Submitted:
15 June 2023
Posted:
16 June 2023
You are already at the latest version
Abstract
Understanding the binding behavior and conformational dynamics of intrinsically disordered proteins (IDPs) is crucial for unraveling their regulatory roles in biological processes. However, their lack of stable 3D structures poses challenges for analysis. To address this, we propose an algorithm that explores IDP binding behavior with protein complexes by extracting topological and geometric features from the protein surface model. Our algorithm identifies a geometrically favorable binding pose for the IDP and plans a feasible trajectory to evaluate its transition to the docking position. We focus on IDPs from Homo sapiens and Mus-musculus, investigating their interaction with the Plasmodium Falciparum (PF) pathogen associated with malaria-related deaths. We compare our algorithm with HawkDock and HDOCK docking tools for quantitative (computation time) and qualitative (binding affinity) measures. Our results indicate that our method outperforms the compared methods in computation performance and binding affinity of experimental conformations.
Keywords:
Intrinsically Disordered Proteins
; protein-protein interaction
; Geometric features
; Binding affinity
; Rigid-body docking
1. Introduction
Intrinsically Disordered Proteins (IDPs) are involved in many biological processes, such as cell regulation and signaling, and their malfunction gets linked to severe pathologies [1,2,3]. Understanding the functional roles of IDPs requires studying their interactions with other proteins, which is very challenging and needs a tight coupling of experimental and computational methods. In contrast to structured/globular proteins, it is not easy to represent IDPs by a single conformation, and their models require ensembles of conformations representing a distribution of states that the protein adopts in solution. Thus, investigation of IDP interaction with structured/globular proteins is indispensable for understanding many biological mechanisms [4]. In terms of applications, understanding such molecular interactions is essential for drug design in pharmacology or protein engineering in biotechnology.
IDPs do not have distinct, well-defined secondary and tertiary structures because of their remarkable backbone flexibility [5]. When an IDP binds to a macromolecule (usually another protein), the large interfaces get involved, resulting in specific but comparatively weak interactions. IDPs common in genomes and proteomes of a living organism have many occurrences in eukaryote groups. They are prevalent in various human diseases and enriched in cardiovascular disease, diabetes, cancer, and neuro-degenerative disease-related proteins [6]. The disordered region can happen spontaneously because millions of copies of proteins get generated during the lifetime of an organism. Making humans become an easy target for many infectious diseases during host-pathogen interactions [7,8]. Pathogen like Plasmodium Falciparum (PF) is a protozoan parasite of humans that inflicts damage to the human immune system and are responsible for most malaria-related deaths [9]. Plasmodium infection of mammals begins with the injection of the sporozoite into the skin of the vertebrate host during the bite of a female Anopheles mosquito. This results in growth and multiplication first in the liver cells and then in the red blood cells leading to kidney failure, severe anemia, and many more [10]. We consider host-pathogen interaction between PF pathogen and the human/mice IDPs to study and analyze the binding behavior of IDPs in structure-based molecular interactions.
The intrinsic disorder poses a challenge for both experimental analyses of the conformation and computational modeling due to the lack of stable structure. In spite of the instability, it is critical to understand the biological functionality during protein-protein interactions. Several rigid-body docking techniques have emerged as helpful tools to assess the prediction of possible interacting pose between two protein bio-molecules for global docking [11]. These docking servers sample conformations of the smaller protein bio-molecule around the larger one and use the scoring functions to determine the top docking predictions. ZDOCK [12], RDOCK [13], and pyDock [14] use Fast Fourier Transforms (FFTs) based algorithms, RosettaDock [15] is built on Monte Carlo (MC) based multi-scale docking algorithm, while FRODOCK [16], and HDOCK [17] use knowledge-based approach to predict the translational and rotational orientation of the interacting proteins. Another tool, HawkDock [18], combines the ATTRACT [19] docking algorithm to predict several binding poses and determines the near-native docking using the HawkRank score. However, these tools have not considered the topology and geometry of the protein to analyze the binding site for structure-based molecular docking.
In this work, we propose a topology-based rigid-body docking algorithm that takes the protein surface models of the globular proteins to predict a binding conformation for the interacting IDPs. Our approach extracts the topological and geometric properties of the protein surface to generate random IDP conformation ensembles around it. It then ranks the conformation ensembles based on the docking score to find the geometrically favorable pose. The algorithm examines the score values to select the geometrically-favorable binding position and plans a feasible trajectory from IDP’s initial location to it. Our method can be used, as a tool, to find the best docking position that is geometrically fitting on the protein surface model when no information other than the individual structures is available. Figure 1 shows an overview of our workflow.
We perform experiments for nine globular proteins interacting with six IDP molecules in the conformation space. We consider the tertiary structure of the globular proteins, ranging between 173-1544 residues, as a stationary object and the rigid body of IDPs as a moving object. Our results show quantitative (i.e., computation time) and qualitative (i.e., binding affinity) analysis of our tool compared to two publicly available tools, i.e., HawkDock and HDOCK. We evaluate the interaction of all IDPs with all proteins over 1250 experiments with values averaged over ten runs in each case and plan a path to the binding pose with the highest score among the top 10 predicted conformations.
2. Background Work
2.1. Protein-protein interactions
An important area of study includes understanding how a protein binds to another protein’s active site and what conformational changes both molecules undergo during docking to the active site or its exit from it. Such information allows for predicting the possibility of an association between protein-protein pairs, the strength of this association, and the protein activity level. Protein function evaluation is a challenging task approached by various sequence-based and structural-based methods [20]. However, the fact that the function of a protein is intrinsically related to its 3D conformation (more than to its primary sequence) motivates the use of structure in predicting protein function [21,22]. During protein-protein interactions, the geometrical structure of the underlying topological manifold play crucial roles that affect specific biologically related functions, such as driving the cellular immune response [23]. To this end, various developed computational approaches predict the 3D conformation for molecular docking [24,25,26,27], where the bio-molecules bind to the protein regions with potential coherence of matching (concave) curvatures. Work in [28] presented an AutoDock-based incremental protocol (DINC) that addresses the limitations of AutoDock’s standard protocol by enabling improved docking of large bio-molecules. DINC performs docking using AutoDock incrementally instead of in one single step by dividing the docking problem into smaller sub-problems.
Another interesting docking conformation prediction tool in [18] integrates the rigid-body docking protocol of ATTRACT [19] docking algorithm to predict several binding poses and determines the near-native docking using HawkRank score. Similarly, HDOCK [17] is a hybrid docking algorithm that combines template-based modeling and template-free docking. The method overcomes misleading templates by switching to a template-free docking protocol and calculates the docking energy score using a knowledge-based iterative scoring function. However, these docking servers are limited to the number of residues or the size of the receptors, which results in failure or degradation of their performance. Our method overcomes this limitation by focusing on the features of the protein surface model independent of its size. Research in [11,29,30] reviewed rigid-body docking methods and experimentally showed that rigid-body docking provides better accuracy than flexible docking. In this work, we perform rigid-body docking to evaluate the binding behavior of IDP on interaction with globular proteins.
Many studies have used a graph representation of the protein, indicative of its geometry and topology, to predict protein function [31]. The topology of protein bio-molecule has shown to be surprisingly effective in simplifying bio-molecular structural complexity attracting attention to a better understanding of bio-molecular behavior during protein-protein interactions. Work in [32] proposed a set of topological methods to examine possible biases introduced in protein-protein interaction network data. Menglun et al. in [33] presented a topology-based network tree to predict PPI using convolutional neural networks (CNN). They characterized PPIs using an element- and site-specific persistent homology. Likewise, the authors in [34] introduced an ensemble learning approach for PPI prediction that integrated multiple learning algorithms and different protein-pair representations. Unlike the discussed strategies, we utilize the topological information of the protein surface to extract the geometric features that help predict the IDP conformation ensembles. Our method benefits from using topological data analysis tools rather than deep learning methods and overcomes the supervised learning time overhead for precise feature extraction.
2.2. Studied biological mechanisms of IDPs
Studying the conformation of highly dynamic IDPs is a challenge in structural biology [35]. Nuclear Magnetic Resonance (NMR), often used in the study of IDPs [36], is a versatile spectroscopy method for studying proteins that, importantly, do not require crystallization. However, NMR spectral data from IDP ensembles have provided conformational constraints. The NMR-constrained molecular dynamics (MD) [37] simulations need multiple copies of the protein (known as replicate exchange MD) to generate possible structural models which fail to ensure the validity of the result regardless of the method used to sample the conformations using NMR data.
