Submitted:
13 July 2023
Posted:
14 July 2023
You are already at the latest version
Abstract
Apple snails of the genus Pomacea (Mollusca: Caenogastropoda: Ampullariidae) are native to the Neotropics and exhibit high species diversity. They hold cultural and ecological significance, serving as an important protein source in Peru. However, this genus has also received broad interest worldwide due to its invasive capacity, especially in Southeast Asia, where some species such as Pomacea canaliculata (Lamarck, 1822) and P. maculata Perry, 1810 are considered significant pests. Consequently, most genetic studies have focused almost exclusively on these invasive species. In contrast, few efforts have been made to understand the evolutionary relationships of the South American native species, especially at the genomic level. In this study, we assembled and annotated the mitochondrial genome of two Pomacea species native to the Peruvian Amazon: Pomacea reevei Ampuero & Ramírez, 2023 and P. aulanieri (Deville & Huppé, 1850). Through a comprehensive comparison with available mitogenomes on NCBI, we found that the length and organization of these mitogenomes fall within the range of what is currently known in Pomacea. Comparisons between these mitogenomes with those previously published revealed differences in the overlapping of adjacent genes, the size of certain protein-coding genes (PCGs) and the secondary structure of some tRNAs which are consistent with the phylogenetic relationships between these species. These findings provide valuable insights into the systematics and genomics of the genus Pomacea.
Keywords:
Ampullariidae
; mitogenome
; phylogeny
; secondary structure
; Control region
1. Introduction
The species of the genus Pomacea (Mollusca: Canogastropoda: Ampullariidae) are commonly known as apple snails and can be found in freshwater habitats such as small ponds, lakes, swamps and canals, extending throughout the Neotropical Region, from Argentina to southeast US and Caribbean Islands [1]. They have significant cultural and economic importance, serving as a traditional food for local populations in the Amazon region since pre-Hispanic times [2,3]. Moreover, due to their protein value and easy cultivation, they offer promising opportunities for aquaculture [4]. However, in the last decades, certain species such as Pomacea canaliculata (Lamarck, 1822) or Pomacea maculata Perry, 1810 have gained growing importance as invasive species, becoming pests of high economic impact on natural habitats and agricultural areas, particularly in China and Southeast Asia [5].
Consequently, numerous genetic studies have focused on the identification [6,7], phylogeography [8] and genetic diversity [9] of these invasive species. Since mitochondrial DNA has proved to be very useful to understand animal systematics and evolution [10,11,12], most genetic studies in Pomacea are based on mitochondrial markers, and mitochondrial genomes are available for invasive species such as P. canaliculata [13,14], P. maculata [15] or P. diffusa Blume, 1957 [16,17]. However, despite comparative studies have been performed on these mitogenomes [18,19], no mitochondrial genome has been published for any native Pomacea species to date.
In Peruvian Amazon, where apple snails are commonly referred to as "churos", up to 20 species of Pomacea have been reported. However, their taxonomy and evolutionary relationships are poorly known [20] and only recently they have received more attention [21,22,23]. Although the phylogenetic relationships of some Peruvian species such as Pomacea aulanieri (Deville & Huppé, 1850) of the P. bridgesii clade and the newly described Pomacea reevei Ampuero & Ramírez 2023 of the P. canaliculata clade have been recently elucidated using mitochondrial markers [22], mitochondrial genome information could offer novel insights into their evolution and phylogenetics.
Therefore, the aim of this study was to assemble and annotate the first mitochondrial genome of two native species of Pomacea from the Peruvian Amazon: Pomacea reevei and Pomacea aulanieri. We analyzed the gene content, nucleotide composition and secondary structure of the ribosomal RNAs (rRNAs), transfer RNAs (tRNAs) and control region, as well as the phylogenetic relationships based on a comparison with other Pomacea mitogenomes to improve our understanding of the molecular evolution in Pomacea.
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
Samples of Pomacea reevei and Pomacea aulanieri were bought from local fishermen from Loreto, Peru (Napo and Huallaga basins, respectively). They were relaxed using ethanol, euthanized by thermal shock (4°C), and then fixed and preserved in 96% ethanol [24]. Genomic DNA (gDNA) was extracted from 1 cm3 of tissue using the E.Z.N.A. Mollusc DNA Kit (OMEGA Bio-tek, Norcross, GA, USA). Concentration and quality of gDNA was measured using Nanodrop Lite Spectrophotometer. DNA integrity was verified with 1% agarose gel electrophoresis. Voucher specimens were deposited in the Museo de Historia Natural of the Universidad Nacional Mayor de San Marcos, Lima, Peru (MUSM).
2.2. Genome Sequencing and Assembly
The whole genome of Pomacea reevei and P. aulanieri was sequenced by Macrogen Inc. (Seoul, South Korea) using an Illumina NovaSeq 6000 platform. Before sequencing, gDNA integrity was verified using TapeStation gDNA Screen Tape on the 4200 TapeStation System. Approximately 5 Gb of raw data from 150 bp paired-end reads were generated for each sample. Raw data quality was evaluated with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and processed using Fastp [25] removing reads with >40% bases with Phred quality < Q15, trimming adapter sequences with >6 bases and trimming polyG tails. Mitogenome assembly were obtained using GetOrganelle [26] with the Pomacea canaliculata mitochondrial genome (KU052865.1) as a starting reference. Bandage [27] was used to verify if the assembly graph was circular.
2.3. Annotation of Mitochondrial Genome and Predictions of Secondary Structures
The MITOS2 web server (http://mitos2.bioinf.uni-leipzig.de/index.py) [28] was used to predict the protein-coding genes (PCGs), transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs). The annotation of the tRNAs was confirmed using ARWEN 1.2 (http://130.235.244.92/ARWEN/index.html) [29]. Protein-coding genes were manually corrected searching for open reading frameworks (ORFs) with the NCBI tool ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). Secondary structure of the RNAs was inferred using R2DT (https://rnacentral.org/r2dt) [30] and manually optimized using the 12S rRNA Eukaryote reference model [31] and the 16S rRNA Mollusca reference model [32]. Potential secondary structure of the control region was predicted using the RNAfold web server (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi). All secondary structures were drawn using Inkscape 1.1 (http://www.inkscape.org/). The mitochondrial genome maps of both species were drawn using the CGView tools [33] available at the web server Proksee (https://proksee.ca/).
2.4. Comparative Analysis
The mitochondrial genome and annotation files of P. canaliculata (KJ739609.1), P. diffusa (MF373586.1), P. maculata (MF401379.1) and P. occulta Yang & Yu, 2019 (KR350466.1) were retrieved from GenBank. We performed a re-annotation of these mitochondrial genomes using MITOS2 Web Server [28] and ORFfinder to verify current annotations. Nucleotide composition was calculated using MEGA X [34] for whole mitogenomes, PCGs, tRNAs, rRNAs and control region. Compositional asymmetry was calculated using the formulas for AT-skew = (A - T)/(A + T) and GC-skew = (G - C)/(G + C) [35]. Relative synonymous codon usage (RSCU) values were calculated using MEGA X [34]. Nucleotide diversity (π) was determined using DnaSP6 [36] with a sliding window analysis of 200 bp and a step size of 25 bp. Overall mean p-distance was calculated for each PCG using MEGA X [34]. The nonsynonymous nucleotide substitutions per nonsynonymous site (Ka), the synonymous nucleotide substitutions per synonymous site (Ks) and the ratio of nucleotide nonsynonymous to synonymous substitutions (Ka/Ks) were calculated for each PCG using DnaSP6 [36].
2.5. Phylogenetic Analysis
The phylogenetic analyses included 35 sequences of Caenogastropoda species and two outgroup species: Haliotis rubra (AY588938) and Aplysia californica (AY569552). Orthologs of the nucleotide sequences of the 13 PCGs, 12S rRNA, and 16S rRNA were individually aligned using Muscle 3.8 [37] implemented in Aliview 1.27 [38]. Ambiguous aligned regions were identified and removed using Gblocks 0.91b [39] under relaxed conditions in PhyloSuite v1.2.3 [40]. Individual gene alignments were concatenated using PhyloSuite v1.2.3 [40]. PartitionFinder2 v2.1.1 [41] in PhyloSuite v1.2.3 [40] was used to determine the best partitioning scheme and substitution model. Maximum Likelihood (ML) analysis was performed using IQ-TREE 2.1.2 [42] on the CIPRES Science Gateway web server [43], with 10000 ultrafast bootstrap replicates. Bayesian Inference (BI) analysis was performed using MrBayes 3.2 [44] on the CIPRES Science Gateway web server [43], with two independent runs of four Markov Chain Monte Carlo (MCMC) chains running for 2 million generations and sampling every 1000 generations with a burn-in of 25%. The resulting phylogenetic trees were visualized through FigTree v1.4.3 [45] and edited with Inskape 1.1. (http://www.inkscape.org/).
3. Results and Discussion
3.1. Organization and Structure of the Mitogenomes
The mitochondrial genomes of Pomacea reevei and P. aulanieri (GenBank accesion numbers OR253802 and OR253803, respectively) contained the typical 37 genes (13 PCGs, two rRNAs and 22 tRNAs), and a large non-coding region known as the control region (Figure 1, Table 1 and Table 2), although circular assemblies were not achieved due to repetitive sequences within this region. Previous studies in Pomacea also failed to recovered the complete control region [19] or have not reported it [18]. Most genes, including all PCGs and rRNAs, where encoded on the heavy strand (H-strand), whereas eigth tRNAs (tRNA-Met, tRNA-Tyr, tRNA-Cys, tRNA-Trp, tRNA-Gln, tRNA-Gly, tRNA-Glu, tRNA-Thr) were located on the light strand (L-strand). The lengths of both mitogenomes were similar: 15,660 bp in P. reevei and 16,096 bp in P. aulanieri. These values closely matched previous reports in other Pomacea species, where lengths between 15,516 (P. occulta) and 16,373 bp (P. diffusa) have been reported [13,14,15,16,17,18,19]. Both mitogenomes showed identical gene arrangements, which were consistent with other Ampullaridae species. Including the control region, 24 intergenic regions were identified in P. reevei, ranging from 2 to 292 bp, for a total of 714 bp (4.6% of mitogenome), whereas 25 intergenic regions were identified in P. aulanieri, ranging from 3 to 524, for a total of 1,081 bp (6.7% of mitogenome). The intergenic regions represented 4.8% of the mitogenome in P. canaliculata [13], 3.5% in P. maculata [19] and 10.1% in P. diffusa [19].
The longest overlapping region was found between ND5/tRNA-Phe in P. aulanieri (21 bp), although this overlapping was only 2 bp in P. reevei. This overlapping has also been reported in P. diffusa [18], with a similar length (20 bp) to P. aulanieri, but no in P. canaliculata, P. maculata nor P. occulta [18,19]. Both P. reevei and P. aulanieri also showed an overlapping region of 7 bp between NAD4L/NAD4 that has been reported in other Ampullariidae such as Marisa cornuaretis [46] and P. diffusa [18], as well as other Caenogastropoda [47,48,49]. The re-annotation of the mitogenomes of P. canaliculata, P. maculata and P. occulta in the present study showed that these species also had this overlapping, although it had not been previously reported [13,14,18,19]. P. aulanieri additionally showed two small overlapping regions between tRNA-Pro/ND6 (1 bp) and tRNA-Ser1/ND2 (1 bp). The overlapping between NAD1/tRNA-Pro reported for P. canaliculata, P. maculata and P. occulta [13,14,18,19] has not been found in P. reevei nor P. aulanieri. The protein-coding genes, rRNAs and tRNAs comprised 71.78%, 14.37% and 9.36% of the whole mitochondrial genome of P. reevei, respectively. Similar values were observed in P. aulanieri, where protein-coding genes, rRNAs and tRNAs represented 69.93%, 14.34% and 9.23%, respectively.
3.2. Nucleotide Composition
The base composition of the mitogenomes of P. reevei (30.10% A, 44.88% T, 13.14% C, 15.87% G) and P. aulanieri (29.48% A, 39.37% T, 14.83% C, 16.32% G) was similar (Table 3), showing a high content of A + T (70.98% in P. reevei and 68.85% in P. aulanieri), a negative AT-skew (-0.15 in P. reevei and -0.14 in P. aulanieri) and a positive GC-skew (0.09 in P. reevei and 0.05 in P. aulanieri). Comparing the different set of elements of the mitogenome, the control region (77.4%) showed the highest A + T content in P. reevei, whereas in P. aulanieri, the tRNAs (71.58%) and the control region (71.56%) showed the highest values. In both mitogenomes, the lowest A+T content was found in the PCGs. The PCGs also showed the lowest AT-skew, whereas the rRNAs had the highest AT-skew in both mitogenomes. Interestingly, although the GC-skew values were positive for the whole mitogenome, PCGs, rRNAs and tRNAs of P. reevei and P. aulanieri, the control region of the first showed a positive value, whereas in the second this region had a negative value. This difference in the GC-skew of the control region can also be observed in the Figure 1.
The comparison of the A+T content (Figure 2), between P. reevei, P. aulanieri and other species of Pomacea, showed that the A+T content of P. reevei (70.98%) was similar to species such as P. canaliculata (71.69%), P. maculata (72.31%) and P. occulta (71.93%), whereas the A+T content values of P. aulanieri (68.85%) was the lowest among the compared species and similar to P. diffusa (69.49%). A similar pattern was found after comparing the A+T content of the PCGs, rRNAs, tRNAs and especially the control region, where P. reevei (77.40%), P. canaliculata (79.39%), P. maculata (85.82%) and P. occulta (86.57%) of the P. canaliculata clade showed high values, in contrast to P. aulanieri (71.56%) and P. diffusa (73.70%) of the P. bridgesii clade, although these values could be affected by the recovered length of the control region in each species.
Negative AT-skew values were observed for the whole mitogenome, protein-coding genes and tRNAs of all Pomacea, whereas positive values were observed in the rRNAs (Figure 3). Although the control region of P. reevei and P. aulanieri showed a negative AT-skew, in other species this value was positive (e.g. P. canaliculata or P. diffusa). A similar pattern was observed in the GC-skew (Figure 4), were the whole mitogenomes and all the sets of elements showed positive values, except for the control region of P. aulanieri and P. diffusa. Perna & Kocher suggested that differences in the number and direction of AT-skew and GC-skew values reflect variations in selective pressures and nucleotide substitution processes [35]. Therefore, the positive AT-skew values of rRNAs and slightly negatives values for tRNAs comparing to the strongly negative values of PCGs could be a signal of the strong selective pressures that force PCGs to eliminate deleterious mutations.
3.3. Protein-Coding Genes and Codon Usage
The length of the PCGs ranged from 159 bp (ATP8) to 1,707 bp (NAD5) in P. reevei and the same was observed in P. aulanieri, but NAD5 was slightly longer (1,728 bp). A comparison of the length of all PCGs among reported Pomacea mitogenomes [13,14,15,16,17,18,19] revealed that the size of COI (1536 bp), COII (687 bp), ATP8 (159 bp), NAD6 (495 bp), CytB (1140 bp), NAD4L (297 bp), COIII (780 bp) and NAD3 (354 bp) is conserved among all species. The ATP6 of most Pomacea showed the same length (699 bp), with the exception of P. diffusa (714 bp). In NAD1, NAD4 and NAD5 we identified three groups: (1) P. reevei, with 939, 1,374 and 1,707 bp, respectively; (2) P. maculata, P. canaliculata and P. occulta, with 960, 1,368 and 1,710 bp, respectively; and (3) P. aulanieri and P. diffusa, with 945, 1,362 and 1,728 bp, respectively. The most conspicous variation in size was found in NAD2: 1,062 bp (P. canaliculata and P. maculata), 1,065 bp (P. occulta), 1,071 bp (P. diffusa) and 1,074 bp (P. reevei and P. aulanieri).
The start codon in all PCGs of both species was the same (ATG), similar to the reported in P. diffusa [16] and Marisa cornuarietis [46], although in P. canaliculata, P. maculata and P. occulta the start codon of COIII was ATA. The most common stop codon was TAA, found in 12 proteins of P. reevei and 9 of P. aulanieri. Only ATP6 in P. reevei used TAG, whereas ATP8, ATP6, NAD3 and NAD2 ended with this stop codon in P. aulanieri. Most Pomacea species also showed a preference for TAA as stop codon [18,19].
