
Submitted:
30 May 2023
Posted:
05 June 2023
You are already at the latest version
Abstract
A 52-y/o asymptomatic male with history of hypertension, was referred to our Heart Failure Clinic due to report of hypertrophic cardiomyopathy, TTE with an increased end-diastolic thick-ness (basal inferoseptal of 23 mm, and basal anteroseptal of 21 mm). CMR demonstrated late gadolinium enhancement at the septum, anterior, inferolateral, and inferior walls with a mid-myocardial distribution, T1 mapping which reported an average T1 of 929 ms. A next-generation sequencing panel was requested. Results demonstrated hemizygosis, in the ga-lactosidase alpha gene, consistent with Fabry Disease. The replacement of the enzyme was start-ed. Extended familial genetic counseling and testing were done.
Keywords:
Cardiomyopathy
; Genetic disorders
; Cardiovascular Imagen
1. Introduction
In hypertrophic phenotype (HP), hypertensive and hypertrophic cardiomyopathy (HCM) are the main two differential diagnoses (1). The latter is defined as an end-diastolic wall thickness ≥ 15mm or ≥ 13mm when there is a positive family history and after ruling out other conditions (2-4).
There is a gap of knowledge regarding the ideal genetic approach in HP due to the phenotype-genotype association inconsistency, the lack of interest of certain clinician and the financial constraints in specific health-care systems in determining genetic work-up (mendelian- non mendelian). Of note, the presence of other phenocopies makes the genetic study a mandatory step in patients with HP.
Myocardial storage disorders is another cause of HP, representing 10% of all patients with HP (5). This diagnosis is challenging due to the clinical and imaging similarity among multiple phenocopies and, also, due to the lack of global availability for genetic testing.
Imaging in HP, particularly cardiac magnetic resonance (CMR) using scar evaluation and parametric mapping, can further guide clinicians for narrowing differential diagnoses in challenging patients. After narrowing the diagnosis, genetic testing ultimately determines the etiology in a large proportion of patients by studying the genetic pedigree and the specific genes associated with the specific phenotypes (4,6,7). In some cases, endomyocardial biopsy is required (5). Having a comprehensive and rationale strategy for the diagnosis of the underlying cause of HP, can allow accuracy in the diagnosis, and the possibility of giving a specific treatment that can modify prognosis in selected patients (5).
2. Case description
52-year-old man with history of hypertension since the age of 35 years, with no other significant personal or family history, under outpatient cardiology treatment, with regular blood pressure control, presented during follow-up studies exercise dyspnea. Studies were done, with an ECG showing left ventricular hypertrophy (LVH), and short PR interval (Figure 1). Transthoracic echocardiogram (TTE) evidenced increased end-diastolic thickness -basal infero-septal wall of 23 mm and basal anteroseptal wall of 21 mm (Figure 2), findings suggestive of HCM. For this reason, he was referred to our cardiac imaging laboratory, with indication of cardiac magnetic resonance, as a complementary study for HCM. CMR showed increased wall thickness, specially compromising the infero-septal wall, reaching 25 mm in the middle segment, without wall motion abnormalities, with normal left ventricle systolic function, LVEF: 64%. There was not systolic anterior motion of the mitral valve, and apical insertion of the papillary muscles was noted. Right ventricle wall thickness and systolic function was normal, RVEF: 72%. After contrast administration, patchy mid-wall late gadolinium enhancement (LGE) was seen in the inferoseptum, as well as in the basal and mid segments of the anterior, inferolateral and inferior wall. Findings suggestive of asymmetric septal HCM. At that time, our center did not have parametric mapping. With these findings, he was referred to our heart failure clinic for complementary studies. As part of the approach to the patient with hypertrophic phenotype, a next-generation sequencing (NGS) panel was requested.
Results demonstrated Hemizygosis of the variant NM_000169.3(GLA): c.1066C>T (p. Arg356Trp), described as pathogenic in the galactosidase alpha (GLA) gene, consistent with Fabry Disease (FD). Alpha-Galactosidase A (GAL) levels, in dried blood spots, were 1.09 nmol/mL / h (reference value - RV: 2.0-21.8 nmol/mL/h)), and Lyso Gb3, 5.3 ng/mL (RV: <2.0 ng/mL); also, decreased GLA activity (0.17 mcmol/L/h (RV:1.68-13.63 mcmol/L/h)), and normal activity of Alpha-Glucosidase (2,11 mcmol/L/h (RV:2.10-29.00 mcmol/L/h)). In addition, the case was reviewed by our cardiovascular imaging group and T1 mapping was ultimately performed (Image 4), which reported an average T1 of 929 ms. Extension studies were done, without renal, pulmonary or neurological comprise.
