[134]
Furthermore, there are significant disparities in access to advanced healthcare technologies across different regions, which could be exacerbated with the advent of gene editing therapies.[135] For instance, patients in low- and middle-income countries may face significant barriers to accessing these therapies due to a lack of healthcare infrastructure, financial constraints, and regulatory challenges.[136]
Addressing these challenges would require concerted efforts from governments, policymakers, healthcare providers, and industry stakeholders. For instance, policies could be developed to incentivize the production of affordable gene editing therapies and facilitate access to needy patients.[137]
Moreover, robust healthcare infrastructure would need to be developed in regions with limited access to advanced healthcare technologies. This would entail investments in healthcare facilities, training healthcare professionals, and evolving regulatory frameworks to ensure gene editing therapies’ safe and effective use.[138]
About 400 million people worldwide with 7,000 diseases caused by mutations in single genes[139] can benefit if GE products are made affordable. Gene editing needs are continuously expanding, such as the recent suggestions to monitor children for prospective genetic disorders and fix them well before they become evident. In terms of the market, the global gene editing market was valued at approximately $3.7 billion in 2019, and it is projected to reach $8.1 billion by 2025. The major players in the market include companies like CRISPR Therapeutics, Intellia Therapeutics, Editas Medicine, and Sangamo Therapeutics.[140]
However, gene editing, with its ability to directly modify the genetic code, has the potential to revolutionize medicine, particularly in genetic diseases that account for most human diseases. The development of tools like CRISPR/Cas9 has made gene editing a practical approach for treating various human diseases caused by single-gene mutations.[141] [142] Despite the enormous potential of gene editing technologies, significant challenges remain, including efficiency, precision, delivery, and safety issues.[143]
Interestingly, gene editing technologies have opened new therapeutic possibilities and brought about a new era of personalized medicine. Therapies can be designed according to the genetic profile of individual patients, thereby improving therapeutic outcomes while minimizing potential adverse effects.[144] For example, gene editing technologies could engineer immune cells to recognize and kill cancer cells more effectively in the context of cancer.[145] Despite these exciting prospects, there are legitimate concerns surrounding gene editing technologies, particularly when it comes to alterations that can be inherited by future generations (germline gene editing).[146]
Ethical considerations are another significant aspect of gene editing technologies, particularly concerning germline editing, i.e., modifications that can be passed on to subsequent generations.[147] The controversy surrounding the announcement of the birth of gene-edited babies in China in 2018 highlights the urgency of addressing these ethical concerns.[148]
Ethicists, scientists, and policymakers worldwide are actively engaged in discussions to develop guidelines and regulations for the ethical use of gene editing technologies. Notably, the World Health Organization has established an expert advisory committee to develop global standards for governance and oversight of human genome editing.[149]
Current Status
A significant novelty in GE efforts comes from the safety and efficacy of CRISPR/Cas9 technology that has been at the forefront of these developments,[150] where clinical trials have demonstrated that gene editing can increase the production of fetal hemoglobin in the red blood cells of patients with β-thalassemia and sickle cell disease.[151] This approach has shown significant promise, with patients demonstrating clinical improvements and decreased need for transfusions.[152] Early successes include using AAV-delivered CRISPR/Cas9 components for treating Leber congenital amaurosis[153] and muscular dystrophy in animal models.[154]
Similarly, promising preclinical results for treating Duchenne Muscular Dystrophy (DMD), a fatal muscle disorder caused by mutations in the DMD gene. CRISPR/Cas9 has been used to restore dystrophin expression in animal models of DMD, offering hope for future clinical applications.[155]
While we have already seen early-stage clinical trials of gene editing therapies for certain diseases, such as sickle cell disease and β-thalassemia,[156] many potential gene editing applications are still in the preclinical or early clinical development stage.[157] This underlines the need for more research to understand and improve the limitations of current technologies, such as improving the precision of gene editing, optimizing delivery systems, minimizing off-target effects, and resolving ethical and safety concerns.[158] [159] The following sections of this review will describe the current status and challenges in the field and discuss the prospects of gene editing for treating human diseases.
Gene editing has also shown promise in treating infectious diseases, particularly in the case of HIV. This retrovirus integrates its genome into host cells, making it extremely difficult to eradicate from the body. However, with gene editing, researchers have been able to excise the integrated HIV genome from infected cells in vitro.[160] Early-phase clinical trials have employed T cells modified by gene editing to resist HIV infection, demonstrating the safety and feasibility of this approach.[161]
Despite much optimism, several challenges must be addressed before these therapies can be broadly and safely applied in the clinic.[162] One such challenge is the potential for off-target effects, i.e., unintended modifications at sites other than the target locus. Such off-target edits could potentially lead to harmful consequences, including the activation of oncogenes or the inactivation of tumor suppressor genes, resulting in cancer.[163]
Methods to improve the specificity of gene editing enzymes and to better detect and predict off-target effects are being actively researched. The development of high-fidelity versions of Cas9, such as eSpCas9 and SpCas9-HF1, represents significant strides in this direction.[164]
Another challenge is the potential immune response to gene editing components, particularly the bacterial-derived Cas9 protein. Pre-existing immunity to Cas9 could reduce the efficacy of gene editing. It could also lead to serious adverse effects [165]Approaches to mitigate this immune response, such as the use of immunosuppressants or the development of humanized versions of Cas9, are being explored.[166]
The efficient and safe delivery of gene editing components to target cells in the body is another critical challenge. Several strategies are being explored, including viral vectors, lipid nanoparticles, and cell-penetrating peptides.[167] However, each approach has limitations, such as immunogenicity, limited cargo capacity, and off-target tissue distribution.[168]
Novel delivery strategies, such as engineered exosomes[169] or gold nanoparticles,[170] are currently under investigation and could potentially overcome some limitations. The future of gene editing, potentially revolutionizing the treatment of a wide range of diseases, including many that currently have no cure,[171] many challenges remain. Technical challenges include improving gene editing tools’ efficiency, precision, and safety. While significant progress has been made in developing more precise and efficient gene editing technologies, further improvements are needed to minimize off-target effects and ensure the safety of these therapies.[172] Developing delivery systems that selectively target specific tissues and cells is another important area of research.[173]
As previously discussed, ethical challenges relate to issues such as germline editing, consent, and the potential use of gene editing for enhancement purposes. These issues will require ongoing discussion and consensus-building among scientists, ethicists, policymakers, and the public.[174]
The rapid advancement of gene editing technologies necessitates equally rapid development of regulatory guidelines to ensure safety and efficacy. Regulatory bodies worldwide are currently grappling with the challenge of developing robust frameworks for gene editing therapies.[175] [176]
There is ongoing research into next-generation gene editing tools that promise increased precision and versatility. Base editors, for instance, enable the direct conversion of one base pair to another without inducing double-strand breaks, potentially minimizing off-target effects.[177] Prime editing, another recently developed technique, allows for the insertion, deletion, and substitution of base pairs with unprecedented precision.[178]
There is also increasing interest in using gene editing tools beyond treating genetic diseases, such as developing advanced cell therapies and creating disease-resistant crops.[179] [180]
The application of gene editing technologies, particularly in the context of human health, raises complex ethical questions. One of the primary ethical issues is the potential for “germline” editing, which involves modifications to the DNA of sperm, eggs, or embryos. These changes would affect the individual they are made to and their descendants.[181] While this could provide an avenue to eliminate genetic diseases from family lines, it also opens a range of ethical considerations, including the potential for unintended consequences and the implications of permanently altering the human gene pool.[182]
In addition to germline editing, “somatic” gene editing – which involves changing the genes in cells of a specific tissue in an individual and does not affect future generations – also raises ethical issues. These primarily revolve around consent, particularly in scenarios where the technology is used in children or other individuals unable to provide informed consent.[183]
The possibility of gene editing being used for enhancement rather than therapeutic purposes – such as altering physical traits or abilities – also presents a significant ethical challenge. It raises questions about the nature of human beings and the acceptability of designing our species.[184]
Lastly, the issue of equity is a crucial ethical concern, given the potential for gene editing to be accessible only to the wealthy, thus exacerbating existing health and social inequalities.[185]
Regulatory
The legal and regulatory landscape surrounding gene editing is complex and rapidly evolving. Current regulatory frameworks worldwide differ significantly in their approach to gene editing, with some countries allowing for broad use in research and others restricting its use, particularly for germline editing.[186]
The Food and Drug Administration (FDA) regulates gene editing in the United States under its broader oversight of gene therapy. The FDA has laid down guidelines for preclinical and clinical development of gene therapy products, which include gene editing therapies. The agency requires rigorous testing for safety and efficacy before such treatments can be approved for use in patients.[187]
The European Medicines Agency (EMA) regulates gene therapies, including gene editing, in the European Union. The EMA has a similar approach to the FDA, requiring comprehensive preclinical and clinical testing for safety and efficacy. Additionally, the EMA considers the potential environmental impact of gene editing, particularly for germline modifications.[188]
In many Asian countries, such as China and Japan, gene editing regulations are less stringent. This has led to rapid advancements in gene editing research in these countries and raised ethical and safety concerns.[189]
Overall, the regulatory framework for gene editing must balance the need for innovation with ethical, safety, and societal considerations. Developing international standards and guidelines could help harmonize the global approach to gene editing and ensure that it is used responsibly and for all benefits.[190]
Regulatory challenges involve creating a regulatory framework that can keep pace with the rapid advancements in gene editing technology while ensuring the safety and efficacy of gene editing therapies. Harmonizing regulatory standards across different countries is another critical challenge to address.[191]
Regulators can do more to promote the standardization of off-target (and on-target) effect measurement, including implementing the proper methods, sample handling practices, quality control measures, data analysis, and clinical interpretation. For example, the EU guidelines for genetically modified cells’ quality, non-clinical requirements, and clinical requirements do not specify methods for determining on- and off-target effects because they are still evolving.[192] The FDA’s guidance is similar in not outlining how off-target effects discovered in pre-clinical studies should be monitored over an extended period.[193]
Since gene editing is a newer field, the regulatory control standards take a high risk-based approach, in the abundance of caution that has significantly hampered the entry of GE products. In certain situations, relying solely on product quality results will not be appropriate, requiring additional non-clinical and clinical data. In addition, release testing may be affected by the personalized nature of gene editing therapies, regulatory batch testing, and release requirements, which can consume a sizable portion of the batch.
Due to the significance of the in vivo cellular environment on gene editing efficiency, for instance, there are difficulties in predicting the clinical effects of gene editing treatments in humans using non-clinical efficacy models. A thorough investigation and attempts to develop such relevant non-clinical efficacy models should be discussed in any case and requested on a case-by-case basis in regulatory interactions, despite the idea that gene editing treatments may not need them or may not have any relevant non-clinical efficacy models.
Numerous unknowns exist regarding the handling and regulatory classification of gene editing products. Some gene editing products, for instance, are subject to GMO regulation and requirements in the EU,[194] where various competent authorities assess an ATMP GMO submission and a Clinical Trial Application (CTA) submission, resulting in timing and content inconsistencies. Several initiatives, such as standard application forms and good practice documents, are being made to clarify and harmonize the requirements.[195]
A Guideline on Assuring the Quality and Safety of Gene Therapy Products has been published by PMDA (not specific to gene editing). However, this raises scientific and policy concerns. Therefore, to maintain the orphan status of potentially curative ATMPs, significant benefit over an authorized ATMP must be demonstrated. Most ex vivo GE products will fulfill the ATMP definition as GTMPs or cell therapies. The US situation is more precise because the FDA classifies all gene editing products (both in vivo and ex vivo) as gene therapy products.
Gene editing products may also have difficulties maintaining orphan designation and proving significant benefits because of their long-lasting or potentially curative effects. In addition, clinical efficacy data to demonstrate superiority may not be attainable for scientific reasons, leaving clinical safety or even only non-clinical data as the primary evidence for a significant benefit claim.
The GE Tools
GE involves homologous recombination (HR), wherein nucleotide sequences between two DNA molecules that are similar or identical are switched; this has long been thought to be a treatment for human genetic illnesses. However, its effectiveness is enhanced by causing DNA double-strand breaks (DSBs) using nucleases, a significant advancement in GE technology.
Nuclease Mediated
In theory, the GE tools (Table 5) substitute one or more bases in any desired gene at any specific position. These technologies used programmable endonucleases and specific DNA target recognition sequences to create DNA double-strand breaks (DSBs), which led to gene replacements, insertions, deletions, and nucleotide substitutions. The repair of DSBs is required to preserve genetic material, but misrepair of DSBs can cause local sequence alteration or gross chromosomal rearrangements. The two main mechanisms to repair DSBs are classical nonhomologous end joining (C-NHEJ) and homologous recombination (HR). HR is generally considered an error-free mechanism because the homologous sister chromosome templates repair in S or G2 phase cells. C-NHEJ involves the direct ligation of DNA ends and can occur with high fidelity or is associated with small alterations at the junctions.[196]
Much of today’s basic science supporting GE tools is built on restriction enzymes to cut DNA, discovered in 1968.[197] Meganucleases (MNs), Zinc Finger Nucleases (ZFNs), Transcription Activator-like Effector Nucleases (TALENs), and Clustered Regularly Interspaced Short Palindromic Repeats-linked proteins (CRISPR-Cas) are the most frequently used methods to correct genetic mutations (Figure 2 and Figure 3). Newer tools evolving out of the CRISPR-Cas system are anticipated in the future.

Table 5.
Comparison of key nuclease-mediated GE technologies currently in use.[198].
Table 5.
