
Submitted:
30 May 2023
Posted:
01 June 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Background: The outbreak of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) resulted in the global COVID-19 pandemic. The urgency for an effective SARS-CoV-2 vaccine has led to the development of a first series of vaccines at unprecedented speed. The discovery of SARS-CoV-2 spike-glycoprotein mutants, however, and consequentially the potential to escape vaccine-induced protection and increased infectivity, demonstrates the persisting importance of monitoring SARS-CoV-2 mutations to enable early detection and tracking of genomic variants of concern.
Results: We developed the CoVigator tool with three components: 1) a knowledge base that collects new SARS-CoV-2 genomic data, processes it and stores its results; 2) a comprehensive variant calling pipeline; 3) an interactive dashboard highlighting the most relevant findings. The knowledge base routinely downloads and processes virus genome assemblies or raw sequenc-ing data from the COVID-19 Data Portal (C19DP) and the European Nucleotide Archive (ENA), respectively. The results of the variant calling pipeline are visualized through the dashboard in the form of tables and customizable graphs, making it a versatile tool for tracking SARS-CoV-2 variants. We put a special emphasis on the identification of intrahost mutations and make available to the community what is, to the best of our knowledge, the largest dataset on SARS-CoV-2 intrahost mutations. In the spirit of open data, all CoVigator results are available for download. The CoVigator dashboard is accessible via covigator.tron-mainz.de.
Conclusion: With increasing demand worldwide in genome surveillance for tracking the spread of SARS-CoV-2, CoVigator will be a valuable resource of up-to-date list of mutations, which can be incorporated into global efforts to sustainably prevent or treat infections.
Keywords:
SARS-CoV-2
; dashboard
; genomic variants
; software
; pipeline
; virus genome assemblies
; knowledge base
& Contributed equally to this work.
1. Introduction
The identification, characterization and monitoring of the pathogen responsible for a novel emerging disease is crucial for the development of a timely public health response. This includes rapid and open sharing of data [1] which has been adapted in past outbreaks to advance research and improve medical support [2,3]. The outbreak of the respiratory disease COVID-19 caused by SARS-CoV-2 demonstrated the increasing value of high-throughput sequencing by enabling the publication of the complete virus genome within one month of sampling [4,5]. The identification of the SARS-CoV-2 spike protein as a valuable target for vaccine design [6,7] led to the development of vaccines at unprecedented speed [8,9] and is still fostering further developments.
Nevertheless, the discovery of SARS-CoV-2 spike-glycoprotein mutants, associated with the potential to escape vaccine-induced protection, demonstrates the importance of monitoring SARS-CoV-2 genomic sequences to enable early detection of these genomic variants of concern. In a first study, we analyzed 1,036,030 genomic assemblies from the Global Initiative on Sharing Avian Influenza Data (GISAID) [10,11,12] and 30,806 Next Generation Sequencing (NGS) datasets from the European Nucleotide Archive (ENA). We reported non-synonymous spike protein mutations and their frequencies and analyzed the effect on known T-cell epitopes [13] . Although we confirmed low mutation rates of the spike protein, we experienced an increase in the number of genomic variants over time. Therefore, we further developed our pipeline to tackle potential escape mutations [14].
There are multiple initiatives to monitor SARS-CoV-2 mutations based on the GISAID dataset: NextStrain [15], CoV-GLUE [16] and Coronapp [17]. A further initiative based on the ENA dataset is the Galaxy project COVID-19 [18] ; other systems use regional data: CLIMB-COVID (COG-UK) [19] and CovRadar [20]. The COVID-19 Data Portal [21] is provided by EMBL-EBI and the European COVID-19 Data Platform to facilitate data sharing and accelerate research by making all the data available in the public domain and encouraging the research community to share SARS-CoV-2 data. Furthermore, there are some open source pipelines to identify mutations on SARS-CoV-2 data implemented in Nextflow: Cecret [22], nf-core viralrecon [23] and ncov2019-artic-nf [24].
Each dataset has its own advantages. While genomic assemblies are easier to share and interpret, raw reads provide granular information about the mutations through access to the pileup of reads supporting each mutation, also allowing the characterization of intrahost mutations. Analyzing both datasets together may be support the identification of potential false positives in the data and the confirmation of trends.
To enable monitoring of SARS-CoV-2 sequences from both sources, we have developed CoVigator, an NGS pipeline and dashboard that allows geographical and temporal navigation through SARS-CoV-2 genomic variants. We automatically download and analyze genomic assemblies from the COVID-19 Data Portal and raw reads from the European Nucleotide Archive (ENA). Furthermore, we screen the literature for studies on SARS-CoV-2 intrahost mutations [25,26] [27,28,29,30,31,32,33,34,35,36,37,38,39,40] and propose a filtering strategy to obtain a high quality set of intrahost mutations in the large and heterogeneous dataset obtained from ENA. Thus, the CoVigator platform supports early detection of variants, which potentially provides guidance for the further adjustment of vaccination strategies or therapeutics.
2. Materials and Methods
The CoVigator knowledge base is implemented in Python version 3.8 and the database for storing data is PostgreSQL (version 13.4).
The CoVigator pipeline (version 0.14.0) is implemented in the Nextflow framework version 19.10.0. All dependencies are managed within conda (version 4.9) environments [41] (see Table 1).
The CoVigator dashboard is also implemented in Python using the visualization framework Dash (version 2.1.0). The computation is distributed through a High Performance Computing cluster with a library that provides advanced parallelism, Dask (version 2022.9.2).
3. Results and discussion
System description
The CoVigator system (Figure 1) has three main components: 1) the knowledge base, 2) the analysis pipeline and 3) the dashboard. The knowledge base orchestrates for every sample the metadata retrieval, raw data download and finally its analysis through the pipeline for the detection of mutations. Furthermore it makes all necessary data available through a database (Postgre-SQL version 13). Finally, the dashboard presents the data to the end user through a set of interactive visualizations.
CoVigator operates via interaction with external systems: a high performance computing (HPC) cluster and the ENA and COVID-19 Data Portal Application Programming Interfaces (APIs).Samples between both original datasets (raw reads and genomic assemblies) may overlap. As recommended, some data providers might automatically upload both data formats. The results presented in the dashboard are stratified by dataset.
Knowledge base
The CoVigator knowledge base collects data from both genomic assemblies and raw reads, orchestrates its processing through the variant calling pipeline, and stores all the metadata, raw data and processed results in a relational database.
The data for both datatypes is fetched via the corresponding API hosted by the European Bioinformatics Institute [55], the metadata is normalized, the FASTQ (raw NGS reads) and FASTA (genomic assemblies) files downloaded and their MD5 checksums are confirmed to ensure data integrity.
Furthermore, the knowledge base iteratively builds a variant co-occurrence matrix (only for the raw reads dataset) and precomputes analyses on the data (binned abundance of mutations, dN/dS ratios per gene and domain, top occurring variants, pairwise co-ocurrence and counts of variants per lineage, country, sample, mutation type, length and nucleotide substitution) that ensure low latency responses.
