
Submitted:
12 July 2023
Posted:
13 July 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Since the first description of SARS-CoV-2 in China in 2019, thousands of variants have emerged worldwide. For some of them, the constellation of mutations caused changes in virus biology, pathogenicity, infectivityity and transmissibility resulting in dissemination throughout the world. Gamma variant (P.1) differs from SARS-CoV-2 Wuhan strain (B.1) by 12 amino acids in the Spike (S) protein, and presented mutations related to greater affinity for the receptor angioten-sin-converting enzyme 2 (ACE-2) and/or immune escape. The Gamma variant and subvariants were responsible for the second wave of COVID-19 in the Brazilian city of Manaus, characterized by high mortality and rapid transmission. The ability of variants to induce cytokine production may be closely related to their pathogenicity. Herein we observed that there was no significant difference in the quantity of cytokines among macrophages or neutrophils infected with P.1 and B.1 strains. Also, no significant difference was observed in the absolute number of macrophages and neutrophils infected with these variants. Furthermore, no evidence of SARS-CoV-2 replication was observed in macrophages when infected by the two analyzed variants. Our findings suggest that the difference in the epidemiological outcome observed during the P.1 variant spread when compared to B.1, it is not explained by differences in the quantity of cytokines and absolute number of macrophages or neutrophils. Through bioinformatics analysis of the S protein, we observed that the physicochemical differences between the variants and subvariants of P.1, probably refer to the degree of infectivity, due to the impact caused in the recognition of antibodies and receptor af-finity
Keywords:
SARS-CoV-2
; P.1 variant
; B.1 strain
; cytokines
; COVID-19
; macrophages
; neutrophils
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a Betacoronavirus, is the etiological agent of COVID-19 [1]. SARS-CoV-2 was detected in 2019, China, and quickly spread around the world, causing a pandemic. Symptoms and clinical signs of COVID-19 range from mild like cough, fever, fatigue and nausea, to more severe presentation like thrombosis, the multisystem inflammatory syndrome in children (MIS-C), which is an uncommon but potentially life-threatening complication of COVID-19, and acute respiratory distress syndrome (ARDS) [2-5]. The infection causes a cytokine storm produced by macrophages and neutrophils recruited to the lung, and characterized by the presence of cytokines such as IL-6, IL-5, IL-10, IL-12p70, IL-17 and IFN-y [3,4].
SARS-CoV-2 has positive-sense single-stranded RNA genomes with nearly 29kb in length [6], and it has structural proteins named Spike (S) protein, envelope (E), membrane (M) and nucleocapsid (N). To enter and infect human cells, the S protein binds the receptor angiotensin-converting enzyme 2 (ACE2), that is found in many mammalian body tissues [7]. The S protein contains two functional domains, the S1 domain contains the receptor-binding domain (RBD) that recognizes ACE2, and S2 contains a lipophilic region that allows the fusion of the viral particle to the cell membrane. The serine protease enzyme TMPRSS2 present on the surface host cell cleaves the S protein allowing the entry of virus [8]. Afterwards, the genome region Open Reading Frames (ORFs) are translated, witch ORF1a and ORF1b encodes a polyproteins pp1a and pp1ab and cleaved into a 16 nonstructural proteins (NSP) that are associated to the virus transcription and replication [9].
Type I and III IFN normally have a rapid response to viral infections, but SARS-CoV-2 infection has been observed to delay the release and production of IFNs [10]. The imbalance in the IFN response would affect the control of viral suppression and consequently a greater production of inflammatory cytokines, causing a hyperinflammation [10,11], characteristic of the cytokine storm [12]. Patients who progress to the severe form of COVID-19 have a high neutrophil/lymphocyte ratio[13], in these critically ill patients, the presence of inflammatory monocytes was identified as being responsible for the production a greater amount of proinflammatory cytokines, characteristics of the cytokine storm and responsible for the ARDS [2]. In ARDS, IL-17 increases lung parenchymal destruction by recruiting neutrophils, producing pro-inflammatory mediators, and preventing apoptosis due to induction of G-CSF expression [14].
SARS-CoV-2 presented several variants of concern (VOCs) such as Alpha, Beta, P.1, Delta and Omicron [15]. At the end of 2020, the P.1 variant was first identified through a man who traveled from the Brazilian state of Amazonas to Japan [16]. The P.1 has three key mutations K417T, E484K, and N501Y in the RBD, that indicate an increase in binding affinity for ACE2, allowing the virus to enter the cell and replicate more efficiently [17-19]. The K417T and E484K mutations interact with hACE2, while E484K is present in the loop region outside the direct human ACE2 interface [20]. Mutations in RBD can also significantly affect the binding and neutralization of the virus, playing a role in decreasing the effectiveness of vaccines [21]. Vaccinated individuals with BNT162b2 (Pfizer/ BioNTech) is less efficient against infection by P.1 variant as compared to convalescent COVID-19 [22].
By the end of 2020, a drastic increase in the number of P.1 variant cases, and consequently in the number of deaths was reported in the city of Manaus, northern Brazil [23,24]. The persistent transmission of SARS-CoV-2 is closely associated with the prevalence of the P.1 variant and the emergence of subvariants, some more transmissible, that have NTD deletions or mutations in the S protein, in the furin cleavage region [25]. The subvariants that have these mutations are called P.1.6 and P.1.7 (P681H), and P.1.8 (P681R). The P.1.3 variant was designated as the one with the deletion in the NTD region and the P.1.4 and P.1.5 variants are the variants with the N679K mutation [25]. Most of the genetic alterations identified in the P.1 subvariants also appear in other VOCs (Alpha, Delta and Omicron), and are related to greater viral infectivity, immune escape, or both [26-28], specifically the P681R mutation also enhances viral replication, viral fusion and viral cell-to-cell spread in vitro [29-31].
The question arose whether the new variant prevalent in Manaus would be more pathogenic than the Wuhan variant as result of increased mortality and contamination. Several studies have shown that the more recently emerged variants have biological and clinical characteristics that are distinct from each other and different from the original Wuhan strain (B.1). These studies indicate greater pathogenicity of the Alpha, Beta, Delta and P.1 variants, with an increase in severe cases and deaths [32-41]. The opposite is observed with the Omicron variant [42-48].
Data on the pathogenicity of the P.1 variant are still unclear, however, some studies indicate a distinct epidemiological outcome, with increased mortality and transmissibility, indicating that it may have relevant biological differences in relation to the B.1 strain [24,41,49]. Considering the participation of neutrophils [50] and macrophages [51] in the pathogeny of severe COVID-19, the main objective of this study was to evaluate whether the P.1 variant has a higher pathogenic biological potential when compared to the B.1 strain regarding their host interaction and cytokine stimulation. Neutrophils and macrophages obtained from severe COVID-19 patients were experimentally infected by both the B.1 strain and the P.1 variant. Additionally, we evaluate, through bioinformatics, the physicochemical characteristics of the S protein of the P.1 variant, its subvariants and of the B.1 strain, which may corroborate possible biological differences among them, such as greater infectivity, transmissibility, escape of antibodies observed in P.1 epidemic.
Data on the pathogenicity of the P.1 variant are still unclear, however, some studies indicate a distinct epidemiological outcome, with increased mortality and transmissibility, indicating that it may have relevant biological differences in relation to the B.1 strain [24,41,49]. Considering the participation of neutrophils [50] and macrophages [51] in the pathogeny of severe COVID-19, the main objective of this study was to evaluate whether the P.1 variant has a higher pathogenic biological potential when compared to the B.1 strain regarding their host interaction and cytokine stimulation. Neutrophils and macrophages obtained from severe COVID-19 patients were experimentally infected by both the B.1 strain and the P.1 variant. Additionally, we evaluate, through bioinformatics, the physicochemical characteristics of the S protein of the P.1 variant, its subvariants and of the B.1 strain, which may corroborate possible biological differences among them, such as greater infectivity, transmissibility, escape of antibodies observed in P.1 epidemic. Data on the pathogenicity of the P.1 variant are still unclear, however, some studies indicate a distinct epidemiological outcome, with increased mortality and transmissibility, indicating that it may have relevant biological differences in relation to the B.1 strain [24,41,49]. Considering the participation of neutrophils [50] and macrophages [51] in the pathogeny of severe COVID-19, the main objective of this study was to evaluate whether the P.1 variant has a higher pathogenic biological potential when compared to the B.1 strain regarding their host interaction and cytokine stimulation. Neutrophils and macrophages obtained from severe COVID-19 patients were experimentally infected by both the B.1 strain and the P.1 variant. Additionally, we evaluate, through bioinformatics, the physicochemical characteristics of the S protein of the P.1 variant, its subvariants and of the B.1 strain, which may corroborate possible biological differences among them, such as greater infectivity, transmissibility, escape of antibodies observed in P.1 epidemic.
2. Materials and Methods
2.1. Human samples
Blood samples from 3 hospitalized severe COVID-19 patients in the acute phase of infection were used in this study. The criteria for confirmed cases with acute SARS-CoV-2 infection included positive result of the nucleic acid sequence of SARS-CoV-2 by real-time RT-qPCR from nasopharyngeal swab samples based on FDA-approved RNA testing. Severe COVID-19 patients were clinically classified as having fever, respiratory infection, respiratory rate of 23 incursions/minute, dyspnea and oxygen saturation < 93% at room air. The research was approved by the Research Ethics Committee (CEP) from Brazilian National Health Council and all patients signed a free and informed consent form in accordance with current legislation and the relevant ethical regulations approved by the Hospital Naval Marcílio Dias (CAAE # 31642720.5.0000.5256).