Comparison of key nuclease-mediated GE technologies currently in use.[198].
| Attribute | Meganucleases | Zinc Finger Nucleases | TALENs | CRISPR/Cas9 |
|---|---|---|---|---|
| Enzyme | Endonuclease | Fok1-nuclease | Fok1-nuclease | Cas9 nuclease |
| Target site | LAGLIDADG proteins | Zinc-finger binding sites | RVD tandem repeat region of TALE protein | PAM/spacer sequence |
| Recognition sequence size | 12–45 bp | 9–18 bp | 14–20 bp | 3–8 bp/20 bp |
| Targeting limitations | MN cleaving site | Difficult to target non-G-rich sites | 5ʹ targeted base must be a T for each TALEN monomer | The targeted site must precede a PAM sequence |
| Advantage | High specificity; Relatively easy to deliver in vivo | Small protein size; Relatively easy in vivo delivery | High specificity; Relatively easy to engineer; target; mitochondrial DNA more efficiently and cause fewer off-target effects than MNs and ZFNs. | Easy to engineer; Easy to multiplex |
| Disadvantage | Target locus must be put into the genome; complex to construct; difficult to multiplex; ineffectiveness and potential genotoxicity. The targeted locus must also contain the unique cleavage site for each endonuclease. | Expensive, time-consuming, labor-intensive, difficult to choose the target sequence, needing the coding gene to be custom-built for each target site, and highly off-target gene editing. All ZF domains must also be active. | Challenging to multiplex; not relevant in the case of DNA methylcytosine; a few in vivo deliveries; We should all be engaged; TALEs are nevertheless constrained by their repetitive sequences, which make it difficult to construct them using polymerase chain reaction (PCR), and by the fact that they are unable to target methylated DNA due to the possibility that cytosine methylation will impede TALE binding and alter recognition by its typical RVD. | Lower specificity; Limited in vivo delivery |
| DNA-recognition mechanism | HR-introduced Protein-DNA interactions | DSB-introduced by Protein-DNA interactions | DSB-introduced by Protein-DNA interactions | DSB introduced by RNA-guided protein-DNA interactions |
| Target specificity | HighPositional mismatches are only occasionally accepted. Protein engineering is necessary for re-targeting. |
Highpreference for G-rich sequences Positional mismatches are only occasionally accepted. Protein engineering is necessary for re-targeting. |
HighRequires a T at each of its target’s five ends. Some positional inconsistencies are accepted. Retargeting necessitates intricate molecular cloning. |
ModerateThe two base pairs that PAM recognizes must come before the RNA-targeted sequence. Positional mismatches are only occasionally accepted. A new RNA guide is necessary for re-targeting. There is no need for protein engineering. |
| Multiplexing | + | + | + | ++++ |
| Delivery | accessible by transduction of viral vectors and electroporation |
accessible by viral vector transduction and electroporation | simple in vitro conception Due to TALEN DNA’s size and the recombination likelihood, it is challenging in vivo. |
simple in vitro The big Cas9’s inadequate packing by viral vectors is the cause of the mild difficulties of distribution in vivo. |
| Use as a gene activator | No | Yes endogenous gene activation minimal impacts off-target To target specific sequences, engineering work may be necessary |
Yes endogenous gene activation minimal impacts off-target There are no time restrictions. |
Yes endogenous gene activation minimal impacts off-target “NGG” PAM is necessary adjacent to the target sequence. |
| Use as gene inhibitor | No | Yes Works by repressing chromatin to prevent transcriptional elongation. minimal impacts off-target To target specific sequences, engineering work may be necessary. |
Yes Works by repressing chromatin to prevent transcriptional elongation. minimal impacts off-target There are no time restrictions. |
Yes Works by repressing chromatin to prevent transcriptional elongation. minimal impacts off-target “NGG” PAM is necessary adjacent to the target sequence. |
| Cost | High | High | High | Reasonable |
| Popularity | Low | Low | Moderate | High |
| Online resources | Database and Engineering for LAGLIDADG Homing Endonucleases (http://homingendonuclease.net/) | The Zinc Finger consortiums include software tools and protocols (http://www.zincfingers.org/) ZFNGenome – resources for locating ZFN target sites (https://bindr.gdcb.iastate.edu/ZFNGenome/) | Mojo Hand (http://www.talendesign.org/) or E-TALEN (http://www.e-talen.org/E-TALEN/) for TALEN designCHOPCHOP (https://chopchop.cbu.uib.no/) target site selection | Guide design: Zlab (https://zlab.bio/guide-design-resources CRISPOR: http://crispor.tefor.net/; Benchling: https://www.benchling.comAddGene: https://www.addgene.org/crispr;; https://crispr.bme.gatech.edu http://www.rgenome.net/cas-offinder/ |
Prime Editing
Prime editing[201] entails small insertions, deletions, and base editing of single bases. Prime editing promises greater specificity by reducing on- and off-target effects by requiring two additional nucleic acid matching steps because it uses CRISPR/Cas in addition to reverse transcriptase. Additionally, because of its capacity to edit farther from the Cas9 nicking site, it promises greater targeting flexibility. The size of the base pairs that can be edited is constrained, though, and more information about reverse transcriptase’s error rate is still needed. Utilizing Cpf-1, a microbial nuclease preferred over Cas9 and only required one CRISPR guide RNA for specificity while producing staggering double-stranded DNA cuts instead of blunt ones is a change.
Base Editing (BE)
Current cytosine or adenine base editors can only accomplish C-to-T (G-to-A) or A-to-G (T-to-C) substitutions in the windows of target genomic sites of organisms; therefore, there is a need to develop base editors that can simultaneously achieve C-to-T and A-to-G substitutions at the targeting site.[202] However, they cannot correct variations other than these six transition mutations or other changes made by Prime editing, like DNA fragment insertions and deletions (PE). PE uses the prime editing guide RNA, a modified sgRNA, and a modified Cas9 nickase fused to reverse transcriptase (RT) enzyme (pegRNA). Clinical trials for base prime editing have yet to begin. However, conventional CRISPR/Cas9 and base editing share some characteristics with PAM restriction, off-targets, and delivery-impairing large molecules present in prime editing.
CRISPR applications have low homology-directed repair (HDR) editing efficiency compared to nonhomologous end joining. The Cytosine Base Editor (CBE), Adenosine Base Editor (ABE), and Glycosylase Base Editors (GBE) theoretically can function in both dividing and non-dividing cells to address several single-nucleotide polymorphisms (SNPs) linked to human diseases; this inefficiency is, however, overcome by these base editors.
BEs are mainly used to target point mutations that may result in an altered DNA sequence with novel or enhanced functions and gene inactivation requiring two main components: a Cas9n fused with a deaminase and a sgRNA that binds to a specific DNA sequence.
The BE system primarily influences the editing window of nearby sites, off-target mutation, and product purity. The editing window should be minimized to increase target base accuracy when only one specific base pair needs to be changed accurately. On the other hand, a large editing activity window is used when the cytosine-based editor (CBE) system is used to introduce premature stop codons, produce large-scale saturation mutations, screen gene function, locate key amino acid positions in protein domains, etc.
Nickase-based genome engineering technologies
Inadequate outcomes, such as p53 activation, translocations, off-target mutations, and complex undesired products, could be linked to DSBs at targeted genomic loci. Since point mutations account for half of all known disease-associated gene variants, the Cas9 nickases have become essential tools with a targetable property. One of the two Cas9 nuclease domains is modified to produce the CRISPR nickase. Nickases can reduce the likelihood of off-target editing by causing a single-strand break instead of a double-strand break when used with two adjacent gRNAs. The Cas9 variants Cas9n D10A and Cas9n H840A mediate the cleavage of a single DNA strand in the complementary or non-complementary DNA strand of gRNA.
Homologous Recombination
The exchange of nucleotide sequences between two DNA molecules that are similar or identical is referred to as homologous recombination in genetics. This is frequently used in mouse genetics. This technique, also known as gene targeting, allows for precisely replacing a gene copy by integrating a gene distinct from the original gene. The development of knockout mice using embryonic stem cells to deliver synthetic genetic material to suppress the target mouse gene is of therapeutic interest. Additionally, this technology enables a reliable and effective knocking of a specific mutation, reporter, or human gene sequence into endogenous loci, producing precise and physiologically more accurate models of human disease.[205]
AAV
Since their discovery about 50 years ago, the replication-defective, non-pathogenic, almost universal single-stranded adeno-associated viruses (AAVs) have gained significance. Recombinant AAVs were created as suitable genetic medicine tools and have now developed into efficient, marketable gene therapies thanks to their distinct life cycle and virus-cell interactions. The capacity of AAVs to accurately modify the genome sets them apart from other types of viruses. Furthermore, AAV only uses the high-fidelity homologous recombination (HR) route and does not require exogenous nucleases for the previous cleavage of genomic DNA, in contrast to all current GE platforms. This leads to an exact editing outcome that preserves genomic integrity without incorporating indel mutations or viral sequences at the target site while preventing off-target genotoxicity. The stem cell-derived AAV (AAVHSCs) was found to mediate precise and efficient HR with high on-target accuracy and at high efficiencies.
Furthermore, AAVHSC editing occurs efficiently in post-mitotic cells and tissues in vivo. Additionally, AAV also has the advantage of an intrinsic delivery mechanism. Thus, this unique GE platform holds tremendous promise for correcting disease-associated mutations without adding to the mutational burden.[206]
Such models significantly impact research experiments and drug development programs spanning activities from target validation to ADMET studies.
ssODNS
Although homologous recombination (HR) can long gene knock-in and single-base alterations, its effectiveness is poor. To increase the effectiveness of knock-in with single-stranded oligo DNA nucleotides (ssODNs), it is necessary to study the best design parameters for ssODNs employing reporter systems for detecting single-base alterations. The knock-in efficiencies of optimized ssODNs are significantly higher than those of unblocked ssODNs. In addition, the number of times the altered sites are cut again can be drastically decreased by the Cas9 protein/sgRNA ribonucleoprotein complexes (Cas9-RNPs).[207]
Synthetic Genomics
In synthetic genomics, new DNA or entire lifeforms are created by manipulating current life forms genetically. Synthetic genomics differs from genetic modification because it does not utilize naturally occurring genes in creating living forms. However, more expansive and unfulfilled, synthetic genomics might use genetic codes that are not constituted of the two base pairs of DNA currently used by life. Instead, it may use a specially constructed base pair series.
Recent technological advancements include creating genomes that don’t exist in nature, assisted by protein folding models, and affordable computational costs.[208]
One of several alternative nucleic acid structures that can take on various three-dimensional formations is peptide nucleic acids (PNAs), synthetic DNA analogs. PNAs enter the double helix and create high-affinity base pairs with DNA. This attracts endogenous DNA repair systems that read desired edits from an ssDNA template. Unusual nucleic acid structures can occur in sequence-specific contexts and are important endogenous repair triggers. These concepts are used by peptide nucleic acids (PNAs) to accomplish non-enzymatic gene editing. PNAs have been used to correct numerous human disease-relevant mutations with little off-target effect by forming high-affinity hetero-triplex structures within the genome. Furthermore, molecular design, chemical modification, and delivery improvements have allowed applying PNAs systemically in vivo.[209]
TFD-ODN Techniques
One option to obstruct a known activated regulatory pathway that promotes disease is to target transcription factors. Therapeutic drug candidates called double-stranded transcription factor decoy (TFD) oligodeoxynucleotides (ODN) specifically target and neutralize the main transcription factors involved in the pathogenesis of a particular disease. The consensus DNA binding site of a particular transcription factor in the promoter region of its target genes is mimicked by these brief double stranded TFD molecules. This nucleic acid-based drug class can treat diseases brought on by the aberrant expression of such target genes, whose byproducts are involved in disease initiation and progression. Specific drug delivery techniques, such as tissue-specific transduction using adeno-associated viral (AAV) vectors or long-term TFD molecule expression in non-dividing cells using ultrasound-targeted microbubble destruction with TFD ODN-coated microbubbles, are also futuristic propositions.
Argonautes
Almost all eukaryotes, bacteria, and archaea contain members of the Argonautes gene family, which is highly conserved. The Argonaute gene family is found in many animal and plant genomes, but the nematode Caenorhabditis elegans stands out because it has at least 26 Argonaute genes. Four conserved domains are found in argonaute proteins: the N-terminal, PAZ (charged with short RNA binding), Mid, and PIWI (which confers catalytic activity). Argonaute proteins function in RNA-based silencing mechanisms by altering protein synthesis and influencing RNA stability. They associate with small non-coding RNAs, such as microRNAs and small interfering RNAs (siRNAs). Piwi-interacting (pi) RNAs, a novel class of small non-coding RNAs that help maintain chromosome integrity and are involved in the maturation of siRNA and miRNA, can also be produced by argonaute proteins.[210]
Integrase
A retrovirus (like HIV) that forms covalent connections between its genetic material with the host cell it infects produces retroviral integrase (IN). This 288 amino acid, 32 kDa viral enzyme mediates the linkage of double-stranded viral DNA into the host cell genome.[211] Retroviral INs should not be confused with phage integrases (recombinases) used in biotechnology, such as phage integrase discussed in site-specific recombination. The IN macromolecule is an essential part of the intasome. This macromolecular complex is bound to the ends of viral DNA and the retroviral pre-integration complex.[212]
Recombinase
In multicellular organisms, DNA recombinases are frequently used to modify genome structure and control gene expression. These enzymes, known as bacteria phages, are derived from bacteria and fungi, and they catalyze DNA exchange reactions between short (30–40 nucleotide) target site sequences specific to each recombinase. Excision, insertion, inversion, translocation, and cassette exchange are the four fundamental functional modules made possible by these reactions. These modules have been used singly or in various combinations to control the expression of genes. Cre recombinase, Hin recombinase, Tre recombinase, and FLP recombinase are examples of recombinase types.
Delivery Tools
While systemic delivery using targeting ligands is a suitable choice, specific delivery methods are disease-dependent; this includes local administration that can be very effective for treating conditions affecting muscle, skin, eye, and ear tissues. Other delivery methods include electroporation, plasmids, and nanovesicles, such as exosomes with different delivery profiles.[213]
Safe and effective intracellular delivery of GE tools is a significant challenge. Electroporation, membrane deformation, and microinjection are physical techniques for delivering genome-editing tools in vitro and ex vivo. In addition, adenoviruses, integrase-defective lentiviral vectors (IDLVs), and adeno-associated viruses frequently are viral vectors to deliver DNA for GE (AAVs). These are most widely used for in vivo applications. Still, they have a limited carrying capacity that often forces drug developers to use modified genes rather than the same human gene with the best corrective potential.
Regarding delivery, CRISPR-Cas can be divided into plasmids, large-sized biomacromolecules, mRNA/gRNA, and RNP. Additionally, numerous nonviral delivery methods successfully deliver CRISPR-Cas into both ex vivo and in vivo target cells. These delivery systems primarily contain cationic lipids, cationic polymers, cationic polypeptides, DNA nanostructures, AuNPs, cell-derived vesicles, and other organic materials, as was previously mentioned.
