Submitted:
24 May 2023
Posted:
26 May 2023
You are already at the latest version
Abstract
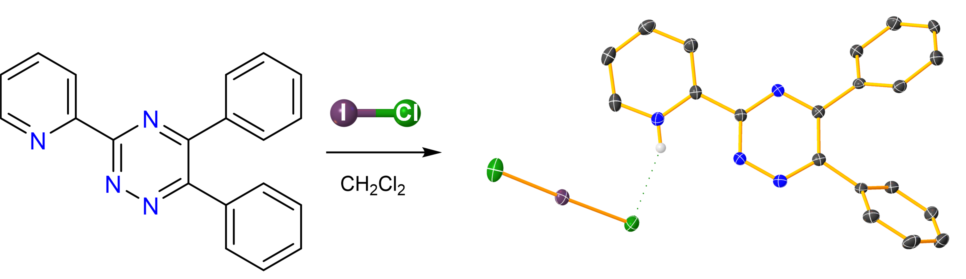
2-(5,6-diphenyl-1,2,4-triazin-3-yl)pyridinium dichloroiodate (I) (1) was synthesized by reacting 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine with ICl in dichloromethane solution. The structural characterization of 1 by SC-XRD analysis was accompanied by elemental analysis, FT-IR, and FT-Raman spectroscopy measurements.
Keywords:
3-(2-pyridyl)-5
; 6-diphenyl-1
; 2
; 4-triazine
; polypyridyl donors
; SC-XRD
; FT-Raman
; DFT
; ICl
1. Introduction
Polypyridine donors react with interhalogens XY and dihalogens X2 (X, Y = Cl, Br, I) to give a variety of products whose nature depend on the nature of the donor, the interhalogen, and the reaction conditions.[1,2,3,4] Among the possible reaction products, Charge-Transfer adducts containing the N···X–Y linear group,[5,6] halonium compounds featuring a N···X+···N moiety5[7,8] or salts where a N-protonated pyridinium cation is counterbalanced by discrete3 or extended[9] (poly)halide anions can be mentioned. FT-Raman spectroscopy can provide useful information on the nature of the resulting products.1,3,9,[10] In simple CT-adducts, the adduct formation results in an elongation of the perturbed X–Y moiety with respect to the free halogen/interhalogen.3,4 When polyhalides are formed, the peculiar stretching vibrations of the interacting synthons, such as neutral (inter)halogens and tri(inter)halides, can be detected in the low-energy region of the FT-Raman spectrum.1–4
3-(2-Pyridyl)-5,6-diphenyl-1,2,4-triazine[11,12] has been often reported as an efficient donor towards a variety of metal ions.[13,14,15,16] This notwithstanding the reactivity of this donor towards halogens or interhalogens has not been described to date. Pursuing our interest towards the reactivity of polypyridyl substrate towards ICl, 1–4 we report here on the synthesis and structural and vibrational characterization of the novel salt 2-(5,6-diphenyl-1,2,4-triazin-3-yl)pyridinium dichloroiodate (I) (1; Scheme 1).
2. Results
The reaction of 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine (L; Scheme 1) with ICl in molar ratios 0.5:1, 1:1, 1:2, and 1:5 in CH2Cl2 yielded products featuring the same microanalytical (elemental analysis, melting point determination) and spectroscopic (FT-IR, FT-Raman) features. Single crystal X-ray diffraction analysis revealed the reaction product to be (HL)[ICl2] (1).
Scheme 1.
Molecular scheme of 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine (L).
Compound 1 crystallizes in the monoclinic space group P21/c with four molecules in the unit cell.