Research in [38] used NMR to characterize the structure and dynamics of IDPs in various functional states and environments. It describes the NMR parameters of the structural ensemble to quantify the conformational propensities of IDPs and the challenges associated with obtaining structural models of dynamic protein-protein complexes involving IDPs. Another survey [39] summarized the recent developments in computational IDP drug design strategies and analyzed the typical properties of reported IDP-binding compounds (iIDPs) as potential drug targets. Researchers have used the combination of molecular dynamics simulations and circuit topology (CT) to analyze the biological behavior of a human androgen receptor with a large N-terminal domain (AR-NTD) [40]. The method constructed the circuit topology of a potentially charged bio-molecule to analyze the fluctuations in the chain using the root-mean-square-fluctuations (RMSF) and root-mean-square-deviations (RMSD) metrics. Although the interaction of IDPs with other bio-molecules is a critical problem that needs a good understanding of IDP’s functionality for drug design, there is little effort devoted to investigating the behavior of IDPs using the surface properties of the binding protein. As a result, we take the first step to evaluate the binding behavior of IDPs through our algorithm using the topological and geometric properties of the bio-molecules.
2.3. Sampling Based Motion Planners (SBMP)
A particular domain of molecular modeling relates to the prediction of the binding structure of protein-protein complexes; this problem is usually addressed with computational methods. The method is required to accurately predict the 3D conformation of the bio-molecule upon binding to the target receptor. A new research area has tried applying robotics-based motion planning techniques to this problem [41,42,43,44], where it randomly samples alternative conformations, in consideration to the position and orientation of the bio-molecule inside the receptor’s binding cleft and plans a feasible path to the binding conformation. The space under which the degrees of freedom (i.e., the number of parameters, like residues or C- atoms, needed to describe the pose) of a bio-molecule explored is called conformation space and the regions free of all internal and external constraints are called space in the conformation space.
Vojtĕch et al. in [45] used Rapidly Exploring Random Tree (RRT) [46] to explore the void space in each frame of the protein dynamics to reconstruct a dynamic tunnel by back-tracking the tree towards the active site. The tunnel paths from an inner protein active site to its surface provide insight into important protein properties (e.g., their stability or activity) in the interaction network. Work in [43] provides a proof-of-concept for mimicking ligand flexibility in rigid body molecular docking that can run efficiently in commodity hardware. The method simulates user search performance with a path optimization algorithm for interactive molecular docking. Research in [47] presented a hybrid algorithm that combines Monte-Carlo sampling and RRT* to explore conformational pathways for large-scale motions. The method improves upon their previous work to produce optimized conformational routes through accurate and efficient search in the conformational space. Recent work in [48] proposed a parallel implementation of a multi-tree variant of the Transition-based Rapidly-exploring Random Tree (TRRT) that globally explores the conformation space for the IDPs. The method performs a randomized exploration of the conformation space to find probable transition paths between stable states of a molecule using the potential energy cost map. Instead, we utilize the topological and geometric properties of the protein structure to generate the IDP conformation ensembles around it.
In this work, taking inspiration from our prior work, we present a new bio-topology algorithm that randomly explores the rotational and translational degrees of freedom of IDPs without exploring its conformational flexibility for rigid docking. The approach utilizes the topological and geometric properties of the protein surface to identify the geometrically suitable structure arrangement of an IDP around a protein receptor and finally plans a feasible path to it.
3. Materials and Methods
3.1. Mathematical Definitions
We discuss some of the mathematical concepts used in our algorithm to extract the topological and geometric features of the protein surface.
Definition 1.
(Abstract Simplicial complex) An abstract simplicial complex K, i.e., a collection of sets closed under the subset operation, is a generalization of a graph useful in representing higher-than-pairwise connectivity relationships.
The elements of the set are called vertices, and the set itself is a simplex. The vertices refer to IDP conformation in the conformation space.
Definition 2.
(Vietoris-Rips complex) Given a set S of points in Euclidean space E, the Vietoris-Rips complex is the abstract simplicial complex whose k-simplices are the subsets of points in S with diameter that is at most ε.
In this work, protein surface models are static objects. S defines the group of all IDP conformations in the simplicial complex . These conformations are generated at a radial distance away from the surface to avoid collisions, such that . We take as the diameter of the circumscribed circle of the IDP bio-molecule. Considering the above parameters, we define the discrete Morse function as follows.
Definition 3.
Let D be the Euclidean distance function that measures the distance between the point and the nearest point y on the protein surface P that is, .
Definition 4.
Let be a density function where 0 and y be the point on the protein surface. The function Γ counts all neighbors close to y in S within distance ϱ.
Definition 5.
Let f be a discrete Morse function on restricted to the vertices of the Vietoris-Rips complex. We formally define f at any point in conformation space by
Please refer to [49] for our expanded definitions and theorems.
Definition 6.
(Critical points) The set of critical points is defined as the set of non-degenerate points on the surface of protein when the given discrete Morse function f reaches its extreme values, i.e., local minima or maxima.
Definition 7.
(Feasible critical points) This set is defined as all possible IDP conformations in S at a radial distance of ϱ from a critical point on the protein surface. In other words, it is the union of intersections of vertices in S within the metric balls of radius ϱ centered at some critical point.
Overall, our method first generates a simplicial complex that captures the topological structure of the globular protein surface, i.e., vertices, edges, and triangles. Then apply the discrete Morse function on the same simplicial complex to extract the critical points information of the surface and identify the feasible critical points (i.e., IDP conformation ensembles) close to the surface. The discrete setting of Morse theory avoids the overhead of differential geometry, thus, reducing the computation complexity for high dimensional structures. The upcoming section discusses the algorithmic details of our method.
3.2. Finding a suitable docking conformation
Algorithm 1 constructs a simplicial complex around the protein surface by sampling and connecting IDP conformations in method ConstructComplex. On satisfying the sampling condition from [50], the algorithm performs topological collapse to remove redundant topological information, i.e., vertices and edges and provides a skeleton of the simplicial complex around the protein surface in line 3, i.e., a surface mesh. Recall that we refer to vertices as the IDP conformations, and the edges are the lines that connect the to/fro movements of IDP between two conformations. It applies discrete Morse function f from [49] to this simplicial complex to identify the local maxima (protrusions) and minima (cavity) curvatures of the protein surface, i.e., critical points, in line 4. The identified critical points are the highest and the lowest peak points on the surface at which function f reaches its extremum.
 |
The algorithm then extracts the feasible critical points at radial distance from the identified critical points of the protein surface in line 5. These feasible critical points are the conformations in close proximity to the protein surface and are part of the simplicial complex , refer to Def.7. Next, we consider the closest conformations as the set of predicted conformations for an IDP and use it to evaluate and determine the ranks of the conformations using Eq. 2. From the predicted conformations, a geometrically favorable binding position of the IDP gets selected such that the conformation is closest to protein surface curvature in lines 9-14. We use the Hausdorff distance to measure the distance between the protein surface (P) and the IDP conformation (I) to find the geometrically suitable docking position, as discussed below.
It takes the conformation with the minimum Hausdorff distance as the final docking position from all the possibly generated conformations. Finally, a path is planned for the IDP from the start conformation to the binding pose conformation taking the other predicted IDP conformations as waypoints (line 14). The process of selecting a binding pose happens internally, where our method ranks and automatically chooses the binding conformation to plan the path during the interaction of protein-protein complexes. As a result, our algorithm outputs an extracted geometric information map consisting of critical points, feasible critical points (predicted IDP conformations), and a pathway from the start conformation to the binding pose conformation.
