Submitted:
27 July 2023
Posted:
28 July 2023
You are already at the latest version
Abstract
Mitochondria are highly dynamic organelles that constantly undergo fusion and fission events to maintain their shape, distribution, and cellular function. Mitofusin 1 and 2 proteins are two dynamin-like GTPases involved in the fusion of outer mitochondrial membranes (OMM). Mitofusins are anchored to the OMM through their transmembrane domain and possess two heptad repeat domains (HR1 and HR2) in addition to their N-terminal GTPase domain. The HR1 domain was found to induce fusion via its amphipathic helix, which interacts with the lipid bilayer structure. The lipid composition of mitochondrial membranes can also impact mitochondrial fusion. However, the precise mode of action of lipids in mitochondrial fusion is not fully understood. In this study, we have examined the role of the mitochondrial lipids phosphatidylethanolamine (PE), cardiolipin (CL) and phosphatidic acid (PA) in membrane fusion induced by the HR1 domain, both in the presence and absence of divalent cations (Ca2+ or Mg2+). Our results show that PE, as well as PA in the presence of Ca2+, effectively stimulate HR1-mediated fusion, while CL has a slight inhibitory effect. By considering the biophysical properties of these lipids in the absence or presence of divalent cations, we infer that the interplay between divalent cations and specific cone-shaped lipids creates regions with packing defects in the membrane, which provides a favorable environment for the amphipathic helix of HR1 to bind to the membrane and initiate fusion.
Keywords:
mitochondria
; membrane
; fusion
; Mifofusin
; amphipathic helix
; divalent cations
; lipid packing defects
1. Introduction
Mitochondria possess a double-membrane structure, consisting of the inner mitochondrial membrane (IMM) that encloses the matrix, and the outer mitochondrial membrane (OMM) that separates the intermembrane space from the cell cytoplasm. These double-membrane organelles form a dynamic network that constantly changes shape through fusion and fission processes. Maintaining a delicate balance between these two processes, collectively known as mitochondrial dynamics, is crucial for mitochondrial function in cellular energy generation and overall cell health [1]. In mammalian cells, the elongation of mitochondria through fusion is orchestrated by the spatio-temporal coordination of two key transmembrane proteins from the dynamin superfamily: Mitofusins (Mfn1 and Mfn2) and OPA1, which mediate the fusion of OMM and IMM, respectively [2]. Mitofusins are composed of an N-terminal GTPase domain, followed by a first heptad repeat domain (HR1), a transmembrane domain (TMD), and a second C-terminal heptad repeat domain (HR2). These different domains are essential for Mitofusin function but their exact mode of action in mitochondrial fusion remains not fully understood [3,4,5,6].
Based on recent structural studies [7,8,9,10], the current working hypothesis for the molecular mechanisms of OMM fusion involves a cis- (i.e., within the same membrane) and/or trans- (i.e., across two different membranes) oligomerization of Mitofusin proteins. This initial event would lead to a long-distance (20–30 nm) membrane docking step [8,11]. Through GTP hydrolysis, membrane-bridging trans-Mitofusin complexes would then transit from an open to a closed conformation bringing OMM in close apposition. The HR2 domain could stabilize this short-distance (5–10 nm) membrane docking step by forming a homodimeric antiparallel coiled-coil complex [5,8,11].
Bringing membranes in close apposition is the first step in membrane fusion, but it is not enough on its own. For fusion to occur, the membrane structure must also be destabilized to facilitate the merging of lipid bilayers. Two recent studies have emphasized the crucial role of amphipathic helices within the Mitofusin sequence in triggering OMM fusion [12,13]. One study found that the HR1 domain mediated liposome fusion in vitro and was essential for mitochondrial fusion in situ [12]. The fusion activity was attributed to a conserved amphipathic helix located at the C-terminus of HR1, suggesting that HR1 induces fusion by interacting with and perturbing the lipid bilayer structure. A similar fusion mechanism has been described for the C-terminal amphipathic tail of Atlastin, another dynamin-like transmembrane protein involved in endoplasmic reticulum (ER) membrane fusion [14,15,16]. Interestingly, another study demonstrated that when the TMD of Mitofusin was replaced with that of Atlastin, the resulting chimeric protein localized to ER membranes and was capable of mediating ER fusion [13]. Furthermore, an amphipathic helix identified between the TMD and the HR2 domain of Mitofusin could effectively replace the C-terminal amphipathic tail of Atlastin in both in vitro liposome fusion and in situ ER fusion.
The lipid composition of mitochondrial membranes also plays a crucial role in facilitating mitochondrial fusion. The successive stages leading to membrane fusion involve the formation of energy-demanding intermediate membrane structures with high curvature [17]. Lipids that can relieve this energy stress by inducing favorable membrane bending therefore facilitate fusion. Mitochondrial membranes contain specific lipids such as phosphatidylethanolamine (PE) and phosphatidic acid (PA), both of which possess a cone-shaped structure with a small headgroup area compared to the cross-sectional area of their hydrophobic chains. As a result, they can induce negative membrane curvatures when present in the outer leaflet of lipid bilayers. Additionally, there is a unique lipid found in mitochondria known as cardiolipin (CL). When bound to divalent cations like Ca2+ or Mg2+, CL can also adopt a conical shape. These three lipids thus have the potential to facilitate mitochondrial fusion events.
Indeed, a previous study found that high concentrations of CL were required for in vitro liposome fusion mediated by OPA1 [18]. CL also allows the generation of PA at the OMM through its cleavage by the mitochondria-localized phospholipase D (MitoPLD) enzyme. Depletion of MitoPLD in mammalian cells through RNA interference (RNAi) led to reduced mitochondrial fusion, suggesting that PA production at the OMM is important for fusion. Similarly, reduction of PE levels in mammalian cells by RNAi silencing of the enzyme phosphatidylserine decarboxylase (Pisd), which converts phosphatidylserine (PS) into PE, resulted in mitochondrial fragmentation, indicating inhibition of mitochondrial fusion [19]. Yeast cells lacking Psd1, the homolog of mammalian Pisd, also exhibited impaired mitochondrial fusion [20]. Additionally, yeast cells which lacked both Psd1 and the CL synthase Crd1 displayed an even more fragmented mitochondrial network [21]. These studies demonstrate the critical importance of PE, CL and PA lipids in promoting efficient mitochondrial fusion. However, the specific molecular mechanisms by which these lipids facilitate mitochondrial fusion events remain to be established.
In this study, we investigate the role of PE, CL and PA lipids in membrane fusion mediated by the HR1 domain of Mitofusin. We also examine the interplay between these lipids and the divalent cations Ca2+ and Mg2+ in facilitating membrane perturbation and fusion via the amphipathic helix of HR1.
2. Materials and Methods
2.1. Chemicals
N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES, OmniPur grade), potassium hydroxide solution 47% (KOH 47%, EMSURE grade for analysis), potassium chloride (KCl, OmniPur grade), calcium chloride dihydrate (CaCl2, OmniPur grade), magnesium chloride hexahydrate (MgCl2, OmniPur grade), glycerol (Molecular Biology grade), tris(2-carboxyethyl)phosphine hydrochloride (TCEP, ≥98% GC), n-octyl-β-D-glucopyranoside (β-OG, ≥98% GC), n-dodecyl β-D-maltoside (DDM, ULTROL grade) and sodium dithionite (Analytical grade) were purchased from Merck. 5-(N-2,3-dihydroxypropylacetamido)-2,4,6-tri-iodo-N,N'-bis-(2,3-dihydroxypropyl)isophthalamide (Nycodenz, ≥98%) was purchased from Proteogenix.
All aqueous solutions were prepared using 18.2 MΩ ultra-pure water and filtered with sterile 0.22 µm polyethersulfone (PES) membranes.