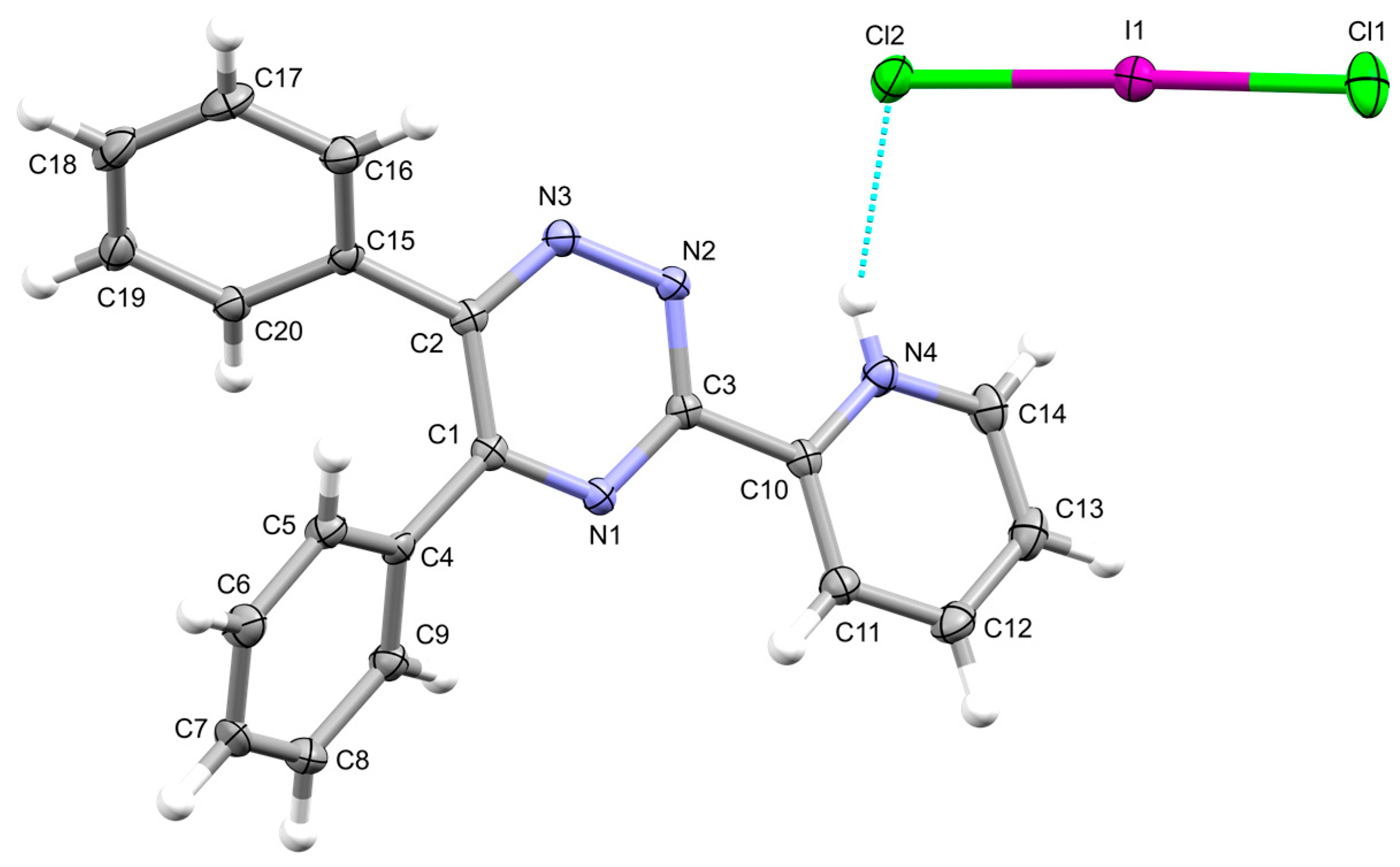
Figure 1.
Ellipsoid plot of compound 1 with the numbering scheme adopted viewed along the b-axis. Displacement ellipsoids are drawn at 50% probability level.
Figure 1.
Ellipsoid plot of compound 1 with the numbering scheme adopted viewed along the b-axis. Displacement ellipsoids are drawn at 50% probability level.
