
Submitted:
22 May 2023
Posted:
24 May 2023
You are already at the latest version
Abstract
Chronic kidney disease (CKD) is actually considered a public health priority according to the increasing number of patients affected by this condition: this casuistry is not only related to specific glomerular, tubular or autoimmune diseases or a consequence of acute kidney injury (AKI) episodes leading to organ failure, but it is tied to the progression of life expectancy and the impact of comorbidities such as cardiovascular disease, diabetes and cancer. CKD and its comorbidities promote low grade inflammatory status with an impact on patients’ clinical conditions, reducing their possibility for kidney transplantation or graft survival and their survival when receiving renal replacement therapies such as hemodialysis (HD) or peritoneal dialysis (PD). CKD is often referred to as an ageing accelerator: innate immune system dysregulation, in the uraemic proinflammatory milieu, is involved in this accelerated senescence phenomena and pentraxins, particularly Pentraxin-3 (PTX-3), are of particular interest in the development of kidney disease. A complete understanding of the mechanism of CKD progression, innate immune system involvement and a proper definition of PTX-3 role in kidney disease, could redefine the approach for diagnosis and a more centered patients’ management to slow down CKD progression over time and reduce its clinical and social impact.
Keywords:
Chronic kidney disease
; Pentraxin-3
; Renal Replacement Therapies
; Inflammaging.
; innate immunity
1. Introduction
The long pentraxin (PTX3) is produced by macrophages, neutrophils, myeloid dendritic cells and nonimmune cells responding to IL-1, tumor necrosis factor (TNF)-α and Toll-like receptor agonists [1,2,3]. Its role in the contest of innate immunity has been described in a recent review [4].
According to its molecular structure, PTX3 presents a unique N-terminal domain [5] and a conserved C-terminal pentraxin-like domain that allow octamer formation of the secreted PTX3 monomers through inter-chain disulfide bonds [6]: a more precise definition of its configuration has been only recently achieved thus indicating the possible binding sites and interactions of this multifaceted pattern recognition molecule (PRM) [7].
PTX3 is suddenly released by neutrophil granulocytes at the local sites of activation, whereas its enduring production is regulated via gene expression in innate immune cells and endothelial cells [8].
In healthy subjects, PTX3 plasma levels are around 2 ng/ml [9] and, according to literature, an association between high PTX3 plasma levels, disease severity and mortality has been observed for kidney diseases [10] and in hemodialysis (HD) patients [11,12]. Chronic low grade inflammation induces accelerated senescence in end stage renal disease (ESRD) [13,14] and is now accepted the strict correlation between this condition of “Inflammaging”, the uraemic milieu and the comorbidities developed in chronic kidney disease (CKD) patients with the consequent increased mortality rate for malignancies, CVD or infections [15] thus giving a new declination of the “frailty” concept in kidney disease [16,17]. In this setting, PTX3 role could employ a pivotal significance.
The purpose of this review is to focus on the possible role of PTX3 as a valid biomarker in the diagnosis and progression of the renal damage, in the setting of chronic and acute kidney diseases, according to its physio pathological role. Farther, its role as therapeutic and diagnostic tool and its possible function as inflammaging biomarker.
2. PTX3 in Chronic Kidney Disease (CKD)
2.1. PTX3 and CKD progression
According to its role in kidney diseases, recent literature suggests that PTX3 could have both protective or causal role in pure CKD progression and, consequently, the observed and increased PTX3 circulating title could be merely due to reduced renal clearance and accumulation of its multimeric form of 440 kDa whose passage through the glomerular membrane is impaired [18]. This hypothesis is not only consistent with the gradual increase of PTX3 levels, according to the progressive decline of glomerular filtration, but also with its enhanced synthesis/release upon inflammatory stimulation in peripheral tissues related to CKD progression [13,14,19].
There are limited researches focused on PTX3 in patients in the early stages of CKD (stages 1 to 2). Sjöberg and coll. investigated the associations, in two independent community-based cohorts of elderly men and women, between PTX3 levels, kidney disease measures and CKD incidence: they showed that PTX3 levels were negatively correlated with glomerular filtration rate (GFR), but not with urinary albumin/creatinine ratio (ACR), after adjusting for age, gender, C-reactive protein and prevalent cardiovascular disease in cross-sectional analyses; in longitudinal analyses, PTX3 levels predicted incident CKD after 5 years in both cohorts [18].