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (PC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (PE), L-α-phosphatidylinositol (Liver, Bovine) (sodium salt) (PI), 1',3'-bis[1,2-dioleoyl-sn-glycero-3-phospho]-glycerol (sodium salt) (CL), 1,2-dioleoyl-sn-glycero-3-phosphate (sodium salt) (PA), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD PS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Rho PE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide] (sodium salt) (MAL), and 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) (NTA-Ni) were purchased from Avanti Polar Lipids as chloroform solutions.
2.2. Peptides
The heptad repeat domain 1 (HR1) of human Mitofusin 1 (Mfn1) used in this study was produced by Fmoc solid-phase peptide synthesis and one-step purification by reverse-phase HPLC (Proteogenix, purity > 95%). The produced sequence was Mfn1-HR1 (T350-L420; C411S, C418S; with a C-terminal Cys or His6 tag). Lyophilized samples (1 mg aliquots) were put on ice and solubilized by slowing adding 1 mL of ice-cold buffer H (25 mM HEPES/KOH, pH 7.4; 150 mM KCl; 10% (v/v) Glycerol) containing 0.25 mM TCEP in the case of Cys-tagged HR1. Peptide solutions were vortexed for 2 min at room temperature followed by 20 sec of sonication on ice to remove any potential aggregates (2 cycles of 10 sec on, at 10 W, and 10 sec off, using the ultrasonic homogenizer UP200St from Hielscher equipped with a 2 mm sonotrode). Samples were snap-frozen in liquid nitrogen and stored at -80 °C as aliquots of 50 µL.
2.3. Liposomes
Liposomes were prepared by the detergent-assisted method [22]. 1.2 µmol of the appropriate lipid mixtures in chloroform were dried in glass tubes for 10 min under a gentle stream of argon, followed by 2 hours under vacuum. The dried lipid films were resuspended in 400 µL of buffer H containing 1% (w/v) β-OG by vigorously vortexing for 30 min at room temperature. The detergent concentration was next reduced below the critical micellar concentration, 0.33% (w/v), by slowly adding 800 µL of buffer H, and then removed by overnight flow dialysis against 4 L of buffer H. Liposomes (final lipid concentration of 1 mM) were stored on ice and protected from light for up to 2–3 weeks.
2.4. Multi-angle dynamic light scattering (MADLS)
5 µL of liposomes at 1 mM and 95 µL of buffer H were mixed in a low volume quartz batch cuvette (ZEN2112, Malvern Panalytical) and their size distribution was determined at 37 °C with the Zetasizer Ultra Red instrument (Malvern Panalytical) using the MADLS mode, which measures the correlation function in three scattering directions: back scatter (173 degrees), side scatter (90 degrees) and forward scatter (13 degrees).
2.5. Cryogenic transmission electron microscopy (cryo-TEM)
A 5 μL drop of the initial sample solution was deposited on “quantifoil” carbon membrane grids (Quantifoil Micro Tools GmbH, Germany). The excess of liquid on the grid was absorbed with a filter paper and the grid was quench-frozen quickly in liquid ethane to form a thin vitreous ice film using an homemade mechanical cryo plonger. Once placed in a Gatan 626 cryo-holder cooled with liquid nitrogen, the samples were transferred in the microscope and observed at low temperature (-180 °C). Cryo-TEM images were recorded on ultrascan 1000, 2k x 2k pixels CCD camera (Gatan, USA), using a LaB6 JEOL JEM2100 (JEOL, Japan) cryo-microscope operating at 200 kV with a JEOL low dose system (Minimum Dose System, MDS, JEOL, Japan) to protect the thin ice film from any irradiation before imaging and reduce the irradiation during the image capture.
2.6. Liposome co-flotation assay
To assess membrane binding of HR1, 100 µL of HR1 at 50 µM and 100 µL of liposomes at 1 mM were incubated together for 1 hour at 37 °C under intermittent gentle mixing (1 min at 300 rpm every 9 min). 50 µL was taken out to serve as input control (unfloated fraction) and the remaining 150 µL was mixed with 150 µL of Nycodenz 80% (w/v) in buffer H. The mixture was transferred to a centrifuge tube (0.8 mL, Open-Top Thinwall Ultra-Clear Tube, 5 x 41 mm, Beckman Coulter) and overlaid with 250 μL of Nycodenz 30% (w/v) in buffer H followed by 50 μL of buffer H alone. The resulting gradient was centrifuged at 192,000 g for 4 hours at 4 °C in a SW 55 Ti Swinging-Bucket rotor (Beckman Coulter) and 37.5 μL of liposomes was collected from the top layer (floated fraction). 12 µL of unfloated or floated fraction was mixed with 4 µL of sample buffer (NuPAGE LDS Sample Buffer 4X, Invitrogen) and separated by electrophoresis on a polyacrylamide gel (NuPAGE 4–12%, Bis-Tris, 1 mm, Mini Protein Gel, Invitrogen) stained with Coomassie G-250 (PageBlue Protein Staining Solution, Thermo Scientific). Images were acquired using a ChemiDoc Touch Imaging System (Bio-Rad). The band intensities in the unfloated and floated fractions were measured with the software ImageJ.
2.7. FRET-based lipid mixing assay
For each condition to be tested, two sub-populations of liposomes with the same lipid composition were generated, except that one sub-population, referred to as the fluorescent donor liposomes, was labeled with the Fluorescence Resonance Energy Transfer (FRET) pair of fluorescent lipids NBD PS and Rho PE. These fluorescent lipids were added at a concentration of 1.5 mol% each at the expense of PC lipids. 27 µL of non-fluorescent acceptor liposomes at 1 mM and 21 µL of buffer H (or 19 µL of buffer H and 2 µL of CaCl2 or MgCl2 at 30 mM in buffer H for experiments performed in the presence of divalent cations) were added to a flat bottom 96-well white polystyrene plate (Thermo Scientific) and pre-warmed at 37 °C for 7 min. 6 µL of fluorescent donor liposomes at 500 µM were carefully added to one side of the well. 6 µL of HR1 at 250 µM were added to another side of the well. The fusion reaction was initiated by shaking the plate in order to mix the three different solutions. Lipid mixing was measured by following fluorescence dequenching of the NBD probes from the donor liposomes resulting from their dilution into the acceptor liposomes. The NBD fluorescence was monitored at 1-min intervals for 90 min (excitation at 460 nm, emission at 535 nm, cutoff at 530 nm) by the SpectraMax M5 microplate reader (Molecular Devices) equilibrated to 37 °C. After 90 min, 10 µL of 2.5% (w/v) DDM was added to completely dissolve the liposomes and thus measure the NBD fluorescence at infinite dilution, Fmax(NBD). The data were then normalized using the following equation that gives the percentage of NBD fluorescence increase at time t, %F(NBD, t):
where Fmin(NBD) is the lowest NBD fluorescence value from all time points.
%F(NBD, t) = [F(NBD, t) - Fmin(NBD)] / [Fmax(NBD) - Fmin(NBD)]
2.8. Sodium dithionite assay
To determine the proportion of hemifused versus fused liposomes, we employed a fluorescence quenching method using sodium dithionite. 5 µL of freshly prepared sodium dithionite solution at 100 mM in buffer H was incubated with 33.3 µL of fluorescent donor liposomes at 1 mM for 15 min at 37 °C. 28.3 µL of buffer H was then added to the mixture to dilute the liposomes to a final concentration of 500 µM. The FRET-based lipid mixing assay was then performed as described above. As sodium dithionite was shown to completely lose its activity after 10 min at 37 °C, this ensures that the fluorescence signal from NBD lipids of the inner leaflets remains unquenched, even when DDM is added to solubilize the liposomes.
The percentage of liposomes that underwent hemifusion at time t, H(t), is given by the equation:
where FT and FI are respectively the normalized fluorescence dequenching signals without and with prior sodium dithionite treatment (total lipid mixing and inner monolayer lipid mixing, respectively), and α is the proportion of lipids residing in the inner monolayer of liposomes.
