Submitted:
13 July 2023
Posted:
14 July 2023
You are already at the latest version
Abstract
Petrosamine (1) – a colored pyridoacridine alkaloid from the Belizean sponge, Petrosia sp. that is also a potent inhibitor of acetylcholine esterase (AChE) – was investigated by spectroscopic and computational methods. Analysis of the petrosamine free energy landscapes, pKa and tautomerism revealed an accurate electronic depiction of the molecular structure of 1 as the di-keto form, with net charge of q = +1, rather than a dication (q = +2) under ambient conditions of isolation-purification. The pronounced solvatochromism (UV-vis) reported for 1, and related analogs, was investigated in detail and is best explained by charge delocalization and stabilization of the ground state (HOMO) of 1 rather than an equilibrium of competing tautomers. Refinement of the molecular structure of 1 by QM methods complements published computational docking studies to define the contact points in the enzyme active site that may improve design of new AChE inhibitors based on the pyridoacridine alkaloid molecular skeleton.
Keywords:
Alkaloid
; pyridoacridine
; acetylcholine esterase
; merocyanine
; marine natural product
; NMR
; UV-vis spectroscopy.
1. Introduction
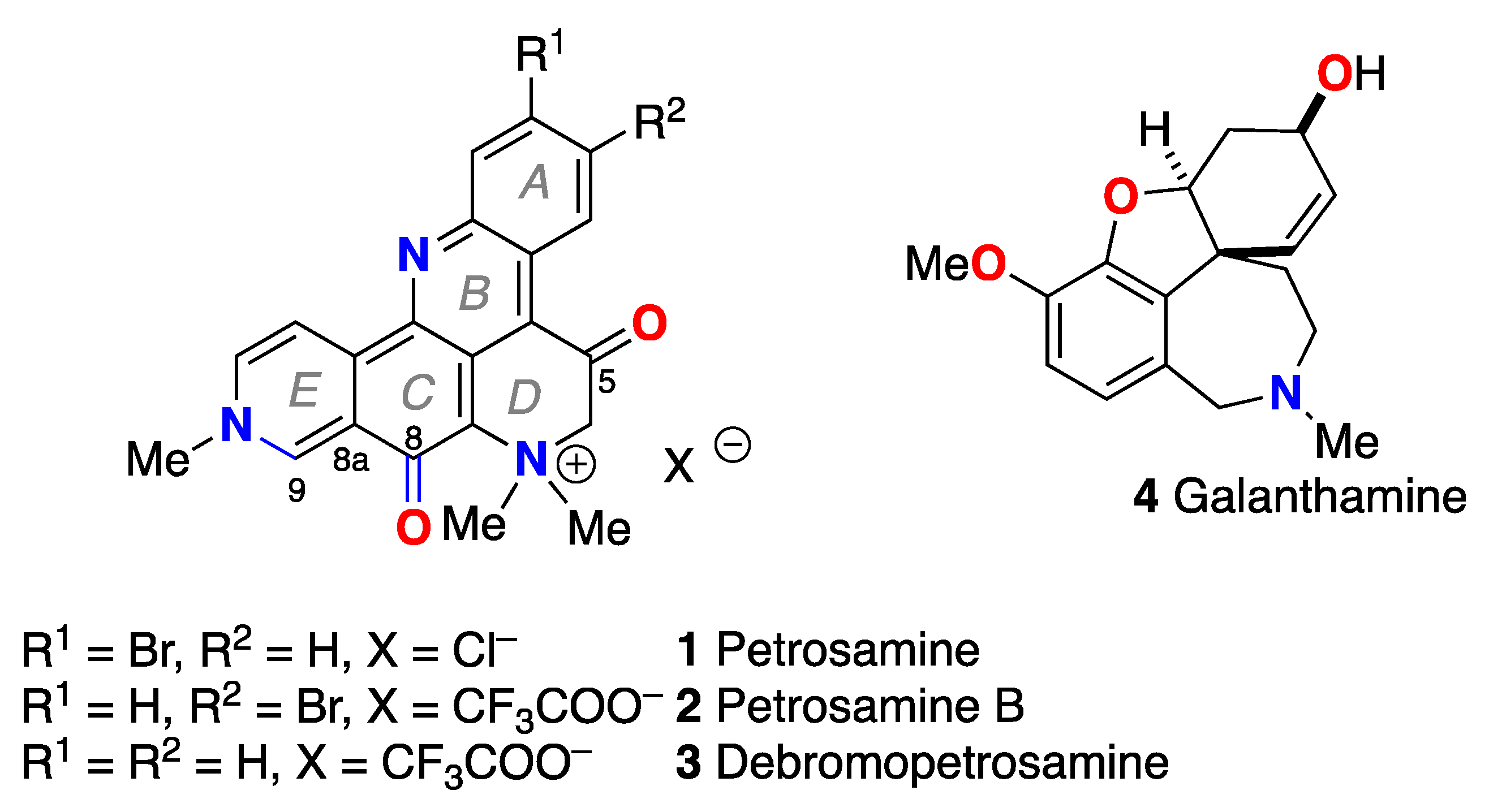
In 1988, Molinski and Faulkner reported the structure of petrosamine (1), a highly condensed pentacyclic alkaloid of intense color that was isolated from the sponge, Petrosia sp., collected at Carrie Bow Cay, Belize [1]. Although no bioactivity of 1was reported at the time, a pronounced solvatochromism – the property of color changes (lmax) in solvents of different polarities – was observed. At the time, 1 represented the most complex structure in a growing class of pyridoacridine alkaloids from marine invertebrates [2]. Structurally, 1 can be considered an methylated and oxidized analog of the pentacyclic amphimedine – the first member of this class of alkaloid, described by Schmitz and co-workers in 1983 [3]. Unlike the structures of most pyridoacridines where the 3 nitrogen atoms are sp2 hybridized, 1 contains a quaternary ammonium salt with an sp3 quaternized N. Pyridoacridines manifest a range of biological activities including cytotoxicity, antineoplastic properties, antibacterial activity, and enzyme inhibition; a subject that has been extensively reviewed [4].
Since its initial report, alkaloid 1 has been reisolated from a Thai species of sponge, Petrosia n sp.[5], and two new analogues have been described; petrosamine B (2) [6] a regioisomer of 1 from the Australian sponge Oceanapia sp. with modest inhibitory activity against aspartate semialdehyde dehydrogenase, and debromopetrosamine (3) from Xestospongia cf carbonaria collected in Palau [7]. Numerous marine natural products have been reported with neuroprotective properties in experimental models for neurodegenerative diseases including acetylcholine esterase (AChE) inhibition [8]. Suwanborirux and coworkers showed that 1 is a potent inhibitor of AChE from the Pacific electric ray, Torpedo californica (IC50 = 91 nM) [5]; approximately 6 times more potent than galanthamine (4), an alkaloid used in the past for treatment of patients with Alzheizmer's disease (AD) to compensate neurotransmitter deficiency.
Preliminary docking studies of 1 with AChE revealed subtle electronic interactions with the putative receptor contacts [5]. Given the global rise of AD within aging populations and the therapeutic importance of AChE inhibitors for treatment, a detailed understanding of the electronic structure and molecular parameters for enzyme-inhibitor molecular contacts of 1 may advance design principles for new AD therapeutics. In this report, we present refined measurements of UV-vis, pKa properties and quantum mechanical calculations of 1 that relate the observed solvatochromism through mapping of dipolar resonance forms to a simple model based on merocyanine dyes, and stabilization of the ground state of the HOMO.
