
Submitted:
15 June 2023
Posted:
16 June 2023
You are already at the latest version
Abstract
The aggregate aftermath of persistent inflammation in patients with inflammatory bowel disease (IBD) places them at increased risk for the advancement to colitis-associated colorectal cancer (CACRC). CACRC is preceded by IBD, the highly heterogenous, pharmacologically incurable, pertinacious, reverting/worsening, and immune-mediated inflammatory pathologies of the colon and rectum. The molecular and immunological basis of CACRC is highly correlated with the length/duration and stringency/severity of colon inflammation predisposed by the exogenous/free hemoglobin alpha chain (HbαC) the byproduct of infiltrating immune cells, extravasated erythrocytes, and macrophage erythrophagocytosis. The exogenous free HbαC prompts oxygen-free radical-arbitrated DNA damage (DNAD) through increased cellular reactive oxygen species (ROS) exacerbated by decreased tissue antioxidant defenses. Mitigation of Fenton reaction via pharmaceutical therapy would attenuate the ROS, promote apoptosis, DNAD repair, and subsequent prevent the incidence of CACRC. Three pharmaceutical options that attenuate hemoglobin toxicity include haptoglobin, deferoxamine, and flavonoids (vitamins C/E). Haptoglobin’s clearance rare from plasma is inversely correlated with its size; the smaller the size, the faster the clearance. Thus, the administration of Hp1-1 may prove to be beneficial. Furthermore, deferoxamine’s hydrophilic structure limits its ability to cross cell membranes. Thus, it may be beneficial if administered intracellularly to avoid the higher plasma concentrations and longer incubation periods associated with extracellular administration. Finally, the effectiveness of flavonoids and natural herb antioxidants is associated with high reactivity of hydroxyl substituents. Multiple analyses are currently underway to assess the clinical context of CACRC and outline the molecular basis of HbαC-induced ROS pathogenesis by exposing colonocytes and/or colonoids to HbαC. These cells are then treated with haptoglobin, deferoxamine (DFO), and flavonoids in order to separate free HbαC and measure their impact on hydroxyl radical formation therapies. The molecular pathogenesis of sporadic colorectal cancer (SCRC) i.e., “inflammation-dysplasia-carcinoma” progression sequence is well described, but the immunopathogenesis of CACRC herein reviewed is broadly still in prodromal stage/phase to be validated and understood. Therefore this timely review outlines the molecular and immunological basis of disease pathogenesis and the pharmaceutical intervention as a protective measure for CACRC.
Keywords:
Inflammatory bowel disease
; Colitis-associated colorectal cancer
; exogenous frees hemoglobin alpha chain
; Fenton reaction
; DNA damage
; haptoglobin
; deferoxamine
; flavonoids
; hydrogen peroxide
; hygiene
; iron
; nanomedicine
; oxidative stress
; polyphenol
; pharmaceutical therapy
1. Core tip
Inflammatory bowel disease-associated colorectal cancer (CACRC) is becoming more prevalent worldwide and presents at a younger age. IBD, as well as CACRC, is evolving worldwide, especially in newly industrialized countries. With an aging population, compound prevalence suggests that CACRC could become an emerging global challenge. Although surveillance and chemoprevention for CACRC exist, sixty percent of patients with CACRC are asymptomatic at detection and over fifty percent present with advanced disease; this eventually turns into a less favorableoutcome compared to sporadic colorectal cancer (SCRC). To understand why, scientist investigators profiled surgical pathology resections of colonic mucosal and submucosal layers from patients with IBD who had undergone pouch surgery, restorative proctocolectomy with ileal pouch-anal anastomosis (RPC-IPAA) [1]. A pool of exogenous/free hemoglobin alpha chain (HbαC) in areas of active colitis was unexpectedly found. Furthermore, the HbαC was produced through the action of immune infiltrated cells (macrophages) that promoted reactive oxygen species (ROS) sequel in epithelial cells deplete of colonic tissue homogenate antioxidants (i.e., nuclear factor erythroid 2–related factor 2 [Nrf2], catalase [CAT] superoxide dismutase [SOD], and glutathione peroxide [GPx]). The antioxidants above are significant regulators of cytoprotective responses to oxidative stress and the primary cellular defense against cytotoxic effects of oxidative stress [2,3,4,5,6]. Intestinal mucosal damage in IBD involves reactive oxygen metabolites (ROMs). Endogenous antixodiant enzymes neutralize ROMs in a carefully balanced two-step pathway. First, SOD converts superoxide anion to hydrogen peroxide (H(2)O(2)). Then, hydrogen peroxide is neutralized to water by CAT or glutathione peroxidase (GPO) [1]. This indicates that exogenous/free HbαC has a physiological role in inducing ROS formation and DNAD and, if not attenuated, can trigger carcinogenesis [1]. Our central focus is that HbαC induces oxygen free radical-mediated DNAD through increased ROS and decreased antioxidant defenses [1,7]. If the Fenton reaction was mitigated by pharmaceutical therapy using haptoglobin, deferoxamine, and/or flavonoids, then this would reduce the ROS, promote apoptosis and DNAD repair, and prevent incidence of CACRC [8].
2. Introduction
Colorectal cancer (CRC) is often described as the “disease no one has to die from”, but approximately 50% of patients with CRC who undergo potentially curative surgery ultimately relapse and die, usually as a consequence of metastatic disease [9,10]. According to GLOBOCAN 2018 data, and the American Cancer Society, for both men and women in the United States of America, colorectal Cancer (CRC) is divulged the third main cause of cancer-related mortality in the world [11,12]. CRC is the deadliest cancer [13,14]. IBD is a known risk factor in developing CACRC [15]. IBD patients are at increased risk of CACRC due to long-standing chronic inflammation, genetic alterations, and epigenetic environmental factors [16,17,18]. Additionally, data indicates that CACRC may have evolved through a pathway of tumorigenesis distinct from that of SCRC.
Predominantly colonic IBD, the “colitides,” which includes ulcerative colitis (UC) and Crohn’s colitis (CC), are two heterogeneous, chronic relapsing and remitting gastrointestinal tract disorders in the colon [18,19,20,21,22]. Currently, both disease affect approximately three million people in the United States. However, the incidence and prevalence of both are increasing worldwide; thus, making them global emergent diseases with significant clinical challenges [22]. The global prevalence of IBD is currently evolving approaching to 90 cases/100,000 people [23], though awareness should be assessed to the geographical locations of the world [24,25]. North America and Canada have the highest rates of IBD in the world [26,27]. However, over the past three decades, the incidence of IBD in low-income countries has steadily risen. [26,28,29,30,31,32,33]. The burden/implication of IBD is discrete in various countries and locations, and especially when contrasted/matched between low-income [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49] andwealthynations [50,51]. Estimated data suggest that 25 to 30 percent of cases of cases with CD and 20 percent of patients with UC present during adolescence and young adulthood of their reproductive age [52,53,54,55,56,57,58,59,60,61]. The extent magnitude of racial/ethnic and regional differences in the prevalence of IBD in the United States remains largely obscure warranting additional research [62,63]. However, IBD has predominantly affected whites, particularly Ashkenazi Jews. But over the last three decades, IBD has "emerged" in minority communities [26,63,64,65,66,67,68]. The genesis of IBD is obscure, but is believed to be multifactorial [18,30,69,70]. It has been hypothesized that intestinal damage in UC and in CC is related both to increased oxygen derived free radical production, mainly resulting from a respiratory burst of infiltrating phagocyte cells, and to a low concentration of endogenous antioxidant defense mechanisms. Indeed, neutrophils and monocytes in patients with active and/or fulminant IBD exhibit higher concentrations of oxygen-derived free radicals than in normal control samples [70,71,72,73]. Compared to other tissues, the gut is potentially more susceptibly exposed to oxidant injury/trauma exacerbated by the low concentration of antioxidant enzymes in epithelial cells, which contributes to the ROS cytotoxicity observed in the colon of patients with IBD [1,74]. IBD has no curative drug often resulting in significant long-term comorbidity [1]. The impact of potential immunosuppressive therapies in IBD aim to achieve long deep remission, but their effect on subsequent CACRC has yet to be established. However, studies have shown that the longer a person has had IBD, the higher chance of developing CACRC [75,76,77]. An extensively referenced comprehensive meta-analysis of 19 longitudinal and cross-sectional studies with age-stratified data reported that the cumulative incidence of CACRC in UC is 2% after 10 years, 8% after 20 years, and 18% after 30 years of disease [78]. In contrast, other studies reported lower incident rates accredited to, among other factors the benefit of endoscopic monitoring surveillance and anti-inflammatory pharmaceutical chemoprophylaxis [79,80,81]. The greatest hope and assurance for cancer prevention in IBD depends to a large extent in broadening our, thus far, insufficient understanding of the molecular pathogenesis link between neoplastic and chronic inflammation pathways. The discovery of exogenous/free HbαC, in IBD produced through the action of immune infiltrated cells and resultant ROS production in epithelial cells is innovative [1]. In this review we summarize the current knowledge and awareness of CACRC genesis, focusing on the fundamental mechanism underlying its pathogenesis, and on the potential implications of the “colonic deposition of exogenous/free HbαC, a previously unknown tissue by-product in IBD as a possible major trigger of CACRC. Herein we discuss “Fenton Reaction” and how exogenous HbαC could be chelated by pharmaceutical intervention to stop the ROS production, promote apoptosis and DNAD repair to prevent the incidence of CACRC carcinogenesis.
3. People with inflammatory bowel disease are at escalated probability for risk for colitis-associated colorectal cancer with a subsequent poor prognosis
People who suffer from colonic IBD are at increased risk for developing CACRC [79,82]. All CACRC are found located in segments with colitis [75]. CACRC is one of the most severe complications of IBD with a mortality rate of 10–15% and the risk is 1.5–2.4-fold way up than in the general population [15,83]. The dysplasia of CACRC develops in a different pathway mechanism in comparison to SCRC [15]. The well-established risk factors for CACRC are time scale and extent of intestinal inflammatory lesions [15,75,84,85,86]. The genetic factors coupled with the longevity of persistent fulminant interdependent inflammatory process in the colonic mucosal layers are believed to play a momentous remarkable role in CACRC carcinogenesis, and consequencing the inflammatory action could decrease this continuous proceedings of inflammation associated carcinogenesis [87,88,89]. The survivability depends on the adherence to colonoscopic surveillance and early elective colectomy is recommended [75,90,91]. However, some oncologic analyses result after curative surgeries in patients with CACRC [89,92]. This warrants continuous surveillance as a quality indicator for postcolectomy safety [75,90,93].
The prevalence of CACRC development is identical for patients with UC and CC [94,95,96,97], so is quantitative exogenous HbαC between the two colitides (1). This timely review was conducted to summarize and determine the efficacy of pharmaceutical safety on Fenton reaction mitigation as a preventive measure over CACRC.
4. Malfunction tight junction protein Caludin-1 is source point of colitis-associated colorectal cancer carcinogenesis
The tight junction is an intercellular junction intricate found in epithelial and endothelial cells that is accountable for the genesis of functional epithelial and endothelial barriers that synchronize the passage of cells and solutes through the paracellular space [98]. Patients with IBD are known to have a dysfunctional claudin-1, an intestinal epithelial tight junction protein, Figure 1 [99,100]. Irregularity functions in claudin-1 leads to change in cell permeability causing blood capillary extravasation (hemorrhage), macrophage erythrophagocytosis, and subsequent release of free HbαC exogenously into the interstitial space, Figure 2 [1]. Within the interstitial space, HbαC is observed to serve as a biological substrate in the Fenton Reaction producing hydroxyl radicals as shown in Figure 3 which leads to DNA damage Figure 4 within normal intestinal mucosa and subsequent tumor formation if the damaged DNA is irreparable [8]. This unveiled molecular understanding of chronic inflammation in patients suffering from IBD inclines to the evolution of CACRC. Inflammation can induce mutagenesis, and the relapsing-remitting nature of this inflammation, coupled with epithelial regeneration, may exert selective pressure acceleration carcinogenesis [101]. In summary, molecular pathogenesis of CACRC sequentially is due to: inflammation, claudin-1 dysfunction, extravasation of erythrocytes, macrophage erythrophagocytosis, and exogenous/free HbαC-ROS-DNAD-carcinogenesis [13,47]. Within the interstitial space, HbαC acts as a substrate in the Fenton Reaction (Fe2+ + H2O2 → Fe3+ + ·OH + OH-), Figure 3 [48]. Production of hydroxyl radicals in the Fenton Reaction as shown in Figure 3 can lead to DNAD within normal intestinal mucosa and subsequent tumor formation if the damaged DNA is not repaired.