Directly introducing sgRNA and Cas9 nuclease can rapidly knock out particular genes using electroporation, lipofectamine therapy, or injection.
Vectors
Many vectors used in gene therapy, which typically emphasizes long-term expression to correct genetic flaws, are rarely appropriate for GE, which only needs temporary delivery of editors. The most popular editors also present additional challenges because of their large sizes (SpyCas9 and TALENs), repetitive sequences, and need to deliver both components of a ribonucleoprotein complex (ZFNs and TALENs), as well as their large sizes (SpyCas9 and TALENs) (RNP; for example, in CRISPR). Accurate tissue targeting is also necessary due to the possibility of on-target or off-target activity in the incorrect tissues.[214]
The AAV vectors demonstrate tissue-specific tropism, immunogenicity, and tumorigenic risks that limit their clinical applications.[215] They produce neutralizing antibodies; it works for vaccines but not for GE products. Although viral vectors are still the main delivery systems of in vivo genome editing, immunogenicity results in less genetic material reaching its destination and prohibit patients from receiving a second (or third) dose. Other vectors include adenoviruses, their associated viruses, and integrase-defective lentiviral vectors (IDLVs).
CRISPR-Cas9 can also be delivered using a system that combines magnetic nanoparticles (MNPs) and recombinant baculoviral vectors (BVs).
The HSV vectors can carry over ten times more genetic material than an AAV and over twice that of a lipid nanoparticle (LNP), making them the preferred route for delivering gene editing drugs. Since HSV vectors are fully neutralized in patients with prior exposure and almost 90% of the population is exposed to HSV in the US and EU, making multiple dosing of HSV is ethical and practical. This allows multiple doses of HSV practical.[216]
Direct
Several difficulties are involved in directly injecting nucleic acids (like plasmid DNA or mRNA encoding Cas9) for in vivo gene editing. Large polynucleotide molecules like DNA and RNA are hydrophobic, negatively charged, and unstable. [217]The cell membranes are attracted to these physiochemical characteristics, preventing them from entering cells independently. They also have a short half-life in circulation because serum nuclease activity makes it impossible for unprotected nucleic acids to reach specific target regions. The kidneys quickly remove these nucleic acids from the body and may stimulate the immune system by interacting with pattern recognition receptors. It calls for sophisticated packaging and delivery systems to overcome these obstacles.
However, these transfection reagents’ cytotoxic and inflammatory effects, including lipofectamine, restrict their use in vivo applications. Many of these obstacles have been removed by creating novel synthetic ionizable cationic lipids and LNP formulations, opening the door to the possibility of LNP-mediated therapeutic gene editing.
LNPs
The arrival of Covid-19 mRNA vaccines brought many breakthroughs, including using lipid nanoparticles to deliver mRNA; it is now highly likely that more GE products will adopt this technology that has matured.[218] However, issues related to the excipients used in lipid nanoparticles, particularly polyethylene glycol, have recently been identified as a new source of anti-PEG antibodies[219] besides the reported skin reactions.[220]These findings would not have been possible had the vaccine not been administered to billions of subjects; it now requires a review of the safety of the LNP formulations developed for GE products. (Schoenmaker, L., et al. 2021)
One of the most well-known and advanced nonviral delivery platforms is LNPs. They have allowed the mRNA-based COVID-19 vaccines created by Pfizer-BioNTech and Moderna and the siRNA drug ONPATTRO (patisiran)[221] to be translated into the clinic.[222]
Newer non-viral delivery techniques like lipid nanoparticles, whose main benefit is their lack of viral components, help minimize safety and immunogenicity concerns and are alternatives to AAV.[223]
The LNPs are a popular therapeutic nucleic acid delivery method. Ionizable cationic lipids, polyethylene glycol (PEG) lipids, zwitterionic phospholipids, and cholesterol are their typical four main lipid constituents. In addition, most LNPs enter cells through the endocytosis pathway. These foundational elements endow LNP systems with special functional elements that cooperate to enable payload encapsulation, transport, and delivery.
LNPs’ capacity to avoid recognition by the innate immune system and longer circulation time make them advantageous as drug carriers.[224] These characteristics are beneficial for delivering hydrophobic medications with brief circulation half-lives, such as proteins and nucleic acids. LNPs containing CRISPR components in nucleic acid or protein forms can effectively induce on-target therapeutic GE in target tissues when given enough circulation time. In addition, ready-to-use LNP formulations can be adopted with little effort. The key features of LNP include:
- Ex vivo GE can be done using delivery methods like Lipofectamine 2000 or unstable PEI in physiological settings. Other in vivo delivery systems, aside from exosomes, which are physiological environments naturally stable, are typically modified with a surface layer of PEG to prevent the adsorption of serum proteins.
- The tailored delivery systems are perfect for effectively enclosing and safeguarding CRISPR-Cas plasmid, RNA, RNP, or other combined forms based on their characteristics. After packaging, plasmid, RNA, and RNP must retain their biological activity and integrity. The delivery systems’ ability to protect CRISPR-Cas from immune cells, nucleases, and proteases are encouraging. For instance, earlier research has shown promise in encasing CRISPR-Cas inside delivery systems and altering a PEG layer on the exterior of delivery systems.
- For each type of CRISPR-Cas to perform as intended inside target intracellular sites, their delivery systems must be capable of moving and releasing them there. For instance, ribonucleoprotein (RNP) -based CRISPR-Cas delivery systems should enable endosomal escape and localize into nuclei for genome editing. The Cas mRNA should be released for translation into the cytoplasm by the RNA-based CRISPR-Cas delivery systems. Endosomes shouldn’t be able to stop plasmid-based CRISPR-Cas delivery systems from translocating into the nucleus for transcription. Cationic polypeptides, such as CPPs and NLS, can facilitate nuclear entry and endosomal escape.
- CRISPR components can be delivered to cells using LNPs in various ways. The three techniques that are most frequently utilized are (1) encasing plasmid DNA (pDNA) encoding both Cas9 protein and gRNA or pDNA encoding Cas9 protein in conjunction with gRNA oligos, (2) Cas9 mRNA and gRNA, and (3) Cas9/sgRNA (protein/RNA) RNP complex. Each technique must apply specific LNP-specific formulation requirements to provide maximal compatibility without compromising function. Each method has advantages and disadvantages.
- The safety of therapeutic GE would be significantly increased by site-specific CRISPR-Cas delivery. Creating delivery systems responsive to CRISPR-Cas stimuli based on plasmids, RNA, and RNPs is also possible. In addition, pCas9 is given tissue- or cell-specific promoters to allow for the site-specific production of gRNA and Cas nuclease.
Nano Particles
One tested method uses nanoparticles endocytosed by the target cell and functions in the nucleus or cytoplasm. Non-viral nanoparticles can deliver genome-editing tools in vitro, ex vivo, and in vivo, occasionally in conjunction with viral vectors. They are frequently made from synthetic, cationic lipid, or polymer delivery materials.
Nanospheres and nanocapsules are two families of nanoparticles that can be distinguished structurally. Nanospheres contain a uniform matrix throughout the particles that store active substances, unlike nanocapsules with a core-shell structure with the payload inside the inner core. Lipid moieties are found in the structures of lipid-based nanoparticles, which have enormous biomedical potential for gene therapy and drug delivery. Lipid-based nanoparticles have several advantages over viral and nonviral nanoparticle systems, including simplicity in formulation, spontaneous self-assembly, high potency, high biocompatibility, a greater payload capacity, and adaptability in design for applications.[227]
For example, after intravenous injection, PEGylated CLAN nanoparticles and LNPs could transfer plasmids or RNA-based CRISPR-Cas into different immune cells or hepatocytes. It is essential to carefully research the in vivo destiny of these systemically administered nanoparticles. Additionally, it is essential to evaluate the safety risks associated with generating GE in healthy cells like hematopoietic stem cells and germ cells. Many non-target cells may endocytose these systemically injected nanoparticles.[228] However, a single dose of nanoparticles can produce effective and long-lasting editing.[229]
Some delivery systems for systemic injection can inject drugs locally to trigger local GE into a tumor, muscle, skin, eye, ear, or brain. For instance, DNA nano clews could deliver the CRISPR-Cas RNP after intertumoral injection to edit tumor genes.[230] After intramuscular injection, AuNPs could also have donor DNA and CRISPR-Cas RNPs to fix the mutated dystrophin gene.[231] Tailoring charge, hydrophilicity, and functional ligands in AuNPs are simple.[232]
Although local GE only has a small number of target cells, this approach has few security concerns. Many vectors used in gene therapy, which typically emphasizes long-term expression to correct genetic flaws, are rarely appropriate for GE, which only needs temporary delivery of editors. The most popular editors also present additional challenges because of their large sizes (SpyCas9 and TALENs), repetitive sequences, and need to deliver both components of a ribonucleoprotein complex (ZFNs and TALENs), as well as their large sizes (SpyCas9 and TALENs) (RNP; for example, in CRISPR). Accurate tissue targeting is also necessary due to the possibility of on-target or off-target activity in the incorrect tissues.[233]
Plasmid
The broad CRISPR-Cas system leaves plasmids with a significant negative charge. As a result, CRISPR-Cas systems must be contained within delivery systems or constrained and compressed into small sizes. Furthermore, delivery techniques that can get beyond in vivo barriers, such as nuclease degradation, immunogenicity, specific cell targeting, and nuclear envelope barriers, might significantly increase GE effectiveness and lessen the possibility of off-target effects. The capacity of GE can also be increased by delivery devices that simultaneously contain ssDNA or dsDNA. More donors of single-strand DNA (ssDNA) or double-strand DNA (dsDNA) are required for gene insertion or repair. Cas mRNA and gRNA are less harmful than plasmids, which carry the risk of genome integration.[234] Cas mRNA has a short half-life and can degrade in as little as 24 hours, which lowers the likelihood of immunogenicity and off-target effects. Using dsDNA templates with sequences derived from pCas and transcription driven by the T7 promoter, Cas mRNA and gRNA are typically produced in vitro. Any CRISPR-Cas on a plasmid can be translated into RNA and delivered. The length of the Cas mRNA varies from 3,000 nucleotides (nt) to over 5,000 nt because of the different types of Cas proteins and their modifiers.
HEK293FT, U2OS, murine ESCs, N2A, and A549 cell lines can be transfected with plasmid-based Cas9/gRNA, RNA mixes of Cas9 and sgRNA, and even RNPs using commercially available transfection reagents designed for plasmid and siRNA delivery. Exosomes can be modified to deliver RNA-based CRISPR-Cas in addition to plasmids.
Ribonucleoproteins (RNP)
RNPs are made up of a big Cas protein and a short gRNA. gRNA can bind to DNA by Watson-Crick base pairing, or the Cas protein can couple to polypeptides, proteins, and PEI. These characteristics allow for the loading of RNP as well. RNP can also be loaded by electrostatic interactions with positively charged objects because of its negative net charge. Metal-organic frameworks (MOFs), polypeptides, PEI, cationic lipids, and others are among these positively charged compounds. RNP can also be delivered through cell vesicles. Since RNPs have a negative net charge, they can be directly transfected without cationic liposomes or LNPs. Positively charged PEI has also been produced for the delivery of RNPs. Cas9 has been conjugated with proteins or polypeptides to make RNP administration easier. In addition to conjugation, it is also possible to complex or encapsulate the RNP with polypeptides. For the delivery of Cas9 RNPs, metal-organic frameworks (MOFs) have been created.[235]
Conclusions
GE will change the map of human destiny when it arrives and if it becomes accessible to billions who need it most. Unlike a biological drug, it has no pharmacology or contact toxicity pattern. The RNA-based CRISPR tool is well-characterized and specific to its target. Unlike biological drugs, there is little variability from batch to batch. It is almost like chemical drugs since the structures of its components are well-defined. With expanding PCR technology, producing a cell-free RNA that goes with Cas9 will be possible. The likely caution is the possibility of off-target genome editing, and the tests created to measure this have proven unreliable. Additionally, as these tools can only be tested in patients, there is a need for regulatory agencies to allow faster testing once GMP compliance is in order. Given the available GMP-grade materials, all off-the-shelf, this should lead to the development of many products without spending billions of dollars.
The future of GE tools will depend on bringing more rationality at the regulatory level and more creativity at the development stage to avoid facing the price structure that is now holding back gene therapies.
