
Submitted:
05 June 2023
Posted:
06 June 2023
You are already at the latest version
Abstract
Additionally, the League’s four observing states, with an additional population of 1.7 billion, if join the group, will form the world’s largest consortium to enhance the accessibility of biotechnology drugs. This effort will also remove the criticism of regulatory agencies that are well-equipped to regulate these products. This should also be a significant economic incentive to consortium states to develop these products, particularly biosimilars, to capture the most critical market anticipated. This plan will also reduce the cost burden on regulatory agencies. The program is divided into two classifications; for products sold in any of the Stringent Regulatory Authority (SRA) states, the registration is automatic but with several requirements that assure ongoing pharmacovigilance; this also applies to copies of reference products or biosimilars. For non-SRA sourcing, a stepwise plan requiring a rapporteur review, as practiced by the EMA and a third-party cGMP audit, is necessary to ensure data and product integrity. However, to reduce the cost burden, approval of these products is based on current scientific findings as presented in this paper.

Keywords:
Biosimilars 1
; Arab states 1
; regulatory guideline 3
; harmonization 4
; centralized approval 5
; EMA 6
; FDA 7
; Global Medicine Authority (GMA) 8.
Introduction
The global spending on medicines is projected to rise to about USD 2 Trillion in 2027 from the current USD 1.5 Trillions [1]; more than half of it is attributed to biological drugs, the oldest class of medicines when humans began using plant and animal products to treat disease for thousands of years. Even modern concepts like vaccination date back hundreds of years–with inoculation against smallpox using powdered scabs being practiced in China as early as the 10th century [2]. The chemical drugs came in the early 20th century, requiring the use of different terminology for biologics such as vaccines, sera, and vitamins began to be mass-produced, resulting in a scramble to standardize their definition, production, and quality–ultimately culminating in the Biologics Control Act enacted by the United States Congress in 1902 [3].
The number and variety of biologics continued to grow through the course of the 20th century, as did the capacity to produce these agents that led to the first successful usage of an in vitro system to produce biologics when Boston Children’s Hospital researchers created Lansing Type II poliovirus using a human tissue cell culture in 1949. This development would set the stage for modern biologics production [4]. But the rise of genetic engineering during the late 1970s and early 1980s opened new avenues for biologics production and development. The ability to modify genetic sequences meant that researchers could modify existing agents to improve their stability, safety, and efficacy. These alterations could also change agent targeting specificity, giving certain agent types, such as antibodies, a significantly greater range of applications. Finally, genetic engineering gave researchers a more extensive portfolio of potential production models. Whereas establishing a cellular factory was previously limited by the genome of the production cell–or, in the case of viral production, its susceptibility to persistent but non-lethal infection–the emergence of transfection and transduction now meant, in theory, that any cell could be made to produce any molecular or protein-based agent. For example, recombinant cell lines can be transformed using CRISPR technology to produce proteins with specific characteristics [5].
Biologics research and production have seen tremendous growth since the 1980s, with considerable progress in developing new therapeutic approaches for cancers, immune disorders, and rare genetic disorders, just to name a few [6]. A field that once was simply the extraction of naturally produced substances has evolved to encompass the conception, engineering, and production of a diverse range of sophisticated designer molecular-, protein-, gene-, cell-, and tissue-based agents capable of highly-selective targeting. Indeed, challenges remain regarding efficacy, specificity, and longevity, but science–and scientists–continually work towards discovering and developing new agents and tweaking existing ones [7].
New drug development costs now run into billions of dollars [8], leading to their high price for at least five years for a new chemical and 12 years for a new biological drug [9]. In addition, the new technology brought new regulatory guidelines, and the cost of a recent biological drug approval rose to billions of USD [10], resulting in the high price of these products (Table 1) to amortize the development over the 12 years of exclusivity.
Motivation
A motivation for the Arab League States to join hands should come from understanding how unaffordable these drugs are to Arab citizens. Just a century ago, the healthcare sector [12,13] in the Arab world struggled to deal with challenges posed by the colonial and military systems that governed its territories at the time. Until the Second World War, hospitals in the region were primarily small, private facilities set up by doctors who had received medical training abroad before returning to their home countries to practice medicine. Although many Arab states had become at least nominally independent states by the 1950s, the legacy of colonialism [14] continued to be felt, including in the healthcare system, where the paternalistic, top-down approach implemented under colonial rule persisted. Often inadequate government facilities were established for the poorest residents. At the same time, those with resources would usually pay for care in private facilities or travel abroad for medical procedures—both trends that continue today.
The region has seen significant turmoil in the many decades since its countries gained independence. As a result, few Arab states have enjoyed the stability, transparency, and prosperity required to build a functional healthcare system. Yet health indicators such as overall life expectancy and infant and maternal mortality rates undeniably improved [15] in the second half of the 20th century, due in large part to a decrease in regional poverty, improvements in water, sanitation, and electricity systems, and a reduction in the mortality burden of infectious diseases. However, due to economic, political, and social factors, health outcomes [16] across the Arab world today differ widely, with the best performance in wealthy states like Bahrain and Oman, and abysmal performance in fragile, low-income countries like Yemen and Somalia.
The healthcare expenditure within the Arab League states varies from the lowest percentage of GDA of 1.8% by Djibouti to 8% by Lebanon [17]. The world’s higher GDP share for healthcare is the US at 18% [18] (Figure 1). Of significance is the total per capita expenditure on healthcare, of which the portion for medicines is less than USD 20 per capita in Somalia, compared to USD 1,400 in the US [19].
The inequity in the healthcare sector in the Arab World is well recognized [20]. But despite much effort by the Arab League, concluding that the health of most citizens across the region is less than optimal [21], especially among the most marginalized, not much has been done.
However, the markets for medical and pharmaceutical products and services have expanded fast in recent decades [23]. Today, the region’s pharmaceutical sector is estimated [24] to reach about USD 60 billion by 2025, up from USD 36 billion in 2016. While this represents just a minor portion of the global pharmaceutical market, it is still a significant leap made over just a few years.
As shown below, there is a need for the League States to engage in manufacturing these products, where possible, such as biosimilars that command high market (Table 2) that can bring remarkable returns on investment.
All these drugs can be made available as biosimilars, reducing their price to more than 80% while keeping a 90% profit margin, based on the WHO finding that all monoclonal antibodies can be manufactured for USD 95-200/gram [26]. Furthermore, these costs will decrease as newer technologies, such as continuous manufacturing [27], are adopted. This perspective should greatly interest companies within the Arab states to consider domestic manufacturing and export to compete in multibillion USD markets.
The market for biosimilars in the Arab World (Middle East and North Africa) region is experiencing significant growth. According to a report by IQVIA, the biosimilar market in the Arab states is projected to reach USD 1.6 billion by 2024, with a compound annual growth rate (CAGR) of 31.6% [28]. The Arab states represent about 2% of the pharmaceutical industry. The biosimilar market for the region is projected to grow from USD 0.71 billion in 2021 to USD 2.17 billion by 2026. This growth results from rising GDP, healthcare expenses, and the demand for cost-effective therapeutic solutions. In addition, biosimilars play a significant role in minimizing public healthcare expenditure on medicines, thus increasing the accessibility of these medicines to more patients.
Saudi Arabia, which had a pharmaceutical sector worth more than USD 10 billion in 2021, expects to grow around 7 percent over the next few years. Many of the world’s major pharmaceutical players, including Pfizer, Sanofi, GSK, and AstraZeneca, operate in Saudi Arabia [29], along with many Saudi and Arab companies, including Jamjoom Pharma, SPIMACO, Sudair, Riyadh, Tabuk Pharmaceuticals, and Jazeera Pharmaceutical Industries. In addition, as part of Saudi Arabia’s Vision 2030 [30] initiative, which emphasizes localization across several industries, multiple pharmaceutical manufacturing agreements have been initiated to increase production within the country.
Tunisia, which established one of the region’s first pharmaceutical warehouses in the late 1930s, is another country with a rapidly growing pharmaceutical sector [31]. From 2014 to 2018, this sector grew more than 45 percent, increasing exports by 7 percent in the same period. This growth resulted from the operations of 120 companies, 33 of which are actively involved in producing medicines for human use. In addition, the country has invested heavily in educating medical and pharmaceutical students, promoting cooperation with pharmaceutical companies worldwide, and signing direct agreements with manufacturers.
Jordan is another major pharmaceutical sector player, contributing largely to the country’s economy. Jordan has a long history in the industry, and aside from providing up to 25 percent of its own population’s medicinal needs—which makes the country less reliant on imports than most of its regional neighbors—Jordan has the distinction of being one of the few countries in the region with significant pharmaceutical exports, primarily of generic drugs. In 2021, Jordan announced [32] that its pharmaceutical exports had reached 1 billion Jordanian dinars (around USD 1.4 billion), making it the nation’s only sector to export more than it imports. Today, around 75 percent of pharmaceutical products produced in Jordan are exported [33]. This remarkable mindset in Jordan is suited for promoting the manufacturing of biological drugs, bringing much higher profit margins and substantially adding to Jordan’s economy.
In recent years, the Arab states market has seen a noticeable increase in the value share of biologics, in line with general industry trends. As a result, biologics’ value share in the Arab states was predicted to rise at a 30 percent annual pace reaching close to 10 billion USD. With over USD 2B in sales, the Kingdom of Saudi Arabia (KSA) dominates the market. The following three countries are Algeria, Egypt, and the United Arab Emirates (UAE), with about 450 million USD in sales [34]. Healthcare costs, the GDP, and the demand for affordable therapies trigger this anticipated market expansion [35].
The Article II of the Charter of the Arab League [36] identifies “health affairs” as its main charter; Article IV describes a mechanism for how the goals of the League are managed. However, there is a need to develop a more formal platform, as proposed in this paper, and it will be possible for the League to consider this suggestion to ensure continuous market growth seriously [37].
A more relevant effort is already initiated to establish the Arabian Medicines Agency (AMA) [38], an initiative that can resolve many issues of affordability of biological drugs. I am promoting this concept and following my discussion with this terminology as the culmination of cooperation among the Arab states.
Recently, the Saudi FDA hosted the 1st meeting of Arab regulatory authorities to enhance Arab integration in medicines control and legislation and promote the exchange of expertise and best practices among Arab medicines control authorities. It was emphasized that coordination and alignment with legal and regulatory requirements for medicines are vital to facilitating the registration and availability of pharmaceutical products and lowering their cost, promoting patient access to these products. The Technical Committee for Arab Medicines also held a simultaneous to monitor the implementation of Resolution No. 17, issued in the 58th session of the Council of Arab Ministers of Health in March 2023.
Gulf Cooperation Council
Saudi Arabia accounts for the largest market share within the Arab states. It is also a member of the Gulf Cooperation Council (GCC) that follows its centralized procedures in which different authorities are involved. The Gulf Health Council (GHC) acquires pharmaceutical products with proven efficacy, quality, and safety. The Gulf Central Committee for Drug Registration (GCC-DR) oversees various procedures- from manufacturing site registration to post-marketing surveillance.
The Saudi Food and Drug Authority (SFDA) reviews and approves biological product and its price before it enters the market. The SFDA regulatory framework follows the United States (U.S.) FDA and the European Medicines Agency (EMA) guidelines with specificities that accommodate the local and regional (GCC) requirements.
Another member of the GCC, the UAE’s requirements for biosimilar registration include data on consistency in the manufacturing process, demonstration of immunogenicity, heterogeneity assessment, safety and efficacy studies, therapeutic equivalence, and a pharmacovigilance plan where the community can directly express their concerns to the Ministry of Health.
Biosimilars produced in the UAE for international markets follow international guidelines set by the EMA and World Health Organization (WHO). However, for local markets, they follow UAE standards and guidance set by the GCC. Changes in the specifications or characteristics of a biosimilar product are considered new products and are required to follow biosimilar procedures.
While there are some similarities regarding the cooperation among states in the GCC, the scope of GMA is very different; it is a global plan that applies only to biotechnology drugs
Misconceptions
The first MENA stakeholder meeting regarding biosimilars was held in Dubai in 2015; the 2nd MENA Stakeholder Meeting on Regulatory Approval, Clinical Settings, Interchangeability, and Pharmacovigilance of Biosimilars was held in Dubai, UAE, on 10 October 2018 suggesting. However, the comments submitted at this meeting show misconceptions that need clarification [39]:
- ❖ “Regulatory bodies need scientists to evaluate a dossier. Others suggested that the region is plenty of scientists, pharmacists, and academicians to serve this duty.”