5. Pharmacological mitigation
5. Pharmacological mitigation of Fenton Reaction to prevent colitis-associated colorectal cancer oncogenes
Ex-vivo studies demonstrated pool of free HbαC (until recently an unknown tissue by-product) in IBD patient mucosal microenvironments modulated by extravasated microphage erythrophagocytosis, Figure 2 [1]. In in-vitro data show HbαC induced high level of ROS production that caused DNAD exacerbated by systemic decreased antioxidant defenses [1,102,103]. The focus is that if the Fenton reaction (Figure 3) were mitigated via pharmaceutical therapy, then this would reduce ROS, promote DNAD repair and apoptosis which can prevent the incidence of CACRC [8].
6. Pharmaceutical approach to prevent colitis-associated colorectal cancer
Colonoscopy surveillance serves as the gold standard for prevention, but it has proved relatively inadequate to ascertain the earliest molecular pathogenic relationship between neoplasia and chronic inflammation. More specifically, Fenton chemistry and its relationship with exogenous/free HbαC, hydroxyl radical (·OH) formation via the Fenton reaction (Fe2+ + H2O2 → Fe3+ + ·OH + OH-), DNA damage (DNAD), and subsequent tumor formation. Meharry-Vanderbilt alliance focuses on understanding iron chelation therapy in mitigating an in-vitro Fenton Reaction through pharmaceutical approach. HbαC removal may be executed and accomplished using chelation therapy with the chelating drugs i.e., deferoxamine (DF), deferiprone (L1) and flavinoids [104,105] to attenuate HbαC toxicity.
6.1. Haptoglobin (Hp)
Free haptoglobin is removed from plasma in 3.5-5 days. On the other hand, the haptoglobin-hemoglobin (Hp-Hb) complex is removed within 20 minutes. This known fact stresses the importance of Hb removal in the presence of Hp. Haptoglobin is a tetrameric protein, a polymer built of four monomer units that contains two light (α) and two heavy (β) chains covalently bound to each other via disulfide bridges. There are three Hp phenotypes: Hp1-1, Hp2-1, and Hp2-2. Haptoglobin polymorphism is due to variations of the α-chain; α-1 chain carries 83 amino acids and α-2 chain accommodates 142 amino acids. The β-chain encompasses 245 amino acids and is not polymorphic. As shown in Figure 5, Hp 1-1 is the smallest haptoglobin protein structure [106,107,108]. Further research proved that the ability of Hp to lay off damage prompted by free radicals is largely phenotype pendent. The various phenotypes have the same binding affinities, but removal of Hp from the extravascular space is size dependent and removal of the Hp1-1:Hb complex occurs more rapidly. While Hp2-2:Hb complex is largest and removal occurs more slowly. Thus, when complexed with Hp2-2, Hb-α stay in circulation prepodently and causes enormous oxidative stress via Fenton chemistry [8,109]. Additionally, the prevalence of Hp2 is higher in IBD patients; thus, contributing to reduced anti-inflammatory effects and increased risk of CACRC development in this population [7,110].
6.2. Deferoxamine (DFO)
Deferoxamine (DFO) is a hydrophilic iron-chelating agent that has shown to inhibit free radical formation [111,112] and polymeric DFO for enhancing iron chelation cancer therapy. However, as shown in Figure 6, its hydrophilic properties limit its ability to cross cell membranes and remain effective in vivo. This feature alone requires higher concentrations and longer incubation periods of DFO in order to yield anti-inflammatory effects (inhibiting Fe-dependent production of hydroxyl radicals) from the agent. The chelation therapy would remove excess exogenous iron from the body and prevent production of hydroxyl radicals [111]. Further, antioxidants may as well play important role. Administering antioxidants would neutralize the free radicals and block their harmful effects on intestinal cells. Salicyaldehyde isonicotinoyl hydrazone (SIH) is a lipophilic iron-chelating agent that crosses cell membranes more effectively when compared to DFO, thus, requiring lower concentrations and incubation periods to produce similar anti-inflammatory effects when compared to DFO.
6.3. Flavonoids
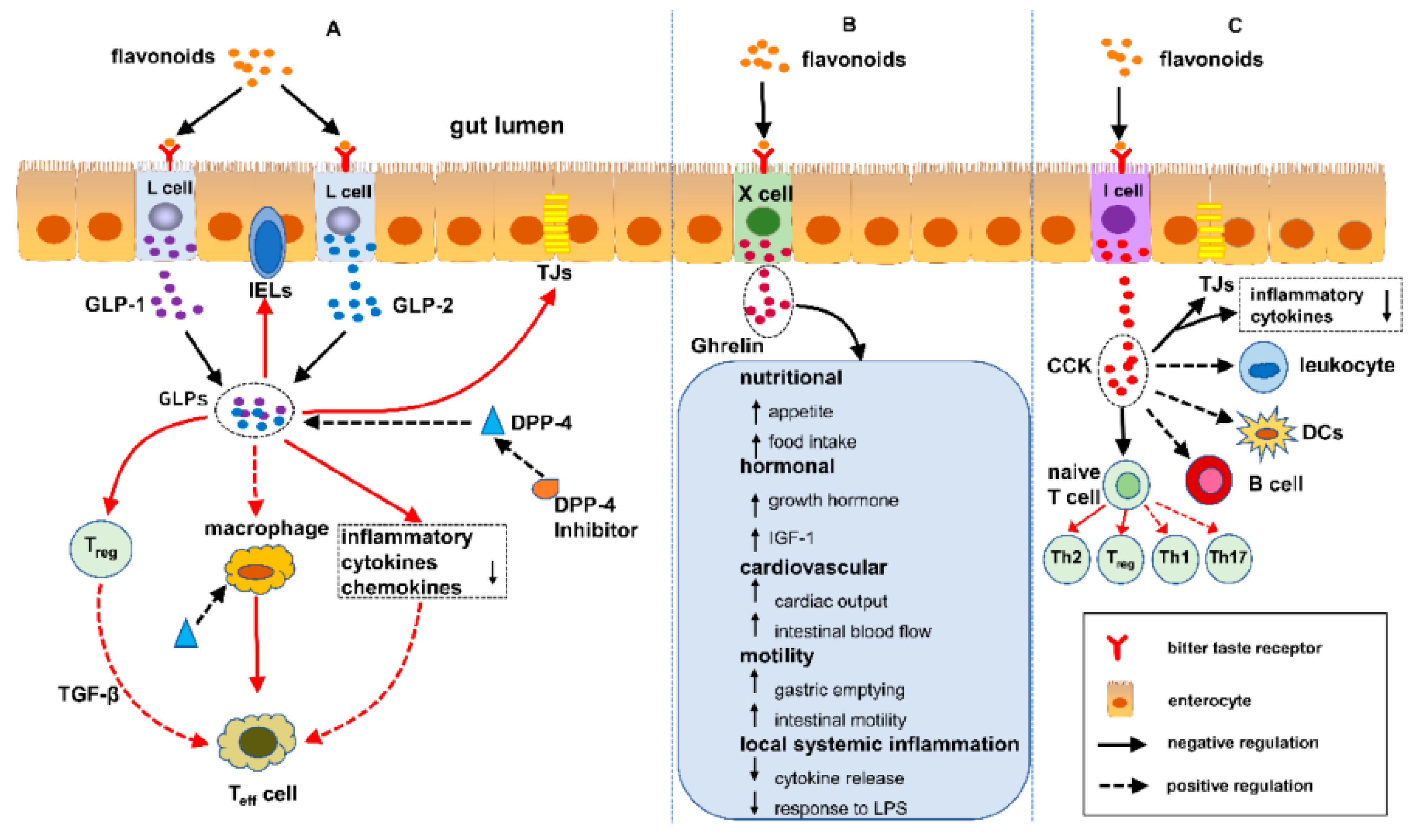
Flavonoids are free radical scavengers and confer a wide variety of antioxidant and anti-inflammatory activities as depicted in Figure 7 [113]. Studies have shown that enteroendocrine system is composed of enteroendocrine cells (EECs) that regulate IBD through the sensing gut microbiota and controling the immune response, thus safegurding the intestinal physical obstacle, as well as the modulation of gut motility [114]. Flavonoids have an impact on the enteroendocrine system to safeguard against IBD, which infers that the alleviation of IBD is possibly associated to the regulation of flavonoids on EECs. Presently, over 4000 multifsriousness of flavonoids have been recognized and ascertain the bright colors of many and various fruits and vegetables [115,116]. Further, a number of studies have reported the effect of flavonoids on enterohormones secretion; however, there is hardly any study demonstrating the association between flavonoids, enterohormones secretion and IBD. So far, the interplay between flavonoids, enterohormones and IBD is herein illuminated in this review. Furthermore, the conclusion can be drawn that flavonoids may safeguard against IBD over regulating enterohormones, such as glucagon-like peptide 1 (GLP-1), GLP-2, dipeptidyl peptidase-4 inhibitors (DPP-4 inhibitors), ghrelin and cholecystokinin (CCK) a possible mechanism of flavonoids protecting/ shielding against IBD [117].
Figure 7.
Pathway of Flavonoids shielding against IBD - regulating enteroendocrine system. (A) Flavonoids attune IBD by the DPP-4/GLPs pathway: (1) safeguarding the gut luminal barrier, (2) regulating Treg and intraepithelial lymphocytes (IELs) by way of controlling their discernment and functional and (3) modifying the task of macrophages and dendritic cells. (B) Flavonoids synchronize IBD by the ghrelin pathway: (1) increased food consumption, (2) increase in growth hormone action, (3) cardiovascular effects, (4) enhanced motility and (5) reduced local and systemic inflammation. (C) Flavonoids control IBD by the cholecystokinin (CCK) pathway: (1) reducing the mucosal production of proinflammatory cytokines and safeguiding the intestinal barrier, (2) decreasing leukocyte migration and impending dendritic cells (DCs) activation and (3) regulating T cells and B cells [112,113,114,115,116]. Flavonoids Free haptoglobin is cleared from plasma in 3.5-5 days. On the other hand, the haptoglobin-hemoglobin (Hp-Hb) complex is removed within 20 minutes. This known fact stresses the importance of Hbα removal in the presence of Hp. However this is a spinning intervention and does not solve the problem without finding a solution of the dysfunctional claudin-1. Reproduced with permission from Li et al. Metabolites, published by MDPI, 2022 under the terms/ conditions of the Creative Commons Attribution license [117]. Abbreviations: DPP-4/GLPs, Dipeptidyl peptidase-4 (DPP-4) inhibitors block the breakdown of GLP-1 and GIP to increase levels of the active hormones; IELs, intraepithelial lymphocytes; CCK, cholecystokinin; DCs, dendritic cells.
Figure 7.
Pathway of Flavonoids shielding against IBD - regulating enteroendocrine system. (A) Flavonoids attune IBD by the DPP-4/GLPs pathway: (1) safeguarding the gut luminal barrier, (2) regulating Treg and intraepithelial lymphocytes (IELs) by way of controlling their discernment and functional and (3) modifying the task of macrophages and dendritic cells. (B) Flavonoids synchronize IBD by the ghrelin pathway: (1) increased food consumption, (2) increase in growth hormone action, (3) cardiovascular effects, (4) enhanced motility and (5) reduced local and systemic inflammation. (C) Flavonoids control IBD by the cholecystokinin (CCK) pathway: (1) reducing the mucosal production of proinflammatory cytokines and safeguiding the intestinal barrier, (2) decreasing leukocyte migration and impending dendritic cells (DCs) activation and (3) regulating T cells and B cells [112,113,114,115,116]. Flavonoids Free haptoglobin is cleared from plasma in 3.5-5 days. On the other hand, the haptoglobin-hemoglobin (Hp-Hb) complex is removed within 20 minutes. This known fact stresses the importance of Hbα removal in the presence of Hp. However this is a spinning intervention and does not solve the problem without finding a solution of the dysfunctional claudin-1. Reproduced with permission from Li et al. Metabolites, published by MDPI, 2022 under the terms/ conditions of the Creative Commons Attribution license [117]. Abbreviations: DPP-4/GLPs, Dipeptidyl peptidase-4 (DPP-4) inhibitors block the breakdown of GLP-1 and GIP to increase levels of the active hormones; IELs, intraepithelial lymphocytes; CCK, cholecystokinin; DCs, dendritic cells.

The most likely way to reduce incidences of oncological transformation related to IBD is by clearance of excess exogenous HbαC from the interstitial space, Figure 8, Point D. However the limitation still remains until the malfunctioned claudin-1, Figure 8, Point C in the extracellular matrix in epithelial endothelium and connective tissue to stop potechial hemorrhage is resolved. This would be the true solid preventive measure to circumvent CACRC development.
7. Closing significance
To date there is still no consensus on colonoscopy surveillance for patients and it is mentioned that few gastroenterologists adhere to the recommended number of biopsy samplings during the procedure. This further proves the point that today's current endoscopic surveillance is inadequate and re-emphasizes the need to look further into the dysfunctional claudin-1 protein; this could hopefully prevent ROS-mediated DNAD and the future need for colonoscopy surveillance that has proved to be inadequate for many patients.