Innate immune system activation occurs through the activation of Toll-like receptors (TLRs), and their expression is upregulated in atherosclerosis, particularly on endothelial cells and macrophages [20]. TLR-4 is considered an early promoter of atherosclerotic disease [21]: its recruitment results in the activation of the NF-kB pathway with a consequent proinflammatory cascade [22]. At the same time, it also well known the strong activation of the NF-kB pathway in conditions of chronic oxidative stress and inflammation, such as CKD [23]. Moreover, TLR4 are also involved in the development of endothelial dysfunction in uremia [22], PTX3 levels do correlate with the risk of developing CKD in elderly [24] and in elderly subjects, PTX3/TLR-4 are down regulated after an 8 weeks aerobic training [25].
The link between PTX3, tubulo-interstitial damage and complement activation in kidneys was shown by Nauta and coll. in a model of human kidney 2 proximal renal tubular epithelial cell (HK-2 PTECs) and cultured cells isolated from kidneys not suitable for transplantation or preimplantation biopsies: PTX3 mRNA expression was detected in tubular cells, mesangial cells and renal fibroblast revealing a strong correlation with IL-1 and TNF-Alfa level. Particularly, PTX3 produced by PTECs was able to bind C1q [26]. In a setting of tubulo-interstitial fibrosis, in order to investigate PTX3 role in epithelial to mesenchymal transition (EMT), using HK-2 cells models and in a unilateral ureteral obstruction model in experimental rats, Hung and coll. bring up to date how PTX3 induces cell migration through a JNK-dependent mechanism [27].
The link between PTX3 and tubulo-interstitial damage, always based upon HK-2 PTCs, has been also investigated in order to evaluate the effects of particulate matter with a diameter less than 2.5 μm (PM 2.5). In vitro studies using HK-2 cells distributed as a monolayer and 3D spheroid cultures showed not only progressive EMT in the observed cell line but was also associated with increased IL-6 production and the activation of signal transducer and activator of transcription 3 (STAT3) which is involved in EMT [28]. The same group, in the same in vitro setting with HK-2 cells, determined also, after PM 2.5 exposure, increased PTX3 mRNA expression levels; moreover, in the absence of PTX3, such as in the previous model of IR injury, IL-6 level were higher and the EMT process more pronounced [29].
2.2. PTX3 and tissue remodelling in CKD
Taking into consideration kidney remodeling in CKD, progressive renal failure is a consequence of disequilibrium between the increased synthesis of extracellular matrix components and their decreased degradation, which occurs as a result of imbalanced matrix metalloproteinase (MMP) activities and their tissue inhibitors (TIMP), leading to nephron loss. So increased inflammation and oxidative stress enhance MMP expression and activity, thus increasing the prevalence of numerous diseases including atherosclerosis, nephritis and fibrosis.Galectin-3, instead, is a molecule with diverse biological activities, might play a role within the complex maze of connections that link inflammation, extracellular matrix remodeling and apoptosis, including anti-adhesion, binding advanced Glycation End Products (AGE), regulation of gene expression and controlling cell proliferation and death. In order to investigate this relationship, Miljković and coll. observed PTX3, MMP-9 and TMP-1 levels in CKD and HD patients in comparison with healthy controls: PTX3 was increased in CKD and RRT patients compared to healthy controls and, at the same time, not only MMP-9 and TIMP-1 level were higher in both CKD groups but the protective Galectin-3 levels were reduced in CKD and RRT patients [30].
Inflammation is a well-known risk factor of morbidity and mortality in CKD and is now established that CKD patients have low to moderate levels of inflammatory mediators in their bloodstream due to imbalanced production, increased release, or impaired renal clearance [31]. This inflammatory status has been linked to serological raise of classical inflammatory markers such as CRP, TNF-α, IL-6, and sVCAM-1: higher PTX3 levels have also been found in the presence of inflammation, cardiovascular disease, and protein-energy wasting [10].
Structural changes involving renal parenchyma determine CKD comorbidities such as hypertension: hypertensive patients present PTX3 levels higher when compared to healthy controls [32]. Through in vitro experiment, Carrizo and coll. detected PTX3 mediated changes in the endothelial layer through a P-selectin/matrix metalloproteinase-1 pathway, with consequent reduced nitrox oxide signaling. In a subsequent rodent model, PTX3 administration was associated with the onset of hypertension, an association between higher PTX3 level and hypertension that was finally also assessed in a small cohort of 31 patients compared to 21 normotensive controls [33].
Interestingly, in a PTX3 deficient mice model, kidney injury is enhanced because P-selectin is reported to be downregulated by PTX3 with a consequent inhibitory effect on leukocyte recruitment in a post ischemic model of kidney injury [34]. Consistent with these data, the lack of PTX3 induced an increase in pro-inflammatory markers and a shift to the more oxidative profile of PTX3-deficient ECs supporting the idea of a protective role for PTX3 in endothelial homeostasis [35].