2.2. Virus strains
Macrophages and neutrophils, in all experiments, were infected by both severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lineages B.1 (hCoV-19/Brazil/RJ-FIOCRUZ-314/2020, GISAID accession number EPI_ISL_414045) and P.1 (hCoV-19/Brazil/AM-FIOCRUZ-3521-1P/2021, EPI_ISL_1402431) in a Biosafety Level 3 (BSL-3) facility.
2.3. Cell cultures
Macrophages and neutrophils were isolated from the collection of 20 mL of heparinized peripheral blood from COVID-19 severe patients and healthy donors. The blood collected was slowly placed in a 50 mL tube containing 1:2 Ficoll, the gradient was centrifuged for 30 min at room temperature without braking and without acceleration (400g). After centrifugation, the upper part containing mononuclear cells was collected for cytokine, microscopy, and viral replication assays. The bottom part containing neutrophils were collected with a Pasteur pipette and, after lysis of the red blood cells, the granulocytes were resuspended in RPMI medium, counted, and adjusted for each experimental condition, microscopy and cytokine assays. Mononuclear cells were also counted and adjusted for each experimental condition, then plated for 2 h in RPMI medium with 1 % nutridoma in 24-well plate containing circular coverslips in the bottom of each well, then after non-adherent cell were removed, the medium was replaced for the next assays.
2.4. Microscopy assay
Macrophages adherent to the coverslips in RPMI medium containing 1% nutridoma adjusted to 1 × 105 macrophages/mL were infected or not with SARS-CoV-2 B.1 virus MOI 9.0 and SARS-CoV-2 P.1 virus MOI 9.0. Samples were then incubated for 24 h at 37 °C/5% CO2. Afterwards, the coverslips were washed with PBS and fixed in 4% paraformaldehyde for 20 min. The coverslips were removed from the plates and stained by the Panoptic method (Laborclin). After staining and fixing the coverslips on slides using Entellan, the macrophages and neutrophils were counted under an optical microscope at 1000x magnification. Cells were counted by visual field and percentage of increase or decrease in cell population was obtained by comparing with the respective experimental control group.
2.5. Detection and quantification of cytokines
Supernatant from the culture of the macrophages and neutrophils from the same experimental conditions of the microscopy assay were collected and then stored at -80 ° in a freezer for posterior analysis. According to the manufacturer’s recommendations (Quantikine Elisa/R&D System), each sample were plated in 96-well plates for the analysis of: MIP-1β, IL-6, IFN-γ, IL-1ra, IL- 5, RANTES, IL-1β, bFGF, PDGF-BB, IL-4, MCP-1, MIP-1α, IL-10, G-CSF, IL-12p70 and IL-17A. Relative concentration of the supernatants ware plotted for each experimental group.
2.6. Detection and quantification of SARS-CoV-2 by RT-PCR
Briefly, a 24-well plate with macrophages (3x105/well) of health donors were infected by both P.1 and B.1. For viral infection, a Multiplicity of Infection (MOI) of 0.01 was used for both SARS-CoV-2 lineages. The control was inoculated with PBS 1X. The macrophages were incubated for 24, 48 or 72 hours, and supernatants were collected for RT-PCR. RNA extractions were performed using the RNA extraction kit - QIAamp Viral RNA Mini Kit (QIAGEN). RT-PCR was performed using the qRT-PCR kit - Molecular SARS-CoV-2 Kit (E/N/RP) (Bio-Manguinhos/IBMP, Brazil).[52]
2.7. Modeling
The sequences of P0DCT2 (Uniprot) were used as a reference sequence for WT-Wuhan. Information on the positions and mutations that occur in the variants was taken from the GISAID [52]. For the construction of variant sequences, mutations that were present in 90% of the sequences sampled by GISAID were used. The search in GISAID was performed through Lineage Comparison with the following parameters: Selected lineages= Gamma (include sublineages), Add a Specific Lineage= B.1.1.28. Through this search, it was possible to identify the mutations that occur in the strains to make the necessary changes in the S protein sequence. The P.1.5 variants have the same mutations as P.1.4, and the P1.6 variant has the same mutations as P.1.7, so the images of these structures were not added to the figures. Mutations were positioned using Jalview. The mutated fasta sequences were modeled with Swissmodel and the quality of the model was evaluated using PROCHECK.
2.8. Calculation of electrostatic potential and solvent accessible surface
The models generated for each variant were used to perform electrostatic surface potential analysis, hydrophobicity and solvent-accessible area analysis calculations. The surface electrostatic potential map was calculated using the Adaptive Poisson-Boltzmann Solver -APBS- https://server.poissonboltzmann.org. Images with information about the position of mutations in the crystals, surface electrostatic potential and hydrophobicity were made with ChimeraX v.1.4. For the calculation of solvent accessible surface area was performed GetArea (Fraczkiewicz et al. 1998- https://curie.utmb.edu/getarea.html). The information for each model was saved and the clusterization was made with pvclust package of RStudio version 4.2 (figure 6a). The webPIPSA was used to compare the interaction properties of the modeled proteins and the results can be seen in the figure 6b and supplementary figure 5. Data referring to the electrostatic map were used to create the surface images with the help of ChimeraX. The generated models were analyzed using GetArea, to identify the solvent accessible areas and the hydrophobicity of the residues.
2.9. Figures with the epitope location frequencies (Immunogenicity Scale)
The Immunome Browser tool disponible in Immune Epitope Database and Analysis Resource –IEDB (www.iedb.org) was used for recovering the figure with experimentally described immunogenicity of the SARS-CoV-2 S protein (figure 5-A and D). The following parameters were used in the search: Assay: B cell, Epitope Source: Organism = SARS-CoV-2 (ID2697049), Antigen= Spyke Glycoprotein P0DTC2, MHC Restriction = No MHC assays, Host = Human. The figures were built with the images of the models generated in ChimeraX. The models were superimposed, and electrostatic surface calculation was applied through the map generated by APBS to allow to see the characteristics of each model in relation to the view of the RBD region and in the lateral position. Images with information about the hydrophobic surface were also generated at the same positions. Regarding the variants, the positions of the mutations were identified in pink and the region of the cleavage site was identified with orange color. The relationship of the variants in relation to the electrostatic surface was verified through the webPIPSA program and can be found in the figure 6b. The webPIPSA evaluates and analyzes the electrostatic potential of the proteins and makes a comparison between the analyzed files, generating a clustering of the analyzed models and an epogram with a color-coded distance matrix. The models were clustered in relation to the ASA residue data and the hydrophobicity of GetArea through the pvclust package of R. The information about the results of webPIPSA and pvclust are found in the supplementary figure 6a.
2.10. Statistical analysis
Results are expressed as mean ± SEM and p ≤ 0.05 was considered significant. Comparisons were made in relation to the control group, using the Student’s T test, and multiple comparisons between the experimental groups, using Two Way ANOVA with Dunnett’s test. Data analysis was performed by using the GraphPad Prism 6.0 software.
3. Results
3.1. Both variants do not change the cytokine profile of macrophages and neutrophils
Cytokine storm, produced by macrophages and neutrophils, is one of the events closely related to the severity of COVID-19 [1-4]. Thus, the ability of variants to induce cytokine production may be closely related to their pathogenicity. Cytokine storm, in COVID-19, is basically promoted by cells present in lung tissue, epithelium, resident macrophage and stromal cells, and is amplified by the recruitment of macrophages and neutrophils into the tissue [53]. In this regard, we assessed the cytokine spectrum released by macrophages and neutrophils interacting with SARS-CoV-2.
To approach this question, we used cells obtained from severe acutely infected COVID-19 patients. The cells were treated with the B.1 or P.1 variants (both with MOI=9). MOCK controls were obtained by culturing with medium only. After 24 hours of infection, the supernatants were collected and the production of cytokines evaluated by ELISA for MIP-1 β, IL-6, IFN-y, IL-1ra, IL-5, RANTES, IL-1β, bFGF, PDGF- BB, IL-4, MCP-1, MIP-1α, IL-10, G-CSF, IL-12p70 and IL-17RA.
Our results showed that, for all cytokines, there is no significant difference between macrophages from patients infected with the P.1 or B.1 variants or MOCK (Figure 1). The same pattern can be observed for the production of cytokines by neutrophils, in all of the evaluated cytokines, we did not observe differences when comparing the P.1 and B.1 variants (Figure 2).
3.2. B.1 variant shows more cytopathogenic than the P.1 variant
Next, we analyzed the survival of macrophages and neutrophils infected with the P.1 or B.1 variants, in order to determine whether P.1 variant could have a greater cytopathogenicity, which could be associated with the induction of a more intense inflammation in the lungs, and consequently greater pathogenicity. To this end, we obtained peripheral blood macrophages and neutrophils from severe acutely infected donors. These cells were infected 24 hours with the P.1 or B.1 variants (both with MOI=9) or MOCK. Cells were stained with Panoptic and counted by light microscopy. Cells were counted by visual field and the percentage of cell population, or the absolute number of cells was obtained (Figure 3).
