
Submitted:
22 June 2023
Posted:
23 June 2023
You are already at the latest version
Abstract
Medulloblastoma (MB) is the second most prevalent brain tumor in children. Although the current cure rate stands at approximately 70%, the existing treatments that involve a combination of radio- and chemotherapy are highly detrimental to the patients’ quality of life. These aggressive therapies often result in a significant reduction in the overall well-being of the patients. Moreover, the most aggressive forms of MB frequently relapse, leading to a fatal outcome in a majority of cases. However, MB are highly vascularized, and both angiogenesis and lymphangiogenesis, are believed to play crucial roles in tumor development and spread. In this context, our objective is to provide a comprehensive overview of the current research progress in elucidating the functions of these two pathways. Specifically, we will focus on the role and mechanism of action of VEGFC, one of the key players in lymphangiogenesis.
Keywords:
Medulloblastoma
; childhood
; angiogenesis
; lymphangiogenesis
; VEGFC
1. Introduction
Medulloblastoma (MB) is a malignant embryonic tumor that develops in the cerebellum. Although it is a relatively rare cancer (400 new cases are diagnosed each year in the United States, and approximately 100 in France), MB is the second most frequent and aggressive intracranial malignant pediatric tumor, accounting for approximately 25% of CNS tumors in children. In contrast, MB is much less common in adults [1]. The median age of patients ranges between 5 and 7 years old, with a higher incidence in males (boy/girl ratio of 1.8/1). MB is a high-grade, rapidly growing tumor that belongs to the category of primary neuroectodermal tumors (PNETs) [2].
MB is not a single disease but rather encompasses a diverse range of pathologies with significant heterogeneity. Initially, the severity of these variations was assessed based on histological criteria. However, with recent advances in sequencing and molecular genetics, our understanding of MB greatly improved. These recent data enabled an update of the World Health Organization (WHO) classification in 2016 defining 4 subgroups, each with unique genetic alterations, epigenetic modifications, transcription profiles and clinical characteristics: Wingless (WNT), Sonic Hedgehog (SHH), non-WNT/non-SHH (group 3 and group 4) [3]. In this classification, SHH tumors were further stratified based on the TP53 gene (wild-type or mutant) status. This mutational status has a significant impact on prognosis and is correlated with distinct clinicopathologic characteristics. This classification partly aligns with the previous histopathological classification: WNT tumors are predominantly characterized by a classic morphology while desmoplastic/nodular MB and MB with extensive nodularity (MBEN) correspond to the SHH group. Anaplastic large cell tumors, which often exhibit MYC amplification, are primarily classified under group 3, with a few cases falling into a molecular subtype of the SHH group [4].
The WNT and SHH subgroups are characterized by aberrant activation of the WNT and SHH signaling pathways, respectively, which play crucial roles in the pathogenesis of these groups. However, no specific signaling pathway appears to play a similar tumorigenic role in the two other groups. Both groups 3 and 4 exhibit distinct molecular characteristics including the overexpression of N-myc and c-myc factors and the inactivation of TP53 [5].
Meta-analyses showed clear distinctions among these four subgroups in terms of histology, chromosomal aberrations, and clinical prognosis [6]. Prognosis prediction is more reliable than with the previous histopathological classification.
The current consensus officially recognizes four MB subgroups, although biological heterogeneity exists both within and between subgroups [7]. An integrated genomic analysis of 194 primary tumors (validated on three independent cohorts) revealed the presence of highly aggressive intermediate tumors, which belong to specific “subsets” of group 3 or 4 [8]. However, these subsets are not yet well characterized.
In 2017, three independent studies identified several molecular subtypes based on DNA methylation profiling assays: i) An integrative analysis of 491 tumors from untreated patients subdivided subgroups 3 and 4 into eight molecularly distinct subtypes (I-VIII) with specific, albeit somehow overlapping, genetic and transcriptional signatures [7]; ii) A study conducted on 740 tumors showed that the initial four subgroups can be further subclassified into twelve different molecular subtypes [9]; iii) A third study identified seven subtypes among 428 primary tumors. These subtypes were validated in an independent cohort consisting of 276 tumors [10]. This further highlights the molecular diversity and complexity within MB.
The 2021 WHO classification of CNS tumors acknowledged the existence of four MB SHH subtypes and eight non-WNT/non-SHH subtypes [11]. These subtypes exhibit distinct clinicopathological characteristics and have diagnostic, prognostic, and predictive impacts on the treatment response (Figure 1). For instance, the SHH-I (or SHH-b) and SHH-II (or SHH-g) subtypes are predominantly found in children under the age of two [12]. The recognition and characterization of these subtypes contribute to a deeper understanding of MB and facilitate tailored therapeutic approaches based on specific molecular profiles.
2. Metastasis and recurrence throughout medulloblastoma subgroups
The current standard multimodal treatment of MB (surgery, radiotherapy, and chemotherapy) results in a five-year overall survival rate of approximately 70%. However, the chances of cure vary depending on the genetic subgroup [13], the stage of the disease, and the patient metastatic status at the time of diagnosis. Unfortunately, one third of patients do not respond to treatment and experience relapse within two years. Unfortunately, these relapses are often fatal, with patients succumbing within five years of diagnosis [1]. The, median survival of relapsed patients is less than one year [14].
The cure rate ranges from 70 to 80% if the tumor remains localized in the cerebellum, compared to 30 to 40% if the disease is metastatic [2,15]. These figures represent overall statistics and do not account for variations observed among different molecular subgroups. Table 1 compiles the molecular and clinical characteristics of the different subgroups, including the proportion of metastasis within each group.
WNT tumors generally exhibit low rates of metastasis and have a favorable long-term prognosis [16,17,18]. However, WNTb tumors are more prone to metastasis compared to WNTa tumors highlighting differences in the activation of the WNT pathway between the two subtypes [9].
WNT tumors generally exhibit low rates of metastasis and have a favorable long-term prognosis [16,17,18]. However, WNTb tumors are more prone to metastasis compared to WNTa tumors highlighting differences in the activation of the WNT pathway between the two subtypes [9].
The SHH subgroup can be categorized into four distinct subtypes: a, b, g, and d, with different age distributions (Table 1). SHHa is primarily observed in children and is characterized by the following features: TP53 mutations; focal amplifications in MYCN, GLI2, and YAP1; loss in 9q, 10q, 17p. SHHb is mainly seen in infants and is associated with a high metastatic rate and PTEN deletion. It has the poorest prognosis [9,12]. SHHg demonstrates a more favorable outcome while SHHd is predominantly present in adults and also displays a favorable outcome.
Group 3 is associated with the worst prognosis among all subgroups, and significant differences exist between its subtypes (Table 1). The characteristics of Group 3 subtypes with respect to Group 4 were recently specified further [22]. Sharma et al suggested that these two groups can be further divided into eight subtypes each with distinct metastatic status and survival outcomes.
Similarly, Group 4 exhibits significant differences in overall survival rates, while the rate of metastasis remains relatively constant across its subtypes (Table 1).
These observations indicate that the rate of metastasis does not necessarily correlate with overall survival in medulloblastoma. This raises the question of whether progression-free survival might serve as a better indicator of MB severity. It also emphasizes the importance of considering the various treatments administered to patients, in addition to the biological and molecular differences among MB. Overall, the complex nature of medulloblastoma underscores the need for a comprehensive understanding of its subtypes, their specific characteristics, and their responses to treatment. This knowledge is crucial for developing more targeted and effective therapeutic approaches for improved patient outcomes.
3. Routes of metastatic dissemination in medulloblastoma
Leptomeningeal metastases are responsible for almost 100% of deaths. While MBs rarely metastasize outside the CNS, they primarily spread to the spinal and intracranial leptomeninges. This observation supports the hypothesis of MB spread through the CSF rather than the bloodstream [23,24].
In the CNS, a lymphatic network has been described, particularly in the meninges (within the dura mater), which facilitates CSF drainage (Figure 2). Part of the CSF (in the subarachnoid space) drains into the cervical lymph nodes connecting with the lymphatic circulation [25,26,27]. This finding suggests that leptomeningeal metastasis occurs not only via the CSF, but also through CNS lymphatics.
In approximately 7% of cases, MB metastases can spread to the lungs, bones, liver, or lymph nodes [28,29,30]. The spread and development of these metastases are probably facilitated by blood and lymphatic networks.
In rare instances, MB metastases can also be found within the spinal cord (vertebral intramedullary metastases) [31,32]. Since the CSF extends along the spinal cord, it is possible that it serves as the primary transport route for these metastases. Thus, metastatic dissemination primarily occurs through the lymphatic route, responsible for local CNS metastases, as well as the blood route, which is more associated with distant metastases outside the CNS.
4. Pathological lymphangiogenesis and cancer
Lymphatic vessels play a crucial role in maintaining tissue fluid homeostasis by draining interstitial fluids throughout the body and towards the venous bloodstream. The entire lymphatic system is also a vital component of the immune defense system. Additionally, it facilitates the absorption of dietary fats, preventing their storage. The development of lymphatic vessels (lymphangiogenesis) originates from a subpopulation of endothelial cells derived from the cardinal vein, which express Prox1 (Prospero homeobox protein 1) and LYVE1 (Lymphatic vessel endothelial hyaluronan receptor 1). The process of lymphangiogenesis relies on the sequential expression of specific lymphatic markers (Prox1, PDPN (podoplanin), VEGFC, VEGFR3). Deregulation of lymphangiogenesis contributes to several pathologies including lymphedema, inflammatory conditions, fibrosis, and cancer [33,34,35,36]. Insufficient lymphangiogenesis disrupts its essential biological functions leading to reduced fluid drainage, chronic tissue edema, compromised immunity, and impaired fat absorption, which can contribute to obesity. Conversely, excessive lymphangiogenesis triggers tissue inflammation, autoimmune diseases, and the metastatic spread of tumors. Furthermore, the abnormal increase in the lymphatic network not only facilitates the transport of tumor cells but also stimulates enhanced antitumor immune response [37]. Administration of lymphangiogenic growth factors or their antagonists might be a potential strategy to target lymphatic vessels in human disease, particularly in cancers, including MB.
4.1. The VEGFC/VEGFR axis in the tumor lymphatic network
Due to their exacerbated proliferation rate, tumor cells generate metabolic waste products that are eliminated through the lymphatic system. However, tumor cells hijack the functions of the lymphatic system to disseminate throughout the body. They secrete growth factors such as VEGFC and VEGFD which stimulate tumor lymphangiogenesis [38,39]. Cells in the tumor microenvironment including stromal cells, fibroblasts [40], activated platelets [41], tumor infiltrating macrophages [42] and other immune cells (as described earlier), also contribute to the initiation of lymphangiogenesis by releasing VEGFC and VEGFD. Overexpression of these factors in tumors promotes the growth of lymphatic vessels and lymph node metastasis [40,43]. A soluble VEGFR3 fusion protein, which inhibits the interaction between VEGFC-VEGFR3, effectively suppresses tumor-associated lymphatic growth and metastasis to regional lymph nodes (although distant metastasis likely occurs via the bloodstream) [44,45]. Inhibition of VEGFR3 with a blocking antibody produces similar effects [46]. Conversely, inhibiting VEGFR2 has no impact on tumor lymphangiogenesis [47]. VEGFA, presumably through its interaction with VEGFR2 or NRP2, decreases tumor lymphangiogenesis and lymph node metastasis. Transgenic mice overexpressing VEGFA exhibit significant proliferation of tumor-associated lymphatic vessels. These neo-vessels overexpress VEGFR2 [48]. These studies collectively indicate that tumors prepare their lymphatic metastatic spread by secreting pro-lymphangiogenic growth factors. These factors act on the surface of LECs through both VEGFR2 and VEGFR3 promoting their migration and proliferation. NRP2, found on lymphatic vessels within and around murine experimental tumors, plays a role in this process. Inhibiting NRP2 using a neutralizing antibody blocks the migration of LECs but not their proliferation, resulting in reduced tumor lymphangiogenesis and a lower incidence of lymph node metastases [49]. Thus, the primary inducers of tumor lymphangiogenesis are the growth factors VEGFC and VEGFD, acting through their interaction with (co)receptors VEGFR2, VEGFR3 and NRP2.
VEGFC also contributes to the dilatation or the widening of lymphatic vessels [50,51]. Notably, the VEGFR2 pathway primarily mediates the widening of these vessels, while VEGFR3 is crucial for lymphatic neo-germination [52]. This hyperplasia resulting from the overexpression of VEGFC in tumors enhances lymphatic flow and increases the proportion of tumor cells invading the lymph nodes [53].
Lymphangiogenesis can occur both at the periphery of the tumor and within the tumor mass. Peritumoral lymphatic vessels facilitate metastatic dissemination while intratumoral vessels are frequently obstructed by infiltrating tumor cells and may not function properly. In a mouse model of melanoma that overexpress VEGFC, only the peri-tumoral lymphatic vessels are necessary for lymphatic metastatic spread [54].
4.2. Molecular mechanisms of tumor lymphangiogenesis
Tumor lymphangiogenesis primarily occurs through the process of lymphatic germination. In response to lymphangiogenic factors, proliferating LECs initiate the sprouting of new lymphatic vessels. Uncontrolled tumor growth and the resulting hypoxia induce the expression of growth factors by tumor cells, stromal cells, and tumor-infiltrating inflammatory cells (Figure 2). Growth factors such as VEGFs, FGFs, PDGFs, angiopoietins 1 and 2 and EGF, promote tumor lymphangiogenesis [48,55,56,57]. Prostaglandins and their receptors expressed by tumor and immune cells increase VEGFC expression, thereby stimulating lymphangiogenesis and metastasis in several types of cancers. In addition, cyclooxygenases (COX), which catalyze the conversion of arachidonic acid into prostaglandin H, enhance VEGFC expression and promote lymph node metastases in breast cancer [58], lung cancer [59], esophageal cancer [60] and head and neck cancers [61].
LEC = lymphatic endothelial cell; M1 = M1 polarized macrophage; HSC = hematopoietic stem cell. Created with BioRender.com.
In addition to sprouting lymphangiogenesis, precursor LECs and bone marrow-derived cells contribute to the formation of lymphatic vessels in cancer. For instance, after transplantation of hematopoietic stem cells expressing GFP into tumor-bearing mice, 3 to 4% of the endothelial cells of tumor lymphatic vessels express GFP [62]. Therefore, hematopoietic stem cells have the potential to differentiate into LECs and integrate into tumor-associated lymphatic vessels. In addition, circulating LEC progenitor cells originating from bone marrow participate in lymphangiogenesis by integrating into new lymphatic vessels within tumors [63].
Differentiated macrophage-like cells derived from the bone marrow integrate into the tumor lymphatic endothelium. In mouse models of pancreatic and prostate cancer, 3% of the newly formed lymphatic vessel cells are derived from the myeloid-monocyte lineage. A depletion of tumor-associated macrophages in these models results in a reduction in the density of peri-tumoral lymphatic vessels [64]. These integrated macrophages acquire the expression of LEC markers. This transdifferentiation is partly regulated by FGF signaling. Macrophages support lymphangiogenesis through two mechanisms: either by differentiating into LECs and incorporating into the lymphatic endothelium, or by stimulating the proliferation of local LECs through the production of VEGFC [65]. In a model of renal fibrosis, macrophages produce VEGFC, which inhibits autophagy in M1 macrophages. Reduced autophagy upregulates M1 marker expression and M1 macrophage polarization. These M1 macrophages can preferentially transdifferentiate into LEC [66]. This process could be similar in another inflammatory context such as cancer. Indeed, the mTOR protein, which inhibits autophagy, also activates lymphangiogenesis. Inhibition of mTOR by rapamycin reduces lymphangiogenesis and cervical lymph node metastasis in head and neck cancers [67]. This suggests that targeting mTOR could be a potential strategy to modulate tumor lymphangiogenesis and metastasis.
Other signaling pathways affect the expression of VEGFC and its receptors, thereby influencing tumor lymphangiogenesis. The WNT1 ligand suppresses VEGFC expression and inhibits lymphangiogenesis and metastasis in melanoma. The precise mechanism of this suppression is not yet understood but it does not depend on downstream GSK3β or b-catenin signaling [68]. This finding may explain why WNT-driven MBs, depending on their signaling mutations, are not highly metastatic. TGFβ also inhibits lymphangiogenesis. Its repression by the type 1 receptor of TGFβ (TβR1) inhibitor, in a xenograft model of pancreatic adenocarcinoma, resulted in increased expression of VEGFC and accelerated lymphangiogenesis [69]. These findings suggest that TGFβ transduces signals within LECs and can enhance lymphangiogenesis in the tumor microenvironment.
4.3. The dual role of cancer-associated lymphangiogenesis
The lymphatic network has two primary roles in cancer:
- In the early stages of tumor development, it has beneficial effects by facilitating the transport of tumor antigens in the lymph to the lymph nodes. These antigens are then presented to naïve LTs to activate the antitumor immune response.
- Later, when the tumors have progressed towards advanced stages, the lymphatic vessels have adverse effects. Aggressive tumors and their associated microenvironment produce significant amounts of VEGFC, which is correlated with an enlarged lymphatic network and tumor dissemination through lymphatic vessels. VEGFC participates in the formation and sprouting of new lymphatic vessels around the tumor [70] as well as in the dilation of existing peri-tumoral lymphatic vessels. The latter carry cancer cells that enter the lymph nodes, where they can survive and proliferate [71]. Consequently, VEGFC-dependent formation of tumor neo-vessels leads to metastatic dissemination.