More to date, PTX3 has a role as an early biomarker of structural changes, demonstrating, in Type 2 diabetic hypertensive patients with proteinuria (organ damage), the improvement in endothelial function following the initiation of therapy with either an angiotensin II receptor blocker (valsartan), a calcium channel blocker (amlodipine), or a combination of both, binding to the normalization of sTWEAK and PTX3. All treatment methods successfully raised flow-mediated dilation (FMD) and decreased proteinuria, with the combined therapy showing the greatest effect. These enhancements were accompanied by a marked decrease in PTX3 [36].
A recent study found that circulating PTX3 levels can be used as a marker of the renal protective effects of atorvastatin. In this study, 117 patients with serum creatinine levels greater than 120 mmol/L were randomly assigned to receive either atorvastatin (10 mg/day, n=56) or placebo (n=61) and were followed for 2.5 years. Results showed that in patients with elevated PTX3 levels at baseline, the decline in GFR during the trial was significantly less in those treated with atorvastatin compared to those in the placebo group [37].
2.3. PTX3, cardiovascular disease and CKD
PTX3 is involved in the formation and progression of atherosclerotic lesions [38] and its levels are increased even in subclinical atherosclerosis [39]. Studies have demonstrated that PTX3 knock-out mice have more severe atherosclerosis compared to wild type, suggesting that PTX3 deficiency increases vascular inflammation [40]. This data is consistent with data from a Japanese clinical study found that the levels of circulating PTX3 were higher in endurance-trained healthy young men compared to sedentary control subjects, highlighting PTX3’s cardio-protective role [41]. Numerous clinical studies have demonstrated that elevated PTX3 levels can independently predict various cardiovascular outcomes, regardless of CRP levels [42]. The results of research on atherosclerosis suggest that high PTX3 levels are associated with a higher risk of acute myocardial infarction [43], increased mortality in patients with heart failure [44], unstable angina pectoris [45] and myocardial infarction [46]. However, the question of whether PTX3 causes or reflects inflammation within blood vessels remains unresolved.
The connection between atherosclerosis, higher risk of cardiovascular disease (CVD) and endothelial dysfunction in CKD patients is challenging to establish due to the presence in this population of multiple cardiovascular risk factors, such as increased oxidative stress, micro-inflammation, lack of physical activity, anaemia, vascular calcifications, endothelial dysfunction, and reduced availability of nitric oxide. Recently, various new biomarkers that can indicate the risk of cardiovascular disease associated with atherosclerosis have been discovered in the general population. Blanco-Colio et al. proposed soluble TNF-like weak inducer of apoptosis (sTWEAK) as a possible novel biomarker of atherosclerosis.
sTWEAK is present in lower amounts in carotid plaque samples of atherosclerotic patients compared to asymptomatic individuals, and its levels are inversely related to carotid intima-media thickness [47]. Recently the decline in kidney function has been linked to a progressive reduction of sTWEAK plasma levels suggesting new connections between sTWEAK and endothelial dysfunction in patients with CKD [48].
More to date, in CKD patient, a decrease in sTWEAK levels is linked to an elevated risk for cardiovascular events and increased circulating levels of PTX3 [49] thus suggesting that in CKD patients, sTWEAK and PTX3 may either act as a failed compensatory mechanism or as a mechanism amplifying inflammation. These findings are in line with previous studies [10,47] showing how both these markers independently predict cardiovascular outcomes and CKD mortality.
All these data, taken together, suggest a possible dual role for PTX3 as enhancer/suppressor of chronic kidney injury and the potential role to identify individuals at risk of cardiovascular disease and progression to ESRD in the general population [50] due the strong association between these clinical conditions and mortality in order to slow, as soon as possible, CKD progression.
2.4. PTX3 and Glomerulonephritis
Glomerulonephritis are one of the major causes leading to CKD and a recent review of literature emphasizes the pivotal role of their immunopathogenesis for a more precise diagnosis and immunotherapy [51]. PTX3 is the only member of the family of long pentraxins whose expression was detected in renal tissues. Expression and production of PTX3 is shown for primary mesangial cells, primary tubular epithelial cells and renal fibroblasts and it is enhanced by IL-1 or TNF-α [52].
PTX3 is strongly expressed in renal biopsies of patients with IgA nephropathy, membranous glomerulonephritis, focal segmental glomerular sclerosis, type I membranoproliferative, diffuse proliferative lupus glomerulonephritis, and it’s suggested a role as modulator of glomerular inflammation and a possible biomarker for these diseases [53].
