
Submitted:
25 July 2023
Posted:
27 July 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The rapid changes in the coronavirus genomes are creating new strains over time after the first variation found in Wuhan in 2019. SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) genotypes need periodically undergo whole genome sequencing. Genome sequencing has been extremely helpful in combating this virus because so many diagnoses, treatments, and vaccinations have been developed against it. To achieve this, we generated 17 high-quality whole-genome sequence data from 48 SARS-CoV-2 genotypes of COVID-19 patients who tested positive by PCR in the Tashkent city of Uzbekistan. We identified nucleotide polymorphisms, including nonsynonymous (missense) and synonymous mutations in the coding regions of the sequenced sample genomes. All whole genome sequences were categorized by phylogenetic analysis into the G clade (or GK sub-clade). A total of 134 mutations were identified, consisting of 65 shared and 69 unique mutations. Collectively, nucleotide changes represented one frameshift mutation, one conservative and disruptive insertion deletion, four upstream region mutations, four downstream region mutations, 39 synonymous mutations, and 84 missense mutations. Furthermore, bioinformatics web tools were used to analyze amino acid changes in virus genomes. Several amino acid mutations were found, which were not found in previously published Delta strain sequences.
Keywords:
SARS-CoV-2
; virus
; transmission
; mutations
; Delta strain
1. Introduction
Considerable research has been conducted on the SARS-Cov-2 genome since 2020. To date, 15,778,185 SARS-CoV-2 genome sequences have been shared via online data platforms such as GISAID [1], which helps track the new variants and mutations. The variants of SARS-CoV-2, the causative agent of COVID-19, continue to change during its travel by human-to-human transmission [2].
The World Health Organization (WHO) officially declared a pandemic on March 11, 2020 [3]. SARS-CoV-2 has scattered globally and is currently a significant problem. Europe and Latin America have suffered more than other nations, particularly in the early stages of the coronavirus epidemic. As of 12 July 2023, WHO had received reports of 767 972 961 confirmed COVID-19 cases, including 6 950 655 fatalities. A total of 13 462 024 421 vaccination doses have been given as of 8 July 2023. From 3 January 2020 to 12 July 2023, Uzbekistan reported 171 083 confirmed cases of COVID-19, with 1 016 fatalities. So far, 80 043 836 vaccination doses have been given as of 3 June 2023 [4].
The established nomenclature systems for naming and tracking SARS-CoV-2 genetic lineages through GISAID, Nextstrain, and Pango are currently being used by scientists in scientific research. The Technical Advisory Group on Virus Evolution was convened by WHO and recommended using letters of the Greek Alphabet, i.e., Alpha, Beta, Gamma, Delta, etc., of the virus strains to make it easier and more practical to be discussed by non-scientific audiences [3].
SARS-Cov-2 belongs to the β-coronavirus genus lineage B, which has large enveloped, positive-sense, single-stranded RNAs (30 kb) and encodes four structural proteins as well as other accessory or non-structural proteins (including the viral pp1a-pp1ab replicase, the 3C-like protease (3CLpro), the papain-like protease (PLpro), and the RNA-dependent RNA polymerase (RdRp) [5,6].
SARS-CoV2 whole genome sequencing is critical for discovering meaningful information on viral lineages, variants of interest, and variants of concern [7]. The Global Initiative on Sharing All Influenza Data (GISAID) [1], the National Center for Biotechnology Information (NCBI) [8], and the Virus Pathogen Database and Analysis Resource (ViPR) [9] have full and partial genome sequences of SARS-CoV-2 obtained from various countries and laboratories.
The worldwide phylogenetic tree of SARS-Cov-2 was constructed using sequenced virus genomes, and the sequences are divided into several clades. Our previous study examined the earliest clades and their characteristics [10]. Many viral variants were later discovered and uploaded to global databases. The B.1.617.2 variant was detected in India in December 2020 [11], and the World Health Organization classified it as VOC Delta on May 11, 2021, according to its increased transmissibility [12]. Delta variants with increased transmissibility have been linked to critical S-protein mutations such as D614G, L452R, P681R, and T478K [13]. The spike protein facilitates virus attachment to host cell-surface receptors and virus-cell membrane fusion. It is also the primary target for neutralizing antibodies produced after infection [14].
For the first time, an infectious epidemic was found in Uzbekistan in March 2020. The government decided to conduct public testing for COVID-19. This incident signaled the beginning of the pandemic in Uzbekistan, necessitating accurate identification and monitoring of coronavirus genotypes that were rapidly spreading throughout the population [10,15].