H(t) = 100 × [FT (t) - FI (t)] / [FT (t) - αFI(t)]
3. Results
3.1. Exploring HR1-mediated fusion with distinct lipid anchors: the influence of phosphatidylethanolamine
To investigate the mechanisms and efficiency of HR1-mediated fusion, we reconstituted HR1 into phosphatidylcholine (PC) liposomes using a lipid-anchorage strategy, and evaluated its fusion activity through a FRET-based lipid mixing assay [22]. We first chose the maleimide lipid-anchorage method [23] (Figure 1a). HR1 was modified to contain a single terminal Cys residue at its C-terminus, which allowed for its specific coupling to liposomes containing 18:1 MPB PE (MAL lipids). This coupling strategy ensures that the orientation of HR1 on the liposomes is consistent with its orientation on mitochondrial membranes. HR1 was introduced at the start of the lipid mixing assay (t=0), and its capacity to mediate liposome fusion was monitored over a duration of 90 minutes. Using this approach, we observed that HR1 induced robust lipid mixing (Figure 1b,c), in agreement with our previous work [12]. In our earlier study, we also noted fusion events between liposomes bearing HR1 and protein-free liposomes, suggesting that HR1 triggered fusion by membrane destabilization. Through detailed bioinformatics analysis of the HR1 sequence, we identified a conserved amphipathic helix at its C-terminal end with the potential to bind and perturb membrane structures. This finding was further supported by circular dichroism experiments, demonstrating an increase in α-helical structure of HR1 upon interaction with liposome membranes. In this new study, we sought to explore the potential impact of HR1 on membrane structural integrity by employing cryo-electron microscopy. We found that after incubation with HR1, many liposomes – appearing to have undergone fusion based on their larger size – exhibited disrupted or disappearing membrane structures, suggesting bilayer structure perturbation by HR1 (Figure 1d).
Specific lipids can also have a significant impact on membrane structure and biophysical properties. The inner and outer mitochondrial membranes are enriched with a high proportion of the cone-shaped non-bilayer forming phosphatidylethanolamine (PE) lipid. In fact, the OMM contains approximately 30 mol% of PE lipids. We thus aimed to explore the effect of PE on HR1-mediated fusion. Surprisingly, when 30 mol% PE was introduced to MAL-containing liposomes, HR1-mediated liposome fusion was completely abolished (Figure 1b,c). Through the analysis of HR1 density on the surface of the liposomes using a liposome co-floatation assay, we discovered that HR1 was unable to bind to MAL-containing liposomes in the presence of PE (Figure 2a,b). This lack of binding can be attributed to the interaction between the amine group of PE and the Maleimide group [24], resulting in the quenching of Maleimide and preventing its interaction with the Cys residue at the C-terminus of HR1.
To overcome this limitation, an alternative lipid-anchorage strategy was employed. HR1 was modified to include a C-terminal His6 tag, enabling its chemical coupling to liposomes functionalized with 18:1 DGS-NTA(Ni) (NTA-Ni lipids) (Figure 1a). In this system, HR1 retained the ability to induce liposome fusion, although to a lesser extent compared to the MAL lipid system (Figures 1b and 1c). This decrease in fusion efficiency can be attributed to the lower reactivity of the NTA-Ni group compared to the MAL group, resulting in a lower surface density of HR1 molecules on the liposomes (Figure 2a,b). The surface density of fusion proteins is, in fact, known to be critical for fusion efficiency [25]. Interestingly, the inclusion of 30 mol% PE in NTA-Ni-containing liposomes resulted in a more than 2-fold increase in HR1-mediated fusion (Figure 1b,c). Importantly, this effect was not due to a higher density of HR1 on the liposome surface in the presence of PE (Figure 2a,b). Since high membrane curvature is known to activate fusion [26,27,28], we also used multi-angle dynamic light scattering to check whether the presence of PE in the liposome membrane affected their size. We observed that the size distribution of liposomes remained unchanged in the presence of PE (Figure S2), which rules out the possibility that fusion was activated by liposomes with high curvature.
In the remainder of the manuscript, we exclusively used PC liposomes functionalized with NTA-Ni lipids as our model system. Specific lipids were added at the expense of PC to modify the liposome composition and examine the role of these lipids in HR1-mediated membrane fusion.
3.2. Cardiolipin inhibits HR1-mediated fusion
Cardiolipin (CL) is an exclusive component of mitochondrial membranes within cells. It is typically found at an average concentration of 5 mol% in the OMM [29]. However, its local concentration can reach up to 20 mol% at sites of contact between outer and inner mitochondrial membranes [29], where fusion might take place [30]. Therefore, to investigate the impact of CL on HR1-mediated liposome fusion, we studied the effects of two concentrations of CL: 5 and 20 mol%. Surprisingly, the addition of CL at either concentration resulted in a slight, concentration-dependent inhibition of HR1-mediated liposome fusion (Figure 3a,b). This was unexpected, as CL is thought to be important for mitochondrial fusion [31]. Since HR1 has a pKa of 6.1, it carries a net negative charge in our working buffer at pH 7.4. Considering that CL is also negatively charged, we investigated whether the fusion inhibition was due to inefficient coupling of HR1 to the liposome surface caused by electrostatic repulsion. The surface density of HR1 on CL-containing liposomes was actually slightly higher than that observed on PC liposomes (Figure S3), ruling out the absence of HR1 on liposome membranes as the reason for the reduction in fusion.
Previous studies have demonstrated that both pure CL liposomes and liposomes composed of an equimolar mixture of PC and CL lipids can undergo fusion when exposed to high concentrations (9 mM or higher) of divalent Ca2+ or Mg2+ ions [32,33]. Here, we aimed to investigate the influence of these cations, when added at a physiological concentration of 1 mM, on HR1-mediated liposome fusion. We found that the addition of 1 mM Ca2+ or Mg2+ ions resulted in a 30–40% increase in HR1-mediated fusion of PC liposomes (Figure 3a,b). In contrast, these ions had no discernible effect on HR1-mediated fusion of liposomes containing either 5 or 20 mol% CL (Figure 3a,b). Of note, an increase of 30–40% in HR1-mediated fusion, similar to that obtained with PC liposomes, was also observed for liposomes containing 30 mol% PE in the presence of 1 mM Ca2+ or Mg2+ ions (Figure S4).
3.3. Phosphatidic acid enhances HR1-mediated fusion in the presence of calcium
Phosphatidic acid (PA) is an important lipid of mitochondria that is present at about 2 mol% on average on mitochondrial membranes [34,35]. PA is primarily synthesized in the ER and transported from the ER to the OMM via contact sites between these two organelles. Alternatively, PA can be generated on the OMM through cleavage of CL by the MitoPLD enzyme. Increase of PA level at the surface of mitochondria by the action of MitoPLD was found to promote close apposition of mitochondrial membranes, which is a crucial step in the fusion process [36]. As MitoPLD interacts directly with Mitofusin, it could create localized regions of high PA concentrations around fusion sites, similar to what has been observed around fission sites previously [37]. Considering that the specific concentration of PA at fusion sites is unknown, we chose to investigate the impact of incorporating 10 or 30 mol% of PA into liposome membranes on HR1-mediated fusion. The selection of 30 mol% PA, despite seemingly high, allowed us to compare the effects of the two cone-shaped lipids, PA and PE, when used at the same concentration on HR1-mediated fusion. As in the case of CL, the addition of PA at either concentration led to a slight, concentration-dependent reduction in HR1-mediated liposome fusion (Figure 4a,b).
