Submitted:
01 August 2023
Posted:
07 August 2023
You are already at the latest version
Abstract
Uncontrolled chronic inflammation results in cardiovascular disease and early death. In this review, we studied the impact of rheumatoid arthritis on the cardiovascular system, including the early and accelerated development of atherosclerosis, and its clinical manifestations, focusing on the inflammatory mechanisms leading to arterial wall damage, rapid atherosclerotic plaque formation, and thrombosis. Furthermore, the influence of medications used to treat rheumatoid arthritis on the cardiovascular system. The influence of chronic inflammation and medicaments on traditional cardiovascular risk factors is not the main subject of this review. We found that uncontrolled chronic inflammation and some medications directly impact all stages of atherosclerosis. In conclusion, reducing inflammation and maintaining long-term remission in rheumatoid arthritis may pre-vent early atherosclerosis. We believe that this review will encourage a better interdisciplinary approach for these patients and further research in this field.
Keywords:
rheumatoid arthritis
; chronic inflammation mechanisms
; cardiovascular risk
; cardiovas-cular mortality
; medications used in rheumatoid arthritis
1. Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory systemic disease that primarily affects the synovial membrane, cartilage, and bones of small- and medium-sized joints, leading to chronic damage and pannus formation; however, blood vessels and various internal organs are often affected [1,2,3,4,5]. The prevalence of RA in the general population is approximately 1% and affects women predominantly, resulting in significant disability, socioeconomic consequences, and a short lifespan of 5 to 18 years, mainly attributable to increased cardiovascular (CV) morbidity and mortality [1,6,7,8,9,10,11,12,13,14,15,16,17]. The causes of RA remain unknown, but female sex and a family history of RA are known risk factors. Known triggers for RA include exposure to bacterial or viral infections, especially bacteria that cause periodontal diseases or Epstein–Barr virus, trauma, bone or joint fractures, cigarette smoking, and obesity. Typically, the symptoms of RA develop slowly over several weeks or months and can range from mild to severe, including articular symptoms such as pain and swelling; morning stiffness or stiffness after prolonged rest lasting >30 minutes; symmetrical involvement and loss of function; general symptoms such as fatigue, fever, and weight loss and extra-articular symptoms such as rheumatoid nodules, cardiopulmonary diseases, eye diseases, Sjogren’s syndrome, rheumatoid vasculitis, neurological manifestations, and Felty’s syndrome [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16].
Atherosclerosis and its complications are the most common cardiovascular manifestations of RA and are the leading causes of death in patients with RA. Moreover, two major mechanisms of chronic inflammation influence CV risk: direct damage to the cardiovascular system, especially the arteries, and indirect influence on traditional CV risk factors. These factors synergistically increase the CV risk, morbidity, and premature mortality. The traditional risk factors include arterial hypertension, cigarette smoking, dyslipidemia, low levels of physical activity, diabetes/insulin resistance [17]. Compared with healthy individuals, traditional CV risk factors in patients with rheumatoid arthritis adversely affect cardiovascular disease (CVD) development: arterial hypertension increases by 53–73% (in most but not all studies), cigarette smoking – increases by 25–50%, dyslipidemia – increases by 73% (difficulty in assessment do to “lipid paradox”), low physical activity – neutral to increased, diabetes/insulin resistance – increased for about two times and obesity – increases by 16% (in the most but not all studies) [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. However, the influence of chronic inflammation in RA and medications used in treating RA on traditional CV risk factors was not the subject of this article. This review focuses on gathering all relevant data and knowledge, analyzing and discussing the role of chronic inflammation and its mechanisms and the impact on the CV system, as well as the influence of medications used in treating RA, adverse or beneficial. We hope that this review will contribute to a better understanding and encourage further research to prevent early atherosclerosis development and its consequences in patients with RA.
2. Materials and Methods
2.1. Objective
To gather, analyze, and systematize all available relevant data regarding the direct effects of chronic inflammation associated with RA and the influence of medications on the cardiovascular system.
2.2. Data sources
Systematic search and review of available relevant literature in the medical databases Web of Science, Scopus, PubMed, and Cochrane Library.
2.3. Keywords used in articles selection process
Rheumatoid arthritis, chronic inflammation mechanisms, cardiovascular risk, cardiovascular mortality, coagulation mechanisms, medications in rheumatoid arthritis, non- steroid anti-inflammatory drugs (NSAIDs), glucocorticoids/corticosteroids, methotrexate, hydroxychloroquine, leflunomide, cyclosporine, azathioprine, biologic medications, infliximab, etanercept, adalimumab, tocilizumab, abatacept, rituximab, certolizumab, tofacitinib, golimumab, sarilumab, anakinra, canakinumab, baricitinib, statins.
2.4. Article selection, inclusion, and exclusion criteria
Data from meta-analyses, large randomized controlled trials, prospective clinical trials, relevant reviews, and data from the European Society of Cardiology and European Alliance of Associations for Rheumatology guidelines were given the highest relevance and prioritized. Smaller studies were included only if no other data were available and we considered them crucial. Only one case report, one expert opinion, and one meta-analysis (critics) were included on a specific topic. All known study limitations are listed in Table S1 (Supplementary Materials). The selected and analyzed articles had to consist of important data regarding the following topics: mechanisms of chronic inflammation in rheumatoid arthritis and its influence on the cardiovascular system, cardiovascular disease development in RA, and the influence of medications used to treat RA on the cardiovascular system. We selected abstracts of published studies according to the inclusion criteria; if suitable, we analyzed the full text. Case reports, pilot projects, studies, meta-analyses, reviews with questionable methodology or reliability, underpowered studies, studies with no full-text available, and studies not directly related to the investigation topics were generally excluded. All important data were extrapolated and copied into pre-prepared tables and analyzed by at least one cardiologist and rheumatologist. Special attention was paid to the statistical data and statistical methods, and reviewed by at least one statistician. The final analysis was performed by all the authors.
2.5. Limitations
We accepted all articles written in English or German to limit the language knowledge. Studies involving populations under 18 years of age were excluded from the review. Some studies did not declare that the number of participants in a meta-analysis or review but were accepted do to important topic, or that only a few articles were available on certain topics. Some limitations of the included studies have been identified.
2.6. Study design
Systematic review.
2.7. Review period
Studies from 1986 to 2022.
3. Results
3.1. Article selection process
More than 500.000 published articles were initially screened in databases using keywords that we pre-selected 375 abstracts by narrowing search parameters and careful combination of key words. From 375 abstracts, including 61 meta-analyses, we extrapolated 144 articles suitable for this review by listing inclusion and exclusion criteria (Figure 1). We divided articles into four groups, with the first two used for introduction: general information about RA and CVD and traditional risk factors, whereas the last two were analyzed for influence on the cardiovascular system of chronic inflammation mechanisms in RA and medications used in RA.
3.2. Statistics
All selected articles were analyzed by two statisticians to confirm the power and reliability of the studies, to find possible limitations, and to assess the statistical study value. The studies were then sorted by addressed topics, analyzed, and compared by the authors. Study characteristics and comments are presented in Table S1 (Supplementary Materials).
4. Discussion
4.1. Chronic inflammation
RA has a complex etiology that involves a combination of genetic susceptibility and environmental triggers. The most prominent genetic risk is the presence of the human leukocyte antigen (HLA) - DRB1*04:01 gene, which encodes a 5-amino acid sequence known as a shared epitope that induces the binding of post-translationally modified (citrullinated) proteins. Another genetic risk factor is PTPN22, which increases citrullination [38]. The major environmental risk factors include tobacco smoking, female sex, advanced age, and certain foods [38,39,40]. Autoimmune processes include the recognition of synovial tissue self-antigens such as type II collagen, proteoglycans, and cartilage protein gp39 [41,42,43]. The activation and combination of these two major mechanisms are crucial for joint destruction and bone erosion in RA. First, the joint intimal lining expands, causing synoviocyte activation and proliferation, which then begins to secrete pro-inflammtory cytokines such as Tumor necrosis factor (TNF), Interleukin-1 (IL-1), Interleukin-6 IL-6), metalloproteinases, prostaglandins, and leukotrienes. Synovial invasion into the adjacent articular structures damages the cartilage and bone, manifesting as joint swelling. Second, synovial layer proliferation contributes to the activation of neutrophils and T- and B-lymphocytes, which infiltrate the joints and secrete cytokines and proteinases that further damage the extracellular matrix. Effector CD4+ T cells play a crucial role in disease progression and are characterized by an imbalance between Th1/Th17 and regulatory T cells [44,45]. As the pathogenic processes of atherosclerosis and synovial inflammation in RA share a common pathway, the understanding of these processes represents a cornerstone of CV risk management in patients with RA. Sustained synovial secretion of inflammatory mediators leads to chronic low-grade activation and dysfunction of the vascular endothelium, promoting accelerated development of atherosclerosis in RA [46].
Citrullinated synovial proteins induce the production of autoantibodies (anti-CCP) specific mostly for RA and are associated with more severe forms of RA [47,48], as well as are the subject of many CV risk studies. According to López-Longo et al., anti-CCP antibodies titer levels of > 25 units/mL carried a higher risk of ischemic heart disease (6.5 vs. 2.6%, odds ratio [OR]: 2.58, 95% CI: 1.17–5.65) without affecting mortality [49]. Previous studies clarified the association of citrullinated proteins and anti-CCP antibodies with early subclinical atherosclerosis and atherosclerotic plaque promotion by targeting citrullinated fibrinogen in plaques with anti-CCP antibodies to induce inflammation and heart failure independent of coronary artery disease by targeting citrullinated sarcomeric proteins, namely, fibrinogen and vimentin [50,51,52]; however, these studies had important limitations, such as the inability to demonstrate a direct anti-CCP complex in plaque [50] and many false positive fluorodeoxyglucose (FDG) uptake results [51]. Several studies addressing anti-CCP positivity reported that it was associated with higher total mortality and increased fatal CV outcomes, but not with heart failure or recurrent ischemia [52,53,54,55]; limitations are listed is Table S1 (Supplementary Materials). However, other large studies did not find a significant association between anti-CCP and rheumatoid factor (RF) positivity and CV morbidity and mortality [56,57], which we consider to have greater relevance. In addition, other antibodies present in RA are possibly associated with CVD risk, including antibodies against carbamylated proteins (anti-CarP) and malondialdehyde-acetaldehyde adducts [53]. Genetic susceptibility and environmental triggers also lead to constant activation and clonal expansion of specific CD4 + CD28null T cell subsets, which play a crucial role in the pathogenesis of RA, especially the loss of CD28, a co-stimulatory molecule required for normal T cell activation, correlate with seropositivity and extra-articular RA manifestations [58]. Increased expression of perforin and killer cell immunoglobulin-like receptors in these cells, with potential direct cytotoxic effects on endothelial cells and their dysfunction, can cause early atherosclerosis, plaque rupture, and thrombosis [59,60,61]. This expression strongly stimulates the activity and recruitment of macrophages and T cells to the plaque, contributing to reactive oxygen species production, inhibiting collagen production, stimulating matrix metalloproteinases, and inducing tissue factor expression [62]. According to Liuzzo et al. [63], the level of CD4 + CD28null T-cells in patients’ blood was an independent predictor of future acute coronary events in patients with RA (OR: 3.01, 95% CI: 1.1–8.25, p = 0.023). The activated endothelium promotes the binding of neutrophils, monocytes, and platelets, which is further potentiated by neutrophils, IL-8, and monocyte CCL2 chemokines. Adherent neutrophils and monocytes promote further activation of the vascular endothelium by PAR-1, creating a vicious cycle that leads to endothelial dysfunction. Neutrophils exposed to activated platelets form intravascular neutrophil extracellular traps which by expression of endothelium-activating proteases, histones, and tissue factor promote the creation of intravascular pro-inflammatory and pro-thrombotic milieus [14]. This finding is supported by the findings of numerous published studies and reviews.