4. Experimental Data
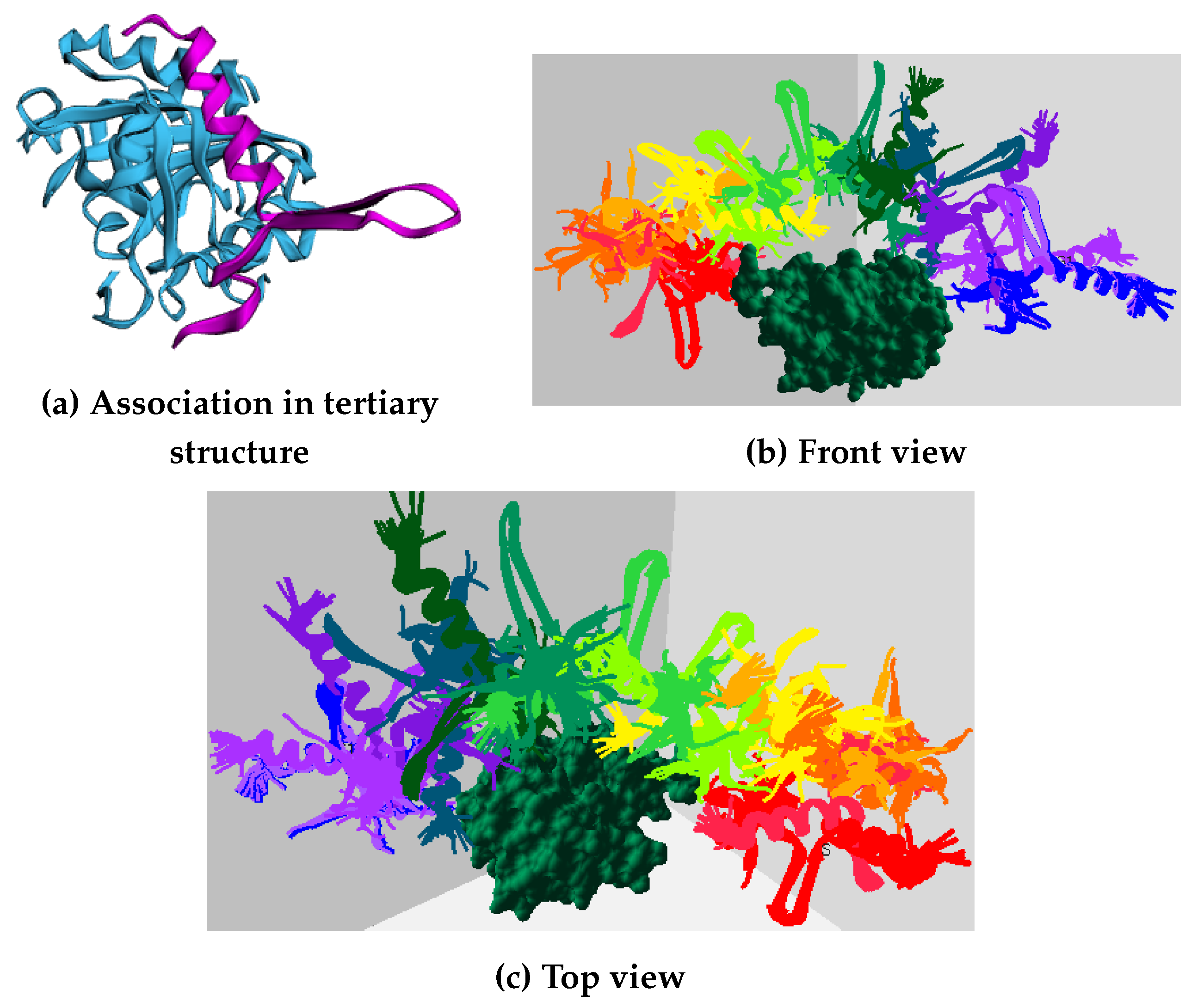
We obtain protein data from the protein data bank (PDB) [52,53] and construct their tertiary structure using CHIMERA [54]. We obtain IDP data from PDB and AlphaFold Protein Structure Database (AlphaFold DB) [55]. We consider nine proteins and six IDP bio-molecules to study and understand the biological binding mechanism of IDPs using protein surface geometries. The high-dimensional surface models of proteins represent a stationary rigid body in the conformation space. Figure 2 shows the tertiary-structure representation of 3SRI protein, its high-dimensional surface model, and the IDP conformation ensembles around it.
The proteins selected include nine Plasmodium Falciparum (PF) pathogen proteins, i.e., 1SQ6, 1TQX, 2MU6, 3NTJ, 3SRI, 4JUE, 4M1N, 6ZRY and 7F9K, as shown in Figure 3 and Figure 4. PF is responsible for most malaria-related deaths and forms part of our ongoing research into identifying feasible protein drug targets. The high mutational capacity, coupled with the changing metabolism of the pathogen, makes the development of malaria drug treatments an evolving problem. In this work, we are interested in studying and analyzing the behavior of PF pathogens in the PPI network. Hence, these proteins were selected as they are the potential targets for malaria inflicts.
We selected 1KRN (88 residues), 2LE3 (42 residues), 5EJW (91 residues), and 7KPI (142 residues) proteins as IDP based on their high disorder behavior shown in the protein feature view plot available on the PDB database. The other IDP bio-molecules, AF-I1E4Y1-F1 (117 residues) and AF-P59773-F1 (190 residues), from AlphaFold DB, are of mus-musculus and homo sapiens species, respectively. The mean per-residue confidence score (pLDDT) for AF-I1E4Y1-F1 is 48, and for AF-P59773-F1 is 59. The pLDDT measure estimates whether the predicted residue has similar distances to neighboring C- atoms (within 15 ) in agreement with the naive structure and scored between 0 and 100. The score assesses the local model quality of the structure, i.e., a lower score refers to the existence of more disordered regions in a bio-molecule. The selected IDPs are bio-molecules of humans and mice susceptible to malaria.
5. Results
We performed experiments on a Dell Alienware Aurora desktop machine running Ubuntu 20.4 LTS operating system and developed algorithms in C++ language using PMPL library [56]. We evaluate performance using quantitative and qualitative measures for all IDPs with each PF protein for geometric feature extraction, path planning to dock position, and binding affinity measure. Overall we executed 1250 experiments and averaged the result values over 10 runs. We compare our method’s performance with two baseline methods, i.e., HawkDock [18] and HDOCK [17].
5.1. Quantitative Analysis
5.1.1. Extracting geometric features of the protein surface
Recall that our method constructs a surface mesh (or simplicial complex) around the considered protein surface models to abstract the topological and geometric information. During the execution, it randomly samples the IDP conformations around the protein surface model, thus, constructing a manifold mesh representation to capture the topology of the protein’s surface. These topological features aid in identifying the geometric properties of the protein surface, i.e., minima or maxima, for better approximation, as shown in Figure 1. Next, our method uses geometric information to find possible binding conformations around the protein surface and apply the scoring function from equation 2 to get the top 10 geometrically-fitting docking conformations for an IDP. Figure 2c shows the top 10 predicted association conformations of 1KRN IDP around the 3SRI protein surface model. We observed that the feature extraction process is independent of the globular protein’s size and has minimal effect on the performance of our algorithm, making it suitable for macro-molecules, as discussed next.
5.1.2. Computational time
We analyze the computation time (in seconds) required for feature extraction and prediction of geometrically favorable docking conformations in these IDP-protein interactions. This study includes all IDPs in nine PF pathogen protein conformation spaces. The feature extraction time measures the duration of extracting topological and geometric features from the globular protein surface, while the ranking time finds the top 10 conformations. To assess our algorithm’s performance efficiency, we compare our total time to output the top 10 docking conformations with HawkDock and HDOCK, as depicted in Figure 5.
Our method demonstrates the faster prediction of IDP binding conformations compared to HawkDock and HDOCK in all PF pathogen protein conformation spaces except for 2LE3. The smaller size of the 2LE3 IDP leads to a longer conformation sampling time necessary for accurate feature capture in large or complex-size proteins. However, this difference does not affect our method’s performance significantly and results in less time overhead compared to the baseline methods. Figure 5 highlights that our method outperforms the baseline methods in most protein conformation spaces, despite minimal time overhead.
In particular, HawkDock fails to find docking conformations for the 3NTJ protein due to its limitation to proteins with fewer than 1000 amino acids.
We analyzed that using the geometric information of protein surface, it is still possible to predict multiple structural arrangements of IDPs around the proteins to find the closest interacting binding pose between two bio-molecules without declining computation performance. Thus, we can conclude that the amount of data assessed by our method does not impact its surface approximation and still provides a quantitatively good performance.
5.2. Qualitative Analysis
5.2.1. Selecting the suitable binding conformation
As initially mentioned, our method predicts the top 10 docking conformations for an IDP across all PF pathogen protein conformation spaces. This process iterates over ten times, and for each iteration, we record the top 10 conformations to assess the likelihood of obtaining the same conformation from ten random iterations. The recorded outputs are then further analyzed to identify the IDP conformation with the highest frequency as the most suitable docking position among the ten experimental runs. This selected conformation is then subsequently utilized as input for path planning. In Figure 4, examples of best binding poses (goal positions) for 3 IDPs are depicted in blue. To validate the quality of the chosen binding pose for protein-protein interactions, we examine the binding affinity before proceeding with path planning, as elaborated in the subsequent discussion.
5.2.2. Binding affinity measure
We compare the binding affinity of our IDP binding conformation with the binding affinity computed for the IDP conformations predicted by HawkDock and HDOCK methods across all PF pathogen proteins. The molar Gibbs free energy G is used to assess the relevance of the binding pose. Gibbs free energy is a thermodynamic potential that quantifies the maximum reversible work capacity of a thermodynamic system under constant temperature and pressure (isothermal, isobaric) conditions [57]. Protein binding occurs when the change in Gibbs free energy G is negative, indicating equilibrium at constant pressure and temperature.