Funding
No funding was received for the preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Berg, P.; Baltimore, D.; Brenner, S.; Roblin, R.O.; Singer, M.F. Studies of the chemical nature of the transforming principle of pneumococcus. Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis 1972, 269, 387–403. [Google Scholar] [CrossRef]
- Shaw, C.H.; Hands, P.; Spence, J.A.; Dyer, T.A. Plant cells contain functional chimeric genes after transformation. Nature 1983, 303, 33–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Malzahn, A.A.; Sretenovic, S.; Qi, Y. The emerging and uncultivated potential of CRISPR technology in plant science. Nat. Plants 2020, 6, 778–794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Q.; Yan, S.; Zhang, S.; Liu, H.; Zhang, Y.; Xia, L. Knockout of the allergenic 27 kDa γ-gliadins in bread wheat (Triticum aestivum L. ) by CRISPR/Cas9. Plant Biotechnology Journal 2020, 18, 1424–1426. [Google Scholar]
- Jiang, X.; Ramsay, J.A.; Ramsay, B.A. Acetone extraction of mcl-PHA from Pseudomonas putida KT2440. J. Microbiol. Methods 2016, 8, 122–128. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Gemmell, N.J. Conservation demands safe gene drive. PLOS Biol. 2017, 15, e2003850. [Google Scholar] [CrossRef]
- Liu, Z.; Alper, H.S. Draft genome sequence of the oleaginous yeast Yarrowia lipolytica PO1f, a commonly used metabolic engineering host. Genome Announcements 2014, 2, e00528–14. [Google Scholar] [CrossRef]
- Green, A.A.; Silver, P.A.; Collins, J.J.; Yin, P. Toehold Switches: De-Novo-Designed Regulators of Gene Expression. Cell 2014, 159, 925–939. [Google Scholar] [CrossRef]
- Guerrero, R.F.; Garcia-Garcia, G.; Delgado-Ramos, L.; Mellado, M. Gene editing for the efficient generation of biopolymers in Yarrowia lipolytica. Journal of Fungi 2019, 5, 69. [Google Scholar]
- Fernandez-Rodriguez, J.; Moser, F.; Song, M.; Voigt, C.A. Engineering RGB color vision into Escherichia coli. Nat. Chem. Biol. 2017, 13, 706–708. [Google Scholar] [CrossRef]
- Bhatnagar, N.; Min, M.K.; Choi, E.H.; Kim, N.; Moon, S.J.; Yoon, I.; Kwon, T.; Jung, K.H.; Kim, B.G. Overexpression of OsARD1 Improves Submergence, Drought, and Salt Tolerances of Seedling Through the Enhancement of Ethylene Synthesis in Rice. Frontiers in Plant Science 2020, 11, 983. [Google Scholar]
- Shapiro, B. Mammoth 2. 0: Will genome engineering resurrect extinct species? Genome Biology 2015, 16, 228. [Google Scholar]
- Li, X.; Kerkhof, L.J. Microbial response to acid and base stress. In Microbiome Convergence; Springer: Singapore, 2019; pp. 43–57. [Google Scholar]
- Kyrou, K.; Hammond, A.M.; Galizi, R.; Kranjc, N.; Burt, A.; Beaghton, A.K.; Nolan, T.; Crisanti, A. A CRISPR–Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol. 2018, 36, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Bess, M. Icarus 2.0: A Historian's Perspective on Human Biological Enhancement. Technol. Cult. 2015, 56, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Mans, R.; Van Rossum, H.M.; Wijsman, M.; Backx, A.; Kuijpers, N.G.; Broek, M.V.D.; Daran-Lapujade, P.; Pronk, J.T.; Van Maris, A.J.; Daran, J.-M.G. CRISPR/Cas9: a molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy012. [Google Scholar] [CrossRef]
- Zhu, C.; Bortesi, L.; Baysal, C.; Twyman, R.M.; Fischer, R.; Capell, T.; Schillberg, S.; Christou, P. Characteristics of Genome Editing Mutations in Cereal Crops. Trends Plant Sci. 2020, 25, 96–100. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, X.; Shan, Q.; Zhang, Y.; Liu, J.; Gao, C.; Qiu, J.-L. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 2016, 32, 947–951. [Google Scholar] [CrossRef]
- Mullard, A. Nature. Gene editing pipeline takes off. Available online: https://www.nature.com/articles/d41573-020-00096-y (accessed on 6 March 2023).
- Alagoz, M.; Kherad, N. Advance genome editing technologies in the treatment of human diseases: CRISPR therapy (Review). Int. J. Mol. Med. 2020, 46, 521–534. [Google Scholar] [CrossRef]
- EMA. Expert meeting on GE technologies used in medicine development. 2017. Available online: https://www.ema.europa.eu/en/events/expert-meeting-genome-editing-technologies-used-medicine-development (accessed on 6 March 2023).
- Ho, B.X.; Loh, S.J.H.; Chan, W.K.; Soh, B.S. In Vivo GE as a Therapeutic Approach. Int J Mol Sci. 2018, 19, 2721. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6163904/ (accessed on 6 March 2023). [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; et al. Applications of GE technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Sig Transduct Target Ther 2020, 5, 1. [Google Scholar] [CrossRef]
- de Buhr, H.; Lebbink, R.J. Harnessing CRISPR to combat human viral infections. Curr. Opin. Immunol. 2018, 54, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; A Gersbach, C. Genome-editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Naldini, L. Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 2011, 12, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef]
- Hong, A. CRISPR in personalized medicine: Industry perspectives in gene editing. Semin. Perinatol. 2018, 42, 501–507. [Google Scholar] [CrossRef]
- Selvakumar, S.C.; Preethi, K.A.; Ross, K.; Tusubira, D.; Khan, M.W.A.; Mani, P.; Rao, T.N.; Sekar, D. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol. Cancer 2022, 21, 1–14. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, Y.; Zhang, L.; Jin, Q.; Wang, L.; Xu, S.; Chen, K.; Li, L.; Zeng, T.; Fan, X.; et al. i-CRISPR: a personalized cancer therapy strategy through cutting cancer-specific mutations. Mol. Cancer 2022, 21, 1–8. [Google Scholar] [CrossRef]
- FDA. Available online: https://www.personalizedmedicinecoalition.org/Userfiles/PMC-Corporate/file/Personalized_Medicine_at_FDA_The_Scope_Significance_of_Progress_in_2021.pdf (accessed on 6 March 2023).
- Hamosh, A.; Scott, A.F.; Amberger, J.; Valle, D.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM). Hum Mutat. 2000, 15, 57–61. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Khedekar, P.; Iqbal, Z. Alkaptonuria, ochronosis, and ochronotic arthropathy. Seminars in Arthritis and Rheumatism 2020, 33, 280–291. [Google Scholar]
- Song, C.; Qi, Q.; Zhang, Y.; Han, Y.; Gao, C.; Gong, H. Targeting DNAJC12 by CRISPR/Cas9 in the rat results in animal lethality and alpha-1 antitrypsin deficiency. Journal of genetics and genomics 2018, 45, 641–644. [Google Scholar]
- Kashtan, C.E.; Michael, A.F. Alport syndrome. Kidney International 2014, 66, 748–758. [Google Scholar]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. New England Journal of Medicine 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome Sequencing,ANGPTL3Mutations, and Familial Combined Hypolipidemia. New Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2020, 30, 16–34. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; A Flinter, F. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med Genet. 1999, 36, 437–446. [Google Scholar] [CrossRef]
- Roberts, A.E.; Nixon, C.; Steward, C.G.; Gauvreau, K.; Maisenbacher, M.; Fletcher, M.; Geva, J.; Byrne, B.J.; Spencer, C.T. The Barth Syndrome Registry: Distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med Genet. Part A 2020, 182, 272–278. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; De La Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Matalon, R.; Michals-Matalon, K. Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings. Frontiers in Bioscience 2000, 5, D307–D311. [Google Scholar] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Flynn, R.; Grundmann, A.; Renz, P.; Hänseler, W.; James, W.S.; Cowley, S.A.; Moore, M.D. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp. Hematol. 2017, 53, 31–41. [Google Scholar] [CrossRef]
- Hodges, C.A.; Conlon, R.A. Delivering on the promise of gene editing for cystic fibrosis. Genes Dis. 2019, 6, 97–108. [Google Scholar] [CrossRef]
- Cherqui, S.; Courtoy, P.J.; Specht, A.; Bukanov, N.; Gahl, W.A.; Igarashi, P. Autophagy induction can improve in vitro and in vivo cystinosis manifestations. Autophagy 2020, 16, 1268–1283. [Google Scholar]
- Ye, L.; Wang, J.; Tan, Y.; Beyer, A.I.; Xie, F.; Muench, M.O.; Kan, Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci. USA 2020, 16, 1268–1283. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Albanese, A.; Bhatia, K.; Bressman, S.B.; DeLong, M.R.; Fahn, S.; Fung, V.S.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and classification of dystonia: A consensus update. Mov. Disord. 2019, 28, 863–873. [Google Scholar] [CrossRef]
- Bonafont, J.; Mencía, Á.; García, M.; Torres, R.; Rodríguez, S.; Carretero, M.; Del Río, M. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-mediated gene editing. Molecular Therapy 2019, 27, 986–998. [Google Scholar] [CrossRef]
- Toomes, C.; Bottomley, H.M.; Jackson, R.M.; Towns, K.V.; Scott, S.; Mackey, D.A.; Craig, J.E.; Jiang, L.; Yang, Z.; Trembath, R.; et al. Mutations in LRP5 or FZD4 Underlie the Common Familial Exudative Vitreoretinopathy Locus on Chromosome 11q. Am. J. Hum. Genet. 2004, 74, 721–730. [Google Scholar] [CrossRef]
- Rio, P.; Baños, R.; Lombardo, A.; Quintana-Bustamante, O.; Alvarez, L.; Garate, Z.; Genovese, P.; Almarza, E.; Valeri, A.; Díez, B.; et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol. Med. 2019, 6, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Marti-Gutierrez, N.; Park, S.-W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N. Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Reports 2015, 13, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Amaral, O.; Alves, S. Therapy of Gaucher disease: past, present and future perspectives. Journal of Inherited Metabolic Disease 2017, 40, 853–867. [Google Scholar]
- Weber, L.; Frati, G.; Felix, T.; Hardouin, G.; Casini, A.; Wollenschlaeger, C.; Dubreil, L. Editing a α1-antitrypsin gene in the liver of newborn mice using zinc finger nucleases. American Journal of Respiratory Cell and Molecular Biology 2020, 62, 114–120. [Google Scholar]
- Endo, M.; Fujii, K.; Sugita, K.; Saito, K.; Kohno, Y.; Miyashita, T. Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am. J. Med Genet. Part A 2012, 158, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Pavani, G.; Laurent, M.; Fabiano, A.; Cantelli, E.; Sakkal, A.; Corre, G.; Monteilhet, V. Treatment of Hemophilia B: focus on gene therapy. Hematology and Medical Oncology 2020, 4, 1–9. [Google Scholar]
- Ramaswamy, S.; et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proceedings of the National Academy of Sciences 2017, 114, E1941–E1950. [Google Scholar] [CrossRef]
- Hosman, A.E.; de Gussem, E.M.; Balemans, W.A.F.; Gauthier, A.; Westermann, C.J.J.; Snijder, R.J.; Post, M.C.; Mager, J.J. Screening children for pulmonary arteriovenous malformations: Evaluation of 18 years of experience. Pediatr. Pulmonol. 2019, 54, 1572–1579. [Google Scholar] [CrossRef]
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; Rosa, A.L.; Lerer, I.; Hamri, A.; Alegria, P.; et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2019, 131, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; A Sharp, P.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, srep02510. [Google Scholar] [CrossRef] [PubMed]
- Bess, M. Icarus 2.0: A Historian's Perspective on Human Biological Enhancement. Technol. Cult. 2015, 56, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. Journal of Clinical Investigation 2017, 127, 2719–2724. [Google Scholar] [CrossRef]
- Wojciak, J.; Płoski, R.; Pollak, A. Application of targeted gene panel sequencing and whole exome sequencing in neonates and infants with suspected genetic disorder. Journal of applied genetics 2020, 61, 341–349. [Google Scholar]
- Whyte, M.P. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nature Reviews Endocrinology 2017, 13, 233. [Google Scholar] [CrossRef]
- Romani, M.; Micalizzi, A.; Valente, E.M. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013, 12, 894–905. [Google Scholar] [CrossRef]
- Nieuwenhuis, M.H.; Vasen, H.F.A. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol. 2007, 61, 153–161. [Google Scholar] [CrossRef]
- den Hollander, A.I.; et al. Leber congenital amaurosis: genes, proteins and disease mechanisms. Progress in Retinal and Eye Research 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt–FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Zelley, K.; Nichols, K.E.; Garber, J. Li-Fraumeni Syndrome. In GeneReviews(R); University of Washington, 2017; pp. 1993–2021. [Google Scholar]
- Giudicessi, J.R.; Ackerman, M.J. Genotype- and Phenotype-Guided Management of Congenital Long QT Syndrome. Curr. Probl. Cardiol. 2013, 38, 417–455. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.; Lynch, P.; Lanspa, S.; Snyder, C.; Lynch, J.; Boland, C. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Brunger, J.M.; Murray, A.; O’Donnell, M.; Usprech, J.; Ding, Q.; Shoichet, M.S. Structural and behavioral interplay of myosin-II and actin in the Drosophila heart. Cell Reports 2017, 19, 507–520. [Google Scholar]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Valstar, M.J.; Ruijter, G.J.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo syndrome: a mini-review. Journal of Inherited Metabolic Disease 2008, 31, 240–252. [Google Scholar] [CrossRef]
- Gilman, S.; Wenning, G.; Low, P.A.; Brooks, D.; Mathias, C.J.; Trojanowski, J.Q.; Wood, N.; Colosimo, C.; Durr, A.; Fowler, C.J.; et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Pinto, B.; Chandran, P.; Bhat, M.; Iarkov, A. Advances in CRISPR/Cas-mediated Gene Therapy in Alzheimer’s Disease. Current Gene Therapy 2020, 20, 253–264. [Google Scholar]
- Evans, D.G.R.; E Baser, M.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med Genet. 2005, 38, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Wang, Y.C.; Chuang, H.Y.; Chen, Y.C.; Lee, Y.S.; Chou, Y.H. Serum levels of bioactive lipids and Niemann-Pick disease severity. Journal of Lipid Research 2021, 62, 100037. [Google Scholar]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2021, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.A.; Hendel, A.; Pruett-Miller, S.M.; Porteus, M.H. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2020, 42, 1365–1378. [Google Scholar] [CrossRef]
- Del Fattore, A.; Cappariello, A.; Teti, A. Genetics, pathogenesis and complications of osteopetrosis. Bone 2008, 42, 19–29. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker's yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. 2020, 114, 201–205. [Google Scholar] [CrossRef]
- Neumann, H.P.; Young, W.F.; Eng, C. Pheochromocytoma and paraganglioma. New England Journal of Medicine 2011, 365, 410–421. [Google Scholar] [CrossRef]
- McGarrity, T.J.; Kulin, H.E.; Zaino, R.J. Peutz-Jeghers syndrome. The American Journal of Gastroenterology 2000, 95, 596–604. [Google Scholar] [CrossRef]
- Rossetti, S.; Consugar, M.B.; Chapman, A.B.; Torres, V.E.; Guay-Woodford, L.M.; Grantham, J.J.; Bennett, W.M.; Meyers, C.M.; Walker, D.L.; Bae, K.; et al. Comprehensive Molecular Diagnostics in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2009, 18, 2143–2160. [Google Scholar] [CrossRef]
- Hordeaux, J.; Dubreil, L.; Deniaud, J.; Iacobelli, F.; Moreau, S.; Ledevin, M.; Cherel, Y. Efficient gene transfer into the CNS and peripheral tissues of newborn primates using AAV9 and a new synthetic AAV9 promoter. Molecular Therapy 2018, 26, 119–134. [Google Scholar]
- Kim, Y.; Cheong, S.-A.; Lee, J.G.; Lee, S.-W.; Lee, M.S.; Baek, I.-J.; Sung, Y.H. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 2019, 34, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid Regeneration from Gene-Corrected SCID-X1 Subject-Derived iPSCs. Cell Stem Cell 2020, 16, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Fernández, O.; Osorio, F.G.; Quesada, V.; Rodríguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R.; et al. Development of a CRISPR/Cas9-based therapy for Hutchinson–Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-H.; Tsai, Y.-T.; Justus, S.; Lee, T.-T.; Zhang, L.; Lin, C.-S.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 2020, 28, 830–840. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. The Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Ananiev, G.; Williams, E.C.; Li, H.; Chang, Q. Isogenic Pairs of Wild Type and Mutant Induced Pluripotent Stem Cell (iPSC) Lines from Rett Syndrome Patients as In Vitro Disease Model. PLOS ONE 2011, 6, e25255. [Google Scholar] [CrossRef]
- Kan, S.-H.; Aoyagi-Scharber, M.; Le, S.Q.; Vincelette, J.; Ohmi, K.; Bullens, S.; Wendt, D.J.; Christianson, T.M.; Tiger, P.M.N.; Brown, J.R.; et al. Delivery of an enzyme-IGFII fusion protein to the mouse brain is therapeutic for mucopolysaccharidosis type IIIB. Proc. Natl. Acad. Sci. 2014, 111, 14870–14875. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. New Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Piguet, F.; Alves, S.; Cartier, N. Clinical Gene Therapy for Neurodegenerative Diseases: Past, Present, and Future. Hum. Gene Ther. 2020, 31, 1264–1282. [Google Scholar] [CrossRef]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. New England Journal of Medicine 2006, 355, 1345–1356. [Google Scholar] [CrossRef]
- A Farrer, L.; Arnos, K.S.; Asher, J.H.; Baldwin, C.T.; Diehl, S.R.; Friedman, T.B.; Greenberg, J.; Grundfast, K.M.; Hoth, C.; Lalwani, A.K. Locus heterogeneity for Waardenburg syndrome is predictive of clinical subtypes. Am. J. Hum. Genet. 1994, 55, 728. [Google Scholar] [PubMed]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: clinical features, pathogenesis and potential therapeutic interventions. Ageing Research Reviews 2016, 33, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2020, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.G.; Bundey, S.E.; Macleod, A.F. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Long, J.D.; Campo-Fernandez, B.; de Oliveira, S.; Cooper, A.R.; Romero, Z.; Kohn, D.B. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell reports 2018, 23, 2606–2616. [Google Scholar] [CrossRef]
- Niu, D.; Wei, H.-J.; Lin, L.; George, H.; Wang, T.; Lee, I.-H.; Zhao, H.-Y.; Wang, Y.; Kan, Y.N.; Shrock, E.; et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science 2017, 357, 1303–1307. [Google Scholar] [CrossRef]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- Blasimme, A. AMA Journal of Ethics. Why Include the Public in Gene Editing Governance Deliberation? 2019. Available online: https://journalofethics.ama-assn.org/article/why-include-public-genome-editing-governance-deliberation/2019-12 (accessed on 6 March 2023).