Crystal data for compound 1: C20H15Cl2IN4, (Mr = 509.16 g mol−1) monoclinic, P21/c (No. 14), a = 17.7427(13) Å, b = 7.4326(5) Å, c = 15.5351(12) Å, β = 100.659(7)°, α = γ = 90°, V = 2013.3(3) Å3, T = 120(2) K, Z = 4, Z’ = 1, µ(Mo Kα) = 1.868 mm−1, 19110 reflections measured, 4624 unique (Rint = 0.0302) which were used in all calculations. The final wR2 was 0.0634 (all data) and R1 was 0.0270 [F2 ≥ 2 σ(F2)].
The asymmetric unit of compound 1 consists of a donor molecule protonated at the N4 pyridine nitrogen atom counterbalanced by a classical[17,18] [ICl2]– anion. Protonation of (poly)pyridyl donors as a result of the reaction with dihalogens or interhalogens is not unusual and it has been attributed to solvolysis or reaction with incipient moisture.1
In the HL+ cation, the two phenyl rings of the donor are rotated by 38.3(3) and 47.7(3)° relative to the plane of the triazine, and the pyridine by 12.6(3)°. Quite surprisingly, notwithstanding no crystal structures featuring the HL+ cation have been deposited to date, these torsion angles can be compared to those featured by the two polymorphs (CSD codes HEWLOL and HEWLOL01) reported for L [phenyl torsions: 32.4(1) and 53.6(1)°; pyridine torsion 1.0(1)° for HEWLOL;11 32.2(2), 56.2(2), and 9.1(2)°, respectively, for HEWLOL0112] and the corresponding values optimized at density functional theory (DFT)[19,20] level for L (phenyl torsion, 29.6 and 34.6°; pyridine torsion, 6.1°) and HL+ (phenyl torsion, 30.9 and 38.1°; pyridine torsion, 4.4°), suggesting that electronic factors rather than crystal packing interactions are responsible for the nonplanar conformation of the ligand.
The Cl2 terminal atom of the [ICl2]– anion is involved in a hydrogen bonding (HB) interaction with the pyridinium moiety [interaction a in Figure 2 and Table 1]. The HB interaction results in a remarkable asymmetry of the [ICl2]– anion [Cl1–I1, 2.4856(8); Cl2–I1, 2.6005(6) Å; Cl1–I1–Cl2, 178.27(2)°]. The Cl1 and Cl2 atoms further interact with the H20i and H13ii protons of different symmetry-related adjacent units (interactions b and c in Table 1 and Figure 2; i = ; ii =). These interactions, along a set of weak C–H···N contacts (Table 1) generate the crystal packing, seen along the b-axis in Figure 2.
The FT-Raman spectrum of compound 1 shows the expected band due to the symmetric stretching mode peculiar to the [ICl2]– ion at 278 cm–1 in the FT-Raman spectrum (Figure 3), in excellent agreement with the corresponding values calculated at hybrid-DFT level [ν(σg) = 257, ν(σu) = 238 cm–1]. The πu bending mode is calculated at 108 cm–1 and can be envisaged both in the FT-Raman and in the FT-FIR spectra within the complex envelope of bands at about 100 cm–1 (Figure S3). Notably the wavenumbers of the stretching and bending modes are very close to those previously reported for different compounds featuring (poly)pyridinium cations balanced by [ICl2]– anions, for which the stretching and the bending modes were reportedly in the range 265–285 cm–1 and 85–90 cm–1, respectively.1–4,[21]
3. Materials and Methods
3.1. General
All the reagents and solvents were purchased from Sigma-Aldrich or Lancaster and used without further purification. Elemental analyses were performed with an EA1108 CHNS-O Fisons instrument. Infrared spectra were recorded on a Bruker IFS55 spectrometer at room temperature using a flow of dried air. Far-IR (500–50 cm−1) spectra (resolution 2 cm−1) were recorded as polythene pellets with a Mylar beam-splitter and polythene windows. Middle IR spectra (resolution 2 cm−1) were recorded as KBr pellets, with a KBr beam-splitter and KBr windows. FT-Raman spectra were recorded (resolution of 2 cm−1) on a Bruker RFS100 FT-Raman spectrometer, fitted with an In–Ga–As detector (room temperature) operating with a Nd:YAG laser (excitation wavelength 1064 nm) with a 180° scattering geometry (excitation powder 5 mW). Melting point was determined on a FALC mod. C apparatus. X-ray diffraction data for compound 1 were collected at 120(2) K by means of combined φ and ω scans with a Bruker Nonius KappaCCD area detector situated at the window of a rotating anode (graphite Mo-Kα radiation, λ = 0.71073 Å). The structure was solved with the ShelXT[22] solution program using dual methods and the model was refined with ShelXL[23] 2018/3 using full matrix least squares minimization on F2. Olex2 1.5[24] was used as the graphical interface. DFT18,19 calculations were carried out both on L, HL+, and [ICl2]– at DFT level with the commercial suite of programs Gaussian 16[25] by adopting the mPW1PW hybrid functional.[26] The def2SVP and def2TZVP basis sets[27,28] were adopted for the atomic species of the L donor and the [ICl2]– ion, respectively. Vibrational frequencies were calculated at the optimized geometries. GaussView[29] and PyFreq[30] were used to investigate the Kohn-Sham molecular orbital composition and the vibrational frequencies.
