Submitted:
16 June 2023
Posted:
19 June 2023
You are already at the latest version
Abstract
Cyclic vomiting syndrome (CVS) is a frequently disabling condition defined by severe, discrete, stereotypical episodes of nausea and vomiting. In our previous report [1], a candidate gene list was evaluated in a group of 80 unrelated participants with CVS with extensive genetic sequencing (whole exome/genome). Twenty-two good candidate genes for CVS association were identified, from which a cellular model of aberrant cation transport and energy metabolism was proposed. Herein, we present additional clinical information on those 80 participants, including eight case reports to highlight general principles and clinical practices. Our participants demonstrate substantial phenotypic and genotypic heterogeneity in CVS, including a propensity for specific co-morbidities in the participants and relatives, evolution towards episodes in which vomiting is less apparent, labeling with “non-organic” diagnoses, multiple genes, polygenic inheritance, and association with variants not designated as Pathogenic. Our case reports illustrate how genetic information can guide clinical management and argue to the high clinical utility of appropriate genetic testing and analysis in CVS. Expression profiling of our 22 candidate genes suggests an anatomical model in a unit defined by vagal afferents and adjacent cells. Our models are consistent with multiple current hypotheses of CVS and the efficacy of commonly-recommended treatments.
Keywords:
case reports
; clinical management
; CVS
; disease mechanisms
; DNA sequencing
; gene expression
; genetic testing
; genetics
; personalized medicine
; vagal afferents
1. Introduction
Cyclic vomiting syndrome (CVS) is defined clinically by Rome IV diagnostic criteria [2] as “two or more periods of unremitting paroxysmal vomiting with or without retching, lasting hours to days within a 6-month period, episodes are stereotypical in each patient, (and) episodes are separated by weeks to months with return to baseline health between episodes of vomiting.” This condition is common, occurring in up to 2% of school-aged children [3]. CVS occurs in all age groups [4,5] and is oftentimes highly disabling. Continued episodes often lead to multiple emergency department or hospital admissions due to intense discomfort and/or dehydration. While treatable in many cases, CVS remains under-recognized by clinicians and many patients suffer from ongoing vomiting episodes for years. Often considered as a migraine variant [6], CVS has also been linked to dysautonomia [7], stress responses [8], energy (mitochondrial) metabolism [9], ion channels [10], neurotransmitters receptors [11], and neuropathy [12,13].
While CVS is essentially an idiopathic condition, there is a strong genetic component as specific co-morbidities are quite common among the first-degree relatives, especially chronic pain, fatigue, and other gastrointestinal and dysautonomic disorders [9,14]. A powerful modern way to identify the genetic components of any idiopathic condition is DNA sequencing. Until recent years, technology and economics limited such sequencing to specific genes based on hypotheses regarding disease pathophysiology. In practice, most studies [15,16] focused on the mitochondrial DNA (mtDNA), a 16-kilobase genome versus the 3-gigabase nuclear genome (chromosomes). However, technology improved dramatically in recent years, allowing for whole exome sequencing (WES: all ~23K genes), or even better, whole genome sequencing (WGS: essentially all the DNA including the vast non-protein coding regions within and between genes).
The current authors recently previously presented a retrospective chart review in 80 unrelated participants meeting the Rome IV criteria for CVS, 75 of which were tested by WES or WGS [1]. Using two essentially invariant features of CVS, its paroxysmal nature and the substantial degree of both discomfort (always nausea, but also pain: abdominal, headache, myalgia) and disability (lethargy, weakness, fatigue) during episodes, 35 genes were identified. Raw sequence data among these genes was queried for variants, especially those particularly apt to be associated with disease. Among the 35 genes, 12 genes were scored as “Highly likely” or “Likely” CVS related. Nine additional genes had sufficient evidence in a literature review for association with CVS, but not from our study participants. Adding the mtDNA (counted as a gene) provided 22 genes as candidates for CVS. All 22 CVS candidate genes are associated with either cation transport or energy metabolism. We proposed a cellular model for CVS in which aberrant ion gradients lead to mitochondrial dysfunction, or vice versa, in a pathogenic vicious cycle of cellular hyperexcitability.
In this paper, we present additional clinical information from those 80 participants [1], including case reports in eight to highlight general principles and clinical practices. We show how our cation-energy model explains the multiple different treatment strategies that have been advocated for CVS, and how it predicts additional therapies not yet reported. We also demonstrate how gene expression among the candidate genes leads the authors to hypothesize an anatomical model for CVS, the vagal afferent unit, which is consistent with previous publications. Altogether, this paper seeks to translate scientific data and understandings about CVS into clinical practice.
2. Materials and Methods
The 80 present participants are the same as were described in Bar et al (2023) [1]. All were evaluated on a clinical basis by the first author and had prior genetic sequencing, including WGS in 35, WES in 40, and panel testing (1,000+ genes, nucSEEK®, Courtagen) in 5 participants. This study was approved by the Advarra IRB (human subjects committee, cirbi@advarra.com) as a retrospective chart review.
As described in Bar et al (2023) [1], the authors presented a list of 35 candidate genes (the paroxysmal gene list) by querying the literature for genes associated with dominant cases of intermittent vomiting or both discomfort and disability; among which the raw genetic sequence was reviewed. “Qualifying” variants were defined as coding, rare, and conserved, as detailed in Bar et al (2023) [1]. Additionally, “Key Qualifying” variants were “Pathogenic/Likely Pathogenic” (e.g., per ClinVar [17]), or “Clinical” based upon the presence of a corresponding diagnosis. Candidate association to CVS (including Highly likely, Likely, Possibly, or No evidence) was based on a point system that considered variants occurring in our participants and cases of paroxysmal vomiting in the literature.
3. Results
3.1. Clinical Manifestations of Our 80 Participants
Clinical parameters of our participant group are listed in Table 1 and Table 2, revealing significant comorbidities inherent in this condition [9]. More detailed information is available in Supplementary Table 1. Among our 80 participants, 46 (58%) were female, and 61 (76%) were of Western Eurasian ancestry (Table 1), while another 10 participants had one parent of Western Eurasian ancestry.
3.2. Our 22 Candidate Genes
3.3. Case Reports
Case reports in 8 of our 80 unrelated CVS participants follow. These eight cases were chosen from among the larger group to highlight various issues of perceived clinical importance. “Pathogenic” with an upper-case P refers to variants that are designated as such by ClinVar or by virtue of the nature of the variant (e.g., frameshift, see [1] for caveats). Such variants are predicted with high certainty to affect protein function. De novo variants were only labeled as “Pathogenic” if they are Key variants (see Methods and [1]). Written informed consent to publish case reports was obtained in all cases. The participant names herein are either the true name or an alias, at the discretion of each patient.
3.3.1. Case 1: Ryan
Ryan (participant #63 in Supplementary Table S1) presented at age 6 years with 5-day episodes whereas the chief complaint was dizziness, also including vomiting, headache, and ketosis. Each episode required 3 days in the hospital for intravenous hydration. Episodes occurred like clockwork every 90 days, with a range of 88 to 92 days, for over 10 years. Ryan failed combined treatment with coenzyme Q10, carnitine, amitriptyline, and propranolol, with target blood levels of the first three [18]. Issues between episodes included mild-moderate cognitive impairment, chronic fatigue, frequent urination, gastroesophageal reflux disease (GERD), eosinophilic esophagitis, constipation, choking episodes, strabismus, and obstructive sleep apnea. He sometimes is unable to move his leg “as if it didn’t belong to him", but no frank incidences of periodic paralysis. Despite the vomiting episodes, the rapid-onset of apparently-spontaneous, highly-aggressive behaviors were more problematic for the family and led to occasional law enforcement intervention and a home institutional placement.
Genetic testing at age 18 years revealed an uncommon, highly-conserved variant of interest in SCN4A (c.2717G>C, p.Ser906Thr) encoding the alpha subunit of the voltage-gated type IV sodium channel. This gene is predominantly expressed in skeletal muscle and is associated with a variety of neuromuscular conditions including periodic paralysis, myasthenia, myotonia congenita, and paramyotonia. Based on this finding, acetazolamide (125 mg BID) and an informal high potassium diet were started, while maintaining the above-listed treatments. Cyclic vomiting episodes resolved within 2 months, and Ryan remains in remission from vomiting episodes over 5 years later. At the same time, aggressive behaviors became rare and no longer spontaneous.
Despite the SCN4A variant being not rare (1.0% allelic frequency, thus present in ~2% of the population), this variant is highly likely to alter channel function. In the presence of p.Ile692Met, a known pathogenic variant in SCN4A, the concomitant presence of p.Ser906Thr “results in longer weakness episodes, more affected muscles, (creatine kinase) elevation, and presence of permanent weakness” [19]. The p.Ile692Met variant is absent in Ryan. The p.Ser906Thr variant is located within the loop joining the second and the third transmembrane domains of the sodium channel, a region important for slow channel inactivation and previously implicated for the paradoxical entities of epilepsy and long QT syndrome [20]. Additionally, p.Ser906Thr likely imparts a “gain-of-function mechanism where muscle tissues may become hyperexcitable due to higher number of channels available to open in the early phase of action potentials” [21]. Of particular interest, the p.Ser906Thr variant is much less common in the “control/biobanks” gnomAD [22] versus the full gnomAD sequence database (917/118,417 versus 2,713/277,801 alleles, P < 0.0001, odds ratio 1.26, 95% C.I. 1.17-1.40). This suggests that this relatively common variant is a significant risk factor for excluding people from the “normal” population in general.
Another variant of interest was identified on genetic testing in Ryan (c.2116C>T, p.Arg706Cys), which is rare and moderately conserved in the MARS gene. This gene encodes for methionyl-tRNA synthetase, the enzyme that attaches methionine to its transfer RNA, and variants in this gene are associated with autosomal dominant, Charcot-Marie-Tooth (CMT)-type, peripheral neuropathy. Of note, five other CMT-associated genes were determined to be Possibly related to CVS in Bar et al (2023) [1].
3.3.2. Case 2: Spencer
Spencer (#72) was noted to have abnormal visual behavior at age 10 years and was diagnosed with chronic progressive external ophthalmoplegia (CPEO) and pigmentary retinopathy. As this phenotype is generally mitochondrial in etiology, mtDNA testing was performed revealing a large heteroplasmic deletion, and the diagnosis of mitochondrial disease was given. He was initially seen by the first author at age 11 years, at which time additional diagnoses noted included growth retardation, paroxysmal abdominal pain, chronic fatigue/exercise intolerance, high-frequency hearing loss, and high-functioning autism. Despite the latter diagnosis, Spencer was in regular classes for his age and doing well. Cardiac examination and an electrocardiogram were unremarkable. Despite the mtDNA variant being absent in the mother, paroxysmal symptomatology was prevalent in her family, including migraine, dizziness, myalgia, and supraventricular tachycardia, present in the mother, an adolescent sister, a maternal aunt, and/or the maternal grandmother.