Elevated levels of PTX3 have been also observed in various autoimmune disorders, including rheumatoid arthritis, psoriasis, scleroderma, Takayasu disease, as well as small-vessel vasculitis seen in Churg-Strauss syndrome, Wegener’s granulomatosis, and microscopic polyangiitis [54,55,56,57].
PTX3 has also been investigated in the pathogenesis of vasculitis. The origin of kidney injury in ANCA-associated glomerulonephritis remains unclear, but neutrophil extracellular traps (NETs) have been proposed as the primary cause of ANCA-related diseases [58]. PTX3 is linked to NETs and is considered an inflammatory marker [59]. Moreover, in ANCA-associated glomerulonephritis with normal CRP levels and severe pathological injury linked to MPO-ANCA, before steroid treatment, elevated PTX3 levels were found in patient’s plasma and in kidney specimens but, after treatment, PTX3 plasma levels dropped thus suggesting how PTX3 could be a significant indicator of activity in ANCA-associated glomerulonephritis [60].
Recent studies have investigated PTX3 role in Diabetic Nephropathy (DN): in renal tissue of diabetic patients, Chen et al. showed that PTX3 levels are closely related to renal injury thus suggesting its ability to quantify the extent of renal injury as early indicator of renal tubular injury but also injury progression on time [61].
The relationship between PTX3 and Systemic Lupus Erythematosus (SLE) has been extensively investigated. PTX3 is actually considered a marker of vascular injury in SLE and its levels could be used as indicator of the activation/dysfunction of vascular endothelium [62].
Circulating and local PTX3 levels are significantly increased in patients with active lupus nephritis (LN), compared to healthy controls, but not in SLE without renal involvement even with comparable Systemic Lupus Erythematosus Disease Activity Index (SLEDAI). More to date, significant correlations appear between serum PTX3 levels and some clinical active LN indices, such as hematuria, non-infectious leucocyturia, serum creatinine value, serum C3 level, and SLEDAI scores. According to renal histopathology, the highest serum PTX3 levels are detected in patients with class IV lupus nephritis and a positive correlation was found between serum PTX3 levels and total activity indices, endocapillary hypercellularity, and leukocyte infiltration. Also immunohistochemistry evaluation showed PTX3 localization in the tubulo-interstitial areas of lupus nephritis patients, but not in the glomeruli. Importantly, serum PTX3 levels decreased significantly during the remission phase. These results suggest that PTX3 expression in the kidneys is linked to clinic histopathological injury indices, particularly tubulo-interstitial features. [63].
As a form of autoimmune disease, LN is a source of autoantigens [64,65], and current evidences supports PTX3 involvement in SLE-driven renal inflammation. PTX3 deposits have been characterized in renal samples and the extent of deposition do correlate with proteinuria and renal fibrosis [63,64,65,66].
Interestingly, when lupus-prone mice undergo immunization with PTX3, they produce anti-PTX3 antibodies with a consequent milder form of lupus-like nephritises thus providing evidence for the ability of anti-PTX3 antibodies to modulate the immune system. This experimental model is consistent with clinical data showing high levels and intensities of anti-PTX3 antibodies in individuals with SLE but not in LN [66,67], thus suggesting a protective role in LN [68].
Taking into account the well-known role of B cells in the development of LN [69], it is tempting to speculate that PTX3-specific B cells (PTX3+ B) may have a regulatory function in LN. Gatto and coll. showed that PTX3+ B cells were present in SLE patients and healthy donors but virtually absent in LN patients. This indicates that these B cells, which are specific to the autoantigen, may represent a layer of regulation that is lost in LN patients [70]. Recent experimental data suggest how immunization with PTX3 prevent the progression from preclinical to the clinical stage of LN thus and preventing renal damage. These data highlight the role of PTX3 as an initiating autoantigen in LN and show that an acquired response against PTX3 hinders ultrastructural lesions which are known to herald an overt LN [71].
2.5. PTX3 in Thrombotic Microangiopathies (TMA)
Thrombotic Microangiopathies (TMA) are a group of diseases characterized by thrombosis in the capillaries and arterioles of numerous vascular beds [72]. TMA causes are different but all lead to endothelial injury and excessive platelet activation/aggregation with consequent progressive kidney damage. The TMAs include three nosological entities: typical haemolytic uremic syndrome (HUS), atypical HUS (aHUS), and thrombotic thrombocytopenic purpura (TTP). These are then added to secondary TMAs that arise in the context of other pathological processes. In aHUS, an abnormality of the complement alternative pathway (60%) is usually present, leading to uncontrolled activation of the AP [73].