We, therefore, started the whole genome sequencing of COVID-19 samples with the primary objective of identifying virus genotypes spread in our territory as well as analyzing genomic diversity, types of mutations, and the emergence of new variations of SARS-CoV-2 [10,15]. In this study, we describe the following whole-genome sequence data from individuals in the Republic of Uzbekistan who were infected with COVID-19. We analyzed 134 mutations with nonsynonymous and synonymous types and successfully assembled the 17 high-quality sample genome sequences for coronavirus genotypes. In the global phylogenetic tree, comparative analysis utilizing SARS-CoV-2 genomes that are known as the Uzbekistan sample genomes into the GK subclade. The sequenced genomic data of coronavirus genotypes were used to develop the edible vaccine in Uzbekistan against coronavirus threats [16].
2. Materials and methods
2.1. Sample collection
Using nasopharyngeal and oropharyngeal swabs sticks (Huachenyang Technology, Shenzhen, China), samples were obtained from 100 symptomatic patients with high fever and intermittent cough and immediately placed in a viral transport medium. Patients suspected of having COVID-19 infection were referred to the diagnostics laboratories of the private BiogenMed COVID-19 testing laboratory in Tashkent, Republic of Uzbekistan. Following SARS-CoV-2 laboratory testing, biological samples were taken randomly from PCR-positive patients.
The Ethics Committee of the Ministry of Health of the Republic of Uzbekistan (#6/20-1582) approved the research investigation. All experiments were conducted in conformity with the applicable standards and legislation. Samples were renumbered and de-identified so that no one, not even researchers, knew who the patients were. Only anonymized data, including age and biological sex, were preserved for reporting purposes. All patients in the sample collection provided verbal consent for voluntary participation. We stated to all participants that the collected samples would be used for a sequencing project exclusively, without disclosing their identities or disturbing them in the future. In this study, verbal consent was preferred over written consent since patients were hesitant to sign any written document due to their concern about COVID-19 infection at the time and a lack of knowledge of the genome sequencing investigation. There were no minors involved in the sample collection. There was no need to do so because the sequencing experiment was non-invasive, participants were not subjected to further downstream clinical procedures, and the samples' identities were anonymized in this investigation.
A MagMax Viral/Pathogen kit was used to extract total RNA from the collected samples (Life Technologies Corporation, Austin, Texas, USA). Using a CFX Connect Real-Time PCR System, each sample was analyzed for the presence of SARS-CoV-2 (threshold - 0,050) (BioRad, Hercules, California, USA). RT-PCR was carried out in 40 ul final volumes. For cDNA synthesis at 35°C for 20 minutes, a first denaturation at 95°C for 5 minutes was followed by 50 cycles of 94°C for 15 seconds and 64°C for 20 seconds (set fluorescence measurement for Fam, Hex, Rox, and Cy5 channels at 64°C) and a SARS-CoV-2/SARS-CoV multiplex real-time PCR assay targeting the nucleocapsid (N), envelope (E), and spike region [10]. Among all evaluated patients, 48 PCR-positive samples (28 females and 20 men) were chosen at random for further research.
2.2. SARS-CoV-2 sequencing
As in our previous study, SARS-CoV-2 sequencing and data analysis were carried out [10]. Variant callers were filtered to remove variants with reading depths of less than 1000 and ion torrent quality scores of less than 400 to keep reliable variants only (Table 2 and Table S1). The filtered variants were used for sample clustering with Maximum Likelihood Tree in Molecular Evolutionary Genetics Analysis (MEGA, https://www.megasoftware.net) software. The consensus for each SARS-CoV-2 genome sequence was then submitted to the GISAID (www.gisaid.org) under the accession numbers of EPI_ISL_3668627 to EPI_ISL_3673673 (available for registered users) and NCBI under the accession numbers GI:2085183815 to GI:2085183892 (or MZ892621.1 to MZ892627.1; NCBI database; Table 1).
3. Results
3.1. Sample selection for sequencing
Over a hundred symptomatic patients with high fever and intermittent cough were chosen randomly from a private COVID-19 testing lab in Tashkent for this investigation. 48 PCR-positive samples were chosen for sequencing. Only 17 high-quality sequences were selected to be submitted to worldwide databases (five males and twelve women attended, with an average age of 34; Table 1).
Because of insufficient sequencing coverage, 31 SARS-CoV-2 samples were excluded from further analysis, resulting in many gaps in the consensus sequence. The remaining 17 samples (five males and twelve women, Table 1) had an average of 779 739 mapped reads per sample. The target reads were 99.61%, and the mean read depth was 3814. The average coverage consistency in chosen samples was 87.8%; (Table 2).
3.2. Identifying unique and reliable mutations in all high-quality sequenced samples
Using the maximum likelihood method, a phylogenetic tree was generated in the MEGA X program based on the 17 viral sequences of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in samples collected from COVID-19 patients in Tashkent city. This maximum likelihood tree was generated using 134 mutations discovered in the 17 sequences and was rooted by the Wuhan reference strain NC 045512.2 (see Table S1, Figure 1). Bootstrap values are illustrated.