We utilize the molar Gibbs free energy G to calculate the binding affinity of the top-ranked IDP conformation ensemble predicted by all three methods. Figure 6 illustrates the binding affinity measure of our predicted IDP binding pose for each protein compared to the binding affinity measures obtained for the IDP conformations predicted by HawkDock and HDOCK methods. HawkDock exhibits a positive binding affinity for the 7KPI IDP conformation when interacting with 1SQ6 and 4JUE proteins. In contrast, our algorithm consistently predicts IDP conformations with negative binding affinities for all IDPs interacting with PF pathogen protein complexes. This evidence indicates a stronger association displayed by our geometrically-favorable docking positions and consistency achieved in identifying favorable binding conformations through our method.
As mentioned previously, HawkDock failed to predict the docking conformation for the 3NTJ protein, resulting in the absence of a binding affinity measure for this case.
Based on the observations in Figure 6, we consistently find that our predicted docking conformations exhibit a negative binding affinity for all IDPs, surpassing the binding affinity of the IDP conformation ensemble generated by the baseline methods. Additionally, we deduce that our method performs well even for macro-molecule proteins, such as 3NTJ, surpassing HDOCK and not being limited to small bio-molecules. Overall, our experimental conformations demonstrate better binding affinity in 95% of the compared cases, highlighting the significance of utilizing protein surface model features in generating conformations with favorable binding affinity outcomes. Consequently, we can conclude that the quality of our binding conformations competes favorably with the binding conformations predicted by existing approaches utilizing coarse-grained force field docking (HawkDock) and knowledge-based template-free docking (HDOCK).
5.3. Path planning to geometrically-favorable binding position
In addition to predicting binding conformations for rigid-body docking, our method also includes feasible trajectory planning toward the selected finalized binding pose during re-scoring. We assess the total time required for path planning to the predicted binding pose for all IDPs in the nine globular protein conformation spaces, as presented in Figure 7. The path planning time represents the duration incurred for an IDP to transition from its initial conformation to the binding conformation while moving closer to the protein surface.
Figure 7 illustrates the distribution of path planning times for all IDPs across different protein conformation spaces. The y-axis represents the averaged path planning time over ten runs, while the x-axis represents the IDPs interacting with the respective proteins. The plot showcases the variability in path planning time, depicting the duration needed to move IDPs from their initial positions to the docking positions around the protein surface. In several protein conformation spaces, the difference between the minimum and maximum planning times is small or negligible for IDPs exhibiting a lower deviation, indicating that the planner consistently finds a similar route majority of times out of the ten runs. However, the 1KRN and 7KPI IDPs in the 1SQ6 protein’s conformation space take longer time spans. This behavior can be attributed to the broader structure of these IDPs, affecting their movement near the 1SQ6 protein surface and resulting in varying time values. Among all the studied IDPs, AF-P59773-F1 demonstrates the vast structure and highest disordered regions, making it challenging to plan its path while considering its structural transformations. Thus, we deduce from Figure 7 that the path planning period for the AF-P59773-F1 IDP is generally higher than other IDPs in most protein conformation spaces.
The unpredictable behavior of IDPs around the studied proteins enables us to analyze the feasibility of their interaction with specific proteins, particularly how easy they align around a protein structure for association. Path planning time provides insights into the locomotion of IDPs around proteins as they search for the most suitable binding pose for rigid-body docking. When used in conjunction with other tools to examine the conformational flexibility of IDPs during their motion around proteins can simplify flexible docking tasks by focusing computational methods solely on the dynamic structure of IDP conformations as they traverse the planned trajectory, facilitating future biological studies.
Figure 8 displays screenshots of the planned path for the 2LE3 IDP around the 1SQ6 protein surface, depicting the motion of the IDP biomolecule from its initial position to the experimentally predicted binding pose conformation. Different view angles illustrate the IDP’s movement around the protein surface, and the intermediate conformations represent the IDP conformations generated during feature extraction, serving as waypoints. These intermediate conformations between the starting and goal positions demonstrate the structural transformations of the 2LE3 IDP as it moves in the vicinity of the 1SQ6 protein surface. Similar movements and structural arrangements occur for remaining IDPs across different protein conformation spaces.
We conclude that our approach successfully captures the geometric features of the protein surfaces and plans a path for IDP bio-molecule to the geometrically favorable binding pose showing a higher affinity compared to affinity measures by baseline methods. Thus, the work showed the significance of our approach for further biological studies.
6. Conclusion
The paper presents a framework that utilizes topological and geometric information from the structured protein surface to investigate the binding behavior of IDPs. The study assesses the performance efficiency and quality of predicted experimental IDP conformations, comparing them to state-of-the-art methods. Additionally, it reports the path planning time required to determine a transition path to the docking position.
The experimental results demonstrate that our method successfully predicts geometrically suitable binding poses for IDPs around protein surface models, specifically for rigid docking. Moreover, our approach outperforms the compared methods in computational performance and the predicted conformation quality. This research serves as an initial step towards further analyzing IDPs and their interactions with other biomolecules, leveraging geometric and topological representations of these entities. The future enhancement will incorporate scoring functions and binding affinity measures in our model by integrating it with computational methods designed to estimate binding affinities to benefit in determining the final association site for dynamically unstable IDPs more effectively. We plan to apply this idea to our future work and provide a prototype accessible to the research community. By achieving a geometrically suitable conformation with the lowest score and a high binding affinity, our approach presents the potential for advancing the development of structure-based vaccine design processes.
Author Contributions
Conceptualization, Aakriti Upadhyay and Chinwe Ekenna; Data curation, Aakriti Upadhyay; Formal analysis, Aakriti Upadhyay and Chinwe Ekenna; Investigation, Aakriti Upadhyay and Chinwe Ekenna; Methodology, Aakriti Upadhyay; Software, Aakriti Upadhyay and Chinwe Ekenna; Supervision, Chinwe Ekenna; Validation, Aakriti Upadhyay and Chinwe Ekenna; Visualization, Aakriti Upadhyay and Chinwe Ekenna; Writing – original draft, Aakriti Upadhyay; Writing – review & editing, Aakriti Upadhyay and Chinwe Ekenna. All authors have read and agreed to the published version of the manuscript.