There is a great misunderstanding among Arab agencies about the qualification of regulatory staff. These include analytical chemists, pharmacokinetics, clinicians, statisticians, quality assurance auditors, lawyers, and others with specific expertise. Unless trained in a specific function, pharmacists, academicians, scientists, and politically appointed heads of the agency should not be part of any dossier evaluation. For this reason, I recommend installing a system of rapporteur evaluation of all biosimilar submissions. In addition, the FDA employs 11,000 full-time scientists [40], so it is not expected of any non-SRI agency to be able to evaluate a regulatory dossier on its own.
- “Discussion between national and international regulatory bodies is needed to ensure biosimilar approval is consistent worldwide.”
This is a broader goal that has never been possible, even for generic chemical drugs; there are agencies like the ICH and WHO that provide guidelines that are often adopted, but to expect that regulatory bodies will agree on issues that take their authority away, is not likely to be happy. However, this is what I am suggesting, but with a narrow goal.
- “The limitations faced by recently established regulatory bodies must be recognized and addressed by mature regulatory bodies worldwide.”
What is meant by a “mature” authority; an “immature” authority should not operate in the first place. Expecting the FDA or EMA support can only be limited to following their guidelines. Suppose it is meant that the region’s authorities have been operating longer to help the newcomers. In that case, this, too, is misleading, for a more extended operation does not necessarily mean maturity.
- “Countries with greater experience must support countries with less experience of biosimilars.”
It is doubtful that any country in the region has the required experience to make them a teacher. Therefore, the right thing to do is to harmonize the registration process, where a single agency approves biosimilars that all member countries will accept.
- “Action should be taken to ensure that all biosimilar products globally are traceable at batch level to ensure adequate pharmacovigilance is upheld. Biosimilar naming will be key to this.”
This is not an issue; all products have registration and batch numbers, and the brand naming system is widely accepted to ensure traceability.
- Strong governmental regulators should be in place to ensure drug products can be tracked.
Traceability is a fundamental process that applies to all drugs, which is the essential function of any regulatory authority.
- “The long-term effects of switching and multi-switching between biosimilars and/or reference products need to be understood and addressed. This requires a concerted international effort to develop an optimal methodological approach.”
This is a misconception; there is no risk in interchangeability that is only an issue in the US, and that, too, is about to be removed. Therefore, there is no need to dwell on this wasteful exercise [41].
- “Biosimilar patient registries could be established and implemented to gather further data on switching.”
It is not necessary. The EMA has recently reasserted this position allowing switching with the reference product and other biosimilars [42]. Most other countries in the rest of the world have already begun this practice that remains in the US due to legislative matters [43].
- “Electronic healthcare records need to be developed and implemented to facilitate pharmacovigilance and gather further data on switching.”
Member countries can’t have electronic health records; pharmacovigilance is a common practice for all drugs. So there is no need to extend this to switching.
- “Encourage meeting with clinicians to explain what biosimilars are and what they are not, to enable them how to decide whether to prescribe biosimilars.”
Clinicians are least qualified to understand the nuances of the regulatory process; they must trust agencies’ decisions. No need to waste time teaching clinicians.
- “Physicians, pharmacists, regulators, patients, and all stakeholders must communicate and share their experiences–challenges, and successes–with biosimilars.”
This wasteful exercise is more of a slogan; there is no need to teach or promote biosimilars. All must accept an approved biosimilar; promoting its safety and efficacy may even cause doubt about its safety and efficacy.
Other misconceptions are summarized as follows:
The WHO is not a regulatory agency; it is an Agency whose decisions are widely criticized since it operates mostly on common consensus. The FDA and EMA guidelines depend on many legislative issues and legal exposure and are slow to change the guidelines as new science teaches otherwise.
- A lack of agreement exists among the Arab states regarding regulatory approval issues, particularly regarding interchangeability and switching. In Saudi Arabia, biosimilars are not automatically interchangeable. For example, ten biosimilars have been approved, of which only two are regarded as interchangeable. It is evident that, in Saudi Arabia, biosimilarity alone is not sufficient for substitution or switching. However, biosimilars approved by EMA are considered interchangeable. Additionally, a clinical trial that involves switching must be run to approve switching- this must happen before the biosimilar is approved.
None of these considerations are needed; as allowed in the EU, all biosimilars are interchangeable, even one biosimilar with another. The issue of interchangeability is indigenous to the US and is up for removal.
- The Egyptian Drug Authority (EDA) “Guideline for registration of Biosimilar products in Egypt” is in place as of March 2020. The applicant must exhibit and compare the biosimilarity of their product to the innovator/reference product by completing and comparing pre-clinical and clinical studies and quality exercises. The EDA adopts the EMA guidelines and refers to the U.S. FDA’s safety and quality considerations, the WHO guidelines for evaluating similar biotherapeutic products, and relevant ICH guidelines. In Egypt, however, the Ministry of Health will make interchangeability decisions, where the patient will not be given a choice.
Misconceptions regarding blanket following a lead to archaic animal toxicology testing and other quality assessments that may not be necessary, as described below. An agency should create its guideline, albeit borrowing from any additional guideline, instead of listing another as the marker. Comments for interchangeability apply as stated above.
- The Jordanian FDA’s guidelines are based on the EMA, where the EMA model has been implemented for quality assessment and comparability. It also authorizes the approval of manufacturing sites as a prerequisite to product approval and filing. Currently, six products have been approved according to Jordan’s biosimilar guidelines. Jordan’s approach to biosimilar regulation can be considered vigilant and strict. Nonetheless, biosimilars manufactured and marketed in reference countries, including but not limited to the UK, USA, Germany, France, Netherlands, Sweden, Australia, Austria, and Japan, are usually given more privileges.
The same comments as offered for the Egyptian guideline apply here. In addition, references to biosimilars from some SRA countries should be expanded and automatically allowed registration without reviewing the dossier.
- Medicines in Tunisia are obtained by centralized pharmacy purchase (PCP). The Biosimilar Specialized Committee makes decisions on a case-by-case basis regarding interchangeability. The committee comprises representatives of pharmaceutical inspection, national control laboratory, regulatory authorities, various clinicians, and experts who utilize biosimilars.
The interchangeability issue is redundant; the approval committee need not include anyone who is not qualified to judge the compliance of a dossier. Moreover, approval should not be based on consensus, a significant weakness in almost all Arab state agencies, as elaborated above.
Biosimilars
A major focus of this paper is to enable faster entry of therapeutic proteins, either as new products or as biosimilars. While new products should be allowed without review of the dossier if it is marketed in an SRA country, as elaborated below, biosimilars from non-SRA sources need a detailed discussion. This section refers to these details.
Therapeutic proteins are produced by recombinant-engineered biological agents, bacteria, mammalian cells, and the like; thus, they are called biological drugs. A new biologic drug is approved based on its extensive safety and efficacy testing; it is characterized but not compared with any other drug. Conversely, a biosimilar is approved based on its similarity with the reference product; if the structure of biological drugs were fixed like the chemical drugs, no analytical assessment would be required, and biosimilars would be approved as chemical generics. The fundamental understanding of biologics lies in their 3D structure, which is responsible for their receptor binding and immunogenicity. So, logically, if we can prove that the 3D structure of a biosimilar is exact (almost identical), that should reduce the testing significantly [46]. However, the expressed protein is subject to post-translational modifications in all expression systems, though more intensely in mammalian cells [47].
An extensive analytical assessment is made in comparison with its reference product to ensure that a biosimilar candidate is highly similar, if not identical. Analytical science has become more sophisticated, now to a point where it is the most robust test of similarity, compared to all other tests like animal toxicology, clinical pharmacology, and even clinical efficacy testing in patients. However, despite this understanding, the regulatory agencies and the developers have maintained a mindset that clinical efficacy testing is needed to assure the safety and efficacy of biosimilars.
In 2006, the EMA approved the first biosimilar guidance and approved the first biosimilar [48]. As of April 2023, 47 biosimilars were approved in the US, including peptides, and 74 in the EU, representing 19 molecules, including peptides, out of more than 260 available recombinant therapeutic protein molecules [49] available as possible choices for biosimilars.
EMA and FDA have modified the biosimilar approval guidelines over time as more evidence about their safety and efficacy became available. The WHO also publishes guidelines to assist its 194 country members [50], but the WHO is not a regulatory agency; many member countries create their guidelines by “cherry picking” the WHO advice [51,52] risking the safety and efficacy of their biosimilars. For example, the Indian guidelines based on the WHO guidance [53] continue to include extensive animal toxicology testing and require efficacy testing in the local population on a fixed number of patients, both redundant and irrelevant.
The first tranche of biosimilar approval guidelines treated biosimilars like new biological drugs for an abundance of caution, including extensive analytical comparisons, animal pharmacology and toxicology, clinical pharmacology, and clinical safety and efficacy studies. The only concession allowed is the extrapolation of indications. A comparative clinical efficacy testing in one indication would be sufficient to qualify for all indications allowed for the reference product. To further assure safety and efficacy, biosimilars must have the same dose, strength, route of administration, and mechanism of action; the formulations may differ. Also, the prescribing information must be the same, and guidelines are available on writing the prescribing information for biosimilars [54].
Over time, the agencies became more convinced of the safety of biosimilars in response to challenges made to the guidelines [55]. It became well accepted that the animal testing of biosimilars is redundant [56] since now even the new biological products may not be required to conduct such testing because the mechanism of action of biological drugs involves receptor binding that is often unavailable in animal species [57]. The value of clinical efficacy testing has also come under criticism for scientific reasons since these studies cannot fail [58] and, if used to overcome a lack of similarity in analytical or clinical pharmacology, create a higher safety risk possibility if these studies are considered for approval. An excellent example of progressive changes to guidelines comes from the MHRA. Last year, as the Brexit transition period ended, the MHRA published its first comprehensive guideline on 14 May 2022 [59] that breaks from all other guidelines by providing clear judgment for not requiring animal and clinical efficacy studies.
Clinical pharmacology studies, including pharmacokinetic and pharmacodynamic comparisons, are part of the analytical methodologies, where we establish similarities in how the body sees the drug and vice versa. These should be enhanced and recommended for newer technologies and approaches to develop structural equivalence.
Several ICH guidelines provide scientific support to developing biosimilars, which should be made part of every guideline [60]. There is a dire need for harmonizing the regulatory guidelines [61], but it is not likely to happen, as evidenced by historical events; for example, the guideline for approving generic chemical drugs remains diversified for more than fifty years since chemical generics were introduced [62]. Moreover, countries do not agree on which oral product should have a waiver of bioequivalence study; Japan denies all. So, it is understandable why harmonization and global concurrence may not be possible for a class or products as complex as biologics. It has little to do with science, but the legislative nature of these guidelines and the perspective held by the agencies are often difficult to convince otherwise.
Now that we have 18 years of experience in using biosimilars and hundreds of published reports on their safety and efficacy, a strong opinion has emerged [63,64] that significant amendments to the approval guidelines for biosimilars must change, not only to reduce the development cost but also to enhance the safety of these products. Furthermore, lowering the development cost is essential to bring more biosimilars, as only nine out of more than 150 possible biosimilar molecule candidates are approved in the US and 14 in the EU. In addition, there are over 200 molecules that could provide excellent accessibility to patients.
Last year, as the Brexit transition period ended, the MHRA published its first comprehensive guideline on 14 May 2022 [65], removing animal testing and reducing clinical efficacy testing requirements. It is anticipated a harmonized ICH guideline will fulfill this gap. Still, the ICH remains a non-regulatory body, like the WHO, so the need for consolidated regulatory guidelines is highly suggested [66]. Furthermore, the consolidation should be based on current scientific understanding to remove unnecessary and irrelevant testing, as the FDA acknowledges [67,68,69]. These steps are essential to reduce the current development cost of biosimilars at USD 100-300 Million [70].
Other misconceptions include the use of animal testing [71] and clinical efficacy testing [72]. At the end of 2022, the US government passed a new law, The FDA Modernization Act 2.0 [73], removing the term “animal toxicology” and replacing it with “nonclinical” to remove all animal testing since animals do not have the receptors to respond to biological drugs. In addition, the MHRA recently announced that animal and clinical efficacy testing might be unnecessary [74]. This will be the first requirement for any universal guideline to remove all animal testing; if used to justify the variability in analytical assessment, as commonly practiced, animal testing creates a risk of approval of unsafe biosimilars.