Link between inflammation and lymphangiogenesis in cancers
During inflammation and cancer, the lymphatic network is remodeled and lymphangiogenesis occurs. Inflammation-induced lymphangiogenesis takes place in both the draining lymph node and in inflamed tissue through the signaling of VEGFA/VEGFR2 and VEGFC/VEGFD/VEGFR3 [37]. In acute inflammation, B lymphocytes express VEGFA which activates VEGFR2 and lymphangiogenesis in the lymph nodes [72]. Activated T cells express INF-g, which suppresses nodal lymphangiogenesis [73]. Macrophages, as they migrate from the inflamed tissue to the draining lymph node, also express VEGFA, thereby inducing nodal lymphangiogenesis [74]. Unlike in lymph nodes, tissue lymphangiogenesis is independent of B cells [75]. In inflamed tissue, lymphangiogenesis is initiated by the infiltration of macrophages that express VEGFA and VEGFC [74]. Inflammation also results in an increase in lymphatic flow [76] promoting faster transport of immune cells. In addition, pro-inflammatory cytokines (TNFa, IL-6, IL-8, IL1-b, INF- g) can increase the permeability of lymphatic endothelial cells [77]. However, while this lymphangiogenesis is associated with inflammation, it can also contribute to cancer progression [78].
4.3.1. Harmful role: metastatic dissemination and immune tolerance
LEC = lymphatic endothelial cell; MDSC = myeloid suppressor cells; LT = T lymphocyte; NK = Natural Killer. Created with BioRender.com.
To disseminate throughout the body, cancer cells must penetrate the vascular network, implant, and proliferate in another organ to form metastases. During the early stages of cancer development, the tumor is avascular. Later, the production of angio- and lymphangiogenic factors leads to tumor neovascularization, where new blood vessels are formed and connect with pre-existing vessels. The presence of blood and/or lymphatic vessels is often associated with high-grade tumors. As the tumor progresses, cancer cells enter the lymphatic vessels and spread through the lymphatic network. Thus, the blood and lymphatic vessels represent the main routes of metastatic dissemination and contributes to the aggressiveness of cancer. Consequently, anti-angiogenic treatments are commonly included in current therapeutic regimens. Despite beneficial effects, they only trigger a modest survival extension of a few months. This limited efficacy could be due to compensatory mechanisms, such as tumor lymphangiogenesis which may counteract the effects of anti-angiogenic therapies [79].
High levels of lymphangiogenic growth factors such as VEGFC (or VEGFD) are released by tumors and the microenvironment including macrophages [80]. These growth factors induce lymphangiogenesis around the tumor and in regional lymph nodes [81]. Tumor lymphangiogenesis promotes tumor growth, invasion to peritumoral lymph nodes and metastasis. It is associated with poor prognosis in melanoma and breast, ovarian, colorectal and lung cancers [82]. For instance, elevated levels of VEGFC in ovarian and breast cancers are correlated with accelerated tumor growth, progression, and dissemination [83]. In mouse models, inhibition of VEGFC, VEGFD or VEGFR3 using monoclonal antibodies or soluble extracellular receptor domains (VEGFC/D traps) decreases the spread of tumor cells to lymph nodes [35,44,45]. Additionally, in mice, tumor cells reaching sentinel lymph nodes can extravasate into blood vessels and disseminate systemically to the lungs [84,85].
To disseminate into the lymphatic vessels, metastatic tumor cells rely on chemokine signaling pathways [86,87]. Under physiological conditions, these pathways regulate the trafficking of APCs that selectively enter lymphatic vessels. LECs express ligands CCL19 and CCL21 (CC-chemokine ligand 19 and 21), which bind to the CCR7 receptor (CC-chemokine receptor 7) on the surface of dendritic cells, LB and LT. LECs also secrete the ligand CXCL12, which binds the CXCR4 receptor on APCs [88]. This chemokine gradient facilitates the entry of cells expressing the corresponding receptors into lymphatic vessels and lymph nodes. Tumor cells exploit this system by expressing the CCR7 and CXCR4 receptors. Under hypoxia, tumor cells overexpress CXCR4 [89] and secrete the cytokines CCL19 and 21 (Figure3A). The expression of CCR7 and CXCR4 is correlated with increased metastasis in patients with breast, lung, gastric or colorectal cancers [90,91,92,93]. Through autocrine and paracrine chemotaxis, tumor cells migrate towards LECs and enter the lymphatic vessels. The activation of CXCR4 and autocrine chemotaxis induced by CXCL12 enhances glioma cell motility, migration, and invasion [94]. VEGFC secreted by tumor cells increases the expression of VEGFR3 by LECs and stimulates the secretion of CCL21 by these LECs. This VEGFC-dependent process promotes paracrine chemotaxis and facilitates tumor invasion [95]. In an experimental model of melanoma in mouse, a soluble CCL21 inhibitor blocks the migration of tumor cells [96]. In addition to its local effect, VEGFC produced by tumor cells induces lymphangiogenesis in the lymph nodes, creating a pre-metastatic niche [97,98]. Nodal lymphangiogenesis increases lymphatic flow and facilitates the entry of metastases. Moreover, these changes in the microenvironment of the lymph node can provide favorable conditions for the survival and growth of tumor cells. Once colonized, the lymph nodes enhance the metastatic potential of the tumor in distant organs [55]. The expression of chemokines and their receptors by tumor cells, as well as the VEGFC-dependent secretion of these cytokines by LECs, accelerates the development of metastases.
Besides providing routes for metastasis, tumor-associated lymphatic vessels also contribute to immune tolerance. The development of a tumor-associated lymphatic network leads to the production of various immunomodulatory signals, including PDL1, IDO (indolamine-2,3-dioxygenase) and TGFb. These factors inhibit the maturation of dendritic cells, impair the cytotoxic function of NKs, dampen the activation of LTs, and promote the activation of regulatory LTs and MDSCs, thus creating an immunosuppressive microenvironment [99]. PDL1 is overexpressed on the surface of LECs upon interaction with specific tumor antigens and is also induced by hypoxia. Blocking PDL1 on LECs that present these tumor antigens enhances the activation of CD8 LT [100]. In several mouse models of melanoma, tumor associated LECs express high levels of PDL1 compared to LECs in normal skin. Expression of PDL1 by these cells prevents the accumulation of CD8 LT in the melanoma environment [101]. VEGFC, produced by tumor cells, not only stimulates tumor growth through an autocrine mechanism but also increases lymph node metastases and promotes immune tolerance. In an immunocompetent mouse model of melanoma, VEGFC produced by tumor cells and the microenvironment leads to the suppression of tumor specific CD8 LTs and an increase in regulatory LTs and MDSCs. In this model, the LECs of the peri-tumoral vessels, activated by VEGFC, also disrupt the response of CD8 LTs (Figure 3B).
In tumor-draining lymph nodes, LECs present tumor antigens complex to MHC-I and induce apoptosis of tumor-specific CD8 T cells. Thus, VEGFC acts as a pro-tumor immunomodulator [102]. T Human studies in melanoma support this findings, as functionally active CD8 T cells remain in circulation, but exhibit a depleted phenotype (weaker presentation of tumor antigens and reduced reactivity) within tumors and patient metastases [103]. In addition, LECs in tumor-associated lymph nodes produce S1P (sphingosine 1-phosphate) which promotes the egress of NK and LT cells from the lymph nodes, facilitating nodal metastatic spread [104,105]. Lymph node LECs also produce nitric oxide in response to inflammatory signals (IFN-g and TNF) produced by LTs, inhibiting LT activation in return [106]. Moreover, the production of IFN- g by CD8 T cells induces the expression of PDL1 on the surface of LECs. Mouse models of melanoma with LECs deficient in IFN-g receptor exhibit an increase in tumor-infiltrating CD8 T cells and improved survival [101]. Thus, during tumor development, a negative feedback loop is set up between the LECs and LTs. PDL1 expression by LECs is increased in response to IFN-g produced by tumor-specific CD8 LTs, subsequently inhibiting their activation and accumulation within tumors (Figure 3C).
In view of data based on these pieces of evidence, targeting tumor lymphangiogenesis emerges as a promising therapeutic approach to prevent metastatic dissemination. Tumor lymphangiogenesis contributes to the limitation of anti-angiogenic treatments in certain cancers. When the blood vasculature is blocked, tumors adapt by increasing the production of lymphangiogenic factors and exploiting the lymphatic vessels for dissemination. To overcome these compensatory mechanisms, future treatments should consider the lymphatic system as a crucial player in the tumor process. Simultaneous targeting of both the blood and lymphatic vasculature networks would deprive the tumor of potential dissemination routes. By addressing both aspects, comprehensive therapeutic strategies can be developed to effectively inhibit tumor progression and metastasis.
4.3.2. Beneficial role and synergy with the immune system
During the early stages of tumor development, lymphangiogenesis serves as an entry route for immune cells that can mount an anti-tumor immune response. Cancer cells or their antigens entering the lymphatic vessels activate immune cells at the inflammatory site or in the draining lymph nodes. APCs present tumor antigens on their surface, initiating an anti-tumor immune response (Figure 4A).
In the context of inflammation, LECs in the lymphatic vessels produce the cytokine CCL21. This cytokine attracts dendritic cells, activated LBs and LTs, expressing the CCR7 receptors in the lymphatic vessels [107] (Figure 4B). The trafficking of leukocyte in lymphatic vessels is also regulated by cell adhesion molecules such as CLEVER-1 (common vascular and lymphatic endothelial receptor-1) and mannose receptor 1 [108]. In tumors, the expression of CCR7 by dendritic cells induces their migration into the tumor-draining lymph nodes, where they activate LTs [109].
LEC = lymphatic endothelial cell; HEV = high endothelial venule; Ag = antigen; DC = dendritic cell; LB = B lymphocyte; LT = T lymphocyte. Created with BioRender.com.
Both activation and infiltration of T cells into tumors are critical steps in antitumor immunity. While infiltration of regulatory LTs is associated with a poor prognosis, the presence of intra-tumor cytotoxic LTs is associated with better clinical outcomes [99,110]. In melanoma or colon cancer patients, the lymphatic network density and the lymphatic gene expression in primary tumors correlate with inflammation and immune cell infiltration [111,112,113,114]. These findings support the role of the lymphatic vascular system in the transport of immune cells.
APCs such as dendritic cells, transport peripheral antigens and deliver them to the lymph nodes, where they present them to LB and LT that constantly enter the lymph nodes [115,116]. These antigens can reach the lymph nodes without the need for central peripheral antigen transport. Once in the lymph nodes, resident follicular dendritic cells and macrophages in the cortical region capture the antigens, leading to the activation of LT and LB within a few hours of antigen presentation [117,118]. Tumor antigens can also locally activate naïve LTs [119,120]. Overall, the lymphatic vascular system plays a crucial role in facilitating the transport of immune cells, activation of T cells, and initiation of anti-tumor immune responses.
Transgenic mice with melanoma that lack lymphatic vessels (or have a disrupted lymphatic system, exhibit impaired tumor drainage, reduced dendritic cell trafficking and a diminished induction of anti-tumor adaptive immune responses [112,121]. Lymphatic vessels are therefore necessary for the initiation of effective antitumor responses. In the early stages of melanoma development, VEGFC initiates the increase in lymphatic intratumoral density and the infiltration of CD8 T cells. However, at metastatic stages, infiltrating lymphocytes are reduced and regulatory T cells (CD19+ FoxP3+) are present, indicating the attraction of immunosuppressive cells [113]. This suggests a shift towards an immunosuppressive microenvironment in advanced melanoma. Similar observations have been made in kidney cancer where overexpression of VEGFC is correlated with increased survival in patients with non-metastatic tumors, but decreased survival in metastatic patients [70]. In addition, VEGFC-deficient tumors exhibit a decrease in activated lymphocyte markers and an increase in the PDL1 marker. These findings suggest that the beneficial activity of VEGFC is transient and limited to the early stages of disease development.
Lymph nodes contain specialized blood vessels called HEVs (high endothelial venules). HEVs express the ligand CCL21, which facilitates the entry of naïve and memory T cells expressing CCR7 into the lymph nodes [122]. Under physiological conditions, HEVs are primarily found within lymphoid tissue. However, they can be generated at sites of chronic inflammation [123] (Figure 4). They have been detected in human tumors and have been associated with a favorable prognosis [124,125,126,127]. They contribute to increased infiltration of LB and LT into both lymph nodes and the tumor itself. This enhanced immune infiltration is linked to an improved antitumor response, particularly in human breast tumors [125]. The density of HEVs in tumors has been correlated with longer metastasis-free survival rates, suggesting that HEVs confer a lower risk of relapse. Therefore, HEVs have emerged as potential targets for the diagnosis and treatment of cancer due to their ability to enhance immune infiltration and potentially improve patient outcomes. However, in advanced stages of the disease, HEVs seem to disappear. The mechanisms underlying the loss of HEVs in advanced tumors are not yet fully understood. Further research is needed to investigate the factors and processes involved in the disappearance of HEVs and their implications for tumor progression and immune responses.
Therapeutically, in melanoma models, induction of lymphangiogenesis by VEGFC in primary tumors promotes the accumulation of CD8 LT and enhances responses to immunotherapy [111]. In a mouse model of glioblastoma, injection of VEGFC into the CSF increases lymph node and tumor infiltration of CD8 LT. VEGFC also enhances the effects of anti-PD1 [128]. These findings highlight the ability of VEGFC to stimulate antitumor immunity and raise the efficacy of immunotherapy, particularly at early stages in certain tumors. Based on these promising results, a pro-lymphangiogenic therapy delivering VEGFC (Lymfactin® or LX-1101) is being evaluated in a phase II clinical trial (NCT03658967) for the treatment of breast cancer patients with secondary lymphedema. By delivering VEGFC, the therapy intends to induce lymphangiogenesis and improve lymphatic function, ultimately relieving lymphedema symptoms.
While the growth of lymphatic vessels can facilitate the spread of metastases and create an immunosuppressive microenvironment in advanced tumors, a functional lymphatic network is necessary to generate appropriate antitumor immune response. It is critical to consider the beneficial effects of tumor lymphangiogenesis when administering anti-angio/lymphangiogenic treatments. The administration of such treatments should be carefully based on specific types of cancer and the stage of the disease. Indiscriminate destruction of the lymphatic vessels involved in the anti-tumor immune response might have unintended consequences, compromising the ability of the immune system to mount an effective antitumor response.
As described previously, VEGFC is involved at several levels: at the systemic level, at the level of the lymphatic network, at the immune level but also in an autocrine manner at the level of tumor cells. VEGFC produced by cancer cells, can act directly on these cells, and induce important biological effects. This autocrine role has been demonstrated in various types of cancer.
In ovarian cancer, VEGFC released by cancer cells stimulates their autocrine migration both in vitro and in vivo through autocrine and paracrine mechanisms [83]. Similarly, VEGFC binding to VEGFR3 induces autocrine proliferation of breast cancer cells [129], in scalp and facial angiosarcomas [130], as well as in airway squamous cell cancer cells – upper digestive tract – [131]. In addition, anti-VEGFC chimeric antibodies inhibit the proliferation and migration of endothelial cells and metastatic kidney cancer cells [132]. Counterintuitively, inactivation of VEGFC in kidney cancer cell lines increases their proliferation and migration. Interestingly, these cells do not form tumors in immunodeficient mice, but develop invasive tumors in immunocompetent mice [70]. These findings highlight the complex interplay between VEGFC, the tumor microenvironment, and the immune system. Inactivation of VEGFC can have differential effects depending on the immune status of the host, indicating the importance of considering the immune context when targeting VEGFC in cancer therapy. Modulating VEGFC signaling pathways may offer potential therapeutic opportunities for suppressing tumor growth, inhibiting metastasis, and improving treatment outcomes in kidney cancer and other malignancies.
5. Tumor angiogenesis: scientific context and therapeutic failure
Angiogenesis involves the formation of new blood vessels from pre-existing ones and its balance is maintained by the interplay between pro-angiogenic and anti-angiogenic factors (Figure 5). In addition to its role in embryogenesis, angiogenesis contributes to organ growth. In physiological conditions, angiogenesis occurs in a controlled manner during specific events such as tissue repair, gestation, the ovarian cycle, or in response to ischemia (lack of blood supply to tissues) [133]. Pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), promote the growth of new blood vessels, while anti-angiogenic factors, such as thrombospondin-1, inhibit angiogenesis, maintaining a delicate balance. However, during tumor progression and metastatic dissemination, this balance is disrupted, leading to abnormal and dysregulated angiogenesis. Tumors require a blood supply to support their growth and metastasis, and therefore, they stimulate angiogenesis to create new blood vessels that can deliver oxygen, nutrients, and growth factors to the tumor cells. This process allows the tumor cells to establish themselves in new locations and promotes the development of metastases. The dysregulated angiogenesis in cancer is driven by the overexpression of pro-angiogenic factors and the downregulation of anti-angiogenic factors. This imbalance promotes the formation of an abnormal tumor vasculature characterized by leaky, disorganized, and tortuous blood vessels. The abnormal tumor vasculature not only supports tumor growth but also contributes to tumor progression by facilitating intravasation of cancer cells into the bloodstream, leading to distant metastasis.
Understanding the molecular mechanisms underlying angiogenesis and its dysregulation in cancer has led to the development of anti-angiogenic therapies, which aim to disrupt the tumor vasculature and inhibit tumor growth. These therapies target pro-angiogenic factors or their receptors to inhibit the formation of new blood vessels, thereby starving the tumor of its blood supply.