- Scheper, A. AMA Policies and Code of Medical Ethics’ Opinions Related to Human Genome Editing. AMA J. Ethic- 2019, 21, E1056–1058. [Google Scholar] [CrossRef]
- Matthew, P.; et al. Gene editing and CRISPR in the clinic: current and future perspectives. Biosci Rep 2020, 40, BSR20200127. [Google Scholar]
- European Commission. European Group on Ethics in Science and New Technologies. Available online: https://research-and-innovation.ec.europa.eu/strategy/support-policy-making/scientific-support-eu-policies/european-group-ethics_en (accessed on 6 March 2023).
- Council of Europe. Ethics and Human Rights must guide any use of GE technologies in human beings. Available online: https://www.coe.int/en/web/bioethics/-/ethics-and-human-rights-must-guide-any-use-of-genome-editing-technologies- in-human-beings (accessed on 6 March 2023).
- Adashi, E.Y.; Cohen, I.G. Selective Regrets: The “Dickey Amendments” 20 Years Later. JAMA Forum Archive 2015. [Google Scholar] [CrossRef]
- NExTRAC NIH. Available online: https://osp.od.nih.gov/policies/novel-and-exceptional-technology-and-research-advisory-committee-nextrac/ (accessed on 6 March 2023).
- NIH Guideline. Final Action Under the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines). Available online: https://www.federalregister.gov/documents/2019/04/26/2019-08462/final-action-under-the-nih-guidelines-for-research-involving-recombinant-or-synthetic-nucleic-acid (accessed on 6 March 2023).
- Wang, D.C.; Wang, X. Off-target genome editing: A new discipline of gene science and a new class of medicine. Cell Biol. Toxicol. 2019, 35, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Fine, E.J.; Bao, G.; Porteus, M.H. Quantifying on- and off-target genome editing. Trends Biotechnol. 2015, 33, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation. Clinical Trials. Available online: https://www.cff.org/research-clinical-trials/types-cftr-mutations (accessed on 6 March 2023).
- FDA Approved Cell and Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 6 March 2023).
- EMA ATMP. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview#advanced-therapies-in-the-product-lifecycle-section (accessed on 6 March 2023).
- Alliance for Regenerative Medicine. Available online: https://www.mednous.com/gene-edited-therapies-produce-first-clinical-data (accessed on 6 March 2023).
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2020, 136, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Drugs. Cost of Gene Therapy Drugs. 2022. Available online: https://www.drugs.com/medical-answers/cost-kymriah-3331548/ (accessed on 6 March 2023).
- Nadat, M. Researchers welcome $3.5-million haemophilia gene therapy — but questions remain. Available online: https://www.nature.com/articles/d41586-022-04327-7 (accessed on 6 March 2023).
- Irvine, A. Paying for CRISPR Cures: The Economics of Genetic Therapies. Available online: https://innovativegenomics.org/news/paying-for-crispr-cures/ (accessed on 6 March 2023).
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Brokowski, C.; Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 2019, 431, 88–101. [Google Scholar] [CrossRef]
- Seoane-Vazquez, E.; Rodriguez-Monguio, R.; Szeinbach, S.L.; Visaria, J. Incentives for orphan drug research and development in the United States. Orphanet J. Rare Dis. 2007, 2, 1–7. [Google Scholar] [CrossRef]
- Khatib, O. Noncommunicable diseases: risk factors and regional strategies for prevention and care. East. Mediterr. Heal. J. 2004, 10, 778–788. [Google Scholar] [CrossRef]
- Cohen, E.R.; Ochalek, J.; Wouters, O.J.; Birger, M.; Chakraborty, S.; Morton, A. Making fair choices on the path to universal health coverage: Applying principles to difficult cases. Health Systems & Reform 2018, 4, 16–23. [Google Scholar]
- Mostaghim, S.R.; Gagne, J.J.; Kesselheim, A.S. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study. BMJ 2017, 358, j3837. [Google Scholar] [CrossRef]
- Glassman, A.; Giedion, U.; Smith, P.C. What’s in, what’s out: designing benefits for universal health coverage; The World Bank, 2016. [Google Scholar]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef]
- Global Gene Editing Market Report (2021). Data Bridge Market Research.
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Juergens, R.A.; et al. The promise of molecularly targeted and immunotherapy against lung cancer. Seminars in radiation oncology 2018, 28, 19–27. [Google Scholar]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. (2017). Human Genome Editing: Science, Ethics, and Governance. National Academies Press.
- Cyranoski, D.; Ledford, H. Genome-edited baby claim provokes international outcry. Nature 2018, 563, 607–608. [Google Scholar] [CrossRef]
- Liang, P.; Xu, Y.; Zhang, X.; Ding, C.; Huang, R.; Zhang, Z.; Lv, J.; Xie, X.; Chen, Y.; Li, Y.; et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2019, 10, 333–344. [Google Scholar] [CrossRef]
- Advisory committee on developing global standards for governance and oversight of Human Genome editing. (2019, December 18). World Health Organization.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; De La Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing ofBCL11Ato Treat Sickle Cell Disease. New Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef] [PubMed]
- FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma. (2017, October 18). U.S. Food and Drug Administration.
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; De La Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; et al. Design and specificity of long ssDNA donors for CRISPR-based knock-in. BioRxiv 2019, 530824. [Google Scholar]
- Wianny, F.; Zernicka-Goetz, M. Specific interference with gene function by double-stranded RNA in early mouse development. Nature 2000, 2, 70–75. [Google Scholar] [CrossRef]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479–479. [Google Scholar] [CrossRef]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. 2014, 111, 11461–11466. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Amini, L.; Wendering, D.J.; Burkhardt, L.-M.; Akyüz, L.; Reinke, P.; Volk, H.-D.; Schmueck-Henneresse, M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019, 25, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef]
- Wang, H.-X.; Li, M.; Lee, C.M.; Chakraborty, S.; Kim, H.-W.; Bao, G.; Leong, K.W. CRISPR/Cas9-Based Genome Editing for Disease Modeling and Therapy: Challenges and Opportunities for Nonviral Delivery. Chem. Rev. 2017, 117, 9874–9906. [Google Scholar] [CrossRef]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.-E.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef]
- Koo, T.; Kim, J.S. Therapeutic opportunities and challenges of CRISPR-Cas9 gene editing: a review. Yonsei Medical Journal 2020, 61, 357–368. [Google Scholar]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 169, 559. [Google Scholar] [CrossRef]
- Yin, H.; Kauffman, K.J.; Anderson, D.G. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 2017, 16, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Brokowski, C.; Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 2019, 431, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef]
- Baylis, F.; McLeod, M. First-in-human phase 1 CRISPR gene editing cancer trials: Are we ready? Current Gene Therapy 2017, 17, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. 2014, 111, 11461–11466. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Y.; Zhang, R.; Zhang, H.; Gao, C. CRISPR/Cas Genome Editing and Precision Plant Breeding in Agriculture. Annu. Rev. Plant Biol. 2019, 70, 667–697. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. (2017). Human genome editing: Science, ethics, and governance. National Academies Press.
- Brokowski, C.; Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 2019, 431, 88–101. [Google Scholar] [CrossRef]
- Nuffield Council on Bioethics. (2016). Genome editing: An ethical review. Nuffield Council on Bioethics.
- Parens, E.; Johnston, J. Does it make sense to speak of “genetic enhancements?”. The Hastings Center Report 2007, 37 (Suppl. S1), S41–S49. [Google Scholar]
- Borry, P.; Bentzen, H.B.; Budin-Ljøsne, I.; Cornel, M.C.; Howard, H.C.; Feeney, O.; Jackson, L.; Mascalzoni, D.; Mendes, Á.; Peterlin, B.; et al. The challenges of the expanded availability of genomic information: an agenda-setting paper. J. Community Genet. 2020, 11, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Ishii, T. International regulatory landscape and integration of corrective genome editing into in vitro fertilization. Reprod. Biol. Endocrinol. 2014, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. (2020). Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). FDA.
- European Medicines Agency. (2018). Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. EMA.
- Zhang, X.; Zhang, Y.; Qiu, Z.Q.; Tian, X.; Wang, H.; Zhang, X.; Yang, H.; Chen, Y. Ethical norms and the international governance of genetic databases and biobanks: findings from an international study. The Kennedy Institute of Ethics Journal 2016, 18, 101–124. [Google Scholar]
- Gómez-Tatay, L.; Hernández-Andreu, J.M.; Aznar, J. Human germline editing: the need for ethical guidelines. The Linacre Quarterly 2017, 84, 275–282. [Google Scholar]
- Araki, M.; Ishii, T. International regulatory landscape and integration of corrective genome editing into in vitro fertilization. Reprod. Biol. Endocrinol. 2014, 12, 108. [Google Scholar] [CrossRef]
- European Commission. Directive 2001/18/ EC on the deliberate release into the environment of GMOs; Directive 2009/41/EC on the contained use of genetically modified micro-organisms. Accessed 6 March 2023.
- FDA. Long Term Follow-Up After Administration of Human Gene Therapy Products; Guidance for Industry, January 2020. Available online: https://www.fda.gov/media/113768/download (accessed on 6 March 2023).
- European Council. Advanced Therapies. Available online: https://ec.europa.eu/health/human-use/advanced-therapies_en#1 (accessed on 6 March 2023).
- Mourby, M.; Morrison, M. Gene therapy regulation: could in-body editing fall through the net? Eur. J. Hum. Genet. 2020, 28, 979–981. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- NIH. First Restriction Enzyme Described. 1968. Available online: https://www.genome.gov/25520301/online-education-kit-1968-first-restriction-enzymes-described (accessed on 6 March 2023).
- https://www.labome.com/method/CRISPR-and-Genomic-Engineering.html.
- https://doi.org/10.1371/journal.pbio.1001877.g001. [CrossRef]
- https://doi.org/10.1371/journal.pbio.1001877.g002. [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021, 39, 35–40. [Google Scholar] [CrossRef]
- Clinical Trias; Base Editors. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=base+editor+&cntry=&state=&city=&dist=&Search=Search (accessed on 6 March 2023).
- CRISPR News Medicine. MegaNuclease Clinical Trials. Available online: https://crisprmedicinenews.com/clinical-trials/meganuclease-clinical-trials/ (accessed on 6 March 2023).