Pharmacological replacement of the enzyme, with agalsidasa-Beta was started since July 2022, receiving it twice a month as specific treatment, continuing with antihypertensive medications. At six months of enzyme replacement, exercise dyspnea was resolved, with functional class NYHA I.
Extended familial genetic counseling and testing were done, demonstrating the presence of the disease in 2 of his brothers whereas 2 daughters turned out to be carriers (Image 5).
3. Discussion
Hypertrophic Phenotype (HP) includes a wide range of pathologies beyond hypertensive heart disease and HCM. Recognizing these etiologies is essential since accurate diagnosis is feasible using an assertive route including: genetic tests when HP is diagnosed by TTE (with global longitudinal strain analysis), CMR (with LGE and parametric mapping) or scintigraphy, and in specific cases endomyocardial byopsy (4-5). Phenocopies are identified in 5-10% of patients who were initially diagnosed with HCM (5-6). A precise diagnosis will allow an oriented treatment for the patient himself and his relatives (when required).
In the case of our patient, after revising the diagnosis process, FD confirmed. It is an X-linked genetic disorder caused by the deficient activity of lysosomal α-galactosidase (α-Gal) due to pathogenic variants in the GLA gene, leading also to a deficiency or reduced activity of the lysosomal progression of globotriasilceramide (Gb3) and soluble globotriaosylsphingosine (lyso-Gb3) (6-7). Cardiac involvement depends on the level of residual α-Gal activity.
Extra-cardiac involvement includes vessels, kidneys, and peripheral nervous system (6-8). ECG findings other than LVH, such as duration of the P wave < 80 ms and PQ interval < 140ms (as in our patient) have an area under the curve of 0.81-0.92, sensitivity of 80-92% and specificity of 70-80%, respectively. They are useful in early detection, even in the absence of LGE (8). Late onset FD phenotype could present with LVH and papillary muscle hypertrophy mimicking HCM and longitudinal myocardial deformation of the basal segments. The multimodality approach in FD patients is essential, so the use of imaging such as CMR not only provides greater accuracy in the measurement of ventricular wall thickness and can visualize myocardial fibrosis typically distributed in the mid-myocardial layer of the posterolateral wall and extent of fibrosis has been correlated with the degree of response to enzyme replacement therapy (7). The use of tools such as T1 mapping allows the detection of early changes in the cardiac muscle interstitium, and low values, especially at the septal level, have been found in patients with FD, and it is believed that this is directly related to the increase in myocardial lipid tissue (10), thus implying earlier recognition of cardiac involvement by FD. In view of this, the systematic use of CMRI in patients with HP may help to reclassify patients. In this patient and in other with HP phenotype, allowing better risk stratification and therapy targeting. An increased left ventricular end-diastolic thickness with a normal or low T1 mapping such as the one noticed in this patient, should make the clinician suspect and test for FD.
The assessment of GLA activity is the first step for the diagnostic approach in men, but sequencing of GLA gene is the recommended test in women with clinical suspicion. Additionally, GLA gene sequencing is recommended in all patients to identify and confirm the presence of a pathogenic or likely-pathogenic variant, to test for amenability to the chaperone therapy; also, for planning family cascade gene screening and prognostic assessment. Plasmatic lyso-Gb3 should be considered for disease severity estimation in FD patients or for patients with GLA gene pathogenic variants of unknown significance (5,7). The gene NM_000169.3(GLA): c.1066C>T(p. Arg356Trp), a nonsense variant, replaces arginine with tryptophan at codon 356 of the GLA protein. It has been described in 10 people with FD, including 2 women. In vitro functional studies demonstrated reduced GLA activity (11).