Variant Caller produced all evaluated mutations in these selected samples. The number of mutations ranged from 36 (samples 38,47, and 36) to 45 (sample 48), altogether unique and shared mutations (Figure 2). Most viral genomes contained between 39 and 44 mutations when compared with the NC_045512.2 reference genome, with mutations in samples 39 and 40 (39 mutations), samples 10, 33, 34 and 43 (40 mutations), samples 36, 37, 41, 44 and 45 (41 mutations), sample 35 (43 mutations) and samples 42 and 46 (44 mutations). The mutations mentioned above were calculated together with shared and unique mutations. Samples 38 and 46 do not carry unique mutations (Figure 2).
The most common nucleotide substitution detected (see Table S1) was from cytosine to thymine (51/134 mutations), followed by guanine to thymine (28/134 mutations) and thymine to cytosine and guanine to adenine (12/134 mutations), and was adenine to guanine (10/134 mutations). All mutations were homozygous. We observed one shared disruptive inframe deletion (c.467_472delAGTTCA), two unique (c.*4300A>G, c.*4352G>T), two shared (c.*4308G>T, c.*4358G>T) downstream gene variant mutations, four unique (c.646C>T, c.2533G>T, c.3761G>T and c.3790G>T) and nine shared missense variant mutations, four unique (c.936C>T, c.1995C>T, c.2259G>A and c.2388T>C) and one shared (c.3183C>T) synonymous variant mutations in the gene encoding the S protein. One unique (c.100C>T and c.463C>T) and one shared (c.245T>C) missense mutation was identified in the M (matrix) region; one shared (c.-3delA) upstream gene variant mutation, three unique (c.200C>T, c.518C>T and c.1085C>T) and six shared (c.188A>G, c.608G>T, c.643G>T, c.1129G>T, c.1152G>C and c.1154G>A) missense mutations and four unique (c.105G>A, c.123G>A, c.987G>A and c.1080C>T) synonymous mutations were found in N (nucleocapsid) region. No mutations were found in E (envelope) region (Tables S1 and S2).
Furthermore, one frameshift, 41 missense mutations, 27 synonymous mutations, and two upstream gene mutations were found in the ORF1ab region. One synonymous mutation and ten missense mutations were detected in the ORF3a region. The ORF6 region showed one missense mutation, while the ORF7a region exhibited six missenses and one synonymous mutation, and the ORF8a region exhibited one missense, one upstream gene, and one conservative insertion-deletion mutation. Finally, one synonymous mutation (c.57C>T) was found in the region of ORF10. We found 134 mutations in total, 65 shared and 69 unique, representing one frameshift mutation, one conservative and disruptive inframe deletion, four upstream region mutations, four downstream region mutations, 39 synonymous mutations, and 84 missense mutations (Table S1).
3.3. The phylogenetic tree was built from viral sequences
We wanted to examine the significant alterations in all sequences to understand what distinguished our cases from others worldwide. MEGA X was used to create a phylogenetic tree based on the 17 viral sequences using the greatest likelihood technique. (Figure 1). Variant caller (v.5.12.0.4) identified 134 mutations from 17 SARS-CoV-2 viral genome sequences in COVID-19 patient samples (Table S1).
Most of the mutations shared with more sequences, for example, eighteen mutations (C3037T, G15451A, C16466T, C21618G, GAGTTCA22028G, T22917G, C22995A, A23403G, C23604G, G24410A, C25469T, AGATTTC28247A, T26767C, TA28270T, A28461G, G28881T, G29402T, and G29742T) with 17 sequences and four mutations (G210T, C241T, C14408T, T27638C, C27752T) with 16 samples, seven mutations (C6402T, C8986T, G9053T, A11201G, A11332G, C19220T and G28916T) with 12 sequences and four mutations (C14925T, A21137G, A24110C and A25439C) with six sequences, accordingly (Table S1).
These variants have grouped with USA, Indian, and England variants. All are considered as Delta strains (B.1.617.2) in the phylogenetic tree provided by genomedetective.com. Interestingly, Beta variant samples are located in the Neighbor Cluster in the tree, whereas Alpha, Gamma, and Omicron variant samples were found in the further branches (Figure 3).
Discussion
Our findings show that the whole-genome sequences of the 17 symptomatic COVID-19 patients represent significant nucleotide diversity (Table 1, Tables S1 and S2). According to the similarity of Delta strain mutations, these 17 sequenced samples have grouped one major clade of SARS-CoV-2 on the GISAID public database named clade G (or GK subclade). The G clade has known for S-protein mutations such as D614G, L452R, P681R, and T478K [13]. These mutations were also found in all 17 samples using the viralvar.org website. However, some other mutations in this region were found in each sample. For example, thirty-four S-protein mutations were identified in sample 39, while 23 mutations were found in sample 47, including known mutations (Table S3).