PTX3 is implicated in the pathogenesis of aHUS. It is known that all pentraxins are involved in complement activation through their opsonizing capacity [74]: acting as antibodies, they opsonize and neutralize pathogens in an antigen-unspecific manner [75]. Moreover, PTX3 interacts with C1q inducing or inhibiting the classical complement pathway activation [76]. In a calcium-independent manner, PTX3 binds complement activator proteins (Clq, L-ficolin, M-ficolin, mannan-binding lectin) or inhibitory proteins (complement factor H, C4b-binding protein) [77,78] and modulate the classical and lectin complement pathways [79].
Trojnar and coll. looked at PTX3 role during the acute phase of TMA: they observed that not only PTX3 levels are elevated during the acute phase of STEC-HUS, aHUS, and secondary TMA, but also reduced in clinical setting of remission from aHUS [80].
PTX3 modulates complement activation also through the interaction with Factor H (FH), thus reducing alternative pathway activation and preventing excessive complement activation and tissue damage. At the same time, FH mutations and autoantibodies in aHUS patients impair PTX3’s recognition of FH, increasing local complement-mediated inflammation [81]. Finally, PTX3 also recruits C4-binding protein (C4BP) to apoptotic cells/ECM to increase C4b inactivation and reduce C5b-9 deposition. PTX3 thus recruits FH and C4BP to limit excessive complement activation on apoptotic cells [77,82].
Csincsi and coll. identified PTX3 as a novel ligand of FH related protein 5 (CFHR5) by observing the interaction on the extracellular matrix exposed during kidney endothelial injury. It was detected that CFHR5, whose binding to PTX3 is of ∼2-fold higher affinity compared with that of FH, dose-dependently inhibited FH binding to PTX3 and increased C1q binding. It also enables the formation of the alternative pathway C3 convertase and supports complement activation. By interfering with FH’s complement-inhibiting function, enhancing C1q binding, and activating complement, CFHR5 can locally boost complement activation, contributing to glomerular disease [83].
In CKD patients, data from literature confirm how PTX3 levels increase on time according to the progression of kidney failure. However, PTX3 multifaceted interactions, even according to specific histological signs of kidney damage in different clinical setting, are still debated and yet to be completely understood. For these reasons, aside its physio pathological role, even if CKD patients could present different starting clinical features, PTX3 represents their common turning point: it follows the course of their underlying pathology, its relapses, the effects of the therapies performed. And as a biomarker, could be feasible and useful for physicians for the long term follow-up of this particular setting of patients and guise therapeutical approaches. Moreover, being a systemic disease and particularly in the setting of inflammaging, aside its role of biomarker for kidney remodelling, glomerular damage or cardiovascular disease progression, it does encompass the progressive loss of response of kidney patients’ biological system and the progression toward the uremic milieu and the consequent need for starting RRT [Figure 1].
3. PTX3 in Acute Kidney Injury (AKI)
Acute ischemia-reperfusion (I/R) injury is a condition that occurs when blood flow to an organ is temporarily restricted, followed by the return of blood flow and re-oxygenation. The transient lack of oxygen and nutrients during ischemia with the following restored basal condition, results in oxidative damage and a significant inflammatory response causing an exacerbation of tissue damage. This kind of injury is typical of acute kidney injury (AKI) [84].
Several studies investigated the immunological and molecular mechanisms involved in ischemia-reperfusion injury and found that the TLR4 signalling pathway plays a dominant role in mediating kidney injury [85,86]. During ischemic AKI, TLR4, a receptor for Damage Associated Molecular Pattern Molecules (DAMPs) and also the lipopolysaccharide (LPS) receptor, is necessary for early activation of the endothelium, resulting in maximum inflammation and injury. In fact, TLR4 is the main receptor for the maladaptive high-mobility group protein B1 (HMGB1) that is released by damaged renal cells. This binding leads to an increase in the expression of pro-inflammatory adhesion molecules. Without the presence of endothelial TLR4, these adhesion molecules are not expressed, leading to decreased inflammation and improvement in injury outcomes. As shown in the murine model of I/R injury of Chen and coll. [87] the kidney-intrinsic TLR4 signalling pathway may be an important positive regulator of PTX3. They demonstrated that PTX3 plays a crucial role in the development of ischemic AKI: after 4 hours of reperfusion, PTX3 was upregulated in plasma and kidneys, mainly in endothelial cells of wild-type mice because of to the effect of HMGB1 on TLR4. The expression of PTX3 was further enhanced by the interaction between reactive oxygen species (ROS) and TLR4 ligands such as HMGB1, LPS or stress fibronectin. PTX3 knockout mice, instead, showed reduced expression of cell adhesion molecules in the endothelium after 4 hours of reperfusion, potentially contributing to a reduction in early maladaptive inflammation in the kidneys of these mice. However, at 24 hours of reperfusion, the expression of endothelial adhesion molecules increased in PTX3 knockout mice as regulatory and reparative leukocytes entered the kidney.