5.1. Tumor neovascularization
As a tumor grows, its demand for oxygen and nutrients exceeds what can be supplied by simple diffusion from nearby blood vessels. The tumor then secretes pro-angiogenic signals including VEGFA to stimulate the formation of new blood vessels from existing vessels. Tumor angiogenesis involves the sprouting and remodeling of nearby blood vessels to supply the growing tumor with a network of blood vessels. Tumor blood vessels are often disorganized and abnormal. They are leaky and inefficient, thus leading to inadequate blood flow, poor oxygenation, and uneven distribution of nutrients within the tumor. Once the tumor has established a network of blood vessels, it gains the ability to invade surrounding tissues and spread to distant sites through the bloodstream. By comparing weakly vascularized quiescent tumors and strongly vascularized fast-growing tumors, Folkman established that initiation of tumor angiogenesis is necessary for tumor progression [134]. He also isolated a tumor-produced factor, responsible for tumor associated angiogenesis, which he named TAF (tumor-angiogenesis factor). He suggested that blocking this factor (and thus angiogenesis) could stop tumor growth [135]. These observations paved the way to understanding the activation of tumor angiogenesis, also called the “angiogenic switch”.
In contrast to the physiological vasculature, tumor blood vessels and their endothelial lining have an abnormal architecture. These tumor vessels are disorganized; they do not present the classical artery-capillary-vein hierarchy. They are more dilated and form arteriovenous shunts that leads to unstable blood flow [136,137,138]. They have many branches, irregular diameter and increased permeability to macromolecules leading to higher interstitial pressure and thus edema, fibrosis, inflammation and local microhemorrhages [139]. The endothelial cells lining tumor vessels arise from the proliferation of normal endothelial cells from surrounding the tissue and are structurally abnormal. They have many fenestrations and enlarged cell junctions. They overlap and migrate into the lumen of the vessel. The phospholipids of the inner membrane layer of tumor endothelial cells is disorganized and shifted to the outer membrane. This redistribution of phospholipids is caused by the oxidative stress of the tumor microenvironment and hypoxia [140]. Moreover, these tumor endothelial cells have a high proliferation rate compared to normal endothelial cells [141]. The basal membrane is discontinuous or absent. The tumor endothelium is sparsely covered with morphologically abnormal pericytes, indicating less maturity [142]. Smooth muscle cells, positive for the a-SMA (smooth muscle actin) marker, are reduced in xenograft models of lung carcinoma [143].
The endothelial junctions of tumor vessels are also aberrant and less cohesive: the glioblastoma secretome provides pro-angiogenic and inflammatory signals (here CXCL8), disrupts the junctions formed by VE-cadherin and promotes the permeability of brain endothelial cells [144]. Thus, abnormalities in the structure and composition of tumor vessels combined with a microenvironment rich in pro-angiogenic and inflammatory factors, are responsible for the abnormally high vascular permeability of tumors [145]. These vascular abnormalities create a hostile environment characterized by hypoxia, low pH, inflammation, and high interstitial pressure that select the most aggressive cancer cells. The resulting vascular leakage contributes to the increase in tumor interstitial pressure and causes vascular edema, which limits the delivery of chemo-therapeutic agents and the anti-tumor immune response [139]. Finally, destruction the endothelium promotes intravasation of tumor cells and metastatic dissemination via the bloodstream [146] (Figure 6).
5.2. Mechanisms of angiogenic hijacking
Angiogenesis rapidly becomes essential for tumor development, which is why the tumor hijacks some physiological mechanisms to its advantage. This angiogenic switch is triggered by several factors. Tumor cells have activating mutations of oncogenes such as the RAS family or inhibitory mutations of tumor suppressor genes such as TP53. These mutations are partly responsible for the dysregulation of physiological angiogenesis: they increase the expression of VEGFA (also inducible by hypoxia (HIF-1a)) and reduce the expression of TSP-1 [136]. Hypoxia induces the overexpression of pro-angiogenic factors, especially VEGFA and PDGF. In addition, defective tumor new vessels create a particular metabolic and immunological microenvironment. Hypoxia, resulting from structural vessel abnormality, reduces the energy metabolism provided by the Krebs cycle, thus leading to an accumulation of succinate. The latter binds to the GPR91 receptor and stimulates the vessel growth [147]. Hypoxia allows the recruitment of bone marrow-derived immune cells to tumor sites, including TAMs, neutrophils, mast cells, and myeloid-derived suppressor cells. These cells release pro-angiogenic signals such as VEGFA or MMPs [148] and participate in immune tolerance (Figure 7). Thus, in addition to the secretion of pro-angiogenic factors by the cancer cells, the microenvironment increases their aggressiveness and angiogenesis.
The endothelial lining of tumor blood vessels arises from the proliferation of normal endothelial cells from the surrounding tissue. Thus, the change in phenotype of these endothelial cells, in a tumor context, is due to the microenvironment and to epigenetic factors. In mouse models, identical human colon adenocarcinoma tumors implanted in the liver or skin show different tumor endothelial phenotypes. The vessels of the hepatic tumor are narrower and more permeable than those of the subcutaneous tumor. The number of leukocytes in the liver tumor and the amount of VEGFA mRNA are decreased compared to the subcutaneous tumor [149]. The phenotype of tumor vessels generated during cerebral or subcutaneous implantation of human glioblastoma, rhabdomyosarcoma and murine mammary carcinoma tumors differs depending on the tissue [150]. These two studies demonstrate the role played by the microenvironment of the receiving tumor tissue. Tumor cell conditioned medium leads to epigenetic alteration of gene expression in cultured endothelial cells [151,152]. The gene expression profiles in primary cultures of isolated glioblastoma endothelial cells differ from those of endothelial cells from normal brain tissue are different [153,154]. The epigenetic profile of tumor endothelial cells is thus influenced by the tumor environment. In addition, the endothelial cells of tumor blood vessels are genetically reprogrammed. Endothelial cells isolated from human melanoma and liposarcoma xenografts exhibit aneuploidy and multiple centrosomes. These CD31+ cells express lower levels of TIE1 and TIE2, proliferate faster, have lower serum requirements, and are more sensitive to FGF and EGF than normal endothelial cells. These important findings suggest that genetic alterations of tumor endothelial cells influence the cell phenotype [155]. Tumors can take up endothelial cells from existing blood vessels and change the phenotype of the endothelium [156]. In some tumors, the vessels are lined by tumor cells instead of endothelial cells. In glioblastoma, a significant proportion of the endothelial cells associated with the tumor vessels are of neoplastic origin. Neural stem cells of glioblastoma promote angiogenesis by releasing VEGFA and differentiating into a tumor endothelial phenotype. These cells connect to tumor vessels and the resulting hybrid vessels are functional [157]. This differentiation mechanism, termed vascular mimicry, is unclear, but the presence of intravascular tumor cells interferes with targeted anti-angiogenic therapies [136,158].
5.3. Anti-angiogenic therapies and their limits
Over the past decades, angiogenesis has emerged as critical strategy in oncology. The aim of anti-angiogenic therapy is to starve tumors by disrupting their oxygen and nutrient supply, ultimately reducing tumor proliferation. Angiogenesis is tightly regulated by a delicate balance of activating and inhibiting signals, as discussed earlier. However, in tumor tissue, VEGFA165 is often overexpressed as the main vascular growth factor. VEGFA165 promotes angiogenesis and tumor growth by binding to and activating VEGFR1 and VEGFR2 thereby initiating a cascade of signaling events. To counteract these effects, a range of anti-VEGFA165/VEGFRs agents have been developed. They demonstrate potent efficacy in inhibiting angiogenesis and suppressing tumor growth in preclinical models. As a result, several anti-VEGFA165/VEGFRs have gained approval for the treatment of various cancers.
Bevacizumab (BVZ, Avastin®) is a humanized monoclonal antibody directed against biologically relevant VEGFA isoforms. It is commonly used as a standalone therapy or in combination with chemotherapy in colorectal and ovarian cancer. The addition of bevacizumab to chemotherapy improves patient survival and response rates in colon [159] and ovarian [160] cancers. In some cases of glioblastomas, bevacizumab is used as monotherapy. It was also combined with with IFN-a-2a for the treatment of metastatic renal carcinomas, [161,162], but its use in this context is no longer prevalent. Bevacizumab is now used for renal cancer in combination with atezolizumab (Tecentriq®), an anti-PDL1 antibody [163]. Other compounds target the tyrosine kinase activity of VEGFA receptors. Sunitinib (Sutent®) inhibits VEGFR1, VEGFR2 and VEGFR3, as well as other receptors with tyrosine kinase activity (PDGFR, CSFR1, c-KIT, etc.), downstream of the signaling of several pro-angiogenic factors. It is approved for the treatment of metastatic renal cell carcinoma and advanced neuroendocrine pancreatic cancer. Other multi-kinase treatments such as pazopanib (Votrient®), vandetanib (Caprelsa®) or sorafenib (Nexavar®) are used for the treatment of various metastatic cancers (kidney, thyroid, liver, etc.).
Despite an initial period of clinical benefit with improved progression-free survival and tumor regression, none of these treatments resulted in complete cure. The treated primary tumors relapse and persistent malignant cells proliferate and disseminate in distant healthy tissues, giving rise to metastases. The mechanisms of resistance in tumors are not fully understood to date. They involve events related to the tumor microenvironment, intrinsic resistance associated with the redundancy of pro-angiogenic factors and acquired resistance leading to tumor revascularization.
Tumor cells use alternative pro-angiogenic factors independent of the VEGFA/VEGFR pathway to resist conventional anti-angiogenic therapies. When the VEGFA/VEGFR pathway is blocked, other pro-angiogenic factors restore tumor angiogenesis. In experimental models of pancreatic cancer in mice, antibodies blocking the VEGFR2 receptor initially inhibit tumor growth. At an advanced stage of the disease, the tumors become resistant and progress. This resistance is correlated with the overexpression of FGF1 and FGF2. However, tumors treated with an FGF inhibitor alongside with the VEGFR2 inhibitor show reduced revascularization and tumor progression compared to tumors treated with the VEGFR2 inhibitor alone [164]. These findings highlight the importance of targeting multiple pro-angiogenic pathways to overcome resistance and improve treatment outcomes.
Kidney cancer cells overexpress several redundant pro-angiogenic factors, including VEGFA and the cytokine CXCL8, compared to healthy tissues. In cellular models of kidney cancer, bevacizumab traps VEGFA and increases the compensatory production of pro-angiogenic ELR+CXCL cytokines. In experimental kidney cancer in mice, anti-VEGFA accelerates tumor growth but when combined with an anti-CXCL8 antibody, tumor growth is inhibited. In animals treated with bevacizumab, the density of tumor blood vessels is decreased while the density of tumor lymphatic vessels is increased. This phenomenon is accompanied by an increase in the levels of the main lymphangiogenic factors VEGFC and VEGFD. Conversely, treatment with anti-CXCL8, either alone or in combination with bevacizumab, reduces the levels of VEGFC. Furthermore, the expression of the receptor-type protein-tyrosine phosphatase-κ (RPTP-κ), an inhibitory EGF receptor (EGFR) phosphatase, is decreased in cells from bevacizumab-treated tumors [165].
MBs, like kidney cancer, are highly vascularized tumors that overexpress several members of the VEGF family and many other markers of angiogenesis (VEGFB, VEGFC, FGF, angiopoietin). However, the response rate of anti-angiogenic treatments in these tumors is low, mainly because of the redundancy of angiogenic factors. Furthermore, these treatments can have detrimental effects on the development of children making their use challenging [166]. Recent experiments have demonstrated the potential of bevacizumab in the treatment of pediatric MB patients who experience relapse. When used in combination with metronomic chemotherapy, bevacizumab has shown promise in improving outcomes for these patients [167]. However, despite these positive results, bevacizumab in this context is not a curative chemotherapy. Another multi-kinase inhibitor, axitinib (Inlyta®), has shown relevant effects on the development of experimental MBs in mice [168,169].
The evasion of tumor from therapies targeting the VEGFA/VEGFR axis may attributed to several factors: increased activation of EGFR; the development of lymphangiogenesis which provides an additional route for metastatic dissemination; the production of compensatory pro-angiogenic factors (FGF, VEGFB, angiopoietins, cytokines of the ELR+CXCL family) to counteract the effects of VEGFA inhibition (Figure 8).
Most anti-angiogenic approaches focus on inhibiting the signaling of pro-angiogenic factors. However, tumor cells have an arsenal of strategies to evade the effects of anti-angiogenics. Due to the limited efficacy of these anti-angiogenic therapies in certain types of cancers including MB, there is a need for the development of new strategies to overcome resistance and improve treatment outcomes.
6. From molecular pathology to targeted therapies
The current approach to cancer treatment lacks the ability to adequately address the inter-tumor heterogeneity observed in different subgroups and subtypes of patients. Therapeutic approaches must therefore be further developed in order to fight tumors more specifically without affecting healthy tissue. The previously described molecular definition of subgroups and subtypes paved the way for the development of more specific therapies in the era of personalized medicine (Figure 1; Table 1; Table 2).
6.1. WNT subgroup medulloblastomas
WNT MBs have a looser BBB than other MBs. This feature contributes to the ability of chemotherapy to penetrate the CNS and these patients have a good prognosis [15]. The WNT pathway plays an important role in tissue regeneration and bone repair during development [170]. Therefore, therapies targeting the WNT pathway are not currently developed for MB. Due to the good prognosis of these patients, clinical trials (NCT02724579, NCT02066220 and NCT01878617) are ongoing that aim to reduce the dose of radio- and chemotherapy (therapeutic de-escalation) and improve the quality of life of these patients without compromising their prognosis.
6.2. SHH group medulloblastomas
Constitutive activation of the SHH pathway is favorable for the development of SMO inhibitors. However, inhibitors such as vismodegib or sonidegib are ineffective when tumors carry SUFU or GLI mutations (see 3.2.2) ([171]; [172]. In addition, vismodegib causes bone and dental problems, disproportionate growth and early puberty that persist long after treatment is discontinued [173]. Patient stratification at diagnosis is therefore essential to avoid unnecessary side effects in patients who do not respond due to the genetic characteristics of their tumor. Efforts are being made in the preclinical phase to get rid of these resistances. Itraconazole and arsenic trioxide, two agents in clinical use, inhibit SMO activity and certain GLI mutations responsible for the resistance to vismodegib and sonidegib. Itraconazole targets the intracellular part of SMO, and arsenic trioxide inhibits GLI2. These inhibitors, alone or in combination, inhibit the growth of MB SHH and prolong the survival of mice that are naïve or resistant to SMO inhibitors [174]. These inhibitors are FDA-approved, and their toxicity is known. Their repositioning in the MB SHH would thus be facilitated and represents hope for patients with mutations downstream of SMO.
6.3. Group 3/4 medulloblastomas
The limited understanding of tumorigenesis in these subgroups limits the development of new targeted therapies. However, there is an urgent need to offer patients new alternatives. One of the first ways is to tailor treatments to patients’ risk. The NCT01878617 clinical trial proposes pemetrexed and gemcitabine for newly diagnosed intermediate- and high-risk patients who have first received radiotherapy and standard chemotherapy. For relapsed MB, prexasertib (inhibitor of checkpoint kinase-1 and -2 proteins involved in cell cycle regulation) is offered in combination with cyclophosphamide (trial NCT04023669) [175].
However, these strategies are not specific to these subgroups. Targeting MYC, which is frequently mutated in these patients, would be particularly important. The bromodomain inhibitor JQ1 blocks MYC activity in mice [176]. Currently, no clinical trials are being conducted in MB. Other bromodomain inhibitors (CPI-0610 and MK-8628) are in phase 1 clinical trials in hematological malignancies, prostate cancer, breast cancer, non-small cell lung cancer and glioblastoma.
MBs with MYC mutations appear to rely on CDK4/6 for their proliferation. In Group 3, palbociclib, a potent CDK4/6 inhibitor in a mouse model [177], has been clinically tested in MB and other brain tumors (NCT02255461). This drug is FDA-approved for breast cancer. However, this treatment does not seem to benefit patients with MB [178].
The development of new, more targeted alternatives for these subgroups of patients remains urgent, given their unfavorable prognosis.
However, given the significant efficacy of current therapies and the lack of prospects for these new targeted therapies, they are only available when patients relapse, regardless of the subgroup. However, they may have fewer side effects for young patients. Moreover, the number of targeted therapies currently on the market is insufficient, despite considerable preclinical efforts. These therapeutic approaches need to be further developed to achieve better efficacy of treatments by reducing their toxicity.
7. What about immunotherapies?
Cancer immunotherapy aims to stimulate and improve the patient's anti-tumor immune response by preserving healthy tissue. It is now considered a new pillar of cancer treatment and is already used clinically for the treatment of lung, melanoma and kidney cancers. However, its use in brain tumors is not yet well known. There are different types of immunotherapies, e.g. immune checkpoint inhibitors, natural killer (NK) cells, CAR-T cells (Chimeric Antigen Receptor T cell), cancer vaccines, oncolytic viruses and immunomodulators (e.g. cytokines or antibodies) [179] (Figure 1; Table 2).
7.1. WNT subgroup medulloblastomas
To escape the response of T lymphocytes, cancer cells up-regulate the expression of immune checkpoints (e.g. PDL1, or B7). This immunosuppressive mechanism, hijacked by the tumor cells, is designed to attenuate the immune response, and prevent autoimmunity. Inhibition of these checkpoints enables reactivation of the antitumor immune system.