3.2. Synthesis of 2-(5,6-diphenyl-1,2,4-triazin-3-yl)pyridinium dichloroiodate (I) (1)
To 2 mL of a CH2Cl2 solution of 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine (26 mg, 8.4×10–5 mol), a 0.054 M solution of ICl in the same solvent was added dropwise in L:ICl molar ratios ranging between 1:05 and 1:5 (0.8 mL, 4.2×10–5 mol; 1.6 mL, 8.4×10–5 mol; 3.1 mL, 1.7×10–4 mol; 7.8 mL, 4.2×10–4 mol). By slow air evaporation of the resulting mixture a crystalline precipitate was isolated and washed with light petroleum ether and dried under reduced pressure. Elemental analysis calcd. for C20H15N4ICl2: 47.18; H, 2.97; N, 11.00%. Found: C, 47.23; H, 2.47; N, 11.28%. M.p. 218 °C. FT-MIR (KBr pellet, 4000–400 cm–1): 3467m, 2362m, 1624m, 1612s, 1608m, 1538m, 1506m, 1445m, 1410s, 1375m, 1306m, 1227s, 1164s, 1005m, 943m, 783m, 766s, 755s, 736m, 692m, 660s, 539m, 534m, 510m, 443m, 416w cm–1. FT-FIR (polytene pellet, 500–50 cm –1): 487m, 444m, 416m, 402s, 391m, 366m, 332m, 319m, 310m, 284m, 273s, 260s, 234s, 218s, 199s, 191s, 163m, 145m, 135s, 129s, 103w, 95m, 85m, 67s cm–1. FT-Raman (500–50 cm–1, 5 mW, relative intensities in parentheses related to the highest peak taken equal to 10.0): 278 (10.0), 163 (3.3), 131 (3.8), 94 (6.2), 73 (7.1) cm–1.
4. Conclusions
2-(5,6-diphenyl-1,2,4-triazin-3-yl)pyridinium dichloroiodate (I) (1) was synthesized and fully characterized. Further studies are ongoing in our laboratories to investigate the reactivity of 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine towards different dihalogens and interhalogens and its potential role as a building block in the formation of extended supramolecular assemblies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1 and S2: FT-IR; Table S1: Crystal data and refinement parameters; Tables S2, S3: bond lengths and angles; Table S4–S5: DFT-optimized orthogonal Cartesian coordinates.
Author Contributions
Conceptualisation: MA, MCA, VL, AM. Data curation: MCA, EP, AP, MA, AM, JBO, SJC. Investigation: MCA, VL, AM, AP, EP, JBO, SJC, MA. Writing (original draft): MA. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge Fondazione di Sardegna (FdS Progetti Biennali di Ateneo, annualità 2018) for financial support and EPSRC (Engineering and Physical Science Research Council) for continued support of the UK’s National Crystallography Service (NCS), based at the University of Southampton.