At age 12 years, episodic, severe back and shoulder pain with allodynia developed, as well as migraine headache and occasional vertigo. Most troublesome were additional paroxysmal events termed “crashes” occurring 3 to 6 per month starting at age 13 years. Crashes were often triggered by environmental heat or sometimes cold, with duration ranging from 1 hour to 2 days (mean 12 hours), and usually resolved with sleep. Crashes consisted of profound fatigue, both mental and physical, during which Spencer was unable to perform any homework, and in one episode was unable to walk. They were stereotypical, well demarcated, and fatigue was only mild between crashes. He was diagnosed with a “conversion disorder” by other providers. However, crashes resolved on reestablishment of mitochondrial-targeted supplements, and have not reappeared since.
Genetic testing confirmed the large heteroplasmic mtDNA deletion (7.7 kb, m.6468-14148, heteroplasmic proportion not determined) as well as identifying a heterozygous variant (c.4003T>A, p.Thr1335Ser (hg19)) in CACNA1S, encoding a slowly-inactivating, voltage-dependent, calcium-channel (L-type, alpha-1S subunit) predominantly expressed in skeletal muscle. Variants in this gene have been reported in paroxysmal conditions including periodic paralysis and malignant hyperthermia. The variant found in Spencer is uncommon and moderately conserved. It is located in the IVS5-S6 extracellular poor-loop region of the encoded calcium channel, whereas it produces significantly faster activation of L-type calcium currents upon depolarization and shows a tendency to increase sensitivity for caffeine-induced calcium release [23]. That same reference reported p.Thr1335Ser to be associated with malignant hyperthermia in a three-generation family.
Episodes of cyclic vomiting syndrome appeared at age 15 years, and Spencer had had four episodes when he presented for follow-up with the first author; he was started on acetazolamide. Vomiting episodes coalesced into a month-long episode at age 16, and then resolved. Spencer has had only a single episode of CVS in the last four years. Pain in the back/shoulders and headaches remain as chronic issues. His therapeutic regiment includes multiple individual mitochondrial-targeted supplements, cannabidiol, amitriptyline, duloxetine, celecoxib, and PRN stress dosing of hydrocortisone for adrenal insufficiency.
3.3.3. Case 3: Lily
Lily (#49) started at age 22 years to have 5 to 6-day episodes, occurring about once a month, not clockwork-like, with triggers including anxiety, stress, and motion sickness. About 50% of the time, these episodes precipitated an emergency department visit due to severe lower chest pain in the area of the diaphragm, requiring dosing with hydromorphone (an opioid narcotic). Episodes also included vomiting, abdominal pain, fatigue, lethargy, and dizziness. Issues between episodes included lower back pain, constipation, insomnia, anxiety, depression, anorexia nervosa, tinnitus, attention deficit hyperactivity disorder (ADHD), and cystic fibrosis. Medical therapies included daily aprepitant, nortriptyline (75 mg qhs), and venlafaxine (225 mg). Family history includes sisters with seizure disorder, learning disability, ADHD, bipolar disorder, depression, anxiety/panic disorders, and/or psychiatric hospitalization.
Genetic testing at age 23 years revealed a rare (0.1%) and extremely-highly conserved variant of interest in SCN4A (c.1430A>G, p.Lys477Arg). Based on the presence of SCN4A variant, a high potassium diet was started, which was increased to prescription-strength potassium chloride 20 mEq TID. On this therapy, CVS episodes became far less frequent (from monthly to once in 6 months) and lessened greatly in intensity without need for narcotics.
A second variant of interest was identified in TRPA1 (c.412G>T, p.Ala138Ser), a non-specific cation transporter and one of the 22 genes associated with CVS. Additionally, an off-target, homozygous Pathogenic variant was identified in CFTR (p.Phe508del, common mutation), thus confirming Lily’s diagnosis of cystic fibrosis.
3.3.4. Case 4: Liliana
Liliana (#47) started to have stereotypical episodes in early infancy. When initially evaluated by the first author at age six years, episodes were monthly, lasting 4-5 days each, and included nausea/anorexia, abdominal pain, headache, dizziness, lethargy, edema, and urticaria. The last two are atypical of CVS yet typical of mast cell activation syndrome (MCAS), for which she was diagnosed based on reactions (edema, urticaria) to multiple foods, additives, and dyes. About 25% of her monthly episodes were more severe, and included blurred vision, ataxia, muscle weakness (symmetrical), leg pain, paresthesia, loss of vision (up to 3-4 minutes), photophobia, and phonophobia. Note that these lists do not include vomiting, and the diagnosis of CVS was not recognized previously. However, vomiting was a significant part of her monthly episodes in years past and was present at age six in about 10% of episodes. Laboratory findings during episodes included metabolic acidosis, ketosis, and hypoglycemia. Clinical manifestations between episodes included migraine headache with aura, leg pain (daily, can last for hours), chronic fatigue with exertion, environmental heat and cold intolerance, hyperhidrosis, urinary hesitancy and frequency, MCAS, and failure-to-thrive from anorexia during episodes. Family history is remarkable for symptoms of POTS, MCAS, and hEDS in the mother, and eosinophilic esophagitis in the father.
Frequent episodes and chronic pain led to extensive school absences and multiple hospitalizations. Liliana had been evaluated by many specialists in multiple academic centers, yet no underlying cause of her episodes was made (except for MCAS, which did not explain many of her manifestations), and legal action was in progress from an allegation of Munchausen-by-proxy (medical child abuse).
Urine organic acids revealed elevated ketones, Krebs cycle intermediates, dicarboxylic acids, and other markers of mitochondrial dysfunction. Additionally, mitochondrial dysfunction was revealed by buccal cell enzymology (MitoSwab Plus, Religen®, Plymouth Meeting, PA) demonstrating deficiency of respiratory complexes I (19% of control enzyme activity) and II (< 2%) with elevated complex IV (439%).
Trio (patient and both parents) WES and mtDNA sequencing was performed, revealing a rare, moderately-conserved, heterozygous missense variant in the SCN10A gene (c.41G>T, p.Arg14Leu), one of our 22 CVS-associated genes. Variants in SCN10A are reported in amplified pain syndromes, and thus this variant is a good candidate for polygenic disease risk in the patient. Additional variants of potential disease association included a common variant in RYR2 (c.5654G>A, p.Gly1885Glu) also one of the 22 CVS genes (see Case #6).
Based on the laboratory results, treatment included a broad-spectrum mitochondrial-targeted supplement with additional coenzyme Q10 in the ubiquinol form (collectively termed mitochondrial cocktail - “mito-cocktail”), without any medications used for CVS treatment. Associated with this intervention, episodes, leg pain, and fatigue all resolved. Interestingly, MCAS-related symptoms also essentially resolved, and the patient is now able to tolerate a wide variety of foods. Liliana gained weight, which became appropriate for height.
Now that her CVS is in remission, Liliana’s current chief complaint is temperature (heat and cold) intolerance as this triggers her migraine headaches. In response to cold, her skin becomes erythematous. The issue is significant enough that she puts insulation pads on her hands before taking anything out of the refrigerator. Of interest, WES also revealed PLCG2 c.3524T>C, p.Ile1175Thr. This gene encodes phospholipase Cγ2, an enzyme responsible for ligand-mediated signaling in cells of the hematopoietic system and plays a key role in the regulation of immune responses. Variants in this gene have been found in autoinflammation, antibody deficiency, immune dysregulation syndrome, and familial cold autoinflammatory syndrome [24]. This is of potential interest because two other genes associated with auto-inflammatory conditions, MEFV and TNFRSF1A, were associated with CVS in Bar et al (2023) [1] and Table 3. Thus, the variant in this gene may be a contributing factor in her CVS.
3.3.5. Case 5: Sydney
Sydney (#74) presented with stereotypical episodes of which the most prominent aspect was severe dizziness (sometimes with vertigo) following E. coli enteritis at age 16 years. Other manifestations of episodes included paresthesia, anesthesia, nausea, palpitations, tinnitus, muscle weakness, and pallor. Most episodes would last under an hour, but some episodes would continue past an hour whereas vomiting and lethargy would appear, sometimes lasting for 2 days and including prominent vomiting. These prolonged episodes always responded rapidly to intravenous (IV) fluids, in particular vomiting stopped, yet symptoms often would return when IV fluids were discontinued. Episode frequency was highly variable, at their peak were 2 to 3 per day, but these were predominately all 30-60 minutes each. Prolonged episodes with vomiting peaked at 3-4 per month, each requiring emergency department visits for IV fluids, and never more than 3 to 4 weeks separated such episodes.
Dizziness, sometimes with vertigo and sometimes labeled as POTS, was chronic and continuously present, but at a severity level (2-3/10 subjective score) far below that occurring during episodes (10/10). There was no nausea or vomiting between episodes. Sydney also had dysphagia, hypotonia, and an abnormal gait. She was performing in school at 2 to 3 grades below grade level in academic subjects; social skills were commensurate with her academic abilities. She had a history of frequent common infections of childhood, including impetigo, otitis media, pharyngitis, and upper (and occasionally lower) respiratory infections, without episodes of life-threatening, sterile-site, or opportunistic infections. The only exception was intensive care for one bout of pneumonia, at which time she received a diagnosis of common variable immunodeficiency (CVID). The family history is remarkable for multiple disease manifestations including migraine, other varieties of chronic pain, chronic fatigue, and gastrointestinal dysmotility in her brother, mother, maternal aunt, and/or maternal grandmother.
Sydney was treated with hemp oil (contains cannabidiol), which was a legal over-the-counter supplement in the state in which she lived and known to her primary care physician, with some degree of relief. She was evaluated at a nearby tertiary-care facility, across a state line whereas this treatment was not legal at that time. A toxicology screen was positive for cannabinoids, and the family was investigated by legal authorities. Munchhausen-by-proxy was alleged due to the perceived lack of an underlying diagnosis to account for her myriad of symptomatology and degree of disability, the usage of medical resources believed to be beyond reasonable, and the alleged lack of an accurate and complete history from the parents (which appeared to focus on the absence of a disclosure of hemp oil therapy). Another concern was that Sydney often appeared to be in no apparent distress upon evaluation not long after arriving for medical care in an emergency department setting (after having been given IV fluids). Furthermore, was the insistence by the family that Sydney had a mitochondrial disorder, a diagnosis that had been given by an internationally-recognized expert, despite being told that this was not the case by another such expert and by the contemporary medical personnel. As a result of the investigation, Sydney was placed in foster care, and the parents were provided monitored, highly limited, and telephone-only contact with her. Following a trial, which included the testimony of the present first author as an expert witness regarding Sydney having CVS, and that the parental behaviors were typical of children with that diagnosis, the judge returned custody to the family.