We next assessed the cell death index, by evaluating the percentage of macrophages and neutrophils infected with B.1 and P.1 variants in relation to the MOCK controls. Our findings showed that there is a reduction in the percentage of macrophages (Figure 3A) infected with the B.1 variant, the same was observed with the percentage of neutrophils (Figure 3B). The infection with B.1variant induces more cell death than the P.1 variant, that is, what this experiment suggests is that the B.1 variant would be more cytopathogenic than the P.1 variant.
We then calculated the absolute number of macrophages and neutrophils after infection with B.1 and P.1 variants. Once we knew the percentage of these cells per coverslip, and the total number of cells plated, we were able to estimate the absolute number of cells after infection. We did not observe any statistical difference between the absolute number of macrophages (Figure 3C) or neutrophils (Figure 3D) after infection with B.1 or P.1. These data together with the cell death index (Figure 3A and 3B) suggest that the P.1 variant does not have greater cytopathogenicity than the original B.1 variant.
3.3. There is absence of viral replication within macrophage cells for both SARS-CoV-2 variants
It has been shown that the SARS-CoV-2 is capable of infecting macrophages in vitro, but this infection is not productive, that is, these cells do not sustain viral replication[54-56]. We then evaluated whether the P.1 variant would have different infective and replicative potential than the B.1 variant (Figure 4). To assess this issue, macrophages were isolated from healthy donors and treated with the variants for 24, 48 or 72 hours, we used a low MOI of 0.01, as we wanted to observe the replicative capacity of each strain. Afterwards, the supernatants were collected, and we performed an RT-qPCR assay to envelope (E) and nucleocapsid (N) genes. We suggest that there was no significant viral replication as no reduction in CT levels was observed over time, regardless of the gene used for detection. Our results suggest absence of viral replication within macrophage cells for both SARS-CoV-2 variants.
3.4. Spike protein structure
When we think about the impact of variants, specifically regarding the potential to increase viral fitness, whether by increasing infectivity, transmissibility or pathogenesis, we must investigate the regions in the viral genome that affect the evasion of the humoral response and gain in affinity with the cell receptors. Therefore, we analyzed the structure of the S protein of the wild-type virus (Spike-WT) and of six variants (B.1.1.28, P.1 and its subvariants), looking for physicochemical and structural variations in immunogenic regions containing the domain that interacts with ACE-2, such as hydrophobicity, Solvent Accessible Surface (SAS) and Electrostatic Potential (EP) (Figure 5).
In Figure 5, we can see a comparison of the Spike protein trimeric structure of all analyzed variants. In columns A, B and C we see a top view of the structure of the Spike protein. In columns D, E, F we have a side view of the Spike protein. Each row will represent an evaluated variant. In column A we observe the position of the mutations for each variant in pink, and in orange the furin cleavage points. In columns B and E we observe the PE surface of the Spike proteins, in columns C and F the hydrophobicity surface.
3.5. Clustering of the analysis of ASA and hydrophobicity of Spike proteins
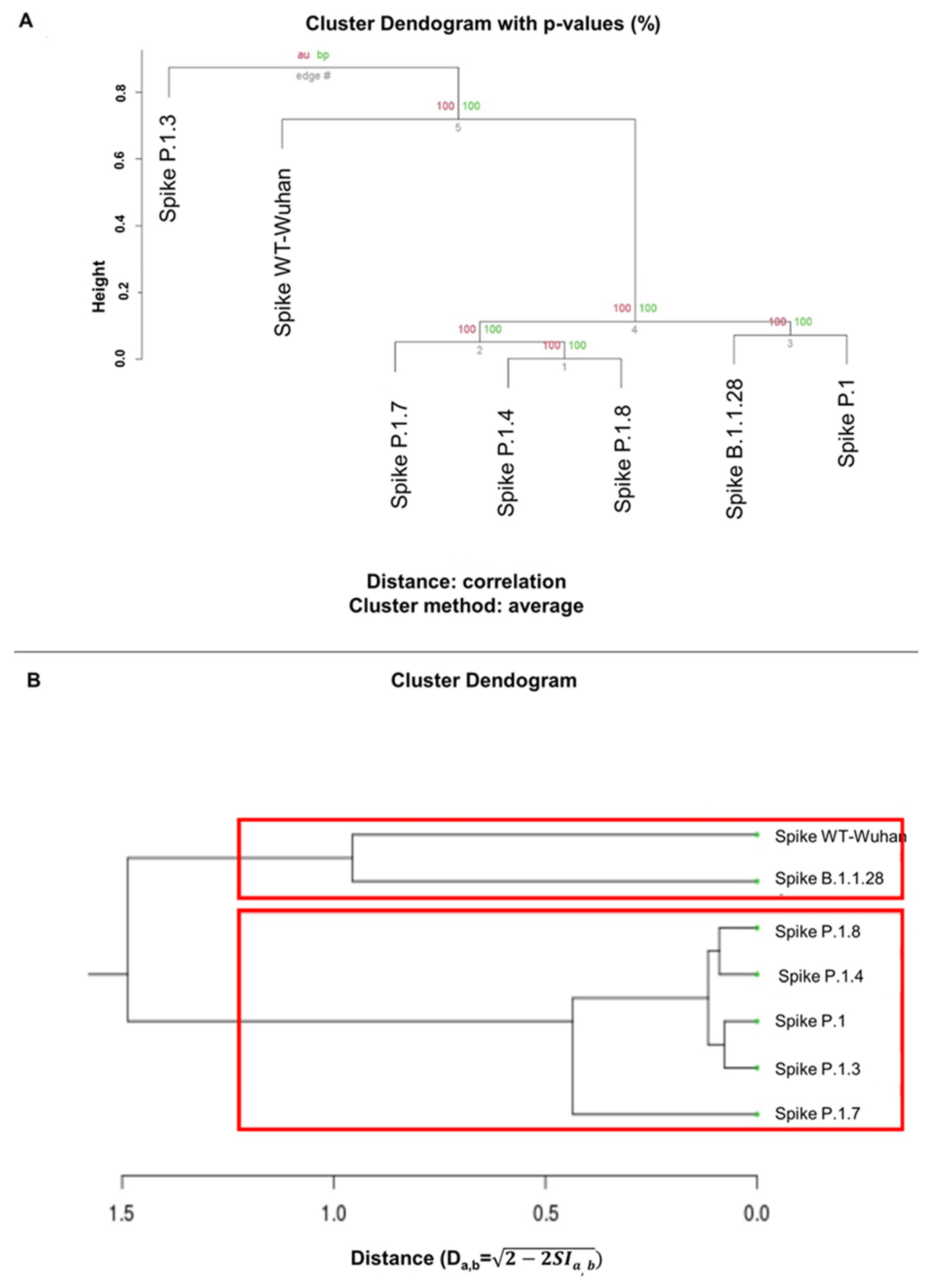
To obtain a more accurate inference, we applied a quantitative approach by two different methods. The comparison of the hydrophobicity and SAS of the proteins (Figure 6A) and similarities of electrostatic interaction properties of the proteins (Figure 6B). For hydrophobic cluster analysis (Figure 6A), clustering was generated between S proteins showing similarities in SAS and hydrophobicity values. In this case, wild-type virus was included as an outgroup, similar to subvariant P.1.3, while B.1.1.28 was grouped together with P.1. In the EP cluster (Figure 6B, Supplementary Figure 1), the distance matrix epogram returned clusters where Spike-WT and B.1.1.28 fall into a distinct group from the remaining variants. Both clusters showed P.1.7 and P.1.8 in close clusters. In both approaches, ancestral S proteins appear to share physicochemical characteristics (WT/B.1.1.28/P.1), while the more derived variants fall into separate groups.
4. Discussion
The VOCs present different viral transmissibility, clinical manifestations, disease severity, and vaccine efficacy, therefore it is important to compare their biological differences and possible clinical impacts. Thus, our main aim was to assess whether the P.1 variant has a different pathogenic biological potential than the original B.1 strain. There is a vast literature linking infection with the Alpha, Beta and Delta variants with a greater severity of COVID-19, increase in mortality and the risk of hospitalization and ICU admission [32,33,57]; [38-40];[34,35]; [36]; [37]. The opposite was observed with Omicron variant, patients infected with this variant are 80% less likely to be hospitalized [44].
Our results indicate that the P.1 variant and the original B.1 strain are comparable in terms of the biological characteristics that we evaluated. There were no significant differences regarding induction of cytokine production by macrophages or neutrophils (Figure 1 and Figure 2), cytopathogenicity (Figure 3), and infectivity (Figure 4). Similar studies corroborate and complement our analyses. In a study, using a hamster intranasal infection model showed that the replicative, infectious and pathogenicity in hamsters of the P.1 variant is similar to the two original strains of SARS-CoV-2 [49]. However, these cells were obtained from patients with severe COVID-19, and perhaps, unlike newly recruited mononuclear phagocytes or neutrophils, they may already be overactivated or even in senescence, unresponsive. Some in vivo and in vitro studies show that these cells in contact with the virus acquire an abnormal, senescent, and even immunosuppressive phenotype [54,58-60].
The evidence also shows an epidemiological outcome distinct from the P.1 variant. Through mathematical models, they estimate that P.1 may be 1.7 to 2.4 times more transmissible than the original strain [24]. The recent European Center for Diseases and Prevention (ECDC) study confirms the increased risk of hospitalization (2.6 times higher) and ICU admission (2.2 times higher) [41]. Official data relate 559,000 deaths to the P.1 variant, but this data could be even higher [61].