Analysis pipeline
In general, the CoVigator pipeline processes FASTQ and FASTA files into annotated and normalized analysis-ready VCF files by two independent workflows (Figure 1 & Figure S1). We implemented the pipeline in the Nextflow framework [56] and managed all dependencies with Conda environments to enable seamless installation. We have embedded the SARS-CoV-2 reference genome ASM985889v3 [5]. Using a different reference, this pipeline could instantly analyze other virus sequences as well.
Dashboard
The dashboard is the user interface to CoVigator. There are two separate views for the raw reads and genomic assemblies datasets. Each view provides a set of tabs that allows the user to explore different aspects of the data held in the database. Each tab provides some interactive visualizations, described below. When applicable, the tabs provide a set of filters on the left side. These have been excluded from the screenshots for the purpose of clarity.
The most relevant tabs are described below and some notable findings are highlighted. The data shown here includes 137,025 samples downloaded from ENA on the 21st October 2022; and 6,165,681 samples downloaded from the COVID-19 Data Portal on the 18th November 2022.
Samples
The samples tab (Figure 2) enables the user to explore the accumulation of samples through time and the evolution of the dN/dS ratio on different genomic regions.
Figure 2A shows the accumulation of samples in each country. The dashboard allows the user to select specific countries and/or lineages.
Figure 2B,C shows the dynamics of the dN/dS ratio through time, over genes and protein domains. The dN/dS ratio aims to estimate the evolutionary pressure on the SARS-CoV-2 proteins and domains. This metric, although originally developed for assessing diverging species, is an imperfect but simple estimation of the evolutionary pressure within the same species [57,58], in this case SARS-CoV-2. There have been recent efforts to develop better alternatives for estimating the evolutionary pressure on SARS-CoV-2 [59]. The traditional interpretation of dN/dS is as follows: dN/dS < 0 indicates purifying selection, dN/dS = 1 indicates neutral evolution and dN/dS > 1 indicates positive selection.
Lineages
The lineages tab enables the user to explore the different lineages through time and geography (Figure 3). Both the accumulation of samples in every lineage worldwide (Figure 3A), and the dominant lineage through time (Figure 3B) can be viewed. In the screenshot, the displacement of B.1 by Alpha (B.1.1.7); then subsequently displaced by the multiple Delta lineages (AY.*); and finally displaced by the three Omicron lineages (BA.1, BA.2 and BA.3) can be seen.
Mutation statistics
The mutation statistics tab provides insights on the variant calling results on the different datasets and genomic regions (Figure 4). Expected trends in the data can be confirmed in these visualizations.
The median number of SNVs per sample in the raw reads dataset is 32, with an interquartile range (IQR) of 30 (Figure 4A). Additionally, the median number of MNVs is two with an IQR of one. The number of deletions is lower (median: 3, IQR: 2) than the number of SNVs and the number of insertions is even lower with few samples having just one insertion. For the genomic assemblies the numbers are slightly different with median SNVs 44 (IQR: 19), MNVs 2 (IQR: 0), deletions 4 (IQR: 3) and again just one insertion in few samples (Figure 4B).
We observe that the base substitution C>T is by large the most frequent, followed by G>T and A>G; the deletion TA>T and the MNV GG>AA is the most frequent in both datasets (Figure 4C,D).
In Figure 4E,F, we confirm that deletions are more frequent than insertions with an insertion-to-deletion ratio of 0.002 and 0.032 for raw reads and genomic assemblies, respectively. We also confirm two previous findings: 1) shorter deletions and insertions are more common than longer ones [60,61] and 2) the deletions and insertions not causing a frameshift are overrepresented as their impact in the resulting protein is more subtle [62]. In the genomic assemblies, we observe a long tail of deletions longer than 8 bp which is not observed in the raw reads results. We suspect this is a technical artifact introduced by our variant calling method. Finally, as shown in Figure 4G,H we observe that the most frequent mutation effect is a missense variant, followed by a synonymous variant. This is coherent between both datasets.
Recurrent mutations
The recurrent mutations tab allows the user to explore the most recurrent mutations by total count of observations through time within their genomic context (Figure 5).
In Figure 5A, the top recurrent mutations and their frequency and counts through time are shown. The size of the table can be parametrized for up to 100 mutations. For instance, the user can explore the most recurrent mutations in the whole genome, a given gene or a given protein domain. Furthermore, the period in which the monthly counts are shown can be parametrized. The gene viewer (Figure 5B) has multiple tracks: i) a scatter plot with the relevant mutations and their frequencies in the virus population, ii) ConsHMM conservation tracks and iii) gene and Pfam protein domains. The table in Figure S5 provides the decline and rise of the Alpha and Delta lineages, respectively, in the counts of mutations between April and July 2021.
Additionally, the mutation statistics tab provides a co-occurrence analysis that points to clusters of co-occurring mutations and their correspondence with virus lineages; or in the case of mutations shared between lineages, these clusters may contain a mixture of different but related lineages. Due to performance limitations, this analysis is only available in the raw reads dataset and at the gene level. In Figure S6, we show the Jaccard index co-occurrence matrix in the spike protein and its clustering results annotated with SARS-CoV-2 lineages in Table S1.
Clonal and intrahost mutations in the raw reads dataset
The FASTQ files provide the pile up of reads across the genome and this gives detailed information into the called variants. In particular, we can count the number of reads supporting each variant and this allows us to identify subclonal variants supported by only a fraction of the reads. These variants likely emerged within the host and are referred to as intrahost variants. The identification of intrahost variants is not possible on the genomic assemblies.
We consider high quality clonal mutations those with a VAF greater than or equal to 80 %, and those with a VAF greater than or equal to 50 % and lower than 80 % as low confidence clonal mutations. Only the high confidence clonal mutations are used to determine a consensus sequence and assign a SARS-CoV-2 lineage (Figure 6).
The remaining dataset of mutations poses a different technical challenge due to the difficulty to separate true low VAF mutations from noise. We first determine those mutations with a VAF below 50 % as raw candidate intrahost mutations.
We observed a large number of low frequency mutations among SARS-CoV-2 genomes. In order to establish a high quality set of intrahost mutations for studying viral evolution, we screened and compared the literature on SARS-CoV-2 intrahost mutations for different filtering approaches and implemented a conservative approach (Table 2).
4. Conclusions
The persistently increasing amount of publicly available SARS-CoV-2 sequencing data calls for robust platforms that allow constant monitoring of genomic SARS-CoV-2 variants in heterogeneous data sets. Our CoVigator pipeline covers the essential steps of preparing the data and calling variants from SARS-CoV-2 raw sequencing data from ENA and genome assemblies from the COVID-19 Data Portal. The pipeline is integrated within the CoVigator knowledge base that orchestrates download, processing and storage of the underlying samples and results. The CoVigator dashboard provides different visualizations and features for selecting clonal variants across all genes from the SARS-CoV-2 genome in a selected period. The dashboard also provides a comprehensive analysis of intrahost variants observed across detected mutations in the raw reads dataset. To this end, we propose a conservative filtering approach based on filtering samples and mutations. The dataset of intrahost mutations derived from public data that we make available through CoVigator is to the best of our knowledge the largest published dataset of SARS-CoV-2 intrahost mutations.