In previous works, it was found that liposomes composed of complex lipid compositions, including 5 mol% PA, exhibited protein-free fusion in the presence of high concentrations of Ca2+ (3 mM or higher) [38,39]. Here, we examined the effect of physiological concentrations of Ca2+ and Mg2+ (1 mM) on HR1-mediated fusion of liposomes with either 10 or 30 mol% PA. We observed a more than 2-fold enhancement in HR1-mediated fusion when liposomes containing 30 mol% PA were exposed to 1 mM Ca2+ (Figure 4a,b), comparable to the fusion levels observed with liposomes containing 30 mol% PE (Figure 1b,c). Importantly, this effect was specific to Ca2+ ions and was not observed in the presence of Mg2+ ions. Furthermore, we did not observe any influence of these cations (Ca2+ or Mg2+) on HR1-mediated fusion of liposomes containing 10 mol% PA (Figure 4a,b).
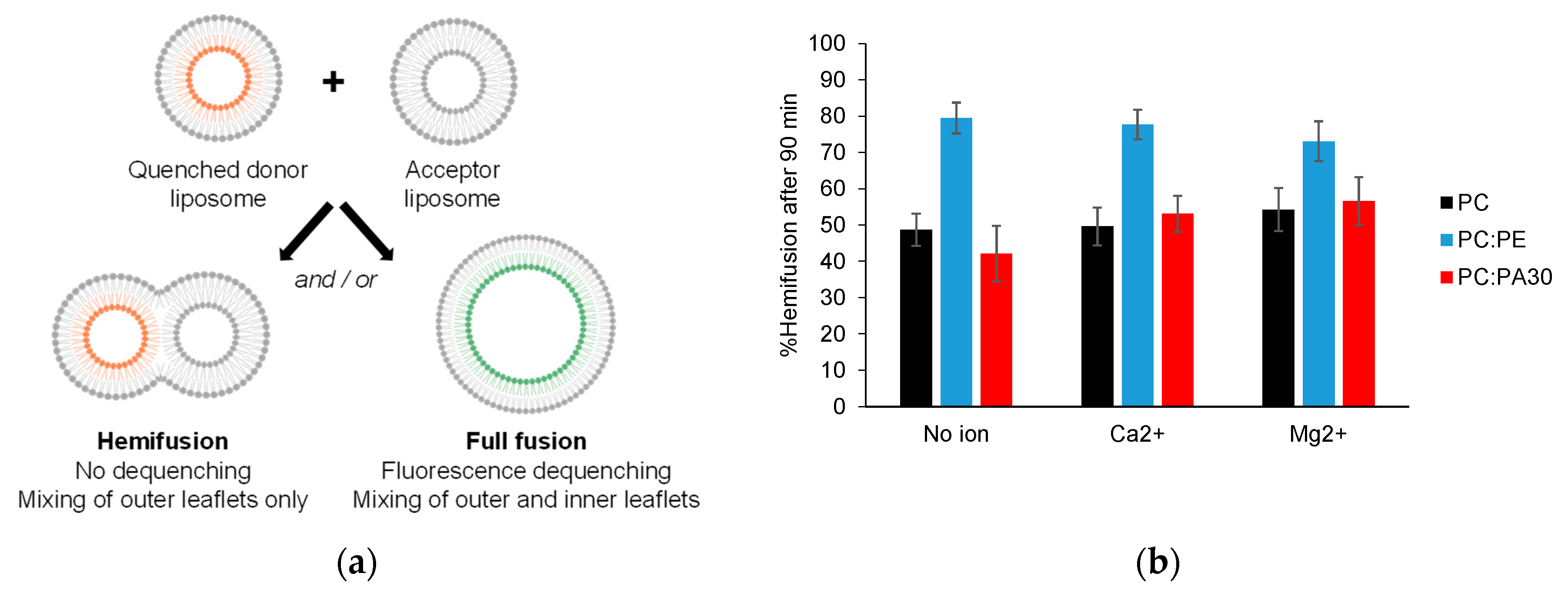
3.4. Phosphatidylethanolamine, but not phosphatidic acid, facilitates hemifusion by HR1
During membrane fusion events, a distinct intermediate stage called a hemifused structure often occurs. This structure is characterized by the fusion of outer leaflets of lipid bilayers, while the inner leaflets and internal contents remain separated [40]. Our previous study using the MAL lipid system revealed that HR1 has the capacity to induce both hemifusion and complete fusion between PC liposomes. In the FRET-based lipid mixing assay, we observed that, at the end of the experiment, 60% of PC liposomes had undergone complete fusion, while 40% had exhibited hemifusion [12]. In this study, we aimed to estimate the capacity of HR1 to induce hemifusion versus complete fusion of PC liposomes functionalized with NTA-Ni lipids and including or not PE or PA lipids, which were identified here as activators of HR1-mediated fusion. To distinguish hemifusion from complete fusion events in the FRET-based lipid mixing assay, we treated the fluorescent donor liposomes with a solution of sodium dithionite before initiating the fusion experiment [41] (Figure 5a). Sodium dithionite exclusively quenches the fluorescence signal of NBD PS lipids residing in the outer leaflet of liposomes. As a result, only complete fusion events involving the mixing of inner leaflets produce an increase in fluorescence during the FRET-based lipid mixing assay. By comparing the lipid mixing curves obtained with liposomes treated or not treated with sodium dithionite, we were able to quantify the hemifusion events between liposomes.
We observed that 40–50% of the PC liposomes functionalized with NTA-Ni lipids underwent hemifusion in the presence of HR1 (Figure 5b), similar to what we observed previously with the MAL lipid system [12]. Interestingly, the incorporation of 30 mol% PE in the liposome membrane promoted hemifusion events, with around 80% of the liposomes that had hemifused by the end of the FRET-based lipid mixing assay (Figure 5b). The introduction of 1 mM Ca2+ or Mg2+ did not have any additional impact on the occurrence of hemifusion. Conversely, while the presence of PA and Ca2+ led to a significant enhancement of HR1-mediated fusion, it did not result in a corresponding increase in hemifusion events, as observed with PE (Figure 5b). This indicates that the activation of fusion by PE, with or without cations (Ca2+ or Mg2+), and by PA in the presence of Ca2+, likely involves partially distinct molecular mechanisms.
4. Discussion
In a previous study, we identified an amphipathic helix at the C-terminal end of the HR1 domain. We proposed that this amphipathic helix induces HR1-mediated fusion by perturbing the lipid bilayer structure, especially in membrane regions presenting lipid packing defects, caused by either high membrane curvature or the presence of lipids with a cone-shaped structure [12]. In this study, through cryo-EM observations, we noticed that pure PC liposomes displayed locally disappearing membrane structure after incubation with HR1 (Figure 1d). This finding is consistent with a perturbation of the lipid bilayer structure when HR1 interacts with the liposome membrane.
The primary objective of this study was to examine the influence of membrane lipid composition, both in the presence and absence of divalent cations (Ca2+ or Mg2+), on HR1-mediated liposome fusion. In the absence of cations, the inclusion of 30 mol% PE lipids in the liposome membrane resulted in a more than 2-fold increase in the extent of lipid mixing mediated by HR1 compared to liposomes composed of pure PC lipids (Figure 1b,c). We attribute this effect to the cone-shaped structure of PE, which induces packing defects in the membrane structure and, consequently, enhances membrane binding and perturbation by the amphipathic helix of HR1. The presence of PE in the liposome membrane also promoted hemifusion events over full fusion mediated by HR1. The percentage of liposomes undergoing hemifusion increased from 50 to 80% when the liposomes contained 30 mol% PE lipids (Figure 5b). This effect can also be associated with the cone-shaped structure of PE, which is known to induce the transition from bilayer to non-bilayer inverted hexagonal phase structures of high local curvature [42], a process believed to take place during the formation of the stalk/hemifused fusion intermediate [43].