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of Gene Knockout Mice. Curr. Protoc. Cell Biol. 2009, 44, 19–12. [Google Scholar] [CrossRef]
- Bijlani, S.; Pang, K.M.; Sivanandam, V.; Singh, A.; Chatterjee, S. The Role of Recombinant AAV in Precise Genome Editing. Front. Genome Ed. 2022, 3, 799722. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, K.; et al. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun 2016, 7, 10431. [Google Scholar] [CrossRef] [PubMed]
- Montague, M.G.; Lartigue, C.; Vashee, S. Synthetic genomics: potential and limitations. Curr. Opin. Biotechnol. 2012, 23, 659–665. [Google Scholar] [CrossRef]
- Zhan, X.; Deng, L.; Chen, G. Mechanisms and applications of peptide nucleic acids selectively binding to double-stranded RNA. Biopolymers 2022, 113, e23476. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Freudenreich, O.; Worth, J.L. Patients with Human Immunodeficiency Virus Infection and Acquired Immunodeficiency Syndrome. 2010, 353–370. [Google Scholar] [CrossRef]
- Masuda, T. Non-Enzymatic Functions of Retroviral Integrase: The Next Target for Novel Anti-HIV Drug DLi, yevelopment. Frontiers in Microbiology 2011, 2, 210. [Google Scholar] [CrossRef]
- Liu, B.Y.; He, X.Y.; Zhuo, R.X.; Cheng, S.X. Reversal of tumor malignization and modulation of cell behaviors through GE mediated by a multi-functional nanovector. Nanoscale 2018, 10, 21209–21218. [Google Scholar] [CrossRef]
- Saha, K.; et al. The NIH Somatic Cell GE program. Nature 2021, 592, 195–204. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef]
- Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., et al., Eds.; Cambridge University Press: Cambridge, 2007. [Google Scholar]
- Yang, W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Making COVID-19 mRNA vaccines accessible: challenges resolved. Expert Rev. Vaccines 2022, 21, 1163–1176. [Google Scholar] [CrossRef]
- Liu, M.-S.; Gong, S.; Yu, H.-H.; Jung, K.; Johnson, K.A.; Taylor, D.W. Engineered CRISPR/Cas9 enzymes improve discrimination by slowing DNA cleavage to allow release of off-target DNA. Nat. Commun. 2020, 11, 3576. [Google Scholar] [CrossRef] [PubMed]
- Pesqué, D.; Pujol, R.M.; Marcantonio, O.; Vidal-Navarro, A.; Ramada, J.M.; Arderiu-Formentí, A.; Albalat-Torres, A.; Serra, C.; Giménez-Arnau, A.M. Study of Excipients in Delayed Skin Reactions to mRNA Vaccines: Positive Delayed Intradermal Reactions to Polyethylene Glycol Provide New Insights for COVID-19 Arm. Vaccines 2022, 10, 2048. [Google Scholar] [CrossRef]
- Akinc, A. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Puri, A. Lipid-Based Nanoparticles as Pharmaceutical Drug Carriers: From Concepts to Clinic. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef]
- Kazemian, P.; Yu, S.-Y.; Thomson, S.B.; Birkenshaw, A.; Leavitt, B.R.; Ross, C.J.D. Lipid-Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Components. Mol. Pharm. 2022, 19, 1669–1686. [Google Scholar] [CrossRef]
- Mirjalili Mohanna, S.Z.; Djaksigulova, D.; Hill, A.M.; Wagner, P.K.; Simpson, E.M.; Leavitt, B.R. LNP-mediated delivery of CRISPR RNP for wide-spread in vivo GE in mouse cornea. J Control Release. 2022, 350, 401–413. [Google Scholar] [CrossRef]
- Qi, F.; Wu, J.; Li, H.; Ma, G. Recent research and development of PLGA/PLA microspheres/nanoparticles: A review in scientific and industrial aspects. Front. Chem. Sci. Eng. 2018, 13, 14–27. [Google Scholar] [CrossRef]
- Huang, K.; Zapata, D.; Tang, Y.; Teng, Y.; Li, Y. In vivo delivery of CRISPR-Cas9 GE components for therapeutic applications. Biomaterials. 2022, 291, 121876. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Jouannet, P. CRISPR-Cas9, germinal cells and human embryo. Biol. Aujourd’hui 2017, 211, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ji, W.; Hall, J.M.; Hu, Q.; Wang, C.; Beisel, C.L.; Gu, Z. Self-Assembled DNA Nanoclews for the Efficient Delivery of CRISPR-Cas9 for Genome Editing. Angew. Chem. Int. Ed. 2015, 54, 12029–12033. [Google Scholar] [CrossRef]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef]
- Duan, L.; Ouyang, K.; Xu, X.; Xu, L.; Wen, C.; Zhou, X.; Qin, Z.; Xu, Z.; Sun, W.; Liang, Y. Nanoparticle Delivery of CRISPR/Cas9 for Genome Editing. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Saha, K.; et al. The NIH Somatic Cell GE program. Nature 2021, 592, 195–204. [Google Scholar] [CrossRef]
- Begley, S. Clever CRISPR Advances. Wang H, Yet. al., (May 2013). “One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering”. Cell 2016, 153, 910–918. [Google Scholar]
- Zheng, Q.; Li, W.; Mao, L.; Wang, M. Nanoscale metal–organic frameworks for the intracellular delivery of CRISPR/Cas9 genome editing machinery. Biomater. Sci. 2021, 9, 7024–7033. [Google Scholar] [CrossRef]
Figure 1.
Genetic disorders. (2023, February 26). In Wikipedia. https://en.wikipedia.org/wiki/Genetic_disorder.
Figure 1.
Genetic disorders. (2023, February 26). In Wikipedia. https://en.wikipedia.org/wiki/Genetic_disorder.
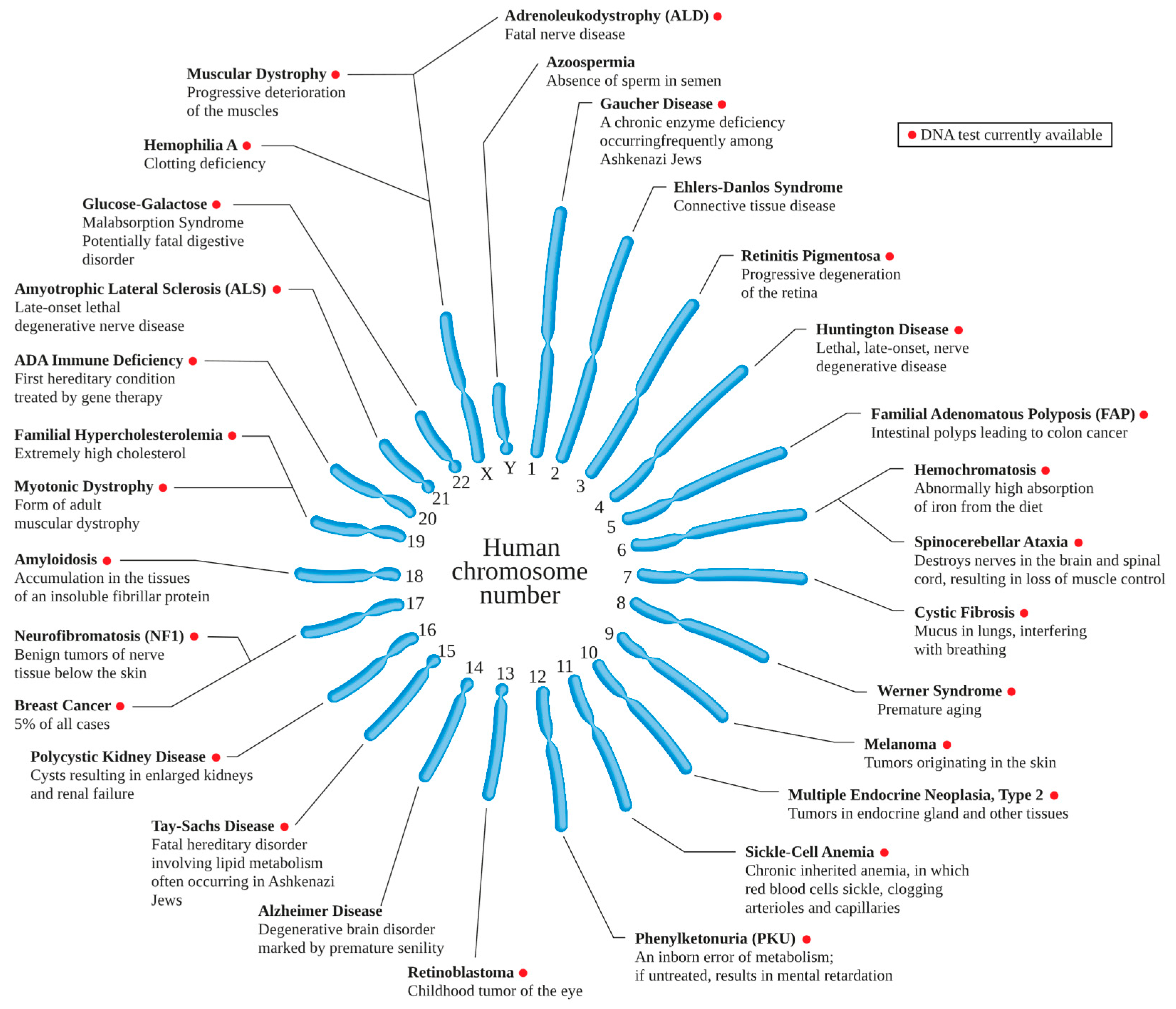
Figure 2.
Schematics of the four classes of sequence-specific nucleases. (A) The meganuclease, I-SceI, is shown to be bound to its DNA target. The catalytic domain, which determines DNA sequence specificity, is shown in red. (B) A ZFN dimer is illustrated bound to DNA. ZFN targets are bound by two zinc-finger DNA binding domains (dark blue) separated by a 5–7-bp spacer sequence. FokI cleavage occurs within the spacer. Each zinc finger typically recognizes 3 bp. (C) Depicted is a TALEN dimer bound to DNA. The DNA binding domains are in dark blue. A 15–20-bp spacer sequence typically separates the two TALEN target sites. Like ZFNs, the TAL effector repeat arrays are fused to FokI. Each TAL effector motif recognizes one base. (D) The CRISPR/Cas9 system recognizes DNA through base pairing between DNA sequences at the target site and a CRISPR-based guide RNA (gRNA). Cas9 has two nuclease domains (shown by red arrowheads) that each cleave one strand of double-stranded DNA.[199] Reproduced under Creative Commons Attribution License”.
Figure 2.
Schematics of the four classes of sequence-specific nucleases. (A) The meganuclease, I-SceI, is shown to be bound to its DNA target. The catalytic domain, which determines DNA sequence specificity, is shown in red. (B) A ZFN dimer is illustrated bound to DNA. ZFN targets are bound by two zinc-finger DNA binding domains (dark blue) separated by a 5–7-bp spacer sequence. FokI cleavage occurs within the spacer. Each zinc finger typically recognizes 3 bp. (C) Depicted is a TALEN dimer bound to DNA. The DNA binding domains are in dark blue. A 15–20-bp spacer sequence typically separates the two TALEN target sites. Like ZFNs, the TAL effector repeat arrays are fused to FokI. Each TAL effector motif recognizes one base. (D) The CRISPR/Cas9 system recognizes DNA through base pairing between DNA sequences at the target site and a CRISPR-based guide RNA (gRNA). Cas9 has two nuclease domains (shown by red arrowheads) that each cleave one strand of double-stranded DNA.[199] Reproduced under Creative Commons Attribution License”.
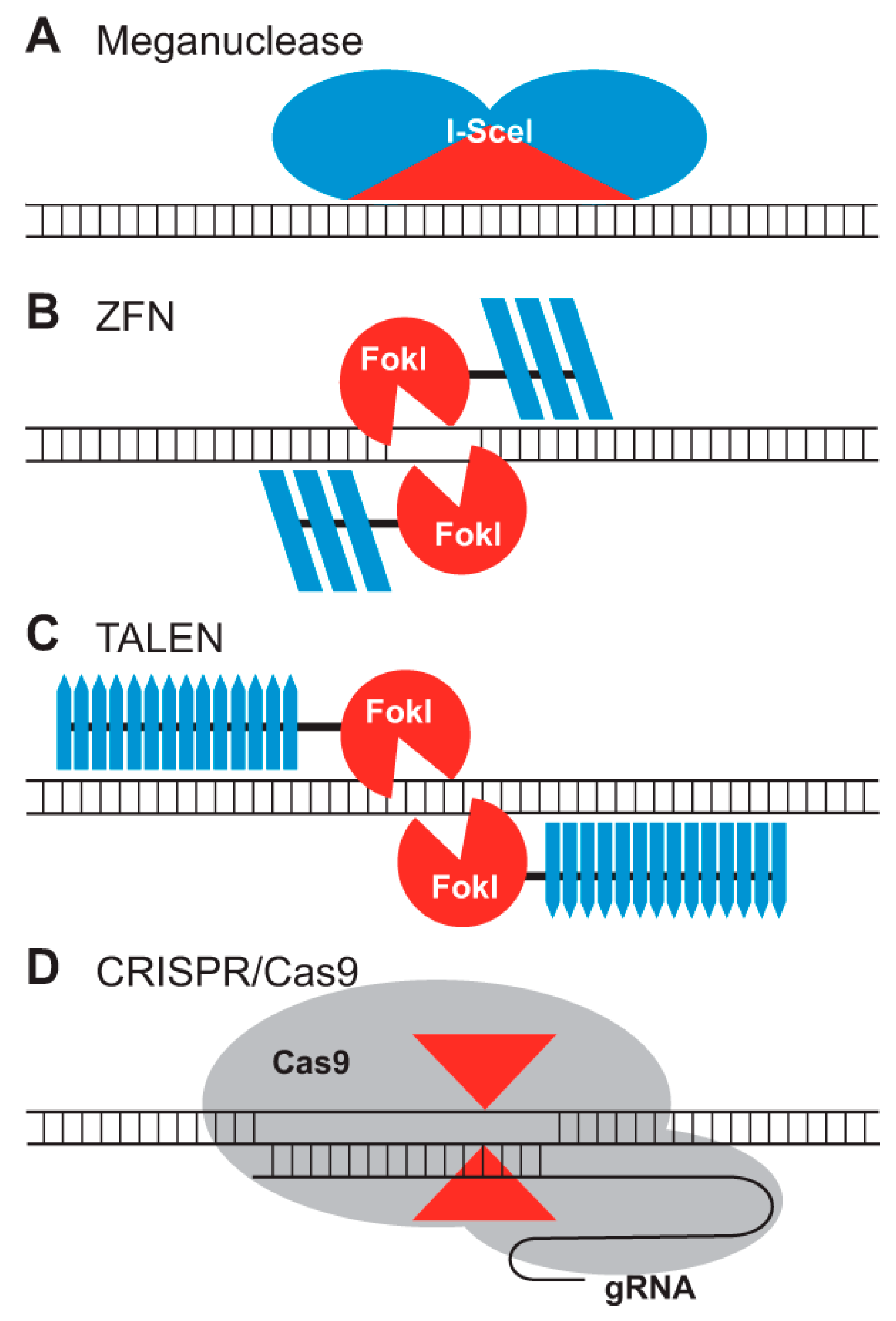
Figure 3.