Complement has a pivotal role in kidney I/R injury. According to a swine model, Divella and coll. [88] investigated the role of PTX3 as possible modulator of complement activation and showed that I/R injury induces early PTX3 deposits in peritubular and glomerular capillary. In normal tissue, PTX3 deposits are very limited. However, following reperfusion, there’s a widespread deposition of PTX3 in the tubulo-interstitial area, at peritubular capillaries and at glomerular levels already at 15 minutes after reperfusion. Even one hour after reperfusion, PTX3 deposits are still detectable at the level of peritubular capillaries. By using confocal laser scanning microscopy, they discovered that PTX3 deposits co-localized with CD31+ endothelial cells. Furthermore, PTX3 was observed to be associated with infiltrating macrophages (CD163), dendritic cells (SWC3a), and myofibroblasts (FSP1). Notably, there was a significant PTX3-mediated activation of the classical (C1q-mediated) and lectin (MBL-mediated) pathways of complement. PTX3 deposits were also co-localized with the activation of the terminal complement complex (C5b-9) on endothelial cells, indicating that PTX3-mediated complement activation mainly occurred at the renal vascular level.
These findings suggest that PTX3 could potentially serve as a therapeutic target to prevent complement-induced I/R injury. In a previous observation, the same workgroup, showed how treatment with recombinant C1-INH (rhC1INH), a potent inhibitor of proteases of the classical and lectin complement pathways, C1r, C1s and MASP2, significantly reduces complement activation leading to a decreased recruitment of inflammatory cells and tubulo-interstitial damage [89]. This finding is consistent with the experience of Delpech and coll., who a few years later, in a swine model of kidney auto transplantation, investigated the benefits of pre-reperfusion treatment with rhC1INH and observed a marked reduction in the deposition of C1q, MASP, and C4d in both glomerular and tubular structures 30 minutes after reperfusion, indicating that C1-INH plays a key role in regulating both classical and lectin pathways [90].
Organ fibrosis caused by AKI is recognized as an important contributor to the development of CKD [91]. In a murine model of AKI, Xiao and coll. [92] showed faster kidney function recovery after PTX3 infusion. Fibrosis was linked to heightened expression of IL-6 and significant activation of Stat3. In vitro, the administration of IL-6 led to increased expression of collagen I and activation of Stat3 in renal epithelial cells undergoing hypoxia-reoxygenation, which was suppressed by PTX3. Furthermore, the reduction in serum creatinine levels and the decreased expression of collagen and smooth muscle actin brought about by PTX3 treatment were reversed by additional administration of IL-6. The decrease in p-Stat3 expression caused by PTX3 was also reversed by the additional IL-6 treatment. These findings indicate that PTX3 counteracts interstitial fibrosis caused by acute kidney injury by suppressing the IL-6/Stat3 pathway.
Even if all data up to here suggest a correlation between AKI injury and PTX3 levels, literature of opposite direction hints a protective role for PTX3.
Always according to a murine model, Lech and coll. found that the absence of PTX3 led to a severe form of post-ischemic AKI, as indicated by widespread tubular necrosis, high levels of TNF and IL-6, and a significant increase in the infiltration of neutrophils and macrophages 24 hours later. This resulted in tubular atrophy, interstitial fibrosis, and shrinkage of the kidney 10 weeks later. In vivo imaging showed increased leukocyte adhesion and migration in post-ischemic micro vessels of mice with a deficiency in PTX3. Administering recombinant PTX3 up to 6 hours after reperfusion prevented kidney inflammation and injury. This suggests that PTX3 release from a group of intra-renal mononuclear phagocytes or late PTX3 treatment can reduce post-ischemic renal inflammation. On the other hand, mutations causing loss of function in PTX3 increase the risk of post-ischemic acute kidney injury and subsequent CKD [93].
Reactive oxygen species (ROS) are deeply involved in AKI progression [94]. In the vitro experience of Lee et coll. exogenous recombinant PTX3 protects kidney cells during ischemia and proinflammatory acute kidney injury, decreasing the activity of caspase-3 and PARP-1, thus preserving the stability of the mitochondrial membrane potential and inhibiting apoptosis [95].
Considering the heterogeneous set of data previously described, PTX3 determinations could helpful not only in determining the beginning of an acute process but, through its serial measurement, particularly in cases of AKI without other comorbidities, be an indicator of “restitutio ad integrum” of the organ or a clinical indicator for effectiveness of performed therapies. Otherwise, as indicator of residual kidney damage, it could shed a light on hindered processes of transition from acute to chronic damage.