Supporting clinicians in their adoption of new screening guidance for colorectal cancer by establishing and fortifying key learning approach may be expected to change as additional research becomes available. The United States Preventive Services Task Force (USPSTF) guidelines recommend the 45–49-year-old cohort begin screening [118,119,120]. This enforcement may help identify high risk populations in primary care settings. Considerations for individuals at dominant liability for poor outcomes due to social determinants of health should be made, and organized screening programs to eliminate barriers to care [121,122,123].
Since there is still no known cure for IBD knowing all the factors that might worsen these diseases is quite important to understand, prevent, and find therapies. More reliable biomarkers of pre-malignancy are required. Such biomarkers should help identify patients who are at increased risk of developing CACRC and these patients should undergo personalized surveillance and treatment. Enhanced detection, particularly the removal of precancerous polyps and dysplasia, and advances in treatment has improved CRC outcomes 124,125]. The standard of care for CRC surveillance is screening starting at age 45 for patients with average risk and earlier and more frequent monitoring for patients with a family history of CRC. Racial minorities, however, receive unequal CRC care and thus experience higher incidence and mortality. African Americans (AA) are less likely to be given a screening recommendation by their provider [126]. Likewise, a study of 5,793 patients found that AA are more likely than White Americans (WA) to report physician non-recommendations as the predominant deterrent to screening (adjusted odds ratio of 1.46) [127]. Patient education, assistance with appointments, as well as enhancement of physician communication and cultural competency have been shown to improve CRC screening in minorities [128,129,130]. Initiating race-specific clinical guidelines for CACRC screening in AA is needed. The implementation of pre-clinical patient navigation and fecal immunochemical testing in the community may increase CRC screening within this population. First, we need to consult the literature on disparities in CRC prevention, detection, and treatment among AA. Next, we must develop clinical guidelines that promote CRC screening in AA and address patient-physician communication and health literacy. Finally, we need to investigate colitis-cancer sequences and their role in reducing the burden of CACRC.
8. Discussion
The primary causative factor for CACRC risk is thought to be the chronic inflammatory condition of the colon and rectum [131,132,133]. CACRC for UC [1925) [134] and CC (1948) [135] is a leading cause of long-term mortality. The prevalence of colorectal cancer development risks for patients with UC and CC is exactly the same [94]. Recent studies have reported that IBD confers a higher risk of CRC in males compared to females [82,136]. CACRC strikes mostly middle age (individuals [136,137]). For almost 30 years, attempts at cancer prevention have been rest on an observational strategy of endoscopic colonoscopy surveillance vigilance with biopsies to substantiate dysplasia, the earliest recognizable precursor of CRC and the most well-founded marker of impending inevitable cancer risk. Ideally, the rationale of surveillance is to permit most patients whose biopsy specimens remain dysplasia-free to avoid unnecessary colectomy surgery, while enabling those with dysplasia to undergo prophylactic removal of the colon before the development of CRC. Although validation of this action plan has been based largely on incidental evidence, surveillance has been widely accepted and widely executed as the standard of care for patients at risk for CRC [137,138]. None the less current, endoscopic surveillance seems to be inadequate in detecting early dysplasia that precedes CACRC. The eminent undertake for cancer prevention in IBD is based greatly in increasing our knowledge of the molecular pathogenesis association between neoplastic and chronic inflammation pathways [95,96,97,139,140].
Despite being “the disease no one has to die from,” CRC is themost deadly cancer among males in three nations and females in five countries [13,14]. Patients with IBD, which constitutes two subclasses i.e., UC, and CD have an increased reliability of progressing CACRC. This is due to prolonged fulminant chronic inflammation in the colon and rectum. CACRC risk increases with pan-colitis as well as prolonged disease duration. One meta-analysis found that the prevalence of CACRC in patients with UC was 3.7% overall compared to 5.4% in patients solely with pan-colitis. Furthermore, the risk of developing CACRC was 2% at 10 years, 8% at 20 years, and 18% at 30 years respectively [78]. Despite endoscopic surveillance and treatment, IBD-associated CACRC is frequently diagnosed at advanced stages. In a retrospective study, Averboukh et al. [141] reviewed medical charts of CACRC patients who underwent RPC-IPAA surgery between 1992-2009. From their review, they discovered that 36% of patients presented at stage III and 17% of patients presented at stage IV. Thus, contributing to the poor prognosis as well as 15% of all IBD-related deaths [141]. Despite this information, further basic research needs to be conducted to implement and ascertain the molecular pathogenic relationship between neoplastic and chronic inflammation. Patients with IBD are known to have a dysfunctional claudin-1, an intestinal epithelial tight junction protein, Figure 1 [99]. Irregularity in claudin-1 can lead to multifunctional cell capillary/ vascular permeability causing blood extravasation, macrophage erythrophagocytosis, and release of exogenous/free HbαC into the interstitial space, Figure 2 [1]. Within the interstitial space, HbαC is observed to serve as a biological substrate in the Fenton Reaction, Figure 3 [142]. Excessive production of hydroxyl radicals in the Fenton Reaction as shown in Figure 3 can lead to DNA damage within normal intestinal mucosa and subsequent tumor formation if the damaged DNA is not repaired.
9. Significance
To date there is no pharmaceutical cure for IBD. Knowing all the factors that might worsen these diseases is quite important to understand symptomatology management. More reliable biomarkers of pre-malignancy are needed to help recognize patients who are at increased risk of getting CACRC and select such patients for personalized surveillance, management and treatment. According to the American Cancer Society, in the United States CRC incidence has doubled in younger adults and is the 3rd leading cause of cancer deaths. The incidence of colorectal cancer (CRC) is rapidly increasing among younger individuals, and the disease is also being diagnosed at more advanced stages in all ages, according to a new report from the American Cancer Society. Diagnoses in people younger than 55 years doubled from 11% (1 in 10) in 1995 to 20% (1 in 5) in 2019. In addition, more advanced disease is being diagnosed; the proportion of individuals of all ages presenting with advanced-stage CRC enhanced from 52% in the mid-2000s to 60% in 2019. The rates are elevating in young people, but it's alarming to see how fast the whole patient population is changing younger, despite decreasing numbers in the overall population (The American Cancer Society)
Enhanced detection, particularly the removal of precancerous polyps and dysplasia, and advances in treatment has improved CRC outcomes [124,125]. The standard of care for CRC surveillance is screening starting at age 45 for patients with average risk and earlier and more frequent screening for patients with a family history of CRC. Racial minorities, however, receive unequal CRC care and thus experience higher incidence and mortality. The study additionally conveys that AAs were less likely to be given a screening recommendation by their provider [126]. Likewise, a study of 5,793 patients found that AAs were more likely than White American (WA) to report physician non-recommendations as the predominant deterrent to screening (adjusted odds ratio of 1.46) [127]. Patient education, assistance with appointments, as well as enhancement of physician communication and cultural competency have been shown to improve CRC screening in minorities [128,129,130]. The implementation of pre-clinical patient navigation and fecal immunochemical testing in the community may increase CRC screening within this population. First, it requires consulting the literature on disparities in CRC prevention, detection, and treatment among AA. Second, develop clinical guidelines that promote CRC screening in AA and address patient-physician communication and health literacy. Third, describe colitis-cancer sequences and mediated conditions characterizing role in reducing the burden of CACRC incidence. Finally, IBD-related health disparities exacerbate the CACRC mortality rate. African American patients with IBD are almost twice as vulnerable to develop CRC when compared to their White Americans (WAs) counterparts. Although early screenings (i.e. endoscopic/ colonoscopy surveillance) have proven to reduce CACRC, AAs have not benefited from such preventative strategies’ secondary to non-compliance [14]. Thus, there is a need to generate alternative preventative measures. If mitigation of the Fenton reaction is successful, then this would - i) reduce the incidence of CACRC and its mortality ii) reduce and/or eliminate the need for endoscopic/ colonoscopy screening for IBD patients which is not favorably viewed by AAs and iii) eliminate non-compliance to screening and thereby reduce CRC morbidity in AA.
10. Ethical Considerations
The study has been managed in affirmation with the ethical proposions of the 1975 Announcement of Helsinki [142]. This project was authorized by Meharry Medical College and Vanderbilt University Medical Center Institutional Review Boards, IRB#s: 100916AM206, 080898 and 100581.
Author Contributions
MAB, WAB and AEM contributed to review conceptualization and design. MAB, and AEM wrote the first draft of the manuscript and drafted figures and tables. MAB and AEM performed literature search, reviewed articles for inclusion, and reviewed, edited, and approved the final manuscript.
Funds acknowledgement
National Institute of Health R21-DK095186, AEM. Vanderbilt-Ingram Cancer Center SPORE in Gastrointestinal P50CA236733, AEM, RJC. Research Foundation, American Society of Colon and Rectal Surgeons-LPG086, AEM. UL1 RR024975-01,GRB. 5P30 DK58404-08/RR/NCRR NIH HHS/USA, AEM.
Data Availability
No new data were generated or analyzed in support of this research.
Acknowledgments
Authors are thankful to Meghan O’Loughlin, Drs. Karen M. Winkfield and Robert J. Coffey.
Abbreviations
HbαC: hemoglobin alpha chain; ROS, reactive oxygen species.
References
- Myers JN, Schaffer MW, Korolkova OY, Williams AD, Gangula PR, M'Koma AE. Implications of the Colonic Deposition of Free Hemoglobin-alpha Chain: A Previously Unknown Tissue By-product in Inflammatory Bowel Disease. Inflammatory bowel diseases. 2014;20(9):1530-47. [CrossRef]
- Kruidenier L, Verspaget HW. Review article: oxidative stress as a pathogenic factor in inflammatory bowel disease--radicals or ridiculous? Aliment Pharmacol Ther. 2002;16(12):1997-2015. [CrossRef]
- D'Odorico A, Bortolan S, Cardin R, D'Inca R, Martines D, Ferronato A, et al. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand J Gastroenterol. 2001;36(12):1289-94. [CrossRef]
- Krzystek-Korpacka M, Neubauer K, Berdowska I, Boehm D, Zielinski B, Petryszyn P, et al. Enhanced formation of advanced oxidation protein products in IBD. Inflammatory bowel diseases. 2008;14(6):794-802. [CrossRef]
- Boehm D, Krzystek-Korpacka M, Neubauer K, Matusiewicz M, Berdowska I, Zielinski B, et al. Paraoxonase-1 status in Crohn's disease and ulcerative colitis. Inflammatory bowel diseases. 2009;15(1):93-9. [CrossRef]
- Koutroubakis IE, Malliaraki N, Dimoulios PD, Karmiris K, Castanas E, Kouroumalis EA. Decreased total and corrected antioxidant capacity in patients with inflammatory bowel disease. Dig Dis Sci. 2004;49(9):1433-7. [CrossRef]
- Babbs, CF. Oxygen radicals in ulcerative colitis. Free Radic Biol Med. 1992;13(2):169-81. [CrossRef]
- Caillet S, Yu, H., Lessard, S., Lamoureux, G., Ajdukovic, D., & Lacroix, M. Fenton reaction applied for screening natural antioxidants. Food Chemistry. 2007;100(2):542-52. [CrossRef]
- de Lacerda TC, Costa-Silva B, Giudice FS, Dias MV, de Oliveira GP, Teixeira BL, et al. Prion protein binding to HOP modulates the migration and invasion of colorectal cancer cells. Clin Exp Metastasis. 2016;33(5):441-51. [CrossRef]
- Nopel-Dunnebacke S, Conradi LC, Reinacher-Schick A, Ghadimi M. [Influence of molecular markers on oncological surgery of colorectal cancer]. Chirurg. 2021;92(11):986-95. [CrossRef]
- Ewing I, Hurley JJ, Josephides E, Millar A. The molecular genetics of colorectal cancer. Frontline Gastroenterol. 2014;5(1):26-30. [CrossRef]
- Rawla P, Sunkara T, Raj JP. Role of biologics and biosimilars in inflammatory bowel disease: current trends and future perspectives. J Inflamm Res. 2018;11:215-26. [CrossRef]
- Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14(2):89-103. [CrossRef]
- M'Koma AE, Moses HL, Adunyah SE. Inflammatory bowel disease-associated colorectal cancer: proctocolectomy andmucosectomy does not necessarily eliminate pouch related cancer incidences. Int J colorect Dis. 2011;26:533-52. [CrossRef]
- Marynczak K, Wlodarczyk J, Sabatowska Z, Dziki A, Dziki L, Wlodarczyk M. Colitis-Associated Colorectal Cancer in Patients with Inflammatory Bowel Diseases in a Tertiary Referral Center: A Propensity Score Matching Analysis. Journal of clinical medicine. 2022;11(3). [CrossRef]
- Lucafo M, Curci D, Franzin M, Decorti G, Stocco G. Inflammatory Bowel Disease and Risk of Colorectal Cancer: An Overview From Pathophysiology to Pharmacological Prevention. Front Pharmacol. 2021;12:772101. [CrossRef]
- Schmitt M, Greten FR. The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol. 2021;21(10):653-67. [CrossRef]
- M'Koma, AE. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointest Disord. 2018;1(1):75-105. [CrossRef]
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979-86. [CrossRef]
- Satsangi J, Silverberg MS, Vermeire S, Colombel JF. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55(6):749-53. [CrossRef]
- Spekhorst LM, Visschedijk MC, Alberts R, Festen EA, van der Wouden EJ, Dijkstra G, et al. Performance of the Montreal classification for inflammatory bowel diseases. World J Gastroenterol. 2014;20(41):15374-81. [CrossRef]
- Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflammatory bowel diseases. 2011;17(1):423-39. [CrossRef]
- Kofla-Dlubacz A, Pytrus T, Akutko K, Sputa-Grzegrzolka P, Piotrowska A, Dziegiel P. Etiology of IBD-Is It Still a Mystery? Int J Mol Sci. 2022;23(20). [CrossRef]
- Pena-Sanchez JN, Osei JA, Marques Santos JD, Jennings D, Andkhoie M, Brass C, et al. Increasing Prevalence and Stable Incidence Rates of Inflammatory Bowel Disease Among First Nations: Population-Based Evidence From a Western Canadian Province. Inflammatory bowel diseases. 2022;28(4):514-22. [CrossRef]
- Jones GR, Lyons M, Plevris N, Jenkinson PW, Bisset C, Burgess C, et al. IBD prevalence in Lothian, Scotland, derived by capture-recapture methodology. Gut. 2019;68(11):1953-60. [CrossRef]
- M'Koma, AE. Inflammatory Bowel Disease: An Expanding Global Health Problem. Clinical Medicine Insights Gastroenterology. 2013(6):33-47. [CrossRef]
- Benchimol EI, Manuel DG, Guttmann A, Nguyen GC, Mojaverian N, Quach P, et al. Changing age demographics of inflammatory bowel disease in Ontario, Canada: a population-based cohort study of epidemiology trends. Inflammatory bowel diseases. 2014;20(10):1761-9. [CrossRef]
- Krzesiek E, Kofla-Dlubacz A, Akutko K, Stawarski A. The Incidence of Inflammatory Bowel Disease in the Paediatric Population in the District of Lower Silesia, Poland. Journal of clinical medicine. 2021;10(17). [CrossRef]
- Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785-94. [CrossRef]
- Ananthakrishnan AN, Kaplan GG, Ng SC. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin Gastroenterol Hepatol. 2020;18(6):1252-60. [CrossRef]
- Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769-78. [CrossRef]
- Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390(10114):2769-78. [CrossRef]
- Ng SC, Tang W, Ching JY, Wong M, Chow CM, Hui AJ, et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn's and colitis epidemiology study. Gastroenterology. 2013;145(1):158-65 e2. [CrossRef]
- Archampong TN, Nkrumah KN. Inflammatory bowel disease in Accra: what new trends. West Afr J Med. 2013;32(1):40-4.