An upper endoscopy revealed eosinophilic esophagitis. MitoSwab Plus demonstrated partial deficiencies of respiratory complexes I (27% of control) II+III (22%) and IV (42%) with elevated complex IV (439%). WES plus mtDNA sequencing revealed two heterozygous variants in the CACNA1A gene (c.6881G>C, p.Arg2294Pro and c.6893G>C, p.Arg2298Pro) corresponding to the C-terminal region of the protein. Both variants are predicted to very likely affect protein function based on being extremely rare, highly conserved, and by the SIFT algorithm (other algorithms did not issue predictions). The CACNA1A gene encodes for alpha-1A subunit of the voltage dependent P/Q-type calcium channel, of which variants predispose towards various forms of migraine, especially with muscle weakness.
In addition, mtDNA sequencing revealed a heteroplasmic variant (m.15740C>T, MT-CYB c.994C>T, p.Leu332Phe). This variant is very rare, but computer predictions of pathogenicity are equivocal and thus uninformative. The heteroplasmic proportions were estimated at 39% in Sydney, 20% in her affected brother, and < 20% (below detection) in their mother. This finding is consistent with the apparent maternal inheritance pattern in family history.
At age 20 years, on broad-spectrum mito-cocktail, dizziness, the previous chief complaint, and vomiting episodes have resolved completely, first with high potassium fluids, then on increasing dosage with prescription-strength potassium chloride.
Autism and learning disabilities were identified recently on neuropsychiatric testing. She graduated from high school, although cognitive functioning has been an issue in employment. She was found to have the same heterozygous Pathogenic variant (c.3448C>T, p.Arg1150*) in the TET3 gene as her brother (autism, epilepsy) and father (learning disabilities only). Disease-related variants in this gene cause Beck-Fahrner syndrome, which is associated with variable degrees of neurodevelopmental disease, including autism, hypotonia, and epilepsy. Dysmorphic features are sometimes noted in the literature yet are not apparent in the present family.
3.3.6. Case 6: Madeline
Madeline (#53) started at 9 years of age to have 6-8 episodes a year, meeting criteria for both CVS and abdominal migraine, decreasing to 0-3 per year after the family learned to abort episodes with ondansetron. In her early 20s, this evolved into severe daily migraine headaches with substantial nausea (no longer with vomiting), triggered by even low levels of stress. Additional comorbidities between episodes included daily severe muscle pain, chronic fatigue, myalgia, irritable bowel syndrome, GERD, and postural orthostatic tachycardia syndrome (POTS). Disease manifestations were severe enough that the patient was on complete disability. Family history was significant for multiple neurological and functional-like disease manifestations among the matrilineal relatives who share the same mtDNA sequence. However, on mtDNA sequencing none of the variants of uncertain significance identified appeared to be excellent candidates for disease association.
At age 26 years, WES revealed a Pathogenic nonsense (stop gain) variant in RYR2 (c.10377T>G, p.Tyr3459*). Propranolol (30 mg QID) was started, and the mito-cocktail was increased. This regimen was associated with reduced migraines somewhat and completely resolved her muscle pain. This enabled the patient to return to full-time work and live a fairly normal life, although moderate chronic fatigue remained and became the chief complaint.
Madeline became pregnant for the first time at age 29 years and developed hyperemesis gravidarum. Her obstetrician agreed to let the patient continue propranolol, mito-cocktail, and PRN ondansetron. Nausea improved substantially on intravenous fluids and L-carnitine. She gave birth at term to a healthy girl. Of interest, a whole-exome association study revealed that RYR2 poses genetic risk for hyperemesis gravidarum [25].
3.3.7. Case 7: Kate
A 36-year-old female (#18) presented to the first author with the chief complaint of longstanding chronic fatigue. Additional comorbidities present since early childhood included hypotonia, parasomnia, high-functioning autism, and learning disabilities. Previous stereotypical episodes occurred monthly, lasted seven days each, and consisted of nausea, vomiting, abdominal pain, lethargy, irritability, photophobia, and drooling. Episodes resulted in over 40 hospitalizations from ages 3 to 16 years, then resolved. As an adolescent, Kate was a participant in a CVS observational study conducted by the first author. At age 36 years, in addition to fatigue, ocular migraine (developed in college), dysautonomia, sensorineural hearing loss, chronic limb pain, and urinary frequency were present. She was not on any CVS or migraine-related treatments except for the ubiquinone form of coenzyme Q10. She has a diagnosis of hypermobile Ehlers-Danlos syndrome (hEDS). Despite these issues, she has a master’s degree and works part-time for Special Olympics.
Trio WES revealed a heterozygous de novo variant (c.1954C>T, p.Cys652Arg) in the POGZ gene. This variant is highly predicted to alter protein function as it is extremely rare in humans, very-highly conserved in vertebrates, and predicted as such by computer algorithms (PolyPhen2, SIFT, and MutationTaster). This variant is Pathogenic by virtue of being de novo and clinical correlation. POGZ encodes for pogo transposable element with zinc finger domain, a protein that binds HP1-alpha, which has an essential role in heterochromatin formation and mitotic progression [26]. De novo variants in the POGZ gene are causal for White-Sutton syndrome, which is an excellent match for Kate’s phenotype including cognitive (learning disabilities, autism, history of hypotonia, poor coordination, friendly demeanor), ocular (nearsightedness, astigmatism, left ptosis), gastrointestinal manifestations (cyclic vomiting), and morphological (facial features, joint laxity) manifestations. Facial morphological markers of that syndrome are present in Kate including a high forehead, tented mouth, anteverted nares, high-arched palate, and pre-auricular skin tags. Of interest, cyclic vomiting was reported in 6/16 (37.5%) of subjects with de novo heterozygous pathogenic variants in POGZ [27]. Despite finding a de novo variant, the family history is positive for migraine, chronic fatigue, gastrointestinal dysmotility, and/or muscle weakness in the mother, a maternal aunt, and/or the maternal grandmother. The sequencing depth of the mtDNA was inadequate for variant identification.
In addition, WES detected two inherited missense variants in the patient that are associated with mitochondrial dysfunction; both of which are in our 22-gene CVS list. The first was the uncommon variant (c.757A>G, p.Ile253Val) in the TRAP1 gene, which encodes for a mitochondrial chaperone, and likely protects mitochondrial proteins in high oxidative situations. This variant is associated with risk for chronic pain, fatigue, and GI dysmotility, including CVS [10]. The second is a rare variant (c.803G>C, p.Gly268Ala) in the POLG gene that encodes for the enzyme that replicates and proofreads mtDNA. This variant is associated with CVS [10] and CPEO [28].
3.3.8. Case 8: Mollie
Mollie (#56) presented with episodes starting at age 1.5 years. Episodes occurred every 6-8 weeks, lasting 2-2.5 days each, and included vomiting (up to every 10 minutes), nausea, photophobia, phonophobia, extreme lethargy, weakness (whole body), drooling, and thirst, but without headache. Amitriptyline was started at age 13 years, with some improvement in episode frequency (to 8-10 weeks) and duration (to 1.5-2 days). Between the ages of 18 and 25 years, her episodes transitioned from classical CVS to migraine-like headaches with little vomiting. In the last 15 years (ages 29-44), episodes have included nausea, lethargy, weakness, and pallor, but only occasionally vomiting. At the age of 41 years, Mollie began seeing an increase in these episodes, including 6-week clusters of headaches, each one-day long, occurring once or twice a week. Clinical manifestations between episodes included chronic fatigue, constipation, POTS, anxiety, and mild intellectual deficits. The family history is consistent with headaches with some vomiting in her sister as an adolescent, hyperemesis gravidarum in all of her mother’s four pregnancies, and migraine in the maternal grandmother and aunt. A comprehensive mito-cocktail was started, with reduction of headache episodes to once every 12 weeks.
Trio WGS revealed a de novo heterozygous, frameshift variant (c.1200delC, p.Tyr401fs) in the PPM1D gene. This variant is Pathogenic by virtue of being both an LOF frameshift and de novo. This gene encodes for a protein phosphatase that regulates a stress-response, DNA-damage response pathway by inhibiting p53 [29]. De novo sequence variants in this gene have been associated with Jansen-de Vries syndrome. This syndrome is an excellent match for Mollie, given the matching features of cyclic vomiting, GERD, longstanding constipation, mild developmental delay, hypotonia, broad-based gait, anxiety, myopia, wide mouth with thin upper lip, small hands and feet, hypoplastic nails, and short stature relative to her family. Per Jansen et al (2017) [29], “eight individuals (62%) had periods of illness with fever and/or vomiting.” Despite finding a de novo variant, note the positive family history in the above paragraph. MtDNA variants of uncertain significance were identified yet none appeared to be excellent candidates for disease association.
4. Discussion
4.1. Clinical Pearls from the Case Reports - Phenotypic Heterogeneity
A major concept throughout the case reports is phenotypical and genotypic heterogeneity in CVS. Regarding the former, none of these cases has isolated CVS, but all have complicated constellations of clinical manifestations which overlap substantially among the cases. Neither the phenotypic complexity nor this overlap is an artifact of our selection of case reports, as can be seen in Table 2 and Supplementary Table 1. Most of our participants with CVS have chronic comorbidities beyond vomiting episodes in terms of pain, gastrointestinal dysmotility, dysautonomia, fatigue, sleep disturbance, mental health conditions, and “other” neurological conditions not otherwise classified in that table. Nearly half of our CVS participants have manifestations in the allergy and immunity realm. Just over a quarter of our participants have neurodevelopmental disorders (which is overrepresented in the case reports selected, although impairment is mild in most of them). Urinary frequency, which can be considered to be a dysautonomic condition, and in many of our participants is clinically bothersome, also appears to be associated with CVS. Overall, our results (Table 2 and Supplementary Table S1) are generally similar to that previously recorded in CVS [6,9].