Among the best-known immune checkpoints, the binding of PDL1 (Programmed cell death ligand 1) to its receptor PD1 (Programmed cell death protein 1) plays an important role. PDL1 is expressed by tumor cells, binds to its PD1 receptor on T lymphocytes (LT) and activates it. PD1 is also expressed by B cells (B cells), activated monocytes, dendritic cells, and NK cells [180]. This binding therefore represents a potential therapeutic target.
Overall, MBs express low levels of PDL1. However, this expression correlates with reduced infiltration of CD8+ T cells and poor prognosis [181]. PDL1 expression seems to be related to the MB subgroups. SHH tumors express high levels, whereas groups 3 and 4 display low levels. An inflammatory microenvironment is required to induce PDL1 expression in these tumors. Interferon gamma (IFN-g) stimulates TH1 cytokines, which induce PDL1 expression in MBs of SHH subgroup [182]. The efficacy of anti-PDL1 in mice depends on the timing of treatment. They are effective from day 7 after transplantation but are ineffective at the time of tumor inoculation. In mice, SHH tumors have distinctly inflammatory microenvironment compared to the tumors of groups 3 and 4. Nevertheless, group 3 tumors respond better to anti-PD1 due to greater infiltration of PD1+ CD8+ T cells and increased survival [183]. The lack of response to anti-PD1 in SHH tumors may be related to the presence of MDSC (Myeloid-Derived Suppressor Cells) and TAM (Tumor Associated Macrophages) involved in immune tolerance via inhibition of the T response [183,184]. Group 3 patients would therefore respond better to immunotherapy, while SHH patients could be resistant to anti-PD1 therapies. The genetic subgroup, the tumor microenvironment and the timing of administration are thus parameters that need to be considered to improve the effect of this therapy.
Clinical trials are currently underway in MB and other CNS tumors. The NCT02359565 phase I clinical trial is investigating the efficacy of the anti-PD1 monoclonal antibody, pembrolizumab (Keytruda®) in children and young adults with recurrent brain tumors including MB. A phase II clinical trial (NCT03173950) is investigating the efficacy of nivolumab (Opdivo®), another anti-PD1 monoclonal antibody, in adults. Finally, the NCT03130959 phase II clinical trial is investigating the effect of nivolumab, alone or in combination with ipilimumab (or Yervoy®, an anti-CTLA4).
The CTLA4 receptor (Cytotoxic T-Lymphocyte Antigen 4 protein) is also an immune checkpoint. It is expressed by LTs and its interaction with B7 (on antigen-presenting cells or tumor cells) transduces an inhibitory signal to lymphocytes. Inhibition of this binding is also a therapeutic way to reactivate the antitumor immune system. B7-H3, a glycoprotein of the B7 family, is overexpressed in all subgroups of MB [185]. Two ongoing clinical trials (NCT04167618 and NCT04743661) are investigating the effect of radiotherapy coupled with anti-B7-H3 immunotherapy (radiolabeled monoclonal antibody) intrathecally in MB [186].
The use of immune checkpoint inhibitors is poorly documented in MB. However, it may be held out as hope, given the urgency to develop less toxic alternative therapies for patients.
Preclinical studies show that the efficacy of new potential treatments must be evaluated and optimally adapted depending on the genetic subgroup of MB. However, in the current clinical trials, this stratification is not performed. In addition, the small number of MB models in immunocompetent mice limits these types of studies.
7.2. Natural Killer NK cells
NK cells are cytotoxic lymphocytes that are able to recognize and lyse damaged “self” and “non-self” cells such as tumor cells. This lysis occurs via the perforin/granzyme and the IFNg pathways. NK cells express germline-encoded activating and inhibitory receptors. Inhibitory receptors (KIR, Killer cell Ig-like Receptors) recognize major histocompatibility complex class I (MHC-I) and make NK cells resistant to healthy tissue and self-proteins. Activation receptors recognize activating signals generated by damaged, infected, or cancerous cells. They also secrete inflammatory and immunosuppressive cytokines that control the adaptive immune response [187].
Autologous NK cells can be harvested, activated, propagated, and genetically modified to increase their antitumor activity, and then returned to the patient. This technique is particularly effective in hematological malignancies and solid tumors [188].
MB cell lines express NK-activating ligands. In particular, the Daoy cell line has high level of NKG2D ligands, an activating NK receptor, responsible for its lytic activity. This property leads to the lysis of MB cells by activated human NK cells in vitro. The lysis is independent of the presence of the CD133 stemness marker [189]. Moreover, NK cells induce apoptosis of human MB tumor cells in the cerebellum of immunodeficient NSG mice. These tumors show a decreased expression of MHC-I, making them more sensitive to lysis by NK [190]. NK-based immunotherapy is therefore an effective approach both on the primary tumor and on the tumor stem cells responsible for self-renewal. However, MB cells also generate immunosuppressive signals (such as TGF-b), so their elimination by NKs is incomplete. Creating NK cells that express a dominant-negative TGF-b allows this inhibiting signal to be ignored [191]. Genetically modified NKs therefore present an advantage and guide the development of an immune strategy.
A phase 1 trial (NCT02271711) is currently attempting to evaluate the efficacy of autologous NK cells activated ex vivo (with artificial antigen-presenting cells) and injected into the cerebellum of children with relapsed or refractory disease [175]. The outcome of this trial will provide a first insight into the effect of NK immunotherapy in patients.
7.3. CAR-T cells
CAR-T cell therapy consists of:
- Collecting autologous or allogeneic T cells by apheresis and genetically modifying them to express tumor antigen-specific receptors (CARs) by viral transduction;
- Amplifying these LTs and reinjecting them into the patient after lymphodepletion, which promotes the expansion and persistence of the CAR-Ts. Thus, these CAR-Ts enable a specific immune response against cancer cells.
The use of CAR-Ts has shown efficacy in hematological malignancies (especially with anti-CD19/CD20 CAR-Ts). However, one of the main side effects is cytokine release syndrome associated with reversible neurological damage after treatment [192,193]. Their efficacy is controversial in solid tumors. Extensive preclinical studies are needed to identify tumor-specific antigens that are not expressed by healthy tissue.
In MB, one of the interesting targets is the receptor tyrosine kinase ERBB2 (HER2). Although all MBs present ERBB2 mRNA, the HER2 protein is expressed by 40% of MBs and is associated with a poor prognosis [194]. However, this receptor is undetectable in normal developing cerebellum [195,196]. It is therefore a relevant target for the development of CAR-Ts. Low-dose anti-HER2 CAR-Ts lead to rapid regression of experimental MB in mice. These immunodeficient mice, treated with CAR-Ts directly in the cerebellum show no systemic toxicity [197]. A phase 1 clinical trial is currently ongoing (NCT03500991) to investigate the effect of anti-HER2 autologous anti-HER2 CD4+ and CD8+ CAR-Ts in patients with relapsed/refractory HER2-positive MB.
Similarly, EPHA2, HER2 and IL13Ra2 targets are expressed by MBs (and ependymomas), but not by the developing healthy brain. CAR-T EPHA2, HER2 and IL13Ra2 cells, alone or in combination into the CSF are effective against primary, metastatic, and recurrent group 3 MB in mouse models (as well as ependymomas) [198].
Adoptive LT therapy is therefore very promising for the treatment of MB. This therapy is tailored to each patient and LTs can pass through the BBB and infiltrate the brain [186]. However, tumors can negatively regulate the antigenic target and patients can develop resistance, as in leukemia [199]. To overcome antigenic escape, multi-variant CAR-Ts can be developed, as previously described for EPHA2, HER2 and IL13Ra2 CAR-Ts. In addition, adverse effects on MB are not known, so implementation of CAR-T strategy should be considered with caution.
8. Conclusions
In conclusion, a significant proportion of patients with MB remain incurable, despite ongoing clinical trials. Treatment resistance and relapse are common due to the inherent heterogeneity of the disease. Relapse is associated with a poor outcome in metastatic patients.
While various therapeutic approaches have been developed, understanding the mechanisms of lymphatic system metastasis in MB could significantly enhance the effectiveness of immune-based therapies such as immune checkpoints and CAR-T cell therapy. Targeting the lymphatic system and its interactions with immune cells may improve immune responses against MB and reduce the incidence of metastases.
Further investigations are necessary to fully understand the complexity of the lymphatic system in MB. By expanding our knowledge in this area, we can potentially enhance treatment strategies and improve outcomes for patients with MB.
Author Contributions
Writing: Original Draft Preparation, S.M.; Review & Editing, M.P-C, G.P., S.M.; Visualization, S.M.; Project Administration, G.P., S.M.; Funding Acquisition, G.P., S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from the Fondation BMS, Rueil-Malmaison (FRANCE), e-mail: fondation@bms.com and from the H2020 TheraLymph Grant project ID: 874708. We also thank the Fondation Flavien and Fondation Mora for their support.
Conflicts of Interest
The authors have no conflict of interest to disclose.
References
- Smoll, N.R. and K.J. Drummond, The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci, 2012. 19(11): p. 1541-4. [CrossRef]
- Bihannic, L. and O. Ayrault, Insights into cerebellar development and medulloblastoma. Bull Cancer, 2016. 103(1): p. 30-40. [CrossRef]
- Louis, D.N., et al., The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol, 2016. 131(6): p. 803-20. [CrossRef]
- Kumar, R., A.P.Y. Liu, and P.A. Northcott, Medulloblastoma genomics in the modern molecular era. Brain Pathol, 2020. 30(3): p. 679-690. [CrossRef]
- Northcott, P.A., et al., Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature, 2012. 488(7409): p. 49-56. [CrossRef]
- Kool, M., et al., Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol, 2012. 123(4): p. 473-84. [CrossRef]
- Northcott, P.A., et al., The whole-genome landscape of medulloblastoma subtypes. Nature, 2017. 547(7663): p. 311-317. [CrossRef]
- Cho, Y.J., et al., Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol, 2011. 29(11): p. 1424-30. [CrossRef]
- Cavalli, F.M.G., et al., Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell, 2017. 31(6): p. 737-754.e6. [CrossRef]
- Schwalbe, E.C., et al., Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol, 2017. 18(7): p. 958-971. [CrossRef]
- Louis, D.N., et al., The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol, 2021. 23(8): p. 1231-1251. [CrossRef]
- Hovestadt, V., et al., Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer, 2020. 20(1): p. 42-56. [CrossRef]
- Shih, D.J., et al., Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol, 2014. 32(9): p. 886-96. [CrossRef]
- Pui, C.H., et al., Challenging issues in pediatric oncology. Nat Rev Clin Oncol, 2011. 8(9): p. 540-9. [CrossRef]
- Phoenix, T.N., et al., Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell, 2016. 29(4): p. 508-522. [CrossRef]
- Clifford, S.C., et al., Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle, 2006. 5(22): p. 2666-70. [CrossRef]
- Ellison, D.W., et al., beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol, 2005. 23(31): p. 7951-7. [CrossRef]
- Fattet, S., et al., Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol, 2009. 218(1): p. 86-94. [CrossRef]
- Juraschka, K. and M.D. Taylor, Medulloblastoma in the age of molecular subgroups: a review. J Neurosurg Pediatr, 2019. 24(4): p. 353-363. [CrossRef]
- Ray, S., et al., Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment. Diagnostics (Basel), 2021. 12(1). [CrossRef]
- Zhou, Z., et al., Research progress in molecular pathology markers in medulloblastoma. Explor Target Antitumor Ther, 2023. 4(1): p. 139-156. [CrossRef]
- Sharma, T., et al., Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol, 2019. 138(2): p. 309-326. [CrossRef]
- Fults, D.W., M.D. Taylor, and L. Garzia, Leptomeningeal dissemination: a sinister pattern of medulloblastoma growth. J Neurosurg Pediatr, 2019: p. 1-9. [CrossRef]
- Li, M., Y. Deng, and W. Zhang, Molecular Determinants of Medulloblastoma Metastasis and Leptomeningeal Dissemination. Mol Cancer Res, 2021. 19(5): p. 743-752. [CrossRef]
- Aspelund, A., et al., A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med, 2015. 212(7): p. 991-9. [CrossRef]
- Absinta, M., et al., Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife, 2017. 6. [CrossRef]
- Louveau, A., et al., Structural and functional features of central nervous system lymphatic vessels. Nature, 2015. 523(7560): p. 337-41. [CrossRef]
- Mobark, N.A., et al., A case of molecularly profiled extraneural medulloblastoma metastases in a child. BMC Med Genet, 2018. 19(1): p. 10. [CrossRef]
- Campbell, A.N., et al., Extracranial metastases in childhood primary intracranial tumors. A report of 21 cases and review of the literature. Cancer, 1984. 53(4): p. 974-81.
- Rochkind, S., et al., Extracranial metastases of medulloblastoma in adults: literature review. J Neurol Neurosurg Psychiatry, 1991. 54(1): p. 80-6. [CrossRef]
- Goyal, A., et al., Surgical Treatment of Intramedullary Spinal Metastasis in Medulloblastoma: Case Report and Review of the Literature. World Neurosurg, 2018. 118: p. 42-46. [CrossRef]
- Jiang, H., et al., Intramedullary metastasis in medulloblastoma: a case report and literature review. Childs Nerv Syst, 2021. 37(6): p. 2091-2095. [CrossRef]
- Kerjaschki, D., et al., Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol, 2004. 15(3): p. 603-12. [CrossRef]
- Petrova, T.V., et al., Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med, 2004. 10(9): p. 974-81. [CrossRef]
- Skobe, M., et al., Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med, 2001. 7(2): p. 192-8. [CrossRef]
- Vollmar, B., et al., Lymph vessel expansion and function in the development of hepatic fibrosis and cirrhosis. Am J Pathol, 1997. 151(1): p. 169-75.
- Tammela, T. and K. Alitalo, Lymphangiogenesis: Molecular mechanisms and future promise. Cell, 2010. 140(4): p. 460-76. [CrossRef]
- Mohammed, R.A., et al., Prognostic significance of vascular endothelial cell growth factors -A, -C and -D in breast cancer and their relationship with angio- and lymphangiogenesis. Br J Cancer, 2007. 96(7): p. 1092-100. [CrossRef]
- Neuchrist, C., et al., Vascular endothelial growth factor C and vascular endothelial growth factor receptor 3 expression in squamous cell carcinomas of the head and neck. Head Neck, 2003. 25(6): p. 464-74. [CrossRef]
- Stacker, S.A., M.E. Baldwin, and M.G. Achen, The role of tumor lymphangiogenesis in metastatic spread. FASEB J, 2002. 16(9): p. 922-34. [CrossRef]
- Wartiovaara, U., et al., Peripheral blood platelets express VEGF-C and VEGF which are released during platelet activation. Thromb Haemost, 1998. 80(1): p. 171-5. [CrossRef]
- Schoppmann, S.F., et al., VEGF-C expressing tumor-associated macrophages in lymph node positive breast cancer: impact on lymphangiogenesis and survival. Surgery, 2006. 139(6): p. 839-46. [CrossRef]
- Mandriota, S.J., et al., Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J, 2001. 20(4): p. 672-82. [CrossRef]
- He, Y., et al., Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J Natl Cancer Inst, 2002. 94(11): p. 819-25. [CrossRef]
- Karpanen, T., et al., Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res, 2001. 61(5): p. 1786-90.
- Shimizu, K., et al., Suppression of VEGFR-3 signaling inhibits lymph node metastasis in gastric cancer. Cancer Sci, 2004. 95(4): p. 328-33. [CrossRef]
- Roberts, N., et al., Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res, 2006. 66(5): p. 2650-7. [CrossRef]
- Hirakawa, S., et al., VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med, 2005. 201(7): p. 1089-99. [CrossRef]
- Caunt, M., et al., Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell, 2008. 13(4): p. 331-42. [CrossRef]
- Dadras, S.S., et al., Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am J Pathol, 2003. 162(6): p. 1951-60. [CrossRef]
- Liang, P., et al., Increased density and diameter of lymphatic microvessels correlate with lymph node metastasis in early stage invasive colorectal carcinoma. Virchows Arch, 2006. 448(5): p. 570-5. [CrossRef]
- Wirzenius, M., et al., Distinct vascular endothelial growth factor signals for lymphatic vessel enlargement and sprouting. J Exp Med, 2007. 204(6): p. 1431-40. [CrossRef]
- Hoshida, T., et al., Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res, 2006. 66(16): p. 8065-75. [CrossRef]
- Padera, T.P., et al., Lymphatic metastasis in the absence of functional intratumor lymphatics. Science, 2002. 296(5574): p. 1883-6. [CrossRef]
- Albrecht, I. and G. Christofori, Molecular mechanisms of lymphangiogenesis in development and cancer. Int J Dev Biol, 2011. 55(4-5): p. 483-94. [CrossRef]
- Bracher, A., et al., Crystal structures of the free and ligand-bound FK1-FK2 domain segment of FKBP52 reveal a flexible inter-domain hinge. J Mol Biol, 2013. 425(22): p. 4134-44. [CrossRef]
- Dadras, S.S., An unexpected role for EGF in lymphangiogenesis-mediated melanoma metastasis to sentinel lymph nodes. J Invest Dermatol, 2013. 133(1): p. 14-6. [CrossRef]
- Zhang, X.H., et al., Coexpression of VEGF-C and COX-2 and its association with lymphangiogenesis in human breast cancer. BMC Cancer, 2008. 8: p. 4. [CrossRef]
- Su, J.L., et al., Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res, 2004. 64(2): p. 554-64.