According to Cherian et al., 2021, newly identified lineages B.1.617.1 and B.1.617.2 dominated the phylogenetic tree. These strains carried the signature mutations L452R, T478K, E484Q, D614G, and P681R in the spike protein, including within the receptor-binding domain (RBD) (Cherian et al., 2021). Those mutations, except E484Q, were also shared with all 17 samples in our result. In addition, mutation T19R was shared with all samples, whereas G142D (in 15 samples), D950N (in 12 samples), I850L (in 6 samples), T95I (in 5 samples), A222V and F797C (in 2 samples) were found (Table S3).
The structural analysis of the RBD mutations L452R, T478K, and E484Q revealed the potential of increasing ACE2 binding, whereas P681R in the furin cleavage site may increase the rate of S1-S2 cleavage, resulting in improved transmissibility. The two RBD mutations, L452R and E484Q, reduced binding to specific monoclonal antibodies (mAbs) and may affect their neutralization potential [17]. Interestingly, the E484Q mutation did not appear in our sequenced samples.
Most of the unique amino acid changes were found in the S-spike region in Sample 39 (26 mutations; F106L, G107V, T108L, T109L, L110stop, D111I, S112R, K113R, T114P, Q115S, S116P, L117Y, I119L, V120L, N121I, N122T, A123L, T124L, N125M, V126L, V127L, I128L, V130S, C131V, E132N, and Q134N) and in Sample 47 (16 mutations; A260V, G261W, A262C, A264S, Y265L, Y266L, V267C, Y269L, L270S, A263C, L277I, Q271S, P272T, R273stop, T274D, and L276S). In contrast, five amino acid changes were found in Sample 33 (Table S3). The virus genome must be sequenced to identify SARS-CoV-2 strains and investigate local and worldwide dissemination. Furthermore, the full-genome sequencing of any virus that causes infection can help to examine outbreak dynamics, such as variations in pandemic size over time, spatial spread, and transmission routes (WHO COVID Report 2021). Indeed, this investigation revealed that many of the diseases originated in the United States, India, and England (GK subclade) (Figure 3). The global distribution of the virus was caused by worldwide travel.
The differences in virus structure between samples demonstrated how virus shape changed over time as environments changed. The genomic sequence data generated from these 17 samples, submitted to global databases such as NCBI and GISAID, should aid public health and research institutions in observing disease propagation patterns in the region. Furthermore, the findings will help in the development of diagnostic assays, medicines, and vaccines. Several institutes, including our center, have begun developing a national vaccine based on the SARS-CoV-2 genome sequences discovered in symptomatic Uzbek patients.
The sequence information reported here should also add fresh sequence and mutational profile information from our area to the COVID sequence database (GISAID), which will be helpful for upcoming molecular epidemiology and evolutionary phylogenetic studies by health and scientific organizations. Our whole-genome sequence data, presented here, should be beneficial for tracing the origin and source of the SARS-CoV-2 variants that are now circulating and for identifying and contrasting newly developing variations in Uzbekistan and elsewhere.
The SARS-CoV-2 genome sequencing obtained from infected Uzbek patients using samples collected at the end of 2021 has been presented here. They were the second wave of the coronavirus disease pandemic, which had spread throughout the country by that period. DNA-based and plant-based edible vaccines have been developed using these sequences of the SARS-CoV-2 spread in Uzbekistan, which are currently in the clinical trial stages [16].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization &Data curation, M.A. (Mirzakamol Ayubov), Z.B., M.M.; Methodology &Visualization, M.A. (Mirzakamol Ayubov), M.M., A.Y., D.U., S.S., K.U.; Funding acquisition: M.A., Z.B.; Supervision, I.A. (Ibrokhim Abdurakhmonov), Z.B.; Writing – original draft preparation, M.A. (Mirzakamol Ayubov), M.M., Z.B.; Writing – reviewing and editing, I.A. (Ibrokhim Abdurakhmonov), M.A.
Institutional Review Board Statement
The research study has been approved by the Ethics Committee under the Ministry of Health of the Republic of Uzbekistan (#6/20-1582). All the experiments were carried out in accordance with the relevant guidelines and regulations. Samples were renumbered and de-identified so no one, even researchers could know the identity of the patients. For the reporting purpose only anonymous data including age and biological sex were kept.
Data Availability Statement
Table S1. One hundred thirty-four mutations were observed in seventeen SARS-CoV-2 viral genome sequences in samples from COVID-19 patients in Tashkent, Uzbekistan. Table S2. Nucleotide mutations of the matrix (M), nucleocapsid (N) and spike (S) regions of Uzbekistan SARS-CoV-2 sequences based on comparison to the reference sequence (GenBank reference sequence accession number NC_045512.2). Table S3. Amino acid mutations of all sequenced samples.
Acknowledgments
We thank the private clinic of BiogenMed, Tashkent, Uzbekistan, including but not limited to Mrs. Larisa E. Alieva, for her collecting the samples from symptomatic patients.
Conflict of Interests
The authors declare that there are no conflicts of interest.
References
- hCoV-19 data sharing via GISAID. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants. (accessed on 20 July 2023).