Petrosamine (1) exhibits several unusual physical and spectroscopic properties. The melting point of 1 is in excess of 300 ˚C suggesting high stabilization within the lattice energy of the crystalline form. Unlike most other pentacyclic pyridoacridines, the color of 1 is strikingly variable depending upon the physical state. Crystals of 1 are deep blue-purple but dilute solutions of 1 show highly complex UV-vis spectra due to the presence of long wavelength absorption bands that manifest pronounced solvatochromism (changes in lmax of 1 when measured in different solvents). For example, solutions of 1 in MeOH appear deep-blue (longest lmax = 595 nm), but aqueous solutions of 1 appear purple lmax = 574 nm) [1]. In dilute THF solutions (sparingly soluble) or DMSO, 1 appears green lmax = 611 nm) [1]. In short, the range ∆lmax of 1 is 36 nm in a range of solvents from H2O to DMSO. Petrosamine also displays pH indicator properties: addition of excess alkali to purple aqueous or blue MeOH solutions of 1, changes the color to green, suggestive of a hypsochromic shift (∆lmax < 0) mediated by the Brønsted acidity of the a-CH2 next to the keto group and deprotonation to the corresponding enolate.
In the report by Quinn and coworkers describing isolation of 2 by "fractionation on C18" and elution with a stepped gradient of acidic (CF3COOH) aqueous-MeOH [6], also reported solvatochromism in 2 very similar to that of 1. Assignment of the structure of 2 was achieved through extensive analysis of 1H, 13C NMR and 2D NMR data (D2O-TFA-1% DMSO-d6). Compound 2 was depicted as a dication different from 1 in the resonance form of an O-protonated vinylogous amide embodying a fully aromatic quinolinium ring E [6]. Additional structural anomalies emerged. The assignment of C-8 in 2 is supported by an HMBC correlation from H-9 to C-8 (3JCH) based on the low 13C NMR chemical shift (d 155.8 ppm), but no inter-ring HMBC correlations were reported for C-5 [6]. In contrast, the X-ray crystal structure of 1 [1] clearly reveals two keto groups in the solid state (d (C-8–O) = 1.256 Å; d (C-5–O) = 1.203 Å), d (C-5–C-6) =1.495(21). The C-O and C-C bond lengths are only compatible with a di-keto structure [9]. As the structures of 1 and 2 are very similar; consequently, the solution properties (pKa, UV-vis) should be also closely matched. Faulkner and Molinski reported the 13C NMR spectra (DMSO-d6) of 1, but not the assignments of the signals [1]. Suwainborix and coworkers, reporting the 13C NMR spectrum of 1 in the latter solvent, assigned the signal at d 161.3, s, the higher-field of the two most deshielded signals (d 187.2, s; 161.3, s) to C-8 [5])
Comparisons of the NMR assignments of 1 and 2 are complicated further by measurements in different solvents, and possible changes in tautomer or enolization states that accompany changes in pH and NMR solvent polarity, and H-bond donor-acceptor properties of the alkaloids. Clearly, the latter factors affect the UV-vis properties of 1 and 2. Surprisingly, neither the Quinn [6] or Suwainborix [5] groups claim to have observed the rapid enolization of 1 in D2O reported by Faulkner and Molinski that resulted in complete deuteriation of C-9 and 'loss' of the C-9 signal [1,10].
In order to reconcile these apparent paradoxes, it is required that a re-examination of 1 be given separate considerations of resonance in the native structure, and possible tautomerism of 1 at the a-CH2 next to the C-5 C=O group. We undertook the task and completed additional solution spectroscopic measurements of 1 [NMR and electronic spectroscopy, UV-vis] and MS time-course studies and augmented by quantum mechanics calculations. The results, presented here, clarify several phenomena of 1 including solvent-dependence of HOMO-LUMO energies, and kinetic and thermodynamic considerations of pKa, and tautomerism.
2. Results and Discussion
The solvatochromism of 1 and 2 is similar to lower-order merocyanine dyes, the canonical substructures of which can be discerned within the molecular framework of both natural products (Figure 1). For example, the same conjugated sub-structure in Brooker's merocyanines [11,12] [generalized by Brooker as the vinylogous amide; neutral ia and ib dipolar (zwitterionic) resonance forms] is embedded in the molecular frameworks of 1 and 2. Brooker merocyanines, for example 5b and 6b (Figure 1) exhibit strongly positive solvatochromism (hypsochromism, or a blue shift in lmax in polar solvents). It is not unreasonable to invoke the same spectroscopic and electronic properties of 5 – zwitterionic form with extended conjugation, and stabilization of the ground state by solvation in polar solvents – as necessary and sufficient conditions to explain the solvatochromism of 1–3.
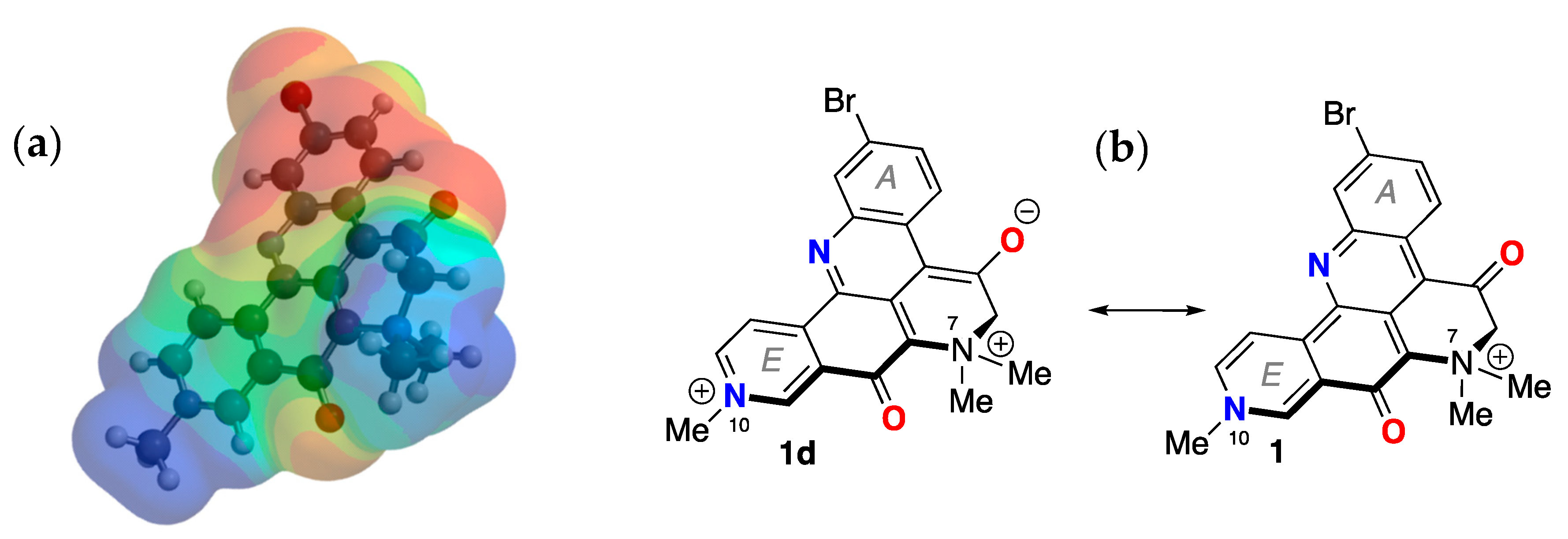
While a complete description of electronic properties of petrosamine (1) may be achieved in rigorous quantum mechanical calculations (see below), it is helpful for visualization purposes to consider Lewis bond formalism and resonance structures [13]. For clarity, only two pairs of resonance forms are depicted for 1 to illustrate the zwitterionic contributions of merocyanine substructures: the non-charged resonance forms 1a,c, and 'zwitterionic' resonance form 1b,d. Forms 1a,b depict the shorter bond path (n = 3, c.f. ia, ib, Figure 4) and dipolar resonance forms 1c,d show a longer bond path in an 'aza-vinylogous amide' (n = 4). The shortest bond path, n = 1 (not shown) would involve only the atoms C-8–C8a–C9–N-10), while the longest path, n = 4, evoking ‘particle-in-a-box’ formalism [14], best explains the long-wavelength UV-vis bands of 1 giving rise to its colors.
Figure 1.
Canonical resonance forms – neutral (a) and dipolar (b) – for merocyanines defined by bond path, n (see [11]). (b) Petrosamine ‘neutral’ and dipolar resonance forms: 1a (n = 3), 1b (n= 2), 1c (n = 4) and 1d (n = 4). (c) Brooker merocyanine resonance forms; neutral (4a, 5a, 6a) and dipolar (4b, 5b, 6b) forms [11].