Many studies have focused on the role of inflammatory cytokines in atherogenesis and CV disease development as well as their use for risk stratification. Newer studies reported that C-reactive protein (CRP) and fibrinogen are less likely to be causally associated with atherogenesis, but pro-inflammatory cytokines, interleukin-6 (IL-6), interleukin-18 (IL-18), and tumor necrosis factor-α (TNF-α) could be directly etiologically associated with atherogenesis by regulation of inflammatory cascades [64,65]. In a prospective study with 1.514 participants and a meta-analysis of 29 studies with approximately 17.000 participants, Kaptoge et al. [66], studied the roles of six pro-inflammatory cytokines, IL-6 and IL-18, matrix metalloproteinase-9 (MMP-9), soluble CD40 ligand (sCD40L), and TNF-α in coronary heart disease and concluded that higher baseline levels of IL-6, IL-18, and TNF-α were associated with a 10–25% higher risk of non-fatal myocardial infarction and CV death, while sCD40L and MMP-9 were not.
Chronic inflammation also has pro-coagulant effects mediated by several mechanisms, including increased expression of adhesion molecules for tissue factors, reduced synthesis of nitrogen oxide and thrombomodulin, and increased pro-coagulant properties of the endothelium [67]. Endothelial dysfunction is further mediated by the induction of NADPH oxidases and dysfunction of antioxidant systems [68]. Significantly increased levels of tissue factor, fibrinogen, von Willebrand factor, factor (F) VIII, activated FXIIa, and markers of thrombin synthesis have been observed in patients with RA having high inflammatory activity [67,69]. Platelets activated by cytokine-sensitized endothelial neutrophils or monocytes or by anti-CCP antibody exposure are key elements in the development of acute cardiovascular syndromes, as well as in atherosclerotic plaque formation, recruiting leukocytes to the sites of endothelial damage and inflammation, activating complement and other inflammatory receptors, and releasing cytokines and chemokines [70]. Together, these mechanisms shift the hemostatic balance to a prothrombotic state in RA [52]. A meta-analysis by Zhou et al. [71] confirmed that platelet counts are elevated in patients with RA and could serve to assess disease activity. CV risk estimation in the general population is based on different risk that underestimates CV risk in patients with RA. It is hypothesized that chronic inflammation is the key determinant to explain those underestimations, Framingham score, or SCORE system and supported by several studies like HOOM and CARRÉ study [72]; however these studies have certain limitations in terms of methods used to estimate CV risk.
IL-6 and TNF-are independently associated with a higher coronary calcium score and increased CV risk, favoring the hypothesis that RA-related increased CV risk is associated with higher levels of inflammatory cytokines and their deleterious effects on endothelial cells [52,73,74]. This finding is supported by several published studies. The effect begins very early in RA, mostly affects the carotid and coronary arteries, and is associated with a significant proportion of acute CV events [75]. High-grade inflammation is associated with increased CV morbidity and mortality, with CRP level and ESR as independent markers, reported in a population-based study during 20 years of follow-up, which is in concordance with several older studies [57,76,77,78]. The use of CRP, or highly sensitive CRP as a predictor of CV risk in modified CV risk calculators, was not adopted in standard cardiology practice [79].
Endothelial dysfunction in RA is the result of complex interactions among modifiable CV risk factors, genetic predisposition, chronic inflammation, pro-oxidative stress, prothrombotic status, and metabolic abnormalities (insulin resistance and dyslipidemia) [79,80]. It is present in very early RA, even before or within one year of the clinical onset of RA, as well as in arterial wall atherosclerosis with an increased risk of coronary heart disease and myocardial infarction [81,82]. According to Gonzalez-Gay et al. [82], endothelial dysfunction is worsened by long-standing RA > 20 years compared with RA < 7 years; however, the success of inflammation control has not been investigated. Endothelial dysfunction in RA can be assessed by measuring circulating soluble adhesive molecules such as E-selectin, P-selectin, intracellular adhesion molecule-1 (ICAM-1), vascular cell type 1 adhesion molecule (VCAM-1), and flow-mediated arterial dilatation, all of which are suggested to be used in CV risk assessment, supported by a meta-analysis of 20 studies including 852 patients with RA [83].
Duplex atherosclerosis screening is most commonly used for detecting atherosclerotic plaques that are predictive of CV disease [84]. Although for assessing cardiovascular burden in RA, carotid intima media thickness (cIMT) measurements were used in previous investigations; it is no longer recommended according to ESC guidelines [17,27], but detection of carotid plaque formation has a predictive value with a pronounced effect in early RA and in male gender with a higher inflammation burden [17,84].
Flow-mediated dilatation, augmentation index, pulse wave velocity, coronary artery calcification score (CAC), SPECT/CT, PET / CT, and PET/MR are also used to assess the atherosclerotic burden; however, non-imaging methods have many limitations and confounding factors [85]. CAC, a measure of coronary artery calcification and subclinical atherosclerosis, is closely related to the degree of atherosclerotic plaque burden and is a strong predictor of CV events [85]. Paccou et al. [86] compared coronary artery calcification and abdominal aorta calcification between asymptomatic patients with RA and healthy controls, and reported that both were more prevalent and severe in patients with RA. Coronary artery calcification was independently associated with older age and hypertension, whereas abdominal aorta calcification was independently associated with older age and erosive arthritis. However, there are some reservations about this method; for example, non-calcified plaques cannot be detected. Accelerated atherosclerosis in RA, in addition to epicardial artery disease, can also cause microvascular dysfunction, which is crucial for the regulation of myocardial perfusion, and further accelerates CV disease development (Figure 2).
4.2. Influence of medicaments
4.2.1. NSAIDs
Although non-steroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 inhibitors (COX2 inhibitors) have good anti-inflammatory and analgesic effects, they can paradoxically increase the risk of acute cardiovascular diseases, particularly stroke and myocardial infarction, which inhibit prostacyclin synthesis, particularly diclofenac and rofecoxib [32,87,88,89,90,91,92,93]. This is supported by strong evidence, particularly by Gargiulo et al. [91]. Other adverse effects, such as an increased risk of atrial fibrillation and heart failure, can induce or aggravate arterial hypertension (due to their effect on sodium and water retention) and can induce acute or chronic kidney damage and gastrointestinal complications, especially in elderly patients with multiple comorbidities [91,92,93,94,95]. Inhibition of different isoenzymes of cyclooxygenase (COX) and a decrease in prostaglandins at inflammatory sites increases thromboxane A2 (COX-1) and decreases prostaglandin I2 (COX-2) production, which may lead to vasoconstriction, platelet activation, hypertension, and accelerated atherosclerosis renal sodium retention with peripheral edema and heart failure [93]. Many trials and meta-analyzes supported those conclusions: COX-2 inhibitors 42% increased risk of CV events [89,95] highly increased risk of CV mortality (adjusted Odds Ratio 0.54, 95%CI 0.34-0.86) [95], increased risk of first-time myocardial infarction within 180 days after the initiation of NSAIDs [96], higher relative risk of myocardial infarction for diclofenac and rofecoxib [97]. Vioxx Gastrointestinal Outcomes Research (VIGOR) and the Adenomatous Polyp Prevention on Vioxx (APPROVe) trials led to the rofecoxib withdrawal due to high risk of thrombotic events supported by meta-analysis of 28 RA studies published by Roubille et al. reported increased risk of all CV events by 18% (RR, 1.18; 95% CI 1.01 to 1.38; p=0.04) and strokes with higher effect of cyclooxygenase-2 (COX-2) inhibitors (RR, 1.36; 95% CI 1.10 to 1.67; p=0.004) rather nonselective NSAIDs (RR, 1.08; 95% CI 0.94 to 1.24; p=0.28) [89,92]. The highest increase of CV evens had rofecoxib (RR, 1.58; 95% CI 1.24 to 2.00; p<0.001), what led to rofecoxib withdrawal but other NSAIDs did not showed significant effect on risk of myocardial infarction, heart failure or major adverse cardiac events but too few events were included in analysis to make strong evidence [90]. Analysis of 19 studies with RA and osteoarthritis patients reported a significantly increased risk of CV events for diclofenac and rofecoxib and an insignificantly increased risk for celecoxib. Etoricoxib and rofecoxib significantly increased the risk of hypertension and naproxen for stroke; however, not all NSAIDs were included in the investigation and cardiovascular side effects were analyzed [98]. NSAIDs should be prescribed at the lowest effective doses and for the shortest possible duration with caution when prescribing to patients with CV disease or in the presence of CV risk factors and should be avoided in patients with treatment-resistant hypertension, high CV risk, and severe chronic kidney disease; naproxen or celecoxib are the preferred choices [72,92,94]. For patients with pre-existing hypertension receiving renin-angiotensin system blockers, dose increases or the addition of a different drug class should be considered. Gastric prophylaxis is also recommended, especially if NSAIDs are combined with glucocorticoids, in the elderly and in patients with a moderate to high risk of peptic ulcer disease [94]. The use of acetylsalicylic acid for the primary prevention of CV disease in patients with RA is not recommended [17,79].
4.2.2. Glucocorticoids
Glucocorticoids have potent anti-inflammatory and immunosuppressive effects and are widely used in RA treatment. The addition of low-dose glucocorticoids (below 7.5 mg/daily prednisone) to DMARDs in early RA slows the radiological progression of bone destruction [93,99]. Long-term use of glucocorticoids at high and low doses significantly increases CV risk, with an unfavorable impact on lipid metabolism, obesity, insulin production, insulin resistance, and blood pressure [22,85,100]. Although a meta-analysis of six randomized controlled trials with 689 patients with RA reported good glucocorticoid safety profiles, numerous studies have found an increased incidence of all-cause and CV mortality, hypertension, hyperglycemia, diabetes, osteoporosis, and myocardial infarction with dose- and time-dependent glucocorticoid usage [22,93,101]. Many registries and large meta-analyses support the conclusions of the effects of GCs on CV risk and mortality with some dose-dependent effects: the Scotland National Health Service RA database, General Practice Research Database, and a meta-analysis of 28 studies by Roubille et al. [89]. An older meta-analysis of 37 studies reported poor associations between low-dose glucocorticoid (< 10 mg/day) use and CV risk factors, beneficial effects on lipid profile, an increase in insulin resistance or glycemia, and no effect on blood pressure, with a trend of increasing major CV events, myocardial infarction, stroke, and mortality [101]. According to current guidelines, glucocorticoids should be used at the lowest possible dose, continuation should be regularly reassessed, and remission withdrawal should be considered [102,103].