Data Availability Statement
Crystallographic data were deposited at CCCD (CIF deposition number 2263970).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rimmer, E.L.; Bailey, R.D.; Pennington, W.T.; Hanks, T.W. The reaction of iodine with 9-methylacridine: Formation of polyiodide salts and a charge-transfer complex. J. Chem. Soc. Perkin Trans. 2 1998, 11, 2557–2562. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Podda, E.; Arca, M.; Pintus, A.; Lippolis, V.; Caltagirone, C.; Bartz, R.H.; Lenardão, E.J.; Perin, G.; Schumacher, R.F.; et al. An unprecedented non-classical polyinterhalogen anion made of [I2Cl]– and I2 at the 2-(p-tolyl)selenopheno[2,3-b]pyridinium cation template. New J. Chem. 2022, 46, 21921–21929. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Hursthouse, M.B.; Huth, S.L.; Isaia, F.; Lippolis, V.; Mancini, A.; Ogilvie, H.R.; Verani, G. Reactions of pyridyl donors with halogens and interhalogens: an X-ray diffraction and FT-Raman investigation. J. Organomet. Chem. 2005, 690, 1923–1934. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Hursthouse, M.B.; Huth, S.L.; Isaia, F.; Lippolis, V.; Mancini, A.; Verani, G. Reactions of Halogens/Interhalogens with Polypyridyl Substrates: The Case of 2,4,6-Tris(2-pyridyl)-1,3,5-triazine. Eur. J. Inorg. Chem. 2008, 3921–3928. [Google Scholar] [CrossRef]
- Kukkonen, E.; Malinen, H.; Haukka, M.; Konu, J. Reactivity of 4-Aminopyridine with Halogens and Interhalogens: Weak Interactions Supported Networks of 4-Aminopyridine and 4-Aminopyridinium. Cryst. Growth Des. 2019, 19, 2434–2445. [Google Scholar] [CrossRef]
- Tuikka, M.; Haukka, M. Crystal structure of the pyridine–diiodine (1/1) adduct. Acta Cryst. 2015, e71, o463. [Google Scholar] [CrossRef]
- Batsanov, A.S.; Lightfoot, A.P.; Twiddle, S.J.R.; Whiting, A. Stereoselective Chloro-Deboronation Reactions Induced by Substituted Pyridine–Iodine Chloride Complexes. Eur. J. Org. Chem. 2005, 1876–1883. [Google Scholar] [CrossRef]
- Batsanov, A.S.; Lightfoot, A.P.; Twiddle, S.J.R.; Whiting, A. Bis(2,6-di methyl pyrid yl)iodonium dibromoiodate. Acta Cryst. 2006, e62, o901–o902. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Hursthouse, M.B.; Huth, S.L.; Isaia, F.; Lippolis, V.; Mancini, A. Square-pyramidal bonding of I2 molecules at the I– nodes of a polyiodide infinite pseudo-cubic 3D-network. CrystEngComm 2004, 6, 540–542. [Google Scholar] [CrossRef]
- Pandeeswaran, M.; Elango, K.P. Electronic, Raman and FT-IR spectral investigations of the charge transfer interactions between ketoconazole and povidone drugs with iodine. Spectrochim. Acta A 2009, 72, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Eltayeb, N.E.; Teoh, S.G.; Ng, S. –L.; Fun, H. –K.; Ibrahim, K. 5,6-Diphenyl-3-(2-pyridyl)-1,2,4-triazine. Acta Crystallogr. 2007, e63, o1041–o1042. [Google Scholar] [CrossRef]
- Eltayeb, N.E.; Teoh, S.G.; Chantrapromma, S.; Fun, H. –K.; Ibrahim, K. A second monoclinic polymorph of 5,6- diphenyl-3-(2-pyridyl)-1,2,4-triazine. Acta Crystallogr., 2007, e63, o3792–o3793. [Google Scholar] [CrossRef]
- Uma, R.; Palaniandavar, M.; Butcher, R.J. Synthesis, structure, spectra and redox interconversions in copper(II) complexes of 5,6-diphenyl-3-(2-pyridyl)-1,2,4-triazine. J. Chem. Soc., Dalton Trans. 1996, 2061–2066. [Google Scholar] [CrossRef]