Therefore, our findings suggest that the P.1 variant does not have substantial biological differences on compared to the B.1 strain. We can explain the differences in the epidemiological outcome of the P.1 variant, regarding the spreading of the variants in relation to the routes and anatomy of the infected respiratory tracts and tissues. Variants that more efficiently infect the lower respiratory tract are associated with greater pathogenicity and disease severity [62,63]. Thus, reduced access to the lower respiratory tract may mean milder symptoms than those experienced with other circulating VOCs [63-65].
Through bioinformatics we can investigate regions of the S protein, which affect the evasion of the humoral response and gain of affinity with cellular receptors, to study the biological differences between the variants of SARS-CoV-2 that arise, including to epidemiological surveillance. In this scenario, we analyzed the S protein from six subvariants of P.1, P.1.3, P.1.4, P.1.5, P.1.6, P.1.7 and P.1.8 and compared them with the wild-type virus (Spike-WT) and the ancestral lineage of P.1, the B.1.1.28. Variant P.1.5 has the same mutations as P.1.4, and variant P.1.6 has the same mutations as P.1.7, so images of these structures have not been added to the figures.
We looked for physicochemical and structural variations in experimentally defined immunogenic regions recognized by antibodies, and RBD that interacts with ACE-2. These physicochemical characteristics include fundamental elements that influence protein interactions, such as: hydrophobicity [66], which is also reflected in SAS and EP, a fundamental element in protein-protein interaction [67,68]. In this sense, all comparisons were focused on these elements, looking for shared patterns or distinct elements that could impact antibody recognition or gain interaction with ACE-2. Qualitative comparisons were based on: immunogenic regions retrieved from the Immune Epitope Database, which are represented in Figure 5 – Spike-WT A and D (A for top view of trimer, and D for side view of trimer containing a monomer); the receptor binding domain that interacts with ACE-2; and furin cleavage site regions, also involved in SARS-CoV-2 transmission [69].
Calculations of hydrophobicity (Figure 6A) and EP (Figure 6B, and Supplementary Figure 1) show subtle changes across the entire surface of the protein between the Spike-WT and P.1 variants. Even considering that this analysis investigated an overview of S proteins (not focusing on specific residues involved in the cleavage of the furin site or RBD domain, for example), we can extract some information about the evaluated variant proteins. In both approaches, ancestral S proteins appear to share physicochemical characteristics (WT/B.1.1.28/P.1), while the more derived variants fall into separate groups. Lineage P.1.3 may have dropped as an outgroup in the hydrophobic group analysis because of three amino acid deletions. This variant has only 29 sequences deposited in GISAID [52], which may reflect its low transmission capacity.
Furthermore, both clusters showed P.1.7 and P.1.8 in close clusters. These mutations have been described as more infectious than the other subvariants and in relation to P.1 [25]. These structural analyzes of the S protein of the P.1 subvariants indicate that the differences presented by these strains refer to the degree of infectivity of all of them, possibly due to the impact caused on antibody recognition and receptor affinity variation.
In the course of evolution and adaptation between pathogen and host, an increase in pathogenicity is expected, followed by avirulence [70]. In this way, it is possible that the set of mutations that the P.1 variant has altered its tropism for certain organs or the greater evasion of the immune response, conferring greater infectivity and pathogenicity in relation to the original strain, thus contributing to the data of higher mortality associated with this variant [24,41,61]. However, there is a lack of experimental data in the literature to support these differences in relation to the P.1 variant.
Some works show exactly the opposite, there is no difference in relation to the infectivity and pathogenicity of the P.1 variant [49,71]. It is also argued that the increase in the number of deaths that occurred in Manaus was due to the collapse of the health system and, consequently, the lack of adequate treatment for patients may have contributed to the high mortality rates, and not necessarily a biological characteristic of the disease. P.1 variant [24].
Despite the limitations of our model, we show that there is not enough evidence to support a higher pathogenicity of the P.1 variant. The main limitation was the lack of healthy donors, to use as controls and to rule out a possible senescent state of cells obtained from patients. The question of how the new mutations is reflected in the distinct pathogenesis between the variants has not yet been answered. The P.1 variant has already been replaced, but new variants continue to emerge, and with them new opportunities to study the virus-host interaction arise, from the perspective of the impact of mutations on the host’s immune response to infection, influencing not only infectivity and transmissibility, but also understanding the pathogenesis and progression of the disease.
Author Contributions
A.M.S., E.C.D., I.D.L., E. S. S.: Conceptualization; Formal analysis; Methodology; Visualization; Writing—original draft, review and editing. S. P. C. B., K. M. C., D. N., C. G. F. L., M. S., A. M.: Investigation; Formal analysis; Validation. C. G. F. L., L. F. L., N. V.: Investigation; Formal analysis; Validation. V. N., A. M., M. A.: Investigation; Formal analysis. G. F. V., A. P. C., C. G. F. L., M. S., A. M.: Project administration; Data curation; Resources Supervision; Validation; Writing review and editing; Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Brazilian National Research Council (CNPq), Rio de Janeiro State Science Foundation (FAPERJ), and Fundação Oswaldo Cruz (FIOCRUZ).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data is contained within this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Viruses, C.S.G.o.t.I.C.o.T.o. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, D.; Hou, J.; Li, H.; Cao, D.; Guo, M.; Ling, Y.; Gao, M.; Zhou, Y.; Wan, Y.; et al. An inter-correlated cytokine network identified at the center of cytokine storm predicted COVID-19 prognosis. Cytokine 2021, 138, 155365. [Google Scholar] [CrossRef] [PubMed]
- Pacha, O.; Sallman, M.A.; Evans, S.E. COVID-19: a case for inhibiting IL-17? Nat Rev Immunol 2020, 20, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Zizza, A.; Recchia, V.; Aloisi, A.; Guido, M. Clinical features of COVID-19 and SARS epidemics. A literature review. J Prev Med Hyg 2021, 62, E13–E24. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J Microbiol Immunol Infect 2021, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Kim, Y.M.; Shin, E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp Mol Med 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N Engl J Med 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, X.; Chen, J.; Jin, Y.; Peng, L.; Wang, H.H.X.; Luo, M.; Chen, L.; Zhao, Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J Infect 2020, 81, e6–e12. [Google Scholar] [CrossRef] [PubMed]
- Muir, R.; Osbourn, M.; Dubois, A.V.; Doran, E.; Small, D.M.; Monahan, A.; O’Kane, C.M.; McAllister, K.; Fitzgerald, D.C.; Kissenpfennig, A.; et al. Innate Lymphoid Cells Are the Predominant Source of IL-17A during the Early Pathogenesis of Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2016, 193, 407–416. [Google Scholar] [CrossRef] [PubMed]
- WHO. Tracking SARS-CoV-2 variants.