Figure 1.
Canonical resonance forms – neutral (a) and dipolar (b) – for merocyanines defined by bond path, n (see [11]). (b) Petrosamine ‘neutral’ and dipolar resonance forms: 1a (n = 3), 1b (n= 2), 1c (n = 4) and 1d (n = 4). (c) Brooker merocyanine resonance forms; neutral (4a, 5a, 6a) and dipolar (4b, 5b, 6b) forms [11].
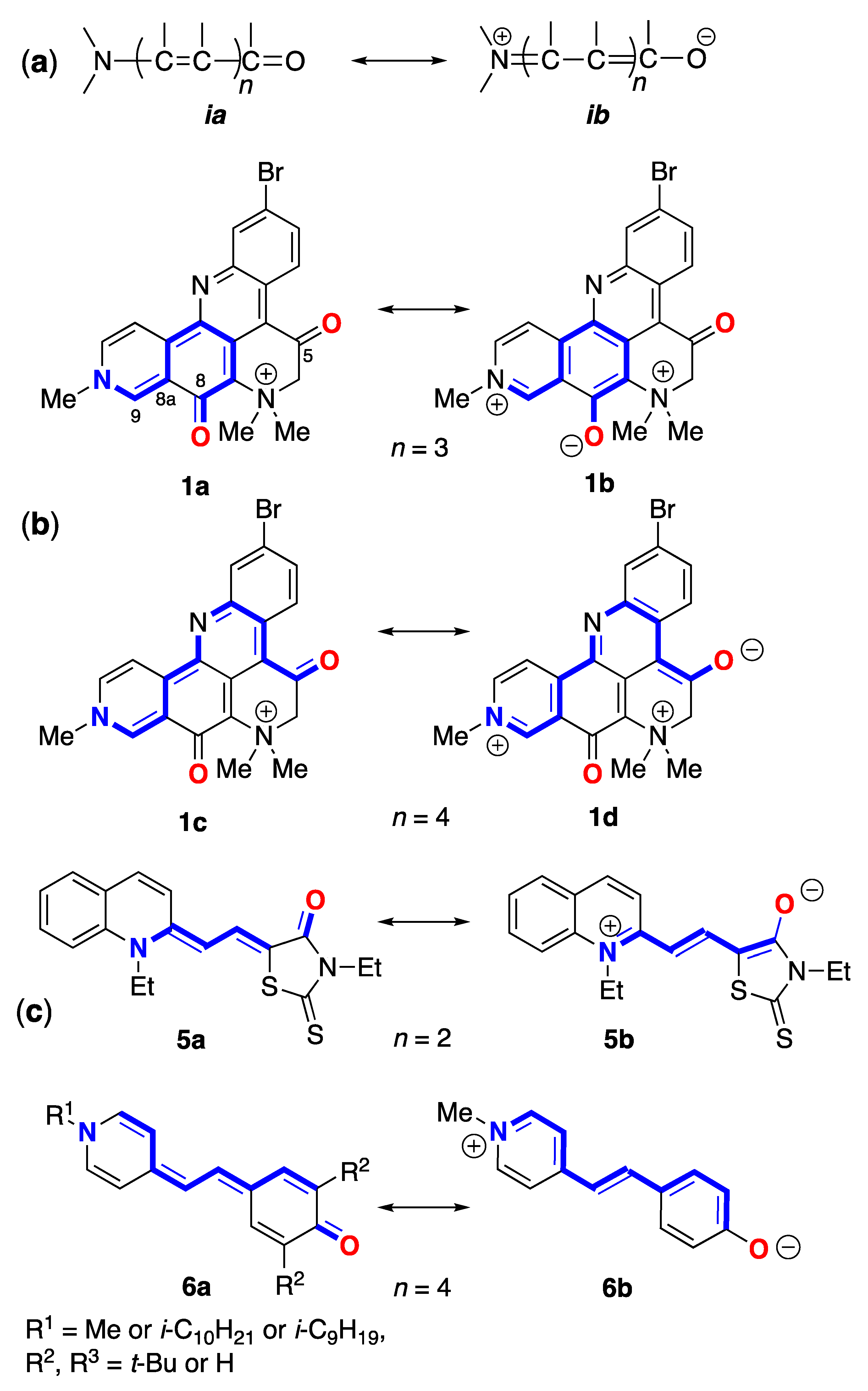
Solvatochromism of Brooker merocyanines has been rationalized [11b], using semi-quantitative valence resonance and frontier orbital theories, as arising from more extensive stabilization of the dipolar form 4a of the ground state relative to the excited state in polar solvents. Accordingly, this differential stabilization gives rise to an increase in the electronic transition energy, ∆E, due to a larger gap between the HOMO and LUMO of the longest wavelength transitions. The HOMO-LUMO gap is predicted to increase (blue shifted absorption) in those structures with higher contributions from the zwitterionic resonance form 4b, leading to more pronounced solvatochromism. More recent semi-empirical calculations (COSMO, PM3) of a different set of substituted Brooker merocyanines (5) by Morley and coworkers [15] supported stabilization of the ground state as mostly responsible for solvatochromism and predicted a larger role for hydrogen bonding in the zwitterionic form 5b over the neutral form 5a.
The structure of petrosamine (1) appears to fulfill the criteria for merocyanine-type solvatochromism. Measurements of the UV-vis spectrum of solutions of 1 prepared in DMSO–H2O solvents of variable composition (Figure 2) exhibit changes in the lmax. Most prominently, the longest wavelength absorption band with the largest hypsochromic shift between 100% DMSO and100% H2O (∆lmax –78 nm) is assigned to the forbidden n–π* transition that lends color to 1, analogous to that of merocyanines.
Figure 2.
Solvatochromism in normalized electronic UV-vis spectra of petrosamine (1) in H2O-DMSO solvents of variable composition (H2O v/v = 0%, 20%, 40%, 60%, 80% and 100%).
Figure 2.
Solvatochromism in normalized electronic UV-vis spectra of petrosamine (1) in H2O-DMSO solvents of variable composition (H2O v/v = 0%, 20%, 40%, 60%, 80% and 100%).