Two therapeutic strategies are currently available, enzyme replacement or chaperone therapy. Enzyme replacement (ERT) therapy in susceptible variants is the current recommended management and is indicated in all symptomatic patients with classical disease (7), Two preparations, agalsidase-β (Fabrazyme®, Sanofi Genzyme, Fabagal®, ISU Abxis) and agalsidase-α (Replagal®, Shire) are currently approved as lifelong options for FD treatment, (7,12). The infusion was well tolerated and improved enzyme activity by 68% (12). Reduce Gb3 endothelial inclusions, improving residual enzyme activity; decreasing inflammation associated with reduced LV mass, with improvement in regional function and stabilizing cardiac disease. However, improvement in cardiac contractility and reduction in ventricular mass is directly related to the degree of fibrosis and the number of segments affected by the disease (7,12). Some trials comparing patients with early vs. late disease showed that at 12 months of enzyme replacement, patients with early disease who had left ventricular hypertrophy had decreased ventricular mass values and almost normalized T1 mapping values (13), as well as CMR based trials showed improvement in segmental contractility disorders (14). When comparing the effectiveness between agalsidase-α and agalsidase-β, one study was able to demonstrate a 30% reduction in Lyso GL3 levels after the switch, so agalsidase-β has been considered as the first line of treatment; on the other hand, one of the recommendations for the use of agalsidase-α is the presence of allergy with the infusion. In addition, Migalastat is the first chaperone therapy that has demonstrated effectiveness and safety, was well tolerated and demonstrated durable, long-term stability of renal function and reduction in left ventricular mass (15). New therapeutic options such as plant derived ERT, substate reduction and gene therapy are under study and results are awaited to determine their suitability.
On the other hand, in patients with heart failure, conventional therapy is necessary, and pacing should be considered in patients with chronotropic incompetence. In patients with QRS prolongation and LV ejection fraction less than 50%, CRT-P is recommended, (5,9). In addition, in patients with persistent or permanent supraventricular tachycardia or pre-excitation syndrome, invasive electrophysiological studies are recommended to guide treatment and determine the need for therapies such as ablation, the presence of a short PR interval as an isolated !nding is not an indication for an electrophysiological study (7).
Figure 3.
CMR imaging. Increased wall thickness, specially compromising the infero-septal wall, reaching 25 mm in the middle segment. After contrast administration, patchy mid-wall late gadolinium enhancement (LGE) was seen in the inferoseptum, as well as in the basal and mid segments of the anterior, inferolateral and inferior wall. (White arrows).
Figure 3.
CMR imaging. Increased wall thickness, specially compromising the infero-septal wall, reaching 25 mm in the middle segment. After contrast administration, patchy mid-wall late gadolinium enhancement (LGE) was seen in the inferoseptum, as well as in the basal and mid segments of the anterior, inferolateral and inferior wall. (White arrows).
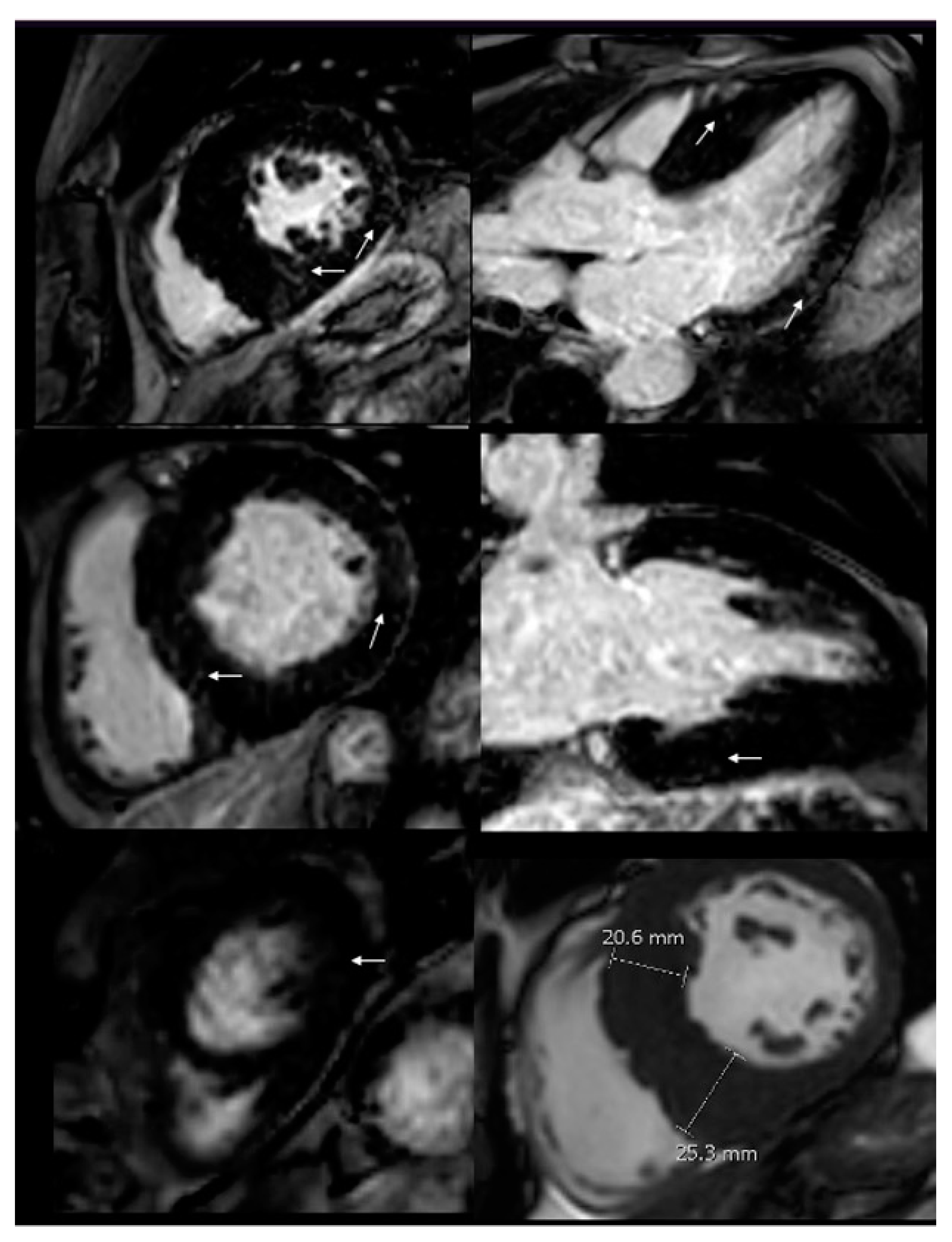
Figure 4.