Abbreviations
The following abbreviations are used in this manuscript:
| IDPs | Intrinsically Disordered Proteins |
| PPI | Protein-Protein Interaction |
References
- Csizmok, V.; Follis, A.V.; Kriwacki, R.W.; Forman-Kay, J.D. Dynamic protein interaction networks and new structural paradigms in signaling. Chemical reviews 2016, 116, 6424–6462. [Google Scholar] [CrossRef]
- Babu, M.M.; van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: regulation and disease. Current opinion in structural biology 2011, 21, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annual review of biophysics 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: emerging interaction specialists. Current opinion in structural biology 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nature reviews Molecular cell biology 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Kulkarni, P.; Uversky, V.N. Intrinsically disordered proteins in chronic diseases, 2019. [CrossRef]
- Casadevall, A.; Pirofski, L.a. Host-pathogen interactions: basic concepts of microbial commensalism, colonization, infection, and disease. Infection and immunity 2000, 68, 6511–6518. [Google Scholar] [CrossRef]
- Tobin, A.R.; Crow, R.; Urusova, D.V.; Klima, J.C.; Tolia, N.H.; Strauch, E.M. Inhibition of a malaria host–pathogen interaction by a computationally designed inhibitor. Protein Science 2023, 32, e4507. [Google Scholar] [CrossRef] [PubMed]
- Holding, P.A.; Snow, R.W. Impact of Plasmodium falciparum malaria on performance and learning: review of the evidence. The Intolerable Burden of Malaria: A New Look at the Numbers: Supplement to Volume 64 (1) of the American Journal of Tropical Medicine and Hygiene 2001. [Google Scholar] [CrossRef]
- Zuck, M.; Austin, L.S.; Danziger, S.A.; Aitchison, J.D.; Kaushansky, A. The promise of systems biology approaches for revealing host pathogen interactions in malaria. Frontiers in microbiology 2017, 8, 2183. [Google Scholar] [CrossRef]
- Sunny, S.; Jayaraj, P. Protein–protein docking: Past, present, and future. The protein journal 2022, 1–26. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, R.; Weng, Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins: Structure, Function, and Bioinformatics 2003, 53, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, B.; Pons, C.; Fernández-Recio, J. pyDockWEB: a web server for rigid-body protein–protein docking using electrostatics and desolvation scoring. Bioinformatics 2013, 29, 1698–1699. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Berrondo, M.; Weitzner, B.D.; Muthu, P.; Bergman, H.; Gray, J.J. Benchmarking and analysis of protein docking performance in Rosetta v3. 2. PloS one 2011, 6, e22477. [Google Scholar] [CrossRef]
- Ramírez-Aportela, E.; López-Blanco, J.R.; Chacón, P. FRODOCK 2.0: fast protein–protein docking server. Bioinformatics 2016, 32, 2386–2388. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein–protein docking. Nature protocols 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: a web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic acids research 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.J.; Schindler, C.E.; de Beauchêne, I.C.; Zacharias, M. A web interface for easy flexible protein-protein docking with ATTRACT. Biophysical journal 2015, 108, 462–465. [Google Scholar] [CrossRef]
- Antunes, D.A.; Abella, J.R.; Devaurs, D.; Rigo, M.M.; Kavraki, L.E. Structure-based methods for binding mode and binding affinity prediction for peptide-MHC complexes. Current topics in medicinal chemistry 2018, 18, 2239–2255. [Google Scholar] [CrossRef]
- Smith, G.R.; Sternberg, M.J. Prediction of protein-protein interactions by docking methods. Current opinion in structural biology 2002, 12, 28–35. [Google Scholar] [CrossRef]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophysical journal 2014, 107, 1785–1793. [Google Scholar] [CrossRef]
- Sable, R.; Jois, S. Surfing the protein-protein interaction surface using docking methods: application to the design of PPI inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials. Signal transduction and targeted therapy 2020, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Ciulli, A. Bifunctional chemical probes inducing protein–protein interactions. Current Opinion in Chemical Biology 2019, 52, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nature communications 2022, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Xiang, J.; Zheng, R.; Wu, F.X.; Li, M. DPCMNE: detecting protein complexes from protein-protein interaction networks via multi-level network embedding. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2021, 19, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Devaurs, D.; Antunes, D.A.; Hall-Swan, S.; Mitchell, N.; Moll, M.; Lizée, G.; Kavraki, L.E. Using parallelized incremental meta-docking can solve the conformational sampling issue when docking large ligands to proteins. BMC molecular and cell biology 2019, 20, 1–15. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Current opinion in structural biology 2008, 18, 178–184. [Google Scholar] [CrossRef]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and its limits in rigid body protein-protein docking. Structure 2020, 28, 1071–1081. [Google Scholar] [CrossRef]
- Fasoulis, R.; Paliouras, G.; Kavraki, L.E. Graph representation learning for structural proteomics. Emerging Topics in Life Sciences 2021, 5, 789–802. [Google Scholar] [CrossRef]
- Nowakowska, A.W.; Kotulska, M. Topological analysis as a tool for detection of abnormalities in protein-protein interaction data. Bioinformatics 2022. [Google Scholar] [CrossRef]
- Wang, M.; Cang, Z.; Wei, G.W. A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nature Machine Intelligence 2020, 2, 116–123. [Google Scholar] [CrossRef]
- Chen, K.H.; Wang, T.F.; Hu, Y.J. Protein-protein interaction prediction using a hybrid feature representation and a stacked generalization scheme. BMC bioinformatics 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Pauwels, K.; Lebrun, P.; Tompa, P. To be disordered or not to be disordered: is that still a question for proteins in the cell? Cellular and Molecular Life Sciences 2017, 74, 3185–3204. [Google Scholar] [CrossRef]
- Jensen, M.R.; Ruigrok, R.W.; Blackledge, M. Describing intrinsically disordered proteins at atomic resolution by NMR. Current opinion in structural biology 2013, 23, 426–435. [Google Scholar] [CrossRef]
- Allison, J.R.; Varnai, P.; Dobson, C.M.; Vendruscolo, M. Determination of the free energy landscape of α-synuclein using spin label nuclear magnetic resonance measurements. Journal of the American Chemical Society 2009, 131, 18314–18326. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Salvi, N.; Blackledge, M.; Jensen, M.R. Characterization of intrinsically disordered proteins and their dynamic complexes: From in vitro to cell-like environments. Progress in nuclear magnetic resonance spectroscopy 2018, 109, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Sun, Q.; Zhang, W.; Liu, Y.; Lai, L. Targeting intrinsically disordered proteins at the edge of chaos. Drug discovery today 2019, 24, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Sheikhhassani, V.; Scalvini, B.; Ng, J.; Heling, L.W.; Ayache, Y.; Evers, T.M.; Estébanez-Perpiñá, E.; McEwan, I.J.; Mashaghi, A. Topological dynamics of an intrinsically disordered N-terminal domain of the human androgen receptor. Protein Science 2022, 31, e4334. [Google Scholar] [CrossRef] [PubMed]
- Al-Bluwi, I.; Siméon, T.; Cortés, J. Motion planning algorithms for molecular simulations: A survey. Computer Science Review 2012, 6, 125–143. [Google Scholar] [CrossRef]
- Ekenna, C.; Thomas, S.; Amato, N.M. Adaptive local learning in sampling based motion planning for protein folding. BMC systems biology 2016, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Adamson, T.; Camarena, J.A.; Tapia, L.; Jacobson, B. Optimizing low energy pathways in receptor-ligand binding with motion planning. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). IEEE, 2019, pp. 2041–2048. [CrossRef]
- Upadhyay, A.; Tran, T.; Ekenna, C. A topology approach towards modeling activities and properties on a biomolecular surface. BIBM: IEEE International Conference on Bioinformatics and Biomedicine. IEEE, 2021. [CrossRef]
- Vonásek, V.; Jurčík, A.; Furmanová, K.; Kozlíková, B. Sampling-based motion planning for tracking evolution of dynamic tunnels in molecular dynamics simulations. Journal of Intelligent & Robotic Systems 2019, 93, 763–785. [Google Scholar] [CrossRef]
- LaValle, S.M.; others. Rapidly-exploring random trees: A new tool for path planning, 1998.
- Afrasiabi, F.; Haspel, N. Efficient exploration of protein conformational pathways using rrt* and mc. Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, 2020, pp. 1–6. [CrossRef]
- Estana, A. Algorithms and comptutational tools for the study of Intrinsically Disordered Proteins. PhD thesis, Toulouse, INSA, 2020.
- Upadhyay, A.; Goldfarb, B.; Wang, W.; Ekenna, C. A new application of discrete morse theory to optimizing safe motion planning paths. International Workshop on the Algorithmic Foundations of Robotics. Springer, 2023, pp. 18–35. [CrossRef]
- Upadhyay, A.; Wang, W.; Ekenna, C. Approximating C free Space Topology by Constructing Vietoris-Rips Complex. 2019 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS). IEEE, 2019, pp. 2517–2523. [CrossRef]
- Upadhyay, A.; Goldfarb, B.; Ekenna, C. Incremental Path Planning Algorithm via Topological Mapping with Metric Gluing. 2022 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS). IEEE, 2022, pp. 1290–1296. [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic acids research 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer Jr, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: a computer-based archival file for macromolecular structures. Journal of molecular biology 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of computational chemistry 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; others. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic acids research 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Lab, P. Parasol Planning Library. https://github.com/parasol-ppl/ppl, 2022.
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef]
Figure 1.
Workflow of our approach.
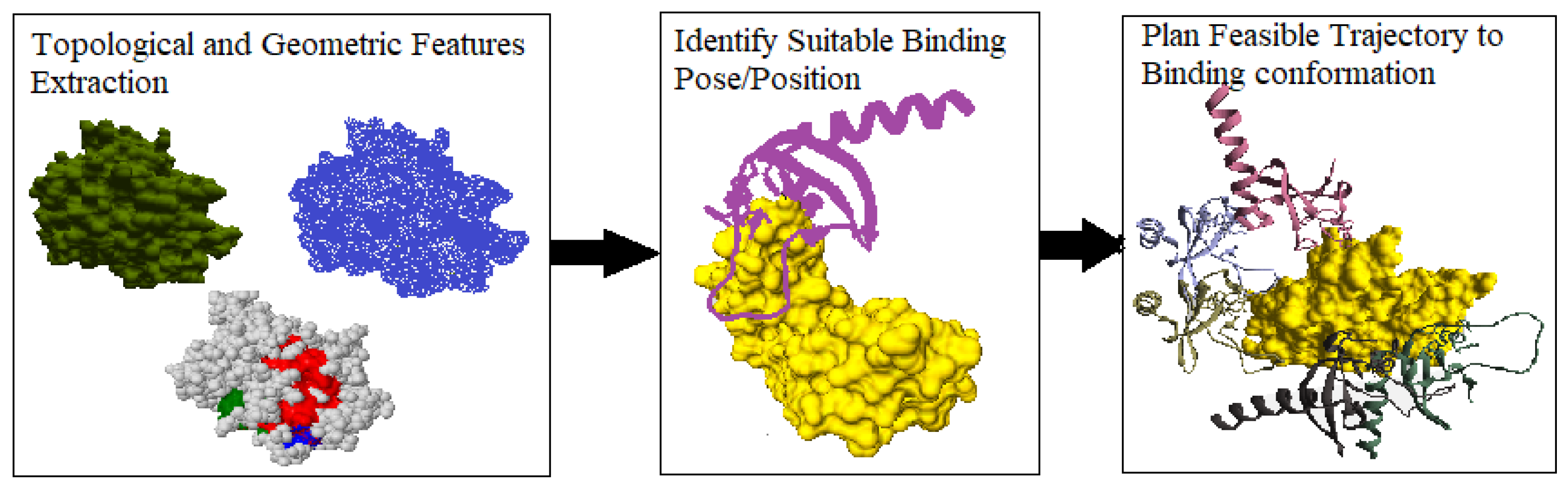
Figure 2.