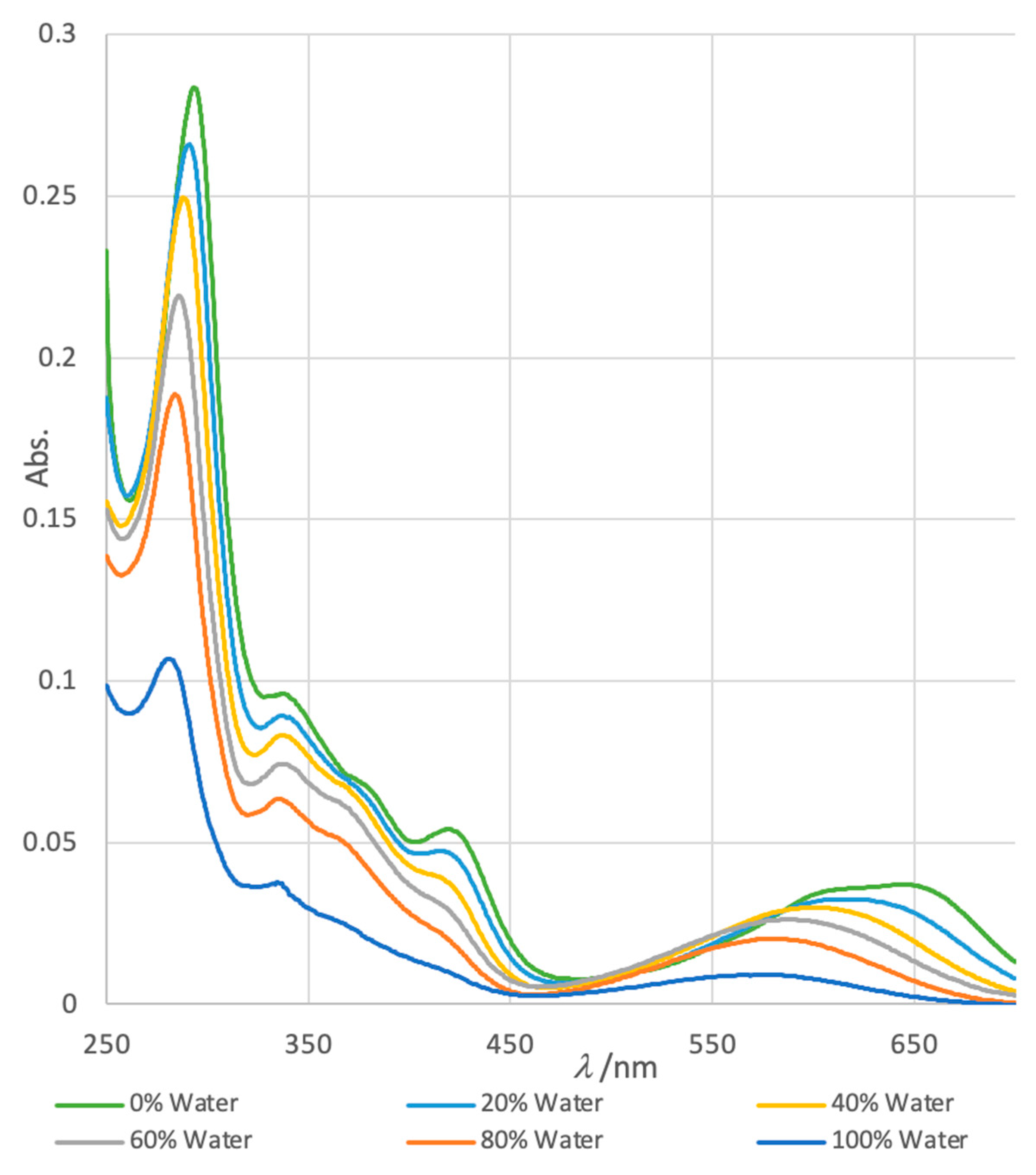
A clear trend emerges: band-1 (lmax1, defined here for convenience, as the dominant π-π* transition) shows a dramatic decrease in e with increasing H2O content of the solvent, and a weak bathochromic shift between 100% H2O to 100% DMSO (∆lmax1 15 nm, Table 1). In contrast, the corresponding changes in band-2 include a strong blue-shift (hypsochromism, lmax2 –78 nm). The band lmax2 is affected most by solvents with increasing H2O content which parallels the reported behavior of Brooker merocyanines.

Table 1.
UV-vis properties of lmax11 and lmax21 in petrosamine (1) in DMSO-H2O.1.
| % H2O v/v) | lmax1 (nm) | e11 | lmax2 (nm) | e21 |
|---|---|---|---|---|
| 0 | 281 | 68,500 | 648 | 24,000 |
| 20 | 282 | 122,252 | 622 | 20,600 |
| 40 | 287 | 141,700 | 604 | 19,400 |
| 60 | 288 | 161,000 | 589 | 16,500 |
| 80 | 290 | 172,000 | 581 | 12,900 |
| 100 | 296 | 184,000 | 570 | 4,700 |
1 See text for definitions. 1.e values normalized from the literature value of lmax2 (e 4,700) [1].
For comparison, we prepared the known merocyanine 6b from 4-methylpyridine by the following sequence adapted from Minch and Sadiq Shah:[16] methylation (MeI, iPrOH, reflux), condensation of the resultant pyridinium methiodide with p-hydroxybenzaldehyde (piperidine, EtOH, reflux), and neutralization of the product 7 (nBu4N+ HO–) to zwitterionic phenolate 6b. Measurements of the UV-vis spectra of 6b (nBu4N+ salt) in mixed solvents (acetone-H2O, see Supporting Information) showed a hypsochromic trend similar to that of 1 in DMSO-H2O. The long-wavelength (band-2) varied from lmax(acetone) 591 nm to lmax (H2O) 444 nm (∆ lmax –69 nm ) while e changed only slightly across the range of solvent mixtures [17].
2.1. QM Calculations
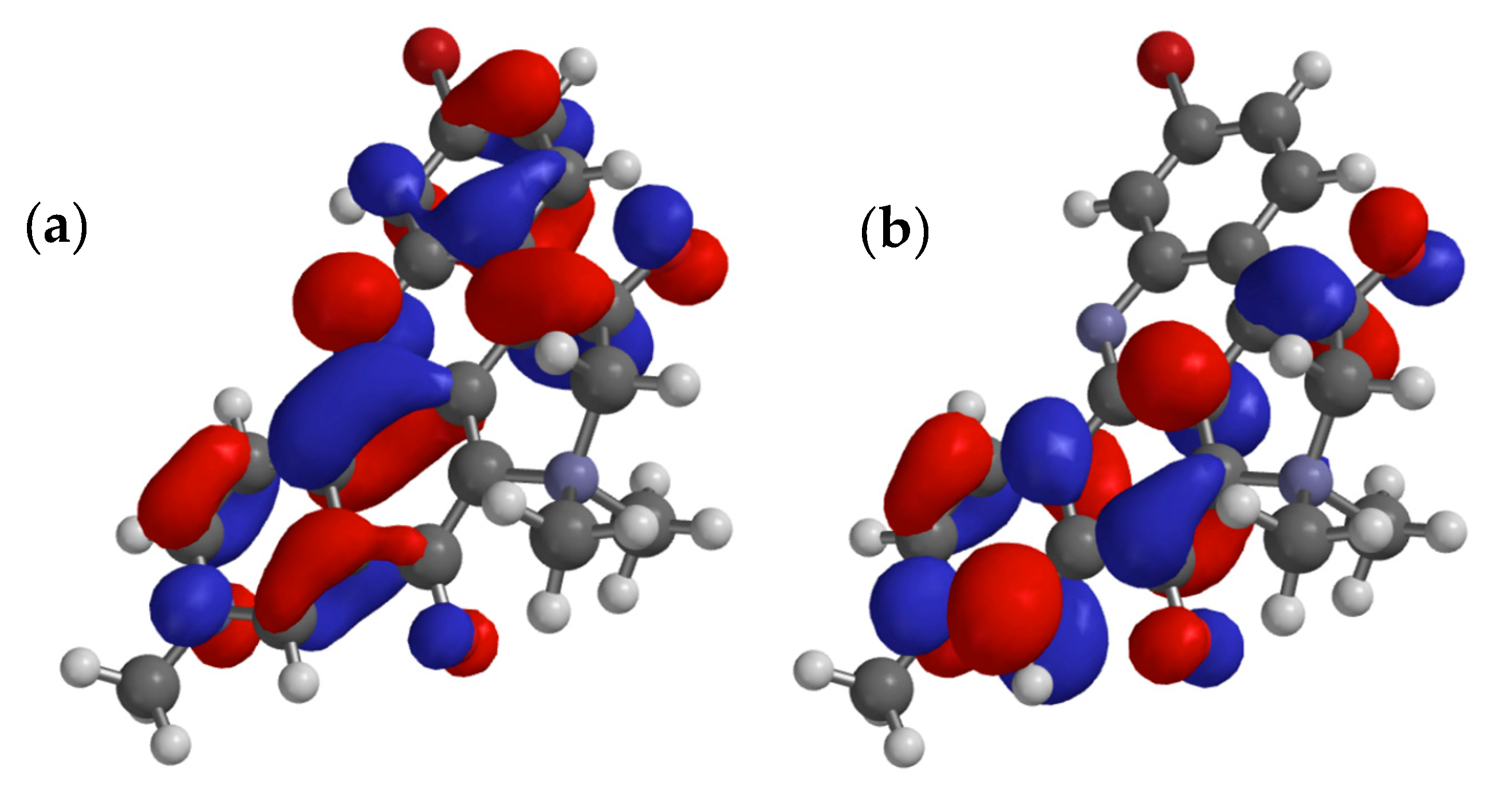
The energies of electronic states of 1 were calculated using QM methods (DFT). Starting with the X-ray coordinates of 1, the geometry of the structure was first minimized using MMFF, then further refined by DFT (B3LYP 6-31G*, polarization continuum model = H2O). The 3D model of 1, overlaid with frontier MOs, is shown in Figure 3. As expected, the ∆E for the HOMO-LUMO gap is relatively small (∆E = xx eV). The orbital coefficients and calculated dipole moment of the excited state (µ = 18.5 D) are consistent with dominant contributions from the resonance form 1b rather than 1a, and the strong donor properties of N-10. At first this may seem anomalous, given the relatively low-field 13C NMR signal measured for C-5 in 1 (d 187.4 [1] or 187.2 in 2 [5]) however, the additional deshielding effect is likely attributed to the inductive effect of the quaternized N-7.