Excluding the stop codons, 3,734 and 3,739 codons were identified in the mitogenome of P. reevei and P. aulanieri, respectively. The codon usage pattern analysis of the PCGs in P. reevei (Table 4, Figure 5) showed that the most frequent amino acids were Leu (16.6%), Ser (14.18%) and Phe (9.1%), and the least used were Cys (1.18%) and Arg (1.5%). The most frequently detected codons in P. reevei were UUA (Leu2) and UUU (Phe), whereas the least common codon was CGG (Arg). Similar values were observed in P. aulanieri (Table 5, Figure 6), although here, CGC (Arg) was the least frequently used codon. In both species, all amino acids have their preferred codons.
Sun et al. indicated that nucleotide compositional asymmetry in the coding strand could affect codon and aminoacid usage [50]. Therefore, we expected that based on the negative AT-skew and positive GC-skew of Pomacea mitogenome, amino acids coded by GT-rich codons might be more frequently used than AC-rich codons. We corroborated this in both species (P. reevei and P. aulanieri), since most abundant aminoacids were Leu2 (TTN) and Phe (TTN). Yang et al. (2018b) found that in Pomacea, codons ending in A or T were more frequent than those ending in C or G. This pattern was found in this study, since in P. reevei and P. aulanieri codons ending in T (47.2% and 43.9%, respectively) and A (35.4% and 33.3%, respectively), collectively represent more than half of all codons.
3.4. Ribosomal and Transfer RNA Genes
Similar to other Pomacea species, the mitochondrial genome of P. reevei and P. aulanieri contained two ribosomal RNA. The 12S rRNA had a length of 909 bp in P. reevei and 960 bp in P. aulanieri, whereas the length of 16S rRNA was 1340 bp in P. reevei and 1348 bp in P. aulanieri. Based on the 12S rRNA and 16S rRNA length in Pomacea, three groups can be identified: (1) P. reevei, with a length of 909 and 1340 bp, respectively; (2) P. maculata, P. canaliculata and P. occulta, with a range of lengths of 929-936 and 1331-1336 bp, respectively; and (3) P. aulanieri and P. diffusa, with a range of lengths of 952-958 and 1346-1348 bp, respectively.
Secondary structures of ribosomal RNAs have been poorly explored among mollusks and the structures presented here for P. reevei and P. aulanieri are the first inferred within Pomacea. The secondary structure of the 12S rRNA of P. reevei (Figure 7) and P. aulanieri (Figure 8) had an identical organization, with four structural domains, although the most studied has been the Domain III as is the most conserved region allowing its successul PCR across a wide variety of taxa [51]. Here we found that the structure of the Domain III in P. reevei and P. aulanieri is similar to previous reports in other mollusks [31,52,53,54]. On the other side, the Domains I and II have been poorly studied and they were difficult to infer in both species, although Simon et al. indicated that these regions showed less homoplasy than Domains III and IV so they could be more phylogenetically informative and particularly useful for resolving ancient evolutionary relationships [51].
The 16S rRNA had a length of 1,340 bp in P. reevei (Figure 9 and Figure 10) and 1,348 bp in P. aulanieri (Figure 11 and Figure 12), showing the same organization, with six structural domains. These secondary structures agree with mollusk consensus structure proposed by Lydeard et al. [32], and they have the same number of stem and loops as the models inferred for the Caenogastropoda Cacozeliana lacertina and Paracrostoma paludiformis (available at https://crw-site.chemistry.gatech.edu/). Smith & Bond [55] observed that among spiders the secondary structure of 16S rRNA could be phylogenetically useful at higher taxonomic levels, whereas the loops, as the most variable regions, could be used at family level relationships. No study has evaluated the phylogenetic signal of the secondary structure of the 16S rRNA among Caenogastropoda.
The length of the tRNAs varied from 62 to 73 bp in P. reevei (Table 1), and from 62 to 74 bp in P. aulanieri (Table 2), a wider range than in P. canaliculata, P. maculata y P. occulta (64-70 bp), but smaller than in P. diffusa (62-76 bp). Comparing the length variation along the whole set of 22 tRNAs in Pomacea, only tRNA-Asp had the same length in all species, whereas the most variable tRNAs were tRNA-Ile (70-76 bp), tRNA-Ala (68-74 bp) and tRNA-Thr (66-71 bp).
Most tRNAs folded into the typical clover leaf secondary structure, with four arms and loops. However, both tRNA for Serine in P. reevei showed a large loop instead of the typical stem-loop structure in the DHU domain (Figure 13), whereas in P. aulanieri only tRNA-Ser2 showed this loop (Figure 14). Yang et al. [19] reported a similar loop in the tRNA-Ser1 of P. maculata, but Yang et al. [18] indicated that all tRNAs in P. canaliculata, P. maculata and P. diffusa had the typical clover leaf structure. However, after inferring with ARWEN 1.2 [29] the secondary structure of the tRNA of P. canaliculata (KJ739609.1), P. maculata (MF401379.1), P. occulta (KR350466.1) and P. diffusa (MF373586.1) we found that in the first three species the tRNA-Ser1 had a loop in the DHU domain, whereas in P. diffusa, tRNA-Ser2 had a loop in the same domain. Therefore, both P. aulanieri and P. diffusa of the P. bridgesii clade had a loop in tRNA-Ser2, whereas P. reevei, P. canaliculata, P. maculata and P. occulta of the P. canaliculata clade had a loop in tRNA-Ser1, although P. reevei also had a loop in tRNA-Ser2.
Figure 13[M1] . Secondary structures of the tRNA genes in the mitogenome of P. reevei.
3.5. Control Region
The incomplete control region of the mitochondrial genome of both species was located between tRNA-Phe and COIII and showed all the specific characteristics for the control region [56,57]: (i), represents the longest intergenic region; (ii), shows a high A+T content; (iii), has a potential secondary structure; and (iv), contains repetitive elements. We retrieved a control region length of 294 bp in P. reevei and 524 bp in P. aulanieri. Both are within the reported range in Pomacea, from 141 bp (P. occulta) to 806 bp (P. diffusa). In another Ampullariid, Marisa cornuarietis, this region is considerably smaller (63 bp) [46]. Brauer et al. also found interspecific differences in the control region length of Conus, with the size of this region in C. consors (698 bp) being five-fold longer than in C. textile or C. borgesi [57]. The control region in C. quercinus is even longer (943 bp) [49]. Within Pomacea, species from P. bridgesii clade (e.g. P. aulanieri) seem to have a longer control region than those of P. canaliculata clade (e.g. P. reevei).
A repetitive unit of 12 bp (ACATACATACAT) was manually identified in the control region of P. reevei, with eigth repetitions on H-strand, and three repetitions on L-strand. A repeat unit was also found in the control region of P. aulanieri, with a length of 23 bp (ATATAATCTCTATATGTGTATGG) and six copies on H-strand. Different repetitive units have been reported in Pomacea: five copies of GATACTATAATATAAA (16 bp) in P. occulta [15]; 13 copies on H-strand and 11 repeats on L-strand of AAGATACTATAATATA (16 bp) in P. maculata [19]; 11 repeats of TAAGATATAAAGAAACTAAGAGA (23 bp) in P. canaliculata [13]; and 19 copies on H-strand and 18 repeats on L-strand of ATCTATACATAC (12 bp) in P. diffusa [16]. Maynard et al. found many AT repeats in Haliotis rubra mitogenome, suggesting that if the number of AT repeats were polymorphic, it could become a marker for individual typing [58]. The variations observed in the number and motif of the repetitive units of the mitochondrial control region of Pomacea suggest it would be valuable to evaluate alternative strategies to resolve this repetitive region, such as long read sequencing [59].
The inferred secondary structure of the control region of P. reevei and P. aulanieri showed differences in the number and organization of stems and loops between both species (Figure 15). Similar differences have been found between the control region of Conus consors, C. borgesi and C. textile [57]. It has been suggested that stem-loop structures has a role to start replication in animal mitochondria [60], but they have been poorly studied in molluscan mitochondrial genome.
3.6. Genetic Variation among Pomacea
The nucleotide diversity along the whole mitogenome was evaluated for six Pomacea species (P. reevei, P. aulanieri, P. canaliculata, P. maculata, P. occulta and P. difusa) using a sliding window approach (Figure 16). The overall mean p-distance calculated for each protein- coding gene ranged from 0.127 (COI) to 0.244 (ATP8), with other genes such as NAD2 (0.213), NAD6 (0.206), NAD5 (0.198) or NAD1 (0.190) also showing elevated values. In contrast, the ribosomal RNAs showed lower variability values: 0.139 (12S rRNA) and 0.136 (16S rRNA), whereas the control region displayed the highest value of the whole mitogenome (0.435). Castellana et al. found among Vertebrates that COI, COII and COIII were the most conserved mitochondrial genes, contrasting with ATP8 and NAD genes that were the most variables, whereas CytB and ATP6 showed intermediate values [61]. Peretolchina et al. compared four mitogenomes among Caenogastropoda family Baicaliidae nad found little variability among COI, recommending instead COII and COIII for phylogenetic studies and CytB and NAD genes for population studies [62]. Here in Pomacea, we found similar patterns, with ATP8, CytB and NAD genes being the most variables, and COI, COII and COIII as the most conserved. Fonseca et al. suggested that the phylogenies inferred from NAD5 are the most topologically concordant with the one produced by the whole mitogenome [63]. Although we have no tested this for Pomacea, the high variability of this gene suggests it could be useful for phylogenetic studies, along with other genes such as CytB or NAD1. We found in Pomacea that all PCGs had Ka/Ks values below 1, indicating that they were evolved under purifying selection. Among the 13 PCGs, COI showed the slowest evolutionary rate, whereas NAD6 had the fastest rate, which is concordant with previous study in the three invasive Pomacea species [18].
3.7. Phylogenetic Analysis
The phylogenetic trees recovered by Maximum Likelihood (ML) and Bayesian Inference (BI) showed an almost identical topology, except for Costapex balwinae within Neogastropoda (Figure 18 and Figure S1). The inference generated with BI exhibited higher support values than ML, and it was used to represent the evolutionary relationships of Caenogastropoda (Figure 18), whereas the ML tree is shown in Figure S1. Most clades on Figure 18 showed a high support (BI: 1.00, ML: >90), with the exception of Ampullarioidea + Viviparoidea (BI: 0.83, ML:61), that have been classified with Obscurella hidalgoi (Cyclophoroidea) within Architaenioglossa, although the monophyly of this group has not been recovered using mitogenomes [64].
Although mitogenome sequence KY008698.1 is identied in GenBank as P. bridgesii, it was published as P. diffusa [17], and posteriorly analyzed with this species name [18,19]. We compared this sequence with P. diffusa MF373586.1, and found that the only difference was on the sizes of the control region, being longer in KY008698.1 due to the higher number of repetitive units. Therefore, we concluded that both represent the mitogenome of P. diffusa.
The phylogenetic relationships within Ampullariidae were recovered with the highest support values, both in the BI and ML trees, with Pomacea recovered as monophyletic. Within Pomacea, we recovered two main clades: (1) P. canaliculata, P. maculata, P. occulta and P. reevei; and (2) P. diffusa and P. aulanieri. Hayes et al. identified four groups within Pomacea based on nuclear and mitchondrial markers: (1) P. canaliculata clade, (2) P. bridgesii clade, (3) Effusa clade, and (4) Flagellata clade [65]. Posteriorly, P. maculata was differentiated within P. canaliculata clade [66]; and Ramírez et al. [22] recovered P. reevei (as Pomacea sp. 1) within P. canaliculata clade, and P. aulanieri within P. bridgesii clade. These phylogenetic relationships are confirmed here based on mitochondrial genomes, and agree with previous results [18,19]. So far, recovered Pomacea mitogenomes have been restricted to P. canaliculata and P. bridgesii clades, but no mitogenome is available for Effusa or Flagellata clades.
Along this study, we have found many differences between the mitogenome structure in Pomacea such as the presence and length of overlapping regions, PCGs lengths, COIII start codon, tRNA length range, secondary structure of tRNAs for Serine and ribosomal RNAs length that are summarized in Table 5. These differences are concordant with the topology of the molecular phylogeny (Figure 18), although P. reevei of the P. canaliculata clade showed some similitudes to P. bridgesii clade species such as P. aulanieri.
4. Conclusions
The comparative analysis of the mitogenomes of P. reevei, P. aulanieri and previously published mitogenomes in Pomacea, has revealed differences in the organization and structure of the mitogenomes, consistent with the phylogenetic relationships between the P. canaliculata and P. bridgesii clades. A similar comparison with other Pomacea clades, as well as other Ampullariidae genera would provide new insights into the evolution of these freshwater snails.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Phylogenetic relationship of Caenogastropoda species based on protein-coding genes and ribosomal RNAs inferred by the maximum likelihood (ML). Numbers on branches are bootstrap values. Pomacea species sequenced in this study are marked with an asterisk.
Author Contributions
Conceptualization, R.R. and A.M.; writing—original draft preparation, A.M.; writing—review and editing, R.R; methodology, validation and formal analyses, R.R., A.M., J.M., J.R., R.S, R.B. F.R, A.A., N.O. and C.C.; project administration and funding acquisition, R.R. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All mitogenome sequences generated in this study were deposited in GenBank under accession numbers OR253802 and OR253803.