- Ukwenya AY, Ahmed A, Odigie VI, Mohammed A. Inflammatory bowel disease in Nigerians: still a rare diagnosis? Ann Afr Med. 2011;10(2):175-9.
- Agoda-Koussema LK, Anoukoum T, Djibril AM, Balaka A, Folligan K, Adjenou V, et al. [Ulcerative colitis: a case in Togo]. Med Sante Trop. 2012;22(1):79-81. [CrossRef]
- Mebazaa A, Aounallah A, Naija N, Cheikh Rouhou R, Kallel L, El Euch D, et al. Dermatologic manifestations in inflammatory bowel disease in Tunisia. Tunis Med. 2012;90(3):252-7.
- Senbanjo IO, Oshikoya KA, Onyekwere CA, Abdulkareem FB, Njokanma OF. Ulcerative colitis in a Nigerian girl: a case report. BMC Res Notes. 2012;5:564. [CrossRef]
- Bouzid D, Fourati H, Amouri A, Marques I, Abida O, Haddouk S, et al. The CREM gene is involved in genetic predisposition to inflammatory bowel disease in the Tunisian population. Human immunology. 2011;72(12):1204-9. [CrossRef]
- O'Keefe EA, Wright JP, Froggatt J, Cuming L, Elliot M. Medium-term follow-up of ulcerative colitis in Cape Town. S Afr Med J. 1989;76(4):142-5.
- O'Keefe EA, Wright JP, Froggatt J, Zabow D. Medium-term follow-up of Crohn's disease in Cape Town. S Afr Med J. 1989;76(4):139-41.
- Segal, I. Ulcerative colitis in a developing country of Africa: the Baragwanath experience of the first 46 patients. Int J Colorectal Dis. 1988;3(4):222-5. [CrossRef]
- Segal I, Tim LO, Hamilton DG, Walker AR. The rarity of ulcerative colitis in South African blacks. Am J Gastroenterol. 1980;74(4):332-6.
- Wright JP, Marks IN, Jameson C, Garisch JA, Burns DG, Kottler RE. Inflammatory bowel disease in Cape Town, 1975-1980. Part II. Crohn's disease. S Afr Med J. 1983;63(7):226-9.
- Wright JP, Marks IN, Jameson C, Garisch JA, Burns DG, Kottler RE. Inflammatory bowel disease in Cape Town, 1975-1980. Part I. Ulcerative colitis. S Afr Med J. 1983;63(7):223-6.
- Brom B, Bank S, Marks IN, Barbezat GO, Raynham B. Crohn's disease in the Cape: a follow-up study of 24 cases and a review of the diagnosis and management. S Afr Med J. 1968;42(41):1099-107.
- Novis BH, Marks IN, Bank S, Louw JH. Incidence of Crohn's disease at Groote Schuur Hospital during 1970-1974. S Afr Med J. 1975;49(17):693-7.
- Sobel JD, Schamroth L. Ulcerative colitis in the South African Bantu. Gut. 1970;11(9):760-3. [CrossRef]
- Giraud RM, Luke I, Schmaman A. Crohn's disease in the Transvaal Bantu: a report of 5 cases. S Afr Med J. 1969;43(21):610-3.
- Ananthakrishnan AN, Kwon J, Raffals L, Sands B, Stenson WF, McGovern D, et al. Variation in treatment of patients with inflammatory bowel diseases at major referral centers in the United States. Clin Gastroenterol Hepatol. 2015;13(6):1197-200. [CrossRef]
- Kappelman MD, Rifas-Shiman SL, Porter CQ, Ollendorf DA, Sandler RS, Galanko JA, et al. Direct health care costs of Crohn's disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135(6):1907-13. [CrossRef]
- Siegmund B, Zeitz M. [Inflammatory bowel disease and pregnancy]. Z Gastroenterol. 2009;47(10):1069-74. [CrossRef]
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46-54 e42; quiz e30. [CrossRef]
- Chouraki V, Savoye G, Dauchet L, Vernier-Massouille G, Dupas JL, Merle V, et al. The changing pattern of Crohn's disease incidence in northern France: a continuing increase in the 10- to 19-year-old age bracket (1988-2007). Aliment Pharmacol Ther. 2011;33(10):1133-42. [CrossRef]
- Shmidt E, Dubinsky MC. Inflammatory Bowel Disease and Pregnancy. Am J Gastroenterol. 2022;117(10s):60-8. [CrossRef]
- Baiocco PJ, Korelitz BI. The influence of inflammatory bowel disease and its treatment on pregnancy and fetal outcome. J Clin Gastroenterol. 1984;6(3):211-6.
- Heetun ZS, Byrnes C, Neary P, O'Morain C. Review article: Reproduction in the patient with inflammatory bowel disease. Aliment Pharmacol Ther. 2007;26(4):513-33. [CrossRef]
- Vermeire S, Carbonnel F, Coulie PG, Geenen V, Hazes JM, Masson PL, et al. Management of inflammatory bowel disease in pregnancy. J Crohns Colitis. 2012;6(8):811-23. [CrossRef]
- Jakobsen C, Paerregaard A, Munkholm P, Faerk J, Lange A, Andersen J, et al. Pediatric inflammatory bowel disease: increasing incidence, decreasing surgery rate, and compromised nutritional status: A prospective population-based cohort study 2007-2009. Inflammatory bowel diseases. 2011;17(12):2541-50. [CrossRef]
- North American Society for Pediatric Gastroenterology H, Nutrition, Colitis Foundation of A, Bousvaros A, Antonioli DA, Colletti RB, et al. Differentiating ulcerative colitis from Crohn disease in children and young adults: report of a working group of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the Crohn's and Colitis Foundation of America. J Pediatr Gastroenterol Nutr. 2007;44(5):653-74. [CrossRef]
- Griffiths, AM. Specificities of inflammatory bowel disease in childhood. Best Pract Res Clin Gastroenterol. 2004;18(3):509-23. [CrossRef]
- Wang YR, Loftus EV, Jr., Cangemi JR, Picco MF. Racial/Ethnic and regional differences in the prevalence of inflammatory bowel disease in the United States. Digestion. 2013;88(1):20-5. [CrossRef]
- Afzali A, Cross RK. Racial and Ethnic Minorities with Inflammatory Bowel Disease in the United States: A Systematic Review of Disease Characteristics and Differences. Inflammatory bowel diseases. 2016;22(8):2023-40. [CrossRef]
- Li D, Collins B, Velayos FS, Liu L, Lewis JD, Allison JE, et al. Racial and ethnic differences in health care utilization and outcomes among ulcerative colitis patients in an integrated health-care organization. Dig Dis Sci. 2014;59(2):287-94. [CrossRef]
- Castaneda G, Liu B, Torres S, Bhuket T, Wong RJ. Race/Ethnicity-Specific Disparities in the Severity of Disease at Presentation in Adults with Ulcerative Colitis: A Cross-Sectional Study. Dig Dis Sci. 2017;62(10):2876-81. [CrossRef]
- Avalos DJ, Mendoza-Ladd A, Zuckerman MJ, Bashashati M, Alvarado A, Dwivedi A, et al. Hispanic Americans and Non-Hispanic White Americans Have a Similar Inflammatory Bowel Disease Phenotype: A Systematic Review with Meta-Analysis. Dig Dis Sci. 2018;63(6):1558-71. [CrossRef]
- Hou JK, El-Serag H, Thirumurthi S. Distribution and manifestations of inflammatory bowel disease in Asians, Hispanics, and African Americans: a systematic review. Am J Gastroenterol. 2009;104(8):2100-9. [CrossRef]
- Ventham NT, Kennedy NA, Nimmo ER, Satsangi J. Beyond gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology. 2013;145(2):293-308. [CrossRef]
- Price, AB. Overlap in the spectrum of non-specific inflammatory bowel disease--'colitis indeterminate'. J Clin Pathol. 1978;31(6):567-77. [CrossRef]
- Farmer M, Petras RE, Hunt LE, Janosky JE, Galandiuk S. The importance of diagnostic accuracy in colonic inflammatory bowel disease. Am J Gastroenterol. 2000;95(11):3184-8. [CrossRef]
- Kader HA, Tchernev VT, Satyaraj E, Lejnine S, Kotler G, Kingsmore SF, et al. Protein microarray analysis of disease activity in pediatric inflammatory bowel disease demonstrates elevated serum PLGF, IL-7, TGF-beta1, and IL-12p40 levels in Crohn's disease and ulcerative colitis patients in remission versus active disease. Am J Gastroenterol. 2005;100(2):414-23. [CrossRef]
- Burczynski ME, Peterson RL, Twine NC, Zuberek KA, Brodeur BJ, Casciotti L, et al. Molecular classification of Crohn's disease and ulcerative colitis patients using transcriptional profiles in peripheral blood mononuclear cells. J Mol Diagn. 2006;8(1):51-61. [CrossRef]
- Fukushima K, Yonezawa H, Fiocchi C. Inflammatory bowel disease-associated gene expression in intestinal epithelial cells by differential cDNA screening and mRNA display. Inflammatory bowel diseases. 2003;9(5):290-301. [CrossRef]
- Shkoda A, Werner T, Daniel H, Gunckel M, Rogler G, Haller D. Differential protein expression profile in the intestinal epithelium from patients with inflammatory bowel disease. J Proteome Res. 2007;6(3):1114-25. [CrossRef]
- M'Koma AE, Moses HL, Adunyah SE. Inflammatory bowel disease-associated colorectal cancer: proctocolectomy andmucosectomy does not necessarily eliminate pouch related cancer incidences. Int J colorect Dis. 2011;26:533-52. [CrossRef]
- Hardy RG, Meltzer SJ, Jankowski JA. ABC of colorectal cancer. Molecular basis for risk factors. Bmj. 2000;321(7265):886-9. [CrossRef]
- Rubin DT, Parekh N. Colorectal cancer in inflammatory bowel disease: molecular and clinical considerations. Curr Treat Options Gastroenterol. 2006;9(3):211-20. [CrossRef]
- Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48(4):526-35. [CrossRef]
- Harpaz N, Talbot IC. Colorectal cancer in idiopathic inflammatory bowel disease. Semin Diagn Pathol. 1996;13(4):339-57.