4.2.3. Classical DMARDs
Considering disease modifying antireumatics (DMARDs) meta-analysis of observational and prospective studies reported beneficial effects of methotrexate (MTX), sulfasalazine, and hydroxychloroquine on CV risk [83,85,101,104,105,106]. MTX is an antifolate immunosuppressive drug that inhibits neutrophil chemotaxis and the synthesis of proinflammatory cytokines and exerts antiatherogenic and cardioprotective effects [103,106]. According to analysis of eight prospective and two retrospective studies and 694 publications, with 66.334 participants and 6.235 events, MTX was associated with a 21% lower overall risk for CV event (95% confidence interval [CI] 0.73-0.87, p <0.001) and an 18% lower risk for myocardial infarction (95% CI: 0.71–0.96, p = 0.01) [104]. A study on coronary artery calcification using computed tomography reported a lower coronary calcification burden with MTX use in patients with RA [86]. Another meta-analysis of 28 studies reported overall 21% CV risk reduction with MTX (RR, 0.72; 95% CI 0.57 to 0.91; p=0.007) as well as 18% myocardial infarction risk reduction, trends towards decreasing risk of heart failure and without effect on strokes and major adverse cardiac events, although the last may be due to the lower number of events resulting in insufficient statistical power to detect significant effect [89]. An Older meta–analysis of 18 studies also reported reduced risk of CV events in MTX threated patients with RA: reduced overall mortality of 41% and 70% in two studies, reduction in CV morbidity of 89, 35, 17%, and 15% in four studies, 18% risk reduction of myocardial infarction in one study (with selection bias due to exclusion of fatal events) and trend towards risk reduction in three studies, one study showed 20% risk reduction of hospitalization with congestive heart failure (significant bias), and 11% risk reduction of stroke in one study and trend towards it in another [107]. Several studies have reported that MTX increases total cholesterol, LDL, HDL, and triglyceride levels in RA, with a possible explanation that reflects the normalization of lipid levels due to suppression of inflammation, without increasing CV risk [74]. Hydroxychloroquine exerts antirheumatic effects by targeting autoantigen processing in macrophages, suppressing T-lymphocytes lymphocytes, and neutralizing the prothrombotic effects of antiphospholipid antibodies [92,105]. Hydroxychloroquine improves lipid and glycemic indices, reduces the risk of thromboembolic events, enhances the elasticity of peripheral arteries and systemic vascular resistance, and reduces CV risk [72,92,105,109]. However, considerations of methodology, transparency, and reproducibility that affect the credibility of the conclusions were asserted in the latter meta-analysis [110]. Several case reports have asserted the consideration of hydroxychloroquine cardiotoxicity (restrictive cardiomyopathy and conduction disorders); however, in those cases, patients had prolonged use of large cumulative doses, but the use of hydroxychloroquine is considered safe at therapeutic doses with periodic ECG monitoring [105]. Combination therapy with MTX, sulfasalazine, and hydroxychloroquine also decreased CV risk by better reducing inflammation, increasing HDL, lowering LDL, and improving the ratio of total cholesterol to HDL compared with MTX monotherapy or combination with etanercept; however, the study included only patients with early RA with high disease activity, naïve to DMARDs [111]. In contrast, azathioprine, cyclosporine, and leflunomide increase the risk of CV events by 80% compared to MTX monotherapy [112,113]. Leflunomide has potent anti-inflammatory and immunomodulatory effects but increases the risk of hypertension, which has been reported in 2.1%-10.6% of cases in different studies [92,114]. Cyclosporine is used for the treatment of severe early RA and has many adverse effects, including vasoconstriction, thrombosis, hypertension, blood pressure, and renal function monitoring is advised when using cyclosporine [92,114]. Azathioprine is a purine analog with rare adverse effects, including angina, renal and subclavian vein thrombosis, hypotension, and cardiogenic shock [92,114].
4.2.4. Biologic agents
Biologics are used to treat many different autoimmune diseases by targeting inflammatory cytokines (TNF-alpha, IL-6) and cytokine receptors interrupting the inflammatory vicious circle and are recommended even in early RA with low-severity inflammatory arthritis [92,113]. By suppressing inflammation and maintaining low disease activity, they significantly reduce the CV risk and the incidence of myocardial infarction, heart failure, and cerebrovascular events in patients with RA [92,114,115,116,117]. Anti-TNF agents neutralize soluble and/or membrane-bound TNF and act as monoclonal antibodies or soluble receptor [92].
In a large cohort study of 2.757 patients with RA treated with infliximab, etanercept, or adalimumab, a significant increase in heart failure was reported in patients with high disease activity and concomitant glucocorticoid or cyclooxygenase inhibitor therapy, whereas anti-TNF therapy itself did not significantly contribute to the risk; only sporadic cases of acute coronary syndromes, arrhythmias, and AV block for infliximab have been reported [116]. In a large, retrospective study, Solomon et al. reported that anti-TNF alpha therapy may be associated with a reduced CV risk compared with an classic DMARD therapy, incidence rates per 100 person-years for the composite cardiovascular end point for classic DMARD and anti-TNF therapy were 3.05 (95% confidence interval [CI], 2.54-3.65) and 2.52 (95% CI, 2.12-2.98) respectively[114]. Jacobsson et al. in a prospective cohort study with 983 participants [117], Ljung et al. in a prospective cohort study with 6.864 patients with RA [118], and an earlier meta-analysis of 20 studies by Westlake et al. reported similar conclusions [107]. Beside effect of anti-TNFα therapy on decreasing inflammation, and consequential improvement of joint function indirectly they may lead to increased levels of physical activity, which will subsequently decrease the incidence of other cardiovascular risk factors such as diabetes mellitus and hypertension. [115]. Karpouzas et al. reported slower non-calcified coronary plaque progression with longer biological usage independent of inflammation, prednisone dose, and statin use [119].
A recently published meta-analysis of 26 longitudinal studies addressing the question of anti-TNF therapy influence on BMI found a small increase in body weight and BMI with anti-TNF therapy, on average 0.90 kg, 2.34 kg and 2.27 kg for infliximab, etanercept, and adalimumab, respectively 4–at 104 weeks of follow up [120]. The ATTACH study reported that the use of anti-TNF therapy increased mortality or worsened heart failure in patients with moderate to severe chronic heart failure, especially those with an ischemic etiology, but the RENAISSANCE and RECOVER clinical trials did not confirm this for etanercept [121]. A possible reduction in insulin resistance with anti-TNF therapy was reported in a meta-analysis of 12 studies; however, the heterogeneity of the studies was high [122]. A meta-analysis of studies considering the impact of biologics (tocilizumab, abatacept, rituximab, and tumor necrosis factor inhibitors) on CV risk and safety reported fewer CV events with rituximab and neutral effects of others compared to classical DMARDs, but significant heterogeneity on CV outcomes was reported [123]. Large meta-analysis of which included 43 biological registers and 27 publications addressed the issue of biologics safety and effect on mortality reported overall mortality and CV events were significant reduction in patients treated with anti-TNFs: RR = 0.60 [95% CI 0.38–0.94] and RR = 0.62 [0.44–0.88], respectively, no effect on neoplasm risk but serious infections were significantly increased during anti-TNF treatment (RR = 1.48 [1.18–1.85]) compared to classical DMARD [124]. Another meta-analysis of patients with RA in 28 studies reported that anti-TNF treatment was significantly associated with an overall CV risk reduction of approximately 30% [89]. Cheung et al. analyzed the effect of anti-TNF therapy on subclinical atherosclerosis, and six studies measured at least one parameter before and after treatment (24th and 52nd weeks): intima media thickness, pulse wave velocity, and augmentation index; they found no effect of anti-TNF therapy on all three parameters at the 24th week as well as intima media thickness at the 52nd week [125]. An older meta-analysis of 32 studies by Daien et al. reported increased total cholesterol and HDL levels and unchanged LDL levels with long-term anti-TNF therapy, with uncertain effects on CV risk [126]; however, the total cholesterol/HDL ratio was not significantly altered by anti-TNF therapy [74]. These anti-TNF therapeutic effects may reflect the normalization of lipid levels to those prior to RA to suppress inflammation [74,115]. Meta-analysis of Zhao et al. which included 6321 subjects with RA from 11 randomized clinical trials reported strong evidence of increased incidence of hypertension associated with some anti-TNF therapy (OR = 1.8896, 95% CI:1.35–2.65) as well as accumulation of hypertension with longer therapy duration, especially for certolizumab but not for etanercept, tofacitinib, infliximab and golimumab [127]. Results from a new Korean observational study with 996 RA subjects found no increased incidence of HA with biologics compared with classical DMARDs in general, whereas MTX had a lower incidence of hypertension, which could be explained by the hypothesis that MTX restores vasodilation-related adenosine levels in the body [128]. Neutral effect of anti-TNF agents on HA incidence was reported by Desai et al. in large cohort study with 4.822 patients using TNF-α inhibitor and 2.400 using classical DMARD [129]. Despite some methodological limitations, these studies and analyses strongly support the increased beneficial effects of anti TNF agents compared to classical RA medications, probably due to more efficient inflammation control and successful achievement of long-term RA remission.
The effect of non-anti-TNF agents, abatacept tocilizumab, sarilumab anakinra, and rituximab on CV risk has also been evaluated; abatacept was found to have 20% greater CV risk reduction compared with anti-TNF agents, tocilizumab had the same effect on CV risk as MTX monotherapy, anakira showed improved vascular and left ventricular function in a small placebo-controlled study, sarilumab and rituximab had a neutral effect on CV events and canakinumab - IL-1 inhibitor given 150 mg every 3 months had a significantly lower rate of myocardial infarction compared to placebo [77,130,131,132,133,134,135]. Several studies have consistently reported that tocilizumab is associated with increased total cholesterol, HDL, LDL, and triglyceride levels [74,92], the MEASURE study found that tocilizumab + MTX treatment did not increase the concentration of proatherogenic small and dense LDL particles, while antiatherogenic small and medium HDL particles were increased and structurally altered to a less inflammatory state than with MTX alone therapy [136]. There have been no reports of an increase in CV events with tocilizumab [92]. These medications, especially abatacept and anakiras, are also strongly recommended for CVD prevention in RA.
4.2.5. Small molecule inhibitors of Janus kinase
Tofacitinib and baricitinib, newer small-molecule inhibitors of Janus kinase (JAK), increase total and LDL cholesterol levels by 10–20% [137]. For baricitinib, a meta-analysis of six studies with 3.552 patients reported significantly increased dose-dependent LDL and HDL with baricitinib users; the mean change was 13.15 mg/dL (95% CI: 8.89–17.42) and 5.40 mg/dL (95% CI: 3.07–7.74), respectively. Although increased relative risk of CV events was not statistically significant, an association may exist [132]. A recently published pooled cohort study of 3.492 patients with RA with over 7.860 patient-years of exposure to baricitinib did not reveal a significant association between baricitinib treatment and the occurrence of CV events or congestive heart failure [138]. The influence of biologic agents and tofacitinib, JAK inhibitor, on the lipid profile of patients with RA was analyzed in a meta-analysis of 20 articles by Soto et al. and reported increased cholesterol levels (odds ratio [OR] 4.64; CI 95% 2.71, 7.95, p < 0.001), HDL (OR 2.25; 95% CI 1.14, 4.44 [p = 0.020]), and LDL (OR 4.80; 95% CI 3.27, 7.05 [p < 0.001]), but despite this effect better inflammation control with those medications appears to have lower mortality and incidence of cardiovascular events, but other biologic or non-biologic agents were not included in analysis [139].
With the exception of the ENTRACTE trial, which compared tocilizumab and etanercept, no head-to-head study has been conducted on the impact of biological agents on CV risk and safety. The ENTRACTE trial reported no significant difference in CV events between the tocilizumab and etanercept groups [86]. Observational studies have reported a higher incidence of myocardial infarction among older patients with anti-TNF initiators than with abatacept and tocilizumab, and no difference in CV risk when comparing tocilizumab with abatacept [77,140].
4.2.6. Statins
The use of statins in the management of CVDs seems to have a neutral effect on RA development, and the risk of RA may be lower in patients with a longer duration or intensity of statin use [141]. Treatment with statins is beneficial in lowering CV risk in patients with RA due to their lipid-lowering and other pleiotropic effects that slow down coronary non-calcified plaque progression and suppress the effects of inflammation on plaque progression and CAC [79,119,142,143]. Meta-analysis of 11 relevant studies reported a standardized mean difference in DAS28 of −0.55 (95% CI: −0.83 to −0.26, p = 0.0002), supporting the positive effect of statins on RA [144]. However, the indiscriminate use of statins is not recommended in all patients with RA, and CV risk assessment and appropriate statin use according to the guidelines for primary and secondary CVD prevention in this population is necessary [17].