Acknowledgments
We are grateful to R. Chujutalli and M. Solis for his help in the collection of material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hayes, K.A.; Cowie, R.H.; Jørgensen, A.; Schultheiß, R.; Albrecht, C.; Thiengo, S.C. Molluscan models in evolutionary biology: apple snails (Gastropoda: Ampullariidae) as a system for addressing fundamental questions. Am. Malacol. Bull. 2009, 27, 47–58. [Google Scholar] [CrossRef]
- Wright, L.E. Identifying immigrants to Tikal, Guatemala: Defining local variability in strontium isotope ratios of human tooth enamel. J. Archaeol. Sci. 2005, 32, 555–566. [Google Scholar] [CrossRef]
- Emery, K.F. Aprovechamiento de la fauna en Piedras Negras: dieta, ritual y artesanía del periodo Clásico maya. Mayab 2007, 19, 51–69. [Google Scholar]
- Llanos, M.; Tito, J.L.; Ruiz, W.; Yraja, P. Manual de Procesamiento de la Carne de Churo. Programa Nacional de Innovación para la Competitividad y Productividad (Innóvate Perú) del Ministerio de la Producción; Universidad Nacional Amazónica de Madre de Dios (UNAMAD): Madre de Dios, Peru, 2008; 12p. [Google Scholar]
- Cowie, R.H.; Hayes, K.A.; Strong, E.E.; Thiengo, S.C. Nonnative apple snails: systematics, distribution, invasion history and reasons for introduction. In Biology and management of invasive apple snails; Joshi, R.C., Cowie, R.H., Sebastian, L.S., Eds.; Philippine Rice Research Institute (PhilRice): Filipinas, 2017; pp. 3–32. [Google Scholar]
- Rawlings, T.A.; Hayes, K.A.; Cowie, R.H.; Collins, .T.M. The identity, distribution, and impacts of non-native apple snails in the continental United States. BMC Evol. Biol. 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Rao, S.R.; Ratnayeke, S.; Yow, Y.Y. The efficiency of universal mitochondrial DNA barcodes for species discrimination of Pomacea canaliculata and Pomacea maculata. PeerJ 2020, 8, e8755. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, Y.; Oulang, S.; Wu, X. Phylogeographic patterns and demographic history of Pomacea canaliculata and Pomacea maculata from different countries (Ampullariidae, Gastropoda, Mollusca). Nat. Conserv. 2019, 36, 71–92. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, M.; Zhang, Z.; Liu, Y.; Deng, Z.; Gong, X. Genetic Diversity of Two Globally Invasive Snails in Asia and Americas in Relation with Agricultural Habitats and Climate Factors. Diversity 2022, 14, 1069. [Google Scholar] [CrossRef]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Pett, W. Animal Mitochondrial DNA as We Do Not Know It: mt-Genome Organization and Evolution in Nonbilaterian Lineages. Genome Biol. Evol. 2016, 8, 2896–2913. [Google Scholar] [CrossRef]
- Ghiselli, F.; Gomes-Dos-Santos, A.; Adema, C.M.; Lopes-Lima, M.; Sharbrough, J.; Boore, J.L. Molluscan mitochondrial genomes break the rules. Philos. Trans. R. Soc. B 2021, 376, 20200159. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Y.; Zhu, S.; Xu, H.; Liu, Y.; Chen, L. The complete mitochondrial genome of Pomacea canaliculata (Gastropoda: Ampullariidae). Mitochondrial DNA Part A 2016, 27, 884–885. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, J.E.; Deng, Z.; Luo, H.; Guo, J.; He, S.; Luo, M.; Zhao, B. The complete mitochondrial genome of the golden apple snail Pomacea canaliculata (Gastropoda: Ampullariidae). Mitochondrial DNA Part B 2016, 1, 45–47. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, S.; Song, F.; Li, H.; Liu, J.; Liu, G; Yu, X.P. The mitochondrial genome of Pomacea maculata (Gastropoda: Ampullariidae). In Mitochondrial DNA Part A; 2016; Volume 27, pp. 2895–2896. [Google Scholar]
- Liu, S.; Yang, Q.; He, C.; Yu, X. The complete mitochondrial genome of Pomacea diffusa (Gastropoda: Ampullariidae). Mitochondrial DNA Part B 2017, 2, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yang, H.; Zhang, C.; Xue, H.; Xia, Y.; Zhang, J.E. Complete mitochondrial genome of the apple snail Pomacea diffusa (Gastropoda, Ampullariidae) with phylogenetic consideration. Mitochondrial DNA Part B 2017, 2, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, J.E.; Xia, J.; Yang, J.; Guo, J.; Deng, Z.; Luo, M. Comparative characterization of the complete mitochondrial genomes of the three apple snails (Gastropoda: Ampullariidae) and the phylogenetic analyses. Int. J. Mol. Sci. 2018, 19, 3646. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, S.; Song, F.; Liu, G.F.; Yu, X.P. Comparative mitogenome analysis on species of four apple snails (Ampullariidae: Pomacea). Int. J. Biol. Macromol. 2018, 118, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, R.; Paredes, C.; Arenas, J. Moluscos del Perú. Rev. De Biol. Trop. 2003, 51 (Suppl. S3), 225–284. [Google Scholar]
- Ramírez, R.; Solis, M.; Ampuero, A.; Morín, J.; Jimenez-Vasquez, V.; Ramirez, J.; Congrains, C.; Temoche, H.; Shiga, Y.B. Identificación molecular y relaciones evolutivas de Pomacea nobilis, base para la autenticación específica del churo negro de la Amazonia peruana. Rev. Peru. De Biol. 2020, 27, 139–148. [Google Scholar] [CrossRef]
- Ramírez, R.; Ramírez, J.L.; Rivera, F.; Justino, S.; Solis, M.; Morín, J.; Ampuero, A.; Mendivil, A.; Congrains, C. 2022. Do not judge a snail by its shell: Molecular identification of Pomacea (Ampullariidae) species, with particular reference to the Peruvian Amazonian giant apple snail. Arch. Für Molluskenkd. 2022, 151, 7–17. [Google Scholar]
- Ampuero, A.; Ramirez, R. Description of two new species of apple snail (Ampullariidae: Pomacea) from Peruvian Amazonia. Zootaxa 2023, 5258, 76–98. [Google Scholar] [CrossRef]
- Sturm, C.F.; Mayhew, R.; Bales, B.R. Field and laboratory methods in malacology. In The mollusks: a guide to their study, collection, and preservation; Sturm, C.F., Pearce, T.A., Valdes, A. Eds, Eds.; American Malacological Society: Pittsburgh, 2006; pp. 9–31. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; dePamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. 2008. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, B.A.; Hoksza, D.; Nawrocki, E.P.; Ribas, C.E.; Madeira, F.; Cannone, J.J.; Gutell, R; Maddala, A.; Meade, C.D.; Williams, L.D.; Petrov, A.S.; Chan, P.P.; Lowe, T.M.; Finn, R.D.; Petrov, A.I. R2DT is a framework for predicting and visualising RNA secondary structure using templates. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Hickson, R.E.; Simon, C.; Cooper, A.; Spicer, G.S.; Sullivan, J.; Penny, D. Conserved Sequence Motifs, Alignment, and Secondary Structure for the Third Domain of Animal 12S rRNA. Mol. Biol. Evol. 1996, 13, 150–169. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, C.; Holznagel, W.E.; Schnare, M.N.; Gutell, R.R. Phylogenetic Analysis of Molluscan Mitochondrial LSU rDNA Sequences and Secondary Structures. Mol. Phylogenetics Evol. 2000, 15, 83–102. [Google Scholar] [CrossRef]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–359. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA Sequence Polymorphism Analysis of Large Dataset. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Nguyen, L.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Computing Environments Workshop (GCE), 2010. [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L. ; Darling, A; Höhna, S.; Larget, B; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 30 May 2022).
- Wang, M.; Qiu, J. Complete mitochondrial genome of the giant ramshorn snail Marisa cornuarietis (Gastropoda: Ampullariidae). Mitochondrial DNA 2014, 27, 1734–1735. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.K; Stevenson, B.J.; Ownby, J.P.; Cady, M.T.; Watkins, M.; Olivera, B.M. The mitochondrial genome of Conus textile, coxI-coxII intergenic sequences and Conoidean evolution. Mol. Phylogenetics Evol. 2008, 46, 215–223. [Google Scholar] [CrossRef]
- Rawlings, T.A.; Macinnis, M.J.; Bieler, R.; Boore, J.L.; Collins, T.M. Sessile snails, dynamic genomes: gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genom. 2010, 11, 440. [Google Scholar] [CrossRef]
- Gao, B.; Peng, C.; Chen, Q.; Zhang, J.; Shi, Q. Mitochondrial genome sequencing of a vermivorous cone snail Conus quercinus supports the correlative analysis between phylogenetic relationships and dietary types of Conus species. PLoS ONE 2018, 13, e0193053. [Google Scholar] [CrossRef]
- Sun, S.; Li, Q.; Kong, L.; Yu, H. Multiple reversals of strand asymmetry in molluscs mitochondrial genomes, and consequences for phylogenetic inference. Mol. Phylogenetics Evol. 2018, 118, 222–231. [Google Scholar] [CrossRef]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved PCR primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Medina, M.; Collins, T.M.; Walsh, P.J. Mtdna Ribosomal Gene Phylogeny of Sea Hares in the Genus Aplysia (Gastropoda, Opisthobranchia, Anaspidea): Implications for Comparative Neurobiology. Syst. Biol. 2001, 50, 7676–688. [Google Scholar] [CrossRef]
- Vogler, R.E.; Beltramino, A.A.; Strong, E.E.; Rumi, A.; Peso, J.G. Insights into the Evolutionary History of an Extinct South American Freshwater Snail Based on Historical DNA. PLoS ONE 2016, 11, e0169191. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, L.B.; Vogler, R.E.; Beltramino, A.A. The mitochondrial genome of the semi-slug Omalonyx unguis (Gastropoda: Succineidae) and the phylogenetic relationships within Stylommatophora. PLoS ONE 2021, 16, e0253724. [Google Scholar] [CrossRef]
- Smith, S.D.; Bond, J.E. An analysis of the secondary structure of the mitochondrial large subunit rRNA gene (16S) in spiders and its implications for phylogenetic reconstruction. J. Arachnol. 2003, 31, 44–54. [Google Scholar] [CrossRef]
- Soroka, M. Characteristics of mitochondrial DNA of unionid bivalves (Mollusca: Bivalvia: Unionidae). II. Comparison of complete sequences of maternally inherited mitochondrial genomes of Sinanodonta woodiana and Unio pictorum. Folia Malacol. 2010, 18, 189–209. [Google Scholar] [CrossRef]
- Brauer, A.; Kurz, A.; Stockwell, T.; Baden-Tillson, H.; Heidler, J.; Wittig, I.; Kauferstein, S.; Mebs, D.; Stöcklin, R.; Remm, M. The Mitochondrial Genome of the Venomous Cone Snail Conus consors. PLoS ONE 2012, 7, e51528. [Google Scholar] [CrossRef]
- Maynard, B.T.; Kerr, L.J.; Mckiernan, J.M.; Jansen, E.S.; Hanna, P.J. Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (Leach). Marine Biotechnology 2005, 7, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Ramón-Laca, A.; Gallego, R.; Nichols, K.M. Affordable de novo generation of fish mitogenomes using amplification-free enrichment of mitochondrial DNA and deep sequencing of long fragments. Mol. Ecol. Resour. 2023, 23, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Vicario, S.; Saccone, C. Evolutionary Patterns of the Mitochondrial Genome in Metazoa: Exploring the Role of Mutation and Selection in Mitochondrial Protein–Coding Genes. Genome Biol. Evol. 2011, 3, 1067–1079. [Google Scholar] [CrossRef]
- Peretolchina, T.E.; Sitnikova, T.Y.; Sherbakov, D.Y. The complete mitochondrial genomes of four Baikal molluscs from the endemic family Baicaliidae (Caenogastropoda: Truncatelloida). J. Molluscan Stud. 2020, 86, 201–209. [Google Scholar] [CrossRef]
- Fonseca, M.M.; Lopes-Lima, M.; Eackles, M.S.; King, T.L.; Froufe, E. The female and male mitochondrial genomes of Unio delphinus and the phylogeny of freshwater mussels (Bivalvia: Unionida). Mitochondrial DNA B 2016, 1, 954–957. [Google Scholar] [CrossRef]
- Osca, D.; Templado, J.; Zardoya, R. Caenogastropod mitogenomics. Mol. Phylogenetics Evol. 2015, 93, 118–128. [Google Scholar] [CrossRef]
- Hayes, K.A.; Cowie, R.H.; Thiengo, S.C. A global phylogeny of apple snails: Gondwanan origin, generic relationships, and the influence of outgroup choice (Caenogastropoda: Ampullariidae). Biol. J. Linn. Soc. 2009, 98, 61–76. [Google Scholar] [CrossRef]
- Hayes, K.A.; Cowie, R.H.; Thiengo, S.C.; Strong, E.E. Comparing apples with apples: clarifying the identities of two highly invasive Neotropical Ampullariidae (Caenogastropoda). Zool. J. Linn. Soc. 2012, 166, 723–753. [Google Scholar] [CrossRef]
Figure 1.
Graphical map of the mitochondrial genomes of P. reevei and P. aulanieri.
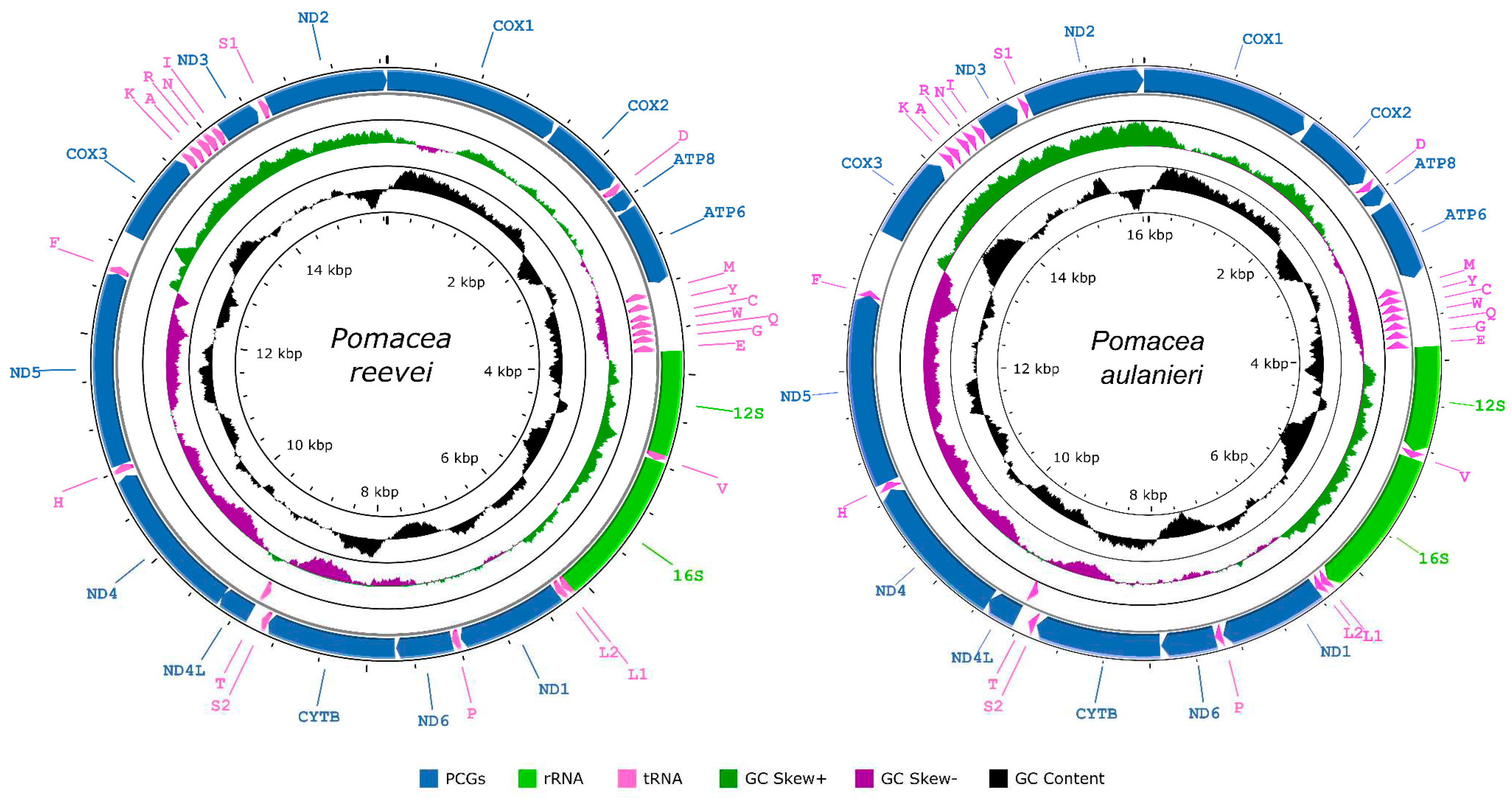
Figure 2.
Figure 2. A+T content of the mitochondrial genome of P. reevei, P. aulanieri, P. canaliculata, P. difusa, P. maculata and P. occulta.
Figure 2.
Figure 2. A+T content of the mitochondrial genome of P. reevei, P. aulanieri, P. canaliculata, P. difusa, P. maculata and P. occulta.
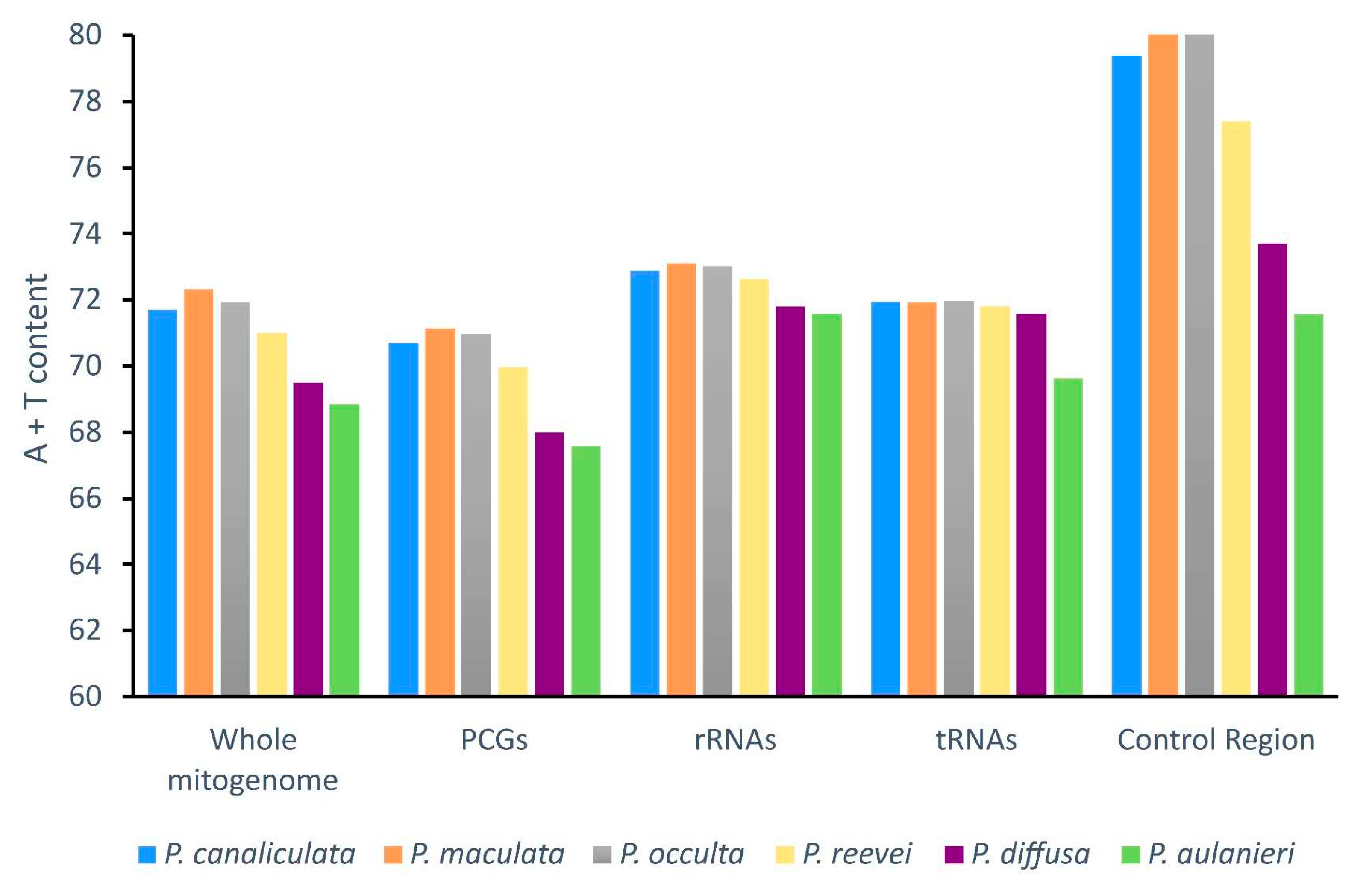
Figure 3.