- Therrien, B.; Säid-Mohamed, C.; Süss-Fink, G. Mononuclear arene ruthenium complexes containing 5,6-diphenyl-3-(pyridine-2-yl)-1,2,4-triazine as chelating ligand: Synthesis and molecular structure. Inorg. Chim. Acta 2008, 361, 2601–2608. [Google Scholar] [CrossRef]
- Anjomshoa, M.; Hadadzadeh, H.; Torkzadeh-Mahani, M.; Fatemi, S.J.; Adeli-Sardou, M.; Amiri Rudbari, H.; Mollica Nardo, V. A mononuclear Cu(II) complex with 5,6-diphenyl-3-(2-pyridyl)-1,2,4-triazine: Synthesis, crystal structure, DNA- and BSA-binding, molecular modeling, and anticancer activity against MCF-7, A-549, and HT-29 cell lines, Eur. J. Med. Chem., 2015, 96, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Pathaw, L.; Khamrang, T.; Kathiravan, A.; Velusamy, M. Synthesis, crystal structure, bovine serum albumin binding studies of 1,2,4-triazine based copper(I) complexes. J. Mol. Struct. 2020, 1207, 127821. [Google Scholar] [CrossRef]
- Sonnenberg, K.; Mann, L.; Redeker, F.A.; Schmidt, B.; Riedel, S. Polyhalogen and Polyinterhalogen Anions from Fluorine to Iodine. Angew. Chem. Int. Ed. Engl 2020, 59, 5464–5493. [Google Scholar] [CrossRef]
- Hallen, H.; Riedel, S. Recent Discoveries of Polyhalogen Anions – from Bromine to Fluorine. Z. Anorg. Allg. Chem. 2014, 640, 1281–1291. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; Wiley-VCH: New York, 2001; ISBN 978-3-527-30372-4. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Bowmaker, G.A.; Tan, K.H.; Taylor, M.J. Vibrational spectra of adducts of iodine monochloride with some pyridine bases. Aust. J. Chem. 1980, 33, 1743–1751. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O. v.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Re-finement and Analysis Program. J Appl Crystallogr 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Gaussian 16 (rev. B01), Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.V.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, J.L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J.A., Jr.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.J.; Heyd, J.J.; Brothers, E.N.; Kudin, K.N.; Staroverov, V.N.; Keith, T.A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.P.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Millam, J.M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B.; Fox, D.J. Gaussian, Inc., Wallingford CT, 2016.
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- GaussView, Version 6, Dennington, Roy; Keith, Todd A.; Millam John M. Semichem Inc., Shawnee Mission, KS, 2016.
- Mancini, A.; Aragoni, M.C.; Bingham, A.L.; Castellano, C.; Coles (née Huth), S.L.; Demartin, F.; Hursthouse, M.B.; Isaia, F.; Lippolis, V.; Maninchedda, G.; et al. Reactivity of Fluoro-Substituted Bis(thiocarbonyl) Donors with Diiodine: An XRD, FT–Raman, and DFT Investigation. Chem. – Asian J. 2013, 8, 3071–3078. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Section of the crystal packing of compound 1 seen along the b-axis. Labelled contacts are described in Table 1.
Figure 2.
Section of the crystal packing of compound 1 seen along the b-axis. Labelled contacts are described in Table 1.