Unlike PE, the inclusion of the cone-shaped PA lipid at either 10 or 30 mol% in the liposome membrane did not activate fusion by HR1 in comparison to pure PC liposomes; instead, it led to a slight concentration-dependent inhibition (Figure 4a,b). This can be explained by the negative charge of the PA headgroup, which hinders the binding of HR1 – carrying a net negative charge at pH 7.4 – with the membrane. Similarly, the reduction of HR1-mediated fusion when liposomes contain CL lipids can be attributed to the electrostatic repulsion between HR1 and the negatively charged CL lipids (Figure 3a,b). Furthermore, the presence of PA in the liposome membrane did not increase the number of hemifusion events by HR1 compared to pure PC liposomes (Figure 5b). This effect is likely due to electrostatic repulsions between PA headgroups, preventing clustering of PA lipids into domains of high local curvature.
In the presence of Ca2+, HR1-mediated lipid mixing of liposomes containing 30 mol% PA was strongly activated. Specifically, we observed a 3-fold increase compared to liposomes with 30 mol% PA in the absence of Ca2+ and a 2-fold increase compared to pure PC liposomes in the presence of Ca2+ (Figure 4a,b). This activation can be attributed to the formation of PA-rich membrane domains facilitated by the presence of Ca2+, which allows the attraction of PA headgroups [44]. At the boundary of these membrane domains, the hydrophobic chains of lipids are exposed, enabling the interaction of the amphipathic helix of HR1 with the membrane structure. This interaction can be further reinforced by electrostatic attractions between the negatively charged PA headgroup and clusters of positive residues within the HR1 sequence. The amphipathic helix of HR1 notably includes a cluster of 3 consecutive positively charged Lysine residues (395-KKK-397). Similar recognition motifs have been identified in PA-binding proteins, including those with an amphipathic helix [45,46,47]. As observed in the absence of Ca2+, hemifusion events were not promoted for liposomes containing 30 mol% PA compared to PC liposomes, suggesting that zones of high curvatures enriched in PA do not form, even when PA-PA headgroup attraction is facilitated by the presence of Ca2+. Surprisingly, in the presence of Mg2+ ions, HR1-mediated fusion of liposomes with 30 mol% PA was not activated. This was unexpected because Ca2+ and Mg2+ ions were found to have a similar capacity to induce protein-free fusion of pure PA liposomes [48,49]. However, Mg2+ ions are known to be more hydrated than Ca2+ ions [50], which may limit their accessibility to PA headgroups that are buried deep below the PC headgroups in mixed PC:PA bilayers [45,51]. Such an accessibility issue would not occur in protein-free fusion of pure PA bilayers. It would also explain why Ca2+ and Mg2+ have comparable effects on HR1-mediated fusion of PC and PC:PE bilayers in our study (Figure S4), as PC and PE headgroups are fully accessible. In these cases, the activation of fusion can be explained by the presence of a small fraction of negative charges in PC bilayers [52], which may be sufficient to induce their interaction with divalent cations and promote the formation of packing defects facilitating HR1-membrane interaction.
In conclusion, we found that the interplay of divalent cations and specific cone-shaped lipids allows the formation of membrane regions with molecular packing defects, which, in turn, facilitates membrane perturbation and fusion mediated by the amphipathic helix of HR1. Future studies will need to address the role of lipids and divalent cations in the mode of action of the other functional domains of Mitofusin and their impact within the context of the full-length protein. It will also be important to consider the contribution of ER-mitochondria contact sites in regulating mitochondrial fusion events [53,54]. These sites, known for facilitating lipid and Ca2+ exchange between the two organelles [55], may act as hotspots, leading to local increases in the concentration of fusogenic lipids and Ca2+ ions, thus promoting efficient Mitofusin-mediated fusion.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figures S1–S4.
Author Contributions
Conceptualization, D.T. and M.C.; methodology, D.T.; validation, A.V., K.N. and D.T.; formal analysis, A.V. and D.T.; investigation, A.V., K.N., H.F., J.-M.G. and D.T.; resources, D.T. and M.C.; writing—original draft preparation, A.V. and D.T.; writing—review and editing, A.V., M.C. and D.T.; visualization, A.V., K.N. and D.T.; supervision, D.T.; funding acquisition, D.T. and M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Agence Nationale de la Recherche (ANR-19-CE11-0018-01) and the Association Française contre les Myopathies (AFM-Téléthon Research Grant 23778).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [PubMed]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Honda, S.; Aihara, T.; Hontani, M.; Okubo, K.; Hirose, S. Mutational analysis of action of mitochondrial fusion factor mitofusin-2. J. Cell Sci. 2005, 118, 3153–3161. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; Gao, S. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Brandt, T.; Cavellini, L.; Kühlbrandt, W.; Cohen, M.M. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. eLife 2016, 5. [Google Scholar] [CrossRef]
- Cohen, M.M.; Tareste, D. Recent insights into the structure and function of Mitofusins in mitochondrial fusion. [version 1; peer review: 2 approved]. F1000Res. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Daste, F.; Sauvanet, C.; Bavdek, A.; Baye, J.; Pierre, F.; Le Borgne, R.; David, C.; Rojo, M.; Fuchs, P.; Tareste, D. The heptad repeat domain 1 of Mitofusin has membrane destabilization function in mitochondrial fusion. EMBO Rep. 2018, 19, e43637. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhou, X.; Hu, X.; Joshi, A.S.; Guo, X.; Zhu, Y.; Chen, Q.; Prinz, W.A.; Hu, J. Sequences flanking the transmembrane segments facilitate mitochondrial localization and membrane fusion by mitofusin. Proc Natl Acad Sci USA 2017, 114, E9863–E9872. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Bian, X.; Sun, S.; Hu, X.; Klemm, R.W.; Prinz, W.A.; Rapoport, T.A.; Hu, J. Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proc Natl Acad Sci USA 2012, 109, E2146–54. [Google Scholar] [CrossRef] [PubMed]
- McNew, J.A.; Sondermann, H.; Lee, T.; Stern, M.; Brandizzi, F. GTP-dependent membrane fusion. Annu. Rev. Cell Dev. Biol. 2013, 29, 529–550. [Google Scholar] [CrossRef] [PubMed]
- Faust, J.E.; Desai, T.; Verma, A.; Ulengin, I.; Sun, T.-L.; Moss, T.J.; Betancourt-Solis, M.A.; Huang, H.W.; Lee, T.; McNew, J.A. The Atlastin C-terminal tail is an amphipathic helix that perturbs the bilayer structure during endoplasmic reticulum homotypic fusion. J. Biol. Chem. 2015, 290, 4772–4783. [Google Scholar] [CrossRef]
- Martens, S.; McMahon, H.T. Mechanisms of membrane fusion: disparate players and common principles. Nat. Rev. Mol. Cell Biol. 2008, 9, 543–556. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef]
- Chan, E.Y.L.; McQuibban, G.A. Phosphatidylserine decarboxylase 1 (Psd1) promotes mitochondrial fusion by regulating the biophysical properties of the mitochondrial membrane and alternative topogenesis of mitochondrial genome maintenance protein 1 (Mgm1). J. Biol. Chem. 2012, 287, 40131–40139. [Google Scholar] [CrossRef]