5. Conclusions
In conclusion, to prevent CV diseases in patients with RA, two main complementary strategies were considered that included strict inflammation control with as few flares as possible and the management of modifiable risk factors.
Since CV morbidity and mortality in RA are alarmingly high, it is crucial to comprehend the mechanisms that cause and control atherosclerosis so that highly specific treatment plans can be created to reduce the burden that patients with RA bear with regard to their CV health. The development of atherogenic foam cells is aided by proinflammatory cytokines including IL-6 and TNF-alpha. The creation of a proatherogenic environment favors the development of atherosclerotic diseases because of endothelial dysfunction and RA-derived autoantibodies, which increase the inflammatory potential of macrophages. Whether and how inflammation and traditional CV risk factors interact to cause atherosclerosis in RA is important and needs to be determined.
Achieving and maintaining long-term RA remission using novel therapeutic agents is crucial. Early recognition and strict control of modifiable risk factors according to these guidelines are paramount. Good patient education, application of these measures, increased surveillance and early active search for risk factors by general practitioners and specialists, interdisciplinary approach, and accessibility of the health care systems play a key role in achieving these goals.
Supplementary Materials
Table S1: Study characteristics and comments included in review.
Author Contributions
D. Bedeković – prime investigator and cardiology data analyzed; I Bošnjak database screening and cardiology data analyzed, D. Kirner cardiology data analyzed, S Novak rheumatology data analyzed
Funding
This research received no external funding
Acknowledgments
Prof. D. Dukić and Prof. M. Dukić for statistical data analyzed
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mitchell, D.M.; Spitz, P.W.; Young, D.Y.; Bloch, D.A.; McShane, D.J.; Fries, J.F. Survival, prognosis, and causes of death in rheumatoid-arthritis. Arthritis and Rheumatism. 1986, 29, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Coulton, B.L.; Symmons, D.P.M. Long-term outcome of threating rheumatoid arthritis – results after 20 years. Lancet, 1987, 1, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; Medsger, T.A.; Mitchell, D.M.; Neustadt, D.H.; Pinals, R.S.; Schaller, J.G.; Sharp, J.T.; Wilder, R.L. The American-rheumatism-association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and Rheumatism. 1988, 31, 315–324. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; Combe, B.; Costenbader, K.H.; Dougados, M.; Emery, P.; Ferraccioli, G.; Hazes, J.M.W.; Hobbs, K.; Huizinga, T.W.J.; Kavanaugh, A.; Kay, J.; Kvien, T.K.; Laing, T.; Mease, P.; Menard, H.A.; Moreland, L.W.; Naden, R.L.; Pincus, T.; Smolen, J.S.; Stanislawska-Biernat, E.; Symmons, D.; Tak, P.P.; Upchurch, K.S.; Vencovsky, J.; Wolfe, F.; Hawker, G. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Annals of the Rheumatic Diseases. 2010, 69, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet, 2001, 358, 903–911. [Google Scholar] [CrossRef]
- Symmons, D.P. M. Epidemiology of rheumatoid arthritis: determinants of onset, persistence and outcome. Best Practice & Research in Clinical Rheumatology. 2002, 16, 707–722. [Google Scholar]
- Sakai, R.; Hirano, F.; Kihara, M.; Yokoyama, W.; Yamazaki, H.; Harada, S.; Nanki, T.; Koike, R.; Miyasaka, N.; Harigai, M. High prevalence of cardiovascular comorbidities in patients with rheumatoid arthritis from a population-based cross-sectional study of a Japanese health insurance database. Modern Rheumatology. 2016, 26, 522–528. [Google Scholar] [CrossRef]
- Koivuniemi, R.; Paimela, L.; Leirisalo-Repo, M. Causes of death in patients with rheumatoid arthritis from 1971 to 1991 with special reference to autopsy. Clinical Rheumatology. 2009, 28, 1443–1447. [Google Scholar] [CrossRef]
- Maradit-Kremers, H.; Nicola, P.J.; Crowson, C.S.; Ballman, K.V.; Gabriel, S.E. Cardiovascular Death in Rheumatoid Arthritis A Population-Based Study. Arthritis and Rheumatism. 2005, 52, 722–732. [Google Scholar] [CrossRef]
- Sokka, T.; Abelson, B.; Pincus, T. Mortality in rheumatoid arthritis: 2008 update. Clinical and Experimental Rheumatology. 2009, 26, S35–S61. [Google Scholar]
- Meune, C.; Touze, E.; Trinquart, L.; Allanore, Y. High risk of clinical cardiovascular events in rheumatoid arthritis: Levels of associations of myocardial infarction and stroke through a systematic review and meta-analysis. Archives of Cardiovascular Diseases. 2010, 103, 253–261. [Google Scholar] [CrossRef]
- Meune, C.; Touze, E.; Trinquart, L.; Allanore, Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: a systematic review and meta-analysis of cohort studies. Rheumatology. 2009, 48, 1309–1313. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of Cardiovascular Mortality in Patients With Rheumatoid Arthritis: A Meta-Analysis of Observational Studies. Arthritis and Rheumatism-Arthritis Care and Research. 2008, 59, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.W. A.; Anderson, R.; Ker, J.A.; Ally, M.T.M. Rheumatoid arthritis and risk of cardiovascular disease. Cardiovascular Journal of Africa. 2018, 29, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Siebert, S.; Lyall, D.M.; Mackay, D.F.; Porter, D.; McInnes, I.B.; Sattar, N.; Pell, J.P. Characteristics of rheumatoid arthritis and its association with major comorbid conditions: cross-sectional study of 502 649 UK Biobank participants. Rmd Open. 2016, 2. [Google Scholar] [CrossRef]
- Van Doornum, S.; McColl, G.; Wicks, I.P. Accelerated atherosclerosis - An extraarticular feature of rheumatoid arthritis? Arthritis and Rheumatism. 2002, 46, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Frank, L.; Visseren, J.; and others. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). European Heart Journal 2021, 42, 3227–3337. [Google Scholar]
- Panoulas, V.F.; Metsios, G.S.; Pace, A.V.; et al. Hypertension in rheumatoid arthritis. Rheumatology (Oxford) 2008, 4, 1286–98. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Sjöberg, C.; Möller, B.; Ljung, L.; Smedby, T.; Södergren, A.; et al. Co-morbidity in patients with early rheumatoid arthritis - inflammation matters. Arthritis Res Ther. 2016, 18, 33. [Google Scholar] [CrossRef]
- Erba, G.; Grosso, G.; Valena, C.A.; Riva, M.; Allevi, E.; Betelli, M.; et al. Cardiovascular risk factor profile in Italian cohort of patients with rheumatoid arthritis: Results of a three years follow-up. J Hypertens. 2015, 33 Suppl 1, e118. [Google Scholar] [CrossRef]
- Gherghe, A.M.; Dougados, M.; Combe, B.; Landewé, R.; Mihai, C.; Berenbaum, F.; et al. Cardiovascular and selected comorbidities in early arthritis and early spondyloarthritis, a comparative study: results from the ESPOIR and DESIR cohorts. RMD Open. 2015, 1, e000128. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.F.; Gourraud, P.A.; Cantagrel, A.; Davignon, J.L.; Constantin, A. Traditional cardiovascular risk factors in rheumatoid arthritis: a meta-analysis. Joint Bone Spine. 2011, 78, 179–83. [Google Scholar] [CrossRef]
- Heliovaara, M.; Aho, K.; Aromaa, A.; Knekt, P.; Reunanen, A. SMOKING AND RISK OF RHEUMATOID-ARTHRITIS. Journal of Rheumatology. 1993, 20, 1830–1835. [Google Scholar] [PubMed]
- Castellanos-De La Hoz, J.; Amaya-Amaya, J.; Molano-Gonzalez, N.; Gutierrez-Infante, F.; Anaya, J.M.; Rojas-Villarraga, A. The influence of cigarette smoking on disease activity and joint erosions in rheumatoid arthritis: a systematic review and meta-analysis. Annals of the Rheumatic Diseases. 2013, 72, 387–387. [Google Scholar] [CrossRef]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Kallberg, H.; Bengtsson, C.; Grunewald, J.; i sur. A new model for an etiology of rheumatoid arthritis. Arthritis and Rheumatism. 2006, 54, 38–46. [Google Scholar] [CrossRef]
- Welsing, P.M. J.; van Gestel, A.M.; Swinkels, H.L.; Kiemeney, L.; van Riel, P. The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis and Rheumatism. 2001, 44, 2009–2017. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; i sur. Task Force 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts). European Journal of Preventive Cardiology. 2016, 23, NP1–NP96. [Google Scholar]
- Baghdadi, L.R.; Woodman, R.J.; Shanahan, E.M.; Mangoni, A.A. The impact of traditional cardiovascular risk factors on cardiovascular outcomes in patients with rheumatoid arthritis: a systematic review and meta-analysis. PLoS One. 2015, 10, e0117952. [Google Scholar] [CrossRef]
- Jiang, P.; Li, H.; Li, X. Diabetes mellitus risk factors in rheumatoid arthritis: a systematic review and meta-analysis. Clinical and Experimental Rheumatology. 2015, 33, 115–121. [Google Scholar]
- Guin, A.; Sinhamahapatra, P.; Misra, S.; Mazumder, S.R.C.; Chatterjee, S.; Ghosh, A. Incidence and effect of insulin resistance on progression of atherosclerosis in rheumatoid arthritis patients of long disease duration. Biomedical Journal, 2019, 42, 394–402. [Google Scholar] [CrossRef]
- Dougados, M.; Soubrier, M.; Antunez, A.; Balsa, A.; Buch, M.H.; Casado, G.; et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: results of an international, cross-sectional study (COMORA). Annals of the Rheumatic Diseases 2020, 73, 62–658. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos-Kalinoglou, A.; Metsios, G.S.; Koutedakis, Y.; Nevill, A.M.; Douglas, K.M.; A. i sur. Redefining overweight and obesity in rheumatoid arthritis patients. Annals of the Rheumatic Diseases. 2007, 66, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.L.; Lanna, C.C.D.; Rocha, M.P.; et al. Recognition and control of hypertension, diabetes, and dyslipidemia in patients with rheumatoid arthritis. Rheumatology International 2018, 38, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Beinsberger, J.; Heemskerk, J.W.M.; Cosemans, J. Chronic arthritis and cardiovascular disease: Altered blood parameters give rise to a prothrombotic propensity. Seminars in Arthritis and Rheumatism. 2014, 44, 345–352. [Google Scholar] [CrossRef]
- Nowak, B.; Madej, M.; Luczak, A.; Malecki, R.; Wiland, P. Disease Activity, Oxidized-LDL Fraction and Anti-Oxidized LDL Antibodies Influence Cardiovascular Risk in Rheumatoid Arthritis. Advances in Clinical and Experimental Medicine. 2016, 25, 43–50. [Google Scholar] [CrossRef]