Given this phenotypic heterogeneity, does our participant group represent CVS specifically, beyond that the authors chose to study and write about this condition? Indeed, could we have based this paper on a group of participants with chronic pain, fatigue, gastrointestinal dysmotility, or dysautonomia, four common conditions in which CVS could be considered as one of their many comorbidities? Can the genetic findings previously reported [1] and discussed below in CVS apply equally to these other conditions? While the short answer is that “more research needs to be done”, there is also a significant degree of asymmetry between CVS and these other conditions. While most individuals with CVS have these four common conditions (Table 2), only a small proportion of those with each of those four conditions have the less-prevalent disorder of CVS. These common conditions are often co-morbid to each other [32,33,34], suggesting the presence of common predisposing genetic factors in their pathogeneses. This is particularly obvious looking at the family history of our participants, in which these four conditions, but generally without CVS, are very common among the first-degree relatives. In 7 of our 8 case reports, the family history demonstrates this phenomenon, being unrevealing in only one of those families (Case 1).
The term “cyclic vomiting syndrome” is unfortunate in that the very name elevates one of multiple clinical manifestations present during stereotypical episodes above the others, although it is the most objective of those manifestations. Can patients have CVS without vomiting? As the diagnostic criteria for CVS as presently defined is predicated on the presence of paroxysmal vomiting, the answer is “no”. However, surely some individuals with the same underlying pathophysiology of disease do not have vomiting, for whatever reason. At a minimum, examples include the close relatives of CVS sufferers with combinations of Table 2-listed conditions, but without vomiting. The natural history of most chronic conditions is that they evolve over time, particularly during important developmental years such as adolescence, and CVS is no exception herein. In CVS clinical practice, one often encounters individuals who met the Rome IV criteria for CVS at some time points, but not at other time points in the natural history of their disease. This is illustrated in many of our cases, including Cases 7-8 which at one point were classical for CVS (with prominent emesis) but evolved into episodes of migraine headache with little or no vomiting, and Case 4 whereas classical CVS evolved into episodes dominated by dizziness. Case 2 had stereotypical paroxysmal episodes dominated by profound fatigue 3 years prior to presenting with classical CVS episodes with vomiting. Case 6 had typical CVS episodes, but also episodes whereas severe pain was more consistent with abdominal migraine. In such cases, the diagnosis of CVS can be challenging. While our case reports were selected in part to illustrate this challenge, this phenomenon is quite common for CVS in general. The first author believes that when a patient meets diagnostic criteria for overlapping paroxysmal conditions, such as CVS, abdominal migraine, and/or migraine headache, that each of these conditions should be coded and considered in terms of treatment ramifications.
Three of these case reports involved substantial allegations of concepts related to “non-organic” disease. Unfortunately, this phenomenon is common in the first author’s practice, at least until the diagnosis of CVS is made. In cases 4 and 5, diagnoses related to medical child abuse were given. Medical child abuse is the present term used when a minor’s caregiver is believed either to give false information or fails to give appropriate information, which leads to inappropriate medical testing and treatments, resulting in harm. Other terms often used for this condition, including Munchausen disorder-by-proxy and fictitious disorder-by-proxy, although they are not fully synonymous. In Case 2, the diagnosis given with that of a conversion disorder. Of note, once the diagnosis of CVS was made and specific treatment was started, clinical symptomatology improved dramatically in all three, each of which are doing well as of this writing. Each of these three individuals was a minor at the time. Among adult patients in this case series (not selected as case reports), allegations of drug seeking were common when obtaining urgent/emergency care services. Regardless of the specific term used, the symptoms in these patients were not considered to be “organic” or “real”, and at times medical treatment for their symptoms was denied.
4.2. Clinical Pearls from the Case Reports - Genotypic Heterogeneity
All eight of our case reports have at least one Qualifying variant among our 22 genes associated with CVS. Indeed, Qualifying variants were identified in at least two of these 22 genes in six of our eight case reports. As detailed in Bar et al (2023) [1], Qualifying variants are coding, rare, and conserved. In addition, Case 1 has a Qualifying variant in a second gene, MARS, which is associated with CMT neuropathy, a condition possibly related to CVS in Bar et al (2023) [1]. This strong polygenic aspect is not an artifact of our selection of case reports, being very common in the full group of 80 participants (Table 2 and Table 3, [1]). At least two Qualifying variants in the larger gene list of 35 genes (our 22 associated with CVS plus 13 Possibly associated) were identified in 41/75 (55%) participants. This suggests that polygenic inheritance is common in CVS. This inheritance model was to be expected as it is the inheritance model present in most human disorders. Additionally, a monogenic Mendelian model would be incompatible with much that is known regarding the complex inheritance of CVS.
Qualifying variants in genes directly involved in cation transport predominate in our case reports (Cases 1-6), with a second focus on genes directly involved in energy metabolism (Cases 2, 5, 7, and 8). This clustering of gene function into cations and energy metabolism is not an artifact of our selection of case reports; our proposed cellular model of CVS is a vicious cycle of aberrant cation transport and energy depletion leading to cellular hyperexcitability [1]. Many of the Qualifying variants in our case reports, as well as in our entire group of 80 participants, are “Key” Qualifying variants in that the sequence variant by itself can be designated as Pathogenic or Likely Pathogenic, or the sequence variant has strong clinical correlation to a non-CVS clinical manifestation in the patients (description in [1], also see Table 2 and Table 3 within). These are the type of sequence variants that are likely to be identified as possibly disease related by a clinical laboratory or identified as such by a clinical geneticist. However, many of the variants in our case reports, and elsewhere in our participants, among the 22 CVS related genes are non-Key variants. Indeed, seven of our eight case reports include such non-Key variants. These variants are unlikely to be identified at this time by a clinical laboratory. However, these variants were often very important in terms of identifying appropriate therapy that improved clinical outcomes, as detailed in some of the case reports and which will be exemplified in the next section. Hopefully, the publication of this report will improve the reporting of Qualifying variants in the 22 CVS-associated genes in patients with CVS or similar paroxysmal conditions.
Two of our case reports (Cases 7 and 8) have CVS as well as neurodevelopmental disease and morphological changes (congenital malformations and/or dysmorphic features). In these two cases, a de novo variant (absent in both parents) was identified. In both cases, as seen in the case reports, the facial morphology and other subtleties about the patient’s phenotype were an excellent fit with the conditions described with variants in the gene (POGZ and PPM1D) whereas the de novo variant was identified. In addition, CVS has been described as common in both conditions [27,29]. While both genes are involved in fundamental genetic machinations of cells, these genes also have indirect associations with cation transport and energy metabolism, respectively [1]. Case 2, with neurodevelopmental disease has a de novo mtDNA variant. Among the remainder of our 80 participants, a boy with neurodevelopmental disease has two de novo variants in POGZ. Neither of the two latter children has known morphological changes. Trio sequencing, including the patient and both parents, is needed to identify de novo variants, but parental sequencing is rarely done in CVS as it generally doubles or triples the sequencing costs. In the first author’s practice, trio sequencing was ordered any time there was significant neurodevelopmental disease or morphological changes (as these are the clinical hallmarks of disease-causal de novo variants in general), and otherwise singleton (patient only) sequencing was performed. Our experience illustrates that de novo variants are likely to be found in cases of CVS and neurodevelopmental disease, with or without morphological changes, whereas trio sequencing is indicated.
In Cases 7 and 8, the presence of a de novo variant might be expected to result in an unremarkable family history, but this was not what we found. In both families, common comorbid phenotypes of CVS, including chronic headaches, fatigue, and gastrointestinal dysmotility, were prominent in either a matrilineal (mtDNA mediated) or an autosomal dominant pattern. How can this be if the disease-causing de novo variant is absent in the relatives? We believe that the answer involves the polygenic nature of CVS. These de novo variants are insufficient in and of themselves to cause CVS, which fits with the literature in that a subset of patients with each syndrome have CVS. Additional risk factors are needed as well, likely genetic factors. The additional genetic factors identified in these two case reports may or may not contribute to this postulated additional genetic factor.
4.3. An Anatomical Model for CVS: Vagal Afferent Unit
As previously reported [1] and as briefly discussed above, our cellular model for CVS is a vicious cycle of anomalous intracellular cation gradients and mitochondrial dysfunction resulting in cellular hyperexcitation. However, in which cell type(s) does this viscous cycle occur? In other words, what is the anatomical location of CVS? For over a century, clinicians and researchers have debated whether CVS is a disorder of the gastrointestinal (GI) or of the nervous system. Of course, nausea and vomiting are protective responses to the intake of toxins or pathogenic organisms, providing both shorter (e.g., vomiting to purge the contents) and longer (e.g., nausea to warn the animal to avoid the same meal thereafter) term solutions. In a bottom-up (GI) model, excitation of cells in the gut and/or of the neurons relaying gut impulses rostrally (mostly via vagal afferents) result in CVS by transmitting false or highly-exaggerated signals [35,36]. In a top-down (neurological/psychiatric) model, CVS is viewed through the exaggerated excitation in one or more of the multiple descending tracts in which “higher functions”, especially severe emotion, can be manifested as nausea and vomiting in the general population, such as those from the limbic system and periaqueductal grey (PAG) [37,38,39,40]. A popular top-down model is that of a “brain-gut disorder” mediated by the neuroendocrine system where corticotropin-releasing factor can cause nausea and delayed gastric emptying as a response to stress [41,42,43]. Beyond the obvious implications for treatment, the model chosen has ramifications in how patients are viewed, as unfortunately the top-down model is often used to dismiss patients as not having “organic” disease and shunting them to mental health services, which in the clinical experiences of the first author is rarely successful.
While further review on this subject is beyond the scope of this study, it is interesting to postulate a response to the anatomical location question by evaluating where the 22 genes associated with CVS are expressed. Gene expression profiles are complicated, in part as most genes are expressed in most cells. However, several of our genes of interest are preferentially expressed in certain cells/tissues relative to others (Table 3). In particular, two of the highest scoring genes (CACNA1S and SCN4A) in our previous publication [1] are expressed at levels far higher in skeletal muscle than in nerve or other tissues. Our case reports 1–3 have qualifying variants in one or the other of these genes. In terms of relevance to CVS, the obvious candidate tissue for these two genes is the diaphragm, a skeletal muscle-containing tissue whereas these genes are highly expressed. In mice carrying an SCN4A variant, the diaphragm generated more force during contraction than wild-type diaphragm [44]. In polymorphic CACNA1S carriers, triggering agents were found to induce excessive calcium efflux from the sarcoplasmic [45]. Thus, variants in either gene may result in diaphragmatic cellular hyperexcitability, potentially resulting in a predisposition towards CVS.