Targeted genome engineering by nonhomologous end-joining or homologous recombination using sequence-specific nucleases. (A) The NHEJ-mediated repair can result in small deletions or insertions at the target sites that can disrupt gene function (knockouts, left). DNA fragments can be inserted via NHEJ-mediated ligation to create targeted insertions (knock-ins, right). (B) When SSNs make two cuts, NHEJ-mediated repair can result in deletions or inversions of large genomic regions (left), targeted gene deletions, or chromosomal translocations (right). (C) HR-mediated repair, involving a homologous DNA template, leads to gene replacement or insertion (). [200]Reproduced under Creative Commons Attribution License”.
Figure 3.
Targeted genome engineering by nonhomologous end-joining or homologous recombination using sequence-specific nucleases. (A) The NHEJ-mediated repair can result in small deletions or insertions at the target sites that can disrupt gene function (knockouts, left). DNA fragments can be inserted via NHEJ-mediated ligation to create targeted insertions (knock-ins, right). (B) When SSNs make two cuts, NHEJ-mediated repair can result in deletions or inversions of large genomic regions (left), targeted gene deletions, or chromosomal translocations (right). (C) HR-mediated repair, involving a homologous DNA template, leads to gene replacement or insertion (). [200]Reproduced under Creative Commons Attribution License”.
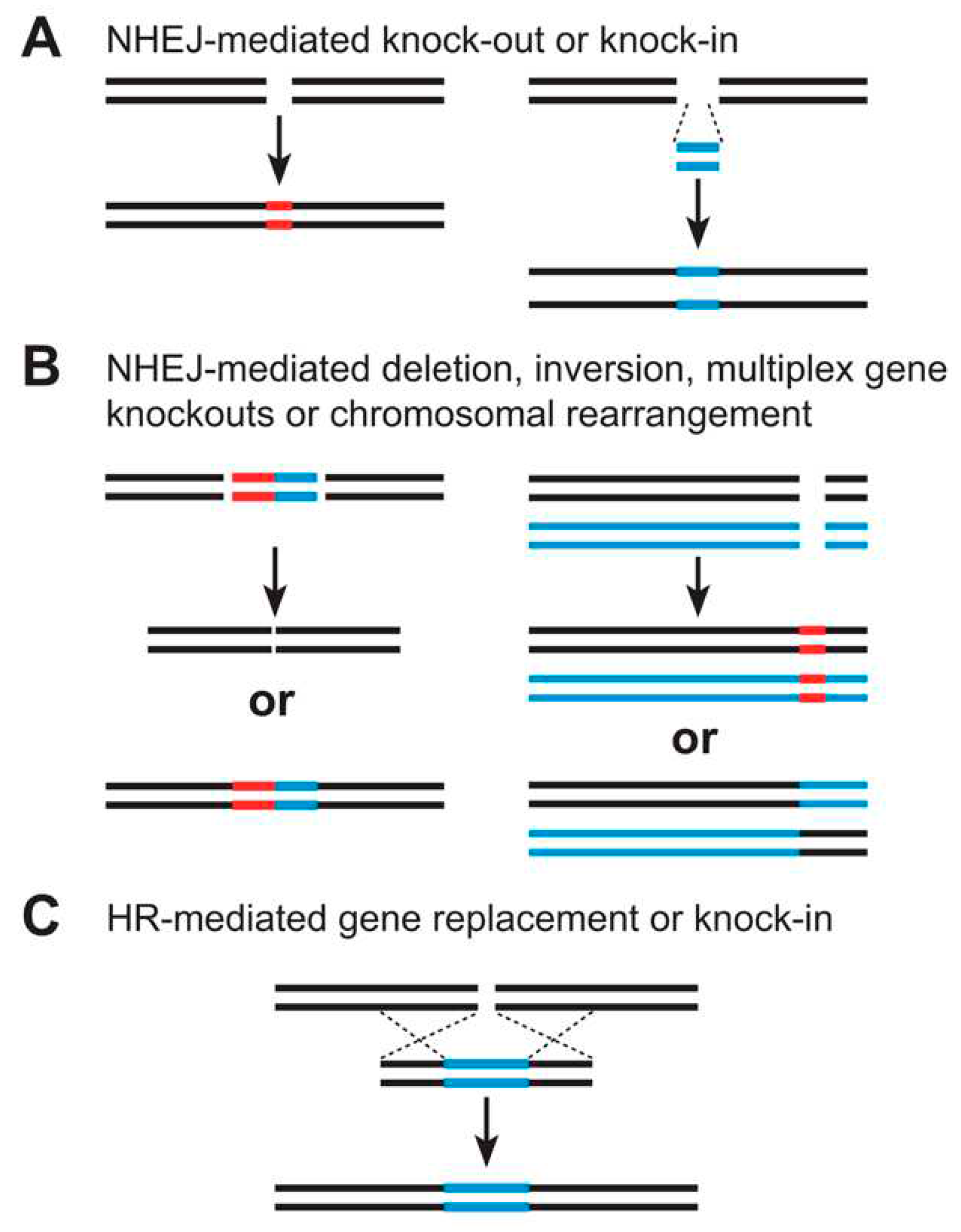
Table 1.
Applications of gene editing.
| Agriculture and Livestock Breeding: Crop improvement and livestock breeding, with the potential to significantly enhance global food security.[3] |
| Allergy-Free Foods: To create versions of common foods that don’t trigger allergic reactions. For example, researchers have used gene editing to create a variety of wheat that does not produce the proteins that cause most wheat allergies.[4] |
| Biodegradable Plastics: Editing the genes of certain bacteria to make them produce biodegradable plastics, offering an environmentally friendly alternative to conventional plastics.[5] |
| Biodiversity Conservation: To bolster conservation efforts by creating white-footed mice immune to the bacteria causing Lyme disease.[6] |
| Biofuel Production: To engineer microbes or plants to produce biofuels more efficiently.[7] |
| Biological Computers: To build biological computers inside living cells to perform complex computations. [8] |
| Biomaterials: Create organisms that produce new kinds of biomaterials with unique properties, opening various industrial and scientific applications.[9] |
| Biosensors Development: Engineer cells to detect specific molecules or conditions, creating biosensors for various applications, from medical diagnostics to environmental monitoring.[10] |
| Climate Change Effects: To genetically engineer crops to withstand better the effects of climate change, such as increased drought or higher salinity.[11] |
| De-extinction of Extinct Species: To bring extinct species back to life, or “de-extinction.” This would involve using DNA from preserved specimens to edit the genes of a closely related existing species.[12] |
| Environmental Decontamination: Genetically edited bacteria could clean up ecological contaminants like oil spills or nuclear waste.[13] |
| Gene Drives: Gene drives using CRISPR/Cas9 systems have been proposed to control disease vectors such as mosquitoes that spread malaria.[14] |
| Human Augmentation: While controversial, gene editing technologies like CRISPR have raised the possibility of enhancing human abilities beyond normal levels, or “human enhancement.”[15] |
| Industrial Yeast Strains: Industrial yeasts produce biofuels and various chemicals. Gene editing can improve the efficiency and versatility of these yeasts.[16] |
| Nutritional Profile of Crops: Gene editing can enhance the nutritional content of food crops, potentially addressing malnutrition problems in areas of the world where specific nutrient deficiencies are common.[17] |
| Plant Disease Resistance: Gene editing can increase the resistance of crops to various diseases, potentially leading to higher yields and food security.[18] |
Table 3.
Potential applications of gene editing to treat human diseases in alphabetical order.
| Alkaptonuria: A metabolic disorder that leads to a buildup of homogentisic acid, causing various symptoms, including dark-colored urine. Gene editing could correct the HGD gene, which causes this condition.[35] |
| Alpha-1 Antitrypsin Deficiency: This genetic disorder can lead to lung and liver disease. Gene editing technologies can potentially correct the faulty SERPINA1 gene that causes it.[36] |
| Alport Syndrome: A genetic disorder characterized by kidney disease, hearing loss, and eye abnormalities. Gene editing could correct the COL4A3, COL4A4, or COL4A5 genes, which cause this condition.[37] |
| Amyotrophic Lateral Sclerosis (ALS): A group of neurological diseases mainly involving the nerve cells (neurons) responsible for controlling voluntary muscle movement. Gene editing might correct the SOD1, TARDBP, FUS, or C9orf72 genes, which can cause this condition.[38] |
| Anti-Aging Research: Genetic modification of senescent cells has been proposed to counteract aging. [39] |
| Antimicrobial Resistance: Gene editing can potentially counteract the growing problem of antibiotic resistance by directly targeting and killing antibiotic-resistant bacteria or by making the bacteria sensitive to antibiotics again.[40] |
| Atherosclerosis: Gene editing can potentially treat atherosclerosis, a disease where plaque builds up inside the arteries. Using gene editing techniques to modify the PCSK9 gene, which controls LDL cholesterol levels, could potentially lower the risk of atherosclerosis.[41] |
| Autoimmune Diseases: With autoimmune diseases like rheumatoid arthritis and lupus, gene editing could modify immune cells and prevent them from attacking the body’s tissues.[42] |
| Bardet-Biedl Syndrome: A disorder that affects many parts of the body and can cause obesity, loss of vision, kidney abnormalities, and extra fingers or toes. Gene editing could correct any 21 genes that cause this condition, such as BBS1, BBS2, or BBS10.[43] |
| Barth Syndrome: A genetic disorder characterized by muscle weakness, delayed growth, and sometimes intellectual disability. Gene editing could correct the TAZ gene, which causes this condition.[44] |
| Beta Thalassemia: Gene editing techniques have been used in attempts to treat beta-thalassemia, a blood disorder that reduces the production of hemoglobin. Researchers have manipulated the BCL11A gene to enhance the production of fetal hemoglobin as a workaround.[45] |
| Brain Function: Gene editing has been used in neuroscience to understand the function of different genes in the brain, which could eventually help us treat or even cure neurological disorders.[46] |
| Canavan Disease: A progressive, fatal neurological disorder that begins in infancy. Gene editing could correct the ASPA gene, which causes this condition.[47] |
| Cancer Therapies: CRISPR has been used to develop novel cancer therapies, such as genetically modifying a patient’s immune cells to target and fight cancer cells. [48] |
| Chronic Granulomatous Disease: Gene editing has been used to correct the genetic mutations that cause chronic granulomatous disease, a disorder that affects the immune system.[49] |
| Cystic Fibrosis: Gene editing technologies could correct the CFTR gene mutation that leads to cystic fibrosis, a disease affecting the lungs and digestive system.[50] |
| Cystinosis: A genetic disorder characterized by an accumulation of the amino acid cystine within cells, leading to various symptoms and complications. Gene editing technologies are being explored to correct the CTNS gene, which causes this condition.[51] |
| Diabetes: In Type 1 diabetes, the immune system destroys insulin-producing cells. Researchers are exploring gene editing as a potential approach to protect these cells from the immune system or to create new insulin-producing cells.[52] |
| Duchenne Muscular Dystrophy: A severe type of muscular dystrophy. Gene editing has shown promise in correcting the gene mutation that causes this condition.[53] |
| Dystonia: A movement disorder in which a person’s muscles contract uncontrollably. Gene editing could correct any of the 20+ known genes that can cause this condition, such as TOR1A, THAP1, or GNAL.[54] |
| Epidermolysis Bullosa: Gene editing technologies can potentially correct the genetic mutations that cause epidermolysis bullosa, a group of genetic conditions that cause the skin to be very fragile and to blister easily.[55] |
| Familial Exudative Vitreoretinopathy: A hereditary disorder that can cause progressive vision loss. Gene editing could correct the FZD4, LRP5, TSPAN12, NDP, or ZNF408 genes, which cause this condition.[56] |
| Fanconi Anemia: A rare genetic disease resulting in bone marrow failure. Gene editing could correct the FANCC gene, one of the known genes that causes this condition when mutated.[57] |
| Fertility Treatments: Gene editing technology may be utilized to treat certain genetic disorders that cause infertility, offering hope to many individuals and couples who wish to have children.[58] |
| Fragile X Syndrome: A genetic disorder causing intellectual disability, behavioral and learning challenges, and various physical characteristics. Gene editing has been proposed as a potential way to correct the FMR1 gene that causes this condition.[59] |
| Gaucher Disease: A genetic disorder that affects the body’s ability to break down fats. Gene editing technologies are being explored to correct the GBA gene mutations that cause this condition.[60] |
| Glycogen Storage Disease Type Ia: A metabolic disorder caused by the deficiency of glucose-6-phosphatase, the enzyme necessary for the final step of gluconeogenesis and glycogenolysis. Gene editing technologies are being explored to correct the G6PC gene mutations that cause this condition.[61] |
| Gorlin Syndrome (Nevoid Basal Cell Carcinoma Syndrome): A genetic condition that affects many parts of the body and increases the risk of developing various cancerous and noncancerous tumors. Gene editing could correct the PTCH1 gene, which causes this condition.[62] |
| Hemophilia A and B: A rare bleeding disorder where a person lacks or has low levels of specific proteins called “clotting factors.” Gene editing might correct the F8 or F9 genes, which cause Hemophilia A and B, respectively.[63] |
| Hereditary Angioedema: A rare genetic disorder characterized by recurrent episodes of severe swelling. Gene editing might correct the SERPING1 gene, which causes this condition.[64] |
| Hereditary Hemorrhagic Telangiectasia: A genetic disorder of blood vessel formation causing multiple direct connections between arteries and veins. Gene editing could correct the ENG, ACVRL1, or SMAD4 gene, which can cause this condition.[65] |
| Hereditary Spastic Paraplegia: A group of inherited disorders characterized by progressive weakness and stiffness of the legs. Gene editing could correct the SPG11 gene, which causes one of the more common types of this disease.[66] |
| Hereditary Tyrosinemia Type 1: A rare genetic disorder characterized by multistep disruptions that break down the amino acid tyrosine. Gene editing could correct the FAH gene mutations causing this condition.[67] |
| HIV/AIDS: Gene editing technology has been used to eradicate HIV from infected cells. This is achieved by targeting the viral DNA integrated into the host genome.[68] |
| Human Augmentation: While controversial, gene editing technologies like CRISPR have raised the possibility of enhancing human abilities beyond normal levels, or “human enhancement.”[69] |
| Huntington’s Disease: This inherited disease causes the progressive breakdown of nerve cells in the brain. Gene editing could potentially correct or deactivate the gene that causes Huntington’s disease.[70] |
| Hypertrophic Cardiomyopathy: A disease in which the heart muscle becomes abnormally thick, making it harder for the heart to pump blood. Gene editing might correct the MYH7 gene, which causes this condition.[71] |
| Hypophosphatasia: A metabolic disease that disrupts mineralization, processes in which minerals such as calcium and phosphorus are deposited in developing bones and teeth. Gene editing could correct the ALPL gene, which causes this condition.[72] |
| Joubert Syndrome: A genetic disorder characterized by decreased muscle tone, difficulties with coordination, abnormal eye movements, and breathing problems. Gene editing could correct any of the 35 genes that cause this condition, such as AHI1, CEP290, or TMEM67.[73] |
| Juvenile Polyposis Syndrome: A genetic condition characterized by multiple polyps in the gastrointestinal tract. Gene editing could correct the BMPR1A or SMAD4 genes, which cause this condition.[74] |
| Leber Congenital Amaurosis: An eye disorder that primarily affects the retina. Gene editing could correct any of the 14 known genes that can cause this condition, such as GUCY2D, RPE65, or CEP290.[75] |
| Leukemia: Certain forms of leukemia are caused by specific genetic mutations. Gene editing could potentially correct these mutations, leading to improved treatment outcomes.[76] |