- Giles, J.T.; Wasko, M.C.M.; Chung, C.P.; Szklo, M.; Blumenthal, R.S.; Kao, A.; i sur. Exploring the Lipid Paradox Theory in Rheumatoid Arthritis: Associations of Low Circulating Low-Density Lipoprotein Concentration With Subclinical Coronary Atherosclerosis. Arthritis & Rheumatology. 2011, 71, 1426–1436. [Google Scholar]
- McGrath, C.M.; Young, S.P. Lipid and Metabolic Changes in Rheumatoid Arthritis. Current Rheumatology Reports. 2015, 17. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis – an approach to understanding the molecular-genetics of susceptibility of rheumatoid arthritis. Arthritis and Rheumatism. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- McInnes, I.B. and Schett, G. MECHANISMS OF DISEASE: The Pathogenesis of Rheumatoid Arthritis. New England Journal of Medicine. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, J.; Lau, J.; Wang, S.H.; Taneja, V.; Matteson, E.L.; Vassallo, R. Mechanisms of lung disease development in rheumatoid arthritis. Nature Reviews Rheumatology. 2019, 15, 581–596. [Google Scholar] [CrossRef]
- Londei, M.; Savill, C.M.; Verhoef, A.; Brennan, F.; Leech, Z.A.; Duance, V.; Maini, R.N.; Feldmann, M. Persistence of collagen type-II-specific T-cell clones in the synovial-membrane of a patient with rheumatoid-arthritis. Proceedings of the National Academy of Sciences of the United States of America. 1987, 86, 636–640. [Google Scholar] [CrossRef]
- Glant, T.T.; Radacs, M.; Nagyeri, G.; Olasz, K.; Laszlo, A.; Boldizsar, F.; Hegyi, A.; Finnegan, A.; Mikecz, K. Proteoglycan-Induced Arthritis and Recombinant Human Proteoglycan Aggrecan G1 Domain-Induced Arthritis in BALB/c Mice Resembling Two Subtypes of Rheumatoid Arthritis. Arthritis and Rheumatism. 2011, 63, 1312–1321. [Google Scholar] [CrossRef]
- Verheijden, G.F. M.; Rijnders, A.W.M.; Bos, E.; deRoo, *!!! REPLACE !!!*; C. J., vanStaveren; Miltenburg, A.M.M.; Meijerink, J.H.; Elewaut, D.; F., deKeyser; Veys, E.; Boots, A.M.H.; C. Human cartilage glycoprotein-39 as a candidate autoantigen in rheumatoid arthritis. Arthritis and Rheumatism. 2011, 40, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; Yamamoto, K. Rheumatoid arthritis. Nature Reviews Disease Primers. 2018, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Feist, E.; Doerner, T. Emerging cell and cytokine targets in rheumatoid arthritis. Nature Reviews Rheumatology. 2014, 10, 77–88. [Google Scholar] [CrossRef]
- Carbone, F.; Bonaventura, A.; Liberale, L.; Paolino, S.; Torre, F.; Dallegri, F.; Montecucco, F.; Cutolo, M. Atherosclerosis in Rheumatoid Arthritis: Promoters and Opponents. Clinical Reviews in Allergy and Immunology. 2020, 58, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mewar, D.; Coote, A.; Moore, D.J.; Marinou, I.; Keyworth, J.; Dickson, M.C.; Montgomery, D.S.; Binks, M.H.; Wilson, A.G. Independent associations of anti-cyclic citrullinated peptide antibodies and rheumatoid factor with radiographic severity of rheumatoid arthritis. Arthritis Research and Therapy. 2006, 8. [Google Scholar]
- Sokolove, J.; Bromberg, R.; Deane, K.D.; Lahey, L.J.; Derber, L.A.; Chandra, P.E.; Edison, J.D.; Gilliland, W.R.; Tibshirani, R.J.; Norris, J.M.; Holers, V.M.; Robinson, W.H. Autoantibody Epitope Spreading in the Pre-Clinical Phase Predicts Progression to Rheumatoid Arthritis. Plos One. 2012, 7. [Google Scholar] [CrossRef]
- Lopez-Longo, F.J.; Oliver-Minarro, D.; de la Torre, I.; de Rabago, E.G.D.; Sanchez-Ramon, S.; Rodriguez-Mahou, M.; Paravisini, A.; Monteagudo, I.; Gonzalez, C.M.; Garcia-Castro, M.; Casas, M.D.; Carreno, L. Association Between Anti-Cyclic Citrullinated Peptide Antibodies and Ischemic Heart Disease in Patients With Rheumatoid Arthritis. Arthritis & Rheumatism-Arthritis Care & Research. 2009, 61, 419–424. [Google Scholar]
- Sokolove, J.; Brennan, M.J.; Sharpe, O.; Lahey, L.J.; Kao, A.H.; Krishnan, E.; Edmundowicz, D.; Lepus, C.M.; Wasko, M.C.; Robinson, W.H. Citrullination Within the Atherosclerotic Plaque: A Potential Target for the Anti-Citrullinated Protein Antibody Response in Rheumatoid Arthritis. Arthritis and Rheumatism. 2013, 65, 1719–1724. [Google Scholar] [CrossRef]
- Geraldino-Pardilla, L.; Zartoshti, A.; Ozbek, A.B.; Giles, J.T.; Weinberg, R.; Kinkhabwala, M.; Bokhari, S.; Bathon, J.M. Arterial Inflammation Detected With F-18-Fluorodeoxyglucose-Positron Emission Tomography in Rheumatoid Arthritis. Arthritis and Rheumatology. 2018, 70, 30–39. [Google Scholar] [CrossRef] [PubMed]
- DeMizio, D.J.; Geraldino-Pardilla, L.B. Autoimmunity and Inflammation Link to Cardiovascular Disease Risk in Rheumatoid Arthritis. Rheumatology and Therapy. 2020, 7, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.P.; Kremers, H.M.; Crowson, C.S.; Snyder, M.R.; Therneau, T.M.; Roger, V.L.; Gabriel, S.E. Autoantibodies and the Risk of Cardiovascular Events. Journal of Rheumatology. 2009, 36, 2462–2469. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, J.H.; van Nies, J.A.B.; Chipping, J.; Marshall, T.; Mil, A.; Symmons, D.P.M.; Verstappen, S.M.M. Rheumatoid factor and anti-citrullinated protein antibody positivity, but not level, are associated with increased mortality in patients with rheumatoid arthritis: results from two large independent cohorts. Arthritis Research and Therapy. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- McCoy, S.S.; Crowson, C.S.; Maradit-Kremers, H.; Themeau, T.M.; Roger, V.L.; Matteson, E.L.; Gabriel, S.E. Longterm Outcomes and Treatment After Myocardial Infarction in Patients with Rheumatoid Arthritis. Journal of Rheumatology. 2013, 40, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Mackey, R.H.; Kuller, L.H.; Deane, K.D.; Walitt, B.T.; Chang, Y.F.; Holers, V.M.; Robinson, W.H.; Tracy, R.P.; Hlatky, M.A.; Eaton, C.B.; Liu, S.M.; Freiberg, M.S.; Talabi, M.B.; Schelbert, E.B.; Moreland, L.W. Rheumatoid Arthritis, Anti-Cyclic Citrullinated Peptide Positivity, and Cardiovascular Disease Risk in the Women’s Health Initiative. Arthritis and Rheumatology. 2015, 67, 2311–2322. [Google Scholar] [CrossRef]
- Innala, L.; Moller, B.; Ljung, L.; Magnusson, S.; Smedby, T.; Sodergren, A.; Ohman, M.L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Research and Therapy. 2010, 13. [Google Scholar] [CrossRef]
- Pawlik, A.; Ostanek, L.; Brzosko, I.; Brzosko, M.; Masiuk, M.; Machalinski, B.; Gawronska-Szklarz, B. The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res Ther. 2003, 5(4), R210–3. [Google Scholar] [CrossRef]
- Winchester, R.; Giles, J.T.; Nativ, S.; Downer, K.; Zhang, H.Z.; Bag-Ozbek, A.; Zartoshti, A.; Bokhari, S.; Bathon, J.M. Association of Elevations of Specific T cell and Monocyte Subpopulations in Rheumatoid Arthritis With Subclinical Coronary Artery Atherosclerosis. Arthritis and Rheumatology. 2016, 68, 92–102. [Google Scholar] [CrossRef]
- Nakajima, T.; Goek, O.; Zhang, X.Y.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circulation Research. 2003, 93, 106–113. [Google Scholar] [CrossRef]
- Dumitriu, I.E.; Baruah, P.; Finlayson, C.J.; Loftus, I.M.; Antunes, R.F.; Lim, P.; Bunce, N.; Kaski, J.C. High Levels of Costimulatory Receptors OX40 and 4-1BB Characterize CD4 (+) CD28 (null) T Cells in Patients With Acute Coronary Syndrome. Circulation Research. 2010, 110, 857–U150. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejias, R.; Castaneda, S.; Gonzalez-Juanatey, C.; Corrales, A.; Ferraz-Amaro, I.; Genre, F.; Remuzgo-Martinez, S.; Rodriguez-Rodriguez, L.; Blanco, R.; Llorca, J.; Martin, J.; Gonzalez-Gay, M.A. Cardiovascular risk assessment in patients with rheumatoid arthritis: The relevance of clinical, genetic and serological markers. Autoimmunity Reviews. 2016, 15, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Biasucci, L.M.; Brugaletta, S.; Digianuario, G.; Pinnelli, M.; Giubilato, G.; Giubilato, S.; Colafrancesco, V.; Rebuzzi, A.G.; Crea, F. An unusual population of T-lymphocytes, (CD4+CD28null) T-cells, is associated with the recurrence of acute coronary events in patients with unstable angina. Circulation. 2005, 112, U586–U586. [Google Scholar]
- Libby, P. Inflammatory mechanisms: The molecular basis of inflammation and disease. Nutrition Reviews. 2007, 65, S140–S146. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, D.I.; Holmes, M.V.; Kuchenbaecker, K.B.; Engmann, J.E.L.; Shah, T.; Sofat, R.; Guo, Y.R.; Chung, C.; Peasey, A.; Ster, R.P.; Mooijaart, S.P.; Ireland, H.A.; Leusink, M.; Langenberg, C.; Li, K.; Palmen, J.; Howard, P.; Cooper, J.A.; Drenos, F.; Hardy, J.; Nalls, M.A.; Li, Y.R.; Lowe, G.; Stewart, M.; Bielinski, S.J.; Peto, J.; Timpson, N.J.; Gallacher, J.; Dunlop, M.; Houlston, R.; Tomlinson, I.; Tzoulaki, I.; Luan, J.; Boer, J.M.A.; Forouhi, N.G.; Onland-Moret, N.C.; van der Schouw, Y.T.; Schnabel, R.B.; Hubacek, J.A.; Kubinova, R.; Baceviciene, M.; Tamosiunas, A.; Pajak, A.; Topor-Madry, R.; Malyutina, S.A.; Baldassarre, D.; Sennblad, B.; Tremoli, E.; de Faire, U.; Ferrucci, L.; Bandenelli, S.; Tanaka, T.; Meschia, J.F.; Singleton, A.; Navis, G.; Leach, I.M.; Bakker, S.J.L.; Gansevoort, R.T.; Ford, I.; Epstein, S.E.; Burnett, M.S.; Devaney, J.M.; Jukema, J.W.; Westendorp, R.G.J.; de Borst, G.J.; van der Graaf, Y.; de Jong, P.A.; der Zee, A.H.M.-V.; Klungel, O.H.; de Boer, A.; Doevendans, P.A.; Stephens, J.W.; Eaton, C.B.; Robinson, J.G.; Manson, J.E.; Fowkes, F.G.R.; Frayling, T.M.; Price, J.F.; Whincup, P.H.; Morris, R.W.; Lawlor, D.A.; Smith, G.D.; Ben-Shlomo, Y.; Redline, S.; Lange, L.A.; Kumari, M.; Wareham, N.J.; Verschuren, W.M.M.; Benjamin, E.J.; Whittaker, J.C.; Hamsten, A.; Dudbridge, F.; Delaney, J.A.C.; Wong, A.; Kuh, D.; Hardy, R.; Castillo, B.A.; Connolly, J.J.; van der Harst, P.; Brunner, E.J.; et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012, 379, 1214–1224. [Google Scholar]
- Kaptoge, S.; Seshasai, S.R.K.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.O.; Jorgensen, T.; Danesh, J. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. European Heart Journal. 2014, 35, 578–U35. [Google Scholar] [CrossRef]
- van den Oever, I.A. M.; Sattar, N.; Nurmohamed, M.T. Thromboembolic and cardiovascular risk in rheumatoid arthritis: role of the haemostatic system. Annals of the Rheumatic Diseases. 2014, 73, 954–957. [Google Scholar] [CrossRef]
- Small, H.Y.; Migliarino, S.; Czesnikiewicz-Guzik, M.; Guzik, T.J. Hypertension: Focus on autoimmunity and oxidative stress. Free Radical Biology and Medicine. 2018, 125, 104–115. [Google Scholar] [CrossRef]