- Joshi, A.S.; Thompson, M.N.; Fei, N.; Hüttemann, M.; Greenberg, M.L. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J. Biol. Chem. 2012, 287, 17589–17597. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.L.; Van Komen, J.S.; Liu, S.; Weber, T.; Melia, T.J.; McNew, J.A. Liposome fusion assay to monitor intracellular membrane fusion machines. Meth. Enzymol. 2003, 372, 274–300. [Google Scholar] [CrossRef]
- McNew, J.A.; Weber, T.; Parlati, F.; Johnston, R.J.; Melia, T.J.; Söllner, T.H.; Rothman, J.E. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 2000, 150, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Jotwani, A.; Richerson, D.N.; Motta, I.; Julca-Zevallos, O.; Melia, T.J. Approaches to the study of Atg8-mediated membrane dynamics in vitro. Methods Cell Biol. 2012, 108, 93–116. [Google Scholar] [CrossRef]
- Ji, H.; Coleman, J.; Yang, R.; Melia, T.J.; Rothman, J.E.; Tareste, D. Protein determinants of SNARE-mediated lipid mixing. Biophys. J. 2010, 99, 553–560. [Google Scholar] [CrossRef]
- Nir, S.; Wilschut, J.; Bentz, J. The rate of fusion of phospholipid vesicles and the role of bilayer curvature. Biochim. Biophys. Acta 1982, 688, 275–278. [Google Scholar] [CrossRef]
- Malinin, V.S.; Frederik, P.; Lentz, B.R. Osmotic and curvature stress affect PEG-induced fusion of lipid vesicles but not mixing of their lipids. Biophys. J. 2002, 82, 2090–2100. [Google Scholar] [CrossRef]
- Lee, J.Y.; Schick, M. Calculation of free energy barriers to the fusion of small vesicles. Biophys. J. 2008, 94, 1699–1706. [Google Scholar] [CrossRef]
- Ardail, D.; Privat, J.P.; Egret-Charlier, M.; Levrat, C.; Lerme, F.; Louisot, P. Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 1990, 265, 18797–18802. [Google Scholar] [CrossRef]
- Fritz, S.; Rapaport, D.; Klanner, E.; Neupert, W.; Westermann, B. Connection of the mitochondrial outer and inner membranes by Fzo1 is critical for organellar fusion. J. Cell Biol. 2001, 152, 683–692. [Google Scholar] [CrossRef]
- Frohman, M.A. Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. 2015, 93, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Wilschut, J.; Holsappel, M.; Jansen, R. Ca2+-induced fusion of cardiolipin/phosphatidylcholine vesicles monitored by mixing of aqueous contents. Biochim. Biophys. Acta 1982, 690, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Killian, J.A.; Verkleij, A.J.; Wilschut, J. Membrane fusion and the lamellar-to-inverted-hexagonal phase transition in cardiolipin vesicle systems induced by divalent cations. Biophys. J. 1999, 77, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 2018, 28, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006, 8, 1255–1262. [Google Scholar] [CrossRef]
- Adachi, Y.; Itoh, K.; Yamada, T.; Cerveny, K.L.; Suzuki, T.L.; Macdonald, P.; Frohman, M.A.; Ramachandran, R.; Iijima, M.; Sesaki, H. Coincident phosphatidic acid interaction restrains drp1 in mitochondrial division. Mol. Cell 2016, 63, 1034–1043. [Google Scholar] [CrossRef]
- Brock, T.G.; Nagaprakash, K.; Margolis, D.I.; Smolen, J.E. Modeling degranulation with liposomes: effect of lipid composition on membrane fusion. J. Membr. Biol. 1994, 141, 139–148. [Google Scholar] [CrossRef]
- Blackwood, R.A.; Smolen, J.E.; Transue, A.; Hessler, R.J.; Harsh, D.M.; Brower, R.C.; French, S. Phospholipase D activity facilitates Ca2+-induced aggregation and fusion of complex liposomes. Am. J. Physiol. 1997, 272, C1279–85. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef]
- Meers, P.; Ali, S.; Erukulla, R.; Janoff, A.S. Novel inner monolayer fusion assays reveal differential monolayer mixing associated with cation-dependent membrane fusion. Biochim. Biophys. Acta 2000, 1467, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M. Structure of the inverted hexagonal (HII) phase, and non-lamellar phase transitions of lipids. Biochim. Biophys. Acta 1990, 1031, 1–69. [Google Scholar] [CrossRef] [PubMed]
- Aeffner, S.; Reusch, T.; Weinhausen, B.; Salditt, T. Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc Natl Acad Sci USA 2012, 109, E1609–18. [Google Scholar] [CrossRef]
- Faraudo, J.; Travesset, A. Phosphatidic acid domains in membranes: effect of divalent counterions. Biophys. J. 2007, 92, 2806–2818. [Google Scholar] [CrossRef]
- Zhukovsky, M.A.; Filograna, A.; Luini, A.; Corda, D.; Valente, C. Phosphatidic acid in membrane rearrangements. FEBS Lett. 2019, 593, 2428–2451. [Google Scholar] [CrossRef]
- Hofbauer, H.F.; Gecht, M.; Fischer, S.C.; Seybert, A.; Frangakis, A.S.; Stelzer, E.H.K.; Covino, R.; Hummer, G.; Ernst, R. The molecular recognition of phosphatidic acid by an amphipathic helix in Opi1. J. Cell Biol. 2018, 217, 3109–3126. [Google Scholar] [CrossRef]
- Tanguy, E.; Kassas, N.; Vitale, N. Protein−phospholipid interaction motifs: A focus on phosphatidic acid. Biomolecules 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Papahadjopoulos, D.; Vail, W.J.; Pangborn, W.A.; Poste, G. Studies on membrane fusion. II. Induction of fusion in pure phospholipid membranes by calcium ions and other divalent metals. Biochim. Biophys. Acta 1976, 448, 265–283. [Google Scholar] [CrossRef]
- Ohki, S.; Ohshima, H. Divalent cation-induced phosphatidic acid membrane fusion. Effect of ion binding and membrane surface tension. Biochim. Biophys. Acta 1985, 812, 147–154. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Importance of metal hydration on the selectivity of Mg2+ versus Ca2+ in magnesium ion channels. J. Am. Chem. Soc. 2013, 135, 17200–17208. [Google Scholar] [CrossRef]
- Putta, P.; Rankenberg, J.; Korver, R.A.; van Wijk, R.; Munnik, T.; Testerink, C.; Kooijman, E.E. Phosphatidic acid binding proteins display differential binding as a function of membrane curvature stress and chemical properties. Biochim. Biophys. Acta 2016, 1858, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Pincet, F.; Cribier, S.; Perez, E. Bilayers of neutral lipids bear a small but significant charge. Eur. Phys. J. B 1999, 11, 127–130. [Google Scholar] [CrossRef]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Cardoso, V.F.; Rochin, L.; Arora, A.; Houcine, A.; Jääskeläinen, E.; Kivelä, A.M.; Sauvanet, C.; Le Bars, R.; Marien, E.; Dehairs, J.; Neveu, J.; El Khallouki, N.; Santonico, E.; Swinnen, J.V.; Tareste, D.; Olkkonen, V.M.; Giordano, F. ORP5/8 and MIB/MICOS link ER-mitochondria and intra-mitochondrial contacts for non-vesicular transport of phosphatidylserine. Cell Rep. 2022, 40, 111364. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef]
Figure 1.
Influence of lipid anchors and PE on HR1-mediated fusion. (a) The peptides used in this study were HR1 fragments of Mfn1 with a Cys or a His6 tag at their C-terminus, allowing their chemical coupling to liposomes functionalized with either 18:1 MPB PE (MAL lipids) or 18:1 DGS-NTA(Ni) (NTA-Ni lipids), respectively; (b) Representative kinetics of a FRET-based lipid mixing assay between liposomes containing 5 mol% of either MAL or NTA-Ni lipids in their membrane, along with 95 mol% PC or 65 mol% PC and 30 mol% PE. The fusion reaction was initiated by adding HR1-Cys or HR1-His6 peptides at t=0, with 25 µM HR1 and 500 µM lipids in the reaction mix. Control experiments in which buffer alone was added in place of HR1 are presented in Figure S1; (c) Average extent of lipid mixing observed after a 90-min period, based on data from n = 7 to 21 independent experiments. The error bars represent the standard errors of the mean. (d) Cryo-EM picture of liposomes composed of 95 mol% PC and 5 mol% MAL after 1-hour incubation at 37 °C with HR1-Cys peptides (12.5 µM of peptides and 500 µM of lipids).
Figure 1.