Diaphragmatic tissues involved in vomiting are innervated by vagal afferents, as are most of the ascending (sensory) fibers leading from the proximal GI tract, such as esophagus, stomach, and small intestine. However, some GI afferents arrive via spinal nerves. The diaphragmatic- and proximal GI-derived vagal afferent neurons have cell bodies in either the jugular or the nodose ganglia and synapse in the nucleus tractus solitarius (NTS) of the medulla (Figure 1). These neurons synapse with neurons in tracts leading caudally (to the dorsal motor nucleus of the vagus, via vagal efferents, to induce vomiting) and rostrally (to cingulate and insular cortex and amygdala, via many tracts, to induce nausea and secondary behavioral aversion). Interestingly, there are also rostral projections from the NTS to brain regions involved in the sleep-wake cycle (e.g., locus ceruleus, PAG), as extreme lethargy, sometimes termed “conscious coma”, is a common feature of CVS episodes.
For illustration, consider the vagal afferent neurons as a unit including the synapsing cells on both their rostral (e.g., afferent and efferent neurons) and caudal (e.g., gut and diaphragm muscle cells) ends. We also include in this unit the associated glial cells which assist in maintaining these neurons, including the enteric glia cells in the gut, satellite glia cells in the jugular/nodose ganglia, and astrocytes in the medulla. It is known that enteric glia cells are vital for proper GI tract motility and secretion and have been implicated with multiple GI conditions [46,47,48,49]. All of the 22 genes our study found to be Highly likely and Likely associated with CVS (Table 3) in our previous paper [1] are expressed in this vagal afferent-centered unit (Figure 1), either highly so or ubiquitously (similar to their expression in most other cell types). Many of the genes with less robust connections to CVS ([1], Table 5 of that reference) are also highly/ubiquitously expressed in this unit, including many of the genes associated with peripheral neuropathy that were identified on post analysis. We hypothesize that genetic predisposition towards hyperexcitability in this vagal afferent-centered unit may be the anatomical location for CVS. This unit also includes the neurons ascending from the NTS to the PAG, a location previously postulated to be a potential site of CVS [50]. Our model requires additional study to confirm or refute. The neurocircuitries are complex, and as mentioned, most genes are expressed ubiquitously. However, the present authors are unaware of another anatomical unit in which all of the 22 CVS-associated genes are highly or ubiquitously expressed.
4.4. Treatment of CVS as Informed by Our Models and Case Reports
How does the information obtained from our cellular model relate to CVS treatment as currently practiced? One obvious answer is that the component of mitochondrial dysfunction in CVS corresponds with the frequent usage of mitochondrial-targeted dietary supplementation. Among these supplements, observational studies have shown efficacy especially for L-carnitine and coenzyme Q10 (5,18, 51-53]. Mitochondrial dysfunction also predicts the efficacy of dextrose-containing intravenous fluids during disease episodes [5,54]. Our Cases 2, 4, and 7 illustrate the association of mitochondrial-targeted dietary supplementation with clinical improvement. The association of CVS with mitochondrial dysfunction, and how this relates to treatment via dietary supplementation, have been discussed in multiple previous reports including the above references. There are a few prescription medications which have significant positive effects in energy metabolism, and thus medications are rarely used to treat mitochondrial dysfunction. However, sumatriptan, a drug often used to abort episodes of CVS and migraine headache, is known to decrease oxidative stress [55,56].
There is a myriad of ways in which the connection of anomalous cation transport in CVS predicts treatment efficacy. Cations are positively charged, and include sodium, potassium, and calcium. Propranolol is a beta-blocker and likely one of two drugs most recommended in CVS prophylaxis [4]. Beta blockade interferes with catecholamine (e.g., stress)-induced release of calcium from the endoplasmic reticulum to the cytosol via the RYR2 calcium channel [57]. Anecdotally, the first author has noted that propranolol has high efficacy in CVS patients with Qualifying RYR2 variants [10], including several of the participants in the previous study [1], including Case 6.
Anticonvulsant medications are also sometimes used in CVS prophylaxis, including topiramate and zonisamide [5], both of which have direct effects on calcium and sodium channel function [58,59,60,61]. Levetiracetam and lamotrigine, additional anticonvulsant medications published as sometimes used in CVS prophylaxis per the same review article [5], also have influences on intracellular calcium and/or sodium homeostasis and neuronal excitability [62,63].
CVS has been associated with peripheral neuropathy [12,23]. In our previous paper, Key Qualifying variants were identified in five genes associated with peripheral neuropathy, and we labeled them as Possibly CVS related ([1], Table 5 of that reference). This suggests that the treatment of peripheral neuropathy might be helpful in CVS. Amitriptyline, possibly the most common drug used for prophylaxis in CVS [4,5] is a standard “go-to drug” for physicians treating peripheral neuropathy. A tricyclic antidepressant, amitriptyline is also a potent blocker of sodium channels [64]. Furthermore, two additional drugs sometimes used in CVS [5], gabapentin and pregabalin, act in part by interacting with calcium channels, and in clinical practice are frequently used to treat peripheral neuropathy. In addition, there are connections between neuropathy and energy metabolism [1,65].
How does the information obtained from our anatomical model relate to CVS treatment as is currently practiced? Aprepitant is an antiemetic agent frequently used and recommended to abort CVS episodes in the early phase. Aprepitant antagonistically binds to neurokinin 1 receptors in the NTS, which mediates the emetic motor reflex [66]. Ondansetron is an antiemetic that acts peripherally in the gastrointestinal tract, where it blocks 5-HT3 receptors on vagal afferent nerve terminals [67]. Some clinicians are using non-invasive vagal stimulation in CVS. As the vagal afferents involved in nausea and vomiting are autonomic fibers, cellular hyperexcitability in CVS may also involve additional autonomic fibers leading to additional autonomic signs and symptoms beyond the GI tract. In fact, abnormal function of the autonomic nervous system (dysautonomia) in general has been documented in CVS [68], and this likely explains at least some of the frequent comorbidities of CVS seen in Table 2.
How does the information obtained from our cellular and anatomical models relate to CVS treatment moving forwards, beyond current practice? The importance of cation transport in terms of CVS therapy is clear from our data and case reports, and indeed is central in our cellular model of CVS. Potassium supplementation, which may interfere with CVS pathophysiology via multiple mechanisms, including membrane ion-exchange proteins involving potassium, sodium, and/or calcium, is a particularly intriguing possibility due to the ease of use with a high potassium diet, as well as with higher dosing possible via prescription. Anecdotally, the first author has found that potassium supplementation, sometimes derived by diet, sometimes by high-dose prescription, has been highly efficacious in many of his CVS patients with ion channelopathies. Our Cases 3 and 5 demonstrated clinical improvement in association with prescription KCl. Additionally, improvement was noted in Case 1 on propranolol, and in Case 2 on acetazolamide (which has many effects on cations). Calcium channel blockers may also warrant investigation in CVS. Additional studies are needed to determine the effects of cation-targeted therapy in CVS beyond anecdotal reports.
Our cellular model proposes that a vicious cycle of aberrant cation transport and energy deficiency result in cellular hyperexcitability. Thus, medications (e.g., memantine, amantadine) and dietary supplements (e.g., magnesium, zinc) aimed at reducing cellular hyperexcitability might be helpful in CVS. The first author routinely uses high dosing of magnesium and zinc in CVS prophylaxis. Anti-inflammatory supplements/medication might be helpful, at least in those harboring Qualifying variants in genes associated with auto-inflammation (MEFV, TNFRSF1A, possibly PLCG2). Sumatriptan, in addition to effects on reactive oxygen species (ROS) release, also decreases TNF-α release, which is the ligand bound by the TNFRSF1A gene encoded receptor.
5. Conclusions
Herein, we present additional clinical information on 80 participants with CVS in which an analysis of extensive DNA sequencing was published recently by our group [1]. These participants demonstrate substantial phenotypic and genotypic heterogeneity in CVS, which we exemplify in eight case reports. Phenotypic heterogeneity includes the strong propensity for chronic inter-episodic manifestations of pain, fatigue, gastrointestinal dysmotility, and/or dysautonomia in the participant, as well as in close relatives. Although all participants met standard diagnostic criteria for CVS and thus had stereotypical episodes including vomiting, in some cases emesis became less apparent or even absent overtime. Many patients receive “non-organic” diagnoses such as medical child abuse, conversion disorder, or drug seeking. Genotypic heterogeneity includes multiple genes, including 22 strong candidates to date, with most patients having variants in more than one of these genes, indicating polygenic inheritance. Many variants associated with CVS in these genes are not designated as Pathogenic or Likely Pathogenic based on variant classifications and are generally not written on reports from clinical diagnostic laboratories, requiring extensive clinical correlation to identify. Our eight case reports illustrate how genetic information can guide clinical management, and strongly argue to the high clinical utility of appropriate genetic testing and analysis in CVS, to include at least WES and mtDNA sequencing. WGS is preferred, and since it generally includes the mtDNA can often be less costly. WGS can also include pharmacodynamic data, which can be useful to dose many medications, such as amitriptyline.
All 22 CVS candidate genes are associated with either cation transport or energy metabolism (14 directly, 8 indirectly). Cation gradients and mitochondrial function are intricately interrelated in a myriad of manners, for example, ion channel gain-of-function variants (“leaky” channels) promote cellular overexcitation and place a strain on energy metabolism. Our findings have suggested a cellular model in which aberrant ion gradients lead to mitochondrial dysfunction, or vice versa, in a pathogenic vicious cycle of cellular hyperexcitability [1]. By virtue of the expression profile for each of our 22 candidate genes, we hereby propose an anatomical model in which the primary location of CVS is likely in a unit defined by vagal afferents and adjacent cells. Our models are consistent with multiple current hypotheses of CVS and the efficacy of commonly recommended treatments. In a clinical context, we show how our models explain the multiple different treatment strategies that have been advocated for CVS, and how they predict additional therapies not yet reported.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Detailed Clinical and Molecular Information on Our 80 Participants.
Author Contributions
Conceptualization, R.B.; methodology, R.B. and O.B.; validation, R.B., O.B., and E.V.; investigation, R.B. and O.B.; resources, R.B. and M.M.; data curation R.B., O.B., L.E. and E.V.; writing—original draft preparation, R.B., O.B. and L.E.; writing—review and editing, R.B., O.B., E.V. and M.M.; supervision, R.B.; project administration, R.B. and O.B.; funding acquisition, R.B. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was internally funded by NeurAbilities Healthcare®.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved as “Exempt” by the Institutional Review Board of Advarra®. An exemption for this study was approved based on adherence to a retrospective chart review format in accordance with national legislation and institutional requirements.
Informed Consent Statement
Written informed consent was obtained from all adult participants or the parent/guardian of minor participants corresponding to all case reports. For the remaining participants, consent was waived by the Institutional Review Board based on study format and applicable law.