AT-skew of the mitochondrial genome of P. reevei, P. aulanieri, P. canaliculata, P. difusa, P. maculata and P. occulta.
Figure 3.
AT-skew of the mitochondrial genome of P. reevei, P. aulanieri, P. canaliculata, P. difusa, P. maculata and P. occulta.
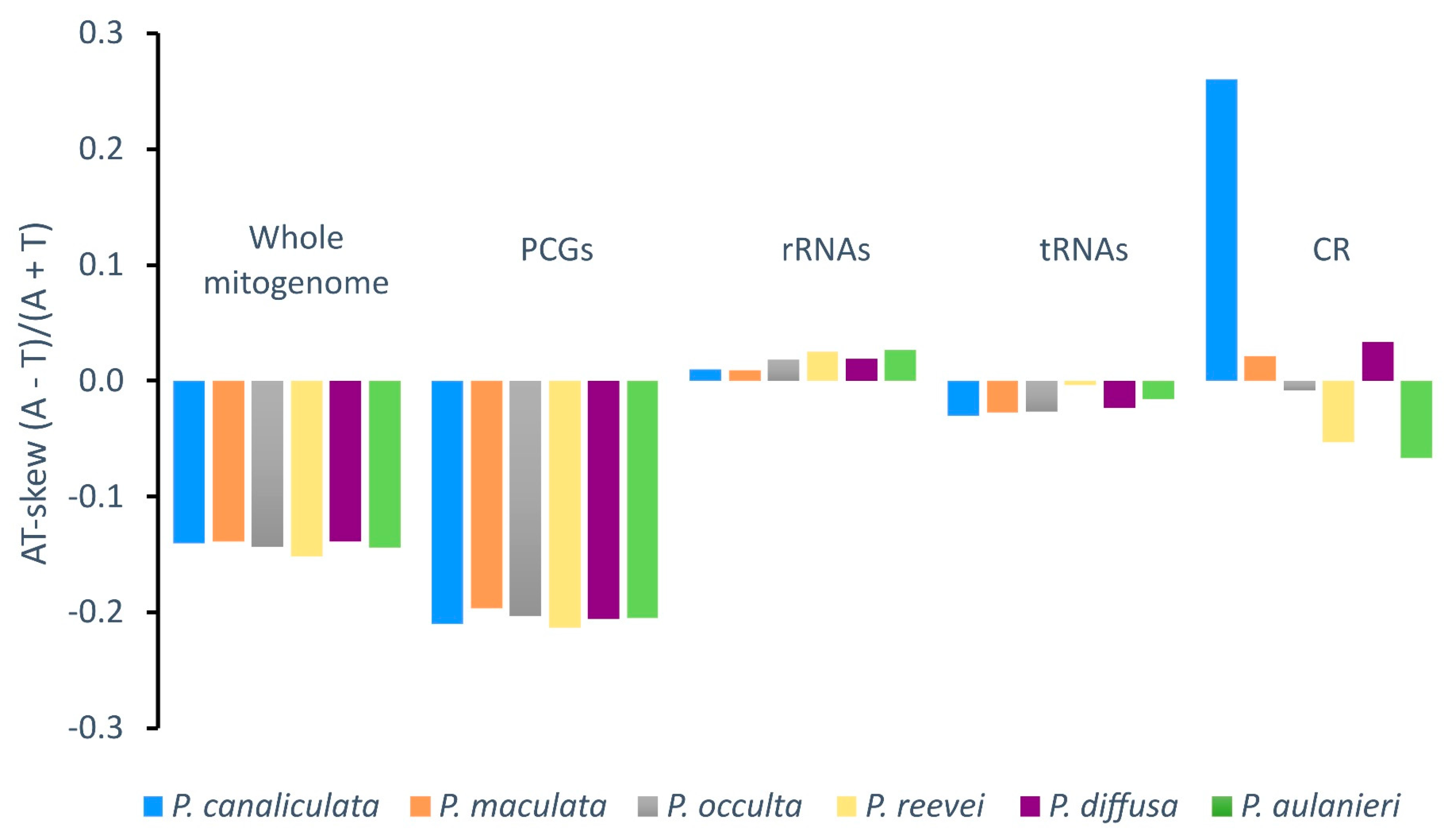
Figure 4.
GC-skew of the mitochondrial genome of P. reevei, P. aulanieri, P. canaliculata, P. maculata, P. occulta and P. difusa.
Figure 4.
GC-skew of the mitochondrial genome of P. reevei, P. aulanieri, P. canaliculata, P. maculata, P. occulta and P. difusa.
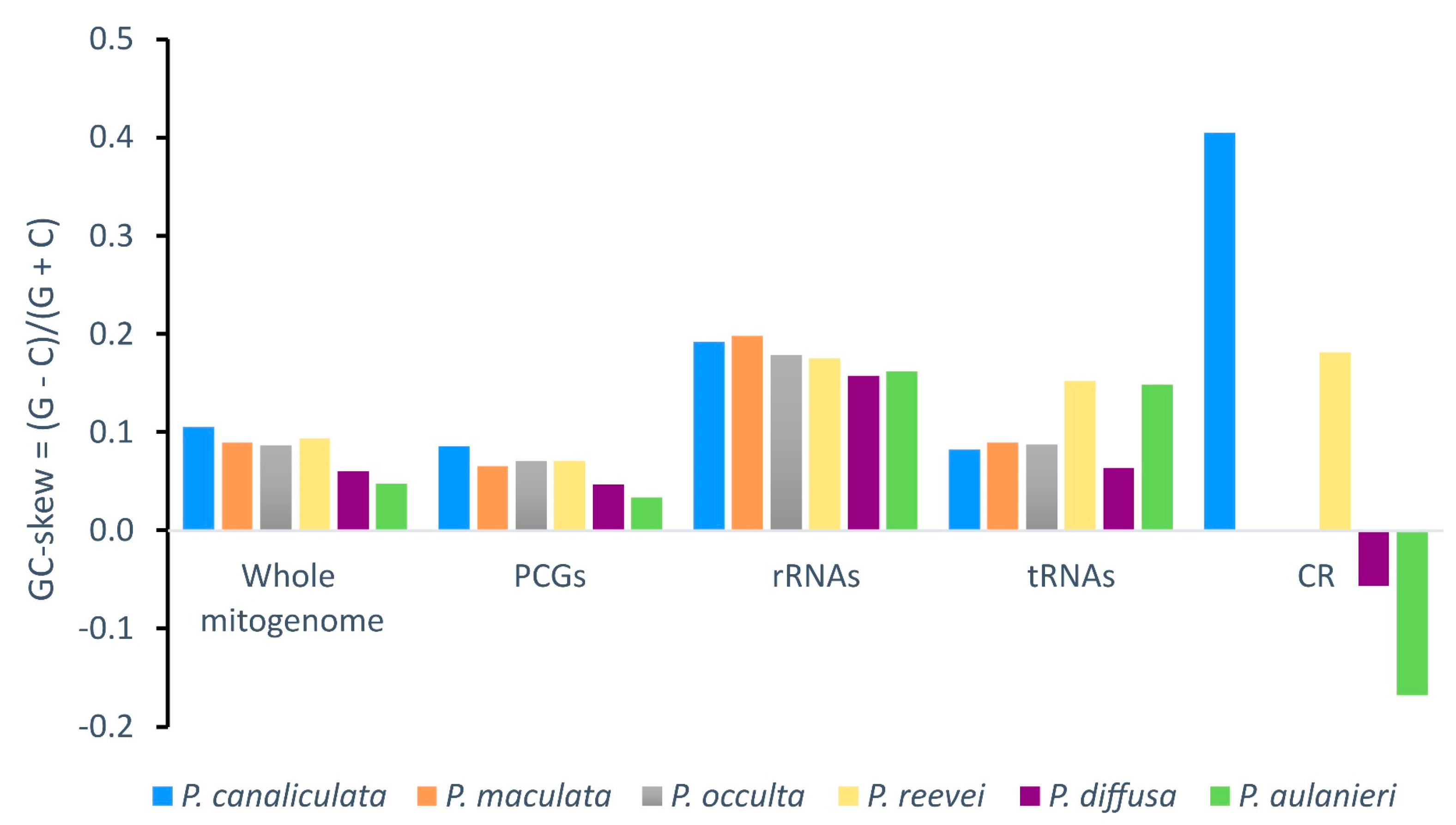
Figure 5.
Relative synonymous codon usages (RSCU) in the mitogenome of P. reevei.
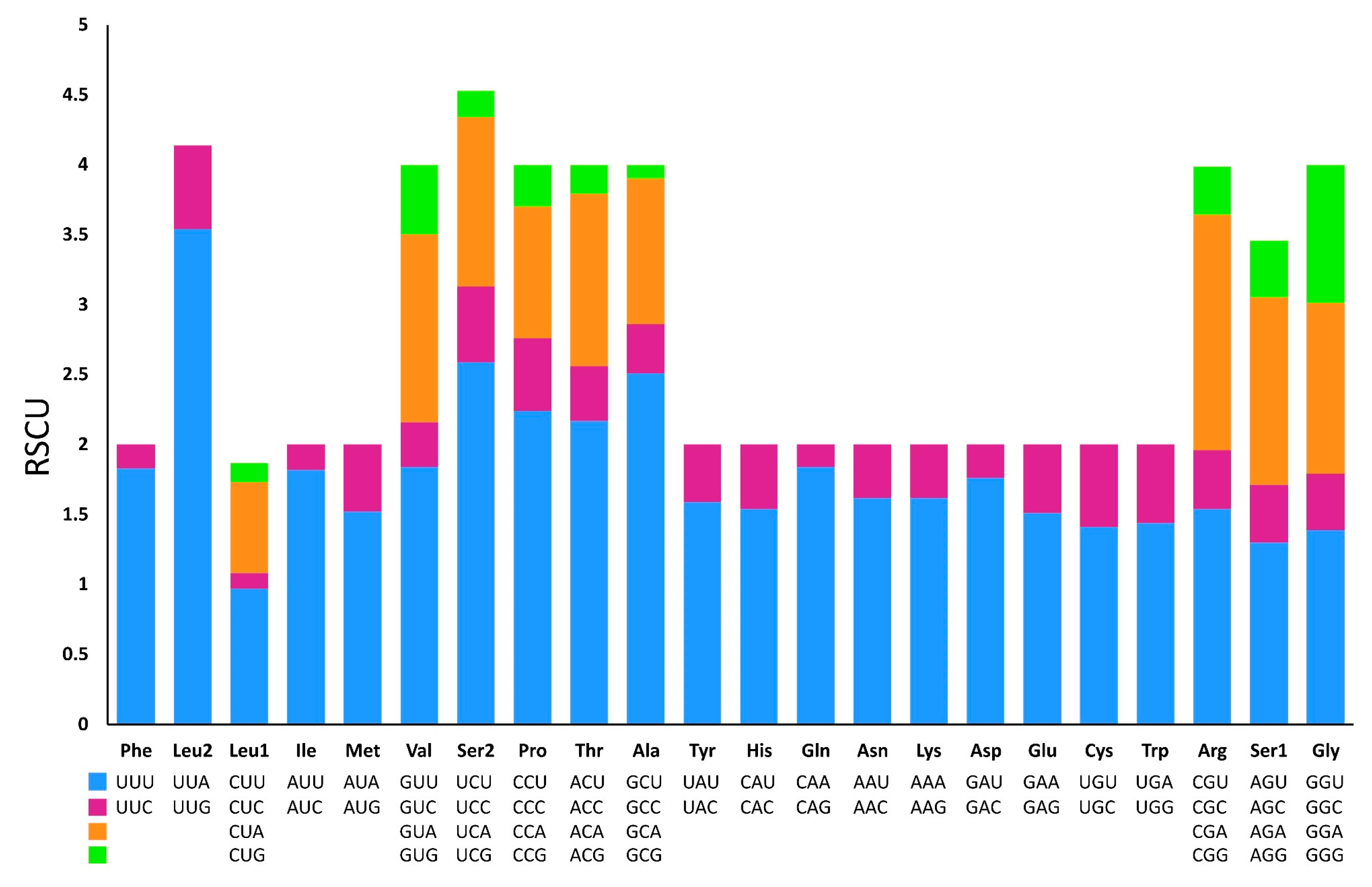
Figure 6.
Relative synonymous codon usages (RSCU) in the mitogenome of P. aulanieri.
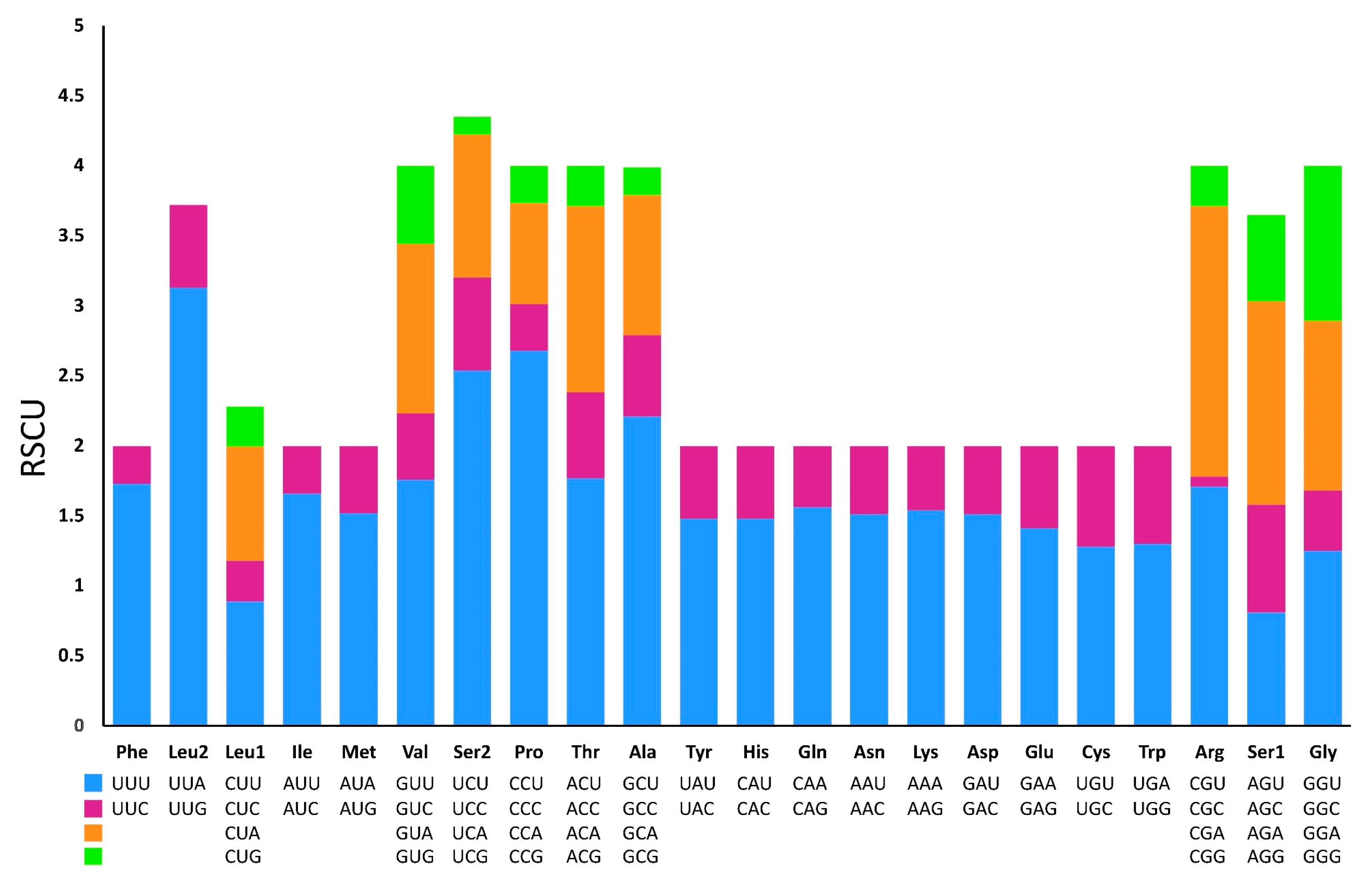
Figure 7.