Understanding the immunogenicity of proteins is crucial in developing biologics and vaccines. Excessive immunogenicity can lead to reduced efficacy or adverse immune reactions, such as allergic responses or the development of neutralizing antibodies. This is the most controversial topic that leads to primary conservative constraints by regulatory agencies. The fact is that all proteins are immunogenic. The T and B lymphocytes (T and B Cells) are involved in the acquired or antigen-specific immune response. The B Cells can transform into plasmocytes and are responsible for producing antibodies (Abs). Thus, humoral immunity depends on the B Cells, while cell immunity depends on the T Cells. From the morphological point of view, T and B lymphocytes are indistinguishable since they are both small cells (8–10 microns in diameter), and each possesses a large nucleus with dense hetero-chromatin and a cytoplasmic border that contains few mitochondria, ribosomes, and lysosomes. When activated by the antigenic stimulus, they may enlarge, thus increasing their cytoplasm and organelle number. Lymphocytes present receptors for antigen (Ag) recognition (TCR and BCR, respectively) with different specificities on their surfaces. The genes that encode these structures undergo a series of DNA recombination, which provides them with immense phenotypic diversity.
If the immunogenicity profile differs but cannot impact the disposition profile, the differences will be meaningless and unnecessary to compare, as in the case of insulins. During the PK trial, data on immunogenicity and safety should be gathered. Some options include anti-drug antibody (ADA) production rate, kinetics, and assessment of their impact on PK (and PD) using a predetermined group study of ADA-negative and ADA-positive participants. Although they wouldn’t replace the immunogenicity assessment in the PK trial, in vitro immunogenicity assays might enhance the functional, analytical assessment. Results of short-term immunogenicity analyses may not reflect real-world experience with biologics, including biosimilars. Rare ADA-related adverse events may not be detected in the premarketing phase due to the limited size of the population exposed and the greater scrutiny of patient care in the clinical trial setting. Therefore, it is recommended to monitor immunogenicity in pharmacovigilance and risk management plans that also monitor other adverse drug reactions.
The limitations of efficacy testing in patients are well-recognized by regulatory agencies. To overcome these concerns, the FDA’s Division of Applied Regulatory Science (DARS) [75] has recently published its recommendations to remove this testing for biosimilars [76] based on comparing pharmacodynamic (PD) properties between a biosimilar candidate and its reference product. It is now labeled as clinical efficacy testing in healthy subjects. A PD biomarker is not required to be a surrogate endpoint or have an established relationship with clinical efficacy outcomes [77,78]. Examples include the absolute neutrophil count area under the effect time curve as a more reliable endpoint than the clinical efficacy endpoint of the duration of severe neutropenia [79]. DARS made these conclusions based on its investigations [80] and clinical studies it has conducted [81,82,83] to define the best practices for characterizing the PD biomarkers for various drug classes. These studies evaluated the use of human plasma proteomic and transcriptomic analysis to find novel biomarkers for the approval of biosimilars [84]. More efforts are underway to remove patient testing of all biological drugs, including monoclonal antibodies that do not show pharmacodynamic markers [85].
The gold standard for evaluating the clinical efficacy of novel medications compared to placebo has come under fire recently. Dr. Janet Woodcock, a past acting commissioner of the FDA, has stated: ‘Why should we put patients through all these different trials just to check a box.’ The FDA has recently questioned this idea of real-time testing, claiming that clinical efficacy testing is “broken” [86]. Following the 21st Century Cures Act, new digital technologies and real-world evidence (RWE) are necessary [87]. Recently, the FDA has announced policies and funding to encourage the development of novel clinical trials and substitute trials with non-clinical methodologies [88].
The clinical effectiveness trials have not revealed any clinically significant differences between a biosimilar and its reference product, according to a review of the published literature. Therefore, they have not led to any product withdrawals or recalls from the market. These data are available in the 96 EPAR files from EMA [89] and 37 approval documents from the FDA [90]. These regulatory submissions all passed their clinical efficacy assessment. In addition, the research published on the clinicaltrials.gov website [91] substantiates that all 141 studies for which the findings are provided complied with the required standards. The PubMed database also provides 435 randomized control clinical trials conducted between 2002 and 2022 that failed to detect a clinically significant difference [92].
The main reason to remove clinical efficacy testing is cost avoidance and ethical and hazardous concerns. The ethical concerns arise from the universal belief that no unnecessary exposure to healthy subjects should be made as codified in the US 21 CFR 320.25(a)(13), the universal belief that “No unnecessary human testing should be performed.” [93]. The hazardous concerns arise from the possibility of justifying critical analytical and pharmacology profiles based on efficacy studies.
The limitations of efficacy testing in patients are well-recognized by regulatory agencies. To overcome these concerns, the FDA’s Division of Applied Regulatory Science (DARS) [94] has recently published its recommendations to remove this testing for biosimilars [95] based on comparing pharmacodynamic (PD) properties between a biosimilar candidate and its reference product. It is now labeled as clinical efficacy testing in healthy subjects. A PD biomarker is not required to be a surrogate endpoint or have an established relationship with clinical efficacy outcomes [96,97]. Examples include the absolute neutrophil count area under the effect time curve as a more reliable endpoint than the clinical efficacy endpoint of the duration of severe neutropenia [98]. DARS made these conclusions based on its investigations [99] and clinical studies it has conducted [100,101,102] to define the best practices for characterizing the PD biomarkers for various drug classes. These studies evaluated the use of human plasma proteomic and transcriptomic analysis to find novel biomarkers for the approval of biosimilars [103]. A joint FDA/Duke Margolis Workshop [104] has covered the study findings that encourage a broader debate on using PD biomarkers to develop biosimilars.
The FDA has also validated that PD biomarker identification can be made using large-scale proteomic approaches and other technologies [105] where PD biomarkers are not readily available. The FDA has also confirmed that the PD biomarkers need not correlate with a clinical response to allow their use to support the claim of biosimilarity. A biosimilar development plan aims to demonstrate similarity to the reference product, not the focus of the reference product, where the safety and effectiveness are established independently. Therefore, the correlation between the PD biomarker and clinical outcomes, while beneficial, is not required [106,107].
Additionally, evaluating PK and PD similarity to detect differences between a proposed biosimilar and its reference product may be more sensitive than evaluating clinical efficacy endpoint (s), should differences exist. For example, quantitative analysis showed that the PD biomarker, the area under the effect-time curve of an absolute neutrophil count, is a more sensitive endpoint than the clinical efficacy endpoint of the duration of severe neutropenia [108].
The standards for surrogate biomarkers used to support the approval of novel drugs are fundamentally different from the standards for PD biomarkers meant to assist a demonstration of biosimilarity [109]. This provides opportunities for biomarkers used as secondary and exploratory endpoints in new drug development programs to support biosimilar testing. In addition, many opportunities are available to identify new PD biomarkers or fill information gaps on existing biomarkers to facilitate using PD biomarker data in clinical pharmacology studies instead of comparative clinical efficacy studies.
Possible examples of drugs that exhibit pharmacodynamic markers and thus are exempt from patient testing are presented in Table 3.
For products that do not display PD biomarkers, such as monoclonal antibodies, other “omic” technologies like transcriptomics and metabolomics may offer a chance to find new, sensitive, and robust candidate biomarkers for further exploration as PD biomarkers [110]. However, a more rational approach will be to take a step back in the testing cycle of biosimilars and examine if ex vivo testing can provide evidence of biosimilarity that is more sensitive and reliable in identifying any “clinically meaningful difference” in the language of the FDA guidelines.
Since the pharmacodynamic response is triggered by receptor binding, cell-based bioassays, or potency assays, such as ELISA, binding assays, competitive assays, cell signaling, ligand binding, proliferation, and proliferation suppression, should provide a good functional comparison of a biosimilar candidate with its reference product. Furthermore, functional tests for the mode of action (MOA), such as testing for apoptosis, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and antibody-dependent cellular cytotoxicity, are generally not required, can be added to provide a higher degree of confidence in safety and efficacy.
Monoclonal antibodies (mAbs) bind to specific protein epitope targets on target cells resulting in a therapeutic response. Characterizing the mAb’s affinity for binding include target antigen and affinity for binding to specific Fc receptors (Fc(RI, Ia, IIa, IIb, IIIa, IIIb; Fc(RN, Effector functions like ADCC and CDC, molecular properties like charge, pI, hydrophobicity, and glycosylation, and off-target binding employing in-silico or in vitro techniques like baculovirus ELISA tools are all robust and objective to establish functional similarity [111,112]. Additional tests can be added based on specific applications such as for TNFα blockers: C1q; CDC; Induction of regulatory macrophage; inhibition of T-Cell proliferation (MLR); LTα; MLR; mTNFα; Off-target cytokines; Reverse signaling; sTNFα; Suppression of cytokine secretion; tmTNF-α. The functional assays form more robust markers to establish efficacy comparisons than the testing in patients, without the necessity to demonstrate any PD response for mABs [113,114]. However, the functional tests (ADCC, ADCP, and CDC) are of little value when the drug targets a soluble antigen [115,116].
A collection of functional assays pertinent to a range of biological activities can be employed for a product having multiple biological activities. For instance, some proteins have a variety of functional domains that express enzymatic and receptor-binding functions. The metric for biological activity is potency. Analytical studies to evaluate these features are easily accessible when immunochemical properties are made part of the activity assigned to the product (for instance, antibodies or antibody-based products). The functional assays form more robust markers to establish efficacy comparisons than the testing in patients, without the necessity to demonstrate any PD response for mABs [117,118].
In May 2023, the FDA issued draft guidance, “Generally Accepted Scientific Knowledge in Applications for Drug and Biological Products: Nonclinical Information,” [119] suggesting that nonclinical testing can be reduced based on GASK, first, where a product contains a substance (either naturally derived or synthesized) that occurs naturally in the body and has known effect on biological processes; second, where a sponsor has demonstrated a drug’s impact on a particular biological pathway to conclude that certain nonclinical studies are not necessary to support approval and labeling of the drug. For example, some drugs have distinct effects on well-known biological pathways, so specific outcomes can be predicted once the drug’s effect is demonstrated on the biological pathway. In addition, in some cases, a drug has either on- or off-target impacts on a biological pathway or molecular mechanism of action that is known to result in adverse effects at clinically relevant exposures based on the operation of the biological pathway. Thus, according to the FDA, it may be appropriate to rely on GASK regarding the impact of the pathway rather than to conduct specific pharmacology and/or toxicology studies intended to measure the impact of the path.
While these concessions may not be available for a new molecule where there is no reported scientific data of a real-world-experience of the class of the product, it will significantly reduce the development cost of copies of approved biological drugs and biosimilars since their mechanism of action receptor binding, can be readily compared with their reference product.
The FDA has also taken significant steps in bringing the real world (REW) data in assessing the safety and efficacy of drugs [120,121], and most recently, it has suggested biosimilar candidates that have known pharmacodynamic markers need not be tested for efficacy in patients, a major change that demonstrates ongoing efforts by the regulatory agencies to bring science ahead of shared beliefs [122]. One such example is the recent FDA Modernization Act [123] that amends the Biological Products Competition and Innovation Act (BPCIA) [124] to remove the term “animal toxicology” and replace it with “nonclinical” testing, to assert that unnecessary testing of biological drugs that act by receptor binding, and thus do not display animal toxicology, is not necessary.
Proposed Guideline
In this paper, I am concentrating on therapeutic proteins since they are most likely to come from non-SRA sources, and as biosimilars, other biotechnology drugs are not allowed copies, so this takes them into the category of SRA new products.
The regulatory process is divided into two for a product developed, approved, and marketed in a Stringent Regulatory Authority (SRA) country [125] and the other for a non-SRA country.
SRA Sourcing
To meet the urgent need to simplify the registration of biological drugs, it is proposed that if a product is approved in one of the SRA countries, either as a novel biological product, or its biosimilars, its approval should be automatic. However, the filer must submit the same dossier, without editing, that resulted in its approval; the product must have the same label (indications and description) as approved in the country of origin, and the batches supplied should come from the same batch distributed; the last condition reduces the burden of pharmacovigilance. Simplifying the registration process and giving access to possibly billions of the population will significantly increase the entrance of new drugs. Large companies who secure new biotechnology drug approval and even the biosimilar companies in the SRA countries find it cumbersome and not worth the expense of obtaining registration in smaller countries with limited markets. The proposed plan will instantly remove such hindrances; the cost of these drugs is not an issue that GMA should deal with, and this can be worked out in each state. However, the dossier submitted will remain limited to the GMA, reducing the concern of the proliferation of technical information to many countries, another major incentive for developers to bring new drugs to the GMA states.