T1 mapping. Average T1 mapping: 929 ms (Measure at basal, mid, and apical LV segments).
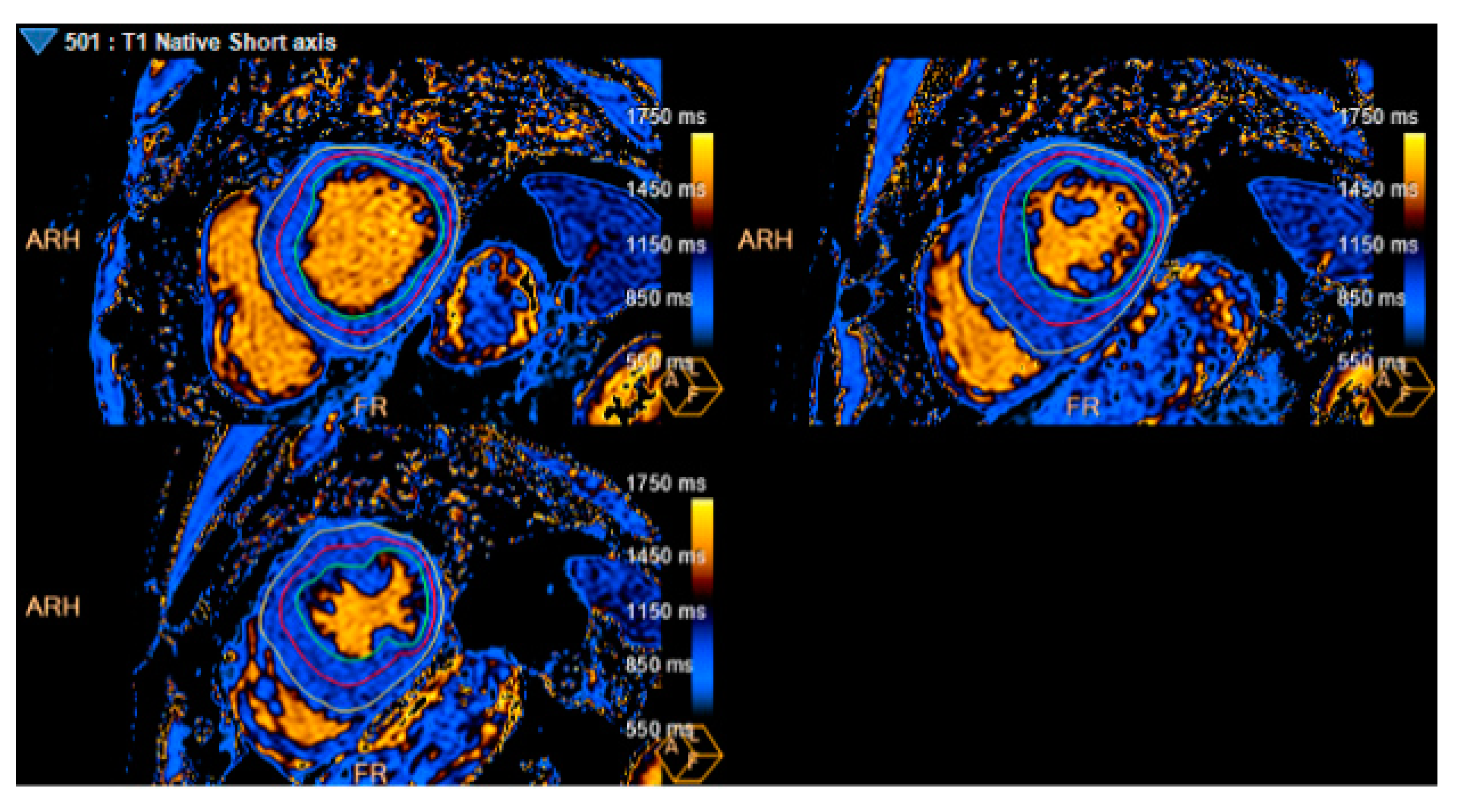
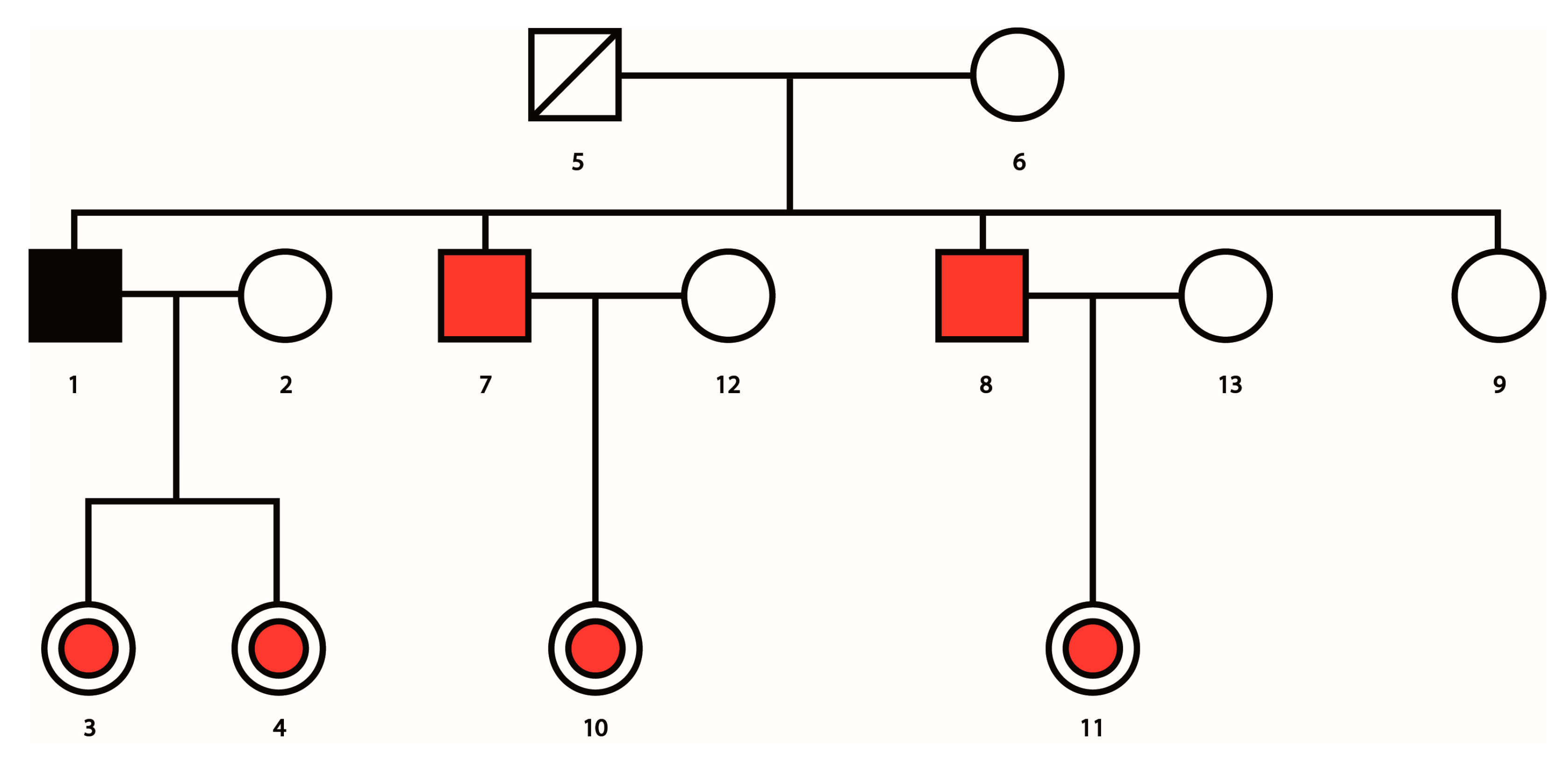
Figure 5.