- Rubio CA, Befrits R, Ljung T, Jaramillo E, Slezak P. Colorectal carcinoma in ulcerative colitis is decreasing in Scandinavian countries. Anticancer Res. 2001;21(4B):2921-4.
- Loftus EV, Jr. Epidemiology and risk factors for colorectal dysplasia and cancer in ulcerative colitis. Gastroenterol Clin North Am. 2006;35(3):517-31. [CrossRef]
- Delaunoit T, Limburg PJ, Goldberg RM, Lymp JF, Loftus EV, Jr. Colorectal cancer prognosis among patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2006;4(3):335-42. [CrossRef]
- Keller DS, Windsor A, Cohen R, Chand M. Colorectal cancer in inflammatory bowel disease: review of the evidence. Tech Coloproctol. 2019;23(1):3-13. [CrossRef]
- Khan MA, Hakeem AR, Scott N, Saunders RN. Significance of R1 resection margin in colon cancer resections in the modern era. Colorectal Dis. 2015;17(11):943-53. [CrossRef]
- Canavan C, Abrams KR, Mayberry J. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn's disease. Aliment Pharmacol Ther. 2006;23(8):1097-104. [CrossRef]
- Stidham RW, Higgins PDR. Colorectal Cancer in Inflammatory Bowel Disease. Clin Colon Rectal Surg. 2018;31(3):168-78. [CrossRef]
- Munkholm, P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18 Suppl 2:1-5. [CrossRef]
- Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. 2016;22(20):4794-801. [CrossRef]
- Baker AM, Cross W, Curtius K, Al Bakir I, Choi CR, Davis HL, et al. Evolutionary history of human colitis-associated colorectal cancer. Gut. 2019;68(6):985-95. [CrossRef]
- Um JW, M'Koma AE. Pouch-related dysplasia and adenocarcinoma following restorative proctocolectomy for ulcerative colitis. Tech Coloproctol. 2011;15(1):7-16. [CrossRef]
- Gallo G, Kotze PG, Spinelli A. Surgery in ulcerative colitis: When? How? Best Pract Res Clin Gastroenterol. 2018;32-33:71-8. [CrossRef]
- Yashiro, M. Ulcerative colitis-associated colorectal cancer. World J Gastroenterol. 2014;20(44):16389-97. [CrossRef]
- Stjarngrim J, Widman L, Schmidt PT, Ekbom A, Forsberg A. Post-endoscopy colorectal cancer after colectomy in inflammatory bowel disease patients: a population-based register study. Eur J Gastroenterol Hepatol. 2023;35(3):288-93. [CrossRef]
- Vleggaar FP, Lutgens MW, Oldenburg B, Schipper ME, Samsom M. [British and American screening guidelines inadequate for prevention of colorectal carcinoma in patients with inflammatory bowel disease]. Ned Tijdschr Geneeskd. 2007;151(50):2787-91.
- M'Koma AE, Seeley EH, Wise PE, Washingtoin MK, Schwartz DA, Herline AJ, Muldon RL, Caprioli RM. Proteomic analysis of colonic submucosa differentiates Crohn's and ulcerative colitis. Gastroenterology. 2009;136(5 Suppl 1):A-349. [CrossRef]
- M'Koma AE, Seeley EH, Washington MK, Schwartz DA, Muldoon RL, Herline AJ, Wise PE, Caprioli RM. Proteomic profiling of mucosal and submucosal colonic tissues yields protein signatures that differentiate the inflammatory colitides. Inflamm Bowel Dis. 2011;17(4):875-83. [CrossRef]
- Seeley EH, Washington MK, Caprioli RM, M'Koma AE. Proteomic patterns of colonic mucosal tissues delineate Crohn's colitis and ulcerative colitis. Proteomics Clin Appl. 2013;7(7-8):541-9. [CrossRef]
- Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010;20(3):142-9. [CrossRef]
- Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest. 2008;88(10):1110-20. [CrossRef]
- Steed E, Rodrigues NT, Balda MS, Matter K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009;10:95. [CrossRef]
- Porter RJ, Arends MJ, Churchhouse AMD, Din S. Inflammatory Bowel Disease-Associated Colorectal Cancer: Translational Risks from Mechanisms to Medicines. J Crohns Colitis. 2021;15(12):2131-41. [CrossRef]
- Zielinska AK, Salaga M, Siwinski P, Wlodarczyk M, Dziki A, Fichna J. Oxidative Stress Does Not Influence Subjective Pain Sensation in Inflammatory Bowel Disease Patients. Antioxidants (Basel). 2021;10(8). [CrossRef]
- Mrowicka M, Mrowicki J, Mik M, Dziki L, Dziki A, Majsterek I. Assessment of DNA damage profile and oxidative /antioxidative biomarker level in patients with inflammatory bowel disease. Pol Przegl Chir. 2020;92(5):8-15. [CrossRef]
- Kontoghiorghes GJ, Pattichi K, Hadjigavriel M, Kolnagou A. Transfusional iron overload and chelation therapy with deferoxamine and deferiprone (L1). Transfus Sci. 2000;23(3):211-23. [CrossRef]
- Szymonik J, Wala K, Gornicki T, Saczko J, Pencakowski B, Kulbacka J. The Impact of Iron Chelators on the Biology of Cancer Stem Cells. Int J Mol Sci. 2021;23(1). [CrossRef]
- Naryzny SN, Legina OK. Haptoglobin as a Biomarker. Biochem Mosc Suppl B Biomed Chem. 2021;15(3):184-98. [CrossRef]
- Wobeto VP, Zaccariotto, T. R., and Sonati, M. D. Polymorphism of human haptoglobin and its clinical importance. Genetics and Molecular Biology. 2008;31(3):602-220. [CrossRef]
- Marquez L, Cleynen, I., Machiels, K., Organe, S., Ferrante, M., Henckaerts. Haptoglobin Poymorphisms are Associated With Ulcerative Colitis and Crohn's Disease. Haptoglobin Poymorphisms are Associated With Ulcerative Colitis and Crohn's Disease. 2010;138(5).
- Tayari M, Afsharzadeh D. Amplification of antioxidant activity of haptoglobin(2-2)-hemoglobin at pathologic temperature and presence of antibiotics. Indian J Clin Biochem. 2012;27(2):171-7. [CrossRef]
- Marquez L, Shen, C., Machiels, K., Perrier, C., Ballet, V., Organe, S. Role of Haptoglobin in Susceptibility of IBD and in Triggering Murine Colitis. Gastroenterology. 2011;140(5). [CrossRef]
- Meczynska S, Lewandowska H, Sochanowicz B, Sadlo J, Kruszewski M. Variable inhibitory effects on the formation of dinitrosyl iron complexes by deferoxamine and salicylaldehyde isonicotinoyl hydrazone in K562 cells.Hemoglobin. 2008;32:157-63. [CrossRef]
- Bruzzese A, Martino EA, Mendicino F, Lucia E, Olivito V, Bova C, et al. Iron chelation therapy. Eur J Haematol. 2023;110(5):490-497. [CrossRef]
- Zhao Q, Liu Y, Wang X, Zhu Y, Jiao Y, Bao Y, Shi W. Cuscuta chinensis flavonoids reducing oxidative stress of the improve sperm damage in 35 bisphenol A exposed mice offspring. Ecotoxicol Environ Saf. 2023;15:255:114831. [CrossRef]
- Yu Y, Yang W, Li Y, Cong Y. Enteroendocrine Cells: Sensing Gut Microbiota and Regulating Inflammatory Bowel Diseases. Inflammatory bowel diseases. 2020;26(1):11-20. [CrossRef]
- Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K, van Leeuwen PA. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr. 2001;74(4):418-25. [CrossRef]
- Ribeiro D, Proenca C, Rocha S, Lima J, Carvalho F, Fernandes E, et al. Immunomodulatory Effects of Flavonoids in the Prophylaxis and Treatment of Inflammatory Bowel Diseases: A Comprehensive Review. Curr Med Chem. 2018;25(28):3374-412. [CrossRef]
- Li M, Weigmann B. A Novel Pathway of Flavonoids Protecting against Inflammatory Bowel Disease: Modulating Enteroendocrine System. Metabolites. 2022;12(1). [CrossRef]
- Hillier TA, Vesco KK, Pedula KL, Beil TL, Whitlock EP, Pettitt DJ. Screening for gestational diabetes mellitus: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2008;148(10):766-75. [CrossRef]
- Lin JS, Piper MA, Perdue LA, Rutter CM, Webber EM, O'Connor E, et al. Screening for Colorectal Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. Jama. 2016;315(23):2576-94. [CrossRef]
- Lin JS, Perdue LA, Henrikson NB, Bean SI, Blasi PR. Screening for Colorectal Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. Jama. 2021;325(19):1978-98. [CrossRef]
- Miller-Wilson LA, Limburg P, Helmueller L, Joao Janeiro M, Hartlaub P. The impact of multi-target stool DNA testing in clinical practice in the United States: A real-world evidence retrospective study. Prev Med Rep. 2022;30:102045. [CrossRef]
- Kim A, Gitlin M, Fadli E, McGarvey N, Cong Z, Chung KC. Breast, Colorectal, Lung, Prostate, and Cervical Cancer Screening Prevalence in a Large Commercial and Medicare Advantage Plan, 2008-2020. Prev Med Rep. 2022;30:102046. [CrossRef]
- Poulson MR, Geary A, Papageorge M, Laraja A, Sacks O, Hall J, et al. The effect of medicare and screening guidelines on colorectal cancer outcomes. Journal of the National Medical Association. 2022. [CrossRef]
- Meester RGS, Peterse EFP, Knudsen AB, de Weerdt AC, Chen JC, Lietz AP, et al. Optimizing colorectal cancer screening by race and sex: Microsimulation analysis II to inform the American Cancer Society colorectal cancer screening guideline. Cancer. 2018;124(14):2974-85. [CrossRef]
- Peterse EFP, Meester RGS, Siegel RL, Chen JC, Dwyer A, Ahnen DJ, et al. The impact of the rising colorectal cancer incidence in young adults on the optimal age to start screening: Microsimulation analysis I to inform the American Cancer Society colorectal cancer screening guideline. Cancer. 2018;124(14):2964-73. [CrossRef]
- Almario CV, May FP, Ponce NA, Spiegel BM. Racial and Ethnic Disparities in Colonoscopic Examination of Individuals With a Family History of Colorectal Cancer. Clin Gastroenterol Hepatol. 2015;13(8):1487-95. [CrossRef]
- May FP, Almario CV, Ponce N, Spiegel BM. Racial minorities are more likely than whites to report lack of provider recommendation for colon cancer screening. Am J Gastroenterol. 2015;110(10):1388-94. [CrossRef]
- Naylor, KB. Addressing Colorectal Cancer Disparities Among African American Men Beyond Traditional Practice-Based Settings. Am J Public Health. 2017;107(9):1356-8. [CrossRef]
- Mehta SJ, Jensen CD, Quinn VP, Schottinger JE, Zauber AG, Meester R, et al. Race/Ethnicity and Adoption of a Population Health Management Approach to Colorectal Cancer Screening in a Community-Based Healthcare System. J Gen Intern Med. 2016;31(11):1323-30. [CrossRef]
- Naylor K, Ward J, Polite BN. Interventions to improve care related to colorectal cancer among racial and ethnic minorities: a systematic review. J Gen Intern Med. 2012;27(8):1033-46. [CrossRef]
- Lin TK, Erdman SH. Double-balloon Enteroscopy: Pediatric Experience. J Pediatr Gastroenterol Nutr. 2010.
- Houghton J, Li H, Fan X, Liu Y, Liu JH, Rao VP, et al. Mutations in Bone Marrow-Derived Stromal Stem Cells Unmask Latent Malignancy. Stem Cells Dev. 2010. [CrossRef]
- Schmidt C, Bielecki C, Felber J, Stallmach A. Surveillance strategies in inflammatory bowel disease. Minerva Gastroenterol Dietol. 2010;56(2):189-201.
- Burrill, B. Crohn BB RH. The Sigmoidoscopic Picture of Chronic Ulcerative Colitis (Non-Specific). Am J Med Sci. 1925(170):220.
- Warren S, Sommers SC. Cicatrizing enteritis as a pathologic entity; analysis of 120 cases. Am J Pathol. 1948;24(3):475-501.
- Soderlund S, Brandt L, Lapidus A, Karlen P, Brostrom O, Lofberg R, et al. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology. 2009;136(5):1561-7; quiz 818-9. [CrossRef]
- Itzkowitz SH, Present DH, Crohn's, Colitis Foundation of America Colon Cancer in IBDSG. Consensus conference: Colorectal cancer screening and surveillance in inflammatory bowel disease. Inflammatory bowel diseases. 2005;11(3):314-21. [CrossRef]
- Barthet M, Gay G, Sautereau D, Ponchon T, Napoleo B, Boyer J, et al. Endoscopic surveillance of chronic inflammatory bowel disease. Endoscopy. 2005;37(6):597-9. [CrossRef]
- Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68(1):31-54. [CrossRef]
- Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145-64. [CrossRef]
- Averboukh F, Ziv Y, Kariv Y, Zmora O, Dotan I, Klausner JM, et al. Colorectal carcinoma in inflammatory bowel disease: a comparison between Crohn's and ulcerative colitis. Colorectal Dis. 2011;13(11):1230-5. [CrossRef]
- Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW. Hemoglobin. A biologic fenton reagent. The Journal of biological chemistry. 1984;259(23):14354-6.