Data Availability Statement
The dataset is provided in Table S1: Detailed Clinical and Molecular Information on Our 80 Participants.
Acknowledgments
We thank Kellee Weiner for participating in data collection and approval of the initial IRB protocol.
Conflicts of Interest
R.B. is an officer and receives equity from NeuroNeeds®, a company that produces dietary supplements for neurological conditions. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Bar, O. , Ebenau, L., Weiner, K., Mintz, M. and Boles, R. (2023). Whole exome/genome sequencing in cyclic vomiting syndrome reveals multiple candidate genes, suggesting a model of elevated intracellular cations and mitochondrial dysfunction. Frontiers in Neurology, 14(1664-2295). [CrossRef]
- Rome IV Criteria - Rome Foundation. Available online: https://theromefoundation.org/rome-iv /rome-iv-criteria/ (Accessed 21 October 2020).
- Abu-Arafeh, I. and Russell, G., 1995. Cyclical vomiting syndrome in children: a population-based study. Journal of pediatric gastroenterology and nutrition, 21(4), pp.454-458. [CrossRef]
- Li, B.U., Lefevre, F., Chelimsky, G.G., Boles, R.G., Nelson, S.P., Lewis, D.W., Linder, S.L., Issenman, R.M. and Rudolph, C.D., 2008. North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition consensus statement on the diagnosis and management of cyclic vomiting syndrome. Journal of pediatric gastroenterology and nutrition, 47(3), pp.379-393. [CrossRef]
- Venkatesan, T., Levinthal, D.J., Tarbell, S.E., Jaradeh, S.S., Hasler, W.L., Issenman, R.M., Adams, K.A., Sarosiek, I., Stave, C.D., Sharaf, R.N. and Sultan, S., 2019. Guidelines on management of cyclic vomiting syndrome in adults by the American Neurogastroenterology and Motility Society and the Cyclic Vomiting Syndrome Association. Neurogastroenterology & Motility, 31, p.e13604. [CrossRef]
- Li, B.U.K., Murray, R.D., Heitlinger, L.A., Robbins, J.L. and Hayes, J.R., 1999. Is cyclic vomiting syndrome related to migraine?. The Journal of pediatrics, 134(5), pp.567-572. [CrossRef]
- Enokizono, T., Nemoto, K., Fujiwara, J., Tanaka, R. and Ohto, T., 2017. Cyclic vomiting syndrome after acute autonomic and sensory neuropathy. Pediatrics International: Official Journal of the Japan Pediatric Society, 59(4), pp.503-505. [CrossRef]
- Sato, T., Igarashi, N., Minami, S., Okabe, T., Hashimoto, H., Hasui, M. and Kato, E., 1988. Recurrent attacks of vomiting, hypertension and psychotic depression: a syndrome of periodic catecholamine and prostaglandin discharge. European Journal of Endocrinology, 117(2), pp.189-197. [CrossRef]
- Boles, R. G., Adams, K. and Li, B. U. K., 2005. Maternal inheritance in cyclic vomiting syndrome. American Journal of Medical Genetics Part A, 133(1), pp.71-77. [CrossRef]
- Lee, J., Wong, S. A., Li, B. U. K. and Boles, R. G., 2015. NextGen nuclear DNA sequencing in cyclic vomiting syndrome reveals a significant association with the stress-induced calcium channel (RYR2). Neurogastroenterology & Motility, 27(7), pp.990-996. [CrossRef]
- Wasilewski, A., Lewandowska, U., Mosinska, P., Watala, C., Storr, M., Fichna, J. and Venkatesan, T., 2017. Cannabinoid receptor type 1 and mu-opioid receptor polymorphisms are associated with cyclic vomiting syndrome. Official journal of the American College of Gastroenterology| ACG, 112(6), pp.933-939. [CrossRef]
- Fleisher, D. R., Gornowicz, B., Adams, K., Burch, R. and Feldman, E. J., 2005. Cyclic vomiting syndrome in 41 adults: the illness, the patients, and problems of management. BMC medicine, 3(1), pp.1-12. [CrossRef]
- Abell, T.L., Adams, K.A., Boles, R.G., Bousvaros, A., Chong, S.K.F., Fleisher, D.R., Hasler, W.L., Hyman, P.E., Issenman, R.M., Li, B.U.K. and Linder, S.L., 2008. Cyclic vomiting syndrome in adults. Neurogastroenterology & Motility, 20(4), pp.269-284. [CrossRef]
- Boles, R. G., Powers, A. L. and Adams, K., 2006. Cyclic vomiting syndrome plus. Journal of child neurology, 21(3), pp.182-189. [CrossRef]
- Wang, Q., Ito, M., Adams, K., Li, B.U., Klopstock, T., Maslim, A., Higashimoto, T., Herzog, J. and Boles, R.G., 2004. Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. American Journal of Medical Genetics Part A, 131(1), pp.50-58. [CrossRef]
- Zaki, E.A., Freilinger, T., Klopstock, T., Baldwin, E.E., Heisner, K.R.U., Adams, K., Dichgans, M., Wagler, S. and Boles, R.G., 2009. Two common mitochondrial DNA polymorphisms are highly associated with migraine headache and cyclic vomiting syndrome. Cephalalgia, 29(7), pp.719-728. [CrossRef]
- Landrum, M.J., Lee, J.M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., Gu, B., Hart, J., Hoffman, D., Hoover, J. and Jang, W., 2016. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research, 44(D1), pp.D862-D868. [CrossRef]
- Boles, R.G., 2011. High degree of efficacy in the treatment of cyclic vomiting syndrome with combined co-enzyme Q10, L-carnitine and amitriptyline, a case series. BMC neurology, 11(1), pp.1-5. [CrossRef]
- Fan, C., Mao, N., Lehmann-Horn, F., Bürmann, J. and Jurkat-Rott, K., 2017. Effects of S906T polymorphism on the severity of a novel borderline mutation I692M in Na v 1.4 cause periodic paralysis. Clinical Genetics, 91(6), pp.859-867. [CrossRef]
- Kuzmenkin, A., Jurkat-Rott, K., Lehmann-Horn, F. and Mitrovic, N., 2003. Impaired slow inactivation due to a polymorphism and substitutions of Ser-906 in the II-III loop of the human Na v 1.4 channel. Pflügers Archiv, 447, pp.71-77. [CrossRef]
- Binda, A., Renna, L.V., Bosè, F., Brigonzi, E., Botta, A., Valaperta, R., Fossati, B., Rivolta, I., Meola, G. and Cardani, R., 2018. SCN4A as modifier gene in patients with myotonic dystrophy type 2. Scientific reports, 8(1), pp.1-10. [CrossRef]
- Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. and Gauthier, L.D., 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), pp.434-443. [CrossRef]
- Pirone, A., Schredelseker, J., Tuluc, P., Gravino, E., Fortunato, G., Flucher, B.E., Carsana, A., Salvatore, F. and Grabner, M., 2010. Identification and functional characterization of malignant hyperthermia mutation T1354S in the outer pore of the Cavα1S-subunit. American Journal of Physiology-Cell Physiology, 299(6), pp.C1345-C1354. [CrossRef]
- Zhou, Y., Fang, L., Jiang, L., Wen, P., Cao, H., He, W., Dai, C. and Yang, J., 2012. Uric acid induces renal inflammation via activating tubular NF-κB signaling pathway. PloS one, 7(6), p.e39738. [CrossRef]
- Fejzo, M.S., Myhre, R., Colodro-Conde, L., MacGibbon, K.W., Sinsheimer, J.S., Reddy, M.P.L., Pajukanta, P., Nyholt, D.R., Wright, M.J., Martin, N.G. and Engel, S.M., 2017. Genetic analysis of hyperemesis gravidarum reveals association with intracellular calcium release channel (RYR2). Molecular and cellular endocrinology, 439, pp.308-316. [CrossRef]
- Nozawa, R.S., Nagao, K., Masuda, H.T., Iwasaki, O., Hirota, T., Nozaki, N., Kimura, H. and Obuse, C., 2010. Human POGZ modulates dissociation of HP1α from mitotic chromosome arms through Aurora B activation. Nature cell biology, 12(7), pp.719-727. [CrossRef]
- Assia Batzir, N., Posey, J.E., Song, X., Akdemir, Z.C., Rosenfeld, J.A., Brown, C.W., Chen, E., Holtrop, S.G., Mizerik, E., Nieto Moreno, M. and Payne, K., 2020. Phenotypic expansion of POGZ-related intellectual disability syndrome (White-Sutton syndrome). American Journal of Medical Genetics Part A, 182(1), pp.38-52. [CrossRef]
- Human DNA Polymerase Gamma Mutation Database. Available online: https://tools.niehs.nih.gov/polg/index.cfm/main/reference (Accessed 01 November 2022).
- Jansen, S., Geuer, S., Pfundt, R., Brough, R., Ghongane, P., Herkert, J.C., Marco, E.J., Willemsen, M.H., Kleefstra, T., Hannibal, M. and Shieh, J.T., 2017. De novo truncating mutations in the last and penultimate exons of PPM1D cause an intellectual disability syndrome. The American Journal of Human Genetics, 100(4), pp.650-658. [CrossRef]
- Breitbart, R.E., Liang, C.S., Smoot, L.B., Laheru, D.A., Mahdavi, V. and Nadal-Ginard, B., 1993. A fourth human MEF2 transcription factor, hMEF2D, is an early marker of the myogenic lineage. Development, 118(4), pp.1095-1106. [CrossRef]
- Rashid, A.J., Cole, C.J. and Josselyn, S.A., 2014. Emerging roles for MEF2 transcription factors in memory. Genes, Brain and Behavior, 13(1), pp.118-125. [CrossRef]
- Clauw, D.J. and Chrousos, G.P., 1997. Chronic pain and fatigue syndromes: overlapping clinical and neuroendocrine features and potential pathogenic mechanisms. Neuroimmunomodulation, 4(3), pp.134-153. [CrossRef]
- Eccles, J.A. and Davies, K.A., 2021. The challenges of chronic pain and fatigue. Clinical Medicine, 21(1), p.19. [CrossRef]
- Steinsvik, E.K., Hausken, T., Fluge, Ø., Mella, O. and Gilja, O.H.., 2023. Gastric dysmotility and gastrointestinal symptoms in myalgic encephalomyelitis/chronic fatigue syndrome. Scandinavian Journal of Gastroenterology, pp.1-8. [CrossRef]
- Andrews, P., Hoyle, C. H. V., Ngoka, T. and Smith, G. E., 2003. Differences in the vagal innervation of the oesophagus may explain the lack of emesis in rodents. Neurogastroenterol Motil, 15, pp.28.