The figure shows the multiscale surface model of the 3SRI protein and the predicted IDP conformations around detected geometric features (critical points) of the protein surface. The conformations viewed in (c) are the top 10 predicted 1KRN bio-molecule conformations around the surface model.
Figure 2.
The figure shows the multiscale surface model of the 3SRI protein and the predicted IDP conformations around detected geometric features (critical points) of the protein surface. The conformations viewed in (c) are the top 10 predicted 1KRN bio-molecule conformations around the surface model.
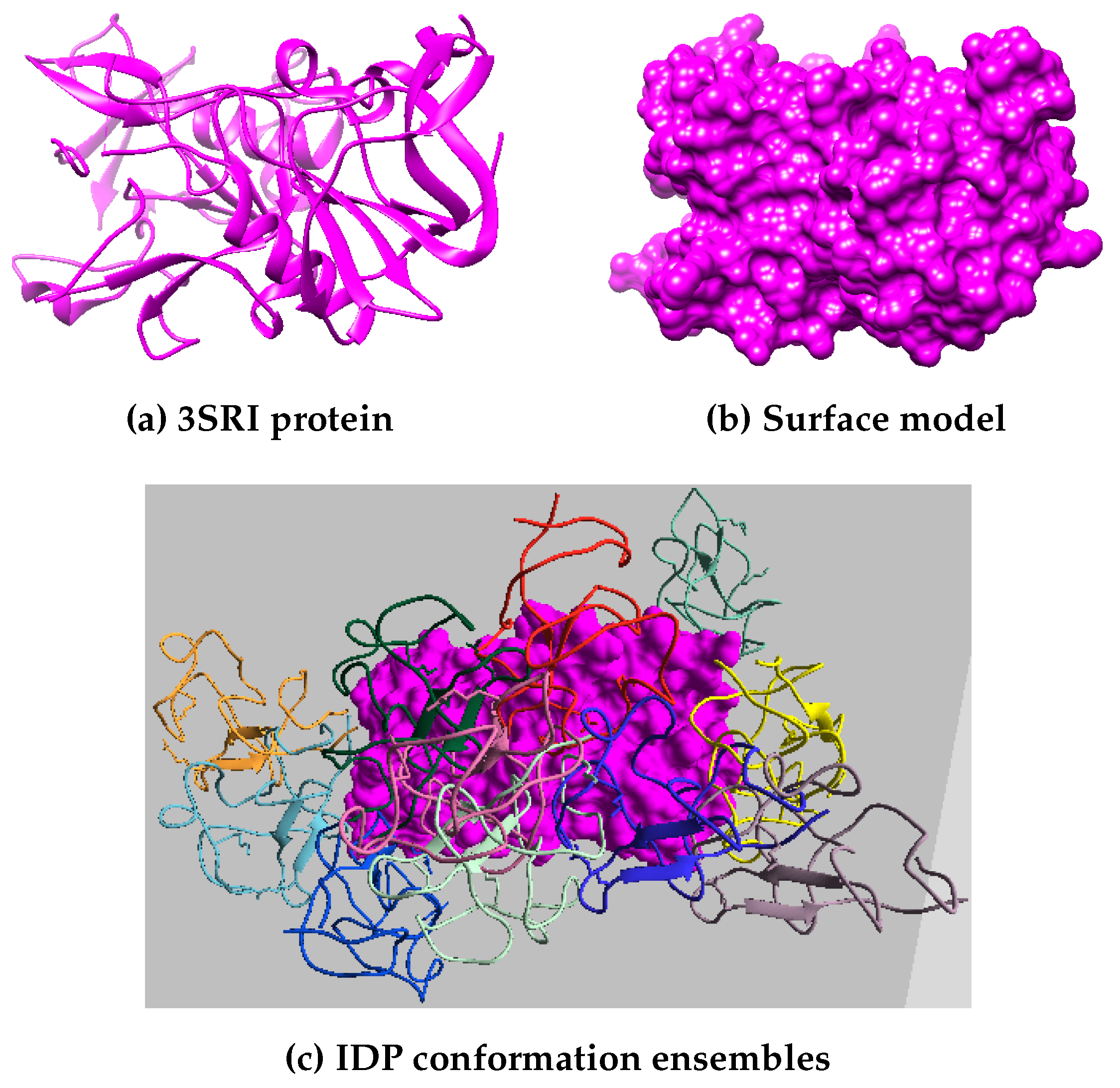
Figure 3.
The figure shows the tertiary structure of PF pathogen proteins taken into consideration for the experiment analysis.
Figure 3.
The figure shows the tertiary structure of PF pathogen proteins taken into consideration for the experiment analysis.
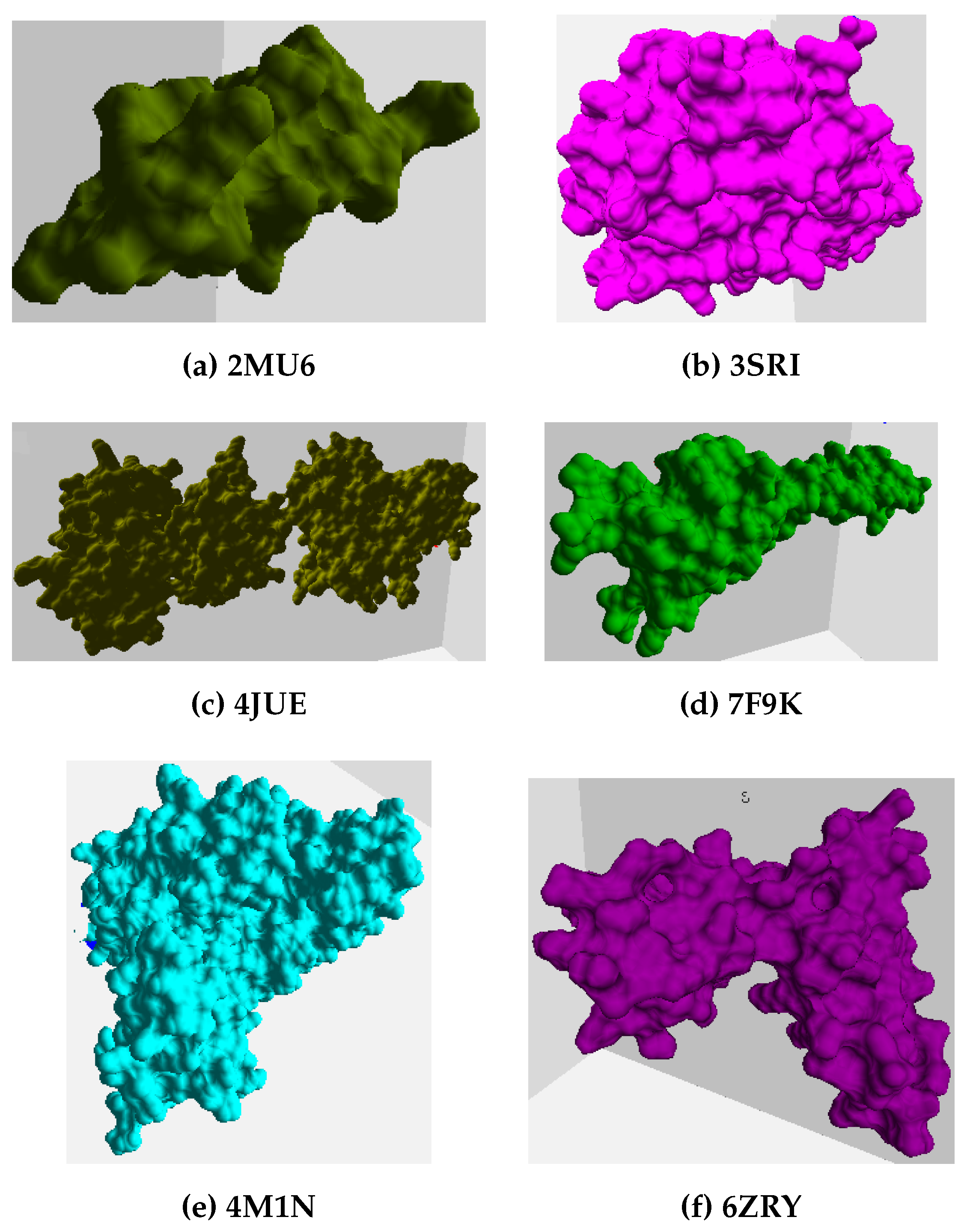
Figure 4.