Influence of lipid anchors and PE on HR1-mediated fusion. (a) The peptides used in this study were HR1 fragments of Mfn1 with a Cys or a His6 tag at their C-terminus, allowing their chemical coupling to liposomes functionalized with either 18:1 MPB PE (MAL lipids) or 18:1 DGS-NTA(Ni) (NTA-Ni lipids), respectively; (b) Representative kinetics of a FRET-based lipid mixing assay between liposomes containing 5 mol% of either MAL or NTA-Ni lipids in their membrane, along with 95 mol% PC or 65 mol% PC and 30 mol% PE. The fusion reaction was initiated by adding HR1-Cys or HR1-His6 peptides at t=0, with 25 µM HR1 and 500 µM lipids in the reaction mix. Control experiments in which buffer alone was added in place of HR1 are presented in Figure S1; (c) Average extent of lipid mixing observed after a 90-min period, based on data from n = 7 to 21 independent experiments. The error bars represent the standard errors of the mean. (d) Cryo-EM picture of liposomes composed of 95 mol% PC and 5 mol% MAL after 1-hour incubation at 37 °C with HR1-Cys peptides (12.5 µM of peptides and 500 µM of lipids).
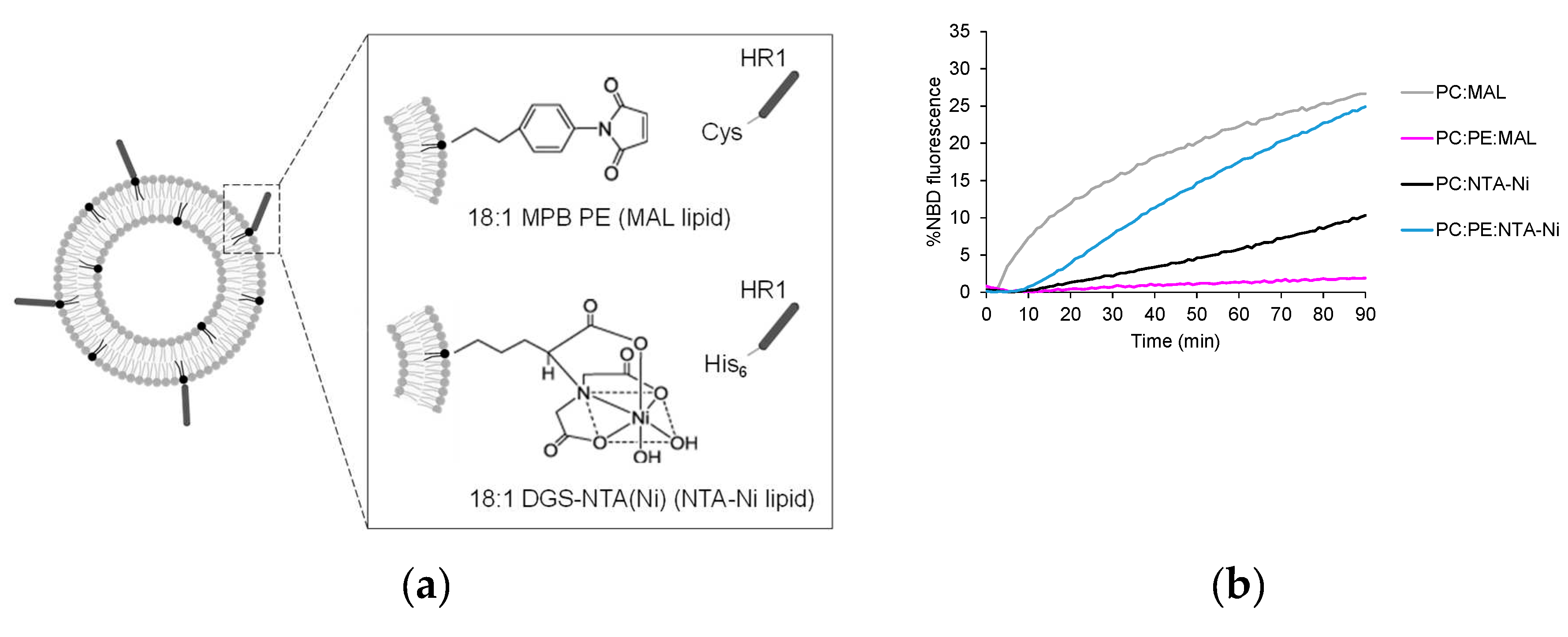
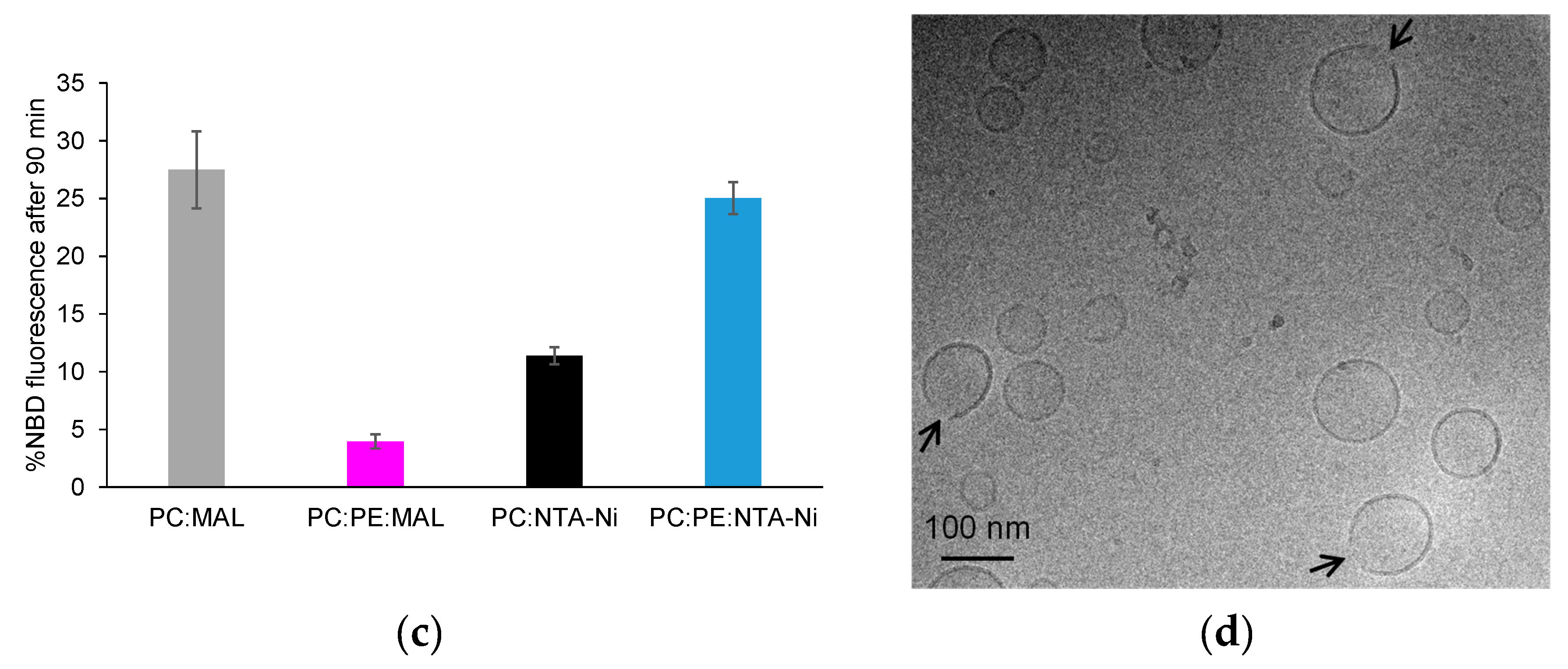
Figure 2.
Surface density of HR1 on the liposome membrane. (a) Liposomes with the same lipid compositions as in Figure 1 were incubated with HR1-Cys or HR1-His6 peptides (500 µM of lipids and 25 µM of peptides) at 37 °C for 1 hour. The reaction mixes were separated using a discontinuous nycodenz gradient to distinguish HR1-bound liposomes from unbound HR1. Protein and lipid recoveries in the floated samples were estimated by SDS-PAGE stained with Coomassie upon comparison with the non-floated samples; (b) HR1-to-lipid ratios in the liposome membrane were estimated from n = 3 to 5 independent experiments. The error bars represent the standard errors of the mean.