- Cui, J.; Li, F.; and Shi, Z. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019, 17(3), 181-192. [CrossRef]
- WHO Director-General's opening remarks at the media briefing on COVID-19 - 11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020.
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 12 July 2023).
- Fehr, A.R. & Perlman, S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 2015, 1282, 1-23. [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270-273. [CrossRef]
- Yavarian, J.; Nejati, A.; Salimi, V.; Shafiei Jandaghi, NZ.; Sadeghi, K.; Abedi, A.; Zarchi, A.S.; Gouya, M.M.; Mokhtari-Azad, T. Whole genome sequencing of SARS-CoV2 strains circulating in Iran during five waves of pandemic. PLoS ONE 2022, 17(5). e0267847. [CrossRef]
- https://www.ncbi.nlm.nih.gov.
- https://www.viprbrc.org.
- Ayubov, M.S.; Buriev, Z.T.; Mirzakhmedov, M.K.; Yusupov, A.N.; Usmanov, D.E.; Shermatov, S.E.; Ubaydullaeva, K.A.; Abdurakhmonov, I.Y. Profiling of the most reliable mutations from sequenced SARS-CoV-2 genomes scattered in Uzbekistan. PLoS One. 2022,,17, (3):e0266417. https://doi.org/10.1371/journal.pone.0266417. [CrossRef]