The identification of mutations over such heterogeneous datasets obtained with different sequencing protocols is challenging. With CoVigator, we observed VAF dilution on mutations identified by targeted amplicon sequencing with overlapping primers, genome edge effects and read edge effects. We aim to address these challenges in the future, e.g. by inferring the primers used in an arbitrary sample. Besides, we implemented a simplistic phasing of clonal mutations occurring in the same amino acid to ensure their correct annotation. However, we identified the need for a phasing method for low VAF mutations that existing germline phasing tools do not cover.
Future versions of CoVigator can be broadened to other use cases, such as other infectious organisms or co-existing infections during pandemic. Additionally, we envision the annotation of detected mutations to facilitate the selection of variants of concern for the development and evolution of preventive vaccines or therapies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1:Workflow of CoVigator pipeline ; Figure S2: screenshot of the lineages tab in the ENA dataset; Figure S3: screenshot of mutation statistics tab in the ENA dataset; Figure S4: recurrent mutations tab for the spike protein in the ENA dataset; Figure S5: top 30 mutations in the spike protein from COVID-19 Data Portal; Figure S6: screenshot of intrahost mutations tab; Figure S7: Jaccard index co-occurrence matrix on the spike protein; Table S1: co-occurrence clusters on the spike protein with its matching lineage information.
Author Contributions
US, ML, BS and TB were involved in conceptualization. TB, PR, PS and JH participated in implementation of dashboard, developing pipeline and hosting webserver. PR and PS were involved in developing documentations and releasing the pipeline. TB, PR, PS, RG, TR and JH were involved in designing of dashboard and pipeline. RG and PR performed additional analysis to show performance of the pipeline. RG, PR, TB prepared the original draft of the manuscript. BS, US, and ML were involved in writing, critical review and editing the final draft of the manuscript.
Funding
BioNTech SE, Mainz, Germany, supports the study. The funder provided support in the form of salary for author U.S., but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of this author is articulated in the ‘author contributions’ section. In addition, the other authors are employees of the non-profit company TRON gGmbH and are supported in the form of salary. TRON gGmbH did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Intel is committed to accelerating access to technology that can combat the current pandemic and enable scientific discovery that better prepares our world for future crises. Funding for this solution was funded in part by Intel’s Pandemic Response Technology Initiative. For more information about healthcare solutions from Intel, visit intel.com/healthcare. For more information about Intel’s COVID-19 response, visit intel.com/COVID-19.
Data Availability Statement
The CoVigator dashboard is accessible via covigator.tron-mainz.de and can be installed via https://github.com/TRON-bioinformatics/covigator. A standalone version of CoVigator pipeline with nextflow is available at https://github.com/TRON-Bioinformatics/covigator-ngs-pipeline. CoVigator documentation is available at https://covigator.readthedocs.io.
Acknowledgments
We thank Franziska Lang, Özlem Muslu, Jonas Ibn-Salem, Jos de Graaf and Rudolf Koopmann for critical discussions. We thank Karen Chu and Paul Kerbs for reviewing, editing and proofreading this article. We gratefully acknowledge the authors from the originating laboratories responsible for obtaining the specimens, as well as the submitting laboratories where the sequence data were generated and shared via the European Nucleotide Archive and the COVID-19 Data Portal.
Conflict of Interest
Author U.S. is co-founder, shareholder and CEO at BioNTech SE. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict interest.
References
- Moorthy, V.S.; Karam, G.; Vannice, K.S.; Kieny, M.-P. Rationale for WHO's new position calling for prompt reporting and public disclosure of interventional clinical trial results. PLoS Med. 2015, 12, e1001819. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.V.; Maia, M.; Bravo-Filho, V.; Góis, A.L.; Belfort, R. Zika virus in Brazil and macular atrophy in a child with microcephaly. The Lancet 2016, 387, 228. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Nirula, A.; Heller, B.; Gottlieb, R.L.; Boscia, J.; Morris, J.; Huhn, G.; Cardona, J.; Mocherla, B.; Stosor, V.; et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID's Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Schrörs, B.; Riesgo-Ferreiro, P.; Sorn, P.; Gudimella, R.; Bukur, T.; Rösler, T.; Löwer, M.; Sahin, U. Large-scale analysis of SARS-CoV-2 spike-glycoprotein mutants demonstrates the need for continuous screening of virus isolates. PLoS One 2021, 16, e0249254. [Google Scholar] [CrossRef] [PubMed]
- Riesgo-Ferreiro, P. VAFator; Github: https://github.com/TRON-Bioinformatics/vafator.git: https://github.com/TRON-Bioinformatics/vafator.git, 2022.
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Gifford, R.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation, 2020.
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A web application to annotate and monitor SARS-CoV-2 mutations. J. Med. Virol. 2021, 93, 3238–3245. [Google Scholar] [CrossRef]
- Maier, W.; Bray, S.; van den Beek, M.; Bouvier, D.; Coraor, N.; Miladi, M.; Singh, B.; Argila, J.R. de; Baker, D.; Roach, N.; et al. Ready-to-use public infrastructure for global SARS-CoV-2 monitoring. Nat. Biotechnol. 2021, 39, 1178–1179. [Google Scholar] [CrossRef]
- Nicholls, S.M.; Poplawski, R.; Bull, M.J.; Underwood, A.; Chapman, M.; Abu-Dahab, K.; Taylor, B.; Colquhoun, R.M.; Rowe, W.P.M.; Jackson, B.; et al. CLIMB-COVID: Continuous integration supporting decentralised sequencing for SARS-CoV-2 genomic surveillance. Genome Biol. 2021, 22, 196. [Google Scholar] [CrossRef] [PubMed]
- Wittig, A.; Miranda, F.; Hölzer, M.; Altenburg, T.; Bartoszewicz, J.M.; Beyvers, S.; Dieckmann, M.A.; Genske, U.; Giese, S.H.; Nowicka, M.; et al. CovRadar: Continuously tracking and filtering SARS-CoV-2 mutations for molecular surveillance, 2021.
- Harrison, P.W.; Lopez, R.; Rahman, N.; Allen, S.G.; Aslam, R.; Buso, N.; Cummins, C.; Fathy, Y.; Felix, E.; Glont, M.; et al. The COVID-19 Data Portal: Accelerating SARS-CoV-2 and COVID-19 research through rapid open access data sharing. Nucleic Acids Res. 2021, 49, W619–W623. [Google Scholar] [CrossRef] [PubMed]
- Cecret; GitHub: GitHub, Accessed 2021.
- Harshil, P.; Sarai, V.; Sara, M.; Jose, E.-C.; Michael, L.H.; Gisela, G.; nf-core bot; Phil, E.; Miguel, J.; Stephen, K.; et al. nf-core/viralrecon; zenodo, 2021.
- connor-lab. ncov2019-artic-nf; GitHub: GitHub, 2022.