The electrostatic potential map of the minimized structure of 1 (Figure 4) clearly shows two loci of positive charge: one centered on the quaternized N-10, as expected, in ring D, and a second associated with the N atom in ring E. The latter supports charge separated forms 1b,d (Figure 1) in which N-10 participates as a donor group. Consequently, the formal bonding electron pair of N-10 is highly delocalized and can confer only weak basicity to 1. Together with the UV-vis properties of 1, the overall electron delocalization consolidates an electronic structure in which a zwitterionic partial structure strongly lends to charge-separation mostly in the ground state.
Figure 3.
Calculated minimized structure and overlaid frontier π-orbitals of petrosamine (1) (DFT B3LYP 6-31G*, polarization continuum model = H2O) (a) HOMO and (b) LUMO.
Figure 3.
Calculated minimized structure and overlaid frontier π-orbitals of petrosamine (1) (DFT B3LYP 6-31G*, polarization continuum model = H2O) (a) HOMO and (b) LUMO.
From an empirical viewpoint, the latter makes sense as the charge-separated forms 1b,d preserve the aromaticity of rings A and E. A result of this delocalization is reduced basicity of N in ring E and, consequently, an expected lower pKa for the conjugate Brønsted acid. Formally, addition of H+ 1 to give a dication should sooner favor the C-8 or C-5 oxygen as an acceptor, rather than N-10 or N-13. In fact, we find no evidence (NMR) for protonation of 1 at pH ~ 2, which attests to the overall poor basicity of 1; an unsurprising finding given the permanent formal charge of +1 in this quaternary ammonium salt in all but the most basic or the most acidic media.
Figure 4.
Minimized energy structure of petrosamine (1, DFT, EDF2 6-31G, dipole moment µ = 18.5 D). (a) electrostatic potential surface and (b) the corresponding molecular framework of 1 (dipolar 1d and ‘charge-minimal’ 1 resonance forms).
Figure 4.
Minimized energy structure of petrosamine (1, DFT, EDF2 6-31G, dipole moment µ = 18.5 D). (a) electrostatic potential surface and (b) the corresponding molecular framework of 1 (dipolar 1d and ‘charge-minimal’ 1 resonance forms).
2.2. pKa of Petrosamine (1). Does the structure of 1 exhibit substantial enol content?
In order to estimate the pKa of petrosamine, the UV-vis spectra of 1 were measured in buffered D2O at different pH. The titration curve of 1 (S1) shows little change in the UV-vis spectra in the range of pH 2 – 10. From the Henderson-Hasselbach relationship (Equation (1))[18] for Brønsted acid HA, the condition pH = pKa is met when concentrations of conjugate species are equal ([HA] = [A–]). We surmise the pKa of 1 lies outside this pH range (pKa >10). Indeed, a bathochromic shift in the UV-vis spectrum of 1 is only observed when a methanolic solution is treated with high concentrations of NaOH (2M, pH >13). Conversely, only when a sample of 1 is dissolved in a very strong Brønsted acid (neat CF3COOH) are conditions met for a reasonable expectation of, diprotonated petrosamine ([1•2H]2+). In the event, when 1 was dissolved in neat TFA, the blue-purple color changed to bright yellow. The latter observation contrasts with the supposition drawn by Quinn and coworkers of doubly-protonated petrosamine B ([2•2H]2+) from their observation that 2 remains "bright blue…when dissolved in methanol”, under the relatively benign conditions used in C18-reversed phase isolation of the alkaloid (5% TFA-MeOH) [6]. In other words, N-10 in the monocationic molecules 1-3 is ‘pyridinium-like’ and non-basic (resembling 1d), and the C-8 C=O is insufficiently Brønsted-basic to be substantially protonated under ordinary isolation conditions.
pH = pKa + log10 [A–]/[HA]
It is evident from the 1H and 13C NMR ( DMSO-d6) that C-5 in petrosamine (1) is in the C8 keto form, a conclusion also reached by both the Suwanborirux and coworkers [5], and independently by Quinn and coworkers for petrosamine B (2) [6]. The single crystal X-ray crystallography of 1 is concordant with the C8 keto form. In fact, solid state 1 is best represented by the di-keto tautomer, e.g. interatomic distances C8-O2, 1.256(17) Å; C5-O1, 1.203(15), and C5-C6, 1.495(21) [1].
2.3. Kinetic Measurements of Hydrogen-Deuterium Exchange of 1
None of the resonance forms of 1a-d (Figure 1)– or more precisely, pathways of electron delocalization – explain the complete exchange of the H-6 protons by deuterium when 1 was dissolved in deuteric solvents (D2O, CD3OD) [1]. The latter can only be rationalized by consideration of the possibility of enolization (Figure 6), either through the positively charged 8a in neutral to weakly acidic pH or the charge-neutral (zwitterionic) enolate 8b under highly basic pH. The kinetic parameters for successive H replacement by D in 1, defined by rate constants k1 and k2, are intrinsic properties as opposed to thermodynamic properties that relate to the equilibrium constants Keq1 and Keq2; the latter are largely dependent upon the strength and concentration of added base, B– (Figure 6).
Figure 6.
Keto-enol tautomerism of 1. Enol 8a (acid -catalyzed), and enolate 8b (base-promoted).
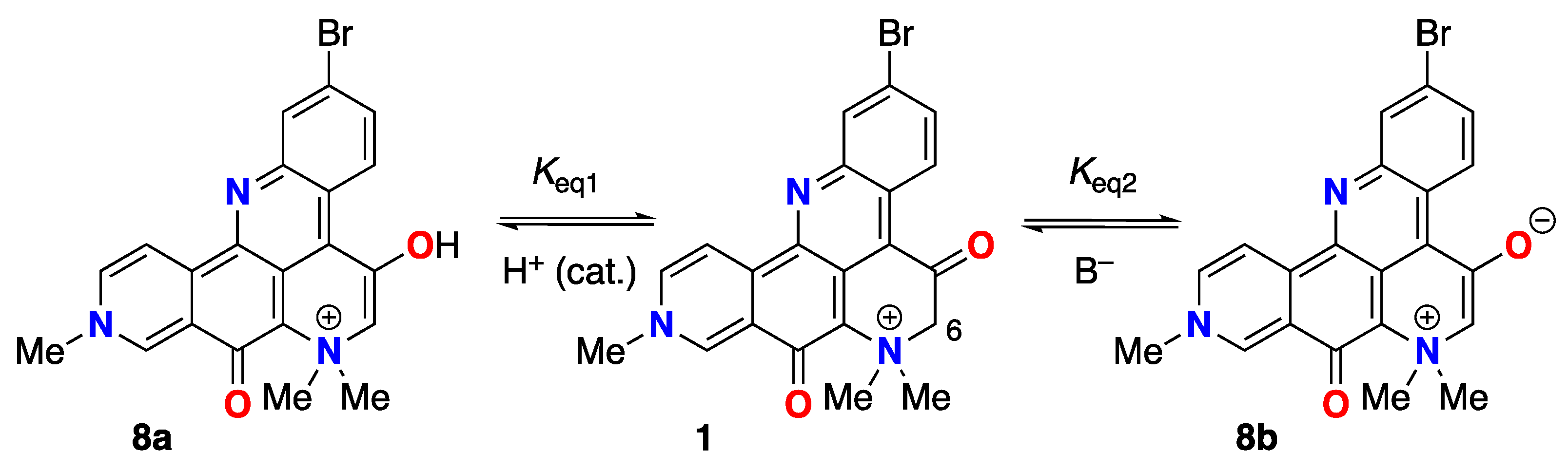
In hydroxylic solvents, 1 also resides largely in the di-keto form, however, we found that in both D2O and CD3OD, the C-6 methylene protons undergo rapid exchange to give the C-6 CD2-isotopomer (1-d2) [1]; a rate so fast that we were unable to detect the C-6 CH2 or the intermediate forms within the time frame between sample preparation and measurement of the 1H NMR spectra. This observation was supported by 1H NMR, HSQC and HMBC measurements of 1 in protic solvent (CD3OH) where the C-6-CH2 group is still observable (HSQC correlation: H-6 to C-6, dH 4.64, s; dC 70.4 ppm). The simplest explanation for both phenomena is catalytic H-D exchange a-to the C-5 C=O group favored by an intermediate; the extensively-conjugated enol tautomer 8a (Figure 6).