- Peters, M.J. L.; Nurmohamed, M.T.; van Eijk, I.C.; Verkleij, C.J.N.; Marx, P.F. Thrombin-activatable fibrinolysis inhibitor and its relation with inflammation in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2009, 68, 1232–1233. [Google Scholar] [CrossRef]
- Habets, K.L. L.; Trouw, L.A.; Levarht, E.W.N.; Korporaal, S.J.A.; Habets, P.A.M.; de Groot, P.; Huizinga, T.W.J.; Toes, R.E.M. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Arthritis Research & Therapy. 2015, 17. [Google Scholar]
- Zhou, Z.W.; Chen, H.M.; Ju, H.X.; Sun, M.Z.; Jin, H. Platelet indices in patients with chronic inflammatory arthritis: a systematic review and meta-analysis. Platelets. 2020, 31, 834–844. [Google Scholar] [CrossRef]
- Agca, R.; Hopman, L.; Laan, K.J.C.; van Halm, V.P.; Peters, M.J.L.; Smulders, Y.M.; Dekker, J.M.; Nijpels, G.; Stehouwer, C.D.A.; Voskuyl, A.E.; Boers, M.; Lems, W.F.; Nurmohamed, M.T. Cardiovascular Event Risk in Rheumatoid Arthritis Compared with Type 2 Diabetes: A 15-year Longitudinal Study. Journal of Rheumatology. 2020, 47, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Marzano, A.V.; Asero, R.; Tedeschi, A. Activation of blood coagulation in chronic urticaria: pathophysiological and clinical implications. Intern Emerg Med. 2010, 5, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Ganeshalingam, K.; Semb, A.G.; Szekanecz, Z.; Nurmohamed, M. Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology. 2014, 53, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Carbone, F.; Bonaventura, A.; Liberale, L.; Paolino, S.; Torre, F.; Dallegri, F.; Montecucco, F.; Cutolo, M. Atherosclerosis in Rheumatoid Arthritis: Promoters and Opponents. Clinical Reviews in Allergy and Immunology. 2020, 58, 1–14. [Google Scholar] [CrossRef]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Roger, V.L.; Fitz-Gibbon, P.D.; Therneau, T.M.; Gabriel, S.E. Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Annals of the Rheumatic Diseases. 2011, 70, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; Kastelein, J.J.P.; Cornel, J.H.; Pais, P.; Pella, D.; Genest, J.; Cifkova, R.; Lorenzatti, A.; Forster, T.; Kobalava, Z.; Vida-Simiti, L.; Flather, M.; Shimokawa, H.; Ogawa, H.; Dellborg, M.; Rossi, P.R.F.; Troquay, R.P.T.; Libby, P.; Glynn, R.J.; Grp, C.T. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. New England Journal of Medicine. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Goodson, N.J.; Brookhart, A.M.; Symmons, D.P.M.; Silman, A.J.; Solomon, D.H. Non-steroidal anti-inflammatory drug use does not appear to be associated with increased cardiovascular mortality in patients with inflammatory polyarthritis: results from a primary care based inception cohort of patients. Annals of the Rheumatic Diseases. 2009, 68, 367–372. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Gonzalez-Juanatey, C.; Martin, J. Rheumatoid arthritis: A disease associated with accelerated atherogenesis. Seminars in Arthritis and Rheumatism. 2005, 35, 8–17. [Google Scholar] [CrossRef]
- Kerola, A.M.; Kerola, T.; Kauppi, M.J.; Kautiainen, H.; Virta, L.J.; Puolakka, K.; Nieminen, T.V.M. Cardiovascular comorbidities antedating the diagnosis of rheumatoid arthritis. Annals of the Rheumatic Diseases. 2013, 72, 1826–1829. [Google Scholar] [CrossRef]
- Södergren, A.; Karp, K.; Boman, K.; Eriksson, C.; Lundström, E.; Smedby, T.; Söderlund, L.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Atherosclerosis in early rheumatoid arthritis: very early endothelial activation and rapid progression of intima media thickness. Arthritis Res Ther. 2010, 12(4), R158. [Google Scholar] [CrossRef]
- González-Gay, M.A.; González-Juanatey, C.; Miranda-Filloy, J.A.; García-Unzueta, M.T.; Llorca, J. Lack of association between flow-mediated endothelium-dependent vasodilatation and biomarkers of endothelial dysfunction in patients with severe rheumatoid arthritis. Rheumatol Int. 2012, 32(12), 4071–2. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.; Ambrosino, P.; Lupoli, R.; Di Minno, A.; Tasso, M.; Peluso, R.; Tremoli, E. Clinical assessment of endothelial function in patients with rheumatoid arthritis: A meta-analysis of literature studies. Eur J Intern Med. 2015, 26(10), 835–42. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Tasso, M.; Lupoli, R.; Di Minno, A.; Baldassarre, D.; Tremoli, E.; Di Minno, M.N. Non-invasive assessment of arterial stiffness in patients with rheumatoid arthritis: a systematic review and meta-analysis of literature studies. Ann Med. 2015, 47(6), 457–67. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.T.; Wasko, M.C.M.; Chung, C.P.; Szklo, M.; Blumenthal, R.S.; Kao, A.; Bokhari, S.; Zartoshti, A.; Stein, C.M.; Bathon, J.M. Exploring the Lipid Paradox Theory in Rheumatoid Arthritis: Associations of Low Circulating Low-Density Lipoprotein Concentration With Subclinical Coronary Atherosclerosis. Arthritis and Rheumatology. 2019, 71, 1426–1436. [Google Scholar] [CrossRef]
- Paccou, J.; Renard, C.; Liabeuf, S.; Kamel, S.; Fardellone, P.; Massy, Z.A.; Brazier, M.; Mentaverri, R. Coronary and Abdominal Aorta Calcification in Rheumatoid Arthritis: Relationships with Traditional Cardiovascular Risk Factors, Disease Characteristics, and Concomitant Treatments. Journal of Rheumatology. 2014, 41, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.; Henry, D. Cardiovascular risk and inhibition of cyclooxygenase - A systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase. Jama-Journal of the American Medical Association. 2006, 296, 1633–1644. [Google Scholar] [CrossRef]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. Bmj-British Medical Journal. 2006, 332, 1302–1305. [Google Scholar] [CrossRef]
- Roubille, C.; Richer, V.; Starnino, T.; McCourt, C.; McFarlane, A.; Fleming, P.; Siu, S.; Kraft, J.; Lynde, C.; Pope, J.; Gulliver, W.; Keeling, S.; Dutz, J.; Bessette, L.; Bissonnette, R.; Haraoui, B. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Annals of the Rheumatic Diseases. 2015, 74, 480–489. [Google Scholar] [CrossRef]
- Cabassi, A.; Tedeschi, S.; Perlini, S.; Verzicco, I.; Volpi, R.; Gonzi, G.; Del Canale, S. Non-steroidal anti-inflammatory drug effects on renal and cardiovascular function: from physiology to clinical practice. European Journal of Preventive Cardiology. 2020, 27, 850–867. [Google Scholar] [CrossRef]
- G. ; Capodanno, D.; Longo, G.; Capranzano, P.; Tamburino, C. Updates on NSAIDs in patients with and without coronary artery disease: pitfalls, interactions and cardiovascular outcomes. Expert Review of Cardiovascular Therapy. 2014, 12, 1185–1203. [Google Scholar] [CrossRef] [PubMed]
- Gasparyan, A.Y.; Ayvazyan, L.; Cocco, G.; Kitas, G.D. Adverse Cardiovascular Effects of Antirheumatic Drugs: Implications for Clinical Practice and Research. Current Pharmaceutical Design. 2012, 18, 1543–1555. [Google Scholar] [CrossRef]
- Szeto, C.C.; Sugano, K.; Wang, J.G.; Fujimoto, K.; Whittle, S.; Modi, G.K.; Chen, C.H.; Park, J.B.; Tam, L.S.; Vareesangthip, K.; Tsoi, K.K.F.; Chan, F.K.L. Non-steroidal anti-inflammatory drug (NSAID) therapy in patients with hypertension, cardiovascular, renal or gastrointestinal comorbidities: joint APAGE/APLAR/APSDE/APSH/APSN/PoA recommendations. Gut. 2020, 69, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.Y.; Du, X.P. Non-steroidal Anti-inflammatory Drugs and Hypertension. Cell Biochemistry and Biophysics. 2014, 69, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, B.; Aldington, S.; Weatherall, M.; Shirtcliffe, P.; Beasley, R. Risk of cardiovascular events and celecoxib: a systematic review and meta-analysis. Journal of the Royal Society of Medicine. 2006, 99, 132–140. [Google Scholar] [CrossRef]
- Helin-Salmivaara, A.; Virtanen, A.; Vesalainen, R.; Gronroos, J.M.; Klaukka, T.; Idanpaan-Heikkila, J.E.; Huupponen, R. NSAID use and the risk of hospitalization for first myocardial infarction in the general population: a nationwide case-control study from Finland. European Heart Journal. 2006, 27, 1657–1663. [Google Scholar] [CrossRef]
- Schneeweiss, S.; Solomon, D.H.; Wang, P.S.; Rassen, J.; Brookhart, M.A. Simultaneous assessment of short-term gastrointestinal benefits and cardiovascular risks of selective cyclooxygenase 2 inhibitors and nonselective nonsteroidal anti-inflammatory drugs - An instrumental variable analysis. Arthritis and Rheumatism. 2006, 54, 3390–3398. [Google Scholar] [CrossRef]
- Fabule, J.; Adebajo, A. Comparative evaluation of cardiovascular outcomes in patients with osteoarthritis and rheumatoid arthritis on recommended doses of nonsteroidal anti-inflammatory drugs. Therapeutic Advances in Musculoskeletal Disease. 2014, 6, 111–130. [Google Scholar] [CrossRef]
- del Rincon, I.; Battafarano, D.F.; Restrepo, J.F.; Erikson, J.M.; Escalante, A. Glucocorticoid Dose Thresholds Associated With All-Cause and Cardiovascular Mortality in Rheumatoid Arthritis. Arthritis and Rheumatology. 2014, 66, 264–272. [Google Scholar] [CrossRef]
- Soubrier, M.; Chamoux, N.B.; Tatar, Z.; Couderc, M.; Dubost, J.J.; Mathieu, S. Cardiovascular risk in rheumatoid arthritis. Joint Bone Spine, 2014, 81, 298–302. [Google Scholar] [CrossRef]
- Ravindran, V.; Rachapalli, S.; Choy, E.H. Safety of medium- to long-term glucocorticoid therapy in rheumatoid arthritis: a meta-analysis. Rheumatology. 2009, 48, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Ruyssen-Witrand, A.; Fautrel, B.; Saraux, A.; Le Loet, X.; Pham, T. Cardiovascular risk induced by low-dose corticosteroids in rheumatoid arthritis: A systematic literature review. Joint Bone Spine. 2011, 78, 23–30. [Google Scholar] [CrossRef]
- Agca, R.; Heslinga, S.C.; Rollefstad, S.; Heslinga, M.; McInnes, B.; Peters, M.J.L.; Kvien, T.K.; Dougados, M.; Radner, H.; Atzeni, F.; Primdahl, J.; Sodergren, A.; Jonsson, S.W.; van Rompay, J.; Zabalan, C.; Pedersen, T.R.; Jacobsson, L.; de Vlam, K.; Gonzalez-Gay, M.A.; Semb, A.G.; Kitas, G.D.; Smulders, Y.M.; Szekanecz, Z.; Sattar, N.; Symmons, D.P.M.; Nurmohamed, M.T. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Annals of the Rheumatic Diseases. 2017, 76, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S.; Bernatsky, S.; Hudson, M. Antirheumatic drug use and the risk of acute myocardial infarction. Arthritis and Rheumatism-Arthritis Care and Research. 2006, 55, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Rempenault, C.; Combe, B.; Barnetche, T.; Gaujoux-Viala, C.; Lukas, C.; Morel, J.; Hua, C. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: a systematic review and meta-analysis. Annals of the Rheumatic Diseases. 2018, 77, 98–103. J1. [Google Scholar] [CrossRef] [PubMed]