4. PTX3 in Kidney Transplantation (KTx)
PTX3 levels in kidney transplant recipients (KTR) are higher when compared to healthy subjects [96] but, patients previously receiving HD and then successfully transplanted, after a one-year follow-up, show a significant reduction of circulating PTX3 levels thus possibly confirming a pivotal role of adequate renal function and reduced inflammatory status [97] in PTX3 removal/production.
Opposite data were collected by Garsu and coll. who evaluated PTX3 role as inflammatory marker in KTR before and after kidney transplant, according to the known changes in inflammatory status after kidney transplant. In order to reduce confounding factors such as induction therapies, HLA mismatch, prolonged ischemia time or previous and prolonged different renal replacement therapies (RRT), their observation focused on a specific pool of 40 patients waiting for kidney transplant from living donation. According to their experience, after kidney transplantation, PTX3 is not a useful biomarker of inflammation when compared to high sensitivity C-reactive protein which showed, instead, a stronger correlation with inflammation. This evidence may be related to immunosuppressive drugs which downregulate IL-6 expression and consequently PTX3 production [98].
Mononuclear cells and dendritic cell are a major source of PTX3 production. Therefore, when corticosteroid therapy is started and kidney transplant is performed, PTX3 production may decrease because of lymphocyte depletion and may increase because of endothelial damage: this hypothesis is consistent with upregulation of PTX3 expression in renal parenchyma after acute kidney rejection episodes [99].
According to histological evaluation of perioperative renal biopsies, acute rejection and follow-up protocol biopsies, not only PTX3 levels are higher in the early phases of AR but significantly decrease after its treatment. Moreover, according to PTX3 deposition along tubulo-interstitial area, peritubular capillaries and glomeruli, it positively correlates with the degree of allograft dysfunction according to 2009 Banff classification thus suggesting a role as biomarker of severity in histological patterns of acute renal allograft rejection [99].
Nephrotoxic effect of immunosuppressors such as calcineurin inhibitors (CNIs) are well known mediators of chronic allograft dysfunction through a sustained, low grade, ischemic damage to epithelial cells [100]. Minimization of these drugs or their avoidance, is considered a first choice for the successful management of transplant recipient [101] with a possible impact on fatal cardiovascular events [102]. In 105 kidney transplant recipients under CNI/mTOR immunosuppression, Infante and coll. found both reduced progression of chronic cardiovascular disease and PTX3 expression [103].
PTX3 is also studied as the main factor, with Fetuin-A, for vascular calcification and inflammation in hemodialysis (HD) and renal transplant (RT) patients: they are associated with increased risk for future cardiovascular morbidity in KTR [104].
Finally, baseline PTX3 levels is an independent predictor of suppression of viral load below level of detection (LOD) at day 21 in solid organ transplant recipients treated for cytomegalovirus (CMV) disease [105].
Therefore, serial PTX3 measurements could impact on kidney transplant recipients’ management because it represents an interesting clinical tool: even if it seems not to correlate with patients’ inflammatory status, in the first phases after kidney transplant it does correlate with acute renal allograft rejection and infectious episodes thus providing a tool for more precise immunosuppressive strategies, diagnostic procedures and histological evaluations. During in KTR, it’s established that PTX3 levels should “physiologically” decrease: excluded histological pattern of graft failure, according to PTX3 association with previous clinically impacting features of patient comorbidities’, it could suggest a worsening of these condition with a possible impact on diagnostic procedures. Finally, from a more speculative point of view, serial PTX3 measurement associated with protocol histological evaluations, could add more information about molecular mechanism of accelerated senescence in KTR.
5. PTX3 in peritoneal Dialysis (PD)
Few data are also present in literature about PTX3 role in Peritoneal Dialysis (PD). Continuous exposure to peritoneal dialysis fluids (PDFs) is associated with pathological responses to a persistent micro-inflammation, which leads to ultrafiltration failure on time [106].
During a single PD exchanges, PTX3 levels tend to increase, because of the contact between peritoneal epithelium and glucose-based PD solutions [107]. This observation was confirmed by a 2016 Japanese study. Ishimatsu and coll. investigated the in vivo PTX3 expression in the peritoneal membrane of a rat model: treatment performed was continuous peritoneal dialysis (PD), with conventional PDF containing 3.86% glucose for 8 weeks. PTX3 was detected in peritoneal mesothelial cells, macrophages and fibroblasts in the thickened sub mesothelial area and glucose was found to induce PTX3 protein expression in cultured rat peritoneal mesothelial cells (RPMCs) as well as macrophage-like cells and fibroblasts. According to this, glucose may be a major driver for PDF-induced local micro-inflammation in the peritoneum [108].