- Woodford, FP. Ethical experimentation and the editor. N Engl J Med. 1972;286(16):892. [CrossRef]
- Abe C, Miyazawa T, Miyazawa T. Current Use of Fenton Reaction in Drugs and Food. Molecules. 2022;27(17). [CrossRef]
- Ruan L, Wang M, Zhou M, Lu H, Zhang J, Gao J, et al. Doxorubicin-Metal Coordinated Micellar Nanoparticles for Intracellular Codelivery and Chemo/Chemodynamic Therapy in Vitro. ACS Appl Bio Mater. 2019;2(11):4703-7. [CrossRef]
- Cao Y, Liu M, Cheng J, Yin J, Huang C, Cui H, et al. Acidity-Triggered Tumor-Targeted Nanosystem for Synergistic Therapy via a Cascade of ROS Generation and NO Release. ACS Appl Mater Interfaces. 2020;12(26):28975-84. [CrossRef]
- Available online: https://www.shutterstock.com/create/editor/CiQ3YTEzMTI1YS0zOTdiLTQzNTAtOTE2ZC0yYWM5N2U0NmRlYWM.
- Bystrom LM, Guzman ML, Rivella S. Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid Redox Signal 2014;20(12):1917-24. [CrossRef]
- Available online: https://pikbest.com/png-images/qianku-drawing-burning-wood-illustration_1963420.html.
Figure 1.
Dynamics and function signaling pathways for tight junctions and the epithelial junction complex: The figure shows a schematic drawing of the malfunctioning tight junction protein claudin-1 due to inflammation in IBD. Tight junctions are an intercellular adhesion complex of epithelial and endothelial cells, and form a paracellular barrier that restricts the diffusion of solutes on the basis of size and charge. Tight junctions are formed by multiprotein complexes containing cytosolic and transmembrane proteins.Reproducedwith permission from Steed et al., Trend Cell Biol, Elsevier, 2010 [98]. Abbreviations: ZO-1 is a tight junction protein establishes a link between the transmembrane protein occludin and the actin cytoskeleton; Occludin, is integral transmembrane component of the tight junction.
Figure 1.
Dynamics and function signaling pathways for tight junctions and the epithelial junction complex: The figure shows a schematic drawing of the malfunctioning tight junction protein claudin-1 due to inflammation in IBD. Tight junctions are an intercellular adhesion complex of epithelial and endothelial cells, and form a paracellular barrier that restricts the diffusion of solutes on the basis of size and charge. Tight junctions are formed by multiprotein complexes containing cytosolic and transmembrane proteins.Reproducedwith permission from Steed et al., Trend Cell Biol, Elsevier, 2010 [98]. Abbreviations: ZO-1 is a tight junction protein establishes a link between the transmembrane protein occludin and the actin cytoskeleton; Occludin, is integral transmembrane component of the tight junction.
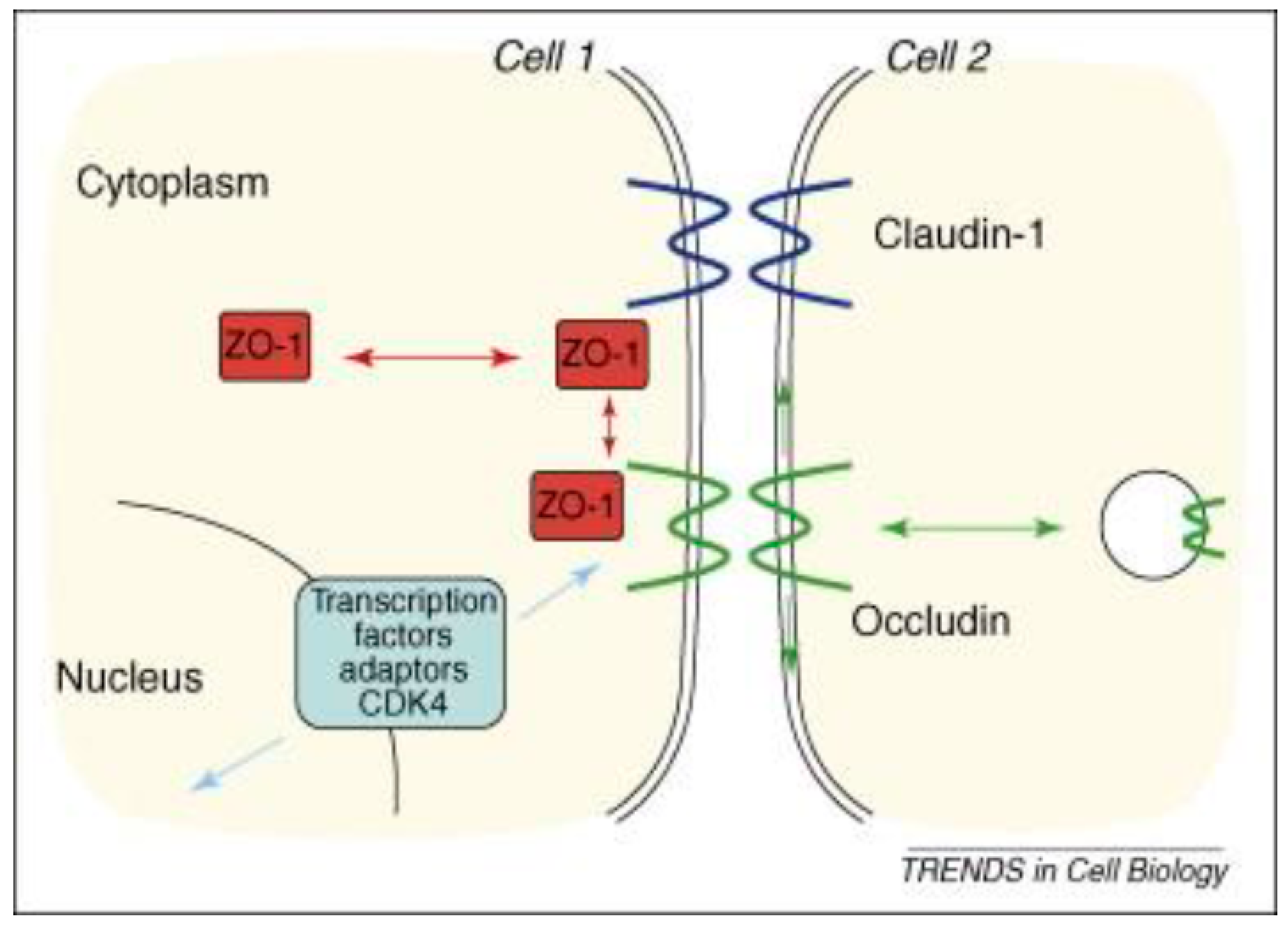
Figure 2.
Depicts extravasated erythrocyte macrophage erythrophagocytosis (EEME): Double immunofluorescent staining for the macrophages marker CD163 (green arrow) in paraffin-embedded sections from a patient with UC. Nuclei were counterstained with DAPI (blue): Red arrows indicate extravasated erythrocytes and white arrow indicate macrophages. Pictures were taken at 60x magnification. A green arrow depicts macrophage erythrophagocytosis - a macrophage engulfed three extravasated erythrocytes (red arrows). Reproduced with permissionfrom Myers et.al, Inflamm Bowel Dis, 2014 [1]. Abbreviations: EEME, extravasated erythrocyte macrophage erythrophagocytosis; UC, ulcerative colitis.
Figure 2.
Depicts extravasated erythrocyte macrophage erythrophagocytosis (EEME): Double immunofluorescent staining for the macrophages marker CD163 (green arrow) in paraffin-embedded sections from a patient with UC. Nuclei were counterstained with DAPI (blue): Red arrows indicate extravasated erythrocytes and white arrow indicate macrophages. Pictures were taken at 60x magnification. A green arrow depicts macrophage erythrophagocytosis - a macrophage engulfed three extravasated erythrocytes (red arrows). Reproduced with permissionfrom Myers et.al, Inflamm Bowel Dis, 2014 [1]. Abbreviations: EEME, extravasated erythrocyte macrophage erythrophagocytosis; UC, ulcerative colitis.
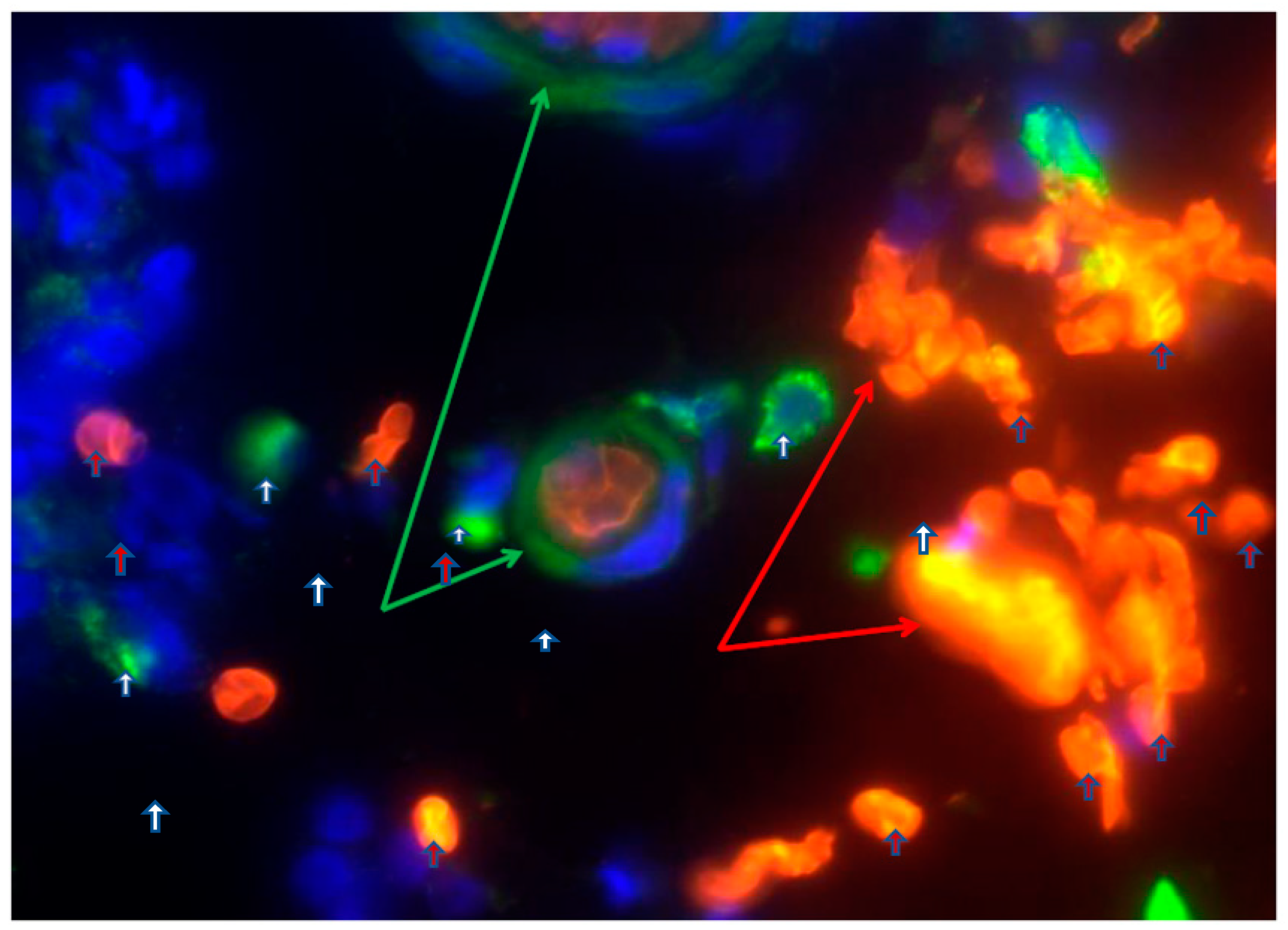
Figure 3.
Comet assay, Hbα causing DNA damage: (A, normal human colon epithelial cells, NCM460, were fed with fresh complete media alone as control, B, with 150µM Hbα for 4hr followed by Comet assay to assess DNAD using denaturing electrophoresis. C, (a) undamaged cells, (b) damaged cells, & (c) severely damaged cells an interpretation of the intensity of DNAD. Reproduced with permission from Myers et.al, Inflamm Bowel Dis, 2014 [1]. Abbreviations: NCM460, normal colonic-epithelial-cell-line; DNAD, Deoxyribonucleic acid–damage; Hbα, hemoglobin alpha.
Figure 3.