Secondary structure of the 12S rRNA of P. reevei.
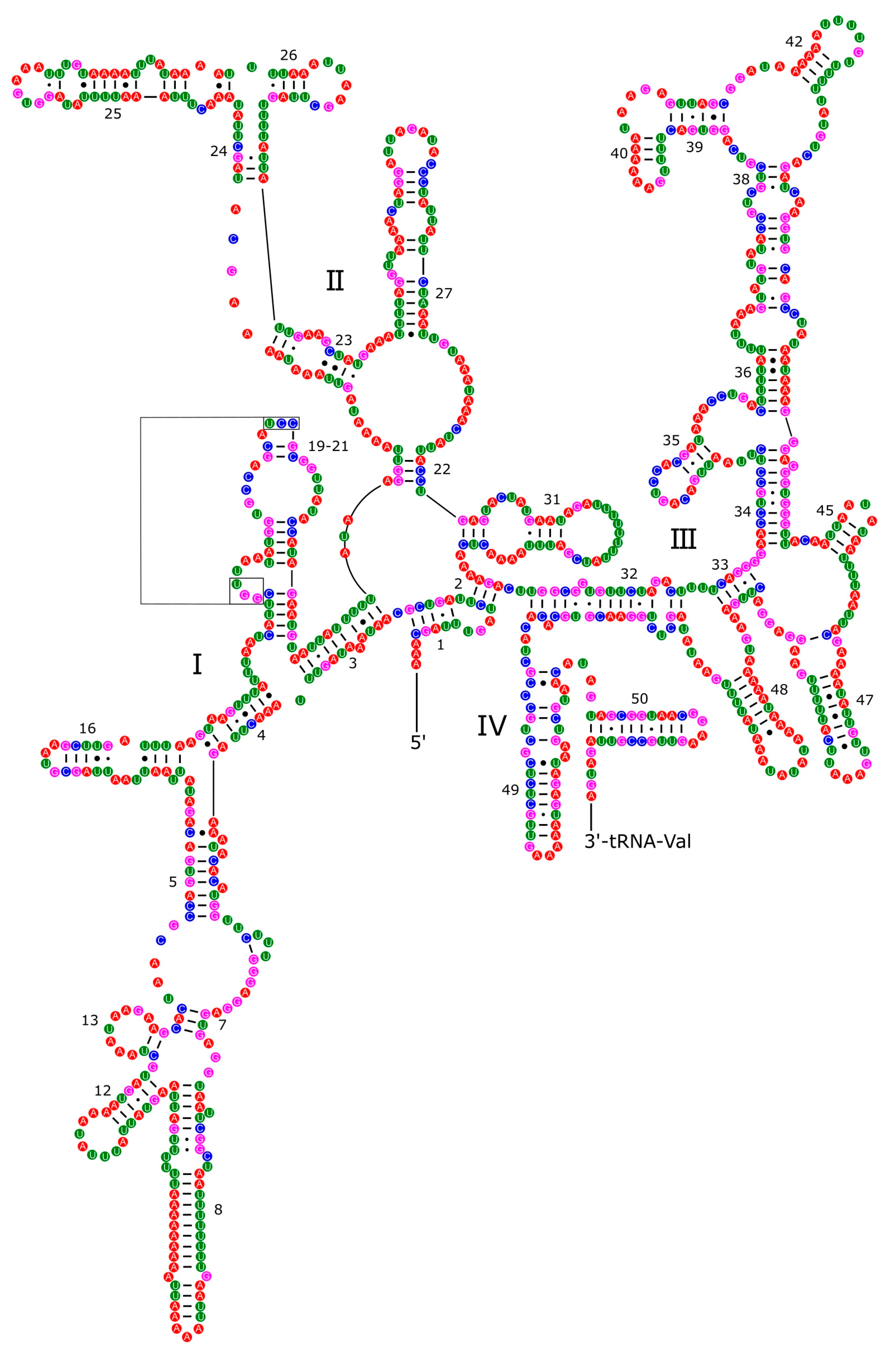
Figure 8.
Secondary structure of the 12S rRNA of P. aulanieri.
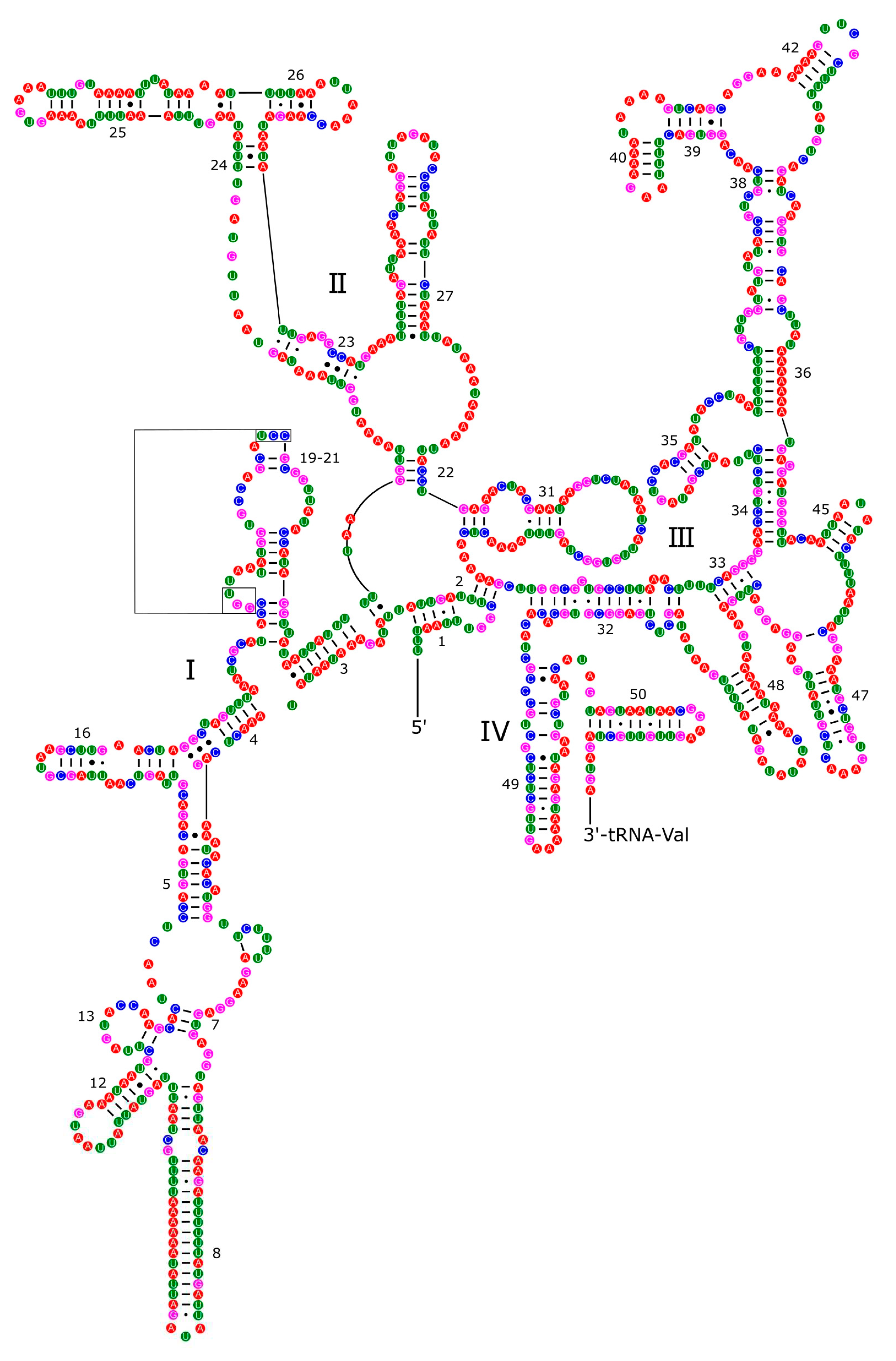
Figure 9.
Secondary structure of the I-III domains of the 16S rRNA of P. reevei.
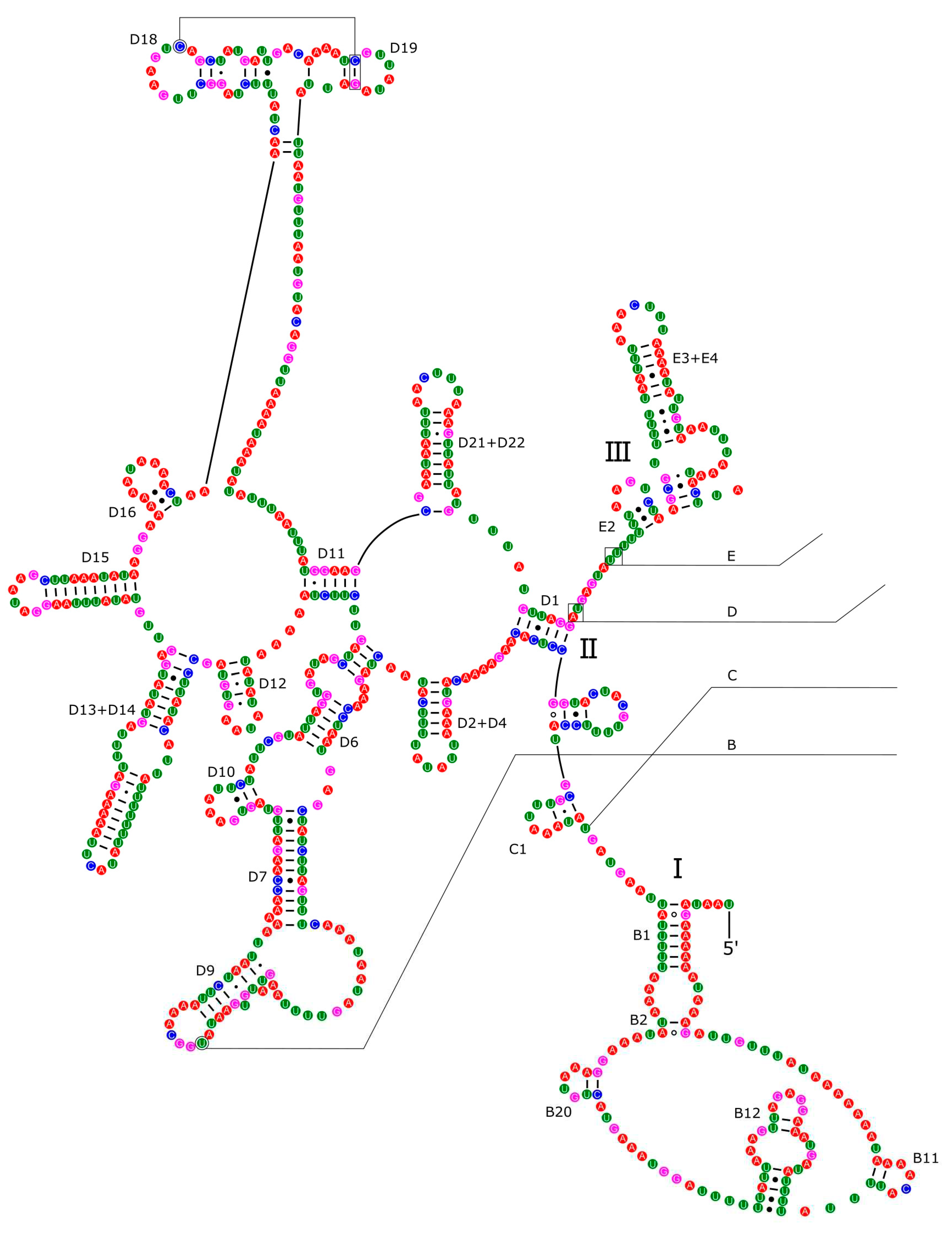
Figure 10.
Secondary structure of the IV-VI domains of the 16S rRNA of P. reevei.
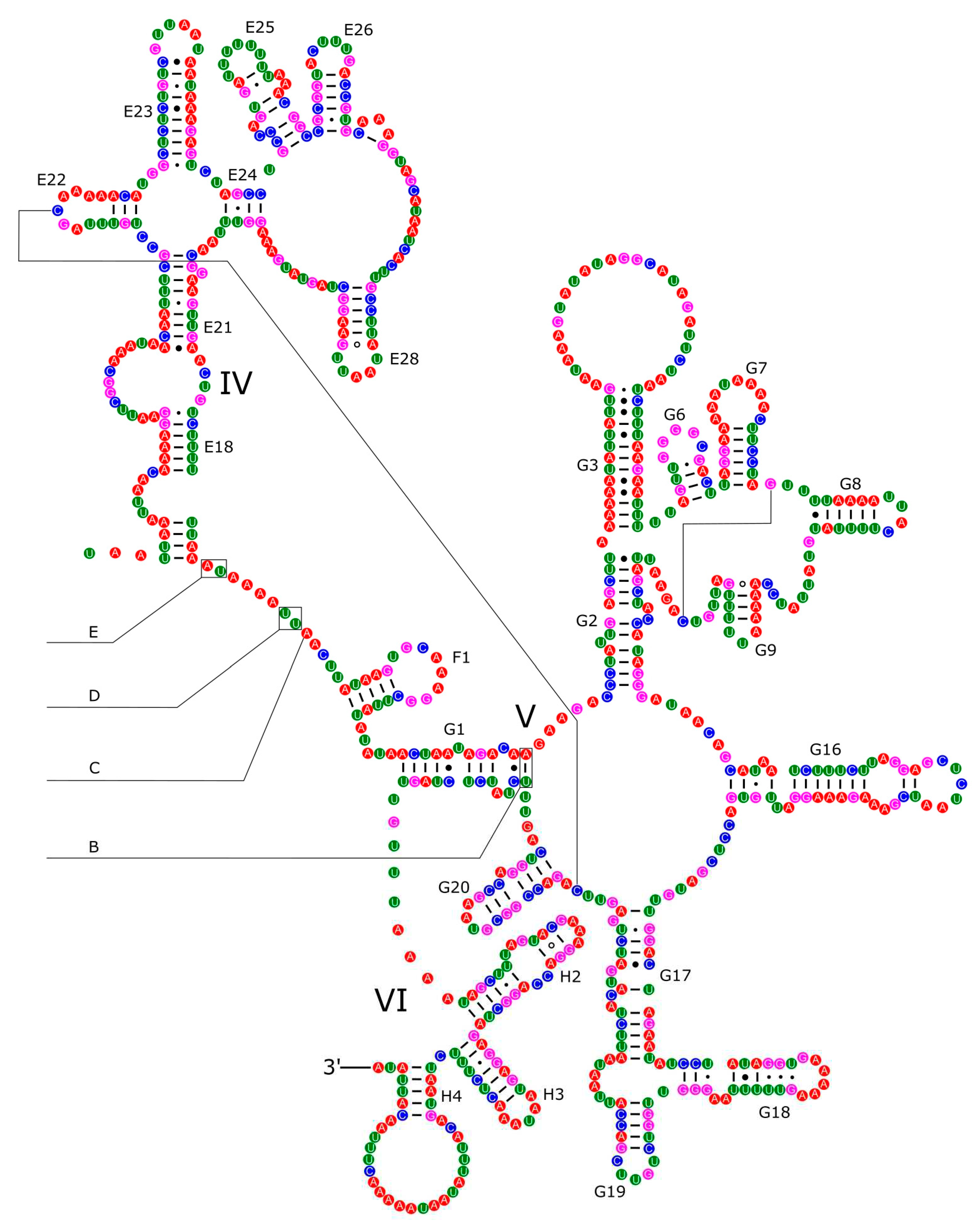
Figure 11.