- Al Khatib, H.A.; Benslimane, F.M.; Elbashir, I.E.; Coyle, P.V.; Al Maslamani, M.A.; Al-Khal, A.; Al Thani, A.A.; Yassine, H.M. Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients With Variable Disease Severities. Front. Cell. Infect. Microbiol. 2020, 10, 575613. [Google Scholar] [CrossRef]
- Armero, A.; Berthet, N.; Avarre, J.-C. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 exhibits intra-host genomic plasticity and low-frequency polymorphic quasispecies. J. Clin. Virol. 2020, 131, 104585. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; Cesare, M. de; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-host diversity and transmission. Science 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Moreno, G.; Katarina M. Braun; Peter J. Halfmann; Trent M. Prall, Kasen K. Riemersma, Amelia K. Haj, Joseph Lalli, Kelsey R. Florek, Yoshihiro Kawaoka, Thomas C. Friedrich, David H. O’Connor. Limited SARS-CoV-2 diversity within hosts and following passage in cell culture 2020. [CrossRef]
- Popa, A.; Genger, J.-W.; Nicholson, M.D.; Penz, T.; Schmid, D.; Aberle, S.W.; Agerer, B.; Lercher, A.; Endler, L.; Colaço, H.; et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Nolan, D.J.; Moot, S.; Feehan, A.; Cross, S.; Garcia-Diaz, J.; Lamers, S.L. Intra-host site-specific polymorphisms of SARS-CoV-2 is consistent across multiple samples and methodologies, 2020.
- Siqueira, J.D.; Goes, L.R.; Alves, B.M.; Carvalho, P.S. de; Cicala, C.; Arthos, J.; Viola, J.P.B.; Melo, A.C. de; Soares, M.A. SARS-CoV-2 genomic and quasispecies analyses in cancer patients reveal relaxed intrahost virus evolution. bioRxiv 2020. [CrossRef]
- Tonkin-Hill, G.; Martincorena, I.; Amato, R.; Lawson, A.R.J.; Gerstung, M.; Johnston, I.; Jackson, D.K.; Park, N.R.; Lensing, S.V.; Quail, M.A.; et al. Patterns of within-host genetic diversity in SARS-CoV-2 2020. [CrossRef]
- Sapoval, N.; Mahmoud, M.; Jochum, M.D.; Liu, Y.; Elworth, R.A.L.; Wang, Q.; Albin, D.; Ogilvie, H.A.; Lee, M.D.; Villapol, S.; et al. SARS-CoV-2 genomic diversity and the implications for qRT-PCR diagnostics and transmission. Genome Res. 2021, 31, 635–644. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Liu, H.; Zhang, Y.-D.; Wu, Y.-Q.; Peng, Min-Sheng: Li, Aimin; Irwin, D.M.; Li, H.; Lu, J.; Bao, Y.; Lu, X.; et al. Worldwide tracing of mutations and the evolutionary dynamics of SARS-CoV-2. bioRxiv 2020. [CrossRef]
- James, S.E.; Ngcapu, S.; Kanzi, A.M.; Tegally, H.; Fonseca, V.; Giandhari, J.; Wilkinson, E.; Chimukangara, B.; Pillay, S.; Singh, L.; et al. High Resolution analysis of Transmission Dynamics of Sars-Cov-2 in Two Major Hospital Outbreaks in South Africa Leveraging Intrahost Diversity. medRxiv 2020. [CrossRef]
- Sashittal, P.; Luo, Y.; Peng, J.; El-Kebir, M. Characterization of SARS-CoV-2 viral diversity within and across hosts. bioRxiv 2020. [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef]
- Valesano, A.L.; Rumfelt, K.E.; Dimcheff, D.E.; Blair, C.N.; Fitzsimmons, W.J.; Petrie, J.G.; Martin, E.T.; Lauring, A.S. Temporal dynamics of SARS-CoV-2 mutation accumulation within and across infected hosts. PLoS Pathog. 2021, 17, e1009499. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, D.; Zhang, L.; Sun, W.; Zhang, Z.; Chen, W.; Zhu, A.; Huang, Y.; Xiao, F.; Yao, J.; et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 2021, 13, 30. [Google Scholar] [CrossRef]
- Conda; Anaconda Software Distribution: https://docs.conda.io/: https://docs.conda.io/, 2022.
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), 2019; pp 314–324.
- van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; Jesus, J.G. de; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Le Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- O'Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Kwon, S.B.; Ernst, J. Single-nucleotide conservation state annotation of the SARS-CoV-2 genome. Commun. Biol. 2021, 4, 698. [Google Scholar] [CrossRef]
- Ensembl annotations SARS-CoV-2; ftp://ftp.ensemblgenomes.org/pub/viruses/json/sars_cov_2/sars_cov_2.json: ftp://ftp.ensemblgenomes.org/pub/viruses/json/sars_cov_2/sars_cov_2.json, Accessed 2021.
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Kryazhimskiy, S.; Plotkin, J.B. The population genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef]
- Spielman, S.J.; Wilke, C.O. The relationship between dN/dS and scaled selection coefficients. Mol. Biol. Evol. 2015, 32, 1097–1108. [Google Scholar] [CrossRef]
- Kistler, K.; Huddleston, J.; Bedford, T. Rapid and parallel adaptive mutations in spike S1 drive clade success in SARS-CoV-2. bioRxiv 2022. [CrossRef]
- Rogozin, I.B.; Saura, A.; Bykova, A.; Brover, V.; Yurchenko, V. Deletions across the SARS-CoV-2 Genome: Molecular Mechanisms and Putative Functional Consequences of Deletions in Accessory Genes. Microorganisms 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Insertions in SARS-CoV-2 genome caused by template switch and duplications give rise to new variants that merit monitoring. bioRxiv 2021. [CrossRef]
- Montgomery, S.B.; Goode, D.L.; Kvikstad, E.; Albers, C.A.; Zhang, Z.D.; Mu, X.J.; Ananda, G.; Howie, B.; Karczewski, K.J.; Smith, K.S.; et al. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 2013, 23, 749–761. [Google Scholar] [CrossRef]
- DeMaio, N.; Conor Walker; Rui Borges; Lukas Weilguny; Greg Slodkowic; Nick Goldman. Issues with SARS-CoV-2 sequencing data. Available online: https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473.
Figure 1.
CoVigator system components. The accessor reads external data and stores it in an SQL database. The processor reads the stored data and distributes the processing of every sample in an HPC cluster via Dask. The pipeline processes FASTA and FASTQ data and finally stores the results back in the database (See Figure S1 for more detailed FASTA and FASTQ processing pipeline). The dashboard reads the results and displays them in a set of interactive plots. The results are also available in raw format.
Figure 1.
CoVigator system components. The accessor reads external data and stores it in an SQL database. The processor reads the stored data and distributes the processing of every sample in an HPC cluster via Dask. The pipeline processes FASTA and FASTQ data and finally stores the results back in the database (See Figure S1 for more detailed FASTA and FASTQ processing pipeline). The dashboard reads the results and displays them in a set of interactive plots. The results are also available in raw format.