Comet assay, Hbα causing DNA damage: (A, normal human colon epithelial cells, NCM460, were fed with fresh complete media alone as control, B, with 150µM Hbα for 4hr followed by Comet assay to assess DNAD using denaturing electrophoresis. C, (a) undamaged cells, (b) damaged cells, & (c) severely damaged cells an interpretation of the intensity of DNAD. Reproduced with permission from Myers et.al, Inflamm Bowel Dis, 2014 [1]. Abbreviations: NCM460, normal colonic-epithelial-cell-line; DNAD, Deoxyribonucleic acid–damage; Hbα, hemoglobin alpha.
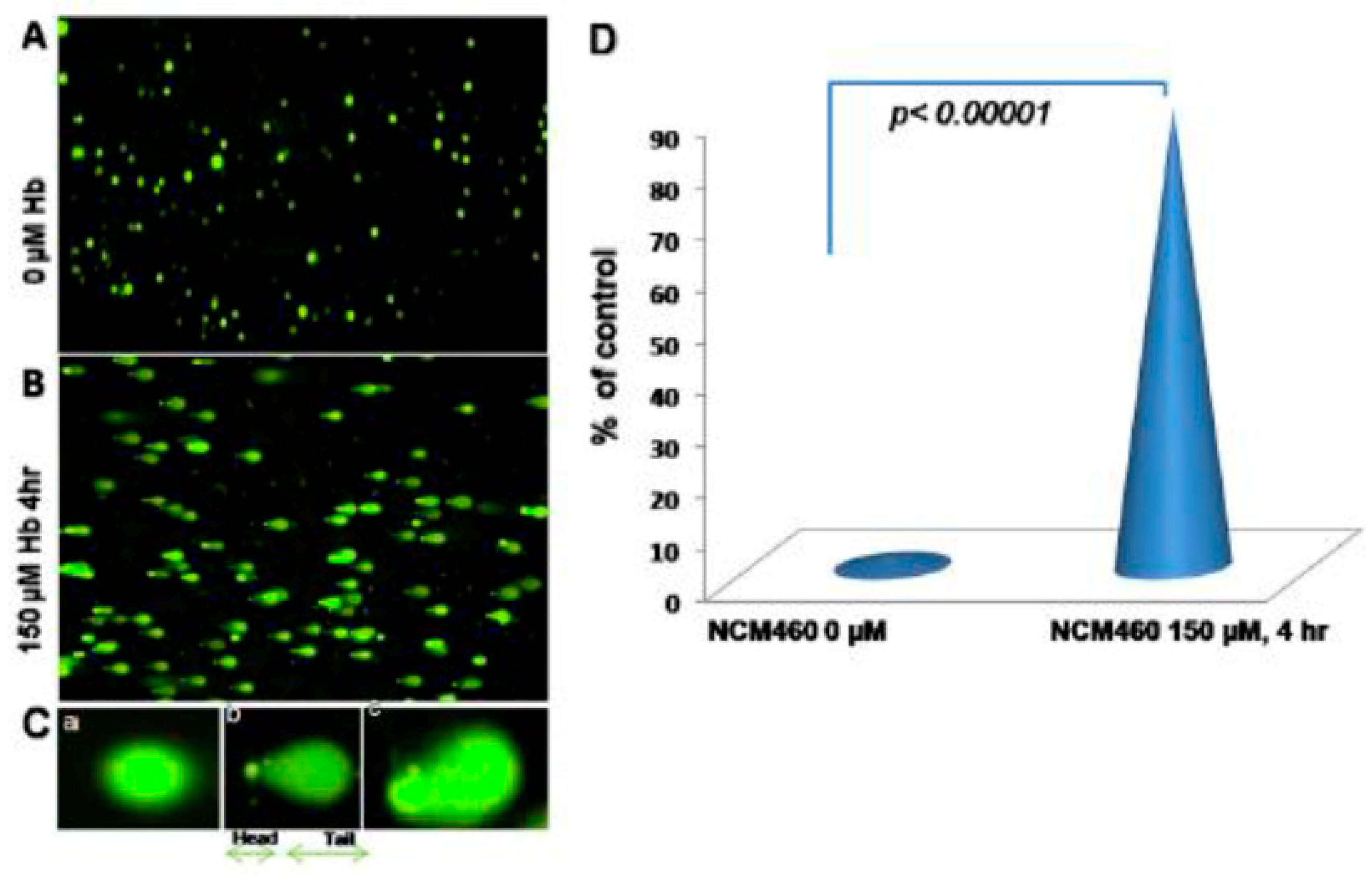
Figure 4.
The pathophysiology of extracellular / exogenous HbαC and subsequent associated with enhanced oxidative reaction "the Fenton Reaction (FR)”: The FR here is the chemical response between exogenous HbαC and hydrogen peroxide, resulting in hydroxyl radical, which is extremely receptive and exceedingly toxic/noxious to living cells and is oncogenic trigger, this can also serve as an aspect that can be a therapeutic target/ strategy for cancer patients. The figure was free downloaded, modified for clarification [148]. Abbreviations: FR, Fenton reaction; HbαC, hemoglobin alpha chain; HO, HO+ is Hydroxide, OH- is Oxyhydride; CO, Carbon Monoxide; Fe2, iron (II); H2O2, Hydrogen peroxide.
Figure 4.
The pathophysiology of extracellular / exogenous HbαC and subsequent associated with enhanced oxidative reaction "the Fenton Reaction (FR)”: The FR here is the chemical response between exogenous HbαC and hydrogen peroxide, resulting in hydroxyl radical, which is extremely receptive and exceedingly toxic/noxious to living cells and is oncogenic trigger, this can also serve as an aspect that can be a therapeutic target/ strategy for cancer patients. The figure was free downloaded, modified for clarification [148]. Abbreviations: FR, Fenton reaction; HbαC, hemoglobin alpha chain; HO, HO+ is Hydroxide, OH- is Oxyhydride; CO, Carbon Monoxide; Fe2, iron (II); H2O2, Hydrogen peroxide.
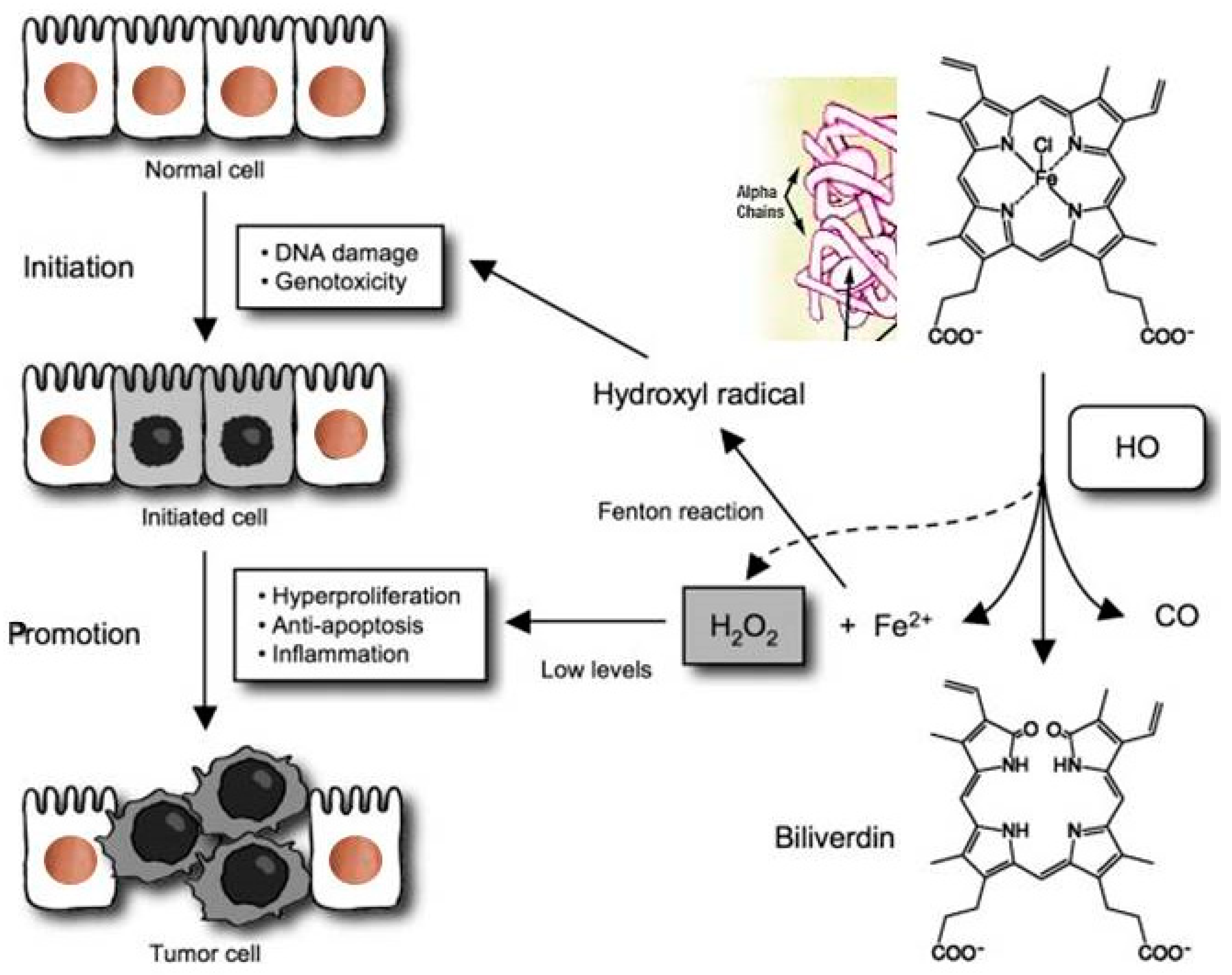
Figure 5.
Haptoglobin phenotype evaluation by a native one-dimensional gel electrophoresis technique. Haptoglobin is the protein made in the liver that in humans is encoded by the HP gene. Three major haptoglobin phenotypes are known to exist: Hp 1-1 – homodimers; Hp 2-1 – liner heterodimers; and Hp 2-2 – cyclic heterodimers. Hp 1-1 is biologically the most effective in binding free hemoglobin and suppressing inflammatory responses associated with free hemoglobin. Hp 2-2 is biologically the least active, and Hp 2-1 is moderately active. In blood plasma, haptoglobin binds with high affinity to free hemoglobin released from erythrocytes, and thereby inhibits its deleterious oxidative activity. Free haptoglobin is removed from plasma in 3.5-5 days. On the other hand, the haptoglobin-hemoglobin (Hp-Hb) complex is removed within 20 minutes. This known fact stresses the importance of Hbα removal in the presence of Hp. Reproduced with permission from Naryzny et al., Biochem Mosc Suppl B Biomed Chem. Springer Nature, 2021 [106]. Abbreviations: Hp 1-1, Hp 2-1, and Hp 2-2. Haptoglobin-hemoglobin complex; Hbα, hemoglobin alpha.
Figure 5.
Haptoglobin phenotype evaluation by a native one-dimensional gel electrophoresis technique. Haptoglobin is the protein made in the liver that in humans is encoded by the HP gene. Three major haptoglobin phenotypes are known to exist: Hp 1-1 – homodimers; Hp 2-1 – liner heterodimers; and Hp 2-2 – cyclic heterodimers. Hp 1-1 is biologically the most effective in binding free hemoglobin and suppressing inflammatory responses associated with free hemoglobin. Hp 2-2 is biologically the least active, and Hp 2-1 is moderately active. In blood plasma, haptoglobin binds with high affinity to free hemoglobin released from erythrocytes, and thereby inhibits its deleterious oxidative activity. Free haptoglobin is removed from plasma in 3.5-5 days. On the other hand, the haptoglobin-hemoglobin (Hp-Hb) complex is removed within 20 minutes. This known fact stresses the importance of Hbα removal in the presence of Hp. Reproduced with permission from Naryzny et al., Biochem Mosc Suppl B Biomed Chem. Springer Nature, 2021 [106]. Abbreviations: Hp 1-1, Hp 2-1, and Hp 2-2. Haptoglobin-hemoglobin complex; Hbα, hemoglobin alpha.