Secondary structure of the I-III domains of the 16S rRNA of P. aulanieri.
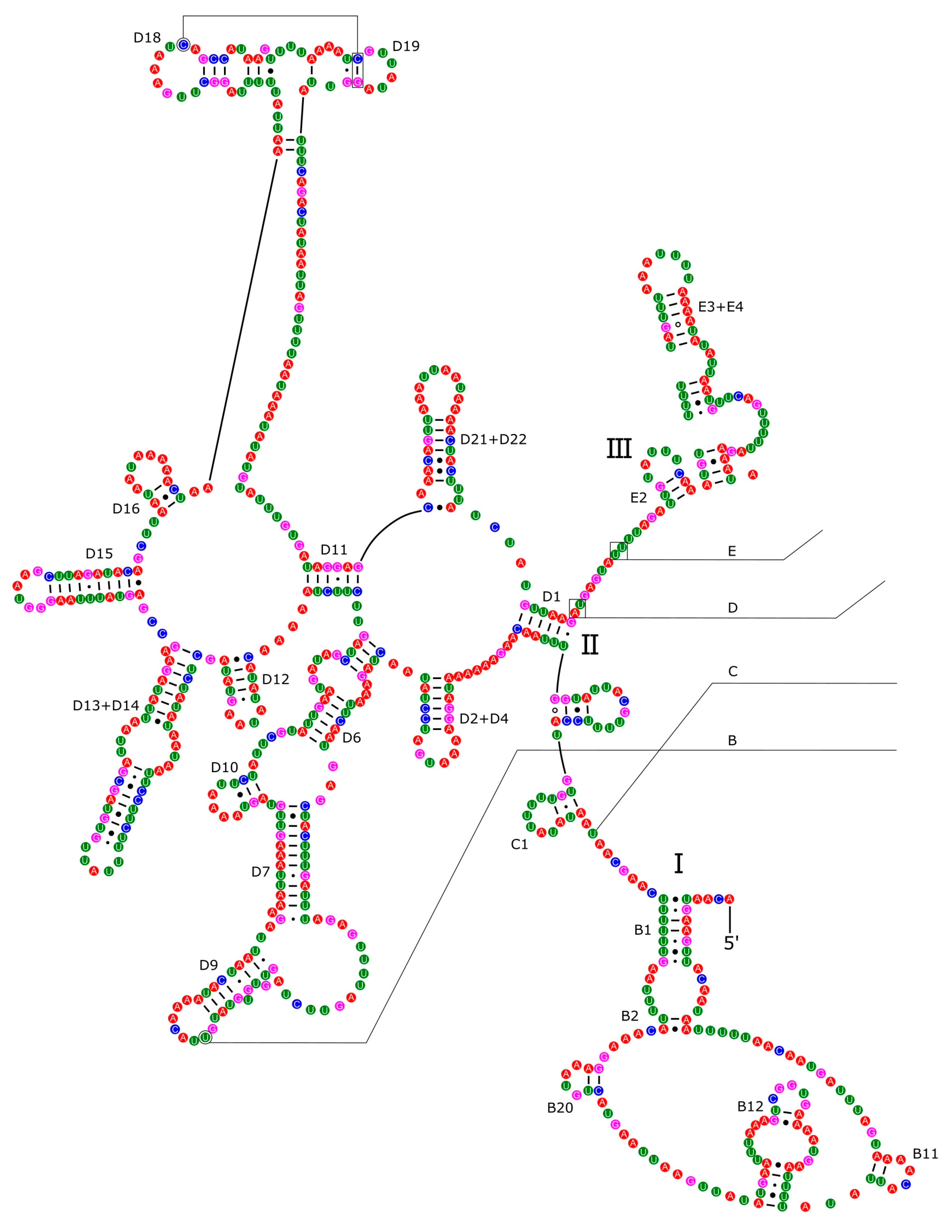
Figure 12.
Secondary structure of the IV-VI domains of the 16S rRNA of P. aulanieri.
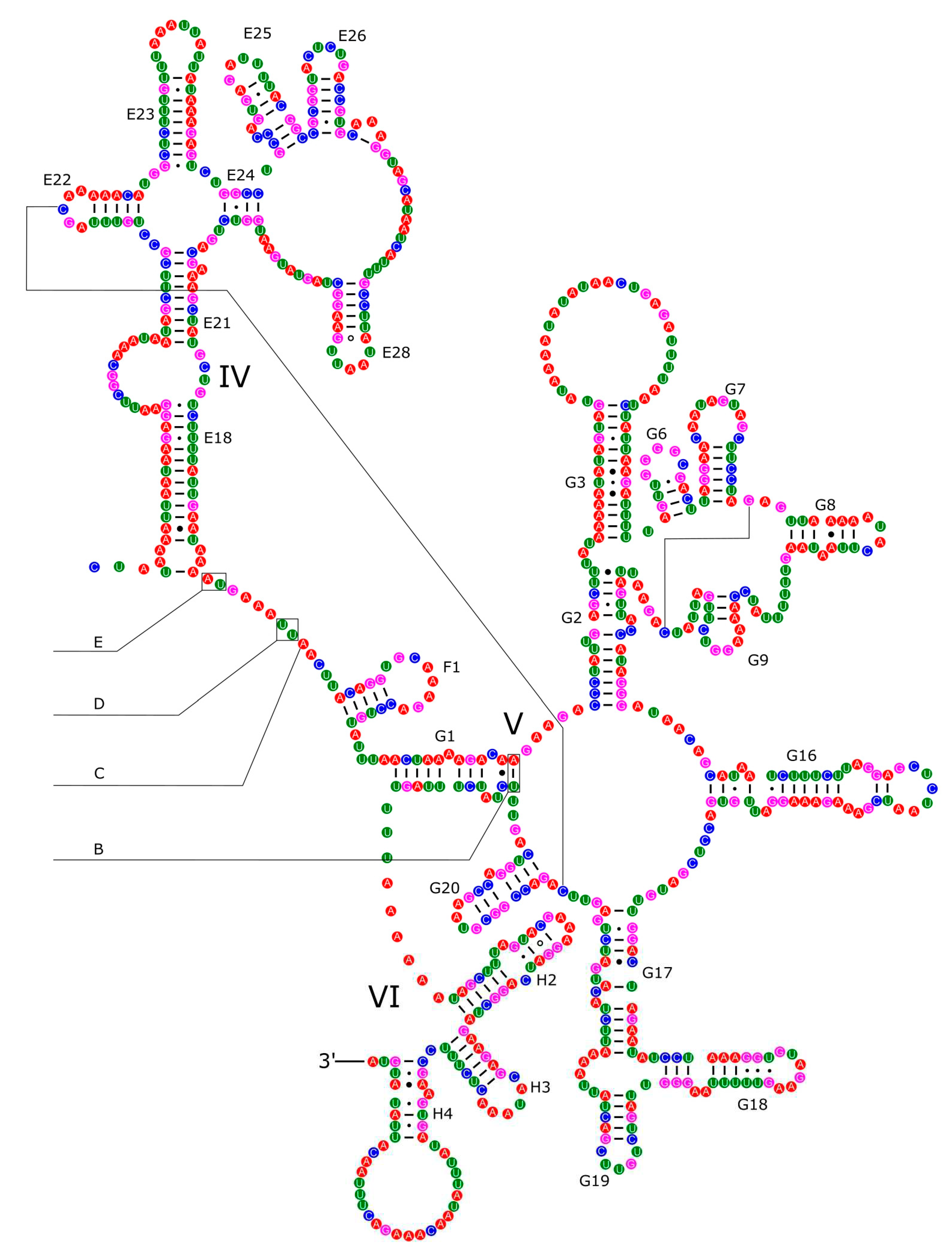
Figure 13.
Secondary structures of the tRNA genes in the mitogenome of P. reevei.
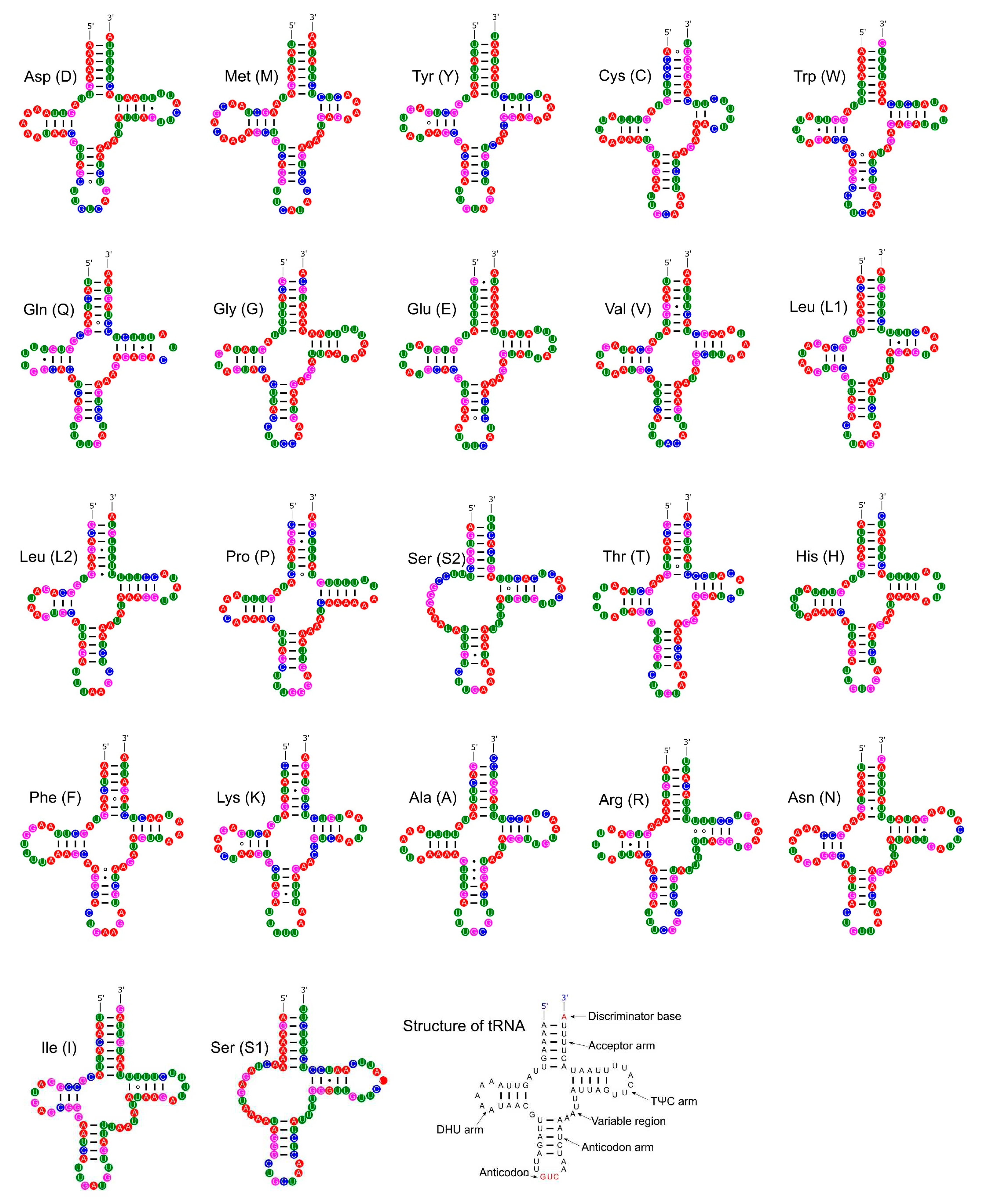
Figure 14.
Secondary structures of the tRNA genes in the mitogenome of P. aulanieri.
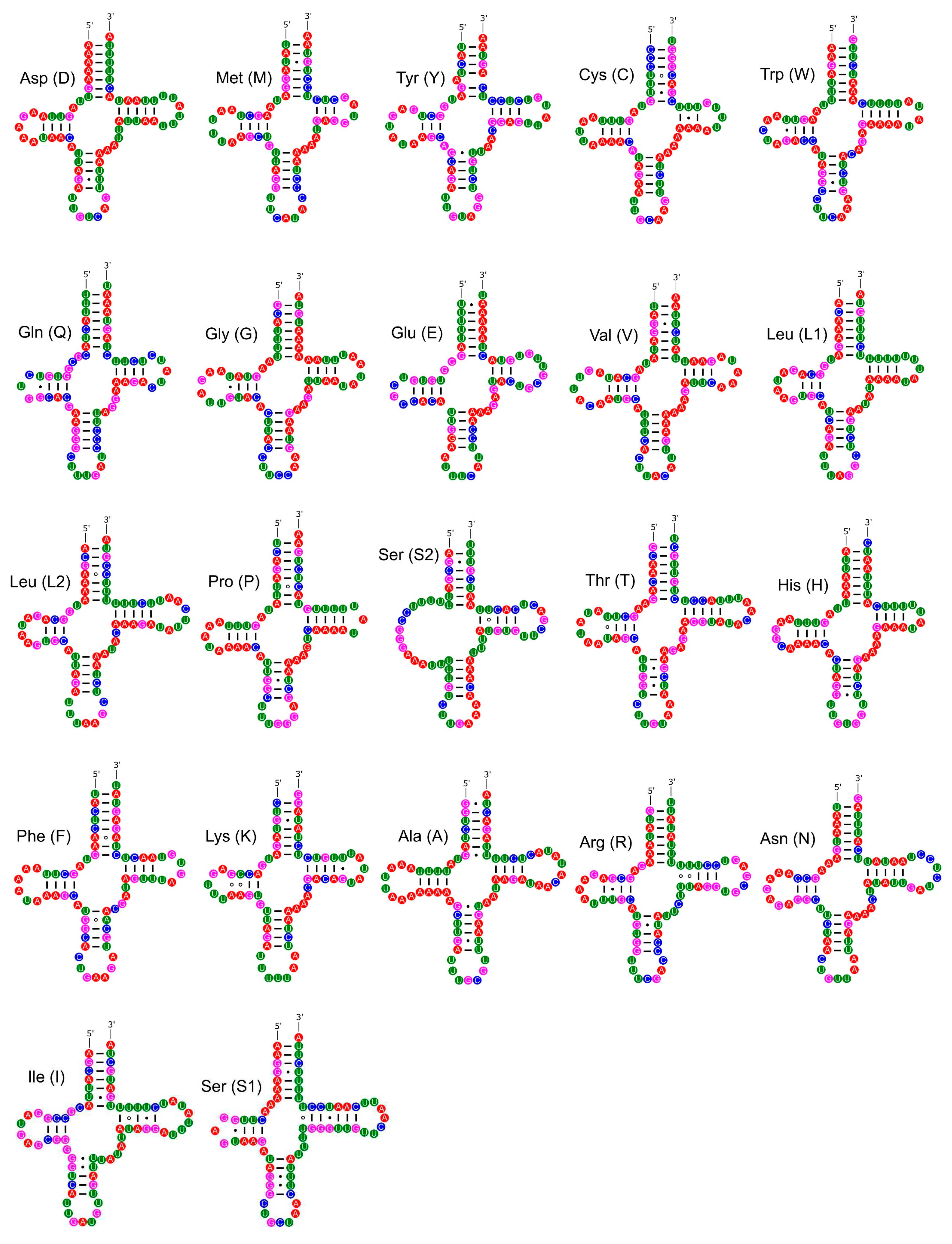
Figure 15.
Secondary structure of the control region of P. reevei and P. aulanieri.