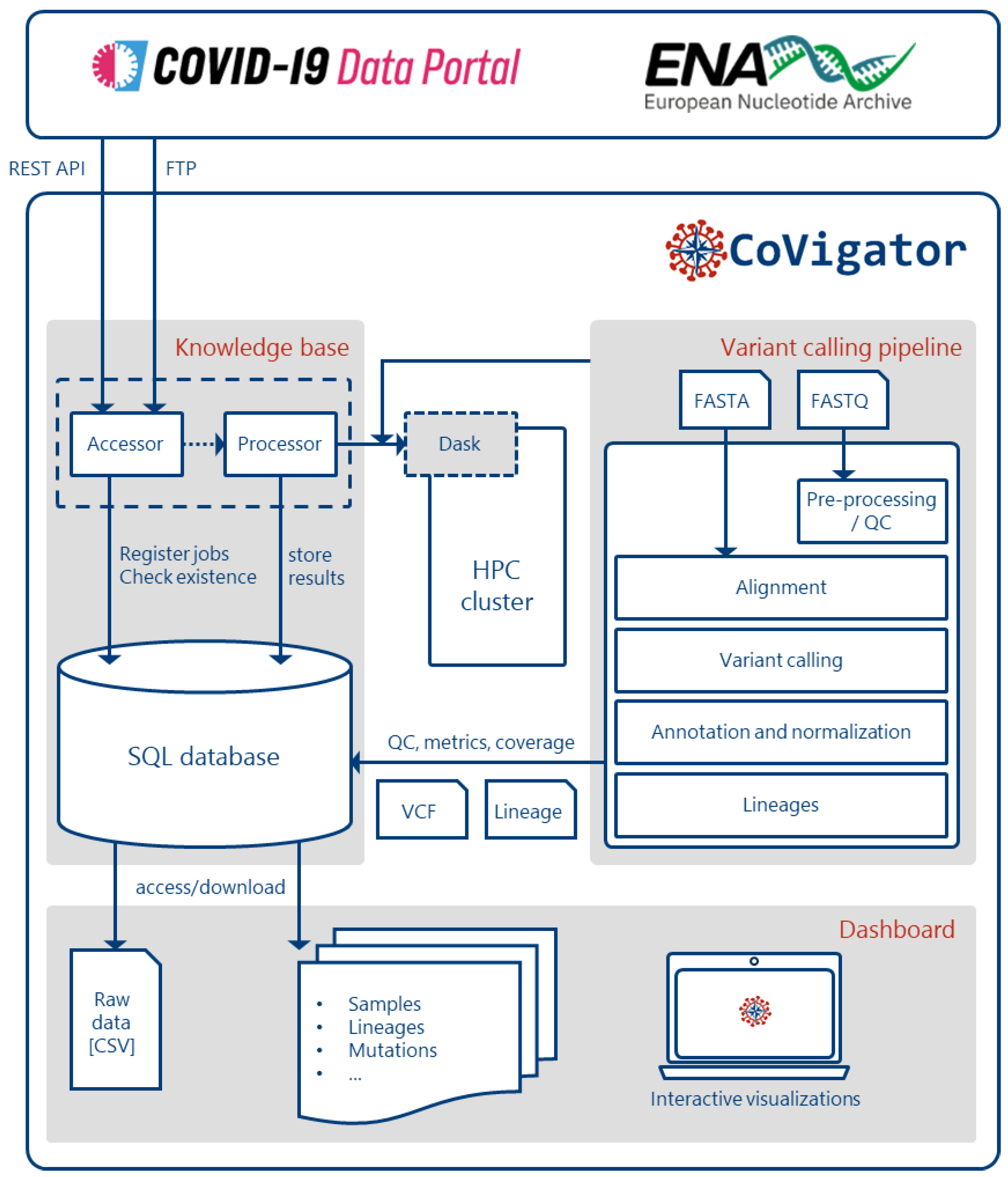
Figure 2.
Samples by country tab plots for raw reads dataset. A) accumulation of samples through time by country; B) dN/dS ratio through time on each SARS-CoV-2 protein; C) dN/dS ratio through time in the domains of the spike protein. See Figure S2 for a screenshot including the filters.
Figure 2.
Samples by country tab plots for raw reads dataset. A) accumulation of samples through time by country; B) dN/dS ratio through time on each SARS-CoV-2 protein; C) dN/dS ratio through time in the domains of the spike protein. See Figure S2 for a screenshot including the filters.
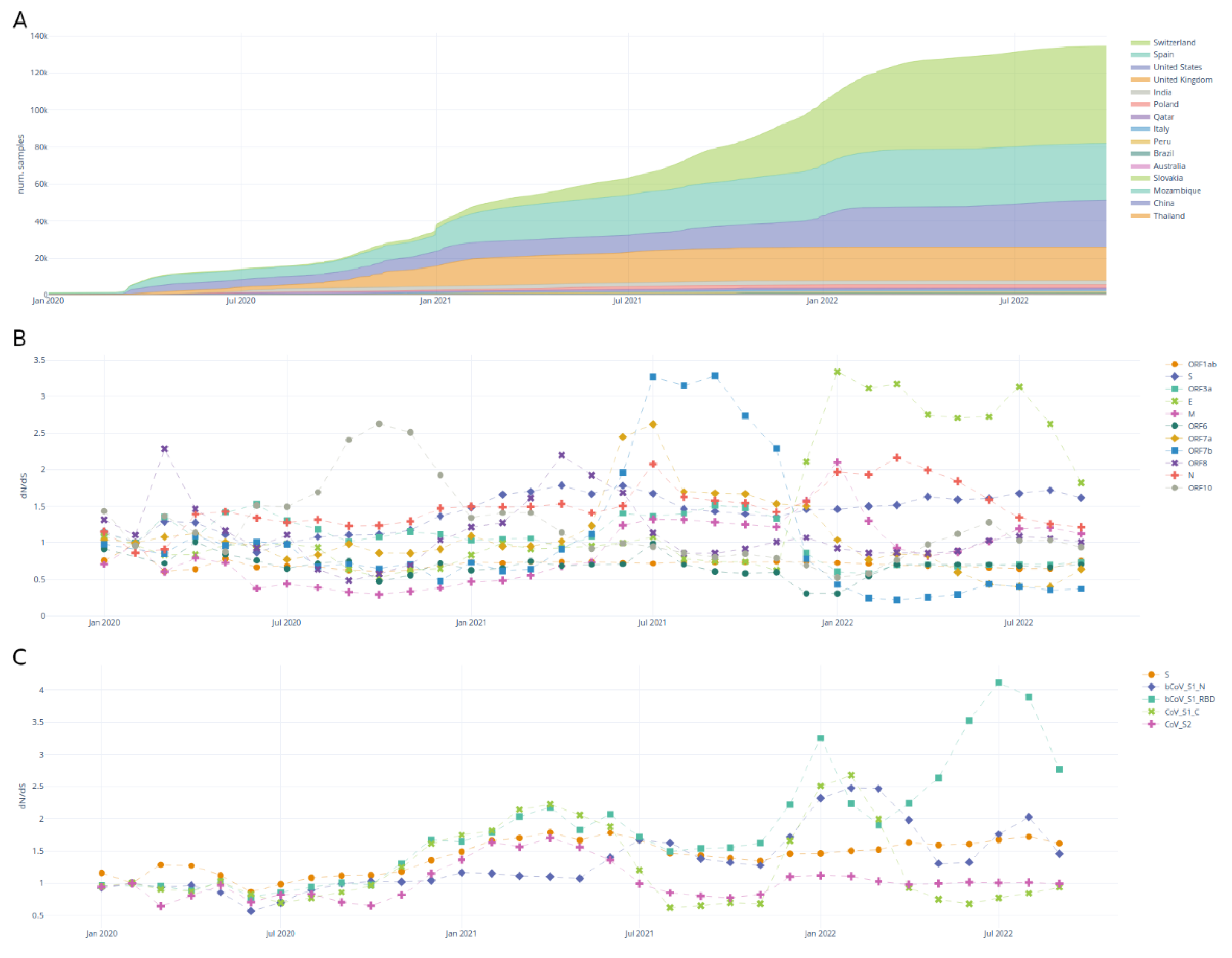
Figure 3.