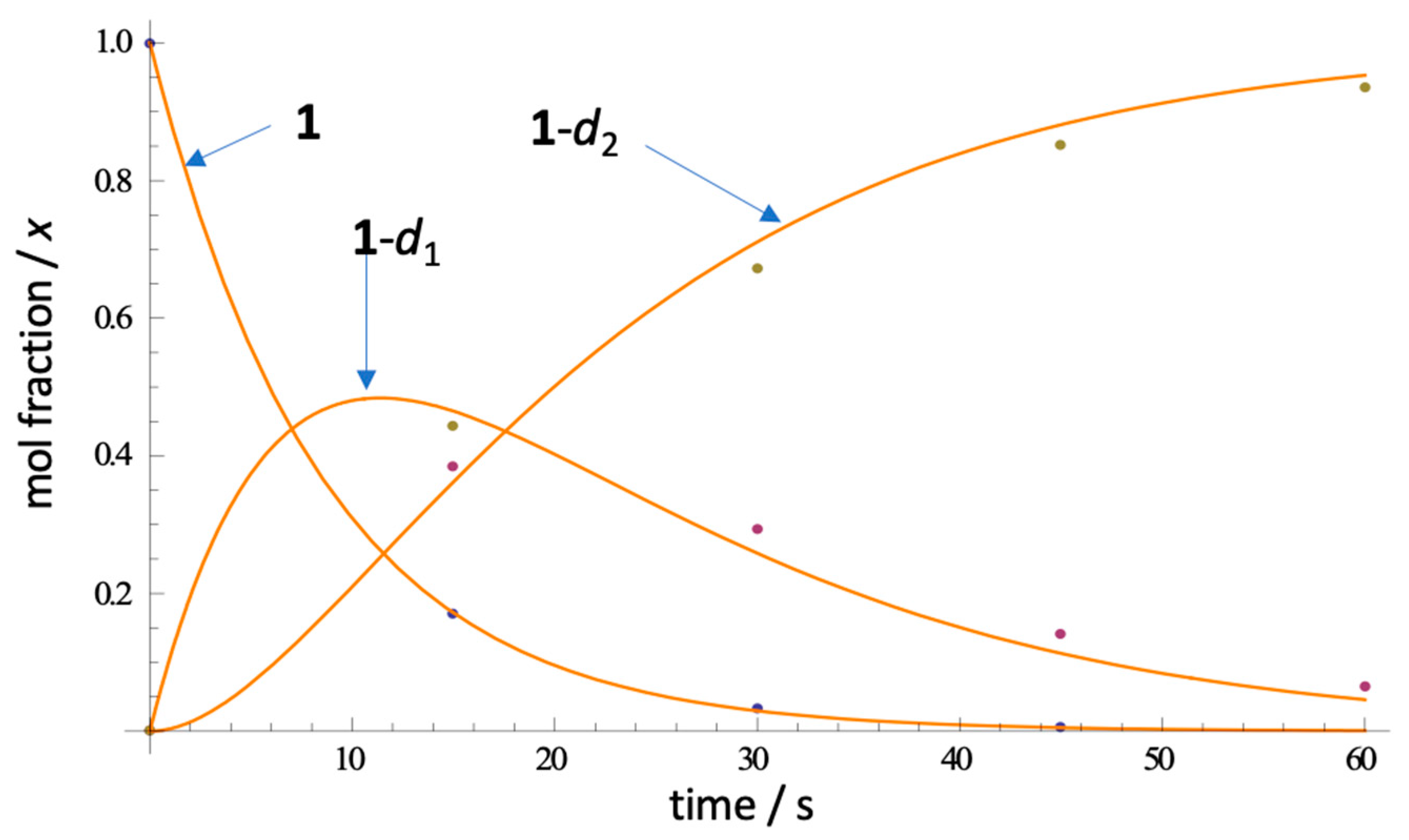
In order to estimate the rate of H-D exchange in 1 and place an upper bound on it, we measured the time-dependent appearance of the CD2-isotopmer by ESI mass spectrometry after rapid dissolution of 1 in CD3OD (Figure 7). Under controlled conditions (23 ˚C, rapid sampling and ESIMS in situ measurement- see Experimental), 1 (C21H1789BrN3O2, m/z 422.0499 [M+]) rapidly disappeared, followed by replacement by ephemeral isotopologue 1-d1 (m/z 423.0561) and finally, convergence upon 1-d2 (m/z 424.0624). Remarkably, complete exchange (>90%) is observed within 90 s of dissolution of 1. From triplicate measurements, and rapid sampling (first measurement at t = 15 sec), we were able to fit the kinetic deuterium exchange data to a first order rate law and estimated the apparent first and second H-D exchange rate constants to be (Table 2), k1= 0.1394(7) s–1 and k2 = 0.0765(47) s–1 (best R2; see Supporting Information). As expected, k2 is about half of k1, consistent with the expected rate laws, and negates involvement of a substantial kinetic isotope effect for the exchange reaction.
Figure 7.
Representative ESIMS measurements of the rate of H-D exchange of petrosamine (1 in CD3OD, 23 ˚C) and fitted curves (non-linear regression). See Supp. Info. for rate law and k determinations.
Figure 7.
Representative ESIMS measurements of the rate of H-D exchange of petrosamine (1 in CD3OD, 23 ˚C) and fitted curves (non-linear regression). See Supp. Info. for rate law and k determinations.
The exchange of 1 to 1-d2, in the absence of added acid, appears to be much faster than H-D exchange rates of other phenones, e.g. propiophenone (pKa 24.4, DMSO [19] a-tetralone (pKa 24.7 [20,21]. The rates of acid-promoted keto-enol equilibration of acetophenone (pKa 18.4±0.03) have been measured. For example, the rate of ketonization of acetophenone enol (1-phenylethen-1-ol) is linearly dependent upon [H3O+] with a catalytic efficiency determined to be kH+ = (1.25±0.02) x 103 M–1s–1 [22]. We conclude that tautomerism of 1 too must be very fast even in the absence of added acid. Either the enol 8a or enolate 8b, although undetectable as a discrete species in the time frame of 1H NMR, present a pathway to the acid-base equilibria of 1 and 1-d2, but enolization of the keto form is likely dominant.
Table 2.
Rate constants k1 and k2 for H-D exchange in 1 in CD3OD (23˚ C, see Figure 5).1.
| Reaction | k1 (s–1) | k2 (s–1) |
|---|---|---|
| 1 –> 1-d1 | 0.1394(7) | – |
| 1-d1 –> 1-d2 | – | 0.0765(47) |
1 See Supporting Information for plotted raw ESIMS data and detailed kinetic analysis.
Some amount of discussion has been given on the keto-enol state of 1. The NMR data for 1 reported by Suwainborirux and coworkers support the C5 keto form in DMSO-d6 [5] and – in agreement with Molinski and Faulkner, the enol form in D2O or CD3OD [1] – but Quinn and coworkers find, “no evidence for this keto-enol isomerism” in 2. [1] Likely, the Keq of keto-enol tautomerism lies on some continuum, dependent upon solvent dialectric and H-bond donor ability.