- Andrews, P. L. and Horn, C. C., 2006. Signals for nausea and emesis: Implications for models of upper gastrointestinal diseases. Autonomic Neuroscience, 125(1-2), pp.100-115. [CrossRef]
- Ricardo, J. A. and Koh, E. T., 1978. Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain research, 153(1), pp.1-26. [CrossRef]
- Hejazi, R. A. and McCallum, R. W., 2011. Review article: cyclic vomiting syndrome in adults–rediscovering and redefining an old entity. Alimentary pharmacology & therapeutics, 34(3), pp.263-273. [CrossRef]
- Ellingsen, D.M., Garcia, R.G., Lee, J., Lin, R.L., Kim, J., Thurler, A.H., Castel, S., Dimisko, L., Rosen, B.R., Hadjikhani, N. and Kuo, B., 2017. Cyclic Vomiting Syndrome is characterized by altered functional brain connectivity of the insular cortex: a cross-comparison with migraine and healthy adults. Neurogastroenterology & Motility, 29(6), p.e13004. [CrossRef]
- Kawai, Y., 2018. Differential ascending projections from the male rat caudal nucleus of the tractus solitarius: an interface between local microcircuits and global macrocircuits. Frontiers in Neuroanatomy, 12, 63. [CrossRef]
- Fleisher, D.R., 1997. Cyclic vomiting syndrome: a paroxismal disorder of brain-gut interaction. Journal of pediatric gastroenterology and nutrition, 25, pp.13-15. [CrossRef]
- Taché, Y., 1999. Cyclic vomiting syndrome: the corticotropin-releasing-factor hypothesis. Digestive diseases and sciences, 44(8 Suppl), pp.79S-86S.
- Taché, Y., Martinez, V., Million, M. and Wang, L., 2001. III. Stress-related alterations of gut motor function: role of brain corticotropin-releasing factor receptors. American Journal of Physiology-Gastrointestinal and Liver Physiology, 280(2), pp.G173-G177. [CrossRef]
- Ammar, T., & Renaud, J. M. (2015). Diaphragm of hyperkalemic periodic paralysis mouse has no contractility abnormality compared to the robust abnormalities in EDL and soleus. The FASEB Journal, 29, 947-8. [CrossRef]
- Schuster, F., Johannsen, S., Isbary, S., Türkmeneli, I., & Roewer, N. (2018). In vitro effects of levosimendan on muscle of malignant hyperthermia susceptible and non-susceptible swine. BMC anesthesiology, 18(1), 1-5. [CrossRef]
- Fung, C., & Vanden Berghe, P. (2020). Functional circuits and signal processing in the enteric nervous system. Cellular and Molecular Life Sciences, 77(22), 4505-4522. 22. [CrossRef]
- Spear, E. T., & Mawe, G. M. (2019). Enteric neuroplasticity and dysmotility in inflammatory disease: key players and possible therapeutic targets. American Journal of Physiology-Gastrointestinal and Liver Physiology, 317(6), G853-G861. [CrossRef]
- Morales-Soto, W. and Gulbransen, B.D., 2019. Enteric glia: a new player in abdominal pain. Cellular and molecular gastroenterology and hepatology, 7(2), pp.433-445. [CrossRef]
- Grundmann, D., Loris, E., Maas-Omlor, S., Huang, W., Scheller, A., Kirchhoff, F., & Schäfer, K. H. (2019). Enteric glia: S100, GFAP, and beyond. The Anatomical Record, 302(8), 1333-1344. [CrossRef]
- Mayer, E. A., Aziz, Q., Coen, S., Kern, M., Labus, J. S., Lane, R., ... & Tracey, I. (2009). Brain imaging approaches to the study of functional GI disorders: a Rome working team report. Neurogastroenterology & Motility, 21(6), 579-596. [CrossRef]
- Van Calcar, Sandra C., Cary O. Harding, and Jon A. Wolff. L-carnitine administration reduces number of episodes in cyclic vomiting syndrome. Clinical pediatrics 41.3 (2002): 171-174. [CrossRef]
- Boles, R. G., Lovett-Barr, M. R., Preston, A., Li, B. U., & Adams, K. (2010). Treatment of cyclic vomiting syndrome with co-enzyme Q10 and amitriptyline, a retrospective study. BMC neurology, 10(1), 1-5. [CrossRef]
- Tillman, E. M., & Harvath, E. M. (2022). Cyclic Vomiting Syndrome in Pediatric Patients: A Review of Therapeutics. The Journal of Pediatric Pharmacology and Therapeutics, 27(1), 12-18. [CrossRef]
- Li, B. U. (2018). Managing cyclic vomiting syndrome in children: beyond the guidelines. European journal of pediatrics, 177(10), 1435-1442. [CrossRef]
- Ala, M., Jafari, R.M., Ala, M., Agbele, A.T., Hejazi, S.M., Tavangar, S.M., Mahdavi, S.R.M. and Dehpour, A.R., 2020. Sumatriptan alleviates radiation-induced oral mucositis in rats by inhibition of NF-kB and ERK activation, prevention of TNF-α and ROS release. Archives of oral biology, 119, p.104919. [CrossRef]
- Gharishvandi, F., Abdollahi, A., Shafaroodi, H., Jafari, R.M., Pasalar, P. and Dehpour, A.R., 2020. Involvement of 5-HT1B/1D receptors in the inflammatory response and oxidative stress in intestinal ischemia/reperfusion in rats. European journal of pharmacology, 882, p.173265. [CrossRef]
- Leenhardt, A., Denjoy, I. and Guicheney, P., 2012. Catecholaminergic polymorphic ventricular tachycardia. Circulation: Arrhythmia and Electrophysiology, 5(5), pp.1044-1052. [CrossRef]
- Taverna, S., Sancini, G., Mantegazza, M., Franceschetti, S. and Avanzini, G., 1999. Inhibition of transient and persistent Na+ current fractions by the new anticonvulsant topiramate. Journal of Pharmacology and Experimental Therapeutics, 288(3), pp.960-968.
- Zhang, X.L., Velumian, A.A., Jones, O.T. and Carlen, P.L., 2000. Modulation of high-voltage–activated calcium channels in dentate granule cells by topiramate. Epilepsia, 41, pp.52-60. [CrossRef]
- Biton, V., 2007. Clinical pharmacology and mechanism of action of zonisamide. Clinical neuropharmacology, 30(4), pp.230-240. [CrossRef]
- Brodie, M.J., Ben-Menachem, E., Chouette, I. and Giorgi, L., 2012. Zonisamide: its pharmacology, efficacy and safety in clinical trials. Acta neurologica scandinavica, 126, pp.19-28. [CrossRef]
- Deshpande, L.S. and DeLorenzo, R.J., 2014. Mechanisms of levetiracetam in the control of status epilepticus and epilepsy. Frontiers in neurology, 5, p.11. [CrossRef]
- Wang, S.J., Sihra, T.S. and Gean, P.W., 2001. Lamotrigine inhibition of glutamate release from isolated cerebrocortical nerve terminals (synaptosomes) by suppression of voltage-activated calcium channel activity. Neuroreport, 12(10), pp.2255-2258. [CrossRef]
- Gerner, P., Haderer, A.E., Mujtaba, M., Sudoh, Y., Narang, S., Abdi, S., Srinivasa, V., Pertl, C. and Kuo Wang, G., 2003. Assessment of differential blockade by amitriptyline and its N-methyl derivative in different species by different routes. The Journal of the American Society of Anesthesiologists, 98(6), pp.1484-1490. [CrossRef]
- Schiavon, C.R., Shadel, G.S. and Manor, U., 2021. Impaired mitochondrial mobility in Charcot-Marie-Tooth disease. Frontiers in Cell and Developmental Biology, 9, p.624823. [CrossRef]
- Massaro, A.M. and Lenz, K.L., 2005. Aprepitant: a novel antiemetic for chemotherapy-induced nausea and vomiting. Annals of Pharmacotherapy, 39(1), pp.77-85. [CrossRef]
- Currow, D.C., Stuart-Harris, R.C. and Noble, P.D., 1995. The clinical use of ondansetron. Medical journal of Australia, 162(3), pp.145-149.
- Chelimsky, T. C. and Chelimsky, G. G., 2007. Autonomic abnormalities in cyclic vomiting syndrome. Journal of pediatric gastroenterology and nutrition, 44(3), pp.326-330. [CrossRef]
Figure 1.
Our Anatomical Model for CVS.
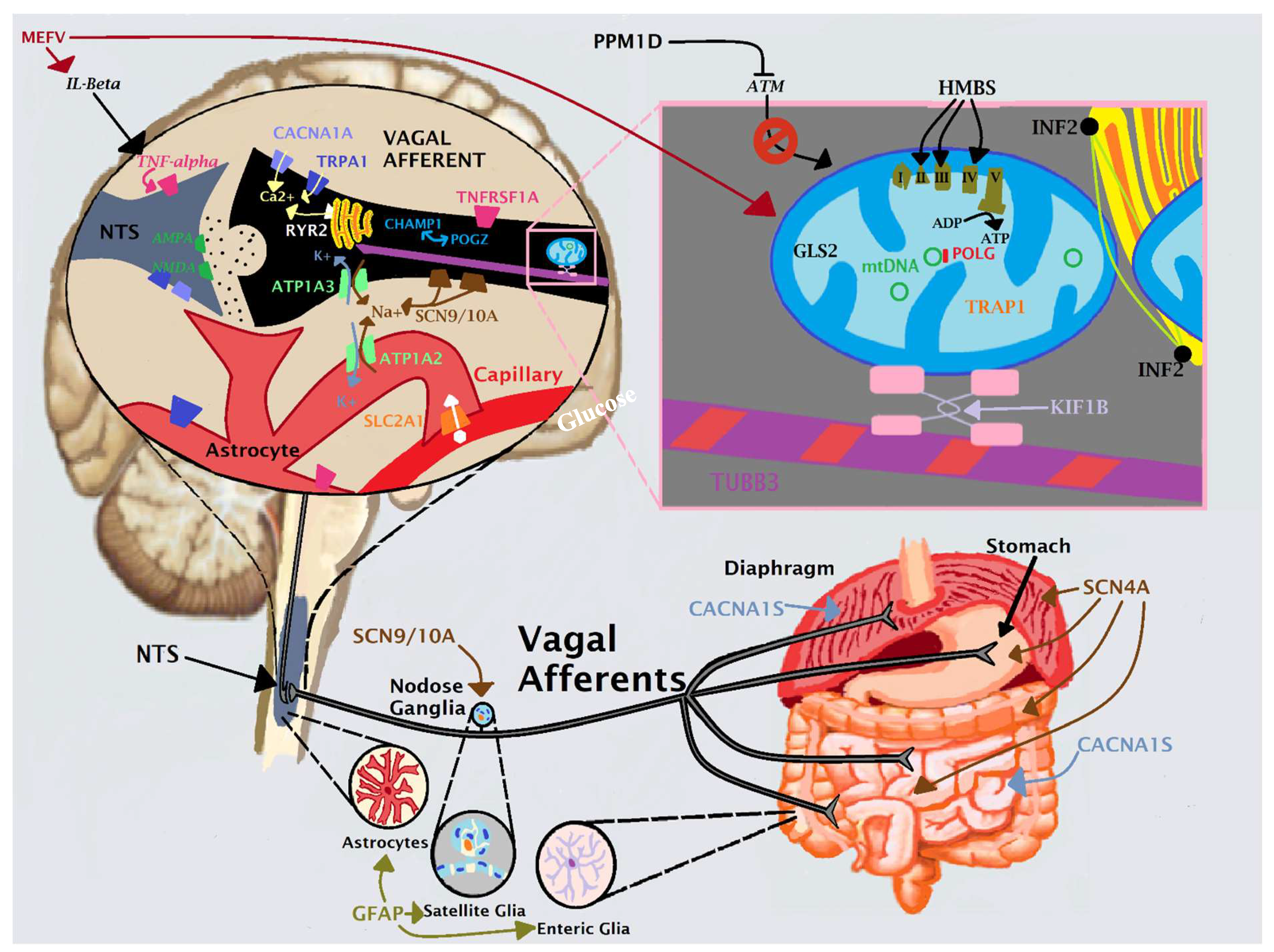

Table 1.