A stringent regulatory authority (SRA) is a national drug regulatory authority that the World Health Organization (WHO) [126] considers to apply stringent standards for quality, safety, and efficacy in its regulatory review of drugs and vaccines for marketing authorization. According to the WHO, it includes members of the ICH and its observer countries. The concept of an SRA was developed by the WHO Secretariat and The Global Fund to Fight AIDS, Tuberculosis, and Malaria to guide decisions regarding procuring medicines for humanitarian assistance. The idea is that countries with non-SRA drug authorities can use an accelerated process to facilitate approval (registration or marketing authorization) of medicines, including vaccines and biologics, which SRAs have already approved. As of 2022, the national regulatory authorities of 36 countries are considered SRAs [127] (Table 4).
Non-SRA Country Biosimilars
The registration dossiers from non-SRA states require extensive scrutiny for compliance as well as data and business practice integrity, as widely reported in the warning letters by the FDA, and to curtail the perception of graft in the registration practice as documented in practice [128], almost globally [129]. Therefore, the proposed plan is presented in Figure 2 to establish a foolproof, practical system.
Qualified product: a product that has a reference product available.
GBA review: when a dossier is submitted, it is reviewed by regulatory experts to ensure that it is complete; at this stage, it is not a scientific review, only a compliance review.
Rapporteur: Using rapporteurs is a standard practice in the EU; the FDA also accepts third-party audits [130]. Rapporteurs are members of the Committee for Medicinal Products for Human Use (CHMP) or the Committee for Medicinal Products for Veterinary Use (CVMP), assigned to assess applications for marketing authorization. They play a critical role in evaluating and monitoring medicines in the EU. The competent national authorities of the EU Member States appoint the rapporteurs. Each medicine under evaluation has a rapporteur and a co-rapporteur responsible for preparing scientific assessments of the medicine, leading to a recommendation by the CHMP or CVMP on whether the medicine should be authorized. The EMA generally identifies the rapporteurs and co-rapporteurs for specific medicines in its assessment reports and other public documents. However, the identities of the rapporteurs and co-rapporteurs might be confidential in certain situations. For example, a list of 61 rapporteurs for biosimilars is available at EMA [131]. In addition, GMA should create its list of rapporteurs from anywhere in the world who are qualified, affordable, and prompt in responding to such requests. The cost of the rapporteur is paid by GMA and charged to clients to avoid any direct contact.
Third-Party cGMP Audit: data and sample integrity: Since the clinical pharmacology testing for biosimilars is conducted in an at-scale cGMP lot, it is imperative that the developer should qualify its cGMP production. The audit is specific to the product and not waived based on previous audits. The audit is conducted by third-party auditors, not any staff of GMA. The auditors also confirm and assure that the samples going out for clinical pharmacology testing are valid and their integrity confirmed.
Validated Samples: The samples used for analytical assessment and clinical pharmacology must be validated for their source, history, and compliance. Generally, an audit will collect these samples and provide them to the third-party testing facility.
Third-Party Analytical Assessment: The final analytical assessment must be conducted by a third party approved by GMA as a qualified testing facility; the facility will also keep the tested samples and report the results based on a pre-approved protocol.
Certified CRO Samples Retained Clinical Pharmacology: CROs should retain the samples if there is an issue regarding an outlier or later inquiry; the time limit is through the product’s shelf-life.
Scope
The main goal of an interactive guideline is to reduce the testing as much as possible, remove redundant testing, and add requirements that would overcome the cGMP issues that are more pertinent to the Arab World.
Definition
A biosimilar product has the same safety and efficacy, mode of action, dose, frequency, route, and concentration (strength) as the reference product. Polypeptides, conjugates, and the products and derivatives that contain them. These proteins and polypeptides can be highly purified and characterized using appropriate analytical methods. Alpha-amino acid polymers composed of 40 or fewer amino acids are considered peptides, not proteins. Glucagon, liraglutide, nesiritide, teriparatide, and teduglutide are peptides. A peptide is regulated as a chemical drug and copied as a generic drug.
Reference Product
A biological product was first approved in one of the SRA countries using a complete dossier and continues to be marketed in the country of origin. Only one source of reference can be used. The lowest strength product should be selected when several strengths or presentations are available for the reference product. To account for the production variability of the reference product, several batches of reference products should be purchased over time (months to years) straight from the relevant market. The reference product batches should be tested during the allotted shelf life and stored according to the label’s suggested storage conditions. Testing batches that have been held for a long time (for example, frozen at -80°C) or beyond their designated shelf life may occasionally be possible if reliable data show that the storage conditions do not affect the relevant quality attributes. The age of the reference product batches (relative to expiry dates) at the testing time should be documented during the analysis.
Characterization
The reference product is characterized by appropriate techniques described in ICH Q6B. These characterizations include determining physicochemical properties, biological activity, immunochemical properties (if any), purity, impurities, contaminants, and quantity. Developers are encouraged to adopt newer technologies as available. Since the quality attributes of the reference product vary from batch to batch, it is essential to establish the ranges of these variations, to allow similar variability in the biosimilar candidate. The variations are either process-related or product-related (the expression system) (the manufacturing system). Generally, a variation in the product-related attributes cannot be resolved, requiring the developer to create a different expression system; the same can be the case for process-related attributes, but these are readily fixed. Both cases cannot submit safety studies to justify a significant difference [132].
Impurities
Impurity profiling is a prerequisite during biosimilar development, and specifications are set vis-à-vis the innovator for product-related variants. For example, a biosimilar may have fewer impurities in type and amount, but there shall be no unmatched impurity; this cannot be justified through any safety study unless this is already reported to be safe in the reference product.
Functional Assays
Analytical and in vitro functional levels should be used to identify critical quality attributes (CQA). Functional assays, such as those that look at apoptosis, complement-dependent cytotoxicity, antibody-dependent cellular phagocytosis, and antibody-dependent cellular cytotoxicity, should be relevant to the potential MOA in all therapeutic indications. A biological occurrence should be considered applicable to the MOA unless sufficient evidence to the contrary is presented. Functional tests (ADCC, ADCP, and CDC) are not required for a reference product mainly targeting a soluble antigen.
Test Methods
Critical product and process-related variants are compared with the reference product to enable suitably, not necessarily validated methods since some test methods cannot be fully validated. Analytical methods must be sensitive, qualified, and sufficiently discriminatory to detect possible differences. The methods used to assess quality attributes for the batch release can also be used for analytical assessment, as detailed in the ICH guidelines (ICH Q2A, Q2B, Q5C, Q6B), where appropriate. In addition, robust data require the application of suitable orthogonal methods.
The number of batches
Generally, eight batches will be tested; one should be the clinical batch. Therefore, the final third-party analytical assessment will include at least three PPQ lots.
Data Evaluation
A visual comparison is adequate for test findings supplied as printed output, such as spectra. The application of quantitative statistics requires data from about ten batches each, and the most effective inference is obtained from the 3Sigma range that is calculated for the reference sample as (μref-3σref, μref + 3σref). The 3Sigma test is accepted if the MinMax range of the test sample is within the 3Sigma range. The 3Sigma approach provides a more practical compromise of error rates, further improving with a larger sample size.
Expression System
The expression system determines the product-related critical quality attributes (CQAs), which include primary structure, higher-order structures (HOS), glycosylation (only in eukaryotic hosts), product-related variations, and process-related variants. The primary structure is further broken down into the secondary structure, tertiary structure, and conformational stability; HOS into the oligosaccharide pattern, glycopeptide mapping, and monosaccharide/sialic acid content; size variants, charge variants, and related proteins resulting from the post-translational modification, as well as product-associated variants (HCD). The expression system should be the same class as the one used to express the reference product. The developers are also advised to select more steady expression systems; generally, high-yielding cell lines produce more variants. Therefore, the cell lines should be qualified according to the ICH Q5D.
Post-translation Modifications
Since the primary sequence of a protein is fixed, it is expected to be precisely the same, except for justified post-translational modifications, such as terminal amino acids that are truncated in the body.
A few examples of heterogeneities produced during the creation, management, and storage of biological products a size-based heterogeneity (aggregates, fragments, and visible/subvisible particles), charge-based heterogeneities (acidic and basic variants), and other product modifications (reduced, oxidized, glycated, misfolded proteins, etc.).
When the environment changes during different stages of the production process, hydrophobic patches of the protein unfurl, causing accumulation or fragmentation. Immunogenic responses could occur. The aggregate size ranges from soluble aggregates to visible residues, depending on the duration of exposure to various stresses such as shear, thermal, chemical, freeze-thaw, etc. Protein loss due to interactions in the stationary phase and salt-induced aggregation or dissociation is common during SEC analysis. To quantitatively evaluate the size distribution, sedimentation velocity-analytical ultracentrifugation (SV-AUC), a matrix-free substitute for SEC, is used.
Charge variations are proteo-forms that arise in various colloidal matrices (such as culture media, in-process buffers, or formulation) at various phases of the manufacturing process and have changing charges. Therefore, several forms of cation exchange (CEX) chromatography are preferable.
Non-enzymatic post-translational modifications (PTMs) include oxidation, phosphorylation, sulfation, acetylation, methylation, and hydroxylation, which are formed during multiple stages of the manufacturing process. Therefore, liquid chromatography is preferred for characterizing PTMs and quantifying related molecular variants and impurities.
Cell substrates, such as HCPs, HCD, cell culture, and downstream processing residuals, are examples of process-related variations or residuals. The preferred HCP and HCD detection and quantitation methods are enzyme-linked immunosorbent assays (ELISA) and real-time or quantitative PCR. Since they are a component of the release specification, these variants are not examined during the drug substance qualification phase.
Release Specification
Reference product characterization allows for establishing release specifications set before the analytical assessment. Characterization of the reference product will include determining its physicochemical properties, biological activity, immunochemical properties, purity, and impurities using suitable testing methods. The test lots can come from the lots used throughout the development process. However, at least one lot tested must be the one used for the first clinical trial, the PK/PD study. In addition, all test methods must be validated or verified if drawn from a pharmacopeia. Injectable products are allowed certain variations based on inevitable variabilities, such as ±3% for protein content, not more than 3% impurity, no single impurity of more than 1%, or ±15% for potency testing. Pharmacopeial specifications for the qualification of the dosage form, such as sterility, fill volume, delivered volume, and physical properties, are also not tested for comparison purposes. Other legacy attributes are independently established, like sterility, invisible particles (a controversial issue with biosimilars to consider as aggregates), protein content, potency, and physical properties specific to the biosimilar candidate. These standards can be used in defining the release specification of the biosimilar candidate.
Formulation
It is acceptable for biosimilars to have a formulation that differs from the formulation of the reference product. Despite any variations in the constituent composition, a formulation with the same number of inactive ingredients or fewer is recommended unless prohibited by intellectual property. Excipients utilized in creating biological products should not be included in another formulation. The integrity, activity, and potency of the active ingredient should be demonstrated, as well as the formulation’s stability, compatibility (i.e., how it interacts with excipients, diluents, and packaging materials), and compatibility. Additional safety tests are needed to ensure there is no unexpected leaching of packaging components into the product if the principal packaging in contact with the product is different. Developers are advised to choose a primary packaging material that is similar instead because these studies would typically be challenging to defend. No unique excipients that have ever been used in a comparable product may be included in the formulation, and all excipients must be free of animal products.
Reference Standards
For biological assay and physicochemical testing of succeeding lots, in-house primary reference material is a suitably described sample created by the manufacturer from a representative lot or lots and calibrated against which in-house working reference material is used. It is the only reference material allowed for reference purposes and its working reference materials. Publicly available reference standards (e.g., Ph. Eur.) cannot be used as the reference product to demonstrate biosimilarity. However, using these standards can be used for method qualification and standardization. No specification in any monograph for a drug substance or drug product can be used to establish a reference product specification or biosimilar candidate. Test methods can be used after verification.
Stability
According to ICH Q5C, the biosimilar candidate’s stability must be assessed. Analytical evaluation is extended through stress stability testing to show that the degradation products are comparable to the reference product. The pharmacopeia’s general monographs include sterility, endotoxins, microbiological limits, volume in the container, uniformity of dosage units, and allowable particle matter. The pharmacopeial standards can be used for these tests because they are release specification tests. In addition, accelerated and stress stability investigations are required to create deterioration profiles and enable a further direct assessment of structural similarity. To decide the requirements for stability studies that give pertinent data to be compared, ICH Q5C and Q1A(R) should be consulted.