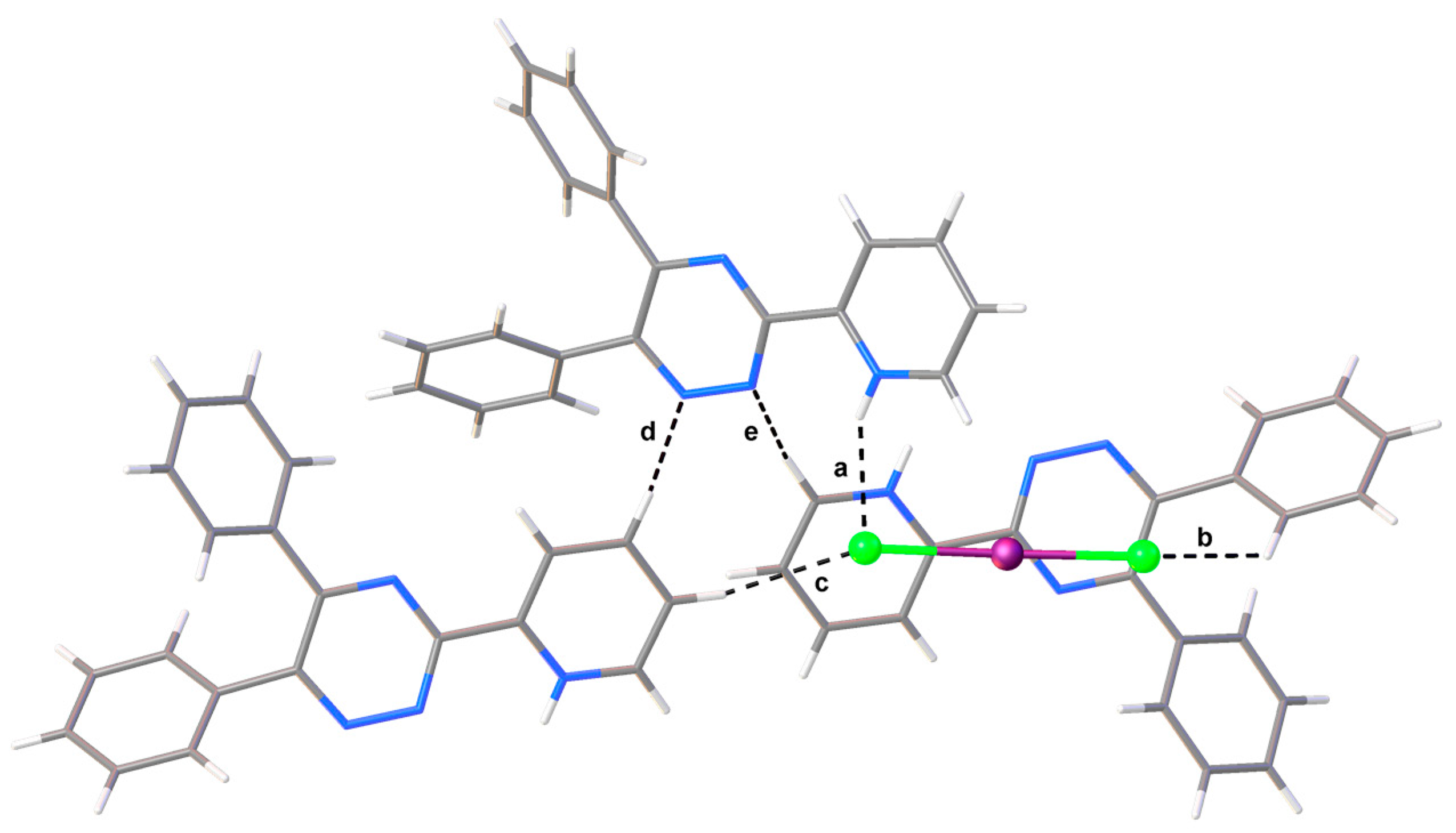
Figure 3.
FT-Raman spectrum recorded for compound 1 in the solid-state.