- von Rahden, B.H., et al., Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res, 2005. 65(12): p. 5038-44. [CrossRef]
- Kyzas, P.A., D. Stefanou, and N.J. Agnantis, COX-2 expression correlates with VEGF-C and lymph node metastases in patients with head and neck squamous cell carcinoma. Mod Pathol, 2005. 18(1): p. 153-60. [CrossRef]
- Jiang, S., et al., Hematopoietic stem cells contribute to lymphatic endothelium. PLoS One, 2008. 3(11): p. e3812. [CrossRef]
- Religa, P., et al., Allogenic immune response promotes the accumulation of host-derived smooth muscle cells in transplant arteriosclerosis. Cardiovasc Res, 2005. 65(2): p. 535-45. [CrossRef]
- Zumsteg, A., et al., Myeloid cells contribute to tumor lymphangiogenesis. PLoS One, 2009. 4(9): p. e7067. [CrossRef]
- Kerjaschki, D., The crucial role of macrophages in lymphangiogenesis. J Clin Invest, 2005. 115(9): p. 2316-9. [CrossRef]
- Zhang, Y., et al., Lymphangiogenesis in renal fibrosis arises from macrophages via VEGF-C/VEGFR3-dependent autophagy and polarization. Cell Death Dis, 2021. 12(1): p. 109. [CrossRef]
- Patel, V., et al., Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res, 2011. 71(22): p. 7103-12. [CrossRef]
- Niederleithner, H., et al., Wnt1 is anti-lymphangiogenic in a melanoma mouse model. J Invest Dermatol, 2012. 132(9): p. 2235-44. [CrossRef]
- Oka, M., et al., Inhibition of endogenous TGF-beta signaling enhances lymphangiogenesis. Blood, 2008. 111(9): p. 4571-9. [CrossRef]
- Ndiaye, P.D., et al., VEGFC acts as a double-edged sword in renal cell carcinoma aggressiveness. Theranostics, 2019. 9(3): p. 661-675. [CrossRef]
- Alitalo, A. and M. Detmar, Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene, 2012. 31(42): p. 4499-508. [CrossRef]
- Angeli, V., et al., B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity, 2006. 24(2): p. 203-15. [CrossRef]
- Kataru, R.P., et al., T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity, 2011. 34(1): p. 96-107. [CrossRef]
- Kataru, R.P., et al., Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood, 2009. 113(22): p. 5650-9. [CrossRef]
- Halin, C., et al., VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood, 2007. 110(9): p. 3158-67. [CrossRef]
- Nemoto, K., et al., Mesenteric lymph flow in endotoxemic guinea pigs. Lymphat Res Biol, 2011. 9(3): p. 129-34. [CrossRef]
- Cromer, W.E., et al., The effects of inflammatory cytokines on lymphatic endothelial barrier function. Angiogenesis, 2014. 17(2): p. 395-406. [CrossRef]
- Liao, S. and P.Y. von der Weid, Inflammation-induced lymphangiogenesis and lymphatic dysfunction. Angiogenesis, 2014. 17(2): p. 325-34. [CrossRef]
- Dufies, M., et al., Sunitinib stimulates expression of VEGFC by tumor cells and promotes lymphangiogenesis in clear cell renal cell carcinomas. Cancer Res, 2017. 77(5): p. 1212-1226. [CrossRef]
- Bieniasz-Krzywiec, P., et al., Podoplanin-Expressing Macrophages Promote Lymphangiogenesis and Lymphoinvasion in Breast Cancer. Cell Metab, 2019. 30(5): p. 917-936.e10. [CrossRef]
- Ma, Q., et al., Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci Adv, 2018. 4(8): p. eaat4758. [CrossRef]
- Vaahtomeri, K. and K. Alitalo, Lymphatic Vessels in Tumor Dissemination versus Immunotherapy. Cancer Res, 2020. 80(17): p. 3463-3465. [CrossRef]
- Decio, A., et al., Vascular endothelial growth factor c promotes ovarian carcinoma progression through paracrine and autocrine mechanisms. Am J Pathol, 2014. 184(4): p. 1050-1061. [CrossRef]
- Brown, M., et al., Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science, 2018. 359(6382): p. 1408-1411. [CrossRef]
- Pereira, E.R., et al., Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science, 2018. 359(6382): p. 1403-1407. [CrossRef]
- Jones, D., E.R. Pereira, and T.P. Padera, Growth and Immune Evasion of Lymph Node Metastasis. Front Oncol, 2018. 8: p. 36. [CrossRef]
- Stacker, S.A., et al., Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer, 2014. 14(3): p. 159-72. [CrossRef]
- Kabashima, K., et al., CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol, 2007. 171(4): p. 1249-57. [CrossRef]
- Staller, P., et al., Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature, 2003. 425(6955): p. 307-11. [CrossRef]
- Balkwill, F., Cancer and the chemokine network. Nat Rev Cancer, 2004. 4(7): p. 540-50. [CrossRef]
- Günther, K., et al., Prediction of lymph node metastasis in colorectal carcinoma by expressionof chemokine receptor CCR7. Int J Cancer, 2005. 116(5): p. 726-33. [CrossRef]
- Koizumi, K., et al., CCL21 promotes the migration and adhesion of highly lymph node metastatic human non-small cell lung cancer Lu-99 in vitro. Oncol Rep, 2007. 17(6): p. 1511-6. [CrossRef]
- Müller, A., et al., Involvement of chemokine receptors in breast cancer metastasis. Nature, 2001. 410(6824): p. 50-6. [CrossRef]
- Munson, J.M., R.V. Bellamkonda, and M.A. Swartz, Interstitial flow in a 3D microenvironment increases glioma invasion by a CXCR4-dependent mechanism. Cancer Res, 2013. 73(5): p. 1536-46. [CrossRef]
- Issa, A., et al., Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res, 2009. 69(1): p. 349-57. [CrossRef]
- Lanati, S., et al., Chemotrap-1: an engineered soluble receptor that blocks chemokine-induced migration of metastatic cancer cells in vivo. Cancer Res, 2010. 70(20): p. 8138-48. [CrossRef]
- Harrell, M.I., B.M. Iritani, and A. Ruddell, Tumor-induced sentinel lymph node lymphangiogenesis and increased lymph flow precede melanoma metastasis. Am J Pathol, 2007. 170(2): p. 774-86. [CrossRef]
- Hirakawa, S., et al., VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood, 2007. 109(3): p. 1010-7. [CrossRef]
- Garnier, L., A.O. Gkountidi, and S. Hugues, Tumor-Associated Lymphatic Vessel Features and Immunomodulatory Functions. Front Immunol, 2019. 10: p. 720. [CrossRef]
- Dieterich, L.C., et al., Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front Immunol, 2017. 8: p. 66. [CrossRef]
- Lane, R.S., et al., IFNγ-activated dermal lymphatic vessels inhibit cytotoxic T cells in melanoma and inflamed skin. J Exp Med, 2018. 215(12): p. 3057-3074. [CrossRef]
- Lund, A.W., et al., VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep, 2012. 1(3): p. 191-9. [CrossRef]
- Baitsch, L., et al., Exhaustion of tumor-specific CD8⁺ T cells in metastases from melanoma patients. J Clin Invest, 2011. 121(6): p. 2350-60. [CrossRef]
- Grigorova, I.L., et al., Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat Immunol, 2009. 10(1): p. 58-65. [CrossRef]
- Pham, T.H., et al., Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med, 2010. 207(1): p. 17-27. [CrossRef]
- Lukacs-Kornek, V., et al., Regulated release of nitric oxide by nonhematopoietic stroma controls expansion of the activated T cell pool in lymph nodes. Nat Immunol, 2011. 12(11): p. 1096-104. [CrossRef]
- Liu, J., et al., Dendritic cell migration in inflammation and immunity. Cell Mol Immunol, 2021. 18(11): p. 2461-2471. [CrossRef]
- Salmi, M. and S. Jalkanen, Cell-surface enzymes in control of leukocyte trafficking. Nat Rev Immunol, 2005. 5(10): p. 760-71. [CrossRef]
- Roberts, E.W., et al., Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell, 2016. 30(2): p. 324-336. [CrossRef]
- Fridman, W.H., et al., The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer, 2012. 12(4): p. 298-306. [CrossRef]
- Fankhauser, M., et al., Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci Transl Med, 2017. 9(407). [CrossRef]
- Lund, A.W., et al., Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J Clin Invest, 2016. 126(9): p. 3389-402. [CrossRef]
- Bordry, N., et al., Lymphatic vessel density is associated with CD8. Oncoimmunology, 2018. 7(8): p. e1462878. [CrossRef]
- Mlecnik, B., et al., The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med, 2016. 8(327): p. 327ra26. [CrossRef]
- Mempel, T.R., S.E. Henrickson, and U.H. Von Andrian, T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature, 2004. 427(6970): p. 154-9. [CrossRef]
- Qi, H., et al., Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science, 2006. 312(5780): p. 1672-6. [CrossRef]
- Alvarez, D., E.H. Vollmann, and U.H. von Andrian, Mechanisms and consequences of dendritic cell migration. Immunity, 2008. 29(3): p. 325-42. [CrossRef]
- Roozendaal, R., R.E. Mebius, and G. Kraal, The conduit system of the lymph node. Int Immunol, 2008. 20(12): p. 1483-7. [CrossRef]
- Peske, J.D., et al., Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun, 2015. 6: p. 7114. [CrossRef]
- Thompson, E.D., et al., Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med, 2010. 207(8): p. 1791-804. [CrossRef]
- Kimura, T., et al., Lymphatic dysfunction attenuates tumor immunity through impaired antigen presentation. Oncotarget, 2015. 6(20): p. 18081-93. [CrossRef]
- Girard, J.P., C. Moussion, and R. Förster, HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol, 2012. 12(11): p. 762-73. [CrossRef]
- Aloisi, F. and R. Pujol-Borrell, Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol, 2006. 6(3): p. 205-17. [CrossRef]
- Coppola, D., et al., Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am J Pathol, 2011. 179(1): p. 37-45. [CrossRef]
- Martinet, L., et al., Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res, 2011. 71(17): p. 5678-87. [CrossRef]
- Martinet, L., et al., High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology, 2012. 1(6): p. 829-839.
- Messina, J.L., et al., 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep, 2012. 2: p. 765. [CrossRef]
- Song, E., et al., VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature, 2020. 577(7792): p. 689-694. [CrossRef]
- Varney, M.L. and R.K. Singh, VEGF-C-VEGFR3/Flt4 axis regulates mammary tumor growth and metastasis in an autocrine manner. Am J Cancer Res, 2015. 5(2): p. 616-28.
- Tokuyama, W., et al., Autocrine and paracrine roles of VEGF/VEGFR-2 and VEGF-C/VEGFR-3 signaling in angiosarcomas of the scalp and face. Hum Pathol, 2010. 41(3): p. 407-14. [CrossRef]
- Matsuura, M., et al., Autocrine loop between vascular endothelial growth factor (VEGF)-C and VEGF receptor-3 positively regulates tumor-associated lymphangiogenesis in oral squamoid cancer cells. Am J Pathol, 2009. 175(4): p. 1709-21. [CrossRef]
- Dumond, A., et al., Anti-Vascular Endothelial Growth Factor C Antibodies Efficiently Inhibit the Growth of Experimental Clear Cell Renal Cell Carcinomas. Cells, 2021. 10(5). [CrossRef]
- Ferrara, N. and R.S. Kerbel, Angiogenesis as a therapeutic target. Nature, 2005. 438(7070): p. 967-74. [CrossRef]
- Folkman, J., Tumor angiogenesis: therapeutic implications. N Engl J Med, 1971. 285(21): p. 1182-6. [CrossRef]
- Folkman, J., et al., Isolation of a tumor factor responsible for angiogenesis. J Exp Med, 1971. 133(2): p. 275-88. [CrossRef]
- Aird, W.C., Endothelial cell heterogeneity. Cold Spring Harb Perspect Med, 2012. 2(1): p. a006429. [CrossRef]
- Jain, R.K., Molecular regulation of vessel maturation. Nat Med, 2003. 9(6): p. 685-93. [CrossRef]
- Nagy, J.A., et al., Heterogeneity of the tumor vasculature. Semin Thromb Hemost, 2010. 36(3): p. 321-31. [CrossRef]
- Azzi, S., J.K. Hebda, and J. Gavard, Vascular permeability and drug delivery in cancers. Front Oncol, 2013. 3: p. 211. [CrossRef]
- Stafford, J.H. and P.E. Thorpe, Increased exposure of phosphatidylethanolamine on the surface of tumor vascular endothelium. Neoplasia, 2011. 13(4): p. 299-308. [CrossRef]
- Denekamp, J. and B. Hobson, Endothelial-cell proliferation in experimental tumours. Br J Cancer, 1982. 46(5): p. 711-20. [CrossRef]
- Morikawa, S., et al., Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol, 2002. 160(3): p. 985-1000. [CrossRef]
- Inai, T., et al., Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol, 2004. 165(1): p. 35-52. [CrossRef]
- Dwyer, J., et al., Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLoS One, 2012. 7(9): p. e45562. [CrossRef]
- Treps, L. and J. Gavard, [Tumor angiogenesis: when the Tree of Life turns bad]. Med Sci (Paris), 2015. 31(11): p. 989-95. [CrossRef]
- Goel, S., et al., Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev, 2011. 91(3): p. 1071-121. [CrossRef]
- Sapieha, P., et al., [Supply and demand: the influence of energy metabolism on angiogenesis]. Med Sci (Paris), 2009. 25(4): p. 346-8. [CrossRef]
- Ferrara, N., Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev, 2010. 21(1): p. 21-6. [CrossRef]
- Fukumura, D., et al., Role of nitric oxide in tumor microcirculation. Blood flow, vascular permeability, and leukocyte-endothelial interactions. Am J Pathol, 1997. 150(2): p. 713-25.
- Roberts, W.G., et al., Host microvasculature influence on tumor vascular morphology and endothelial gene expression. Am J Pathol, 1998. 153(4): p. 1239-48. [CrossRef]
- Hellebrekers, D.M., et al., Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res, 2006. 66(22): p. 10770-7. [CrossRef]
- Hellebrekers, D.M., et al., Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res, 2007. 67(9): p. 4138-48. [CrossRef]
- Bussolati, B., et al., Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J, 2003. 17(9): p. 1159-61. [CrossRef]
- Unger, R.E., et al., Isolation and molecular characterization of brain microvascular endothelial cells from human brain tumors. In Vitro Cell Dev Biol Anim, 2002. 38(5): p. 273-81.