Process Qualification
Upstream and downstream processes must be validated before conducting any analytical assessment for similarity. Bridging studies are required to validate if the production size changes; however, once the clinical pharmacology studies are completed, no batch size change is allowed; the developer may do this under ICHQ5E, which applies only post-approval.
Animal Toxicology
No animal toxicology study is required for biosimilars.
Clinical Pharmacology
Pharmacokinetic and pharmacodynamic studies are an extension of analytical assessment reflecting how the body sees the molecule and vice versa. Even though a product is administered intravenously, PK studies are required to assess the extent and strength of receptor binding that might change pharmacokinetic parameters like the distribution volume and clearance. This applies to all biosimilars even if they are not administered by the parenteral route, like the biological drugs injected into the eye. It is noteworthy that the purpose of PK/PD studies is to compare the profile, not characterize the profile of the reference product and the biosimilar candidate; the testing can be conducted in a local population to reduce the inter- and intra-subject variability, thus reducing the study size. All studies must conform to the standards associated with bioequivalence testing. Ideally, the PK experiment should be planned and powered to demonstrate equivalence to the reference product in healthy volunteers. Crossover or parallel designs should be supported by a robust design. Although a crossover strategy is superior at identifying changes, it may not be suitable for reference products with solid immune responses or lengthy half-lives. If suitable population PK or PK-PD models for the reference product are available in the literature, modeling and simulation should be considered to optimize the study design, such as choosing the most sensitive dose(s), study population, and sample size PK differences. Consideration should be given to linear (nonspecific) clearance and nonlinear (target-mediated) clearance, for instance, through dosage selection and evaluation of partial areas under the curve (AUCs). Body weight adjustments or other factors (such as subject sex) to be employed in the statistical analysis of a parallel group experiment should be predefined in the statistical analysis strategy. The equivalence margins must be pre-specified, with an interval of 80.00–125.00% generally acceptable. The PK trial should demonstrate equivalence of the primary PK parameters, usually AUC0-∞ and Cmax. If the extrapolated portion of AUC0-∞ makes up >20% of the total AUC0-∞ in >20% of observations, this requires a discussion of the study’s validity. A root cause analysis should be carried out, and the results should be appropriately taken into account in the planning and execution of a new PK study if a PK study is unsuccessful (i.e., the 90% confidence intervals for the main PK parameters do not entirely fall within the pre-specified acceptance limits).In most cases, the cause of failure is the subject variability that can be reduced by choosing narrow criteria for qualification regarding gender and age. The PK trial can be used to test PD parameters, and descriptive results should be provided to support a finding of biosimilarity.
Immunogenicity
Immunogenicity is an inherent property of proteins, and it is best tested in healthy subjects in clinical pharmacology profiling. However, it’s important to note that the immunogenicity of a specific protein can be assessed through preclinical and clinical studies during drug development. These studies evaluate the protein’s potential to elicit an immune response, including producing antibodies against the protein.
Clinical Efficacy
No clinical efficacy and safety testing is required.
Naming
Biosimilars should have a brand name and share the same International Nonproprietary Name (INN) as the reference product and any additional designations required in the local jurisdiction. Biosimilars should also have a different brand name.
Label
The label must state all risks associated with the reference product, have the same indications, and be formatted and detailed as described in this guidance without exception. Once a biosimilar candidate is proven highly similar to the reference product, all indications granted to the reference product are allowed, provided they are not protected by market exclusivity or patents. The developer may not request fewer or additional indications.
Substitution
Biosimilars can be substituted or interchanged with the reference product or other biosimilars approved using the same reference product [133]. The EMA has recently confirmed it.
Pediatrics
No pediatric compliance studies are required for biosimilars [134].
Human Factor Studies
These studies are required to ensure that the correct dose is administered when a patient administers a product. However, if the device used is highly similar to the device used by the reference product, these studies are waived. In addition, no such studies are required when the product is administered by a healthcare professional.
Risk Management Plan
The risk management plan (RMP) for a biosimilar product is the same as for the reference product. Furthermore, brand name and batch number must ensure precise biosimilar traceability. There is no need for post-market surveillance data submission.
Conclusions
Biotechnology drugs remain unaffordable to most around the world. A novel approach is presented involving the establishment of a Global Medicine Agency (GMA), a regulatory approval platform for biotechnology drugs that will allow instant distribution to all member countries, starting with the 22 states in the League of Arab States, expanding further the four observers and likely to all non-SRA countries. Such a large market with a single approval will motivate the entry of new products and encourage manufacturing by non-SRA candidates. The GMA’s function is well-defined as a supervisory agency that outsources all confirmatory work to ensure ethical compliance. To establish a role model, it is suggested that the League of Arab States develop this model because of their homogeneity and the dire need; it can then expand across the globe. An excellent example for the Arab League to follow is the African Vaccine Manufacturing Initiative (AVMI) which includes biological drugs and biosimilars [135]. However, to achieve this status, GMA must have high standards but practical requirements that comprise the modern understanding of the safety and efficacy of biosimilars, the only class of biotechnology products that allow making a copy of the reference product.
I anticipate many objections to the proposal made here; first, giving the rights to others like rapporteurs or third-party auditors is not a sign of weakness of a regulatory agency; it is a sign of greater transparency and assuring consistency, as well as removing any sign of discrimination and graft. Second, states agreeing to cooperate are not given up their rights to distribute or price these products. And finally, understand that this is not a replication of the EMA guidelines; the guidelines are different, and so is the jurisdiction. It is anticipated that if this plan is adopted and becomes functional, many countries will join, creating the largest global consortium to make biological drugs accessible; the focus and scope of this plan can be expanded to include future medicines like gene therapy, CRISPR-Cas9, mRNA therapeutics [136], CART therapy, and many more. Furthermore, this proposal is not to compare with the role of the WHO or ICH, which are not regulatory agencies; however, their guidelines, along with the guidelines of the FDA and EMA [137], were adopted in creating the current plan. The affordability issue is critical, as more than 80% of the world remains suffering; it will take a drastic and bold move to break this financial monotony; I am also encouraging developing countries to become self-sufficient by making these drugs locally. The manufacturing science has evolved significantly, making manufacturing establishment and cost of goods affordable to any small to mid-size company.
Conflicts of Interest
The author is an advisor to regulatory agencies and a consultant and developer of biotechnology drugs.
References
- https://www.statista.com/statistics/280572/medicine-spending-worldwide/.
- C.P. Gross and K.A. Sepkowitz, “The myth of the medical breakthrough: smallpox, vaccination, and Jenner reconsidered,” Int J Infect Dis, 3(1):54-60, 1998.
- A. von Schwerin et al., “Biologics: an introduction,” in Biologics, a History of Agents Made From Living Organisms in the Twentieth Century, A. von Schwerin, H. Stoff, B. Wahrig, ed., London: Pickering & Chatto, 2013, pp. 1-33.
- A. von Schwerin et al., “Biologics: an introduction,” in Biologics, a History of Agents Made From Living Organisms in the Twentieth Century, A. von Schwerin, H. Stoff, B. Wahrig, ed., London: Pickering & Chatto, 2013, pp. 1-33.
- Kalkan AK, Palaz F, Sofija S, Elmousa N, Ledezma Y, Cachat E, Rios-Solis L. Improving recombinant protein production in CHO cells using the CRISPR-Cas system. Biotechnol Adv. 2023 May-Jun;64:108115. [CrossRef] [PubMed]
- L. Andrews et al., “A snapshot of biologic drug development: Challenges and opportunities,” Hum Exp Toxicol, 34(12):1279-1285, 2015.
- N. Casadevall et al., “Evolution of biological agents: how established drugs can become less safe,” BMJ. 357:j1707, 2017.
- Wouters OJ, et. Al. , Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA. 2020 Mar 3;323(9):844-853. Erratum in: JAMA. 2022 Sep 20;328(11):1110. Erratum in: JAMA. 2022 Sep 20;328(11):1111. [CrossRef]
- https://www.fda.gov/drugs/development-approval-process-drugs/frequently-asked-questions-patents-and-exclusivity#howlongexclusivity.
- Schlander, M. , Hernandez-Villafuerte, K., Cheng, CY. et al. How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. PharmacoEconomics 39, 1243–1269 (2021). [CrossRef]
- www.pharmaceutical-technology.com/features/most-expensive-drugs-us/.
- Kronfol, N. Eastern Mediterranean Health Journal. Vol. 18 No. 11, 2012. https://apps.who.int/iris/bitstream/handle/10665/118493/EMHJ_2012_18_11_1151_1156.pdf;jsessionid=A6C226BFF2849E550596A7F50848641E?sequence=1.
- McCall SJ, Semaan A, Altijani N, Opondo C, Abdel-Fattah M, Kabakian-Khasholian T. Trends, wealth inequalities and the role of the private sector in caesarean section in the Middle East and North Africa: A repeat cross-sectional analysis of population-based surveys. PLoS One. 2021 Nov 16;16(11):e0259791. [CrossRef]
- Goffman, LF. Medicine and Health in the Modern Middle East and North Africa. 2019. https://www.jadaliyya.com/Details/40332.
- Mate K, Bryan C, Deen N, McCall J. Review of Health Systems of the Middle East and North Africa Region. International Encyclopedia of Public Health. 2017:347–56. [CrossRef]
- WHO. The global health observatory. https://www.who.int/data/gho/publications/world-health-statistics.
- https://data.worldbank.org/indicator/SH.XPD.CHEX.GD.ZS?locations=1A.
- https://www.cms.gov/research-statistics-data-and-systems/statistics-trends-and-reports/nationalhealthexpenddata/nhe-fact-sheet#:~:text=NHE%20grew%202.7%25%20to%20%244.3,Gross%20Domestic%20Product%20(GDP).
- https://www.reutersevents.com/pharma/commercial/middle-east-pharma-market-making#:~:text=The%20total%20regional%20market%20is,imported%20generics%20and%20branded%20drugs.
- https://arabcenterdc.org/resource/advancement-and-inequity-in-the-arab-worlds-medical-and-pharmaceutical-sectors/.
- https://arabcenterdc.org/resource/health-care-in-the-arab-world-outcomes-of-a-broken-social-contract/.
- Arab Language International Council. https://alarabiah.org/.
- https://www.emergobyul.com/news.
- https://www.pharmasolutions-int.com/rise-growth-of-pharma-distributors-in-the-middle-east/.
- https://www.drugdiscoverytrends.com/50-of-2022s-best-selling-pharmaceuticals/.
- https://www.who.int/news-room/articles-detail/call-for-consultant-on-monoclonal-antibodies-for-infectious-diseases#:~:text=The%20cost%20of%20goods%20to,US%2095%2D200%20per%20gram.
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q13-continuous-manufacturing-drug-substances-and-drug-products.
- https://www.coherentmarketinsights.com/market-insight/mena-biologics-and-biosimilars-market-4393#:~:text=The%20Arab World%20biologics%20%26%20biosimilars%20market,period%20(2020%2D2027).
- https://www.coherentmarketinsights.com/market-insight/saudi-arabia-pharmaceuticals-market-3557.
- https://www.vision2030.gov.sa/.
- https://tia.gov.tn/storage/app/media/ARGUMENTAIRES/TIA_TUNISIA_PHARMA/AG%20PHARMA%20ANG.pdf.
- https://www.jordantimes.com/news/local/jordans-pharmaceutical-exports-reach-jd1b-%E2%80%94-japm.
- https://www.marketdataforecast.com/market-reports/mea-biosimilars-market.
- Bassil N, Sasmaz S, M El S, Akalankam A. Realizing biosimilar potential in the Middle East & Africa. the Middle East and Africa perspective white paper [Internet]. IQVIA; 2020 [cited August 24, 2022]: 28. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/mea/white-paper/biosimilar_iqvia-whitepaper_final.pdf. Accessed September 20, 2022.
- Market Data Forecast. MEA biosimilars market size, trends, growth | 2022 to 2027 https://www.marketdataforecast. com/market-reports/mea-biosimilars-market. Accessed September 20, 2022.
- https://www.refworld.org/docid/3ae6b3ab18.html.
- Market Data Forecast. MEA biosimilars market size, trends, growth | 2022 to 2027 https://www.marketdataforecast. com/market-reports/mea-biosimilars-market. Accessed September 20, 2022.
- https://www.sfda.gov.sa/en/news/88233.