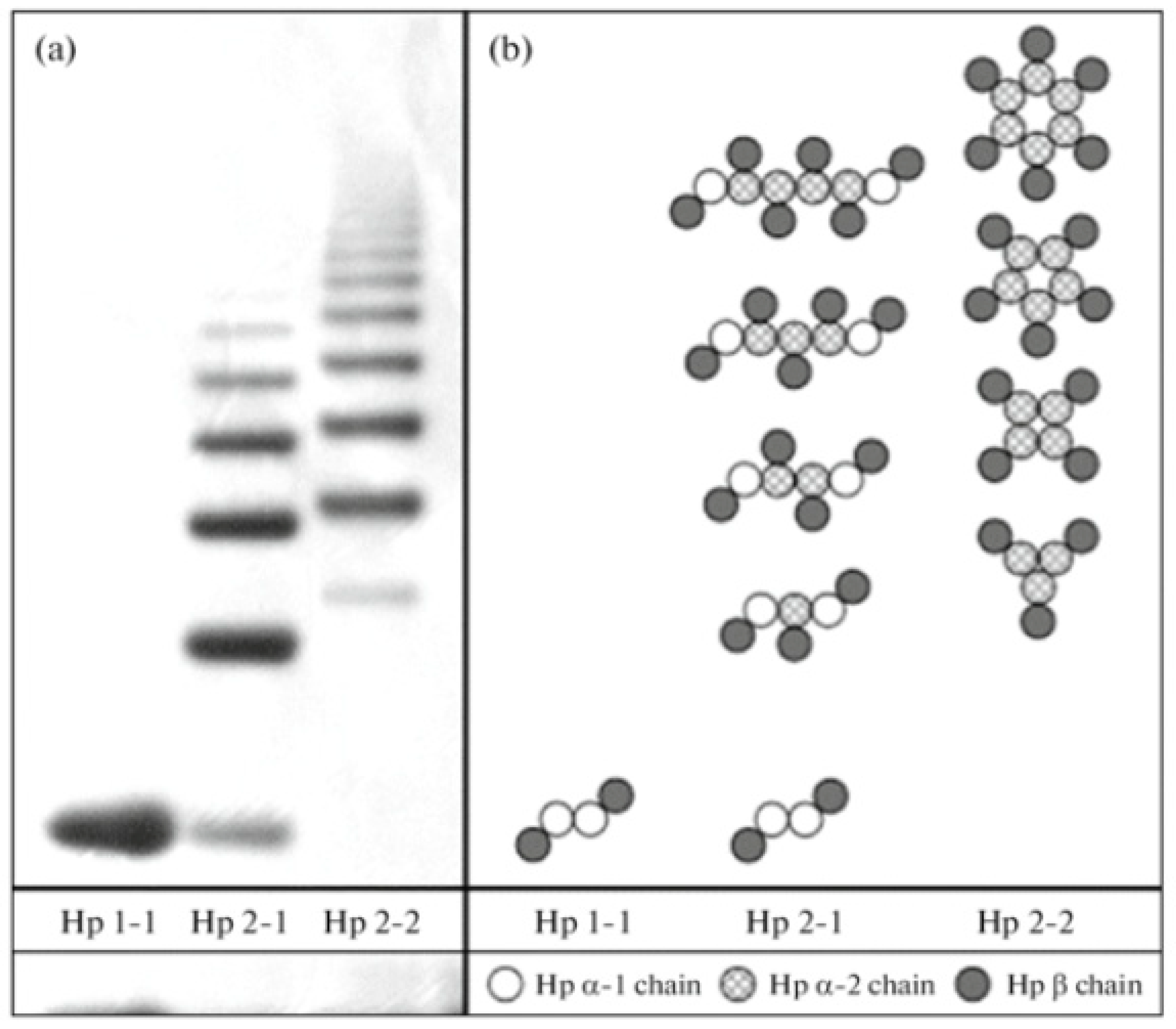
Figure 6.
Deferoxamine, Figure 6. Deferoxamine, alias desferrioxamine or desferal, a chelating agent: utilized to clear away unwanted excess iron or aluminum from the body. It reacts by confining exogenous free iron or aluminum in the bloodstream and reinforce its elimination in the urine. Reproduced with permission from Cao et al., American Chemical Society, 2020 [146]. Abbreviations: C25H48N6O8, Deferoxamine.
Figure 6.
Deferoxamine, Figure 6. Deferoxamine, alias desferrioxamine or desferal, a chelating agent: utilized to clear away unwanted excess iron or aluminum from the body. It reacts by confining exogenous free iron or aluminum in the bloodstream and reinforce its elimination in the urine. Reproduced with permission from Cao et al., American Chemical Society, 2020 [146]. Abbreviations: C25H48N6O8, Deferoxamine.
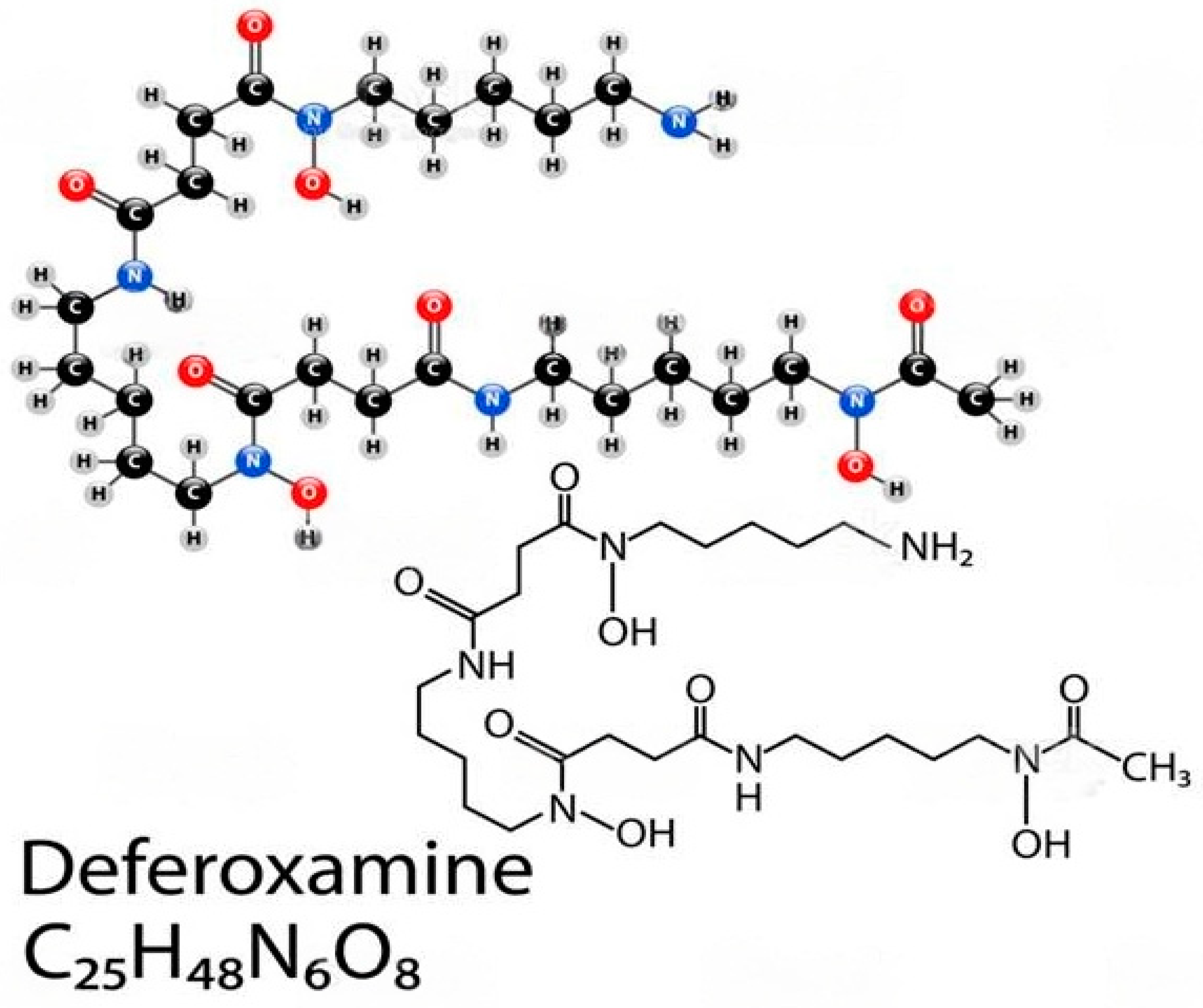
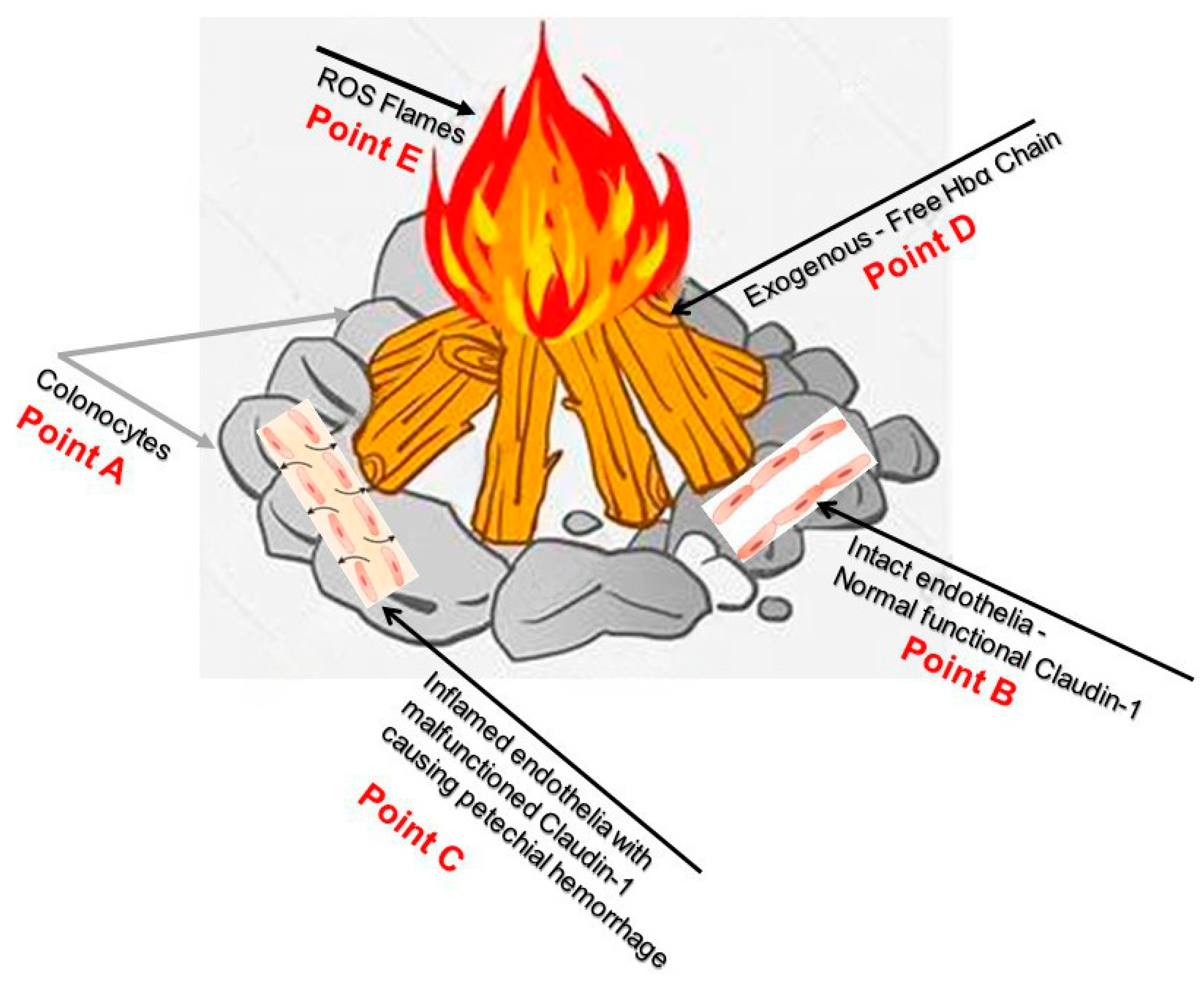
Figure 8.
Point A demonstrates colonocytes of the colonic mucosal epithelium, while Point B illustrates intact healthy capillary endothelia with normal functional Caludi-1. Point C depicts active IBD and dysfunctional Claudin-1, the source of potential hemorrhage, Point D show exogenous free HbαC that cause ROS flames in Point E. Current endoscopic surveillance is inadequate and re-emphasizes the need to look further into the dysfunctional claudin-1 protein; this could hopefully prevent ROS-mediated DNA damage. The figure was an imagination of the pathophysiology of CACRC, A-E. Reproduced with permission from Pinbest.com [149]. Abbreviations: HbαC, hemoglobin alpha chain; ROS, reactive oxygen species; CACRS, colitis-associated colorectal cancer.
Figure 8.
Point A demonstrates colonocytes of the colonic mucosal epithelium, while Point B illustrates intact healthy capillary endothelia with normal functional Caludi-1. Point C depicts active IBD and dysfunctional Claudin-1, the source of potential hemorrhage, Point D show exogenous free HbαC that cause ROS flames in Point E. Current endoscopic surveillance is inadequate and re-emphasizes the need to look further into the dysfunctional claudin-1 protein; this could hopefully prevent ROS-mediated DNA damage. The figure was an imagination of the pathophysiology of CACRC, A-E. Reproduced with permission from Pinbest.com [149]. Abbreviations: HbαC, hemoglobin alpha chain; ROS, reactive oxygen species; CACRS, colitis-associated colorectal cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