| Li-Fraumeni Syndrome: A rare, hereditary disorder that greatly increases the risk of developing several types of cancer, particularly in young adults and children. Gene editing could correct the TP53 gene, which causes this condition.[77] |
| Long QT Syndrome: A disorder of the heart’s electrical activity that can cause sudden, uncontrollable, and irregular heartbeats (arrhythmias), which may lead to premature death. Gene editing could correct any of the 17 known genes that can cause this condition, such as KCNQ1, KCNH2, or SCN5A.[78] |
| Lynch Syndrome: A genetic condition that increases the risk of many types of cancer, particularly colorectal cancers. Gene editing could correct the MSH2, MLH1, MSH6, or PMS2 genes, which cause this condition.[79] |
| Marfan Syndrome: A genetic disorder affecting the body’s connective tissue. Gene editing has been proposed to correct the faulty gene that causes this syndrome.[80] |
| Mitochondrial Diseases: Mitochondrial diseases often result from mutations in the mitochondrial DNA. Scientists have used gene editing techniques to eliminate mutated mitochondrial DNA and prevent these diseases selectively.[81] |
| Mucopolysaccharidosis: A group of metabolic disorders caused by the absence or malfunctioning of lysosomal enzymes needed to break down molecules called glycosaminoglycans. Gene editing could correct any of the 11 known genes that cause these conditions.[82] |
| Multiple System Atrophy: A rare neurodegenerative disorder characterized by autonomic dysfunction, parkinsonism, and ataxia. Gene editing could investigate and possibly correct the underlying genetic contributors to this condition, which are not yet fully understood.[83] |
| Muscular Dystrophy: Scientists have used gene editing to correct the mutation that causes Duchenne muscular dystrophy in animal models, and clinical trials are in the works.[84] |
| Neurodegenerative Disorders: Gene editing can be employed to study and potentially treat neurodegenerative disorders like Parkinson’s, Alzheimer’s, and Huntington’s disease by targeting the specific genes involved in these conditions.[85] |
| Neurofibromatosis: Genetic disorders that cause tumors to form on nerve tissue. Gene editing could correct the NF1 or NF2 genes that cause these conditions.[86] |
| Niemann-Pick Disease: A group of severe inherited metabolic disorders in which sphingomyelin accumulates in cell lysosomes. Gene editing could correct the SMPD1 gene, which causes type A and B of this disease.[87] |
| Oculocutaneous Albinism: A group of conditions that affect the coloring (pigmentation) of the skin, hair, and eyes. Gene editing could correct the OCA2 gene, causing some forms of this condition.[88] |
| Osteogenesis Imperfecta: This group of genetic disorders mainly affects the bones, resulting in bones that break easily. Gene editing could correct or compensate for the faulty genes causing these conditions.[89] |
| Osteopetrosis: A group of rare, genetic bone disorders that result in the bone being overly dense. Gene editing could correct the TCIRG1, CLCN7, or SNX10 genes, which cause this condition.[90] |
| Pantothenate Kinase-Associated Neurodegeneration (PKAN): A type of neurodegeneration with brain iron accumulation. Gene editing could correct the PANK2 gene, which causes this condition.[91] |
| Paraganglioma and Pheochromocytoma: Rare neuroendocrine tumors originating in the adrenal glands or near specific nerves and blood vessels. Gene editing could correct the SDHA, SDHB, SDHC, SDHD, SDHAF2, VHL, RET, NF1, TMEM127, or MAX genes, which can cause these conditions.[92] |
| Peutz-Jeghers Syndrome: A genetic condition characterized by the development of noncancerous growths called hamartomatous polyps in the gastrointestinal tract and a significantly increased risk of developing certain types of cancer. Gene editing could correct the STK11 gene, which causes this condition.[93] |
| Polycystic Kidney Disease: A genetic disorder characterized by the growth of numerous cysts in the kidneys. Gene editing might correct the PKD1 or PKD2 genes, which cause this condition.[94] |
| Pompe Disease: A metabolic disorder caused by the buildup of a complex sugar called glycogen within cells. Gene editing could correct the GAA gene, which causes this condition.[95] |
| Prader-Willi Syndrome: This is a complex genetic condition that affects many parts of the body, causing weak muscle tone, feeding difficulties, poor growth, and delayed development. Using gene editing to reactivate the silenced paternal copy of the genes could provide a cure.[96] |
| Primary Immunodeficiencies: Gene editing may provide treatments for primary immunodeficiencies, like severe combined immunodeficiency (SCID), where gene alterations affect the immune system’s development and function.[97] |
| Progeria: Gene editing technology has shown promise in treating Progeria (also known as Hutchinson-Gilford Progeria Syndrome), a rare, fatal genetic disorder characterized by an appearance of accelerated aging in children. Gene editing can potentially correct the mutation in the LMNA gene associated with this disease.[98] |
| Restoring Sight: Researchers have used gene editing to restore sight in blind mice, which could eventually lead to treatments for certain forms of inherited blindness in humans.[99] |
| Retinal Diseases: Gene editing holds promise in treating inherited retinal diseases. Scientists have successfully used gene editing techniques to correct a mutation causing Leber congenital amaurosis, inherited blindness—breakdown, and loss of cells in the retina. Gene editing might correct any of the 60+ known genes that can cause this condition, such as RHO, RPGR, or USH2A.[100] |
| Rett Syndrome: A rare genetic disorder causing severe cognitive and physical impairments. Gene editing could potentially reactivate the silenced MECP2 gene that causes Rett syndrome.[101] |
| Sanfilippo Syndrome: A type of Mucopolysaccharidosis, Sanfilippo syndrome is characterized by the body’s inability to break down certain sugars properly. Gene editing technologies are being developed to correct the SGSH gene, which causes this condition.[102] |
| Sickle Cell Disease: Gene editing has shown promise in correcting the genetic mutation responsible for sickle cell disease, which causes misshapen red blood cells.[103] |
| Spinocerebellar Ataxias: These are a group of genetic diseases characterized by degenerative changes in the part of the brain related to the control of movement. Gene editing techniques have been applied in experimental models to correct the associated genetic mutations.[104] |
| Tuberous Sclerosis Complex: A genetic disorder characterized by the growth of numerous noncancerous (benign) tumors in many body parts. Gene editing could correct the TSC1 or TSC2 genes that cause this condition.[105] |
| Waardenburg Syndrome: A group of genetic conditions that can cause hearing loss and changes in coloring (pigmentation) of the hair, skin, and eyes. Gene editing could correct the PAX3 or MITF genes, which cause this condition.[106] |
| Werner Syndrome: A disorder characterized by the premature appearance of features associated with normal aging. Gene editing could correct the WRN gene, which causes this condition.[107] |
| Wilson Disease: A condition in which copper builds up in the body, potentially leading to life-threatening organ damage. Gene editing might correct the ATP7B gene mutations causing Wilson’s disease.[108] |
| Wolfram Syndrome: A genetic disorder characterized by diabetes mellitus and progressive vision loss. Gene editing could correct the WFS1 or CISD2 genes, which cause this condition.[109] |
| X-Linked Agammaglobulinemia: Gene editing can potentially correct mutations in the BTK gene, which cause X-linked agammaglobulinemia, an immune system disorder that leaves the body prone to infections.[110] |
| Xenotransplantation: Researchers have used gene editing to remove retroviruses from pig genomes, bringing us one step closer to pig-to-human organ transplants.[111] |
| Zellweger Spectrum Disorder: A group of conditions that can affect many body parts. Gene editing could correct any 12 PEX genes known to cause these conditions.[112] |
Table 2.
FDA-approve Gene Therapy products.
| Product | Developer | Indication |
| ABECMA (idecabtagene vicleucel) | Celgene Corporation | Adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. |
| ADSTILADRIN | Ferring Pharmaceuticals A/S | Adult patients with high-risk Bacillus Calmette-Guérin (BCG)-unresponsive non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors |
| BREYANZI | Juno Therapeutics, Inc. | Adult patients with large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B, who have: Refractory disease to first-line chemoimmunotherapy or relapse within 12 months of first line chemoimmunotherapy; or Refractory disease to first-line chemoimmunotherapy or relapse after first line chemoimmunotherapy and are not eligible for hematopoietic stem cell transplantation (HSCT) due to comorbidities or age; or Relapsed or refractory disease after two or more lines of systemic therapy. |
| CARVYKTI (ciltacabtagene autoleucel) | Janssen Biotech, Inc. | Adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody. |
| ELEVIDYS delandistrogene moxeparvovec | Sarepta Therapeutics, Inc. | Ambulatory pediatric patients aged 4 through 5 years with Duchenne muscular dystrophy (DMD) with a confirmed mutation in the DMD gene. |
| GINTUIT (Allogeneic Cultured Keratinocytes and Fibroblasts in Bovine Collagen) | Organogenesis Incorporated | It is an allogeneic cellularized scaffold product indicated for topical (non-submerged) application to a surgically created vascular wound bed in mucogingival conditions in adults. |
| HEMGENIX | CSL Behring LLC | HEMGENIX is an adeno-associated virus vector-based gene therapy indicated for adults with Hemophilia B (congenital Factor IX deficiency) who: Currently use Factor IX prophylaxis therapy or have current or historical life-threatening hemorrhage or Have repeated, spontaneous severe bleeding episodes. |
| IMLYGIC (talimogene laherparepvec) | BioVex, Inc. | For the local unresectable cutaneous, subcutaneous, and nodal lesions in patients with recurrent melanoma after initial surgery. |
| KYMRIAH (tisagenlecleucel) | Novartis Pharmaceuticals Corporation | KYMRIAH is a CD19-directed genetically modified autologous T-cell immunotherapy indicated for adult patients with relapsed or refractory follicular lymphoma after two or more lines of therapy |
| LANTIDRA (donislecel) | CellTrans Inc. | Adults with Type 1 diabetes who cannot approach target hba1c because of repeated episodes of severe hypoglycemia despite intensive diabetes management and education. |
| LAVIV (Azficel-T) | Fibrocell Technologies | Improvement of the appearance of moderate to severe nasolabial fold wrinkles in adults. |
| LUXTURNA | Spark Therapeutics, Inc. | Patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. |
| MACI (Autologous Cultured Chondrocytes on a Porcine Collagen Membrane) | Vericel Corp. | Repair single or multiple symptomatic, full-thickness cartilage defects of the knee with or without bone involvement in adults. MACI is an autologous cellularized scaffold product. |
| OMISIRGE (omidubicel-onlv) | Gamida Cell Ltd. | Adults and pediatric patients 12 years and older with hematologic malignancies are planned for umbilical cord blood transplantation following myeloablative conditioning to reduce the time to neutrophil recovery and the incidence of infection. |
| PROVENGE (sipuleucel-T) | Dendreon Corp. | Asymptomatic or minimally symptomatic metastatic castrate-resistant (hormone refractory) prostate cancer. |
| RETHYMIC | Enzyvant Therapeutics GmbH | For immune reconstitution in pediatric patients with congenital athymia. |
| ROCTAVIAN (valoctocogene roxaparvovec-rvox) | BioMarin Pharmaceutical Inc | Adults with severe hemophilia A (congenital factor VIII deficiency with factor VIII activity <1 IU/dl) without pre-existing antibodies to adeno-associated virus serotype 5 detected by an FDA-approved test. |
| SKYSONA (elivaldogene autotemcel) | bluebird bio, Inc. | To slow the progression of neurologic dysfunction in boys 4-17 years of age with early, active cerebral adrenoleukodystrophy (CALD). |
| STRATAGRAFT | Stratatech Corporation | Adults with thermal burns containing intact dermal elements for which surgical intervention is clinically indicated (deep partial-thickness burns). |
| TECARTUS (brexucabtagene autoleucel) | Kite Pharma, Inc. | Adult patients with relapsed or refractory mantle cell lymphoma (MCL). New Indication for this supplement: Adult patients with relapsed or refractory (r/r) B-cell; precursor acute lymphoblastic leukemia (ALL) |
| VYJUVEK | Krystal Biotech, Inc. | Wounds in patients 6 months of age and older with dystrophic epidermolysis bullosa with mutation(s) in the collagen type VII alpha 1 chain (COL7A1) gene |
| YESCARTA (axicabtagene ciloleucel) | Kite Pharma, Incorporated | Adult patients with large B-cell lymphoma refractory to first-line chemoimmunotherapy or that relapse within 12 months of first-line chemoimmunotherapy. Axicabtagene ciloleucel is not indicated in patients with primary central nervous system lymphoma. |
| ZOLGENSMA (onasemnogene abeparvovec-xioi) | Novartis Gene Therapies, Inc. | Adult and pediatric patients with ß-thalassemia who require regular red blood cell (RBC) transfusions |
| ZYNTEGLO (betibeglogene autotemcel) | bluebird bio, Inc. | Spinal muscular atrophy (type I) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