- http://gabi-journal.net/2nd-mena-stakeholder-meeting-on-biosimilars-2018-report.html.
- https://www.fda.gov/science-research/about-science-research-fda/fda-science-forum#:~:text=The%20Forum%20offers%20an%20exciting,efforts%20of%20FDA’s%2011%2C000%20scientists.
- https://www.lee.senate.gov/2022/11/sen-lee-introduces-biosimilar-red-tape-elimination-act. 2022.
- https://www.ema.europa.eu/en/news/biosimilar-medicines-can-be-interchanged.
- Niazi, SK. Biosimilars: Harmonizing the Approval Guidelines. Biologics. 2022; 2(3):171-195. [CrossRef]
- de Mora, F. Biosimilars: a value proposition. BioDrugs. 2019;33(4):353–356. [CrossRef]
- Bassil N, Sasmaz S, M El S, Akalankam A. Realizing biosimilar potential in the Middle East & Africa. the Middle East and Africa perspective white paper [Internet]. IQVIA; 2020 [cited August 24, 2022]: 28. Available from: https://www.iqvia.com/-/media/iqvia/pdfs/mea/white-paper/biosimilar_iqvia-whitepaper_final.pdf. Accessed September 20, 2022.
- Niazi, SK. Molecular Biosimilarity-An AI-Driven Paradigm Shift. Int J Mol Sci. 2022 Sep 14;23(18):10690. [CrossRef]
- Beygmoradi A, Homaei A, Hemmati R, Fernandes P. Recombinant protein expression: Challenges in production and folding related matters. Int J Biol Macromol. 2023 Apr 1;233:123407. [CrossRef] [PubMed]
- EMA Centrally Approved Biosimilars. https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar; (Accessed on 8 June 2022).
- Inxight Drug Database. https://drugs.ncats.io/substances?facet=Development%20Status%2FUS%20Approved%20Rx&facet=Substance%20Class%2Fprotein&facet=Substance%20Form%2FPrincipal%20Form&page=1 (accessed on 25 April 2023).
- WHO Guidelines on Evaluation of Biosimilars. Replacement of Annex 2 of WHO Technical Report Series, No. 977 https://www.who.int/publications/m/item/guidelines-on-evaluation-of-biosimilars WHO. https://www.who.int/publications/i/item/9789240021853; (Accessed on 8 June 2022).
- Niazi, S. The WHO Biosimilar guidance is based on weak science. https://www.centerforbiosimilars.com/view/who-biosimilar-guidance-is-based-on-weak-science.
- Niazi, S. Opinion: One step forward, half step back. https://www.centerforbiosimilars.com/view/opiniononestepforwardhalfastepbackforwhobiosimilarguidance; (Accessed on 8 June 2022).
- CDSCO, India. https://cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadAlertsFiles/BiosimilarGuideline2016.pdf; (Accessed on 8 June 2022).
- https://www.fda.gov/files/drugs/published/Labeling-for-Biosimilar-Products-Guidance-for-Industry.pdf.
- https://www.forbes.com/sites/nicolefisher/2018/07/25/one-mans-mission-to-fix-the-fdas-biosimilar-problem/?sh=2386ed9d2380.
- Niazi, SK. End animal testing for biosimilar approval. Science. 2022 Jul 8;377(6602):162-163. [CrossRef] [PubMed]
- Li J, Florian J, Campbell E, et al. Advancing Biosimilar Development Using Pharmacodynamic Biomarkers in Clinical Pharmacology Studies. Clin Pharmacol Ther. 2020;107(1):40-42. [CrossRef]
- Niazi, S. Scientific Rationale for Waiving Clinical Efficacy Testing of Biosimilars. Drug Des Devel Ther. 2022 Aug 24;16:2803-2815. [CrossRef]
- MHRA. Biosimilar Guidance. https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products; (Accessed on 8 June 2022).
- Global Biosimilar Guidelines. https://www.gabionline.net/reports/Guidelines-for-biosimilars-around-the-world.
- Niazi, SK. Biosimilars: Harmonizing the Approval Guidelines. Biologics. 2022; 2(3):171-195. [CrossRef]
- Garcia-A, A, et al., J Pharm Pharm Sci (www.cspsCanada.org). 2021; 24, 113–126.
- Niazi, S.K. The Coming of Age of Biosimilars: A Personal Perspective. Biologics 2022, 2, 107–127, https://www.mdpi.com/2673-8449/2/2/9. [Accessed 8 June 2022]. [Google Scholar] [CrossRef]
- Niazi, SK. (2022) Biosimilars: A futuristic fast-to-market advice to developers, Expert Opinion on Biological Therapy, 22:2, 149-155. [CrossRef]
- MHRA. Biosimilar Guidance. https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products; (Accessed on 8 June 2022).
- Niazi, SK. Biosimilars: Harmonizing the Approval Guidelines. Biologics. 2022; 2(3):171-195. [CrossRef]
- https://www.fda.gov/drugs/development-resources/advancing-real-world-evidence-program.
- Niazi, S.K. The Coming of Age of Biosimilars: A Personal Perspective. Biologics 2022, 2, 107–127 https://wwwmdpicom/2673. [Google Scholar] [CrossRef]
- Niazi, SK. (2022) Biosimilars: A futuristic fast-to-market advice to developers, Expert Opinion on Biological Therapy, 22:2, 149-155. [CrossRef]
- Chen Y, Monnard A, Jorge Santos Da S, An inflection point for biosimilars, McKinsey & Co. [cited 2021 Oct 12]. Available from: https://www.mckinsey.com/industries/life-sciences/our-insights/an- inflection-point-for-biosimilars; (Accessed on 8 June 2022).
- Niazi, SK. End animal testing for biosimilar approval. Science. 2022 Jul 8;377(6602):162-163. [CrossRef] [PubMed]
- Niazi, S. Scientific Rationale for Waiving Clinical Efficacy Testing of Biosimilars. Drug Des Devel Ther. 2022 Aug 24;16:2803-2815. [CrossRef]
- https://www.congress.gov/bill/117th-congress/senate-bill/5002.
- MHRA. Biosimilar Guidance. https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products; (Accessed on 8 June 2022).
- https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/division-applied-regulatory-science.
- Chiu, K. , et al., (2023). New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Frontiers in Medicine, 9. [CrossRef]
- US Food and Drug Administration. FDA Guidance: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product https://www.fda.gov/media/88622/ download (2016).
- Chiu K, et al., New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front Med (Lausanne). 2023 Jan 19;9:1109541. [CrossRef]
- Li, L. et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for GCSF in breast cancer patients. Clin Pharmacol Ther 104, 742–748 (2018).
- Li J, Florian J, Campbell E, Schrieber S, Bai J, Weaver J, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology studies. Clin Pharmacol Ther. (2020) 107:40–2. [CrossRef]
- Sheikhy M, Schrieber S, Sun Q, Gershuny V, Matta M, Bai J, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development- (I) a randomized trial with PCSK9 inhibitors. Clin Pharmacol Ther. (2022). [CrossRef]
- Gershuny V, Sun Q, Schrieber S, Matta M, Weaver J, Ji P, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development–(II) a randomized trial with IL-5 antagonists. Clin Pharmacol Ther. (2022). [CrossRef]
- Florian JG, Sun Q, Schrieber S, Matta M, Hazel A, Sheikhy M, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development–(III) a randomized trial with interferon beta-1a products. Clin Pharmacol Ther. (2022). [CrossRef]
- Hyland P, Chekka L, Samarth D, Rosenzweig B, Decker E, Mohamed E, et al. Evaluating the utility of proteomics for the identification of circulating pharmacodynamic biomarkers of IFNbeta-1a biologics. Clin Pharmacol Ther. (2022). [CrossRef]
- Niazi, S.K. Functional Biosimilarity to Replace Clinical Efficacy Testing of mAb Biosimilars: Advancing the FDA Perspective. Preprints.org 2023, 2023041141. [Google Scholar] [CrossRef]
- Woodcock. https://www.biopharmadive.com/news/fdas-woodcock-the-clinical-trial-system-is-broken/542698/. 5426.
- US Congress. https://www.govinfo.gov/content/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdf.
- https://www.fda.gov/drugs/biosimilars/biosimilars-science-and-research.
- EMA Biosimilar EPAR Program. https://www.ema.europa.eu/en/search/search/field_ema_web_categories%253Aname_field/Human/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar?search_api_views_fulltext=epar%20biosimilar.
- FDA. Biosimilars Approval Documents. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.
- Clinical Trial Database. https://clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=With&Search=Apply.
- Biosimilar Clinical Trials @PubMed. https://pubmed.ncbi.nlm.nih.gov/?term=biosimilar+clinical+trial).
- US Congress. BPCIA. https://www.congress.gov/bill/111th-congress/house-bill/3590.
- https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/division-applied-regulatory-science.
- Chiu, K. , et al., (2023). New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Frontiers in Medicine, 9. [CrossRef]
- US Food and Drug Administration. FDA Guidance: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product https://www.fda.gov/media/88622/ download (2016).
- Chiu K, et al., New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front Med (Lausanne). 2023 Jan 19;9:1109541. [CrossRef]
- Li, L. et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for GCSF in breast cancer patients. Clin Pharmacol Ther 104, 742–748 (2018).
- Li J, Florian J, Campbell E, Schrieber S, Bai J, Weaver J, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology studies. Clin Pharmacol Ther. (2020) 107:40–2. [CrossRef]
- Sheikhy M, Schrieber S, Sun Q, Gershuny V, Matta M, Bai J, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development- (I) a randomized trial with PCSK9 inhibitors. Clin Pharmacol Ther. (2022). [CrossRef]
- Gershuny V, Sun Q, Schrieber S, Matta M, Weaver J, Ji P, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development–(II) a randomized trial with IL-5 antagonists. Clin Pharmacol Ther. (2022). [CrossRef]
- Florian JG, Sun Q, Schrieber S, Matta M, Hazel A, Sheikhy M, et al. Considerations for use of pharmacodynamic biomarkers to support biosimilar development–(III) a randomized trial with interferon beta-1a products. Clin Pharmacol Ther. (2022). [CrossRef]
- Hyland P, Chekka L, Samarth D, Rosenzweig B, Decker E, Mohamed E, et al. Evaluating the utility of proteomics for the identification of circulating pharmacodynamic biomarkers of IFNbeta-1a biologics. Clin Pharmacol Ther. (2022). [CrossRef]
- Florian JS, et al., Pharmacodynamic biomarkers for biosimilar development and approval: a workshop summary. Clin Pharmacol Ther. (2022). [CrossRef]
- Wang YM, Strauss DG. Advancing Innovations in Biosimilars. Clin Pharmacol Ther. 2023 Jan;113(1):11-15. [CrossRef] [PubMed]
- US Food and Drug Administration. FDA Guidance: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (2015). https://www.fda.gov/media/82647/download. (accessed on 25 April 2023).
- US Food and Drug Administration. FDA Guidance: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product (2016). https://www.fda.gov/media/88622/download. (accessed on 25 April 2023).
- Li, L. et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G-CSF in breast cancer patients. Clin Pharmacol Ther 104, 742–748 (2018).
- US Food and Drug Administration. FDA draft guidance: Biomarker Qualification: Evidentiary Framework (2018). https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM628118.pdf. (accessed on 25 April 2023).
- Strauss DG, Wang YM, Florian J, Zineh I. Pharmacodynamic Biomarkers Evidentiary Considerations for Biosimilar Development and Approval. Clin Pharmacol Ther. 2023 Jan;113(1):55-61. [CrossRef]
- Hotzel, I. , et al., A strategy for risk mitigation of antibodies with fast clearance mAbs, 4 (2012), pp. 753-760.
- Sharma, TW. , et al., In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci, 111 (2014), pp. 18601-18606.
- Cymera F, Becka H, Rohde A, Reusch D. Therapeutic monoclonal antibody N-glycosylation—structure, function and therapeutic potential. Biologicals. 2018;52:1–11.
- Prior S, et al., International standards for monoclonal antibodies to support pre- and post-marketing product consistency: evaluation of a candidate international standard for the bioactivities of rituximab. MAbs. 2018;10(1):129–42.
- Ryding J, Stahl M, Ullmann M. Demonstrating biosimilar and originator antidrug antibody binding comparability in antidrug antibody assays: a practical approach. Bioanalysis. 2017 Sep;9(18):1395-1406. [CrossRef] [PubMed]
- Wang X, An Z, Luo W, Xia N, Zhao Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell. 2018 Jan;9(1):74-85. [CrossRef]
- Cymera F, Becka H, Rohde A, Reusch D. Therapeutic monoclonal antibody N-glycosylation—structure, function and therapeutic potential. Biologicals. 2018;52:1–11.