Interactive plots in the lineages tab for the raw reads dataset. A) Accumulation of samples in each lineage through time; B) Dominant lineages through time. See Figure S3 for a screenshot including the filters.
Figure 3.
Interactive plots in the lineages tab for the raw reads dataset. A) Accumulation of samples in each lineage through time; B) Dominant lineages through time. See Figure S3 for a screenshot including the filters.
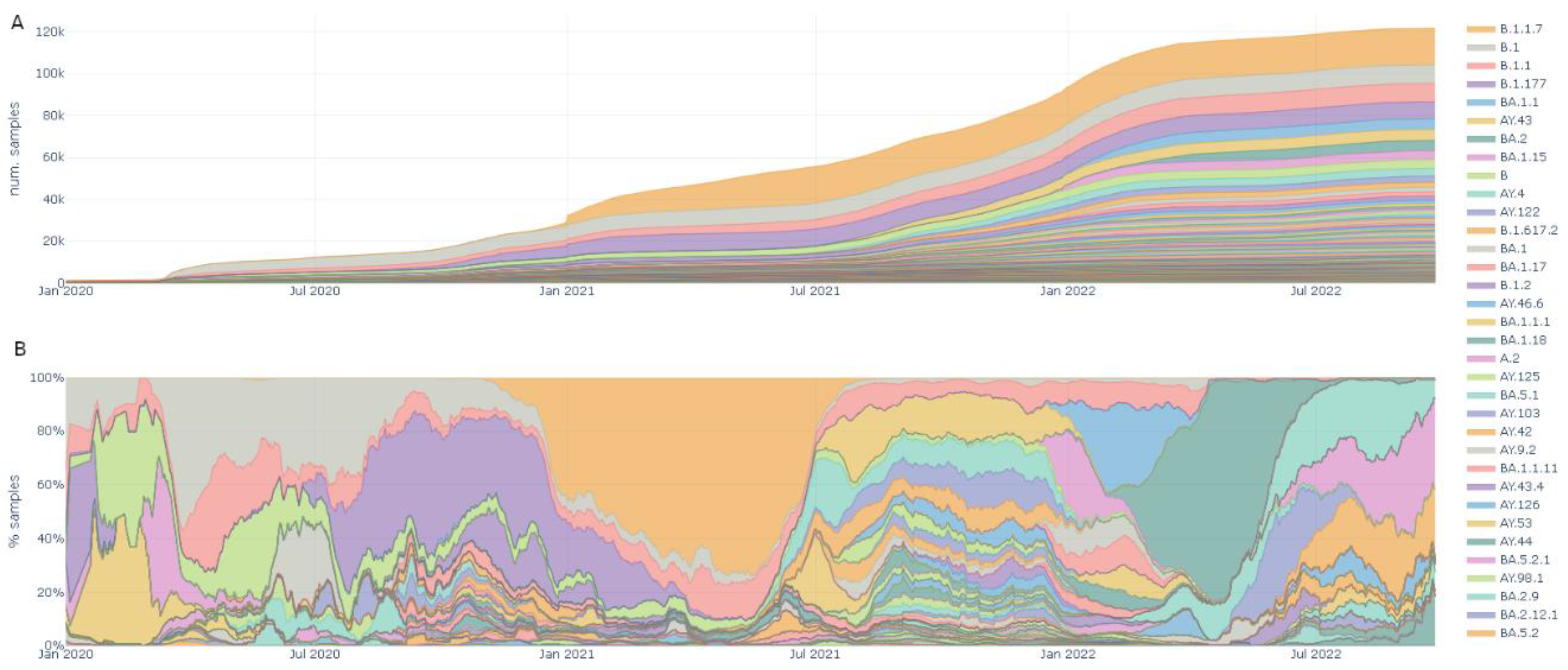
Figure 4.
Interactive plots on mutation statistics tab showing results for raw reads and genomic assemblies datasets. A) ENA distribution of the number of mutations per sample; B) C19DP distribution of the number of mutations per sample; C) ENA frequency of base substitutions, D) C19DP frequency of base substitutions; E) ENA indel length distribution; F) C19DP indel length distribution; G) ENA frequency of mutation effect on the protein; H) C19DP frequency of mutation effect on the protein. See Figure S4 for a screenshot including the filters.
Figure 4.
Interactive plots on mutation statistics tab showing results for raw reads and genomic assemblies datasets. A) ENA distribution of the number of mutations per sample; B) C19DP distribution of the number of mutations per sample; C) ENA frequency of base substitutions, D) C19DP frequency of base substitutions; E) ENA indel length distribution; F) C19DP indel length distribution; G) ENA frequency of mutation effect on the protein; H) C19DP frequency of mutation effect on the protein. See Figure S4 for a screenshot including the filters.