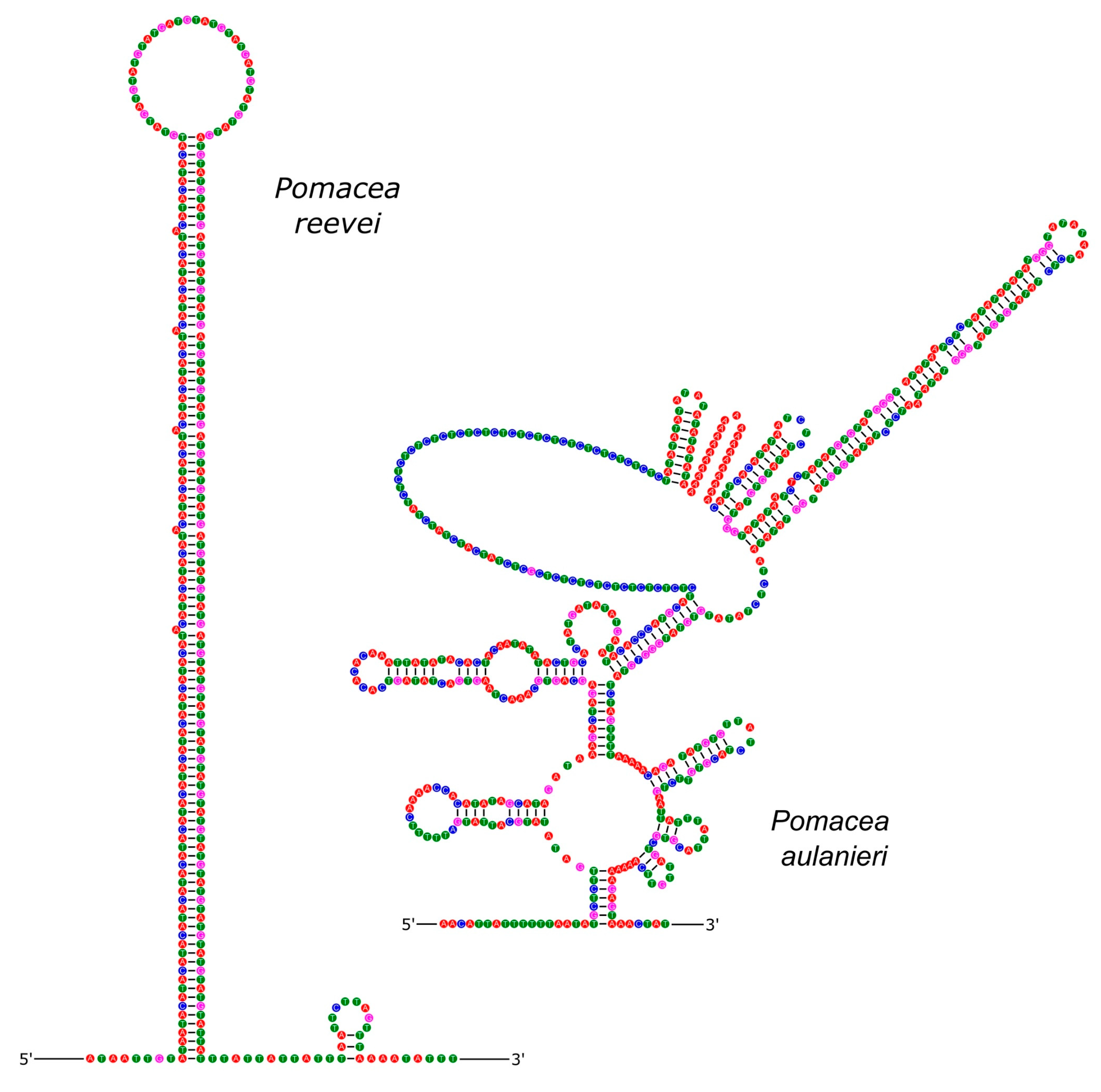
Figure 16.
Nucleotide diversity (π) among the Pomacea mitogenomes available in GenBank and the mitogenomes included in this study.
Figure 16.
Nucleotide diversity (π) among the Pomacea mitogenomes available in GenBank and the mitogenomes included in this study.
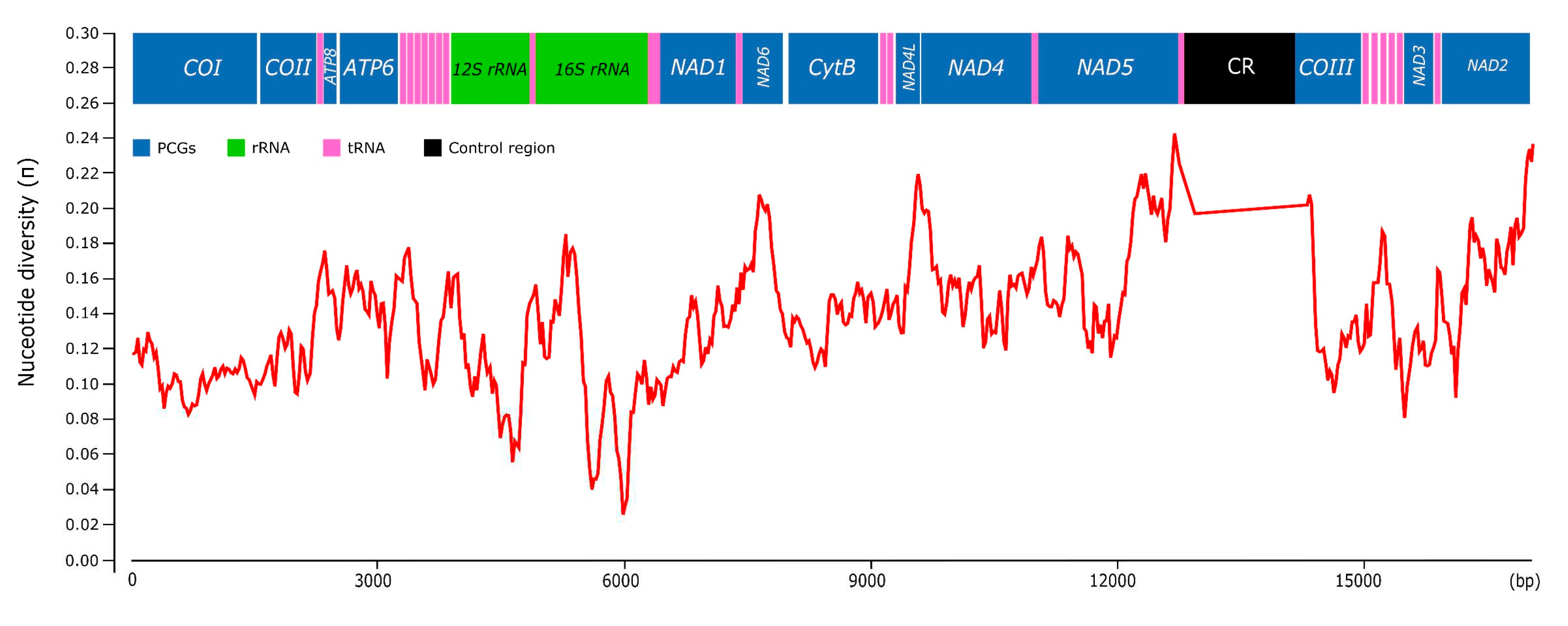
Figure 17.
[M2] Evolutionary rate of the Pomacea mitogenomes published in GenBank and those from this work. Ka is the nonsynonymous nucleotide substitutions per nonsynonymous site, Ks is the synonymous nucleotide substitutions per synonymous site, and Ka/Ks is ratio of nucleotide nonsynonymous to synonymous substitutions.
Figure 17.
[M2] Evolutionary rate of the Pomacea mitogenomes published in GenBank and those from this work. Ka is the nonsynonymous nucleotide substitutions per nonsynonymous site, Ks is the synonymous nucleotide substitutions per synonymous site, and Ka/Ks is ratio of nucleotide nonsynonymous to synonymous substitutions.
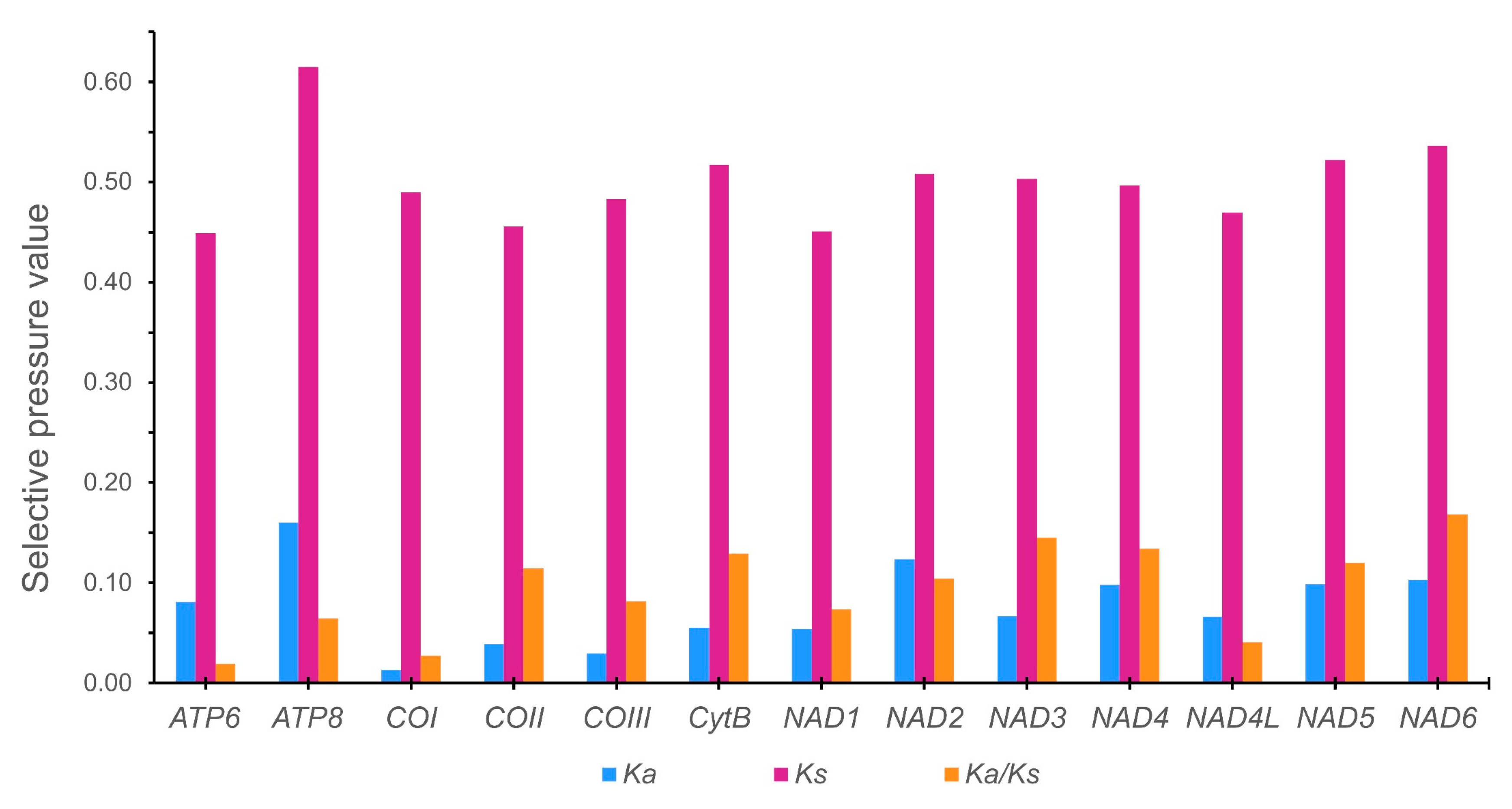
Figure 18.
Phylogenetic relationship of Caenogastropoda species based on protein-coding genes and ribosomal RNAs, inferred with BI. Numbers on branches are ML bootstrap values (left) and posterior probabilities from BI (right). Pomacea species sequenced in this study are marked with an asterisk.
Figure 18.
Phylogenetic relationship of Caenogastropoda species based on protein-coding genes and ribosomal RNAs, inferred with BI. Numbers on branches are ML bootstrap values (left) and posterior probabilities from BI (right). Pomacea species sequenced in this study are marked with an asterisk.
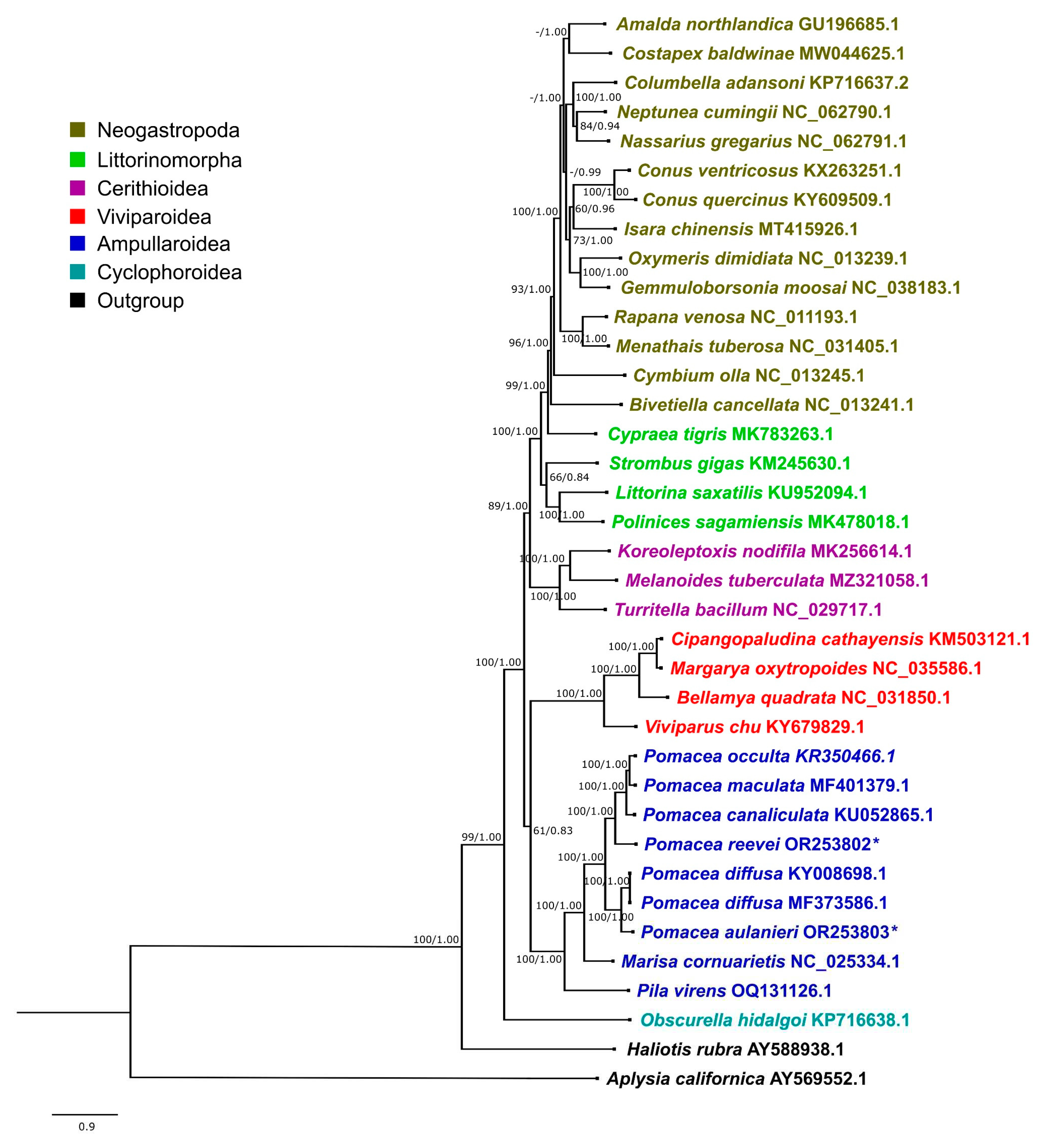

Table 1.