Clinical Characteristics of Participants and Manifestations of Vomiting Episodes.
| Sex | Female: 46, Male: 34 |
|---|---|
| Race/Ethnicity | Western Eurasian: 61 (European (EU, 1 patient had distant Finnish ancestry): 51, Ashkenazi (AS): 3, Middle Eastern: (ME) 1, EU/AS: 5, EU/ME: 1), South Asian (SA): 3, African American (AA): 2, East Asian (EA): 2, Latin American (LA): 1, EU/LA: 6, EU/SA: 2, EU/EA: 2, AA/LA: 1 |
| Age of Onset | < 1 year: 14, 1-3 years: 11, 3-6: 7, 6-12: 12, 12-18: 21, > 18: 7, NR: 8 |
| Episode Triggers | None: 30, Stress/anxiety: 26, Fatigue/exhaustion: 26, Illness: 22, Temperature: 9, Food: 7, Menses: 6, Overexcitement: 5, Other: 141 |
| Prodrome2 | None: 50, Pain: 15, Sensory: 6, Behavioral: 5, Other: 181 |
| Manifestations During Episodes | Nausea and Vomiting 803, None: 7, Fatigue/Lethargy: 62, Pain: 48, Sensory: 36, Dysautonomia: 28, GI: 22, Muscular: 22, Neuro: 12, Behavioral: 4, Other: 231 |
| Episode Frequency | < 2 weeks: 16, 2-5 weeks: 28, 5-9: 9, 9-13: 8, 13-26: 8, > 26: 2, Varies substantially: 6, NR: 3 |
| Episode Duration | < 4 hours: 10, 4-12 hours: 10, 12-24: 5, 24-48: 11, 48-96: 11, 96-168: 13, > 168: 5, Varies: 11, NR: 4 |
NR: not reported; 1Some participants have more than one; 2Manifestations prior to onset of nausea and vomiting; 3By definition of CVS and inclusion criteria.
Table 2.
Clinical Manifestations Between Vomiting Episodes1.
| Pain (60/80, 75%) | Headache: 51 (migraine 39/51), Joints: 22, Abdomen: 20, Extremities: 20, Neck/back: 18, Muscles: 10, Throat/lymph nodes: 7, Chest: 7, Pelvic: 32 |
|---|---|
| Gastrointestinal (58/80, 72.5%) | Bowel3: 42, Nausea: 30, GERD: 10, Other: 202 |
| Dysautonomia (57/80, 71.25%) | Temperature intolerance4: 35, POTS-like manifestations5: 32, Unexplained fevers: 5, Other: 82 |
| Allergy & Immunity (33/80, 41.25%) | Immunodeficiency6: 16, Pruritis: 15, Rash: 12 (urticaria 5/12), Asthma: 9, MCAS: 4, Other: 32 |
| Sleep (45/80, 56%) | Parasomnia: 28, Insomnia: 21, Hypersomnia: 3, Other: 12 |
| Psychological (54/80, 68%) | Anxiety: 51, Depression: 22 (suicide attempt 2/22), Mood instability: 3, Other: 52 |
| Neurological7 (47/80, 68%) | Muscular: 18 (hypotonia 8, muscle weakness 5, other 53), Tinnitus: 13, Abnormal movements: 14 (tics 7, tremors 5, other 43), Sensory 13 (photophobia 8, aversions 1, other 2), Ophthalmological: 8, Paresthesia: 7, Seizures: 7, Ataxia: 7, Neuropathy: 5, Periodic paralysis: 5, Other: 172 |
| Neurodevelopmental (31/80, 40%) | ADHD: 14, ASD: 10, Learning disability: 8, Intellectual disability: 7, Other: 22 |
| Fatigue (56/80, 70%) | |
| Urinary (20/80, 25%) | Frequency: 11, Hesitancy: 4, Enuresis: 4, Dysuria: 3, Other: 82 |
| Connective (9/80, 11%) | Hypermobility: 6, Ehlers-Danlos syndrome: 3 |
| Endocrine (10/80, 12.5%) | Thyroid: 5, Other: 52 |
| Other (21/80, 26.25%) | Heme: 8, Syndromic: 3, Cardiac: 3, Other: 72 |
ADHD: attention deficit hyperactivity disorder; ASD: autism spectrum disorder; GERD: gastrointestinal reflux disease; MCAS: mast cell activation syndrome; POTS: postural orthostatic tachycardia syndrome; 1Stereotypical episodes of nausea and vomiting; 2Numbers do not add up as participants can have more than one. 3Includes constipation, diarrhea, irritable bowel syndrome, etc. 4Heat: 16, cold: 7, both: 13; 5Palpitations: 12, orthostatic hypotension: 9, dizziness: 7, POTS: 7, syncope: 6, presyncope: 5, lightheadedness: 1b; 6Frequent infections: 12, CVID (common variable immunodeficiency): 3, Hypogammaglobulinemia: 3b; 7A "waste basket" category of neurological manifestations not fitting well into the other categories.
Table 3.
Candidate Genes at Least Likely to Be Related to CVS Expression and Function.
| Gene | Points by Our Study | Points by Literature | Composite | Expression, with Emphasis per Our Model | Protein Function |
| ATP1A2 | 1 | 21 | Muscle, brain, and glia (mainly astrocytes) | Maintains the electrochemical gradients of Na and K ions across the plasma membrane; requires ATP | |
| ATP1A3 | 0 | 26 | Most prominently in GABA interneurons; in vagal afferents | Maintains the electrochemical gradients of Na and K ions across the plasma membrane; requires ATP | |
| CACNA1A | 19 | 14 | In most CNS synapses including the NTS; also in vagal afferents | Transmembrane pore-forming subunit of a voltage-gated calcium channel | |
| CACNA1S | 17 | 0 | Skeletal muscle (e.g., diaphragm) | Transmembrane pore-forming subunit of a voltage-gated calcium channel | |
| CHAMP1 | 1 | 6 | Ubiquitous (essentially uniform in all cell types) | Enables chromosome segregation. Associated with expression of genes involving cations and neurotransmitter transport | |
| GFAP | 3 | 18 | Glial cells only | Intermediate filaments in glia. Related to regulation of voltage-gated Na+, K+ and Ca2+ channels | |
| HMBS | 0 | 6 | Ubiquitous | Intermediate step in heme production, which is necessary for cytochrome production in the mitochondrial respiratory chain | |
| MEFV | 12 | 2 | Ubiquitous, yet higher in blood cells | Modulates innate immunity. Promotes inflammasome formation and maturation of IL-beta, which can induce TNF signaling | |
| OTC | 0 | 54 | Ubiquitous, yet higher in liver | Mitochondrial matrix enzyme involved in the urea cycle to detoxify ammonia into urea for excretion | |
| POGZ | 7 | 10 | Ubiquitous | Regulates mitotic progression. Interacts with CHAMP1 and thus may regulate cation transport | |
| POLG | 9 | 2 | Ubiquitous, highest in muscle & nerve | Replication and proof-reading of mitochondrial DNA | |
| PPM1D | 3 | 8 | Ubiquitous, including nervous tissue | Regulates the DNA damage response through p53 & ATM. ATM affects mitochondrial homeostasis & modulates mitophagy. | |
| RYR2 | 15 | 4 | Highest in heart and nerve, including brain and vagus | Stress-activated calcium channel on the endoplasmic reticulum | |
| SCN4A | 20 | 0 | Skeletal muscle (e.g., diaphragm), also in brain | Transmembrane pore-forming subunit of a voltage-gated sodium channel | |
| SCN9A | 8 | 0 | Ubiquitous, high in sensory neurons (e.g., in viscera, vagus) | Transmembrane pore-forming subunit of a voltage-gated sodium channel | |
| SCN10A | 9 | 0 | Ubiquitous, including in sensory neurons (e.g., vagus) | Transmembrane pore-forming subunit of a voltage-gated sodium channel | |
| SLC2A1 | 0 | 11 | Brain astrocytes, placenta, and erythrocytes | Major glucose transporter across the blood-brain barrier | |
| TNFRSF1A | 10 | 0 | Ubiquitous including brain, vagus, and glia | Binds TNF-alpha, important in inflammation; involved in mitophagy and glial/neuronal excitation; binds TRAP1 | |
| TRAP1 | 14 | 2 | Mitochondria; ubiquitous, including brain, vagus, and glia | Mitochondrial chaperone protein induced in times of oxidative stress; involved in mitophagy | |
| TRPA1 | 7 | 0 | Ubiquitous, highest in gut, also present in nerve (e.g., vagus) | Non-specific cation channel involved in perception of pain, cold, itch, sound, and oxygen concentration | |
| TUBB3 | 0 | 9 | Highest in brain, present in vagus | A beta tubulin protein that forms microtubules in neurons | |
| mtDNA | - | - | Ubiquitous, highest in nerve and muscle | Encodes subunits of the respiratory chain |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