The observed 1H and 13C NMR spectra of petrosamine (1), and the C6 exchange to CD2 in CD3OD/NaOD, support the enol form in hydroxylic solvents. It’s likely fast exchange hybrid structure of 1 and 8a,b with an equilibrium constant Keq1 largely in favor of 1 in DMSO-d6, moving to the dominant 8a in hydroxylic solvents. The enolate 8b may be favored as the catalytically important intermediate, for reasons of charge neutralization, but insufficient data can support this hypothesis with more certainty. In either case – enol or enolate intermediate – we surmise the electron-withdrawing quaternized N+Me2 group in 1 and the related petrosamines, 2, 3, plays an significant role in accelerating the rates of H-D exchange and lowering the pKa of the C-6 CH2 group.
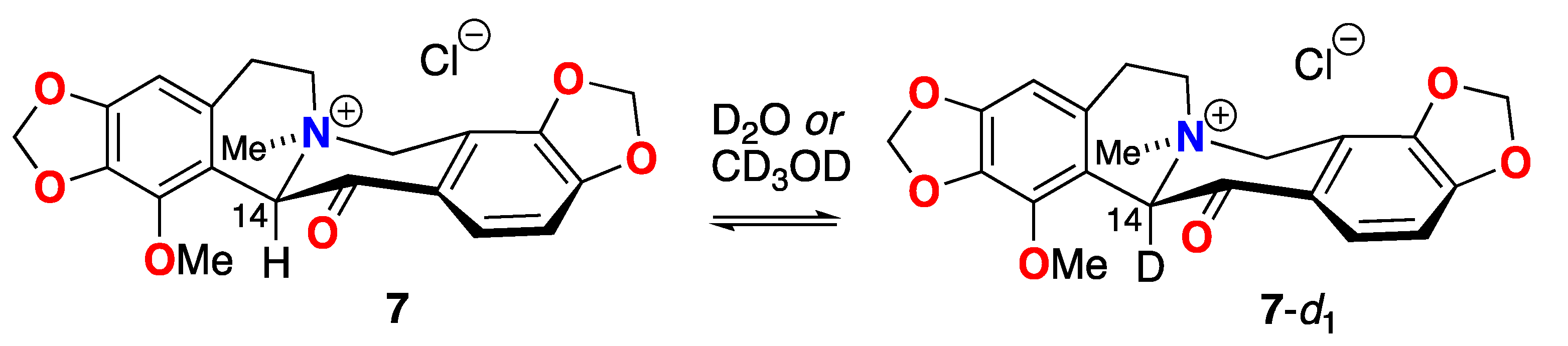
To the best of our knowledge, rapid H-D exchange of a substituted b-quaternary ammonium ketone within a natural product has been observed only in one other instance, coulteroberbinone (7, Figure 8 [23]), an N-quaternary ammonium isoquinoline alkaloid from the leaves of Romneya trichocalyx. The authors note the C-14 C-H in 7, assigned to the α-proton between the carbonyl and quaternized nitrogen (dH 5.64, s), underwent rapid proton-deuterium exchange in D2O or CD3OD to C-D (7-d1) under ambient conditions (supported by 1H NMR and ESIMS data).
Figure 8.
Rapid H-D exchange of coulterberbinone (7) in deuteric solvent under ambient conditions.
Two major factors most likely explain the relatively low pKa of 1 and 7: the electron withdrawing (inductive) effect of the -N+Me2 quaternary ammonium group (N-7) and stabilization of enol 5 (or enolate 6) through extensive conjugation of the heteroaromatic core, not unlike the stabilization of the enolates of alkyl phenones, e.g. acetophenone, determined by UV-vis (pKa = 18.4). Indirect determinations of the pKa of the enol of acetophenone has also be obtained from kinetics of reactions of acetophenone: e.g. a-chlorination [24], and aminolysis of the corresponding enol acetate [25]. In contrast, 1 appears to undergo rapid enolization in the absence of added Brønsted acid, suggesting the possibility that this tautomerization that may even be autocatalytic.
Is the enol 8a of petrosamine (1) present in substantial concentrations? From the 1H NMR spectrum of 1, (DMSO-d6) we detect no signals attributable to 8a. It can be ascertained from 1H NMR that Keq1 (Figure 7) is very small (estimated from the limits of integration, Keq1 < 0.05) and the equilibrium of tautomers lies well towards the keto form 1. As noted above, in highly-basic aqueous solutions of 1, the charge-minimal enolate 8b begins to appear in substantially concentrations placing a lower-boundary of pKa ~15 for 1. In the absence of base, the mechanism of H-D exchange 1 to 1-d2 in CD3OD most likely engages substantial equilibrium concentrations 8a to allow rapid exchange of the CH2 to CD2 in deuteric solvents within less than 2 minutes at 23 ˚C. Estimation of an accurate pKA of 1 is not accessible from measurements in aqueous solvents and securing the same will likely require measurements in a suitable aprotic solvent (e.g. titration in DMSO [20,26][2]).
3. Materials and Methods
3.1. General Experimental Procedures
Inverse detected 2D NMR spectra were measured on a Jeol ECA (500 MHz) spectrometer, equipped with a 5 mm 1H{13C} 5 mm probe, or a Bruker Avance III (600 MHz) NMR spectrometer with a 1.7 mm 1H{13C} microcryoprobe. 13C NMR spectra were collected on a Varian NMR spectrometer (125 MHz) equipped with a 5 mm Xsens 13C{1H} cryoprobe. NMR spectra were referenced to residual solvent signals, (CD3)2CO (dH 2.05, dC 29.8). High-resolution ESITOF analyses were carried out on an Agilent 1200 HPLC coupled to an Agilent 6350 TOFMS. Low-resolution MS measurements were made on a Thermoelectron Surveyor UHPLC coupled to an MSD single-quadrupole detector. HPLC was performed on an Agilent 1200 HPLC. UV-vis spectra were measured on a Jasco V-630, spectrometer in quartz cells (1.00 cm pathlength, Helma). FTIR spectra were collected on thin film samples using a Jasco FTIR-4100 fitted with an ATR accessory (ZnSe plate). Optical densities (OD, l nm) in microplate wells were measured using a Molecular Devices Spectramax 384 Plus. Measurements of pH were made with a digital pH meter (Denver Instrument, Model 220), calibrated against standard solutions (NaH2PO4-Na2HPO4).
3.2. UV-vis Measurements
Standard solutions of accurately-weighed 1 and 6b were prepared in volumetric flasks and used for serial dilutions to the final working concentrations in specified media, either pure HPLC grade solvent (DCM, DMSO, acetone, DMF – see Supporting Information for 6b in acetone) or mixtures of aqueous HPLC solvents of defined H2O composition.
3.3. Synthesis of Merocyanine Dye (6b) [16]
Iodomethane (3.11 mL, 50 mmol) was slowly added to a cold mixture of 4-methylpyridine (4.86 mL, 50 mmol) and 2-propanol (5 mL). The stirred mixture was heated at reflux overnight. Removal of the solvent gave the crude 4-methylpyridinium methiodide (10.23 g) as an off-while solid. A portion of the latter salt (3.00 g, 12.8 mmol), 4-hydroxybenzaldehyde and piperidine (1.06 mL, 10.7 mmol) were dissolved in anhydrous EtOH (16 mL) and heated at reflux overnight, with stirring. Upon cooling, a red precipitate was deposited. The red solid was filtered (Büchner funnel) and dissolved in KOH solution (0.2 M, 17 mL, 15 mmol) and heated with stirring for 30 min. Blue-red shiny crystals were recovered by filtration, washed with a little cold water, and dried to give of 6b (2.00 g, 65%). The 1H NMR of the sample was consistent with the expected product.
3.4. H-D Exchange Measurements of 1 by ESIMS
A sample of petrosamine (1) was rapidly dissolved in CD3OD (23 ˚C). An aliquot of the solution was immediately introduced into the inlet of the TOF mass spectrometer and the ratios of 1-d0, 1-d1 and 1-d2 were measured ‘on the fly’ from the corresponding molecular ion intensities [M] + normalized to the [M] + of a solution of 1 in CH3OH at the same concentration. Subsequent measurements were made at 15 s intervals over a total reaction time of 90 s. See Figure 7 and S6.