- Hida, K., et al., Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res, 2004. 64(22): p. 8249-55. [CrossRef]
- Holash, J., et al., Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science, 1999. 284(5422): p. 1994-8. [CrossRef]
- Ricci-Vitiani, L., et al., Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature, 2010. 468(7325): p. 824-8. [CrossRef]
- Naito, H., T. Iba, and N. Takakura, Mechanisms of new blood-vessel formation and proliferative heterogeneity of endothelial cells. Int Immunol, 2020. 32(5): p. 295-305. [CrossRef]
- Hurwitz, H., Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin Colorectal Cancer, 2004. 4 Suppl 2: p. S62-8. [CrossRef]
- Burger, R.A., et al., Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med, 2011. 365(26): p. 2473-83. [CrossRef]
- Escudier, B., et al., Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet, 2007. 370(9605): p. 2103-11. [CrossRef]
- Escudier, B., et al., Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol, 2010. 28(13): p. 2144-50. [CrossRef]
- Rini, B.I., et al., Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet, 2019. 393(10189): p. 2404-2415. [CrossRef]
- Casanovas, O., et al., Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell, 2005. 8(4): p. 299-309. [CrossRef]
- Grepin, R., et al., Acceleration of clear cell renal cell carcinoma growth in mice following bevacizumab/Avastin treatment: the role of CXCL cytokines. Oncogene, 2012. 31(13): p. 1683-94. [CrossRef]
- Huber, H., et al., Angiogenic profile of childhood primitive neuroectodermal brain tumours/medulloblastomas. Eur J Cancer, 2001. 37(16): p. 2064-72. [CrossRef]
- Slavc, I., et al., Improved Long-Term Survival of Patients with Recurrent Medulloblastoma Treated with a "MEMMAT-like" Metronomic Antiangiogenic Approach. Cancers (Basel), 2022. 14(20). [CrossRef]
- Pagnuzzi-Boncompagni, M., et al., Antiangiogenic Compound Axitinib Demonstrates Low Toxicity and Antitumoral Effects against Medulloblastoma. Cancers (Basel), 2021. 14(1). [CrossRef]
- Schwinn, S., et al., Cytotoxic effects and tolerability of gemcitabine and axitinib in a xenograft model for c-myc amplified medulloblastoma. Sci Rep, 2021. 11(1): p. 14062. [CrossRef]
- Houschyar, K.S., et al., Wnt Pathway in Bone Repair and Regeneration - What Do We Know So Far. Front Cell Dev Biol, 2018. 6: p. 170. [CrossRef]
- Quaglio, D., et al., Hedgehog signaling pathway inhibitors: an updated patent review (2015-present). Expert Opin Ther Pat, 2020. 30(4): p. 235-250. [CrossRef]
- Robinson, G.W., et al., Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J Clin Oncol, 2015. 33(24): p. 2646-54. [CrossRef]
- Robinson, G.W., et al., Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget, 2017. 8(41): p. 69295-69302. [CrossRef]
- Kim, J., et al., Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell, 2013. 23(1): p. 23-34. [CrossRef]
- Menyhárt, O. and B. Győrffy, Molecular stratifications, biomarker candidates and new therapeutic options in current medulloblastoma treatment approaches. Cancer Metastasis Rev, 2020. 39(1): p. 211-233. [CrossRef]
- Bandopadhayay, P., et al., BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res, 2014. 20(4): p. 912-25. [CrossRef]
- Hanaford, A.R., et al., DiSCoVERing Innovative Therapies for Rare Tumors: Combining Genetically Accurate Disease Models with In Silico Analysis to Identify Novel Therapeutic Targets. Clin Cancer Res, 2016. 22(15): p. 3903-14. [CrossRef]
- Archer, T.C., et al., Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell, 2018. 34(3): p. 396-410.e8. [CrossRef]
- Esfahani, K., et al., A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol, 2020. 27(Suppl 2): p. S87-S97. [CrossRef]
- Keir, M.E., et al., PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol, 2008. 26: p. 677-704. [CrossRef]
- Murata, D., et al., High programmed cell death 1 ligand-1 expression: association with CD8+ T-cell infiltration and poor prognosis in human medulloblastoma. J Neurosurg, 2018. 128(3): p. 710-716. [CrossRef]
- Martin, A.M., et al., PD-L1 expression in medulloblastoma: an evaluation by subgroup. Oncotarget, 2018. 9(27): p. 19177-19191. [CrossRef]
- Pham, C.D., et al., Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin Cancer Res, 2016. 22(3): p. 582-95. [CrossRef]
- Gabrilovich, D.I. and S. Nagaraj, Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol, 2009. 9(3): p. 162-74. [CrossRef]
- Gregorio, A., et al., Small round blue cell tumours: diagnostic and prognostic usefulness of the expression of B7-H3 surface molecule. Histopathology, 2008. 53(1): p. 73-80. [CrossRef]
- Voskamp, M.J., et al., Immunotherapy in Medulloblastoma: Current State of Research, Challenges, and Future Perspectives. Cancers (Basel), 2021. 13(21). [CrossRef]
- Zhang, Y. and B. Huang, The Development and Diversity of ILCs, NK Cells and Their Relevance in Health and Diseases. Adv Exp Med Biol, 2017. 1024: p. 225-244. [CrossRef]
- Shimasaki, N., A. Jain, and D. Campana, NK cells for cancer immunotherapy. Nat Rev Drug Discov, 2020. 19(3): p. 200-218. [CrossRef]
- Castriconi, R., et al., Both CD133+ and CD133- medulloblastoma cell lines express ligands for triggering NK receptors and are susceptible to NK-mediated cytotoxicity. Eur J Immunol, 2007. 37(11): p. 3190-6. [CrossRef]
- Kennis, B.A., et al., Monitoring of intracerebellarly-administered natural killer cells with fluorine-19 MRI. J Neurooncol, 2019. 142(3): p. 395-407. [CrossRef]
- Powell, A.B., et al., Medulloblastoma rendered susceptible to NK-cell attack by TGFβ neutralization. J Transl Med, 2019. 17(1): p. 321. [CrossRef]
- Gauthier, J. and I. Yakoub-Agha, Chimeric antigen-receptor T-cell therapy for hematological malignancies and solid tumors: Clinical data to date, current limitations and perspectives. Curr Res Transl Med, 2017. 65(3): p. 93-102. [CrossRef]
- Mohanty, R., et al., CAR T cell therapy: A new era for cancer treatment (Review). Oncol Rep, 2019. 42(6): p. 2183-2195. [CrossRef]
- Gilbertson, R.J., ERBB2 in pediatric cancer: innocent until proven guilty. Oncologist, 2005. 10(7): p. 508-17. [CrossRef]
- Orentas, R.J., et al., Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front Oncol, 2012. 2: p. 194. [CrossRef]
- Press, M.F., C. Cordon-Cardo, and D.J. Slamon, Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene, 1990. 5(7): p. 953-62.
- Nellan, A., et al., Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer, 2018. 6(1): p. 30. [CrossRef]
- Donovan, L.K., et al., Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med, 2020. 26(5): p. 720-731. [CrossRef]
- Tasian, S.K. and R.A. Gardner, CD19-redirected chimeric antigen receptor-modified T cells: a promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther Adv Hematol, 2015. 6(5): p. 228-41. [CrossRef]
Figure 1.
Representation of the main MB classifications associated with the techniques that led to their discovery. This figure provides an overview of the histopathological classification of MB. It encompasses the ancestral classification, the 2016 WHO classification with four subgroups, and the updated 2021 WHO classification with 12 subtypes (four subtypes for the SHH group and eight subtypes for the non-WNT/non-SHH group). The subtypes belonging to group 3 are represented in yellow, while those from group 4 are depicted in orange. Notably, subtypes I, V, and VII display characteristics of both group 3 and group 4. Created with BioRender.com.
Figure 1.
Representation of the main MB classifications associated with the techniques that led to their discovery. This figure provides an overview of the histopathological classification of MB. It encompasses the ancestral classification, the 2016 WHO classification with four subgroups, and the updated 2021 WHO classification with 12 subtypes (four subtypes for the SHH group and eight subtypes for the non-WNT/non-SHH group). The subtypes belonging to group 3 are represented in yellow, while those from group 4 are depicted in orange. Notably, subtypes I, V, and VII display characteristics of both group 3 and group 4. Created with BioRender.com.
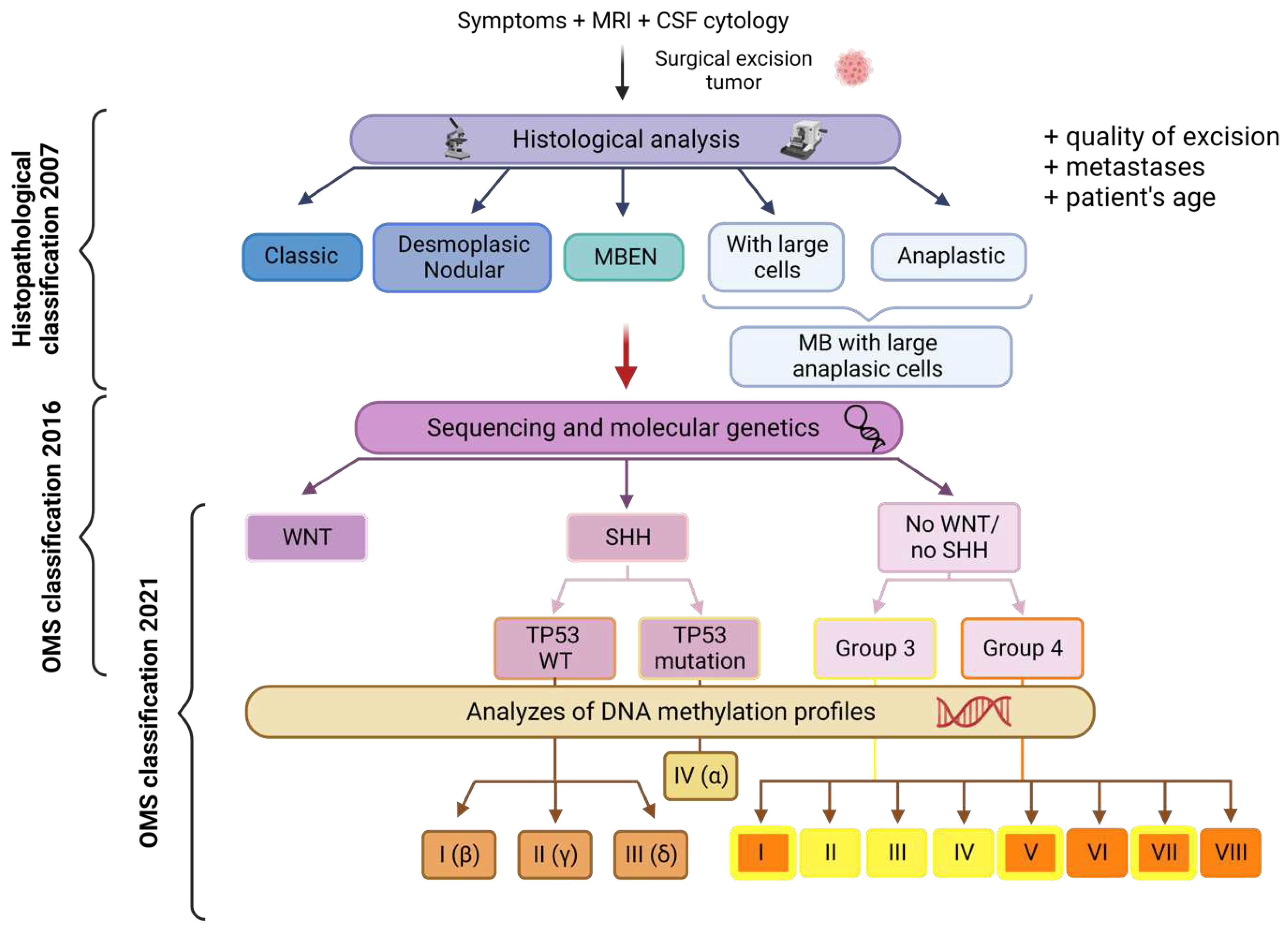
Figure 2.
Molecular mechanisms of tumor lymphangiogenesis. In response to hypoxia and other signals, tumor, stromal, and tumor-infiltrating inflammatory cells secrete lymphangiogenic factors such as VEGFC, VEGFD, VEGFA, FGF, PDGF, Angiopoietin 1 and 2, and EGF. These factors act on existing lymphatic vessels present at the tumor site, thus triggering vessel sprouting, LEC proliferation, and generating new lymphatic vessels both within and around the tumor (intra- and peri-tumoral lymphangiogenesis). The newly formed lymphatic vessels facilitate the transport of tumor cells to nearby lymph nodes. Bone marrow derived LEC precursors and myeloid cells can integrate into these new tumor lymphatic vessels or stimulate LEC proliferation, further supporting the process of tumor lymphangiogenesis.
Figure 2.
Molecular mechanisms of tumor lymphangiogenesis. In response to hypoxia and other signals, tumor, stromal, and tumor-infiltrating inflammatory cells secrete lymphangiogenic factors such as VEGFC, VEGFD, VEGFA, FGF, PDGF, Angiopoietin 1 and 2, and EGF. These factors act on existing lymphatic vessels present at the tumor site, thus triggering vessel sprouting, LEC proliferation, and generating new lymphatic vessels both within and around the tumor (intra- and peri-tumoral lymphangiogenesis). The newly formed lymphatic vessels facilitate the transport of tumor cells to nearby lymph nodes. Bone marrow derived LEC precursors and myeloid cells can integrate into these new tumor lymphatic vessels or stimulate LEC proliferation, further supporting the process of tumor lymphangiogenesis.
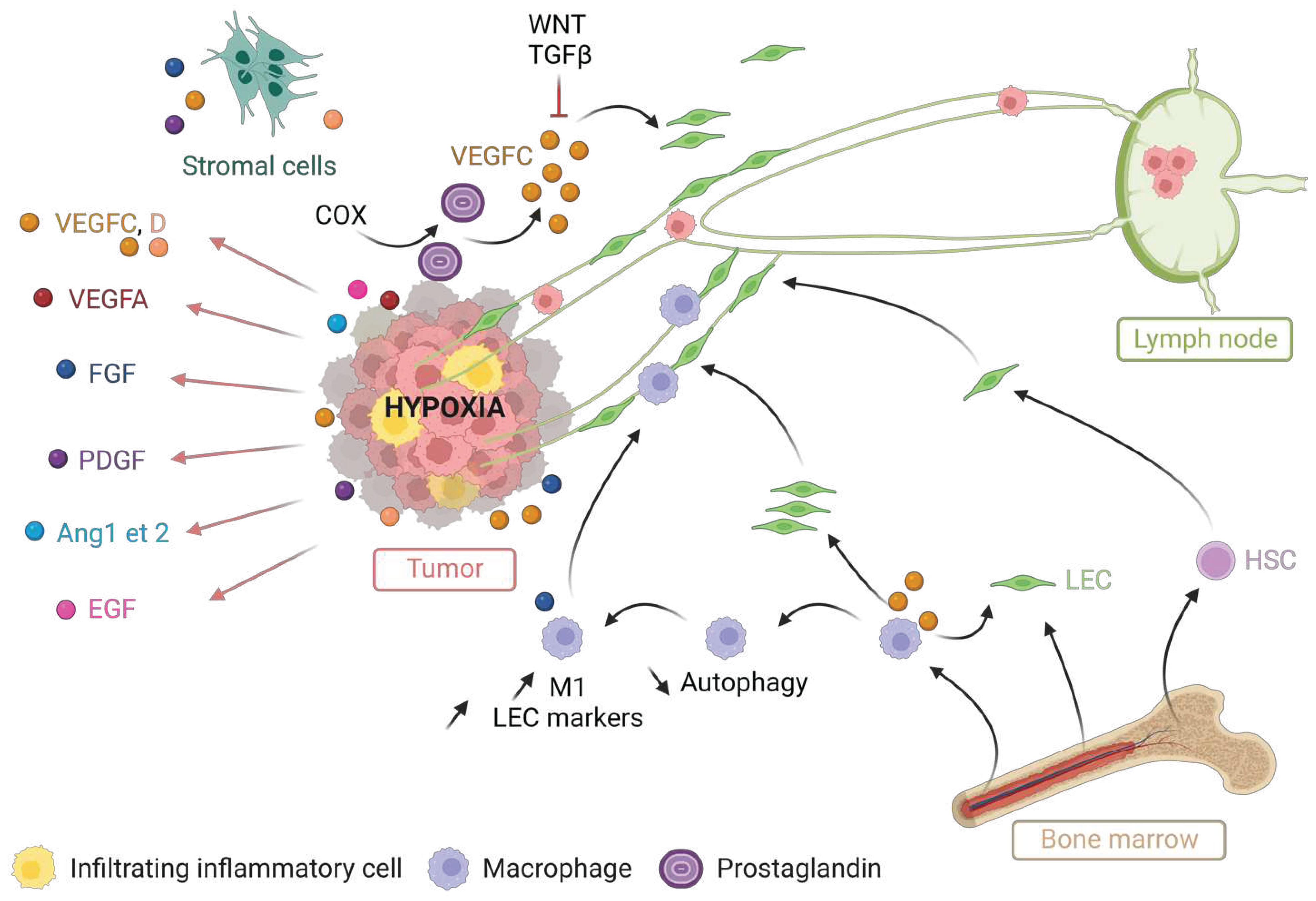
Figure 3.
Adverse roles of tumor lymphangiogenesis: metastatic dissemination and immune tolerance. A. The cytokines CXCL12 and CCL19/21, synthesized by hypoxic tumor and LECs, respectively bind the CXCR4 and CCR7 receptors on the surface of tumor cells. The cells then intravasate into the lymphatic vessels and leading to an increased metastatic dissemination. Tumor-synthesized VEGFC further amplifies this process by enhancing the secretion of CCL21 by the LECs, thereby promoting metastatic dissemination. B. Intra- and peri-tumoral LECs secrete immunoregulatory factors that activate MDSCs and regulatory LTs, and inhibit the activity of CD8 LTs, NKs and dendritic cells. VEGFC is involved in this immune tolerance. C. Within the lymph node, LECs activated by tumor antigens overexpress PDL1. This overexpression is further increased in response to TNF and INF-γ secreted by LTs. These signals induce the secretion of nitric oxide by LECs, which subsequently reduces the activity of CD8 LT. PDL1 also inhibits the activation of LT, NK and dendritic cells expressing PD1.
Figure 3.
Adverse roles of tumor lymphangiogenesis: metastatic dissemination and immune tolerance. A. The cytokines CXCL12 and CCL19/21, synthesized by hypoxic tumor and LECs, respectively bind the CXCR4 and CCR7 receptors on the surface of tumor cells. The cells then intravasate into the lymphatic vessels and leading to an increased metastatic dissemination. Tumor-synthesized VEGFC further amplifies this process by enhancing the secretion of CCL21 by the LECs, thereby promoting metastatic dissemination. B. Intra- and peri-tumoral LECs secrete immunoregulatory factors that activate MDSCs and regulatory LTs, and inhibit the activity of CD8 LTs, NKs and dendritic cells. VEGFC is involved in this immune tolerance. C. Within the lymph node, LECs activated by tumor antigens overexpress PDL1. This overexpression is further increased in response to TNF and INF-γ secreted by LTs. These signals induce the secretion of nitric oxide by LECs, which subsequently reduces the activity of CD8 LT. PDL1 also inhibits the activation of LT, NK and dendritic cells expressing PD1.