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan. Emerg Infect Dis 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e1220. [Google Scholar] [CrossRef]
- Villoutreix, B.O.; Calvez, V.; Marcelin, A.G.; Khatib, A.M. In Silico Investigation of the New UK (B.1.1.7) and South African (501Y.V2) SARS-CoV-2 Variants with a Focus at the ACE2-Spike RBD Interface. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Singh, J.; Samal, J.; Kumar, V.; Sharma, J.; Agrawal, U.; Ehtesham, N.Z.; Sundar, D.; Rahman, S.A.; Hira, S.; Hasnain, S.E. Structure-Function Analyses of New SARS-CoV-2 Variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Ramesh, S.; Govindarajulu, M.; Parise, R.S.; Neel, L.; Shankar, T.; Patel, S.; Lowery, P.; Smith, F.; Dhanasekaran, M.; Moore, T. Emerging SARS-CoV-2 Variants: A Review of Its Mutations, Its Implications and Vaccine Efficacy. Vaccines (Basel) 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Levi, J.E.; Oliveira, C.M.; Croce, B.D.; Telles, P.; Lopes, A.C.W.; Romano, C.M.; Lira, D.B.; de Resende, A.C.M.; Lopes, F.P.; Ruiz, A.A.; et al. Dynamics of SARS-CoV-2 Variants of Concern in Brazil, Early 2021. Front Public Health 2021, 9, 784300. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; Souza, V.; Corado, A.L.; Nascimento, F.; Silva, G.; Mejía, M.C.; Brandão, M.J.; Costa, Á.; Duarte, D.; et al. Spread of Gamma (P.1) Sub-Lineages Carrying Spike Mutations Close to the Furin Cleavage Site and Deletions in the N-Terminal Domain Drives Ongoing Transmission of SARS-CoV-2 in Amazonas, Brazil. Microbiol Spectr 2022, 10, e0236621. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.; Ge, J.; Ren, W.; Zhang, R.; Lan, J.; Ju, B.; Su, B.; Yu, F.; Chen, P.; et al. Analysis of SARS-CoV-2 variant mutations reveals neutralization escape mechanisms and the ability to use ACE2 receptors from additional species. Immunity 2021, 54, 1611–1621.e1615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep 2022, 39, 110829. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Irie, T.; Suzuki, R.; Maemura, T.; Nasser, H.; Uriu, K.; Kosugi, Y.; Shirakawa, K.; Sadamasu, K.; Kimura, I.; et al. Enhanced fusogenicity and pathogenicity of SARS-CoV-2 Delta P681R mutation. Nature 2022, 602, 300–306. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ 2021, 372, n579. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H.; Group, C.C.-W. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.P.H.S.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and virological features of SARS-CoV-2 variants of concern: a retrospective cohort study comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta). Clin Infect Dis 2021. [Google Scholar] [CrossRef]
- Loconsole, D.; Centrone, F.; Morcavallo, C.; Campanella, S.; Accogli, M.; Sallustio, A.; Peccarisi, D.; Stufano, A.; Lovreglio, P.; Chironna, M. Changing Features of COVID-19: Characteristics of Infections with the SARS-CoV-2 Delta (B.1.617.2) and Alpha (B.1.1.7) Variants in Southern Italy. Vaccines (Basel) 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Shoji, K.; Akiyama, T.; Tsuzuki, S.; Matsunaga, N.; Asai, Y.; Suzuki, S.; Iwamoto, N.; Funaki, T.; Ohmagari, N. Comparison of the clinical characteristics and outcomes of COVID-19 in children before and after the emergence of Delta variant of concern in Japan. J Infect Chemother 2022, 28, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Huang, X.; Zhang, J.; Fu, S.; Ding, D.; Tao, Z. Differences in Clinical Characteristics Between Delta Variant and Wild-Type SARS-CoV-2 Infected Patients. Front Med (Lausanne) 2021, 8, 792135. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.; Pharris, A.; Spiteri, G.; Bundle, N.; Melidou, A.; Carr, M.; Gonzalez, G.; Garcia-Leon, A.; Crispie, F.; O’Connor, L.; et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Euro Surveill 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Fisman, D.N.; Tuite, A.R. Evaluation of the relative virulence of novel SARS-CoV-2 variants: a retrospective cohort study in Ontario, Canada. CMAJ 2021, 193, E1619–E1625. [Google Scholar] [CrossRef]
- Veneti, L.; Seppälä, E.; Larsdatter Storm, M.; Valcarcel Salamanca, B.; Alnes Buanes, E.; Aasand, N.; Naseer, U.; Bragstad, K.; Hungnes, O.; Bøås, H.; et al. Increased risk of hospitalisation and intensive care admission associated with reported cases of SARS-CoV-2 variants B.1.1.7 and B.1.351 in Norway, December 2020 -May 2021. PLoS One 2021, 16, e0258513. [Google Scholar] [CrossRef]
- Cantón, R.; De Lucas Ramos, P.; García-Botella, A.; García-Lledó, A.; Gómez-Pavón, J.; González Del Castillo, J.; Hernández-Sampelayo, T.; Martín-Delgado, M.C.; Martín Sánchez, F.J.; Martínez-Sellés, M.; et al. New variants of SARS-CoV-2. Rev Esp Quimioter 2021, 34, 419–428. [Google Scholar] [CrossRef]
- Bouzid, D.; Visseaux, B.; Kassasseya, C.; Daoud, A.; Fémy, F.; Hermand, C.; Truchot, J.; Beaune, S.; Javaud, N.; Peyrony, O.; et al. Comparison of Patients Infected With Delta Versus Omicron COVID-19 Variants Presenting to Paris Emergency Departments : A Retrospective Cohort Study. Ann Intern Med 2022. [Google Scholar] [CrossRef] [PubMed]
- Maslo, C.; Friedland, R.; Toubkin, M.; Laubscher, A.; Akaloo, T.; Kama, B. Characteristics and Outcomes of Hospitalized Patients in South Africa During the COVID-19 Omicron Wave Compared With Previous Waves. JAMA 2022, 327, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Wolter, N.; Jassat, W.; Walaza, S.; Welch, R.; Moultrie, H.; Groome, M.; Amoako, D.G.; Everatt, J.; Bhiman, J.N.; Scheepers, C.; et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: a data linkage study. Lancet 2022, 399, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Modes, M.E.; Directo, M.P.; Melgar, M.; Johnson, L.R.; Yang, H.; Chaudhary, P.; Bartolini, S.; Kho, N.; Noble, P.W.; Isonaka, S.; et al. Clinical Characteristics and Outcomes Among Adults Hospitalized with Laboratory-Confirmed SARS-CoV-2 Infection During Periods of B.1.617.2 (Delta) and B.1.1.529 (Omicron) Variant Predominance - One Hospital, California, July 15-September 23, 2021, and December 21, 2021-January 27, 2022. MMWR Morb Mortal Wkly Rep 2022, 71, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Houhamdi, L.; Gautret, P.; Hoang, V.T.; Fournier, P.E.; Colson, P.; Raoult, D. Characteristics of the first 1119 SARS-CoV-2 Omicron variant cases, in Marseille, France, November-December 2021. J Med Virol 2022, 94, 2290–2295. [Google Scholar] [CrossRef]
- Kim, M.K.; Lee, B.; Choi, Y.Y.; Um, J.; Lee, K.S.; Sung, H.K.; Kim, Y.; Park, J.S.; Lee, M.; Jang, H.C.; et al. Clinical Characteristics of 40 Patients Infected With the SARS-CoV-2 Omicron Variant in Korea. J Korean Med Sci 2022, 37, e31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, N.; Zhao, D.; Hu, Z.; Tao, Z. Clinical Characteristics of COVID-19 Patients Infected by the Omicron Variant of SARS-CoV-2. Front Med (Lausanne) 2022, 9, 912367. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Halfmann, P.J.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Chiba, S.; Watanabe, T.; Nakajima, N.; Ito, M.; Kuroda, M.; Kiso, M.; et al. Characterization of a new SARS-CoV-2 variant that emerged in Brazil. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; Dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decoté-Ricardo, D.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep 2020, 10, 19630. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Rotondi, M. Interleukin-6, CXCL10 and Infiltrating Macrophages in COVID-19-Related Cytokine Storm: Not One for All But All for One! Front Immunol 2021, 12, 668507. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Vanderbeke, L.; Van Mol, P.; Van Herck, Y.; De Smet, F.; Humblet-Baron, S.; Martinod, K.; Antoranz, A.; Arijs, I.; Boeckx, B.; Bosisio, F.M.; et al. Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat Commun 2021, 12, 4117. [Google Scholar] [CrossRef] [PubMed]
- Niles, M.A.; Gogesch, P.; Kronhart, S.; Ortega Iannazzo, S.; Kochs, G.; Waibler, Z.; Anzaghe, M. Macrophages and Dendritic Cells Are Not the Major Source of Pro-Inflammatory Cytokines Upon SARS-CoV-2 Infection. Front Immunol 2021, 12, 647824. [Google Scholar] [CrossRef] [PubMed]
- Percivalle, E.; Sammartino, J.C.; Cassaniti, I.; Arbustini, E.; Urtis, M.; Smirnova, A.; Concardi, M.; Belgiovine, C.; Ferrari, A.; Lilleri, D.; et al. Macrophages and Monocytes: "Trojan Horses" in COVID-19. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Boumaza, A.; Gay, L.; Mezouar, S.; Bestion, E.; Diallo, A.B.; Michel, M.; Desnues, B.; Raoult, D.; La Scola, B.; Halfon, P.; et al. Monocytes and Macrophages, Targets of Severe Acute Respiratory Syndrome Coronavirus 2: The Clue for Coronavirus Disease 2019 Immunoparalysis. J Infect Dis 2021, 224, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Liu, Y.; Tang, X.; He, D. The Disease Severity and Clinical Outcomes of the SARS-CoV-2 Variants of Concern. Front Public Health 2021, 9, 775224. [Google Scholar] [CrossRef]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Baßler, K.; Schlickeiser, S.; Zhang, B.; Krämer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e1423. [Google Scholar] [CrossRef]
- Chevrier, S.; Zurbuchen, Y.; Cervia, C.; Adamo, S.; Raeber, M.E.; de Souza, N.; Sivapatham, S.; Jacobs, A.; Bachli, E.; Rudiger, A.; et al. A distinct innate immune signature marks progression from mild to severe COVID-19. Cell Rep Med 2021, 2, 100166. [Google Scholar] [CrossRef]
- Burnett, C.E.; Okholm, T.L.H.; Tenvooren, I.; Marquez, D.M.; Tamaki, S.; Munoz Sandoval, P.; Willmore, A.; Hendrickson, C.M.; Kangelaris, K.N.; Langelier, C.R.; et al. Mass cytometry reveals a conserved immune trajectory of recovery in hospitalized COVID-19 patients. Immunity 2022, 55, 1284–1298.e1283. [Google Scholar] [CrossRef]
- Ferrante, L.; Duczmal, L.; Steinmetz, W.A.; Almeida, A.C.L.; Leão, J.; Vassão, R.C.; Tupinambás, U.; Fearnside, P.M. How Brazil’s President turned the country into a global epicenter of COVID-19. J Public Health Policy 2021, 42, 439–451. [Google Scholar] [CrossRef]
- Yang, N.; Wang, C.; Huang, J.; Dong, J.; Ye, J.; Fu, Y.; Xu, D.; Cao, G.; Qian, G. Clinical and Pulmonary CT Characteristics of Patients Infected With the SARS-CoV-2 Omicron Variant Compared With Those of Patients Infected With the Alpha Viral Strain. Front Public Health 2022, 10, 931480. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R. SARS-CoV-2 Omicron spike mediated immune escape and tropism shift. Res Sq 2022. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Iida, S.; Iwatsuki-Horimoto, K.; Maemura, T.; Kiso, M.; Scheaffer, S.M.; Darling, T.L.; Joshi, A.; Loeber, S.; Singh, G.; et al. SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature 2022, 603, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.P.Y.; Ho, J.C.W.; Cheung, M.C.; Ng, K.C.; Ching, R.H.H.; Lai, K.L.; Kam, T.T.; Gu, H.; Sit, K.Y.; Hsin, M.K.Y.; et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Rego, N.B.; Xi, E.; Patel, A.J. Identifying hydrophobic protein patches to inform protein interaction interfaces. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Shashikala, H.B.M.; Chakravorty, A.; Alexov, E. Modeling Electrostatic Force in Protein-Protein Recognition. Front Mol Biosci 2019, 6, 94. [Google Scholar] [CrossRef]
- Vascon, F.; Gasparotto, M.; Giacomello, M.; Cendron, L.; Bergantino, E.; Filippini, F.; Righetto, I. Protein electrostatics: From computational and structural analysis to discovery of functional fingerprints and biotechnological design. Comput Struct Biotechnol J 2020, 18, 1774–1789. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Alizon, S.; Sofonea, M.T. SARS-CoV-2 virulence evolution: Avirulence theory, immunity and trade-offs. J Evol Biol 2021, 34, 1867–1877. [Google Scholar] [CrossRef]
- Nicolete, V.C.; Rodrigues, P.T.; Fernandes, A.R.J.; Corder, R.M.; Tonini, J.; Buss, L.F.; Sales, F.C.; Faria, N.R.; Sabino, E.C.; Castro, M.C.; et al. Epidemiology of COVID-19 after Emergence of SARS-CoV-2 Gamma Variant, Brazilian Amazon, 2020-2021. Emerg Infect Dis 2022, 28, 709–712. [Google Scholar] [CrossRef]
Figure 1.