- Danner, C.; Rosa-Aquino, P. What we know about the dangerous COVID B.1.617.2 (Delta) variant NY Intelligencer. Available online: https://nymag.com/intelligencer/article/covid-b-1-617-2-delta-variant-what-we-know.html. Accessed 17 June 2021.
- Ng, A. 11 May 2021. WHO labels a COVID strain in India as a “variant of concern”—here’s what we know. CNBC Africa, Gauteng, South Africa.
- Dhawan, M.; Sharma, A.; Priyanka, Thakur, N.; Rajkhowa, T.K.; Choudhary, O.P. Delta variant (B.1.617.2) of SARS-CoV-2: Mutations, impact, challenges and possible solutions. Hum Vaccin Immunother. 2022, 18(5), 2068883. [CrossRef]
- Harvey, W.T., Carabelli, A.M., Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 2021, 19, 409–424. [CrossRef]
- Abdullaev, A.; Abdurakhimov, A.; Mirakbarova, Z.; Ibragimova, S.; Tsoy, V., Nuriddinov S.; Dalimova D.; Turdikulova S.; Abdurakhmonov I. Genome sequence diversity of SARS-CoV-2 obtained from clinical samples in Uzbekistan. PLOS ONE. 2022, 17(6), e0270314. [CrossRef]