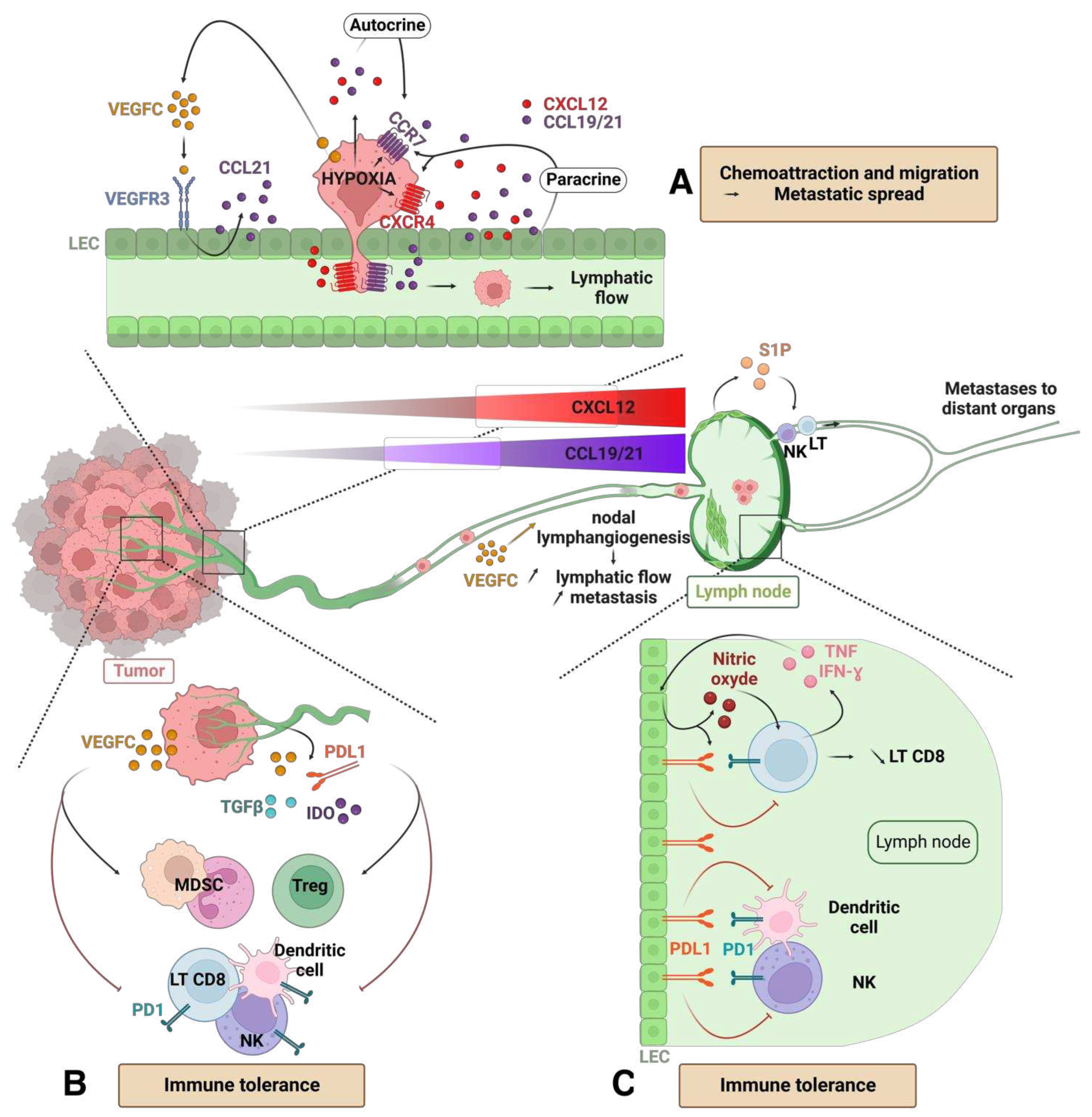
Figure 4.
Beneficial roles of tumor lymphangiogenesis: recruitment and activation of the immune system. A. At inflammatory sites, antigens released by the tumor are captured by dendritic cells (DCs) and presented on their surface (1 and 2). These DCs migrate into the lymphatic vessels and are activated, leading to the presentation of tumor antigen by MHCs on their surface (2). B. The synthesis of CCL21 by LECs and high endothelial venules (HEVs) facilitates the migration of DC, LB and LT in the lymphatic vessels (3). These immune cells express the CCL21 receptor CCR7. C. Tumor antigen presenting DCs activate lymphocytes in the lymph nodes (4). Once activated, these lymphocytes undergo expansion and accumulation (5). Upon the action of VEGFC, lymphatic density and flow increase, allowing for better infiltration of antitumor lymphocytes in the lymph nodes. These lymphocytes then migrate through the lymphatic vessels towards the tumor (6) and induce a specific antitumor response (7).
Figure 4.
Beneficial roles of tumor lymphangiogenesis: recruitment and activation of the immune system. A. At inflammatory sites, antigens released by the tumor are captured by dendritic cells (DCs) and presented on their surface (1 and 2). These DCs migrate into the lymphatic vessels and are activated, leading to the presentation of tumor antigen by MHCs on their surface (2). B. The synthesis of CCL21 by LECs and high endothelial venules (HEVs) facilitates the migration of DC, LB and LT in the lymphatic vessels (3). These immune cells express the CCL21 receptor CCR7. C. Tumor antigen presenting DCs activate lymphocytes in the lymph nodes (4). Once activated, these lymphocytes undergo expansion and accumulation (5). Upon the action of VEGFC, lymphatic density and flow increase, allowing for better infiltration of antitumor lymphocytes in the lymph nodes. These lymphocytes then migrate through the lymphatic vessels towards the tumor (6) and induce a specific antitumor response (7).
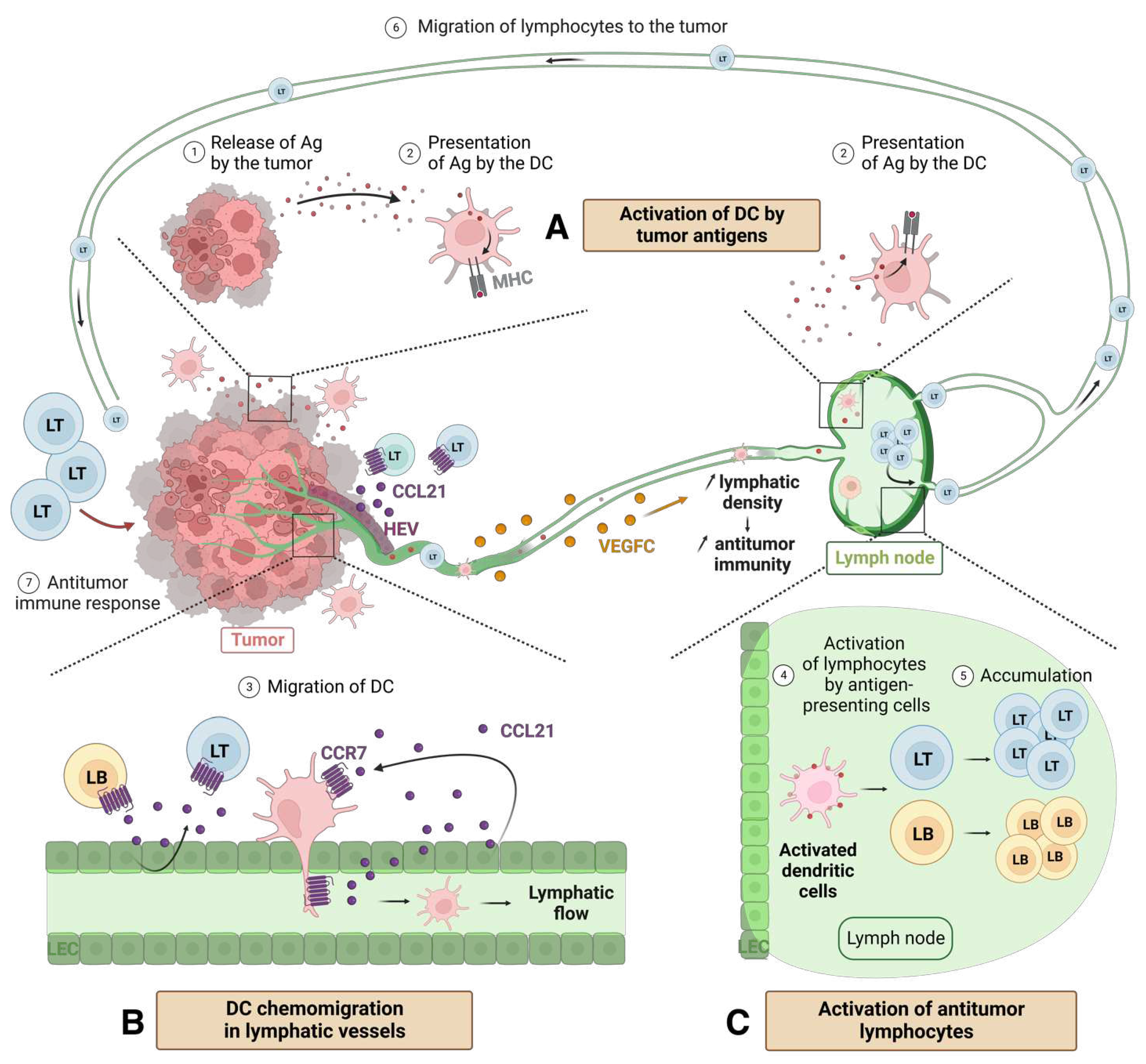
Figure 5.
Control of angiogenesis by pro- and anti-angiogenic factors. A. Physiological angiogenesis: balance between pro-angiogenic factors (VEGFA, VEGFB, HIF, FGF, low concentration TGFb, MMP, PDGF, IL-8, Ang1 and 2) and anti-angiogenic factors (derived from hyaluronic acid , angiostatin, INF-g, thrombospondin (TSP), high concentration TGFb and Ang1 and 2). B. Activation of angiogenesis by overexpression of pro-angiogenic factors (++) and repression of anti-angiogenic factors (-): case of embryogenesis, healing, or cancers. Created with BioRender.com.
Figure 5.
Control of angiogenesis by pro- and anti-angiogenic factors. A. Physiological angiogenesis: balance between pro-angiogenic factors (VEGFA, VEGFB, HIF, FGF, low concentration TGFb, MMP, PDGF, IL-8, Ang1 and 2) and anti-angiogenic factors (derived from hyaluronic acid , angiostatin, INF-g, thrombospondin (TSP), high concentration TGFb and Ang1 and 2). B. Activation of angiogenesis by overexpression of pro-angiogenic factors (++) and repression of anti-angiogenic factors (-): case of embryogenesis, healing, or cancers. Created with BioRender.com.
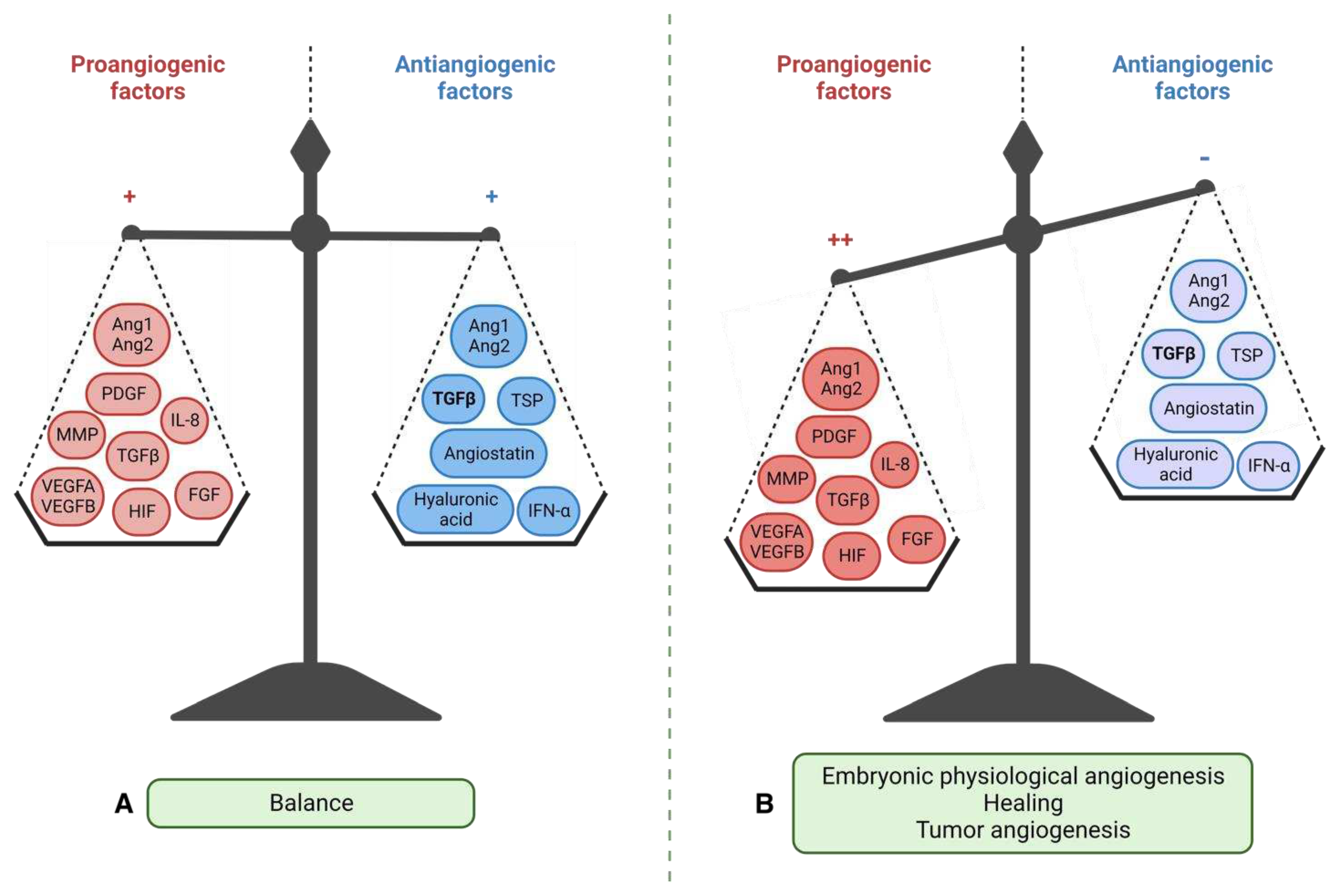
Figure 6.
Disorganization of the tumor vascular network compared to the normal vasculature. A. Normal blood vessel with contiguous endothelial cells, a continuous basement membrane, and a layer of smooth muscle cells and pericytes. The circulation of immune cells and macromolecules is normal. B. Tumor blood vessel showing many branches, irregular vessels, interrupted endothelial cells (EC) and basement membrane, few pericytes and smooth muscle cells, causing limited supply of chemotherapy molecules (CT) and antitumor immune cells but favoring the spread of tumor cells. Created with BioRender.com.
Figure 6.
Disorganization of the tumor vascular network compared to the normal vasculature. A. Normal blood vessel with contiguous endothelial cells, a continuous basement membrane, and a layer of smooth muscle cells and pericytes. The circulation of immune cells and macromolecules is normal. B. Tumor blood vessel showing many branches, irregular vessels, interrupted endothelial cells (EC) and basement membrane, few pericytes and smooth muscle cells, causing limited supply of chemotherapy molecules (CT) and antitumor immune cells but favoring the spread of tumor cells. Created with BioRender.com.
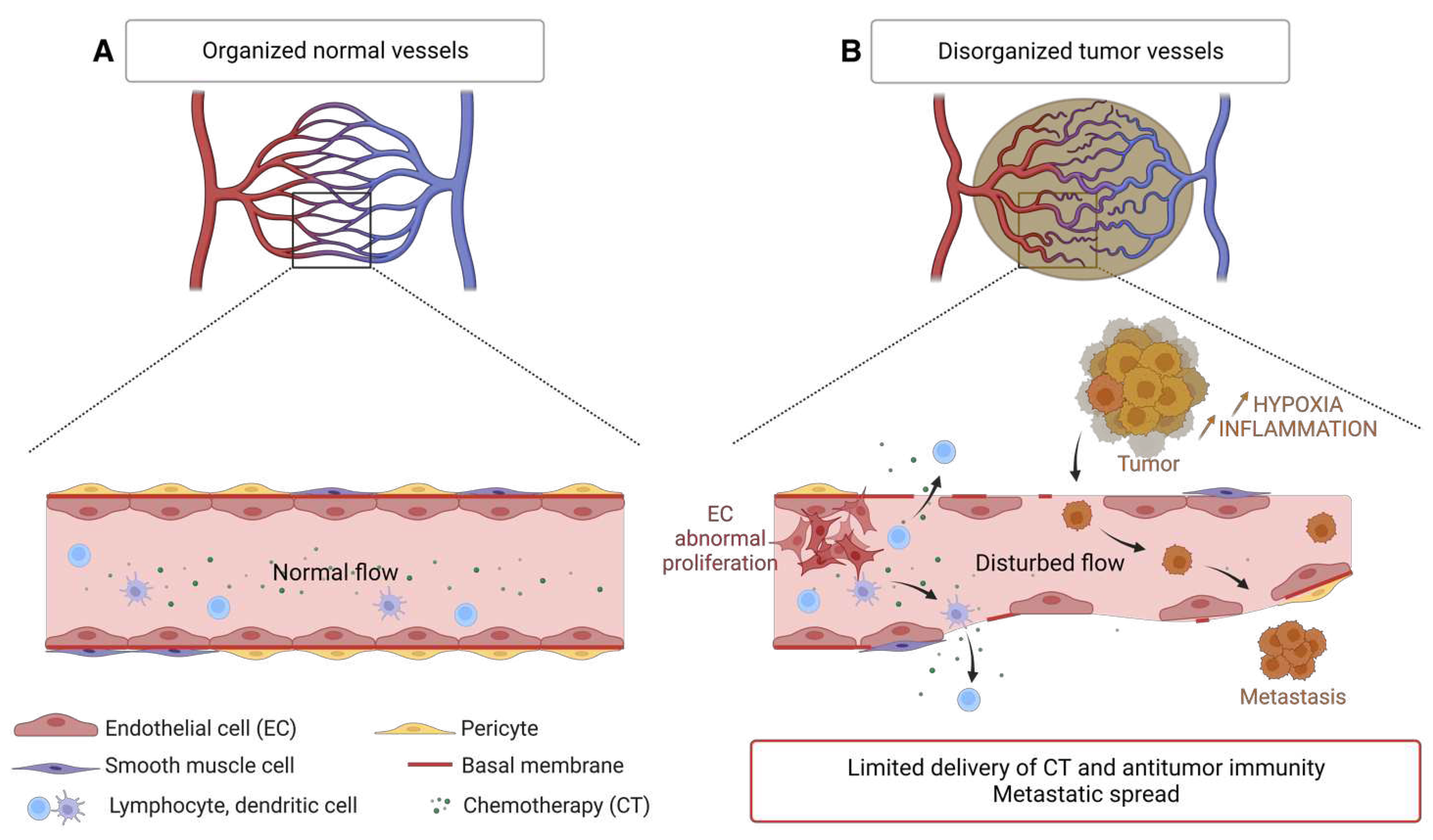
Figure 7.