A genealogical tree showing index patient (black square), siblings affected by the disease (red squares), and daughters carrying the disease (red circles.
Figure 5.
A genealogical tree showing index patient (black square), siblings affected by the disease (red squares), and daughters carrying the disease (red circles.
4. Conclusions
We describe a complex HP case in a patient with an initial diagnosis of HCM due to the severe asymmetric end-diastolic wall thickness by TTE and CMR, which demonstrates the importance of a comprehensive clinical, multimodal imaging approach, and genetic diagnostic tests. All these are necessary to reach a timely and accurate diagnosis. Without forgetting the importance of studying the family group, in a potentially treatable disease. Of note, the T1 mapping (done after the genetic testing) was low (929 ms) for the extent of increased wall thickness, which may have been an initial hint that other conditions such as FD needed to be included in the differential diagnoses. Finally, we suggest some future directions in patients with HP: 1. HP involves a high number of entities with different backgrounds but the same phenotype, so the importance of having some other techniques as multiparametric evaluation can guide us more often to diagnosis. 2. To be alert to the initial red flags in the first steps of the diagnosis of FD for example the use of the ECG. 3. Genetic tests are necessary in the HP to differentiate the phenocopies and to guide the genetic cascade.
Author Contributions
Conceptualization, A.F.B., J.A.G, H.M.M and M.J.R.; writing—original draft preparation, A.F.B., J.A.G, C.P.J, M.C.M, H.M.M and M.J.R; writing—review and editing, A.F.B., J.A.P, H.M.M and M.J.R.; visualization, J.A.P, M.F.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Investigation Department of Fundación Cardioinfantil-LaCardio.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Lakdawala, N.K.; Ware, J.S.; Helms, A.S.; Colan, S.D.; et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic cardiomyopathy: Clinical update. JACC Heart Fail. 2018, 6, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017, 121, 749–770. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar,, J.A.; Kantor, P.; et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: A report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. J Am Coll Cardiol. 2020, 76, e159–e240. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Ciabatti, M.; Saletti, E.; Tavanti, V.; Santangeli, P.; Martinese, L.; Liistro, F. , Olivotto, I.; Bolognese, L. Beyond sarcomeric hypertrophic cardiomyopathy: How to diagnose and manage phenocopies. Curr Cardiol Rep 2022, 24, 1567–1585. [Google Scholar] [CrossRef] [PubMed]
- Militaru, S.; Ginghina, C.; Popescu, B.A.; Saftoiu, A.; Linhart, A.; Jurcut, R. Multimodality imaging in Fabry cardiomyopathy: From early diagnosis to therapeutic targets. Eur Heart J Cardiovasc Imaging 2018, 19, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur J Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Steffel, J.; Vidovic, M.; Brunckhorst, C.B.; Holzmeister, J.; Lüscher, T.F.; Jenni, R.; Duru, F. Electrocardiographic changes in early recognition of Fabry disease. Heart 2011, 97, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Vardarli, I.; Weber, M.; Rischpler, C.; Führer, D.; Herrmann, K.; Weidemann, F. Fabry Cardiomyopathy: Current Treatment and Future Options. J Clin Med. 2021, 10, 3026. [Google Scholar] [CrossRef] [PubMed]
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging 2013, 6, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Submissions for variant NM_000169.3(GLA): C.1066C>T (p. Arg356Trp) (rs104894827). Utah.edu. Available in: https://clinvarminer.genetics.utah.edu/submissions-by-variant/NM_000169.3%28GLA%29%3Ac.1066C%3ET%20%28p.Arg356Trp%29.
- Umer, M.; Kalra, D.K. Treatment of Fabry disease: Established and emerging therapies. Pharmaceuticals 2023, 16, 320. [Google Scholar] [CrossRef] [PubMed]
- Nordin, S.; Kozor, R.; Vijapurapu, R.; Augusto, J.B.; Knott, K.D.; Captur, G.; Treibel, T.A.; Ramaswami, U.; Tchan, M.; Geberhiwot, T.; et al. Myocardial storage, inflammation, and cardiac phenotype in Fabry disease after one year of enzyme replacement therapy. Circ Cardiovasc Imaging 2019, 12, e009430. [Google Scholar] [CrossRef] [PubMed]
- Koeppe, S.; Neubauer, H.; Breunig, F.; Weidemann, F.; Wanner, C.; Sandstede, J.; Machann, W.; Hahn, D.; Köstler, H.; Beer, M. MR-based analysis of regional cardiac function in relation to cellular integrity in Fabry disease. Int J Cardiol 2012, 160, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol Genet Metab 2020, 131, 219–228. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
ECG: Sinus rhythm, normal axis, PQ interval of 82 ms. Wide and deep negative T waves in the anteroseptal and lateral walls. R wave in aVL of 21 mm, consistent with LVH.