Figure 2.
Surface density of HR1 on the liposome membrane. (a) Liposomes with the same lipid compositions as in Figure 1 were incubated with HR1-Cys or HR1-His6 peptides (500 µM of lipids and 25 µM of peptides) at 37 °C for 1 hour. The reaction mixes were separated using a discontinuous nycodenz gradient to distinguish HR1-bound liposomes from unbound HR1. Protein and lipid recoveries in the floated samples were estimated by SDS-PAGE stained with Coomassie upon comparison with the non-floated samples; (b) HR1-to-lipid ratios in the liposome membrane were estimated from n = 3 to 5 independent experiments. The error bars represent the standard errors of the mean.

Figure 3.
Effect of CL and divalent cations on HR1-mediated fusion. (a) Representative kinetics of a FRET-based lipid mixing assay between liposomes containing 5 mol% NTA-Ni lipids in their membrane, along with different lipid compositions: 95 mol% PC (black), 90 mol% PC and 5 mol% CL (light green), or 75 mol% PC and 20 mol% CL (dark green). The fusion reaction was initiated by adding HR1-His6 peptides at t=0 in the absence or presence of the divalent cations Ca2+ or Mg2+ (500 µM of lipids, 25 µM of peptides, and 1 mM of cations). Control experiments with buffer alone instead of HR1 are presented in Figure S1; (b) Average extent of lipid mixing observed after a 90-min period, based on data from n = 3 to 21 independent experiments. The error bars represent the standard errors of the mean.
Figure 3.
Effect of CL and divalent cations on HR1-mediated fusion. (a) Representative kinetics of a FRET-based lipid mixing assay between liposomes containing 5 mol% NTA-Ni lipids in their membrane, along with different lipid compositions: 95 mol% PC (black), 90 mol% PC and 5 mol% CL (light green), or 75 mol% PC and 20 mol% CL (dark green). The fusion reaction was initiated by adding HR1-His6 peptides at t=0 in the absence or presence of the divalent cations Ca2+ or Mg2+ (500 µM of lipids, 25 µM of peptides, and 1 mM of cations). Control experiments with buffer alone instead of HR1 are presented in Figure S1; (b) Average extent of lipid mixing observed after a 90-min period, based on data from n = 3 to 21 independent experiments. The error bars represent the standard errors of the mean.

Figure 4.
Effect of PA and divalent cations on HR1-mediated fusion. (a) Representative kinetics of a FRET-based lipid mixing assay between liposomes containing 5 mol% NTA-Ni lipids in their membrane, along with different lipid compositions: 95 mol% PC (black), 85 mol% PC and 10 mol% PA (yellow), or 65 mol% PC and 30 mol% PA (red). The fusion reaction was initiated by adding HR1-His6 peptides at t=0 in the absence or presence of the divalent cations Ca2+ or Mg2+ (500 µM of lipids, 25 µM of peptides, and 1 mM of cations). Control experiments with buffer alone instead of HR1 are presented in Figure S1; (b) Average extent of lipid mixing observed after a 90-min period, based on data from n = 7 to 21 independent experiments. The error bars represent the standard errors of the mean.
Figure 4.
Effect of PA and divalent cations on HR1-mediated fusion. (a) Representative kinetics of a FRET-based lipid mixing assay between liposomes containing 5 mol% NTA-Ni lipids in their membrane, along with different lipid compositions: 95 mol% PC (black), 85 mol% PC and 10 mol% PA (yellow), or 65 mol% PC and 30 mol% PA (red). The fusion reaction was initiated by adding HR1-His6 peptides at t=0 in the absence or presence of the divalent cations Ca2+ or Mg2+ (500 µM of lipids, 25 µM of peptides, and 1 mM of cations). Control experiments with buffer alone instead of HR1 are presented in Figure S1; (b) Average extent of lipid mixing observed after a 90-min period, based on data from n = 7 to 21 independent experiments. The error bars represent the standard errors of the mean.
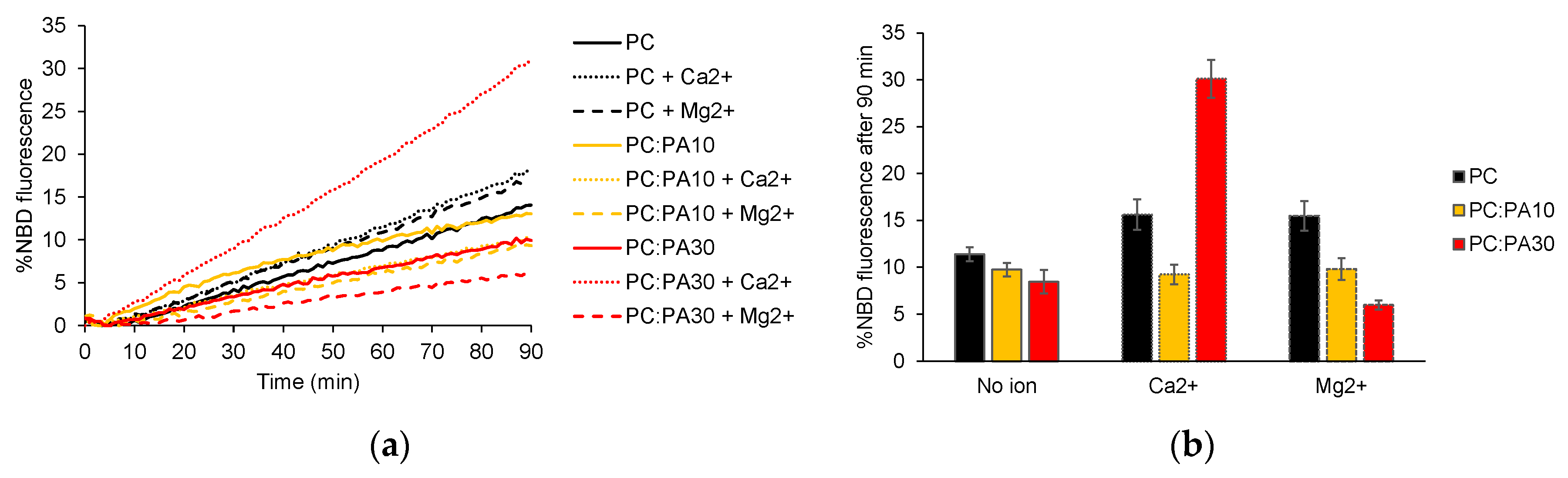
Figure 5.
Effect of PE and PA on HR1-mediated hemifusion. (a) Hemifusion events were quantified using the sodium dithionite assay. Fluorescent donor liposomes were pre-treated with sodium dithionite to eliminate the NBD fluorescence of their outer leaflet, allowing only full fusion events to lead to fluorescence dequenching in the FRET-based lipid-mixing assay; (b) The percentage of liposomes that underwent hemifusion after 90 min was determined by comparing the fluorescence dequenching signals obtained with or without prior sodium dithionite treatment (n = 3 to 11 independent experiments; error bars represent standard errors of the mean).
Figure 5.
Effect of PE and PA on HR1-mediated hemifusion. (a) Hemifusion events were quantified using the sodium dithionite assay. Fluorescent donor liposomes were pre-treated with sodium dithionite to eliminate the NBD fluorescence of their outer leaflet, allowing only full fusion events to lead to fluorescence dequenching in the FRET-based lipid-mixing assay; (b) The percentage of liposomes that underwent hemifusion after 90 min was determined by comparing the fluorescence dequenching signals obtained with or without prior sodium dithionite treatment (n = 3 to 11 independent experiments; error bars represent standard errors of the mean).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