- Widdifield, J.; Abrahamowicz, M.; Paterson, J.M.; Huang, A.J.; Thorne, J.C.; Pope, J.E.; Kuriya, B.; Beauchamp, M.E.; Bernatsky, S. Associations Between Methotrexate Use and the Risk of Cardiovascular Events in Patients with Elderly-onset Rheumatoid Arthritis. Journal of Rheumatology. 2019, 46, 467–474. [Google Scholar] [CrossRef]
- Westlake, S.L.; Colebatch, A.N.; Baird, J.; Curzen, N.; Kiely, P.; Quinn, M.; Choy, E.; Ostor, A.J.K.; Edwards, C.J. Tumor necrosis factor antagonists and the risk of cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology. 2011, 50, 518–531. [Google Scholar] [CrossRef]
- Micha, R.; Imamura, F.; von Ballmoos, M.W.; Solomon, D.H.; Hernan, M.A.; Ridker, P.M.; Mozaffarian, D. Systematic Review and Meta-Analysis of Methotrexate Use and Risk of Cardiovascular Disease. American Journal of Cardiology. 2011, 108, 1362–1370. [Google Scholar] [CrossRef]
- Morris, S.J.; Wasko, M.C.M.; Antohe, J.L.; Sartorius, J.A.; Kirchner, H.L.; Dancea, S.; Bili, A. Hydroxychloroquine Use Associated With Improvement in Lipid Profiles in Rheumatoid Arthritis Patients. Arthritis Care and Research. 2011, 63, 530–534. [Google Scholar] [CrossRef]
- Li, H.Z.; Xu, X.H.; Lin, N.; Lu, H.D. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: a systematic review and meta-analysis. Annals of the Rheumatic Diseases. 2019, 78, E21–E21. [Google Scholar] [CrossRef]
- Charles-Schoeman, C.; Wang, X.Y.; Lee, Y.Y.; Shahbazian, A.; Navarro-Millan, I.; Yang, S.; Chen, L.; Cofield, S.S.; Moreland, L.W.; O’Dell, J.; Bathon, J.M.; Paulus, H.; Bridges, S.L.; Curtis, J.R. Association of Triple Therapy With Improvement in Cholesterol Profiles Over Two-Year Followup in the Treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis and Rheumatology. 2016, 68, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; J. Avorn; Katz, J.N.; Weinblatt, M.E.; Setoguchi, S.; Levin, R.; Schneeweiss, S. Immunosuppressive medications and hospitalization for cardiovascular events in patients with rheumatoid arthritis. Arthritis and Rheumatism. 2006, 54, 3790–3798. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewe, R.; Breedveld, F.C.; Buch, M.; Burmester, G.; Dougados, M.; Emery, P.; Gaujoux-Viala, C.; Gossec, L.; Nam, J.; Ramiro, S.; Winthrop, K.; de Wit, M.; Aletaha, D.; Betteridge, N.; Bijlsma, J.W.J.; Boers, M.; Buttgereit, F.; Combe, B.; Cutolo, M.; Damjanov, N.; Hazes, J.M.W.; Kouloumas, M.; Kvien, T.K.; Mariette, X.; Pavelka, K.; van Riel, P.; Rubbert-Roth, A.; Scholte-Voshaar, M.; Scott, D.L.; Sokka-Isler, T.; Wong, J.B.; van der Heijde, D. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Annals of the Rheumatic Diseases. 2014, 73, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Curtis, J.R.; Saag, K.G.; Lii, J.; Chen, L.; Harrold, L.R.; Herrinton, L.J.; Graham, D.J.; Kowal, M.K.; Kuriya, B.; Liu, L.; Griffin, M.R.; Lewis, J.D.; Rassen, J.A. Cardiovascular Risk in Rheumatoid Arthritis: Comparing TNF-alpha Blockade with Nonbiologic DMARDs. American Journal of Medicine. 2013, 126. [Google Scholar] [CrossRef]
- Toussirot, E. Effects of TNF alpha inhibitors on adiposity and other cardiovascular risk factors: implications for the cardiovascular prognosis in patients with rheumatoid arthritis. Expert Opinion on Drug Safety. 2015, 14, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Barnabe, C.; Martin, B.J.; Ghali, W.A. Systematic Review and Meta-Analysis: Anti-Tumor Necrosis Factor alpha Therapy and Cardiovascular Events in Rheumatoid Arthritis. Arthritis Care and Research. 2011, 63, 522–529. [Google Scholar] [CrossRef]
- Jacobsson, L.T. H.; Turesson, C.; Gulfe, A.; Kapetanovic, M.C.; Petersson, I.F.; Saxne, T.; Geborek, P. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. Journal of Rheumatology. 2005, 32, 1213–1218. [Google Scholar]
- Ljung, L.; Rantapaa-Dahlqvist, S.; Jacobsson, L.T.H.; Askling, J.; Study, G.A. Response to biological treatment and subsequent risk of coronary events in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2016, 75, 2087–2094. [Google Scholar] [CrossRef]
- Karpouzas, G.A.; Ormseth, S.R.; Hernandez, E.; Budoff, M.J. Impact of Cumulative Inflammation, Cardiac Risk Factors, and Medication Exposure on Coronary Atherosclerosis Progression in Rheumatoid Arthritis. Arthritis and Rheumatology. 2020, 72, 400–408. [Google Scholar] [CrossRef]
- Singh, S.; Fumery, M.; Singh, A.G.; Singh, N.; Prokop, L.J.; Dulai, P.S.; Sandborn, W.J.; Curtis, J.R. Comparative Risk of Cardiovascular Events With Biologic and Synthetic Disease-Modifying Antirheumatic Drugs in Patients With Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Arthritis Care and Research. 2020, 72, 561–576. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; Investigators, A. Randomized, double-blind, placebo-controlled, trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure - Results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003, 107, 3133–3140. [Google Scholar] [PubMed]
- Leporini, C.; Russo, E.; D’Angelo, S.; Arturi, F.; Tripepi, G.; Peluso, R.; Grembiale, R.D.; Olivieri, I.; De Sarro, G.; Ursini, F. Insulin-Sensiting Effects of Tumor Necrosis Factor Alpha Inhibitors in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Reviews on Recent Clinical Trials. 2018, 13, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Castagne, B.; Viprey, M.; Martin, J.; Schott, A.M.; Cucherat, M.; Soubrier, M. Cardiovascular safety of tocilizumab: A systematic review and network meta-analysis. Plos One. 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Divonne, M.D.; Gottenberg, J.E.; Salliot, C. Safety of biologic DMARDs in RA patients in real life: A systematic literature review and meta-analyses of biologic registers. Joint Bone Spine. 2017, 84, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.T.; Tsoi, M.F.; Cheung, B.M.Y. Effect of TNF inhibitors on subclinical atherosclerosis in patients with rheumatoid arthritis: A meta-analysis. Annals of the Rheumatic Diseases. 2015, 74, 685–685. [Google Scholar] [CrossRef]
- Daien, C.I.; Duny, Y.; Barnetche, T.; Daures, J.P.; Combe, B.; Morel, J. Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: a systematic review with meta-analysis. Annals of the Rheumatic Diseases. 2012, 71, 862–868. [Google Scholar] [CrossRef]
- Zhao, Q.W.; Hong, D.S.; Zhang, Y.; Sang, Y.L.; Yang, Z.H.; Zhang, X.G. Association Between Anti-TNF Therapy for Rheumatoid Arthritis and Hypertension A Meta-Analysis of Randomized Controlled Trials. Medicine. 2015, 94. [Google Scholar]
- Kim, S.K.; Kwak, S.G.; Choe, J.Y. Association between biologic disease modifying antirheumatic drugs and incident hypertension in patients with rheumatoid arthritis Results from prospective nationwide KOBIO Registry. Medicine. 2020, 99, 9. [Google Scholar]
- Desai, R.J.; Solomon, D.H.; Schneeweiss, S.; Danaei, G.; Liao, K.P.; Kim, S.C. Tumor Necrosis Factor- Inhibitor Use and the Risk of Incident Hypertension in Patients with Rheumatoid Arthritis. Epidemiology. 2016, 27, 414–422. [Google Scholar] [CrossRef]
- Jin, Y.Z.; Kang, E.H.; Brill, G.; Desai, R.J.; Kim, S.C. Cardiovascular (CV) Risk after Initiation of Abatacept versus TNF Inhibitors in Rheumatoid Arthritis Patients with and without Baseline CV Disease. Journal of Rheumatology. 2018, 45, 1240–1248. [Google Scholar] [CrossRef]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; van Vollenhoven, R.F. Integrated safety in tocilizumab clinical trials. Arthritis Research and Therapy. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Lin, Y.; John, G.S.; van der Heijde, D.; Qiu, C.F.; Gomez-Reino, J.J.; Maldonado-Cocco, J.A.; Stanislav, M.; Seriolo, B.; Burmester, G.R. Long-term safety with Sarilumab plus conventional synthetic disease modifying antirheumatic drugs and Sarilumab monotherapy in rheumatoid arthritis: An integrated analysis with 9000 patient-years of follow up. Annals of the Rheumatic Diseases. 2019, 78, 1130–1131. [Google Scholar]
- Ikonomidis, I.; Lekakis, J.P.; Nikolaou, M.; Paraskevaidis, I.; Andreadou, I.; Kaplanoglou, T.; Katsimbri, P.; Skarantavos, G.; Soucacos, P.N.; Kremastinos, D.T. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008, 117, 2662–2669. [Google Scholar] [CrossRef]
- van Vollenhoven, R.F.; Emery, P.; Bingham, C.O.; Keystone, E.C.; Fleischmann, R.M.; Furst, D.E.; Tyson, N.; Collinson, N.; Lehane, P.B. Long-term safety of rituximab in rheumatoid arthritis: 9.5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Annals of the Rheumatic Diseases. 2013, 72, 1496–1502. [Google Scholar] [CrossRef]
- Day, A.L.; Singh, J.A. Cardiovascular Disease Risk in Older Adults and Elderly Patients with Rheumatoid Arthritis: What Role Can Disease-Modifying Antirheumatic Drugs Play in Cardiovascular Risk Reduction? Drugs and Aging. 2019, 36, 493–510. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Thompson, L.; Giles, J.T.; Bathon, J.M.; Salmon, J.E.; Beaulieu, A.D.; Codding, C.E.; Carlson, T.H.; Delles, C.; Lee, J.S.; Sattar, N. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Annals of the Rheumatic Diseases. 2015, 74, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Suzuki, M.; Nakamura, H.; Toyoizumi, S.; Zwillich, S.H.; Tofacitinib, I. Study Phase II Study of Tofacitinib (CP-690,550) Combined With Methotrexate in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate. Arthritis Care and Research. 2011, 63, 1150–1158. [Google Scholar] [CrossRef]
- Taylor, P.C.; Weinblatt, M.E.; Burmester, G.R.; Rooney, T.P.; Witt, S.; Walls, C.D.; Issa, M.; Salinas, C.A.; Saifan, C.; Zhang, X.; Cardoso, A.; Gonzalez-Gay, M.A.; Takeuchi, T. Cardiovascular Safety During Treatment With Baricitinib in Rheumatoid Arthritis. Arthritis and Rheumatology. 2019, 71, 1042–1055. [Google Scholar] [CrossRef]
- Souto, A.; Salgado, E.; Maneiro, J.R.; Mera, A.; Carmona, L.; Gómez-Reino, J.J. Lipid profile changes in patients with chronic inflammatory arthritis treated with biologic agents and tofacitinib in randomized clinical trials: a systematic review and meta-analysis. Arthritis Rheumatol. 2015, Jan;67(1), 117-27.