Figure 1.
ECG: Sinus rhythm, normal axis, PQ interval of 82 ms. Wide and deep negative T waves in the anteroseptal and lateral walls. R wave in aVL of 21 mm, consistent with LVH.
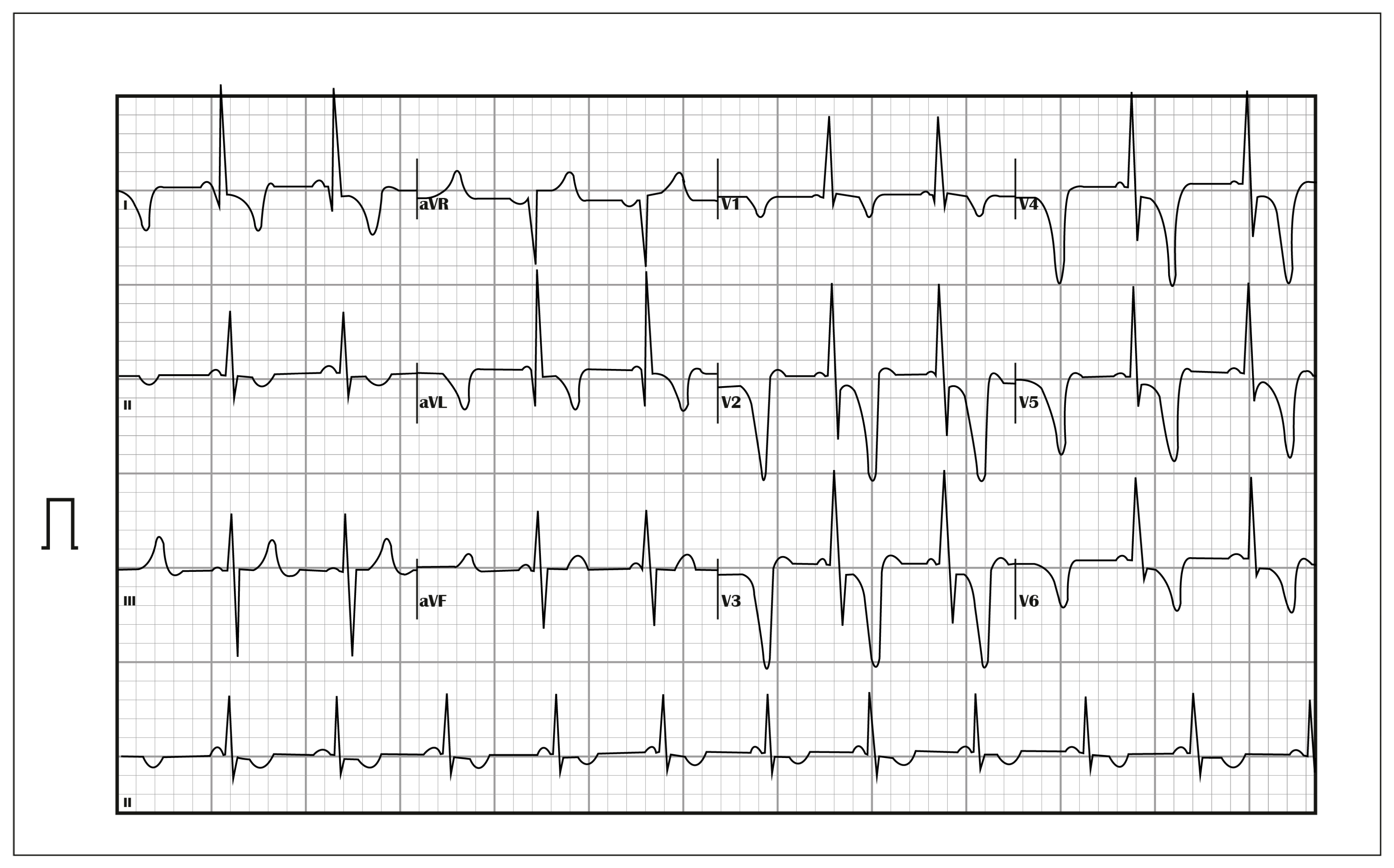
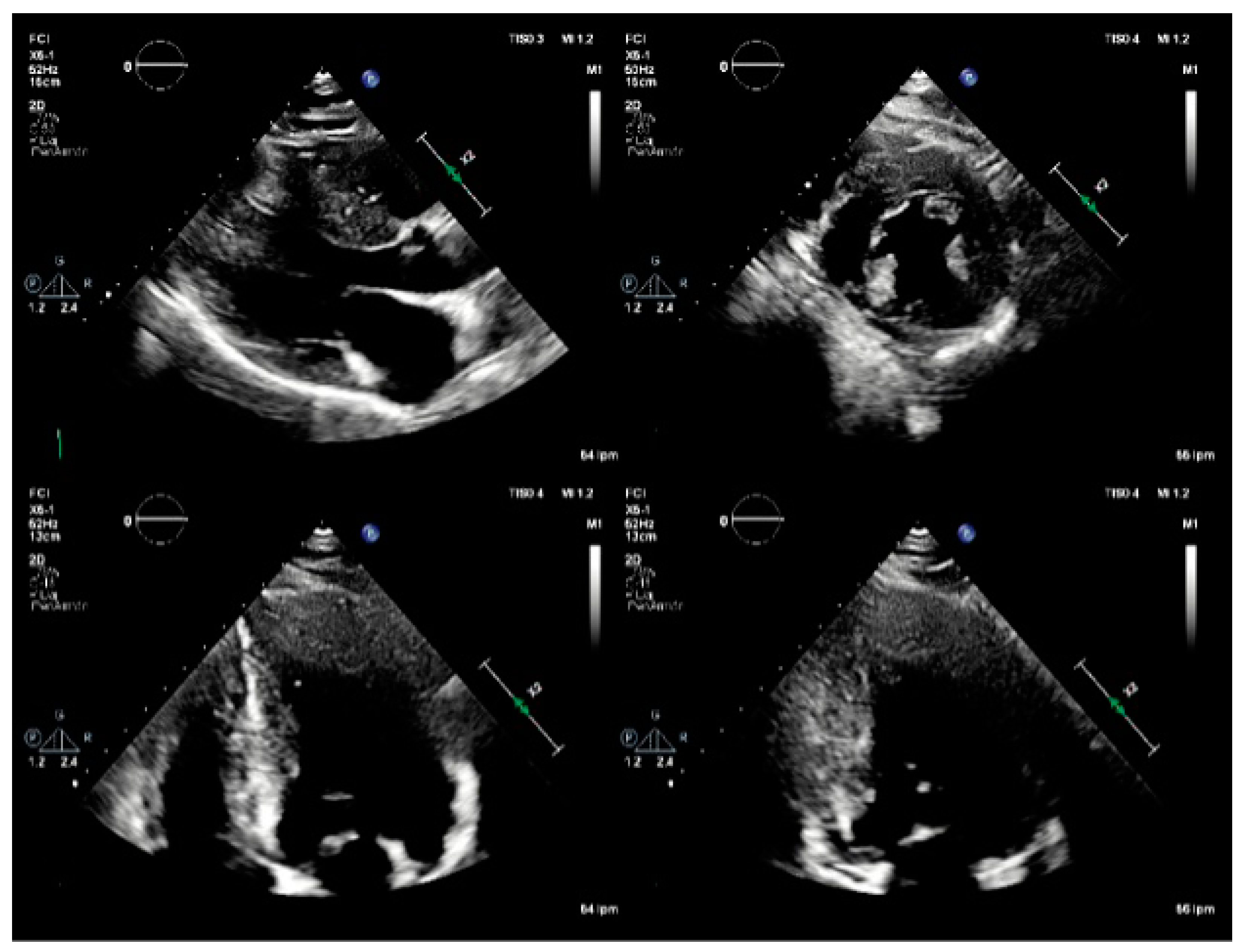
Figure 2.
TTE: Parasternal long and short axis, apical 4-chamber, and apical 2-chamber views As. Basal inferoseptal thickness of 23 mm, and basal anteroseptal thickness of 21 mm. Normal LVEF (60%). Decreased LV global longitudinal strain, affecting mainly the inferoseptal wall, and the apical segments.
Figure 2.
TTE: Parasternal long and short axis, apical 4-chamber, and apical 2-chamber views As. Basal inferoseptal thickness of 23 mm, and basal anteroseptal thickness of 21 mm. Normal LVEF (60%). Decreased LV global longitudinal strain, affecting mainly the inferoseptal wall, and the apical segments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