- Prior S, et al., International standards for monoclonal antibodies to support pre- and post-marketing product consistency: evaluation of a candidate international standard for the bioactivities of rituximab. MAbs. 2018;10(1):129–42.
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/generally-accepted-scientific-knowledge-applications-drug-and-biological-products-nonclinical.
- https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence.
- Corrigan-Curay J, Sacks L, Woodcock J. Real-World Evidence and Real-World Data for Evaluating Drug Safety and Effectiveness. JAMA. 2018;320(9):867–868. [CrossRef]
- Chiu, K. , et al., (2023). New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Frontiers in Medicine, 9. [CrossRef]
- https://www.congress.gov/bill/117th-congress/senate-bill/5002.
- https://www.congress.gov/bill/110th-congress/senate-bill/1695.
- https://www.who.int/initiatives/who-listed-authority-reg-authorities/SRAs.
- World Health Organization & WHO Expert Committee on Specifications for Pharmaceutical Preparations (2018). Fifty-second report of the WHO Expert Committee on Specifications for Pharmaceutical Preparations (PDF). Geneva, Switzerland: World Health Organization. p. 355–6. ISBN 978-92-4-121019-5. OCLC 1039407367.
- No authors listed. “WHO | List of Stringent Regulatory Authorities (SRAs)”. World Health Organization. Retrieved 2022-08-30.
- https://www.outlookindia.com/business/biocon-biologics-biocon-biologics-bribery-cbi-arrests-joint-drug-controller-for-alleged-bribery-to-favour-biocon-biologics-medicine-news-203753.
- https://www.financierworldwide.com/bribery-and-corruption-in-the-pharmaceutical-sector#.Y1AWiOzMKlM.
- https://www.fda.gov/files/drugs/published/Data-Integrity-and-Compliance-With-Current-Good-Manufacturing-Practice-Guidance-for-Industry.pdf.
- https://www.ema.europa.eu/en/search/search/field_ema_web_categories%253Aname_field/Human/field_ema_web_topics%253Aname_field/Biosimilars?search_api_views_fulltext=rapporteur.
- Niazi, SK. Molecular Biosimilarity-An AI-Driven Paradigm Shift. Int J Mol Sci. 2022 Sep 14;23(18):10690. [CrossRef]
- Niazi, SK. No two classes of biosimilars: Urgent advice to the US Congress and the FDA. J Clin Pharm Ther. 2022 Sep;47(9):1352-1361. [CrossRef] [PubMed]
- Niazi, SK. BioRationality. https://www.centerforbiosimilars.com/view/biorationality-a-dr-sarfaraz-niazi-column-the-ema-declares-biosimilars-interchangeable.
- https://www.avmi-africa.org/projects/current-projects/#1513773628836-0f0598a8-ec17.
- Niazi, SK. Making COVID-19 mRNA vaccines accessible: challenges resolved. Expert Rev Vaccines. 2022 Sep;21(9):1163-1176. [CrossRef] [PubMed]
- Niazi SK, Al-Shaqha WM, Mirza Z. Proposal of International Council for Harmonization (ICH) Guideline for the Approval of Biosimilars. J Mark Access Health Policy. 2022 Nov 17;11(1):2147286. [CrossRef]
Figure 1.
Per capita income and healthcare expenditure correlation within the 22 Arab States (Algeria; Bahrain; Chad; Comoros; Djibouti; Egypt; Iraq; Jordan; Kuwait; Lebanon; Libya; Mauritania; Morocco; Oman; Palestine; Qatar; Saudi Arabia; Somalia; Sudan; Syria; Tunisia; United Arab Emirates; and Yemen) with a population of over 400 million [22].
Figure 1.
Per capita income and healthcare expenditure correlation within the 22 Arab States (Algeria; Bahrain; Chad; Comoros; Djibouti; Egypt; Iraq; Jordan; Kuwait; Lebanon; Libya; Mauritania; Morocco; Oman; Palestine; Qatar; Saudi Arabia; Somalia; Sudan; Syria; Tunisia; United Arab Emirates; and Yemen) with a population of over 400 million [22].
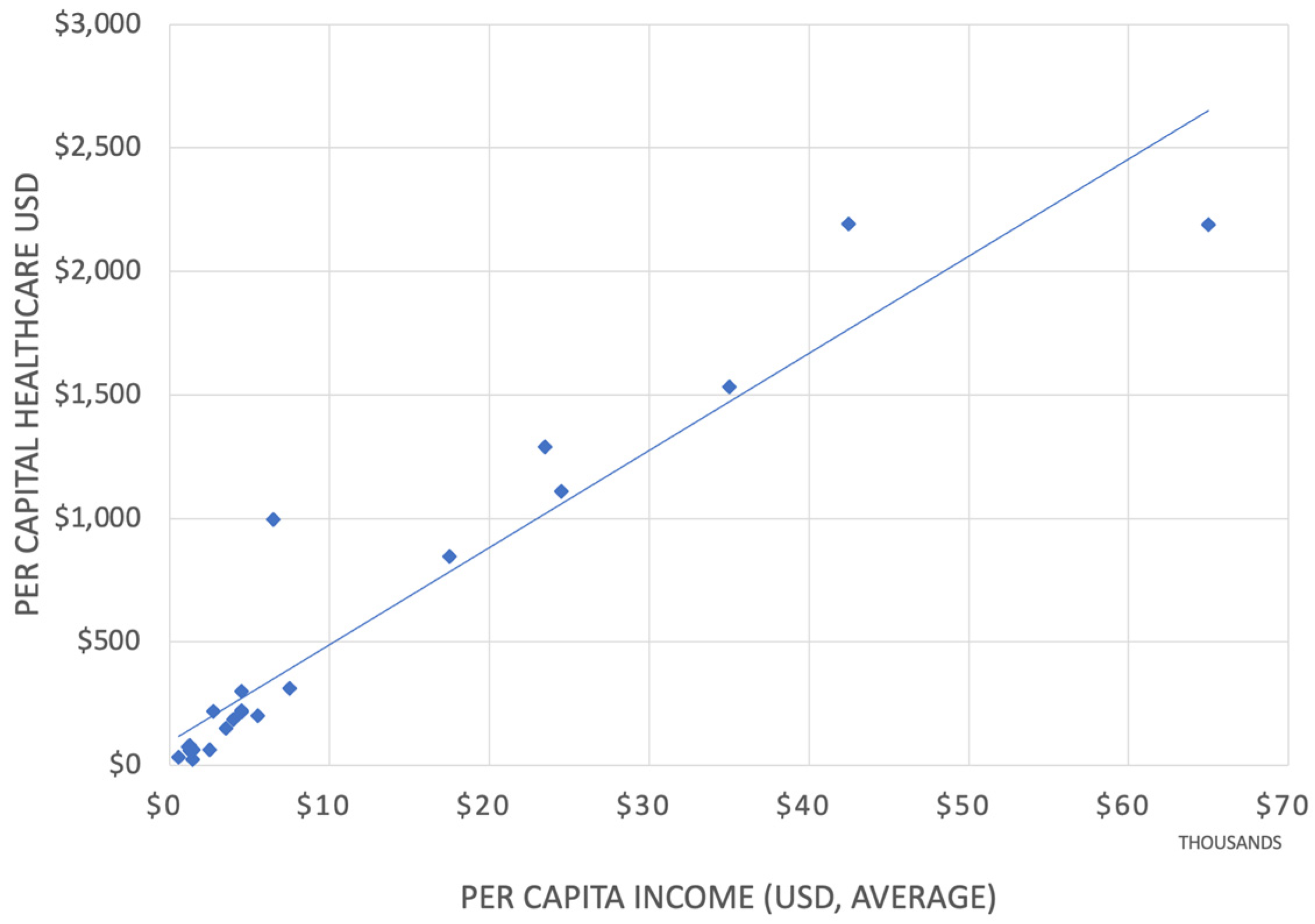
Figure 2.
The approval process of registration dossiers from non-SRA states.
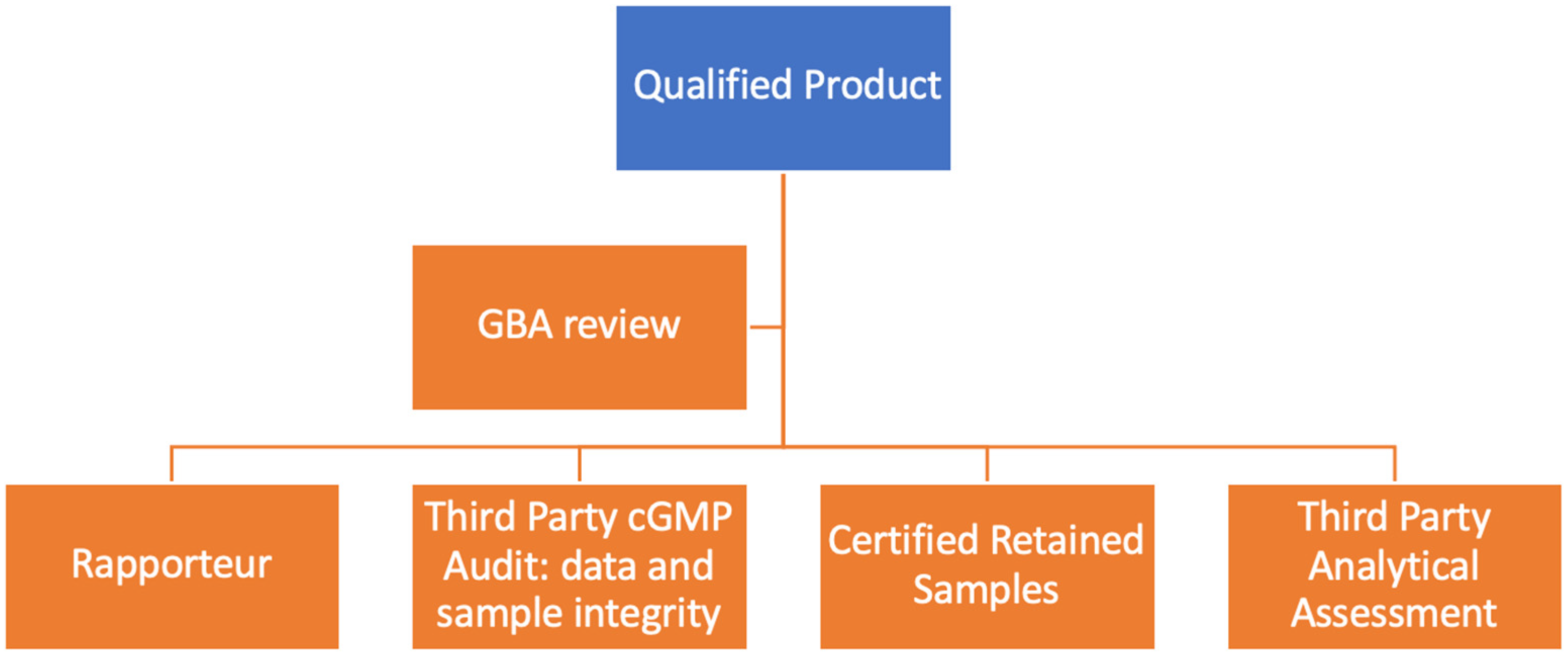

Table 1.
Most expensive treatment costs: therapeutic proteins are marked in bold [11]..
Table 1.