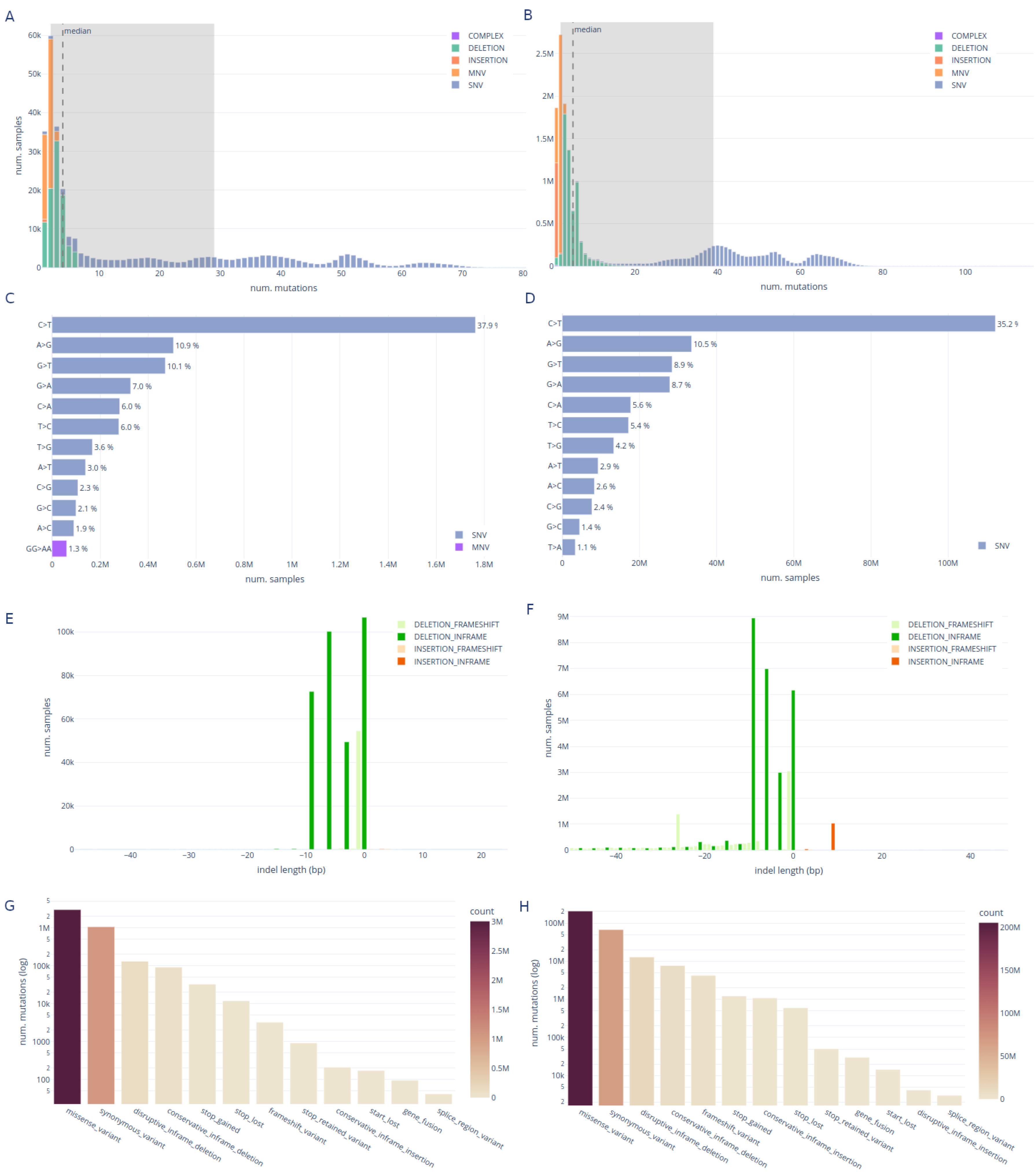
Figure 5.
Gene view for the spike protein on the raw reads dataset. A) Table of the top 30 recurrent mutations with the frequency segregated by month between September 2021 and September 2022; B) gene view showing mutations (synonymous and unique mutations excluded) in the spike protein and their frequencies in the virus population, the ConsHMM conservation tracks in grey and the Pfam protein domains in tones of purple. See Figure S4 for a screenshot including the filters.
Figure 5.
Gene view for the spike protein on the raw reads dataset. A) Table of the top 30 recurrent mutations with the frequency segregated by month between September 2021 and September 2022; B) gene view showing mutations (synonymous and unique mutations excluded) in the spike protein and their frequencies in the virus population, the ConsHMM conservation tracks in grey and the Pfam protein domains in tones of purple. See Figure S4 for a screenshot including the filters.
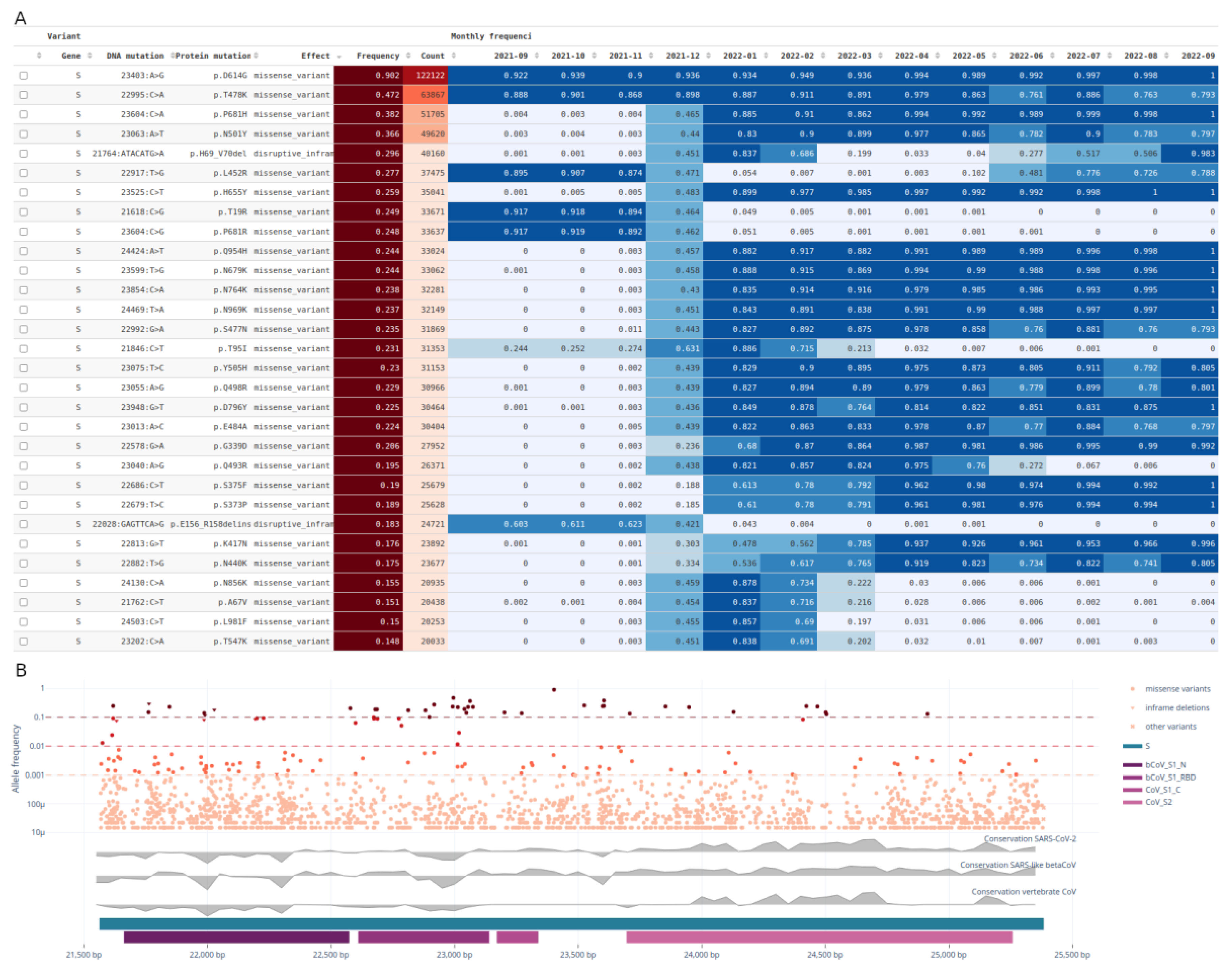
Figure 6.