The figure captures a random combination of a globular protein surface model and an IDP from the experimental analysis, with IDP names mentioned in the brackets. The red color conformation refers to the start position, and the docking position is in blue.
Figure 4.
The figure captures a random combination of a globular protein surface model and an IDP from the experimental analysis, with IDP names mentioned in the brackets. The red color conformation refers to the start position, and the docking position is in blue.
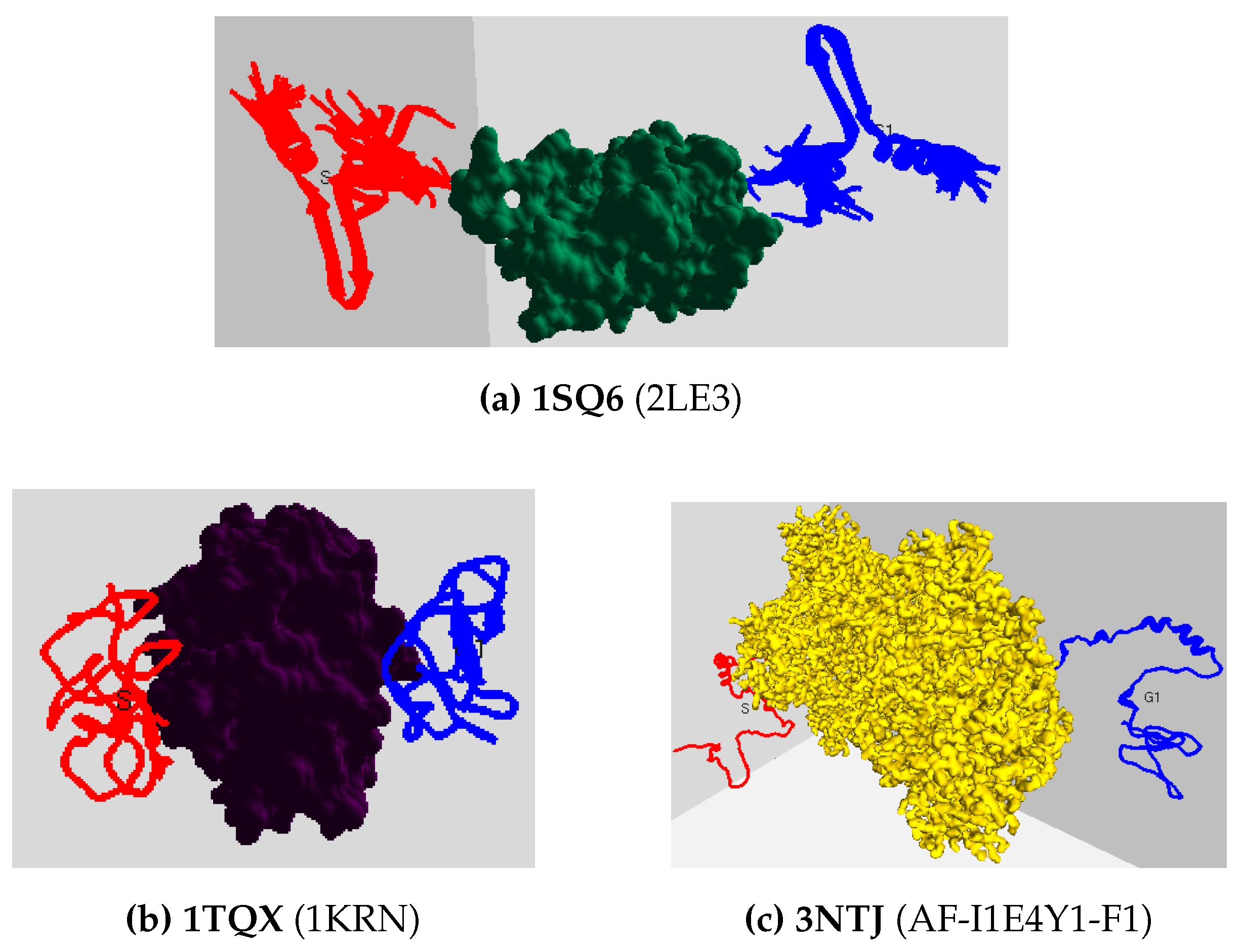
Figure 5.
The plots show the total computation time taken (in seconds) by all three methods to predict the top 10 IDP docking conformation ensembles around the protein surface model.
Figure 5.
The plots show the total computation time taken (in seconds) by all three methods to predict the top 10 IDP docking conformation ensembles around the protein surface model.
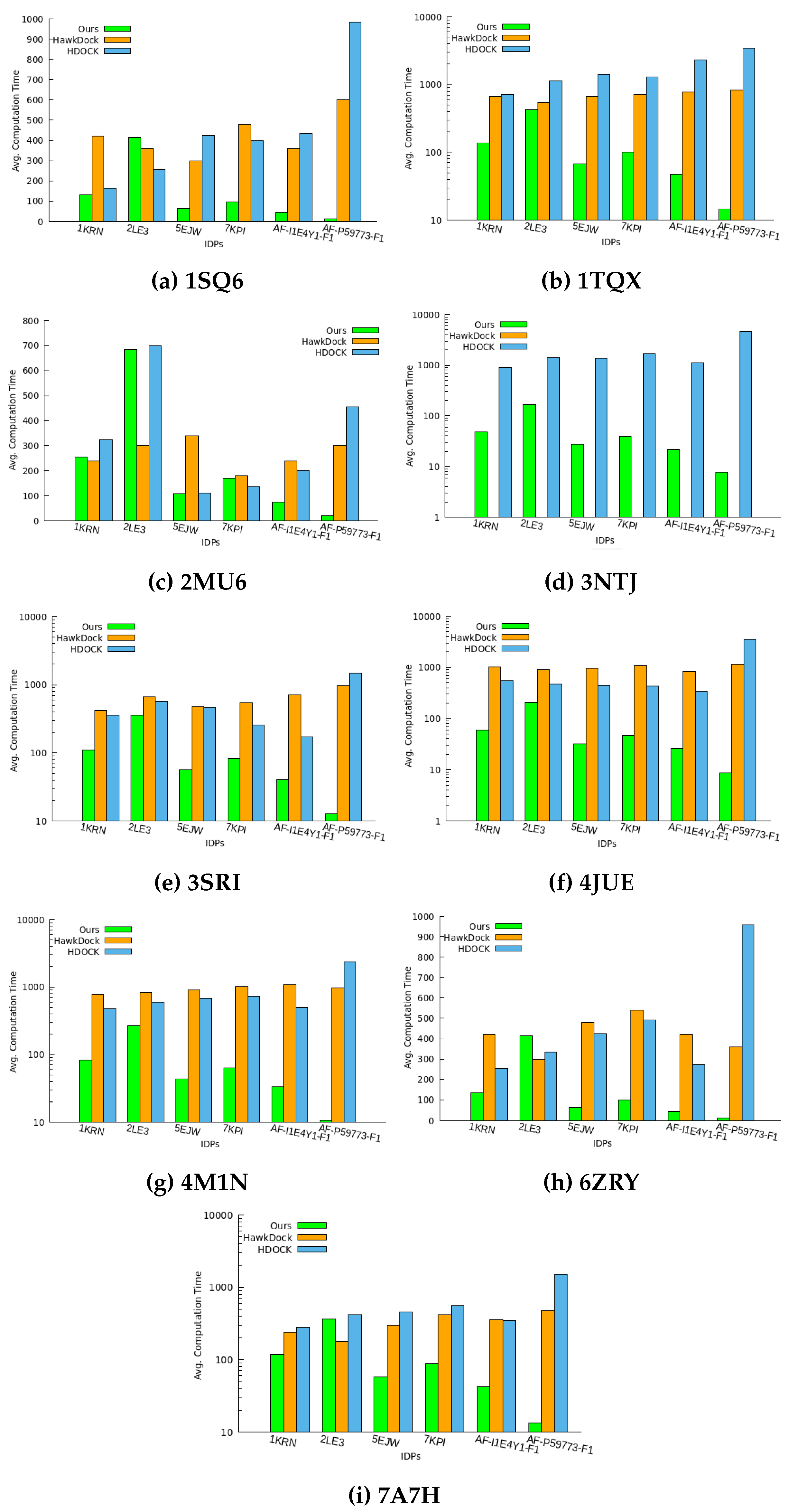
Figure 6.