- Abdurakhmonov, I.; Buriev, Z.; Shermatov, S.; Usmanov, D.; Mirzakhmedov, M.; Ubaydullaeva, K.; Kamburova, V.; Rakhmanov, B.; Mirzakamol Ayubov, M. et al. The edible tomato COVID-19 vaccine, TOMAVAC, induces neutralizing IgGs. Research Square. 2023, preprint. [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; Team, N. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms. 2021, 9(7), 1542. [CrossRef]
Figure 1.
The Uzbek SARS-CoV-2 sequences clustered into two major groups.
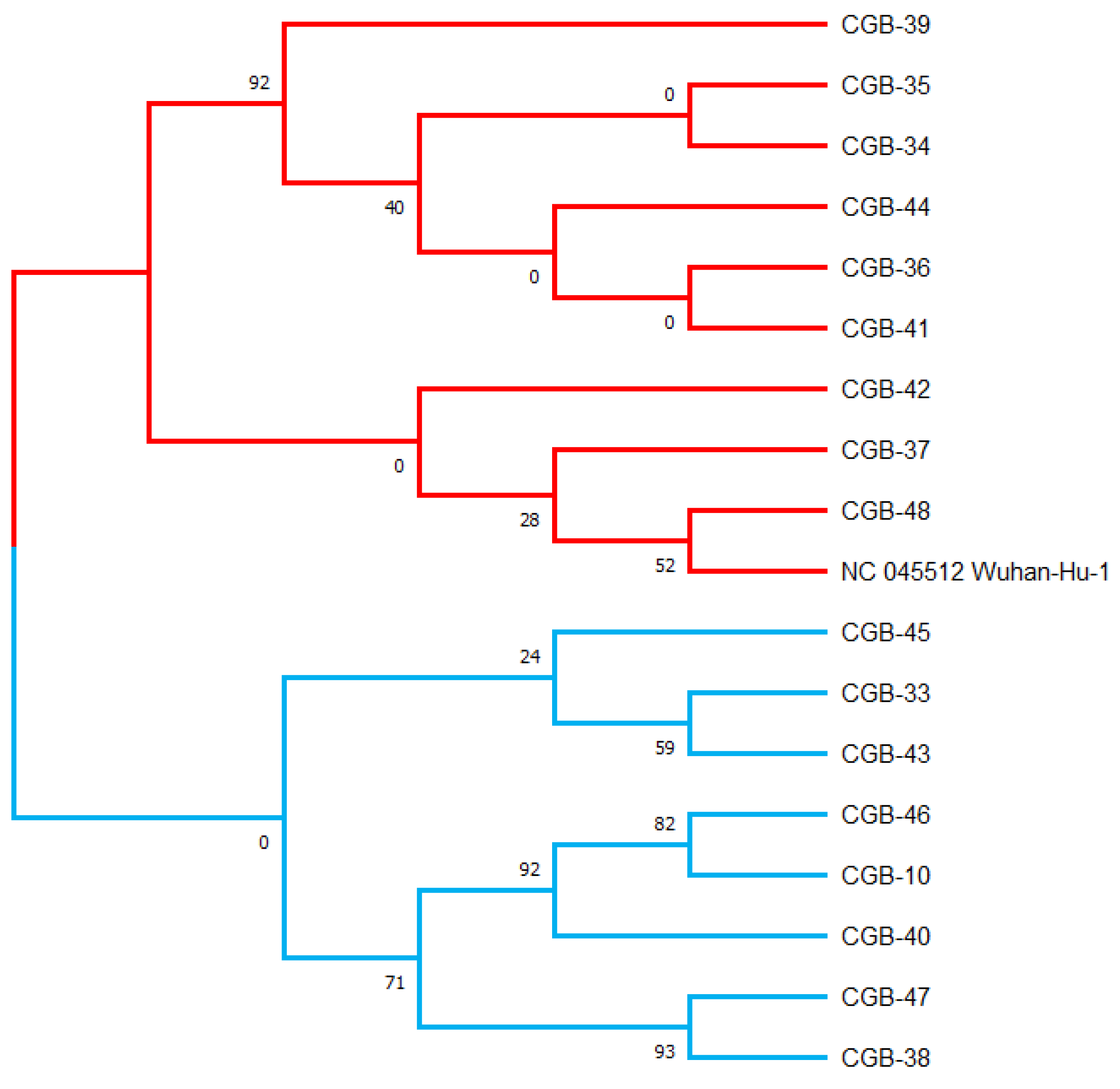
Figure 2.
The number of mutations in sequenced SARS-CoV-2 genomes. The number of shared mutations is calculated along with unique mutations per individual sample.
Figure 2.
The number of mutations in sequenced SARS-CoV-2 genomes. The number of shared mutations is calculated along with unique mutations per individual sample.
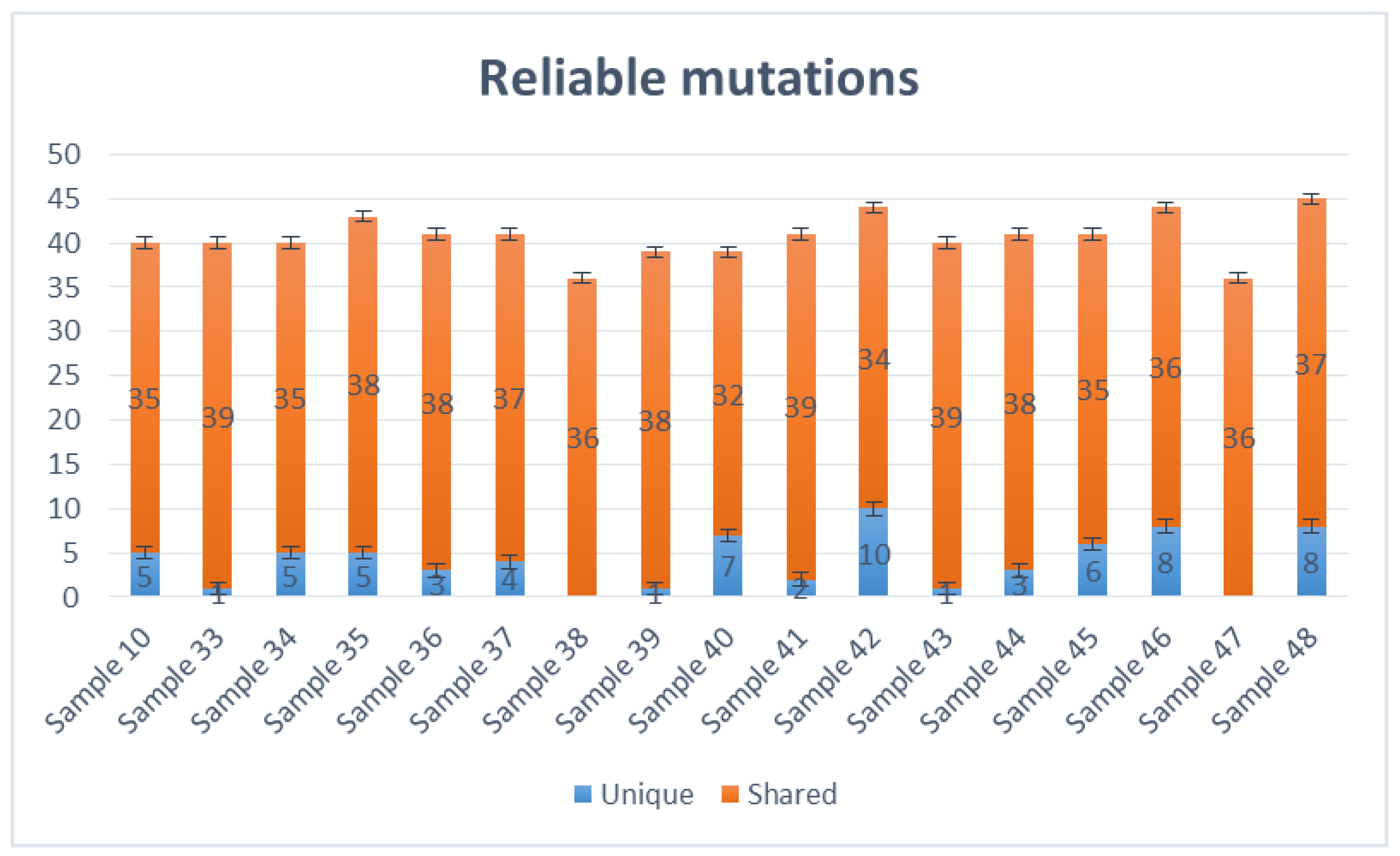
Figure 3.
Coronavirus typing tool analysis result. This phylogenetic tree provided by genomedetective.com using our sequenced samples.
Figure 3.
Coronavirus typing tool analysis result. This phylogenetic tree provided by genomedetective.com using our sequenced samples.
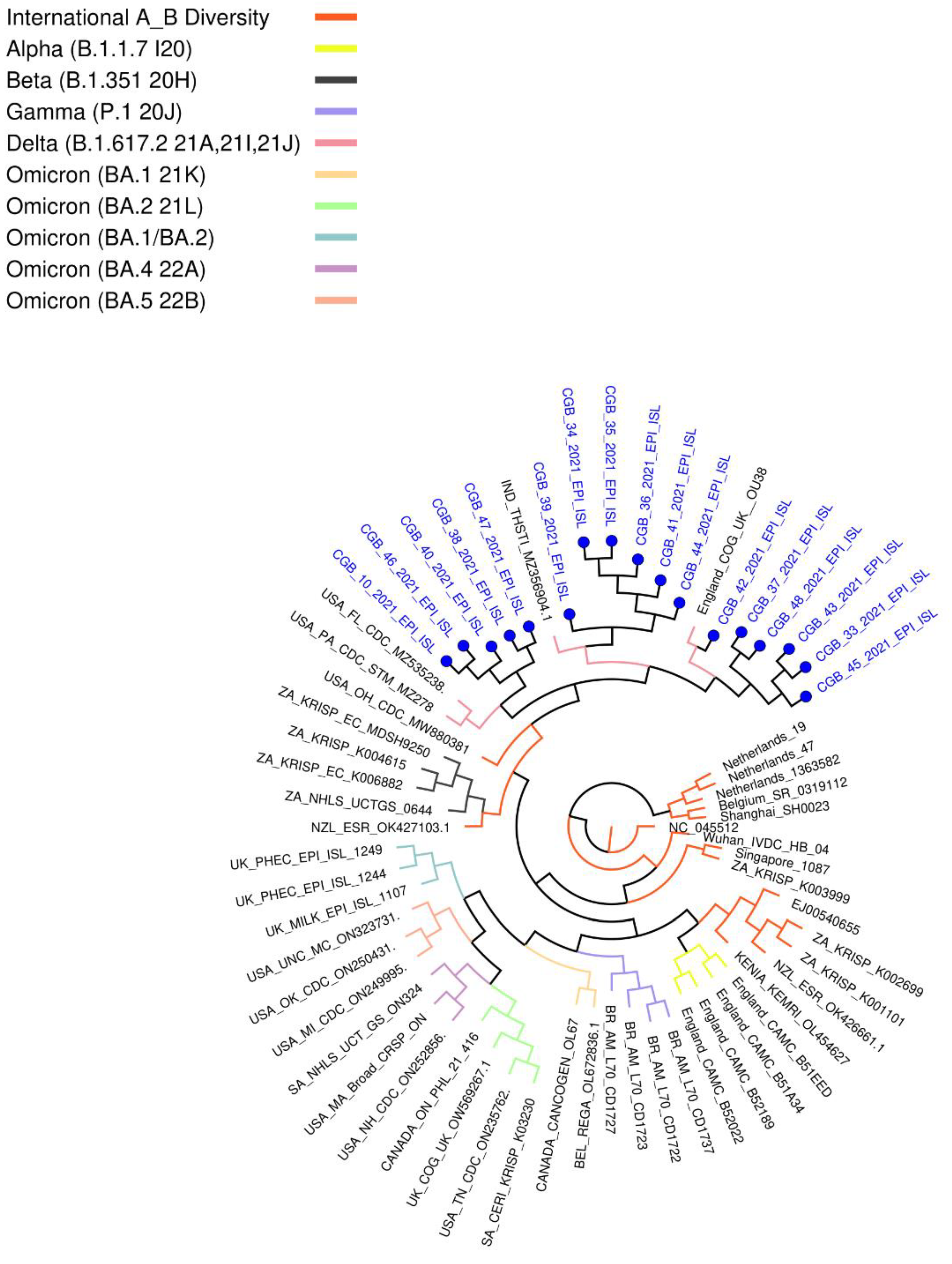

Table 1.