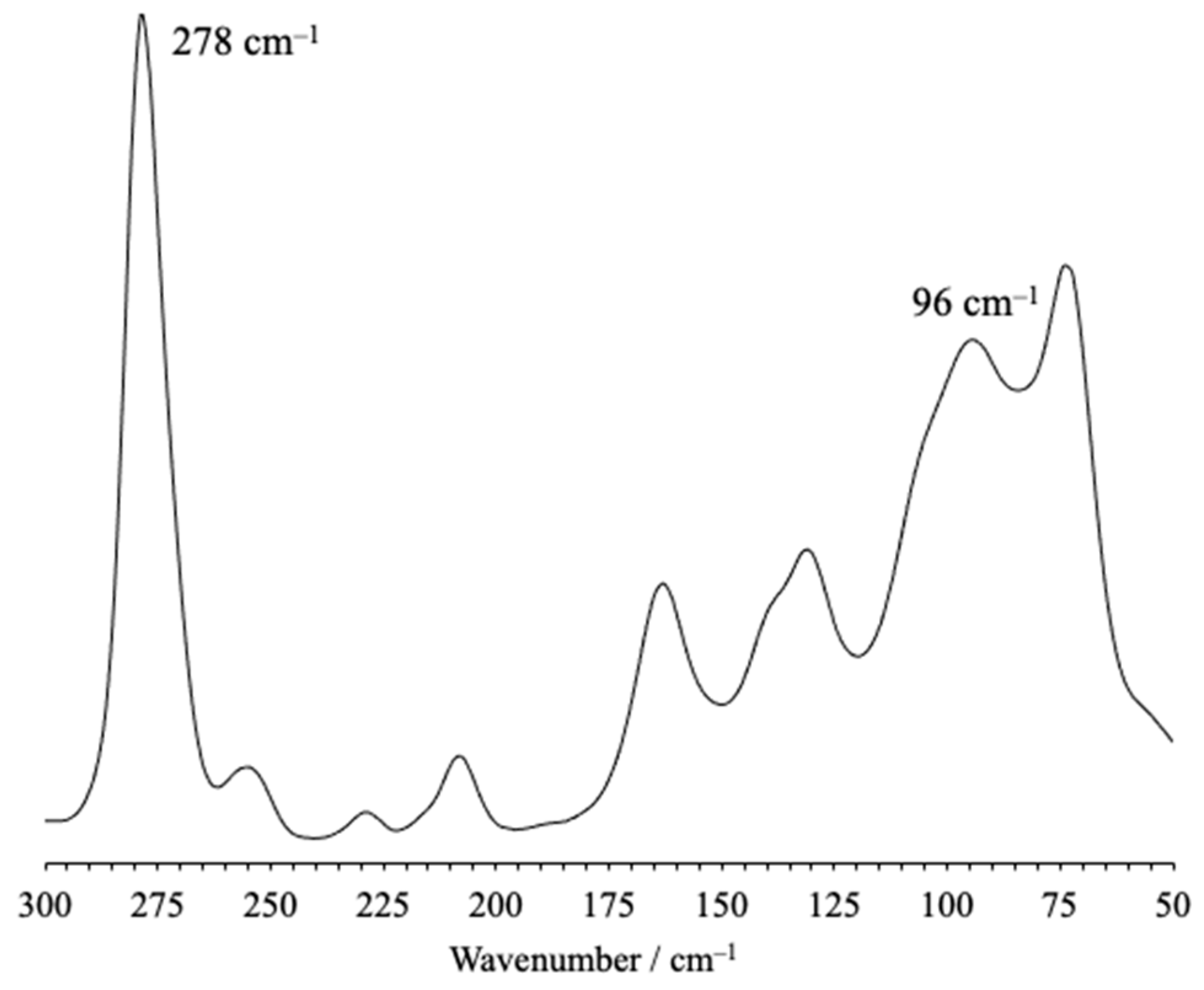

Table 1.
Intermolecular interactions of compound 1.
| Interaction | dD–H (Å) | dH∙∙∙A (Å) | dD∙∙∙A (Å) | αD–H···A (°) | |
| a | N4‒H4∙∙∙Cl2 | 0.85(3) | 2.39(2) | 3.174(2) | 153(2) |
| b | C20i‒H20i∙∙∙Cl1 | 0.95 | 2.88 | 3.541(2) | 128.2 |
| c | C13ii‒H13ii∙∙∙Cl2 | 0.95 | 2.70 | 3.610(3) | 160.9 |
| d | C12ii ‒H12ii ∙∙∙N3 | 0.95 | 2.66 | 3.291(3) | 124.5 |
| e | C14i–H14i···N2 | 0.95 | 2.62 | 3.425(3) | 143.2 |
i = ; ii =.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