- Zhang, J.; Xie, F.; Yun, H.; Chen, L.; Muntner, P.; Levitan, E.B.; Safford, M.M.; Kent, S.T.; Osterman, M.T.; Lewis, J.D.; Saag, K.; Singh, J.A.; Curtis, J.R. Comparative effects of biologics on cardiovascular risk among older patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 2016, 75, 1813–1818. [Google Scholar] [CrossRef]
- Myasoedova, E.; Karmacharya, P.; Garcia, A.D.; Davis, J.; Murad, M.H.; Crowson, C. What Is the Effect of Statins on the Risk of Rheumatoid Arthritis? Results of a Systematic Review and Meta-Analysis. Arthritis and Rheumatology. 2019, 71. [Google Scholar]
- Soulaidopoulos, S.; Nikiphorou, E.; Dimitroulas, T.; Kitas, G.D. The Role of Statins in Disease Modification and Cardiovascular Risk in Rheumatoid Arthritis. Frontiers in Medicine. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Danninger, K.; Hoppe, U.C.; Pieringer, H. Do statins reduce the cardiovascular risk in patients with rheumatoid arthritis? International Journal of Rheumatic Diseases. 2014, 17, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Yin, Y.-F.; Zhao, L.-D.; Wang, L.; Zheng, W.-J.; Chen, H.; Wu, Q.-J.; Tang, F.-L.; Zhang, F.-C.; Shan, G.; Zhang, X. Effect of 3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Inhibitor on Disease Activity in Patients With Rheumatoid Arthritis A Meta-Analysis. Medicine. 2015, 94. [Google Scholar]
Figure 1.
Article selection process for review - schematics.
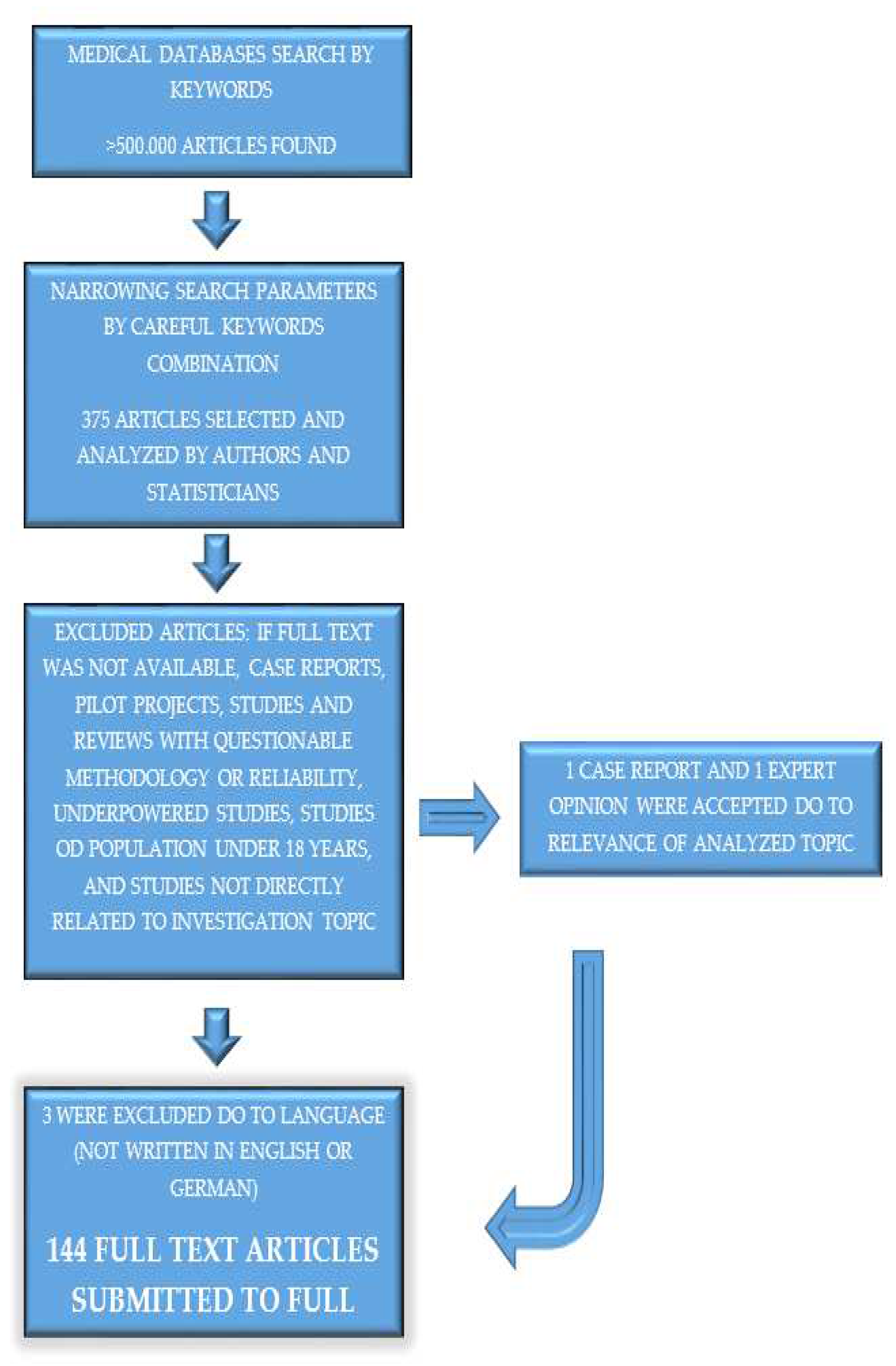
Figure 2.
proinflammatory cytokines interleukin 1 (IL-1), tumour necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) are interconnected in signalling pathways and they are targets for drugs that are used to treat rheumatic diseases and have gained interest as potential drugs for secondary prevention of ASCVD in RA.
Figure 2.
proinflammatory cytokines interleukin 1 (IL-1), tumour necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) are interconnected in signalling pathways and they are targets for drugs that are used to treat rheumatic diseases and have gained interest as potential drugs for secondary prevention of ASCVD in RA.
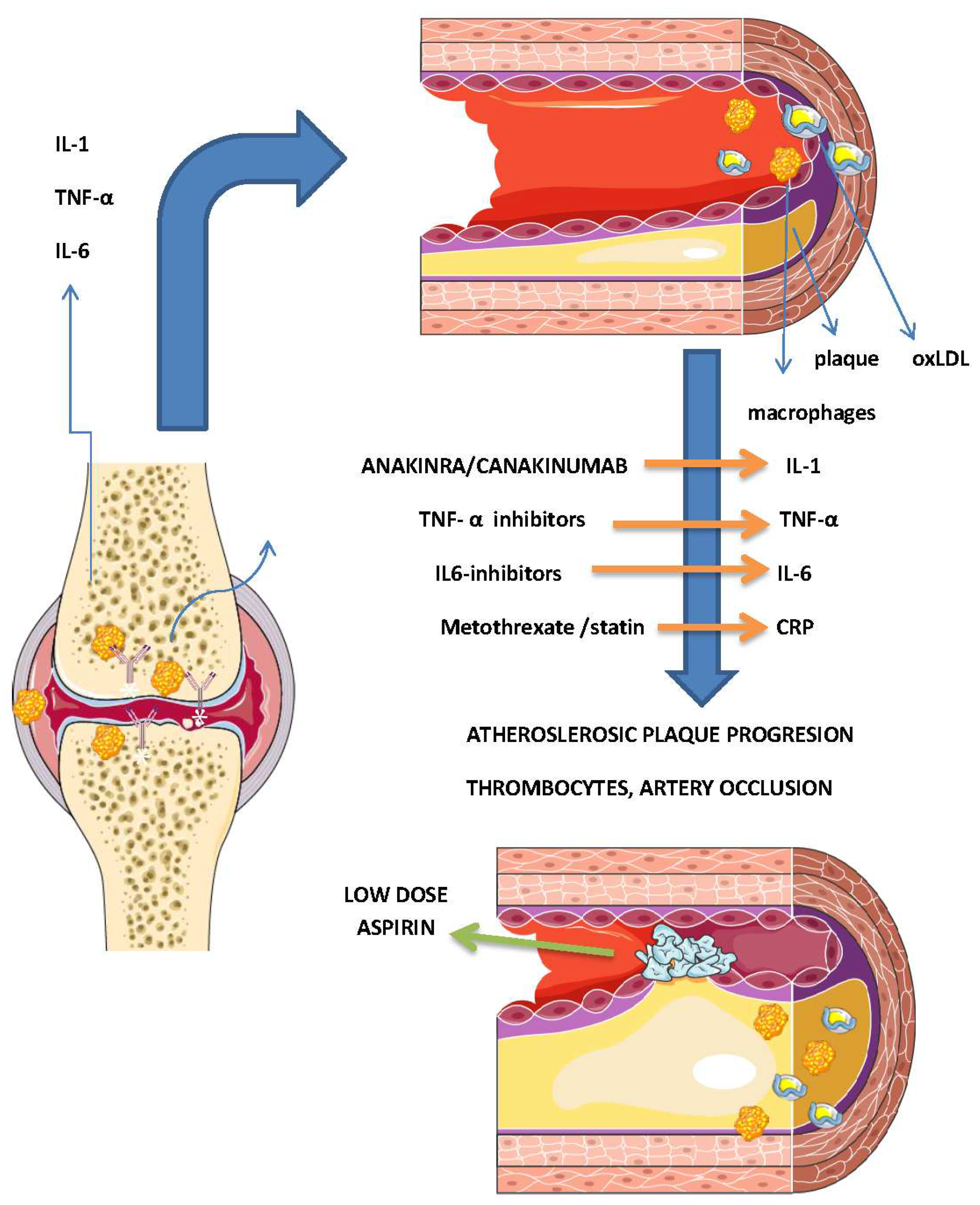
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