Distribution of VAF across all mutation calls (4,665,192 with VAF >= 0.8; 222,297 with VAF >= 0.5 and < 0.8; and 26,231,409 with VAF < 0.5) in 135,347 samples. High confidence clonal mutations overlapping the same amino acid are merged into MNVs or complex variants. See Figure S7 for a screenshot of intrahost mutations tab including the filters.
Figure 6.
Distribution of VAF across all mutation calls (4,665,192 with VAF >= 0.8; 222,297 with VAF >= 0.5 and < 0.8; and 26,231,409 with VAF < 0.5) in 135,347 samples. High confidence clonal mutations overlapping the same amino acid are merged into MNVs or complex variants. See Figure S7 for a screenshot of intrahost mutations tab including the filters.
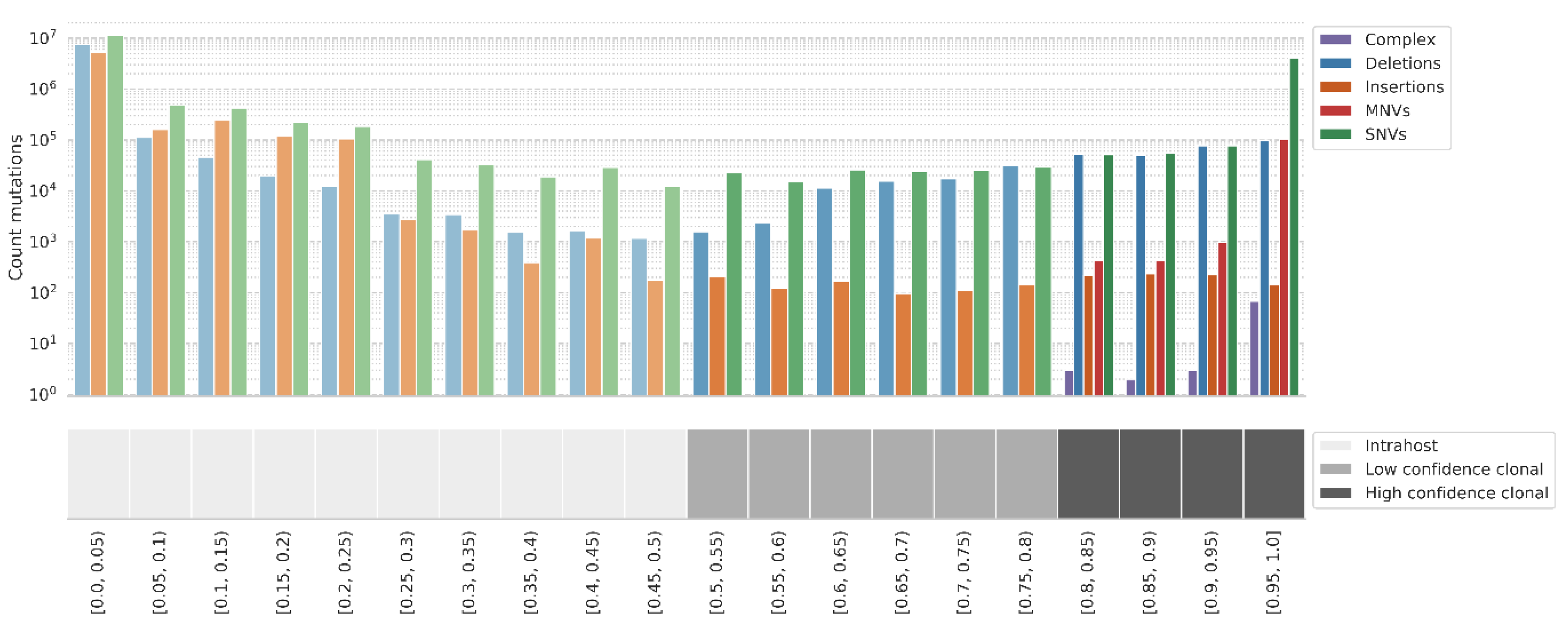

Table 1.
Tools employed in the pipeline specific versions and settings.
| Tool | Purpose | Settings | References | Version | FASTQ | FASTA |
|---|---|---|---|---|---|---|
| fastp | Adapter trimming | [42] | 0.20.1 | X | ||
| BWA mem 2 | Alignment | Default | [43] | 2.2.1 | X | |
| GATK | Variant calling and alignments preprocessing | MQ>=20, BQ>=20, ploidy=1 | [44] | 4.2.0.0 | X | |
| sambamba | Read deduplication | MQ>=20, BQ>=20, ploidy=1 | [45] | 0.8.2 | X | |
| samtools | Coverage analysis | [46] | 1.12 | X | ||
| LoFreq | Variant calling | MQ>=20, BQ>=20 | [47] | 2.1.5 | X | |
| BCFtools | Variant calling, normalization and annotation | MQ>=20, BQ>=20 | [48] | 1.14 | X | X |
| iVar | Variant calling | MQ>=20, BQ>=20 | [49] | 1.3.1 | X | |
| Biopython | Custom variant calling on assemblies sequences based on Needleman-Wunsch global alignment | aligner.mode = 'global'aligner.match = 2aligner.mismatch = -1aligner.open_gap_score = -3aligner.extend_gap_score = -0.1aligner.target_end_gap_score = 0.0aligner.query_end_gap_score = 0.0 | [50] | 1.79 | X | |
| SnpEff | Functional annotations | [51] | 5.0 | X | X | |
| VAFator | Technical annotations | MQ>0, BQ>0 | [14] | 1.2.5 | X | |
| Pangolin | Lineage calling | [52] | 4.1.2 | X | X | |
| ConsHMM | Conservation annotations | [53] | NA | X | X | |
| Pfam | SARS-CoV-2 protein domains | [54] | NA | X | X |
Table 2.
Published and implemented filtering approaches for intrahost variants.
| Approach | Sample filters | Variant filters |
|---|---|---|
| Valesano-like [39] | >= 50,000 mapped reads>= 29,000 bp horizontal coverage | VAF >= 2%, VAF < 50 %DP >= 100>= 10 supporting reads |
| Sapoval-like [34] | >= 20,000 mapped reads | VAF >= 2%, VAF < 50 %DP >= 10Mask extremes of genome + homoplasmic positions [63] |
| Tonkin-Hill-like [33] | Excessive number iSNVs (99.9th percentile)Outlier number of iSNVs with mid-VAFs, between 40% and 80 % | VAF >= 5%, VAF < 50 %DP >= 100>= 5 supporting reads |
| CoVigator approach | >= 50,000 mapped reads>= 29,000 bp horizontal coverageExcessive number iSNVs (99.9th percentile)Outlier number of iSNVs with mid-VAFs, between 40% and 80% | VAF >= 2%, VAF < 50 %DP >= 100>= 10 supporting readsMask extremes of genome + homoplasmic positions [63] from indels <= 10 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