Summarized data for the COVID-19 samples used in this study.
| ID | Collection date | Sex | Age | Coverage | GISAID accession # |
NCBI accession # | Clade |
|---|---|---|---|---|---|---|---|
| 10 | 23/07/2021 | Female | 54 | 5943 | EPI_ISL_3668627 | MZ892621.1 | GK |
| 33 | 23/07/2021 | Female | 36 | 2854 | EPI_ISL_3668631 | * | GK |
| 34 | 23/07/2021 | Female | 24 | 17146 | EPI_ISL_3673667 | * | GK |
| 35 | 23/07/2021 | Female | 22 | 854,5 | EPI_ISL_3673670 | MZ892622.1 | GK |
| 36 | 23/07/2021 | Female | 6 | 5729 | EPI_ISL_3673672 | * | GK |
| 37 | 23/07/2021 | Male | 58 | 3305 | EPI_ISL_3668628 | MZ892623.1 | GK |
| 38 | 23/07/2021 | Female | 11 | 3167 | EPI_ISL_3668632 | MZ892624.1 | GK |
| 39 | 23/07/2021 | Male | 33 | 1813 | EPI_ISL_3673666 | * | GK |
| 40 | 23/07/2021 | Female | 59 | 2856 | EPI_ISL_3668633 | * | GK |
| 41 | 23/07/2021 | Male | 31 | 3134 | EPI_ISL_3673668 | * | GK |
| 42 | 23/07/2021 | Female | 38 | 1874 | EPI_ISL_3673671 | * | GK |
| 43 | 23/07/2021 | Female | 13 | 1807 | EPI_ISL_3668629 | MZ892625.1 | GK |
| 44 | 23/07/2021 | Male | 35 | 3442 | EPI_ISL_3673673 | * | GK |
| 45 | 23/07/2021 | Male | 50 | 1339 | EPI_ISL_3668634 | * | GK |
| 46 | 23/07/2021 | Female | 29 | 3448 | EPI_ISL_3668630 | MZ892626.1 | GK |
| 47 | 23/07/2021 | Female | 47 | 2819 | EPI_ISL_3673669 | * | GK |
| 48 | 23/07/2021 | Female | 28 | 3309 | EPI_ISL_3668635 | MZ892627.1 | GK |
* - GISAID only has accession numbers for these samples, but not in NCBI. Only registered users have access to the sequencing data provided to the GISAID database.
Table 2.
Selected high-quality sequenced samples along with high coverage.
| # | Sample Name | Mapped Reads | Target Reads | Mean Depth | Uniformity |
|---|---|---|---|---|---|
| 1 | CGB-10 | 1121237 | 99.64% | 5943 | 87.93% |
| 2 | CGB33 | 518399 | 99.84% | 2854 | 87.54% |
| 3 | CGB-34 | 3133616 | 99.76% | 17146 | 90.83% |
| 4 | CGB-35 | 201758 | 99.46% | 854.5 | 92.67% |
| 5 | CGB-36 | 1151910 | 99.76% | 5729 | 80.04% |
| 6 | CGB-37 | 620105 | 99.83% | 3305 | 80.50% |
| 7 | CGB-38 | 789833 | 99.41% | 3167 | 89.13% |
| 8 | CGB-39 | 442640 | 99.48% | 1813 | 90.51% |
| 9 | CGB-40 | 506837 | 99.81% | 2856 | 78.00% |
| 10 | CGB-41 | 714637 | 99.47% | 3134 | 92.57% |
| 11 | CGB-42 | 436501 | 99.52% | 1874 | 91.55% |
| 12 | CGB-43 | 431153 | 99.48% | 1807 | 91.85% |
| 13 | CGB-44 | 643427 | 99.88% | 3442 | 73.85% |
| 14 | CGB-45 | 279054 | 99.67% | 1339 | 89.57% |
| 15 | CGB-46 | 806289 | 99.45% | 3448 | 90.32% |
| 16 | CGB-47 | 665638 | 99.43% | 2819 | 91.42% |
| 17 | CGB-48 | 792525 | 99.44% | 3309 | 93.81% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