Cytokine profile of SARS-CoV-2 infected macrophages. Cytokine analysis of the supernatant from non-infected macrophages (MOCK), B.1 strain infected macrophages (B1) and P.1 variant infected macrophages (P1). (A) MIP-1β, (B) IL-6, (C) IFN-γ, (D) IL-1ra, (E) IL-5, (F) RANTES, (G) IL-1β, (H) bFGF, (I) PDGF-BB, (J) IL-4, (K) MCP-1, (L) MIP-1α, (M) IL-10, (N) G-CSF, (O) IL- 12p70 and (P) IL-17A. Data do not present significative difference between groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
Figure 1.
Cytokine profile of SARS-CoV-2 infected macrophages. Cytokine analysis of the supernatant from non-infected macrophages (MOCK), B.1 strain infected macrophages (B1) and P.1 variant infected macrophages (P1). (A) MIP-1β, (B) IL-6, (C) IFN-γ, (D) IL-1ra, (E) IL-5, (F) RANTES, (G) IL-1β, (H) bFGF, (I) PDGF-BB, (J) IL-4, (K) MCP-1, (L) MIP-1α, (M) IL-10, (N) G-CSF, (O) IL- 12p70 and (P) IL-17A. Data do not present significative difference between groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
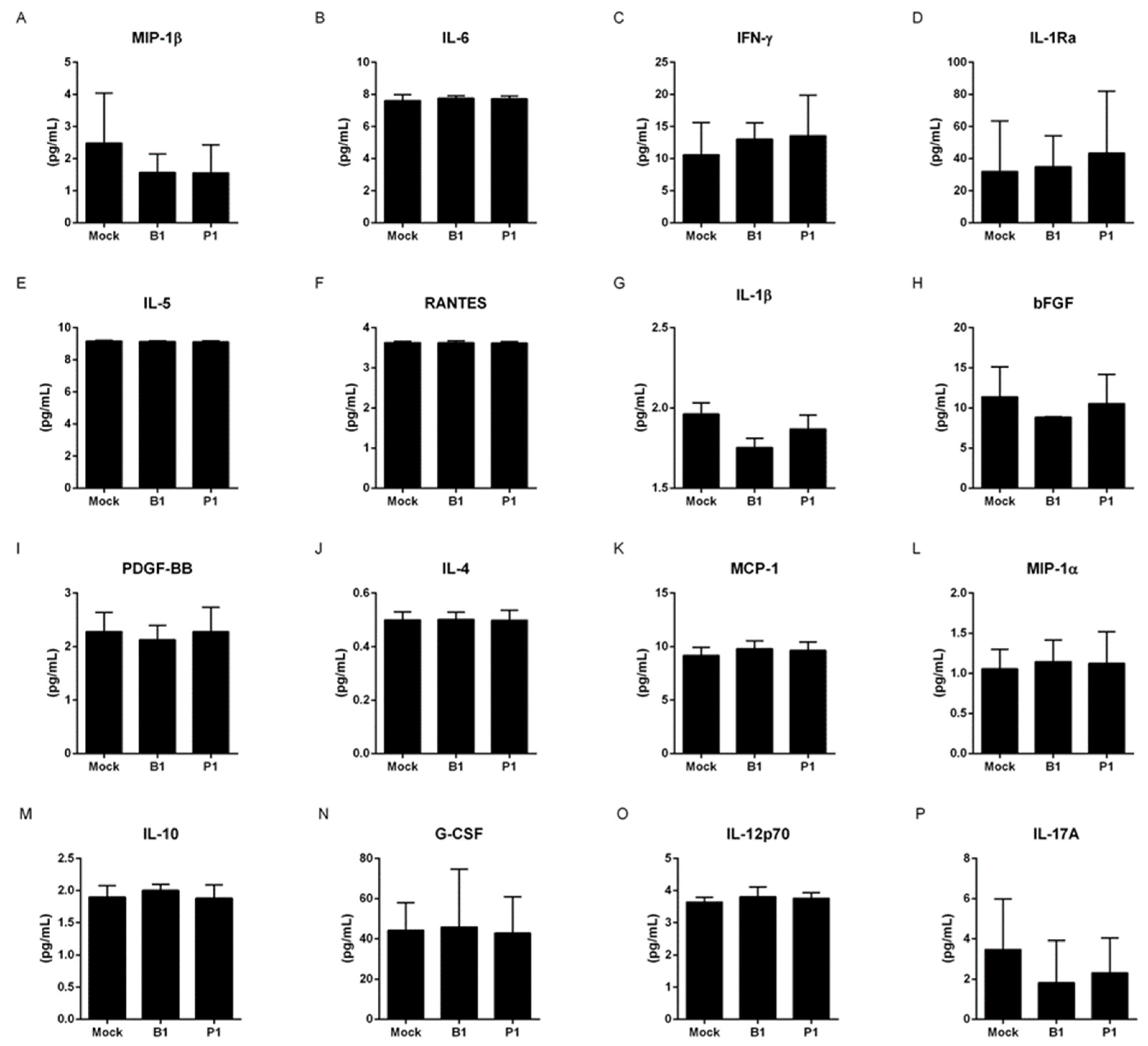
Figure 2.
Cytokine profile of SARS-CoV-2 infected neutrophils. Cytokine analysis of the supernatant from non-infected neutrophils (MOCK), B.1 strain infected neutrophils (B1) and P.1 variant infected neutrophils (P1). (A) IL-6, (B) IFN-γ, (C) IL-1ra, (D) IL-5, (E) RANTES, (F) IL-1β, (G) bFGF, (H) PDGF-BB, (I) IL-4, (J) MCP-1, (K) MIP-1α, (L) IL-10, (M) G-CSF, (N) IL-12p70 and (O) IL-17A. Data do not present significative difference between groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
Figure 2.