Most expensive treatment costs: therapeutic proteins are marked in bold [11]..
| Drug | Active | Indication | Cost, USD |
|---|---|---|---|
| Actemra | Tocilizumab | Rheumatoid arthritis and cytokine release syndrome | 6,000–9,000 per month. |
| Acthar Gel | Repository corticotropin | Multiple sclerosis, infantile spasms, and nephrotic syndrome | 40,000–60,000 per vial. |
| Actimmune | Interferon gamma-1b | Chronic granulomatous disease and severe, malignant osteopetrosis | 157,000/yr |
| Alecensa | Alectinib | Non-small cell lung cancer (NSCLC) | 159,000–178,000/yr |
| Almita | Olorinab | Breast cancer | 150,000/yr |
| Amondys 45 | Casimersen | Duchenne muscular dystrophy | 300,000/yr |
| Blenrep | Belantamab mafodotin | Multiple myeloma | 400,000/yr |
| Blincyto | Blinatumomab | Acute lymphoblastic leukemia (ALL) | 178,000/trt |
| Braftovi | Encorafenib | Melanoma | 174,000/yr |
| Brineura | Cerliponase alfa | Late infantile neuronal ceroid lipofuscinosis type 2 (CLN2) | 350,000/yr |
| Bylvay | Lumasiran | Bile acid synthesis disorders | 300,000/yr |
| Calquence | Acalabrutinib | Chronic lymphocytic leukemia (CLL) | 175,000/yr |
| Ceredase | Alglucerase | Gaucher disease | 200,000–300,000/yr |
| Cerezyme | Imiglucerase | Gaucher disease | 350,000/yr |
| Darzalex | Daratumumab | Multiple myeloma | 150,000–170,000/yr |
| Elaprase | Idursulfase | Hunter syndrome (MPS II) | 375,000/yr |
| Erwinaze | Asparaginase Erwinia chrysanthemi | Acute lymphoblastic leukemia (ALL) | 14,000–28,000 per vial. |
| Evrysdi | Risdiplam | Spinal muscular atrophy | 340,000/yr |
| Firdapse | Amifampridine | Lambert- Eaton myasthenic syndrome (LEMS) | $140,000/yr |
| Gattex | Teduglutide | Short bowel syndrome | 350,000–400,000/yr |
| Gilenya | Fingolimod | Multiple sclerosis | 90,000–100,000/yr |
| Glybera | Alipogene tiparvovec | Lipoprotein lipase deficiency | 1,000,000/trt |
| Harvoni | Ledipasvir + Sofosbuvir | Chronic hepatitis C | 94,500/trt |
| Hemlibra | Emicizumab | Prevention of bleeding episodes in hemophilia A with factor VIII inhibitors | 482,000/yr |
| Ilaris | Canakinumab | Cryopyrin- associated periodic syndromes (CAPS) and systemic juvenile idiopathic arthritis | 200,000–300,000/yr |
| Imfinzi | Durvalumab | Certain types of cancer, including lung cancer | 150,000–170,000/yr |
| Isturisa | Osilodrostat | Cushing’s disease | 295,000/yr |
| Jakafi | Ruxolitinib | Myelofibrosis and polycythemia vera | 12,000–14,000 per month. |
| Kalydeco | Ivacaftor | Cystic fibrosis | 330,000–360,000/yr |
| Keytruda | Pembrolizumab | Various types of cancer, including melanoma and lung cancer | 150,000–170,000/yr |
| Kymriah | Tisagenlecleucel | Certain types of non-Hodgkin lymphoma and acute lymphoblastic leukemia | $475,000/trt |
| Kyprolis | Carfilzomib | Multiple myeloma | 180,000–200,000/yr |
| Lumakras | Sotorasib | Non- small cell lung cancer (NSCLC) | $17,000 per month. |
| Luxturna | Voretigene neparvovec | Inherited retinal diseases causing blindness | 850,000/trt |
| Mavenclad | Cladribine | Multiple sclerosis | 99,000/yr |
| Monjuvi | Tafasitamab | Diffuse large B- cell lymphoma | $160,000/yr |
| Myalept | Metreleptin | Leptin deficiency in generalized lipodystrophy | 700,000/yr |
| Naglazyme | Galsulfase | Mucopolysaccharidosis VI (MPS VI) | 375,000/yr |
| Nerlynx | Neratinib | Breast cancer | 150,000/yr |
| Ocrevus | Ocrelizumab | Multiple sclerosis | 65,000/yr |
| Olysio | Simeprevir | Chronic hepatitis C | 66,000–84,000/trt |
| Opdivo | Nivolumab | Various types of cancer, including melanoma and lung cancer | 150,000–170,000/yr |
| Opdivo plus | Nivolumab + Ipilimumab | Certain types of cancer, including melanoma and lung cancer | 250,000–270,000/yr |
| Opsumit | Macitentan | Pulmonary arterial hypertension (PAH) | 200,000–220,000/yr |
| Orfadin | Nitisinone | Hereditary tyrosinemia type 1 | 275,000/yr |
| Orkambi | Lumacaftor + Ivacaftor | Cystic fibrosis | 260,000–300,000/yr |
| Orladeyo | Berotralstat | Hereditary angioedema | 470,000/yr |
| Orlissa | Elagolix | Endometriosis and uterine fibroids | 30,000/yr |
| Padcev | Enfortumab vedotin | Urothelial cancer | 16,000 per month. |
| Pomalyst | Pomalidomide | Multiple myeloma | 160,000–180,000/yr |
| Pulmozyme | Dornase alfa | Cystic fibrosis | 311,000/yr |
| Ravicti | Glycerol phenylbutyrate | Chronic management of urea cycle disorders | 350,000–400,000/yr |
| Remicade | Infliximab | Rheumatoid arthritis, Crohn’s disease, and other autoimmune conditions | 30,000–40,000/yr |
| Rozlytrek | Entrectinib | Solid tumors with NTRK gene fusion | 450,000/yr |
| Sandostatin LAR | Octreotide | Acromegaly and neuroendocrine tumors | 15,000–20,000 per month. |
| Signifor | Pasireotide | Cushing’s disease and acromegaly | 200,000–300,000/yr |
| Soliris | Eculizumab | Paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (ahus) | 500,000–700,000/yr |
| Sovaldi | Sofosbuvir | Chronic hepatitis C | 84,000/trt |
| Spinraza | Nusinersen | Spinal muscular atrophy | 375,000 for the first year and 375,000/yr after that. |
| Sprycel | Dasatinib | Chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) | 120,000–130,000/yr |
| Stelara | Ustekinumab | Psoriasis, psoriatic arthritis, and Crohn’s disease | 30,000–40,000/yr |
| Strensiq | Asfotase alfa | Hypophosphatasia | 300,000/yr |
| Synagis | Palivizumab | Prevention of respiratory syncytial virus (RSV) in infants | 9,000–15,000 per month during RSV season. |
| Takhzyro | Lanadelumab | Hereditary angioedema | 488,000/yr |
| Tecentriq | Atezolizumab | Certain types of cancer, including bladder cancer | 150,000–170,000/yr |
| Translarna | Ataluren | Duchenne muscular dystrophy | 262,000/yr |
| Trikafta | Elexacaftor + Tezacaftor + Ivacaftor | Cystic fibrosis | 311,000/yr |
| Ultomiris | Ravulizumab | Paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (ahus) | 498,000/yr |
| Vimizim | Elosulfase alfa | Morquio A syndrome | 375,000/yr |
| Vitrakvi | Larotrectinib | Solid tumors with NTRK gene fusion | 400,000/yr |
| Xalkori | Crizotinib | Small cell lung cancer (NSCLC) | $149,000–167,000/yr |
| Xolair | Omalizumab | Severe asthma and chronic idiopathic urticaria | 32,500/yr |
| Xpovio | Selinexor | Multiple myeloma | 160,000/yr |
| Xtandi | Enzalutamide | Prostate cancer | 129,000–144,000/yr |
| Xyrem | Sodium oxybate | Narcolepsy with cataplexy | 50,000–75,000/yr |
| Yervoy | Ipilimumab | Certain types of cancer, including melanoma | 150,000–170,000/yr |
| Yescarta | Axicabtagene ciloleucel | Certain types of non-Hodgkin lymphoma | $373,000/trt |
| Zolgensma | Onasemnogene abeparvovec | Spinal muscular atrophy | 2,100,000/trt |
| Hemgenix | Viral gene therapy | Hemophilia B gene | 4,3000,000/dose |
Table 2.
Fifty top-selling drugs in 2022 [25]. Total sales USD 186.40 B.
Table 2.
Fifty top-selling drugs in 2022 [25]. Total sales USD 186.40 B.
| Drug name | 2022 Sales, USD B |
|---|---|
| Actemra/RoActemra (tocilizumab) | USD 2.58 |
| Darzalex (daratumumab) | USD 7.98 |
| Dupixent (dupilumab) | USD 17.42 |
| Enbrel (etanercept) | USD 4.12 |
| Eylea (aflibercept) | USD 12.72 |
| Hemlibra (emicizumab) | USD 3.65 |
| Humira (adalimumab) | USD 21.24 |
| Imfinzi (durvalumab) | USD 2.78 |
| Lantus (insulin glargine) | USD 2.38 |
| Ocrevus (ocrelizumab) | USD 5.76 |
| Opdivo (nivolumab) | USD 8.25 |
| Perjeta (pertuzumab) | USD 3.90 |
| Prolia (denosumab) | USD 3.63 |
| Remicade (infliximab) | USD 2.34 |
| Skyrizi (risankizumab) | USD 5.17 |
| Stelara (ustekinumab) | USD 9.72 |
| Taltz (ixekizumab) | USD 2.48 |
| Tecentriq (atezolizumab) | USD 3.55 |
| Tremfya (guselkumab) | USD 2.67 |
| Trulicity (dulaglutide) | USD 7.44 |
Table 3.
Biosimilars with PD markers are exempted from clinical efficacy testing in patients.
| Drug | Patent Expiry |
|---|---|
| Interferon beta-1b | 2004 |
| Parathyroid hormone | 2004 |
| Interferon alfa-2b | 2004 |
| Chorionic gonadotropin | 2007 |
| Interferon alfa-n3 | 2011 |
| Etanercept | 2012 |
| Menotropins | 2015 |
| Urofollitropin | 2015 |
| Peginterferon alfa-2b | 2015 |
| Interferon beta-1a | 2020 |
| Insulin regular | 2025 |
| Insulin lispro | 2014 |
Table 4.
List of SRA countries.
| Country | Authority | The criterion for consideration as SRA |
|---|---|---|
| Australia | Therapeutic Goods Administration | Mutual recognition agreement with ICH members |
| Austria | Austrian Agency for Health and Food Safety (AGES) | EC member |
| Belgium | Federal Agency for Medicines and Health Products (FAMHP) | EC member |
| Bulgaria | Bulgarian Drug Agency | EC member |
| Canada | Health Canada | ICH observer |
| Croatia | Agency for Medicinal Products and Medical Devices of Croatia (HALMED) | EC member |
| Cyprus | Ministry of Health — Pharmaceutical Services | EC member |
| Czech Republic | State Institute for Drug Control (SUKL) | EC member |
| Denmark | Danish Medicines Agency | EC member |
| Estonia | State Agency of Medicines (Ravimiamet) | EC member |
| Finland | Finnish Medicines Agency (Fimea) | EC member |
| France | National Agency for the Safety of Medicine and Health Products (ANSM) | EC member |
| Germany | Federal Institute for Drugs and Medical Devices | EC member |
| Greece | National Organization for Medicines | EC member |
| Hungary | National Institute of Pharmacy and Nutrition (OGYEI) | EC member |
| Iceland | Icelandic Medicines Agency | EFTA member/mutual recognition agreement |
| Ireland | Health Products Regulatory Authority | EC member |
| Italy | Italian Medicines Agency (AIFA) | EC member |
| Japan | Ministry of Health, Labour and Welfare/Pharmaceuticals and Medical Devices Agency | ICH member |
| Latvia | State Agency of Medicines | EC member |
| Liechtenstein | Office of Health / Department of Pharmaceuticals | EFTA member/mutual recognition agreement |
| Lithuania | State Medicines Control Agency (VVKT) | EC member |
| Luxembourg | Ministry of Health | EC member |
| Malta | Medicines Authority | EC member |
| Netherlands | Health and Youth Care Inspectorate (IGZ) | EC member |
| Norway | Norwegian Medicines Agency | EFTA member/mutual recognition agreement |
| Poland | Chief Pharmaceutical Inspectorate | EC member |
| Portugal | National Authority of Medicines and Health Products (Infarmed) | EC member |
| Romania | National Agency for Medicines and Medical Devices | EC member |
| Slovakia | State Institute for Drug Control (SIDC) | EC member |
| Slovenia | Agency for Medicinal Products and Medical Devices (JAZMP) | EC member |
| Spain | Spanish Agency of Medicines and Medical Devices (AEMPS) | EC member |
| Sweden | Medical Products Agency | EC member |
| Switzerland | Swiss Agency for Therapeutic Products (Swissmedic) | ICH observer/EFTA member |
| United Kingdom | Medicines and Healthcare Products Regulatory Agency (MHRA) | EC member (as of 23 October 2015) |
| United States of America | Food and Drug Administration | ICH member |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