The mechanisms of the angiogenic switch. Illustration of some mechanisms hijacked by tumors to increase their angiogenesis. Release of pro-angiogenic factors (VEGFA, PDGF) and suppression of anti-angiogenic factors (TSP-1) under the influence of genomic instability of tumors (activating RAS mutation or inhibiting TP53) and hypoxia. Recruitment of immune cells from the bone marrow to tumor sites and secretion of pro-angiogenic factors (VEGFA, MMP) under hypoxia. Induction of hypoxia by abnormal tumor vessels and decrease in energy metabolism. Development of angiogenesis by accumulation of succinate and binding to its GPR91 receptor. TAM = Tumor Associated Macrophages, Neutro = Neutrophils, Masto = Mast Cells, MDSC = Myeloid Derived Suppressive Cells, MMP = Metalloproteinases, SMC = Smooth Muscle Cells. Created with BioRender.com.
Figure 7.
The mechanisms of the angiogenic switch. Illustration of some mechanisms hijacked by tumors to increase their angiogenesis. Release of pro-angiogenic factors (VEGFA, PDGF) and suppression of anti-angiogenic factors (TSP-1) under the influence of genomic instability of tumors (activating RAS mutation or inhibiting TP53) and hypoxia. Recruitment of immune cells from the bone marrow to tumor sites and secretion of pro-angiogenic factors (VEGFA, MMP) under hypoxia. Induction of hypoxia by abnormal tumor vessels and decrease in energy metabolism. Development of angiogenesis by accumulation of succinate and binding to its GPR91 receptor. TAM = Tumor Associated Macrophages, Neutro = Neutrophils, Masto = Mast Cells, MDSC = Myeloid Derived Suppressive Cells, MMP = Metalloproteinases, SMC = Smooth Muscle Cells. Created with BioRender.com.
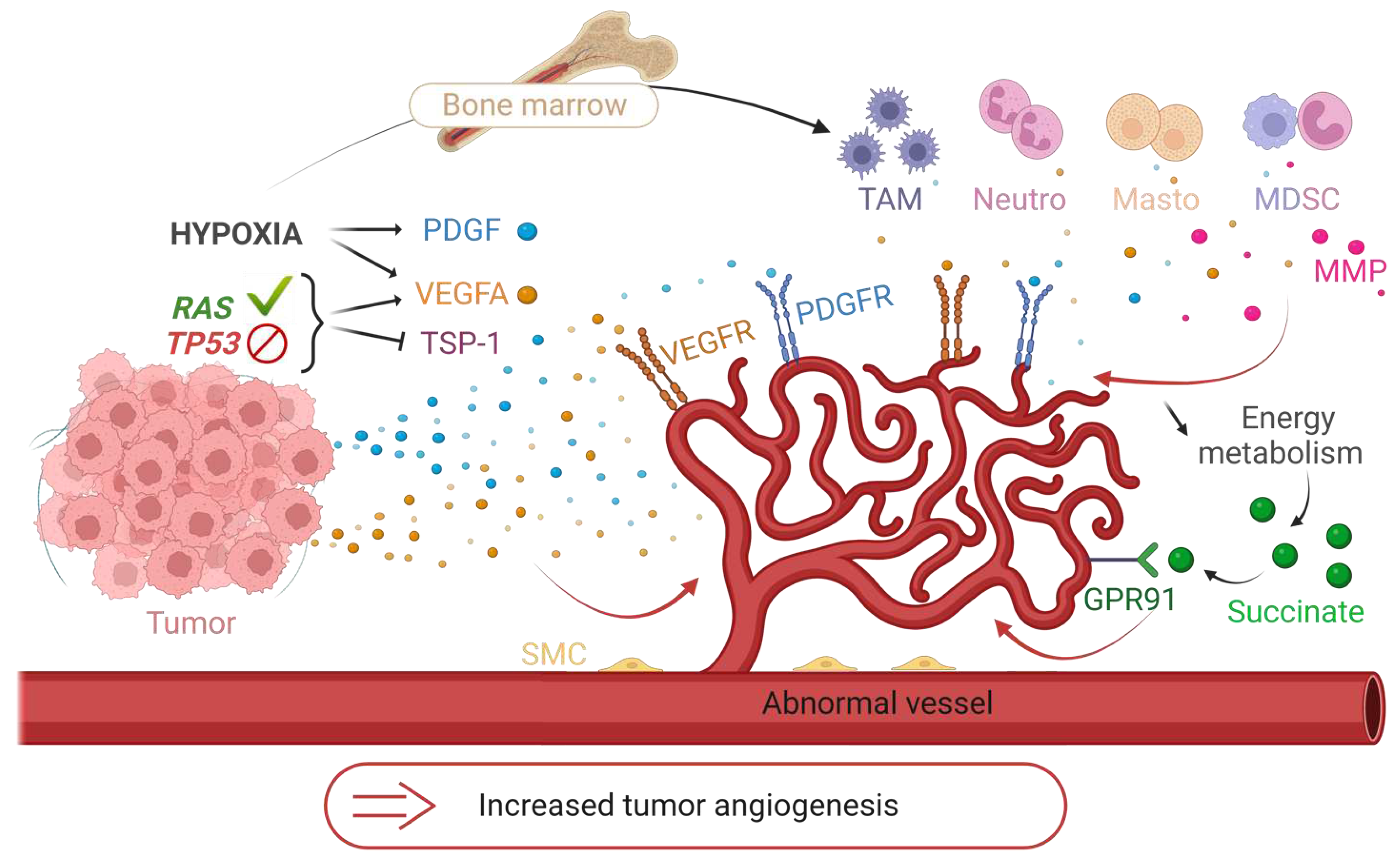
Figure 8.
Expression of multiple tumor pro-angiogenic factors responsible for evasion of anti-VEGFA/VEGFR treatments. Illustration of the expression of pro-angiogenic factors that compensate VEGFA by the tumor, thus explaining the limitation of conventional therapies (such as bevacizumab or sunitinib) targeting the VEGFA/VEGFR axis. These redundant factors are the other members of the VEGF family, FGFs, the cytokine CXCL8, angiopoietins (Ang). Created with BioRender.com.
Figure 8.
Expression of multiple tumor pro-angiogenic factors responsible for evasion of anti-VEGFA/VEGFR treatments. Illustration of the expression of pro-angiogenic factors that compensate VEGFA by the tumor, thus explaining the limitation of conventional therapies (such as bevacizumab or sunitinib) targeting the VEGFA/VEGFR axis. These redundant factors are the other members of the VEGF family, FGFs, the cytokine CXCL8, angiopoietins (Ang). Created with BioRender.com.
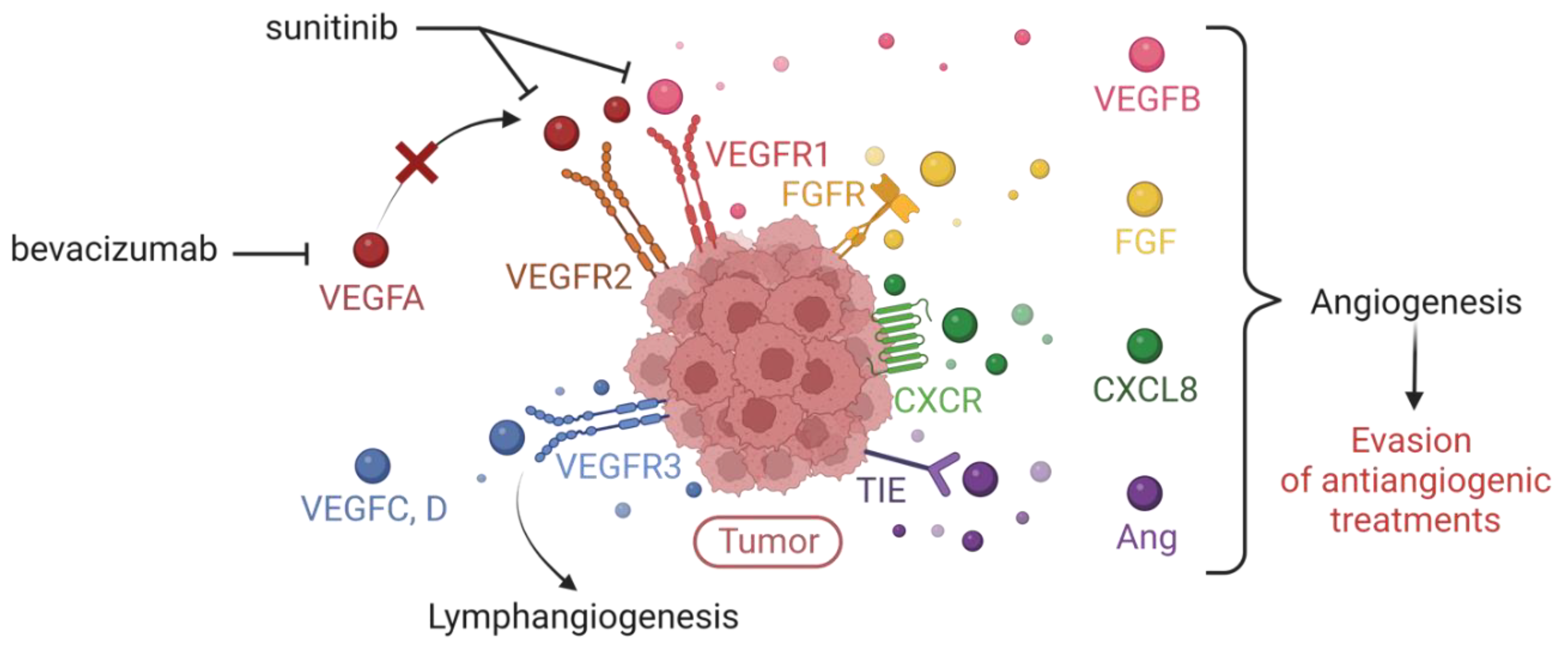

Table 1.
Molecular and clinical features of medulloblastoma subgroups. Data retrieved from [9,12,19,20,21,22]
| Subgroup | Subtype | Frequency | Demography | Main genetic events | Metastasis rate | 5-year overall survival |
|---|---|---|---|---|---|---|
| WNT | WNTa | 70% | Infants - adolescents |
CTNNB1, TP53, DDX3X, MLL2/3 mutation Monosomy chromosome 6 |
8.6% | 97% |
| WNTb | 30% | Children – young adults | 21.4% | 100% | ||
| SHH | SHHa | 29% | Children - adolescents | Loss of 9q, 10q, 17p MYCN, GLI2, YAP1 amp; TP53 mutation |
20% | 69.8% |
| SHHb | 16% | Infants | PTEN loss | 33% | 67.3% | |
| SHHg | 31% | Infants | Low copy number alterations | 8.9% | 88% | |
| SHHd | 24% | Young adults | TERT promoter mutation | 9.4% | 88.5% | |
| Group 3 | Group 3a | 47% | Infants - children | i17q; loss of 8q and 17p | 43.4% | 66.2% |
| Group 3b | 26% | Children - adolescents | OTX2 gain and DDX31 loss; activation of GFI1 and GFI1B oncogenes | 20% | 55.8% | |
| Group 3g | 28% | Infants - children | i17q; 8q gain and MYC amplification | 39.4% | 41.9% | |
| Group 4 | Group 4a | 30% | Children - adolescents | i17q; loss of 8p; 7q gain; MYCN and CDK6 amplification | 40% | 66.8% |
| Group 4b | 33% | Children - adolescents | i17q; 17p loss; SNCAIP duplication | 40.7% | 75.4% | |
| Group 4d | 37% | Children - adolescents | i17q; loss of 8p; 7q gain; CDK6 amplification | 38.7% | 82.5% |
Table 2.
Clinical trials referencing new treatments under study against pediatric MB.
| Reference | Title | Phase | Enrollment | Intervention/treatment type |
|---|---|---|---|---|
| NCT00602667 | Risk-Adapted Therapy for Young Children with Embryonal Brain Tumors, Choroid Plexus Carcinoma, High Grade Glioma or Ependymoma | 2 | 293 | Drug: Induction Chemotherapy Drug: Low-Risk Therapy Drug: High-Risk Therapy Drug: Intermediate-Risk Therapy |
| NCT01878617 | A Clinical and Molecular Risk-Directed Therapy for Newly Diagnosed Medulloblastoma | 2 | 660 | Radiation: Craniospinal Irradiation with boost to the primary tumor site Drug: Cyclophosphamide Drug: Cisplatin Drug: Vincristine Drug: Vismodegib Drug: Pemetrexed Drug: Gemcitabine Other: Aerobic Training Other: Neurocognitive Remediation |
| NCT02017964 | Combination Chemotherapy in Treating Younger Patients With Newly Diagnosed, Non-metastatic Desmoplastic Medulloblastoma |
2 | 26 | Drug: Carboplatin Other: Cognitive Assessment Drug: Cyclophosphamide Drug: Etoposide Other: Laboratory Biomarker Analysis Drug: Methotrexate Drug: Vincristine Sulfate |
| NCT02066220 | International Society of Paediatric Oncology (SIOP) PNET 5 Medulloblastoma |
2; 3 | 360 | Radiation: Radiotherapy without Carboplatin Drug: Reduced-intensity maintenance chemotherapy Radiation: Radiotherapy with Carboplatin Drug: Maintenance chemotherapy Radiation: WNT-HR < 16 years Radiation: WNT-HR >= 16 years Drug: Induction Chemotherapy Radiation: SHH-TP53 M0 Radiation: SHH-TP53 M+ (germline) Radiation: SHH-TP53 (somatic) Drug: Vinblastin Maintenance |
| NCT02238899 | Multicenter Register for Children and Young Adults With Intracranial Localized Medulloblastoma, CNS-PNET or Ependymoma | 354 | ||
| NCT02255461 | Palbociclib Isethionate in Treating Younger Patients With Recurrent, Progressive, or Refractory Central Nervous System Tumors | 1 | 35 | Drug: palbociclib isethionate Other: pharmacological study Other: laboratory biomarker analysis |
| NCT02271711 | Expanded Natural Killer Cell Infusion in Treating Younger Patients With Recurrent/Refractory Brain Tumors | 1 | 12 | Other: Laboratory Biomarker Analysis Biological: Natural Killer Cell Therapy |
| NCT02359565 | Pembrolizumab in Treating Younger Patients With Recurrent, Progressive, or Refractory High-Grade Gliomas, Diffuse Intrinsic Pontine Gliomas, Hypermutated Brain Tumors, Ependymoma or Medulloblastoma |
1 | 110 | Procedure: Diffusion Tensor Imaging Procedure: Diffusion Weighted Imaging Procedure: Dynamic Contrast-Enhanced Magnetic Resonance Imaging Procedure: Dynamic Susceptibility Contrast-Enhanced Magnetic Resonance Imaging Other: Laboratory Biomarker Analysis Procedure: Magnetic Resonance Spectroscopic Imaging Biological: Pembrolizumab Procedure: Perfusion Magnetic Resonance Imaging |
| NCT02724579 | Reduced Craniospinal Radiation Therapy and Chemotherapy in Treating Younger Patients With Newly Diagnosed WNT-Driven Medulloblastoma | 2 | 45 | Drug: Cisplatin Drug: Cyclophosphamide Other: Laboratory Biomarker Analysis Drug: Lomustine Radiation: Radiation Therapy Drug: Vincristine Drug: Vincristine Sulfate |
| NCT03130959 | A Study to Evaluate the Safety and Efficacy of Nivolumab Monotherapy and Nivolumab in Combination With Ipilimumab in Pediatric Participants With High Grade Primary Central Nervous System (CNS) Malignancies (CheckMate 908) | 2 | 166 | Biological: Nivolumab Biological: Ipilimumab |
| NCT03500991 | HER2-specific CAR T Cell Locoregional Immunotherapy for HER2-positive Recurrent/Refractory Pediatric CNS Tumors | 1 | 48 | Biological: HER2-specific chimeric antigen receptor (CAR) T cell |
| NCT04023669 | Evaluation of LY2606368 Therapy in Combination With Cyclophosphamide or Gemcitabine for Children and Adolescents With Refractory or Recurrent Group 3/Group 4 or SHH Medulloblastoma Brain Tumors | 1 | 21 | Drug: Prexasertib Drug: Cyclophosphamide Drug: Gemcitabine Biological: filgrastim Biological: peg-filgrastim |
| NCT04743661 | 131I-Omburtamab, in Recurrent Medulloblastoma and Ependymoma | 2 | 62 | Drug: Irinotecan Drug: Temozolomide Drug: Bevacizumab Drug: Omburtamab I-131 Drug: Liothyronine Drug: SSKI Drug: Dexamethasone Drug: Antipyretic Drug: Antihistamine Drug: anti-emetics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