Organization of the mitochondrial genome of P. reevei.
| Gene | Strand | Position | Size (bp) | Intergenic Length | Anticodon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|
| COI | H | 1-1536 | 1536 | 18 | - | ATG | TAA |
| COII | H | 1555-2241 | 687 | 14 | - | ATG | TAA |
| tRNA-Asp | H | 2256-2323 | 68 | 0 | GTC | - | - |
| ATP8 | H | 2324-2482 | 159 | 8 | - | ATG | TAA |
| ATP6 | H | 2491-3189 | 699 | 33 | - | ATG | TAG |
| tRNA-Met | L | 3223-3288 | 66 | 39 | CAT | - | - |
| tRNA-Tyr | L | 3328-3393 | 66 | 38 | GTA | - | - |
| tRNA-Cys | L | 3432-3493 | 62 | 3 | GCA | - | - |
| tRNA-Trp | L | 3497-3562 | 66 | 8 | TCA | - | - |
| tRNA-Gln | L | 3571-3633 | 63 | 5 | TTG | - | - |
| tRNA-Gly | L | 3639-3704 | 66 | 26 | TCC | - | - |
| tRNA-Glu | L | 3731-3797 | 67 | 0 | TTC | - | - |
| 12S rRNA | H | 3798-4706 | 909 | 0 | - | - | - |
| tRNA-Val | H | 4707-4772 | 66 | 0 | TAC | - | - |
| 16S rRNA | H | 4773-6112 | 1340 | 0 | - | - | - |
| tRNA-Leu1 | H | 6113-6175 | 63 | 0 | TAG | - | - |
| tRNA-Leu2 | H | 6176-6241 | 66 | 0 | TAA | - | - |
| ND1 | H | 6242-7180 | 939 | 13 | - | ATG | TAA |
| tRNA-Pro | H | 7194-7260 | 67 | 0 | TGG | - | - |
| ND6 | H | 7261-7755 | 495 | 10 | - | ATG | TAA |
| CytB | H | 7766-8905 | 1140 | 3 | - | ATG | TAA |
| tRNA-Ser2 | H | 8909-8973 | 65 | 35 | TGA | - | - |
| tRNA-Thr | L | 9009-9074 | 66 | 7 | TGT | - | - |
| ND4L | H | 9082-9378 | 297 | -7 | - | ATG | TAA |
| ND4 | H | 9372-10745 | 1374 | 38 | - | ATG | TAA |
| tRNA-His | H | 10784-10849 | 66 | 0 | GTG | - | - |
| ND5 | H | 10850-12556 | 1707 | -2 | - | ATG | TAA |
| tRNA-Phe | H | 12555-12622 | 68 | 0 | GAA | - | - |
| Control region | - | 12623-12914 | 292 | 0 | - | - | - |
| COIII | H | 12915-13694 | 780 | 27 | - | ATG | TAA |
| tRNA-Lys | H | 13722-13787 | 66 | 17 | TTT | - | - |
| tRNA-Ala | H | 13805-13872 | 68 | 21 | TGC | - | - |
| tRNA-Arg | H | 13894-13963 | 70 | 2 | TCG | - | - |
| tRNA-Asn | H | 13966-14038 | 73 | 15 | GTT | - | - |
| tRNA-Ile | H | 14054-14123 | 70 | 0 | GAT | - | - |
| ND3 | H | 14124-14477 | 354 | 40 | - | ATG | TAA |
| tRNA-Ser1 | H | 14518-14584 | 67 | 0 | GCT | - | - |
| ND2 | H | 14585-15658 | 1074 | 2 | - | ATG | TAA |
Table 2.
Organization of the mitochondrial genome of P. aulanieri.
| Gene | Strand | Position | Size (bp) | Intergenic Length | Anticodon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|
| COI | H | 1-1536 | 1536 | 35 | - | ATG | TAA |
| COII | H | 1572-2258 | 687 | 27 | - | ATG | TAA |
| tRNA-Asp | H | 2286-2353 | 68 | 0 | GTC | - | - |
| ATP8 | H | 2354-2512 | 159 | 30 | - | ATG | TAG |
| ATP6 | H | 2543-3241 | 699 | 28 | - | ATG | TAG |
| tRNA-Met | L | 3270-3332 | 63 | 21 | CAT | - | - |
| tRNA-Tyr | L | 3354-3418 | 65 | 22 | GTA | - | - |
| tRNA-Cys | L | 3441-3502 | 62 | 22 | GCA | - | - |
| tRNA-Trp | L | 3525-3590 | 66 | 25 | TCA | - | - |
| tRNA-Gln | L | 3616-3681 | 66 | 26 | TTG | - | - |
| tRNA-Gly | L | 3708-3774 | 67 | 34 | TCC | - | - |
| tRNA-Glu | L | 3809-3873 | 65 | 0 | TTC | - | - |
| 12S rRNA | H | 3874-4833 | 960 | 0 | - | - | - |
| tRNA-Val | H | 4834-4900 | 67 | 0 | TAC | - | - |
| 16S rRNA | H | 4901-6248 | 1348 | 0 | - | - | - |
| tRNA-Leu1 | H | 6249-6312 | 64 | 0 | TAG | - | - |
| tRNA-Leu2 | H | 6313-6381 | 69 | 0 | TAA | - | - |
| ND1 | H | 6382-7326 | 945 | 9 | - | ATG | TAA |
| tRNA-Pro | H | 7336-7402 | 67 | -1 | TGG | - | - |
| ND6 | H | 7402-7896 | 495 | 10 | - | ATG | TAA |
| CytB | H | 7907-9046 | 1140 | 15 | - | ATG | TAA |
| tRNA-Ser2 | H | 9062-9127 | 66 | 29 | TGA | - | - |
| tRNA-Thr | L | 9157-9227 | 71 | 10 | TGT | - | - |
| ND4L | H | 9238-9534 | 297 | -7 | - | ATG | TAA |
| ND4 | H | 9528-10889 | 1362 | 10 | - | ATG | TAA |
| tRNA-His | H | 10900-10964 | 65 | 0 | GTG | - | - |
| ND5 | H | 10965-12692 | 1728 | -21 | - | ATG | TAA |
| tRNA-Phe | H | 12672-12740 | 69 | 0 | GAA | - | - |
| Control region | - | 12741-13264 | 524 | 0 | - | ||
| COIII | H | 13265-14044 | 780 | 65 | - | ATG | TAA |
| tRNA-Lys | H | 14110-14176 | 68 | 10 | TTT | - | - |
| tRNA-Ala | H | 14187-14260 | 74 | 45 | TGC | - | - |
| tRNA-Arg | H | 14306-14374 | 69 | 10 | TCG | - | - |
| tRNA-Asn | H | 14385-14455 | 71 | 27 | GTT | - | - |
| tRNA-Ile | H | 14483-14552 | 70 | 5 | GAT | - | - |
| ND3 | H | 14558-14911 | 354 | 39 | - | ATG | TAG |
| tRNA-Ser1 | H | 14951-15020 | 70 | -1 | GCT | - | - |
| ND2 | H | 15020-16093 | 1074 | 3 | - | ATG | TAG |
Table 3.
Nucleotide composition of the mitogenome of P. reevei and P. aulanieri.
| Feature | Length | A% | T% | C% | G% | %A+T | %G+C | AT-skew | GC-skew |
|---|---|---|---|---|---|---|---|---|---|
| Whole mitogenome | |||||||||
| P. reevei | 15660 | 30.10 | 40.88 | 13.14 | 15.87 | 70.98 | 29.02 | -0.152 | 0.094 |
| P. aulanieri | 16096 | 29.48 | 39.37 | 14.83 | 16.32 | 68.85 | 31.15 | -0.144 | 0.048 |
| PCGs | |||||||||
| P. reevei | 11241 | 27.52 | 42.45 | 13.95 | 16.08 | 69.98 | 30.02 | -0.213 | 0.071 |
| P. aulanieri | 11256 | 26.87 | 40.70 | 15.67 | 16.76 | 67.57 | 32.43 | -0.205 | 0.033 |
| rRNAs | |||||||||
| P. reevei | 2251 | 37.23 | 35.41 | 11.28 | 16.08 | 72.63 | 27.37 | 0.025 | 0.175 |
| P. aulanieri | 2308 | 36.74 | 34.84 | 11.92 | 16.51 | 71.58 | 28.42 | 0.027 | 0.162 |
| tRNAs | |||||||||
| P. reevei | 1465 | 35.77 | 36.04 | 11.95 | 16.25 | 71.81 | 28.19 | -0.004 | 0.153 |
| P. aulanieri | 1485 | 34.28 | 35.35 | 12.93 | 17.44 | 69.63 | 30.37 | -0.015 | 0.149 |
| Control region | |||||||||
| P. reevei | 292 | 36.64 | 40.75 | 9.25 | 13.36 | 77.40 | 22.60 | -0.053 | 0.182 |
| P. aulanieri | 524 | 33.40 | 38.17 | 16.60 | 11.83 | 71.56 | 28.44 | -0.067 | -0.168 |
Table 4.
Codon count and relative synonymous codon usage in the mitochondrial genome of P. reevei. The asterisk (*) in the table indicates the stop codon.
Table 4.
Codon count and relative synonymous codon usage in the mitochondrial genome of P. reevei. The asterisk (*) in the table indicates the stop codon.
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 311 | 1.83 | UCU(S) | 120 | 2.59 | UAU(Y) | 108 | 1.59 | UGU(C) | 31 | 1.41 |
| UUC(F) | 29 | 0.17 | UCC(S) | 25 | 0.54 | UAC(Y) | 28 | 0.41 | UGC(C) | 13 | 0.59 |
| UUA(L) | 367 | 3.54 | UCA(S) | 56 | 1.21 | UAA(*) | 12 | 1.85 | UGA(W) | 75 | 1.44 |
| UUG(L) | 62 | 0.6 | UCG(S) | 9 | 0.19 | UAG(*) | 1 | 0.15 | UGG(W) | 29 | 0.56 |
| CUU(L) | 100 | 0.97 | CCU(P) | 74 | 2.24 | CAU(H) | 61 | 1.54 | CGU(R) | 22 | 1.54 |
| CUC(L) | 10 | 0.11 | CCC(P) | 17 | 0.52 | CAC(H) | 18 | 0.46 | CGC(R) | 6 | 0.42 |
| CUA(L) | 67 | 0.65 | CCA(P) | 31 | 0.94 | CAA(Q) | 69 | 1.84 | CGA(R) | 23 | 1.68 |
| CUG(L) | 14 | 0.14 | CCG(P) | 10 | 0.3 | CAG(Q) | 6 | 0.16 | CGG(R) | 5 | 0.35 |
| AUU(I) | 281 | 1.82 | ACU(T) | 83 | 2.17 | AAU(N) | 112 | 1.62 | AGU(S) | 60 | 1.3 |
| AUC(I) | 28 | 0.18 | ACC(T) | 16 | 0.39 | AAC(N) | 26 | 0.38 | AGC(S) | 19 | 0.41 |
| AUA(M) | 162 | 1.52 | ACA(T) | 47 | 1.23 | AAA(K) | 81 | 1.62 | AGA(S) | 62 | 1.34 |
| AUG(M) | 51 | 0.48 | ACG(T) | 8 | 0.21 | AAG(K) | 19 | 0.38 | AGG(S) | 19 | 0.41 |
| GUU(V) | 114 | 1.84 | GCU(A) | 146 | 2.51 | GAU(D) | 65 | 1.76 | GGU(G) | 81 | 1.39 |
| GUC(V) | 19 | 0.32 | GCC(A) | 19 | 0.35 | GAC(D) | 9 | 0.24 | GGC(G) | 23 | 0.4 |
| GUA(V) | 83 | 1.34 | GCA(A) | 60 | 1.04 | GAA(E) | 62 | 1.51 | GGA(G) | 70 | 1.22 |
| GUG(V) | 31 | 0.5 | GCG(A) | 6 | 0.1 | GAG(E) | 19 | 0.49 | GGG(G) | 57 | 0.99 |
Table 5.
Codon count and relative synonymous codon usage in the mitochondrial genome of P. aulanieri. The asterisk (*) in the table indicates the stop codon.
Table 5.
Codon count and relative synonymous codon usage in the mitochondrial genome of P. aulanieri. The asterisk (*) in the table indicates the stop codon.
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 297 | 1.73 | UCU(S) | 120 | 2.54 | UAU(Y) | 109 | 1.48 | UGU(C) | 25 | 1.28 |
| UUC(F) | 47 | 0.27 | UCC(S) | 31 | 0.66 | UAC(Y) | 38 | 0.52 | UGC(C) | 14 | 0.72 |
| UUA(L) | 321 | 3.13 | UCA(S) | 48 | 1.02 | UAA(*) | 9 | - | UGA(W) | 68 | 1.3 |
| UUG(L) | 61 | 0.59 | UCG(S) | 7 | 0.13 | UAG(*) | 4 | - | UGG(W) | 36 | 0.7 |
| CUU(L) | 91 | 0.89 | CCU(P) | 89 | 2.68 | CAU(H) | 60 | 1.48 | CGU(R) | 23 | 1.71 |
| CUC(L) | 30 | 0.29 | CCC(P) | 10 | 0.33 | CAC(H) | 21 | 0.52 | CGC(R) | 1 | 0.07 |
| CUA(L) | 83 | 0.81 | CCA(P) | 23 | 0.72 | CAA(Q) | 61 | 1.56 | CGA(R) | 27 | 1.93 |
| CUG(L) | 30 | 0.29 | CCG(P) | 9 | 0.27 | CAG(Q) | 17 | 0.44 | CGG(R) | 4 | 0.29 |
| AUU(I) | 248 | 1.66 | ACU(T) | 73 | 1.77 | AAU(N) | 102 | 1.51 | AGU(S) | 38 | 0.81 |
| AUC(I) | 51 | 0.34 | ACC(T) | 25 | 0.61 | AAC(N) | 33 | 0.49 | AGC(S) | 36 | 0.77 |
| AUA(M) | 150 | 1.52 | ACA(T) | 55 | 1.33 | AAA(K) | 75 | 1.54 | AGA(S) | 68 | 1.45 |
| AUG(M) | 48 | 0.48 | ACG(T) | 12 | 0.29 | AAG(K) | 23 | 0.46 | AGG(S) | 29 | 0.62 |
| GUU(V) | 109 | 1.76 | GCU(A) | 130 | 2.21 | GAU(D) | 59 | 1.51 | GGU(G) | 72 | 1.25 |
| GUC(V) | 29 | 0.47 | GCC(A) | 34 | 0.58 | GAC(D) | 20 | 0.49 | GGC(G) | 25 | 0.43 |
| GUA(V) | 76 | 1.21 | GCA(A) | 59 | 1 | GAA(E) | 55 | 1.41 | GGA(G) | 70 | 1.21 |
| GUG(V) | 35 | 0.56 | GCG(A) | 12 | 0.2 | GAG(E) | 23 | 0.59 | GGG(G) | 64 | 1.11 |
Table 5.
[M3] Comparison of mitogenome features in Pomacea, where (+) indicates presence and (-) indicates absence of the feature.
Table 5.
[M3] Comparison of mitogenome features in Pomacea, where (+) indicates presence and (-) indicates absence of the feature.
| Feature | Pomacea reevei | Pomacea aulanieri | Pomacea diffusa | Pomacea canaliculata | Pomacea maculata | Pomacea $occulta |
|---|---|---|---|---|---|---|
| Overlapping NAD5/tRNA-Phe | + | + | + | - | - | - |
| Overlapping NAD1/tRNA-Pro | - | - | - | + | + | + |
| ATP6 length (bp) | 699 | 699 | 714 | 699 | 699 | 699 |
| NAD1 length (bp) | 939 | 945 | 945 | 960 | 960 | 960 |
| NAD2 length (bp) | 1074 | 1074 | 1071 | 1062 | 1062 | 1065 |
| NAD4 length(bp) | 1374 | 1362 | 1362 | 1368 | 1368 | 1368 |
| NAD5 length (bp) | 1707 | 1728 | 1728 | 1710 | 1710 | 1710 |
| Start codon COIII | ATG | ATG | ATG | ATA | ATA | ATA |
| tRNA length range (bp) | 12 | 13 | 15 | 7 | 7 | 7 |
| Tallo D en tRNA-Ser1 | - | + | + | - | - | - |
| Tallo D en tRNA-Ser2 | - | - | - | + | + | + |
| 12S rRNA length (bp) | 909 | 960 | 952 | 929 | 936 | 934 |
| 16S rRNA length (bp) | 1340 | 1348 | 1346 | 1334 | 1336 | 1331 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