4. Conclusions
Evidence supports that the major tautomer of petrosamine is the keto form 1, both in solution and solid states. While the enol form 8a was not detectable by 1H NMR in DMSO-d6, it is nevertheless likely to be responsible for H-D exchange of 1 in deuteric solvents. The exceptional solvatochromism of 1 is best attributable to charge delocalized resonance forms 1a and 1d – partial structures seen within 1 that are analogous to Brooker merocyanine dyes [11]. As with the latter, the hyposochromic solvents effects mostly correlate with polar-solvent stabilization of the ground state (HOMO) and attribution to the color changes of 1. The complete isotopic exchange of the C6 CH2 group in 1 to CD2 in deuterated solvent under ambient conditions is supported by 1H NMR and fast-sampled ESIMS measurements. The exchange mechanism likely proceeds through rapid acid-catalyzed keto-enol tautomerism.
Refinement of the molecular structure of 1 by QM methods maps the electron delocalization and accompanying charge distribution in 1. These and other refinements complement computational docking studies that can lend more precise definition of host-guest interactions of 1 in its cognate enzyme active site. In turn, these observations may assist in the design and synthesis of new AChE inhibitors based on the pyridoacridine skeleton: a privileged alkaloid molecular framework produced exclusively by marine invertebrates.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. FTIR of merocyanine 6b, pH dependent UV-vis spectra of 1 FTIR of merocyanine 6b and raw data (ESIMS) for H-D exchange rate studies of 1.
Author Contributions
C.G. carried out the experimental work with consultation from T.F.M. The manuscript was co-written by C.G. and T.F.M.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Original data will be made available upon reasonable request.
Acknowledgments
We are most grateful for a generous gift of petrosamine (1) from P. Stout (Sirenas Marine Discovery, LLC, San Diego, CA). We thank C. Perrin (UCSD) for insightful discussions and N. Goodyear for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Molinski, T. F.; Faulkner, D. J. Petrosamine, a Novel Pigment from the Marine Sponge Petrosia sp. J. Org. Chem. 1988, 53, 1341–1343. [Google Scholar] [CrossRef]
- Molinski, T. F. Marine Pyridoacridine Alkaloids: Structure, Synthesis, and Biological Chemistry. Chem. Rev. 1993, 93, 1825–1838. [Google Scholar] [CrossRef]
- Schmitz, F. J.; Agarwal, S. K.; Gunasekera, S. P.; Schmidt, P. G.; Shoolery, J. N. Amphimedine, New Aromatic Alkaloid from a Pacific Sponge, Amphimedon sp. Carbon Connectivity Determination from Natural Abundance 13C-13C Coupling Constants. J. Am. Chem. Soc. 1983, 105, 4835–4836. [Google Scholar]
- Marshall, K. M.; Barrows, L. R. Biological activities of pyridoacridines. Nat. Prod. Rep. 2004, 21, 731–751. [Google Scholar] [PubMed]
- Nukoolkarn, V. S.; Saen-oon, S.; Rungrotmongkol, T.; Hannongbua, S.; Ingkaninan, K.; Suwanborirux, K. Petrosamine, a potent anticholinesterase pyridoacridine alkaloid from a Thai marine sponge Petrosia n. sp. Bioorg. Med. Chem. 2008, 16, 6560–6567. [Google Scholar]
- Carroll, A. R.; Ngo, A.; Quinn, R. J.; Redburn, J.; Hooper, J. N. A. Petrosamine B, an Inhibitor of the Helicobacterpylori Enzyme Aspartyl Semialdehyde Dehydrogenase from the Australian Sponge Oceanapia sp. J. Nat. Prod. 2005, 68, 804–806. [Google Scholar]
- Wei, X.; Bugni, T. S.; Harper, M. K.; Sandoval, I. T.; Manos, E. J.; Swift, J.; Van Wagoner, R. M.; Jones, D. A.; Ireland, C. M. Evaluation of Pyridoacridine Alkaloids in a Zebrafish Phenotypic Assay. Mar. Drugs 2010, 8, 1769–1778. [Google Scholar]
- Choi, D-Y. ; Choi, H. Natural products from marine organisms with neuroprotective activity in the experimental models of Alzheimer’s disease, Parkinson’s disease and ischemic brain stroke: their molecular targets and action mechanisms. Arch. Pharm. Res. 2015, 38, 139–170. [Google Scholar]
- See Supporting information of Ref.
- Exchange of 1H-13C for 2H-13C results in dramatic attenuation of the 13C signal due to quadrupolar relaxation, and absence of the heteronuclear nOe, among other effects.
- Brooker, L. G. S.; Heyes, G. H.; Sprague, R. H.; Van Dyke, R. H.; Van Lare, E.; Van Zandt, G.; White, F. L. Studies in the Cyanine Dye Series. XI. The Merocyanines. J. Am. Chem. Soc. 1951, 73, 5326–5332. [Google Scholar]
- Brooker, L. G. S.; Heyes, G. H.; Sprague, R. H.; Van Dyke, R. H.; Van Lare, E.; Van Zandt, G.; White, F. L.; Cressman, H. W. J.; Dent, S. G. Color and Constitution. X. Absorption of the Merocyanines J. Am. Chem. Soc. 1951, 73, 5332–5350. [Google Scholar]
- Other possible resonance forms, for example invoking electron donation from Br, are considered only minor contributors.
- Simpson, W.T. A Mathematical Treatment of the Color of the Merocyanine Dyes. J. Am. Chem. Soc. 1951, 73, 5359–5356. [Google Scholar] [CrossRef]
- Morley, J. O.; Morley, R. M.; Docherty, R.; Charlton, M. H. Fundamental Studies on Brooker’s Merocyanine. J. Am. Chem. Soc 1997, 119, 10192–10202. [Google Scholar] [CrossRef]
- Minch, M.J.; Sadiq Shah, S. A Merocyanin Dye Preparation for the Introductory Organic Laboratory. J. Chem. Educ. 1977, 54, 709. [Google Scholar] [CrossRef]
- We confirm the solvatochromism of one example, i, by synthesis and its solvent-dependent UV-vis properties. See Supporting Information.
- Hasselbalch, K.A. Die Berechnung der Wasserstoffzahl des Blutes aus der freien und gebundenen Kohlensäure desselben, und die Sauerstoffbindung des Blutes als Funktion der Wasserstoffzahl. Biochem. Z. 1917, 78, 112–114. [Google Scholar]
- Bordwell, F.K.; Harrelson Jr., J. A. Acidities and homolytic bond dissociation energies of the αC—H bonds in ketones in DMSO. Can. J. Chem. 1990, 68, 1714–1718. [Google Scholar]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-reich-bordwell.
- Chiang, Y.; Kresge, J.; Wirz, J. Flash-Photolytic Generation of Acetophenone Enol. The Keto-Enol Equilibrium Constant and pKa of Acetophenone in Aqueous Solution. J. Am. Chem. Soc. 1984, 106, 6392–6395. [Google Scholar] [CrossRef]
- Valpuesta, M.; Díaz, A.; Suau, R. Coulteroberbinone, a quaternary isoquinoline alkaloid from Romneya coulteri. Phytochemistry 1999, 51, 1157–1160. [Google Scholar] [CrossRef]
- Guthrie, J. P.; Cossar, J.; Klym, A. pKa values for substituted acetophenones: values determined by rates of halogenation. Can. J. Chem. 1987, 65, 2154–2159. [Google Scholar] [CrossRef]
- Novak, M.; Marc Loudon, G. Aminolysis of Acetoxystyrenes. The pKa of Acetophenones in Aqueous Solution. J. Am. Chem. Soc. 1976, 98, 3591–3597. [Google Scholar] [CrossRef]
- Steiner, E.C.; Gilbert, J.M. The Acidities of Weak Acids in Dimethyl Sulfoxide. J. Am. Chem. Soc. 1963, 85, 3054–3055. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