The plot shows the Binding Affinity measure for the top-most IDP docking conformation predicted by the three methods.
Figure 6.
The plot shows the Binding Affinity measure for the top-most IDP docking conformation predicted by the three methods.
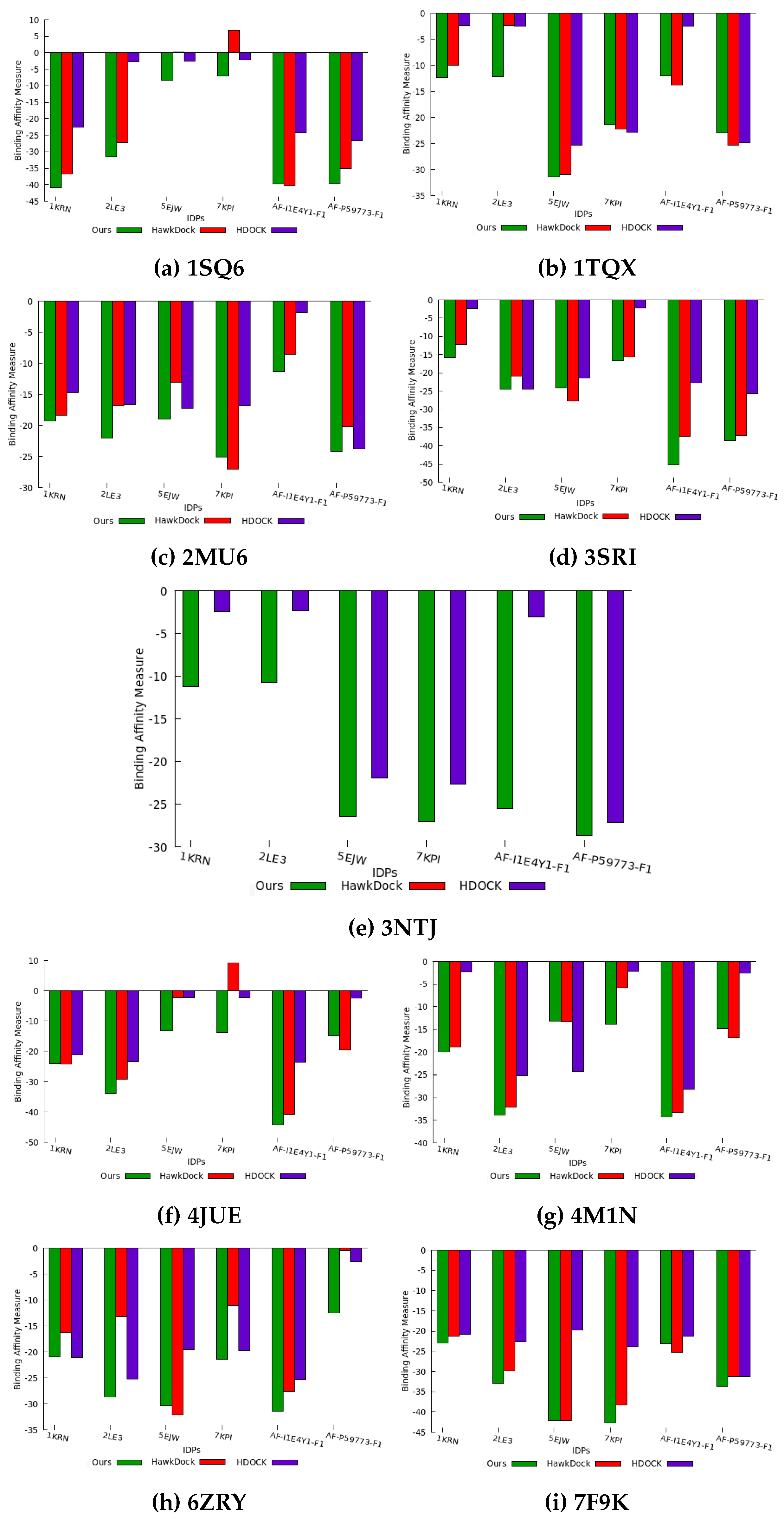
Figure 7.
The total time taken (in seconds) to plan a path for all IDPs in each protein’s conformation space.
Figure 7.
The total time taken (in seconds) to plan a path for all IDPs in each protein’s conformation space.
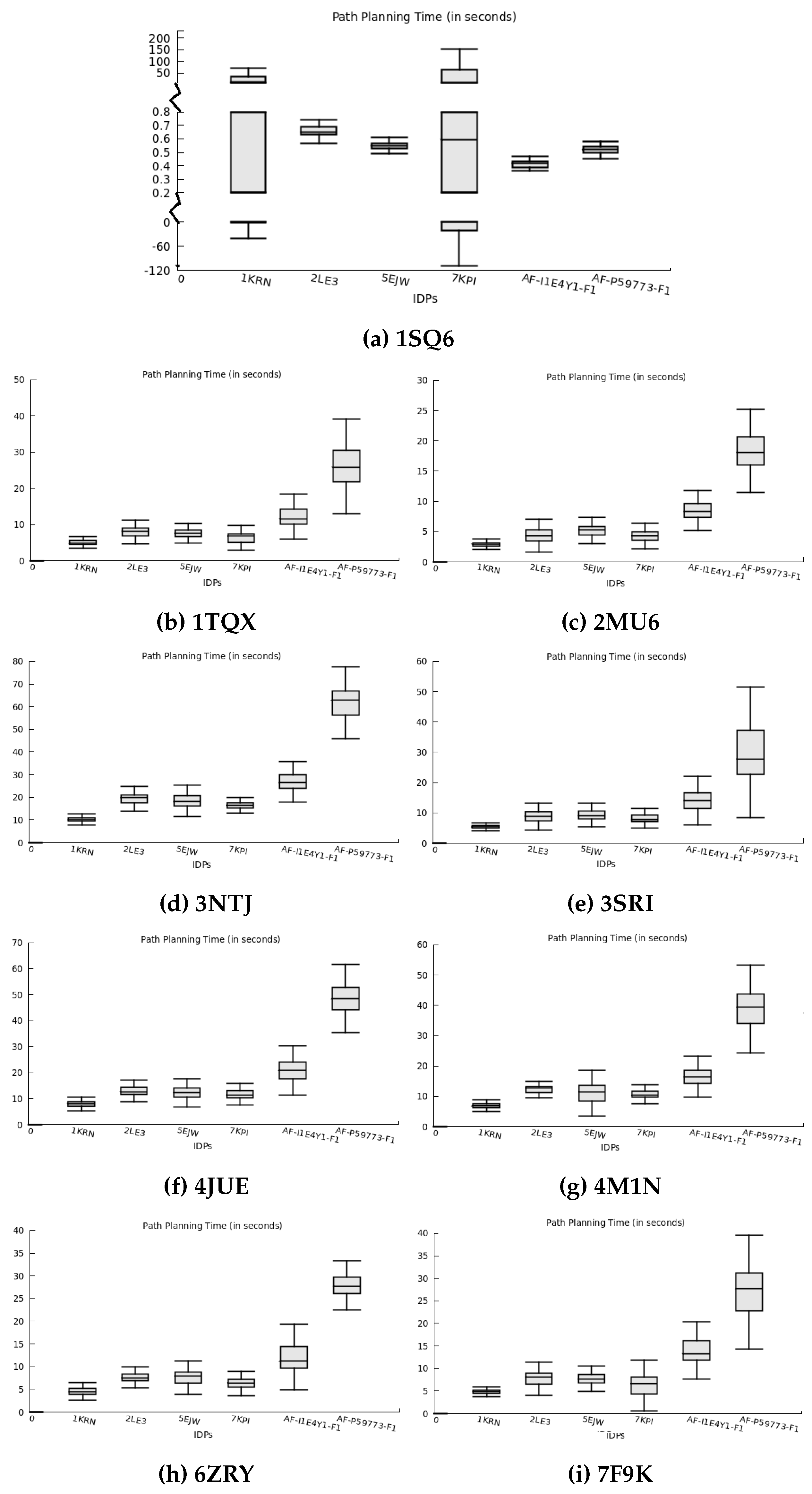
Figure 8.
The figures display a path planned for 2LE3 IDP around the 1SQ6 protein surface model using the geometrically favorable conformation ensembles. The start conformation is in red, and the binding goal position is in dark blue.
Figure 8.
The figures display a path planned for 2LE3 IDP around the 1SQ6 protein surface model using the geometrically favorable conformation ensembles. The start conformation is in red, and the binding goal position is in dark blue.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