Cytokine profile of SARS-CoV-2 infected neutrophils. Cytokine analysis of the supernatant from non-infected neutrophils (MOCK), B.1 strain infected neutrophils (B1) and P.1 variant infected neutrophils (P1). (A) IL-6, (B) IFN-γ, (C) IL-1ra, (D) IL-5, (E) RANTES, (F) IL-1β, (G) bFGF, (H) PDGF-BB, (I) IL-4, (J) MCP-1, (K) MIP-1α, (L) IL-10, (M) G-CSF, (N) IL-12p70 and (O) IL-17A. Data do not present significative difference between groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
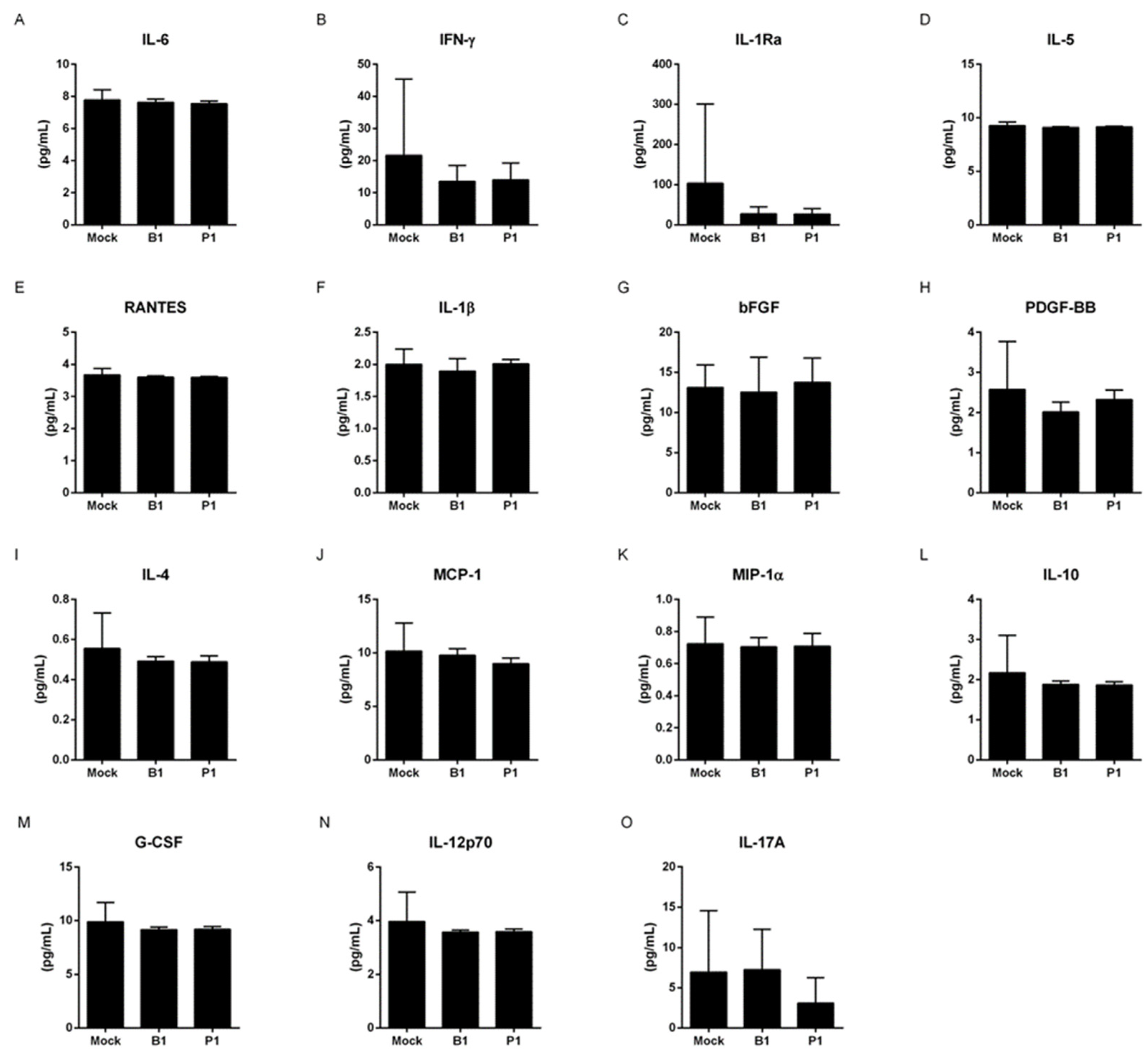
Figure 3.
Microscopy analysis of macrophage and neutrophil percentage after SARS-CoV-2 infection. Microscopy analysis of (A, C) MOCK macrophages, B.1 strain infected macrophages (B1) and P.1 variant infected macrophages (P1) from COVID- 19 severe patients and (B, D) MOCK neutrophils, B.1 strain infected neutrophils (B1) and P.1 variant infected neutrophils (P1) from COVID-19 severe patients. (* p<0,05; ** p<0,01; **** p<0,0001) for comparison with the MOCK group. (+ p<0,05; ++++ p<0,0001) comparison between indicated groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
Figure 3.
Microscopy analysis of macrophage and neutrophil percentage after SARS-CoV-2 infection. Microscopy analysis of (A, C) MOCK macrophages, B.1 strain infected macrophages (B1) and P.1 variant infected macrophages (P1) from COVID- 19 severe patients and (B, D) MOCK neutrophils, B.1 strain infected neutrophils (B1) and P.1 variant infected neutrophils (P1) from COVID-19 severe patients. (* p<0,05; ** p<0,01; **** p<0,0001) for comparison with the MOCK group. (+ p<0,05; ++++ p<0,0001) comparison between indicated groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
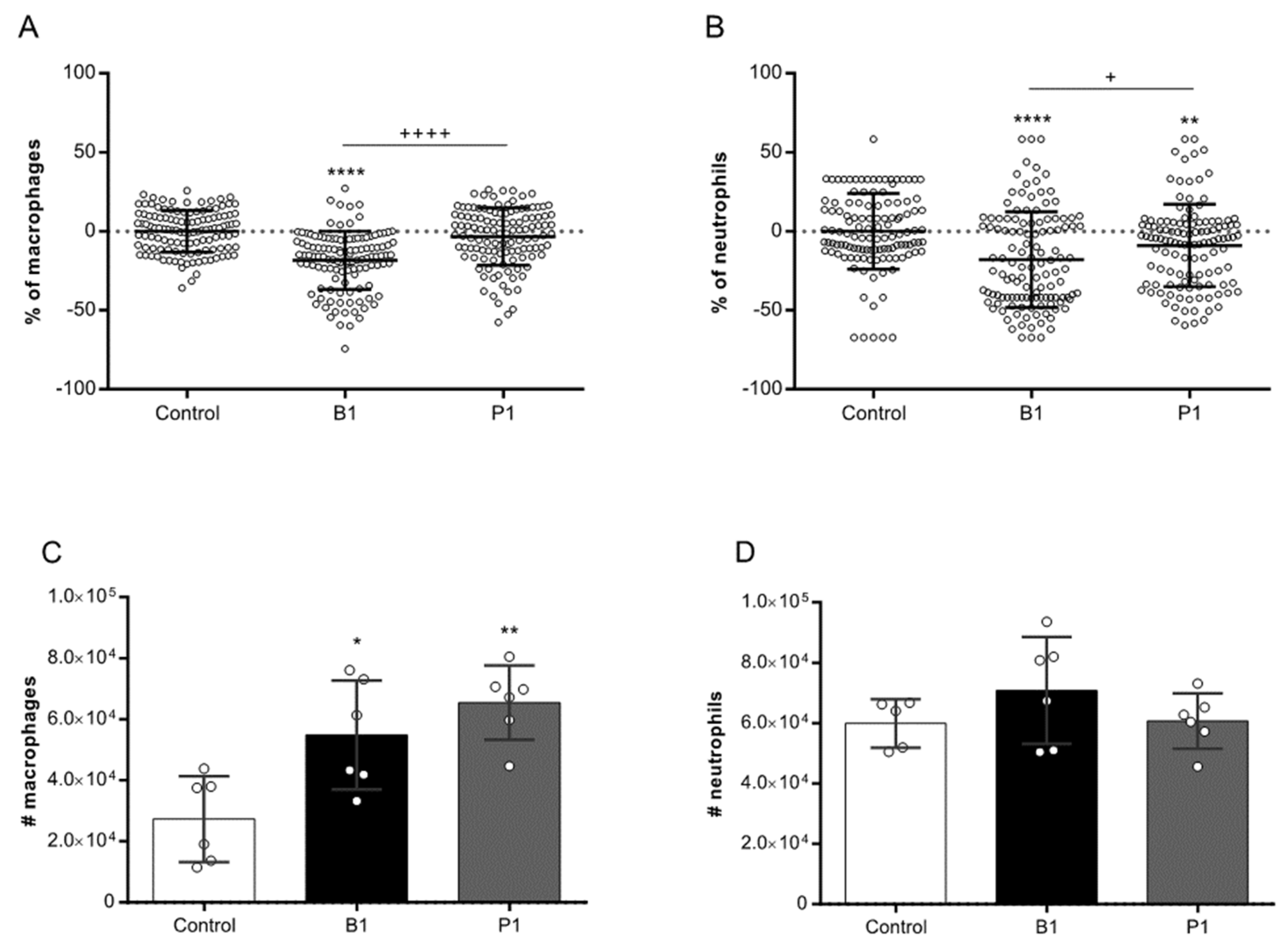
Figure 4.
SARS-CoV-2 infection of macrophages. SARS-CoV-2 B.1 and P.1 replication cycles (CT) in terms of (A) envelope genes and (B) nucleocapsids genes, after 24 h, 48 h and 72 h of incubation period. Data do not present significative difference between groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
Figure 4.
SARS-CoV-2 infection of macrophages. SARS-CoV-2 B.1 and P.1 replication cycles (CT) in terms of (A) envelope genes and (B) nucleocapsids genes, after 24 h, 48 h and 72 h of incubation period. Data do not present significative difference between groups. Comparisons were made to the control group, using the Student’s T test. Multiple comparisons between the experimental groups were obtained using Two Way ANOVA with Dunnett’s test.
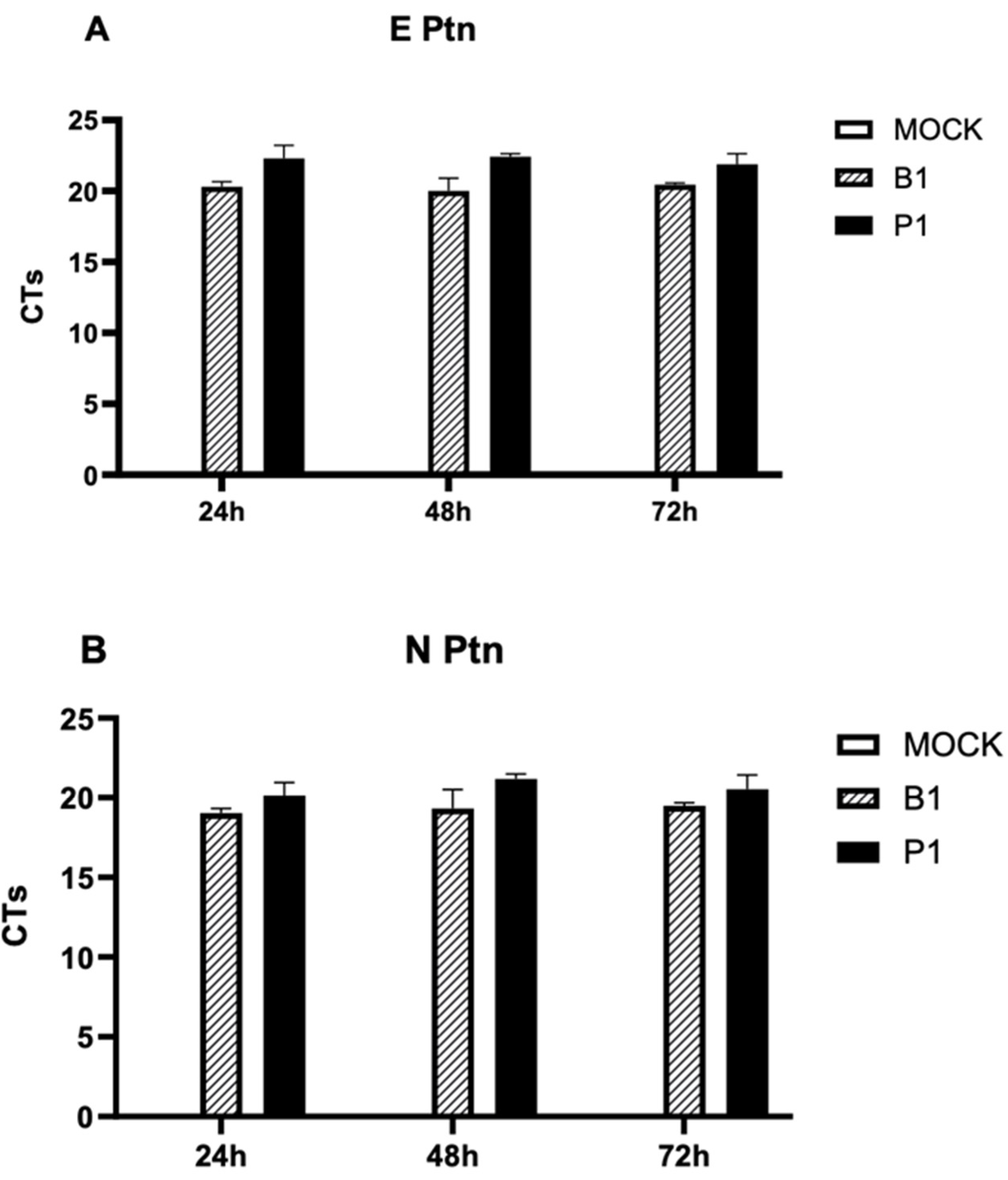
Figure 5.
At the top we have the images referring to the Wuhan sequence. A: in the first line is a heat map of epitopes found in the IEDB database for S Glycoprotein of the SARS-CoV-2 epitopes found in the IEDB database (normalized values) in the RBD view and in the subsequent lines, an RBD view of the mutation position map for each variant, with the cleavage site in orange and mutations in pink. B: electrostatic potential surface in the RBD view; C: hydrophobicity surface in the RBD view; D: in the first line is a heat map of the epitopes found in the IEDB, side view of S Glycoprotein and in subsequent lines a side view map to mutation position for each variant; E: electrostatic potential surface in side view; F: hydrophobic surface in side view. Each line over a variant, whose modeled S protein is below the same position to allow a comparison between variants P.1.3, P.1.4, P.1.7, Wuhan.1.8 with an original information S protein and with B.1.1.28, which is the parental lineage of P1.
Figure 5.
At the top we have the images referring to the Wuhan sequence. A: in the first line is a heat map of epitopes found in the IEDB database for S Glycoprotein of the SARS-CoV-2 epitopes found in the IEDB database (normalized values) in the RBD view and in the subsequent lines, an RBD view of the mutation position map for each variant, with the cleavage site in orange and mutations in pink. B: electrostatic potential surface in the RBD view; C: hydrophobicity surface in the RBD view; D: in the first line is a heat map of the epitopes found in the IEDB, side view of S Glycoprotein and in subsequent lines a side view map to mutation position for each variant; E: electrostatic potential surface in side view; F: hydrophobic surface in side view. Each line over a variant, whose modeled S protein is below the same position to allow a comparison between variants P.1.3, P.1.4, P.1.7, Wuhan.1.8 with an original information S protein and with B.1.1.28, which is the parental lineage of P1.
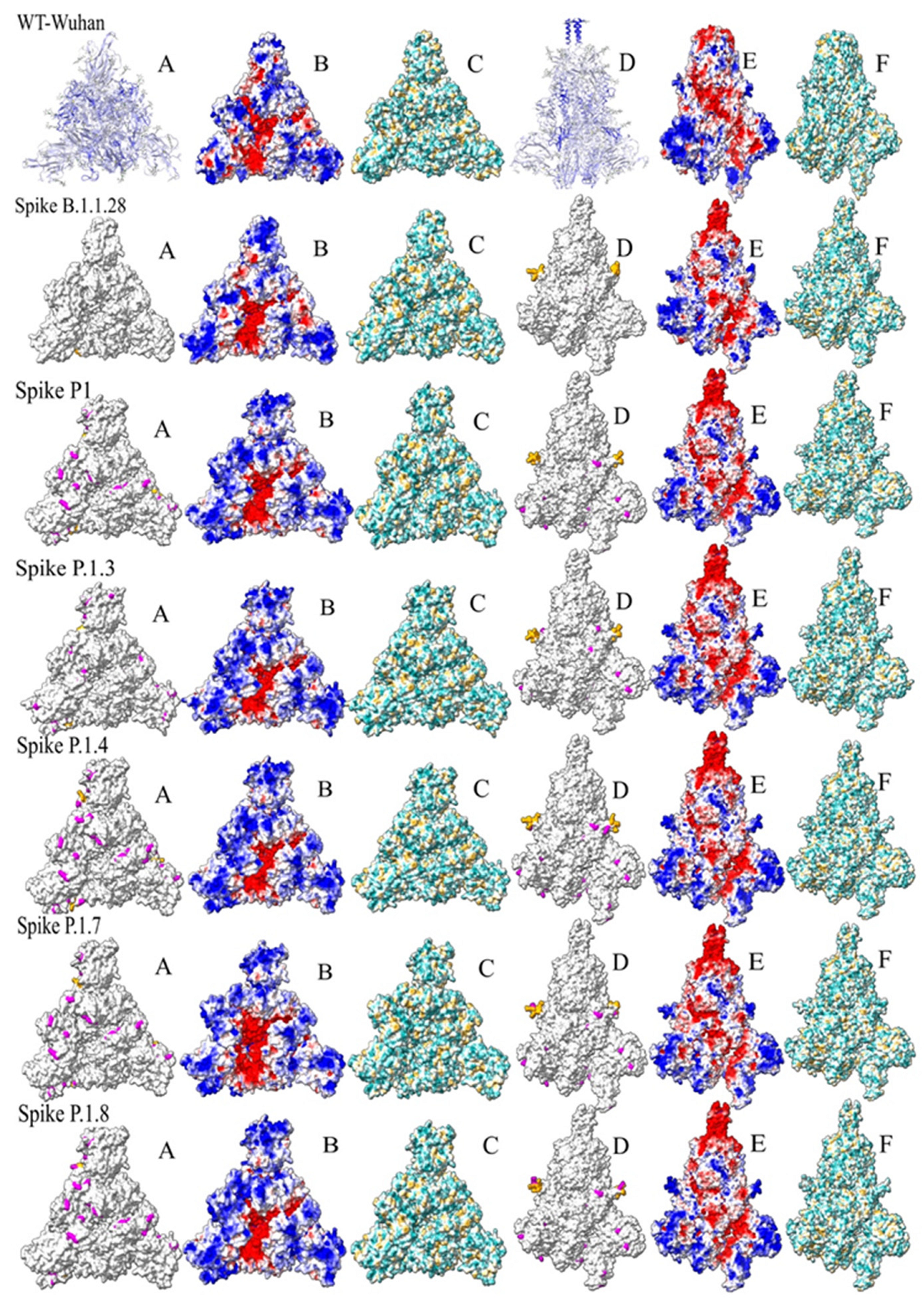
Figure 6.
A- Clustering of the analysis of ASA and hydrophobicity of the residues of the models through the pvclust package executed in Rstudio. The models of P.1.4 and P.1.8 and P1 and B.1.1.28 have smaller distances on their surfaces, and the model of P.1.7 has a smaller distance in relation to these two clusters. The P.1.3 and WT-Wuhan models have a greater distance in relation to the analyzed models. B- Cluster dendogram of webPIPSA analysis. The analysis of electrostatic surfaces indicates the clustering of the S protein from WT-Wuhan with an ancestral variant B.1.1.28 of Gamma lineage. The other variants are grouped together, therefore having smaller distances between them.
Figure 6.
A- Clustering of the analysis of ASA and hydrophobicity of the residues of the models through the pvclust package executed in Rstudio. The models of P.1.4 and P.1.8 and P1 and B.1.1.28 have smaller distances on their surfaces, and the model of P.1.7 has a smaller distance in relation to these two clusters. The P.1.3 and WT-Wuhan models have a greater distance in relation to the analyzed models. B- Cluster dendogram of webPIPSA analysis. The analysis of electrostatic surfaces indicates the clustering of the S protein from WT-Wuhan with an ancestral variant B.1.1.28 of Gamma lineage. The other variants are grouped together, therefore having smaller distances between them.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