As seen before, also in PD patients PTX3 could exert a role as biomarker, particularly specific for peritoneal inflammation and progressive fibrosis leading to functional failure of the replacement treatment. In a 50 patients’ cohort of PD patients, Kanda and colleagues observed PTX3 expression in peritoneal endothelial cells, fibroblasts, mesothelial cells before and after a single PD session. PTX3 levels in peritoneal effluent (PE), at the cessation of PD, was significantly higher than that at the initiation of PD. Of particular interest, effluent PTX3 levels in patients with a history of peritonitis or a PD duration of more than 8 years were significantly higher than those in patients without peritonitis or patients with a PD duration of <8 years thus suggesting an association with chronic inflammation. More to date, PTX3 levels do correlate with matrix metalloproteinase-2 (MMP-2) and interleukin-6 (IL-6) levels in PE, as well as the thickness of the sub mesothelial compact (SMC) zone, small vessel vasculopathy and the loss of mesothelial cells [109].
According to these data, PTX3 could effectively flank clinical evaluations routinely performed in order to define a more comprehensive evaluation of patients conditions and forewarn frequent complications such as peritonitis or peritoneal sclerosis progression.
6. PTX3 in Hemodialysis (HD)
PTX3 pre-dialysis levels above 2.3 ng/ml are associated with arterial stiffness, a mortality predictor in hemodialysis patients [110] but HD patients show higher PTX levels (about 6 ng/ml) with even more increased PTX3 levels in HD patients with severe cardiovascular disease [97,111,112]. A 3 years, prospective, observational cohort study enrolled 135 HD patients, looking for a correlation between plasma level of PTX3 and arteriovenous fistula (AVF) failure: higher PTX3 levels were associated with higher risks of AVF functional patency loss in chronic HD patients [113]. These data strengthen PTX3 role as a serological biomarker independently associated with CVD [114] even in the absence of a precise molecular mechanism [38].
More than in CKD, in fact, PTX3 levels in HD patients are increased because of the HD patients’ uraemic milieu and frailty status: CVD is a highly common complication and the first cause of death in patients receiving HD and their mortality rate, due to CVD, is 20 times higher compared to general population, considering not only ventricular hypertrophy but also non-traditional risk factors, such as chronic volume overload, anemia, inflammation, oxidative stress, chronic kidney disease–mineral bone disorder or protein energy wasting conditions [115].
According to its possible role as biomarker, PTX3 is pointed out as the most sensitive inflammatory predictor of all-cause mortality after adjustment for the main confounding factors in HD patients [116] even according to nutritional, inflammatory and oxidative status [117].
PTX3 levels do also correlate with obesity, which is a common feature in HD patients. Data from literature point out a survival benefit for HD patients with high BMI, particularly those older than 65 years [118] suggesting the hypothesis of the “Obesity paradox” in CKD [119]. Several works show a unique and inverse association between BMI and PTX3 and not for classical inflammatory biomarkers: non-overweight patients, and particularly PEW (protein energy wasting) patients, present not only higher IL-6 and TNF-α concentrations compared to overweight patients but also higher PTX3 levels, [120,121] and this association is confirmed even by “in vivo” analysis of fat tissue in CKD and HD patients [97].
Aside patients’ frailty status related to uraemia, RRT itself promotes inflammation: PTX3 levels could be higher because of the contact with the dialyzer, according to biocompatibility issues, but also for inefficient removal because of its molecular weight, thus mimicking the condition previously described for CKD.
PTX3 levels increase during a single 4-hour treatment reaching basal levels at the end of the intradialytic period with associated, reduced, intracellular PTX3 content in neutrophils [112,122,123,124]. Neutrophil PTX3 over-expression, instead, happens during a single HD: after a 4-hours treatment not only PTX3 but also ROS production is increased. More to date, flow mediated dilation (FMD) of the brachial artery inversely correlates with PTX3 levels with a worst performance at the end of the dialysis treatment [125].
More recently, Fukushi and coll. added another piece to the possible role of neutrophil, suggesting that increased PTX3 release could be due to progressive apoptosis of white blood cells because of the combining effects of CKD related chronic inflammation and biocompatibility issues during a 4-hours HD session even if performed with new generation dialyzers [126].
Recent insight from literature also suggest the possibility that single nucleotide polymorphism (SNP) in ESRD patients could mimic PTX3 deficiency: even not affecting mortality, the PTX3 (rs2305619) polymorphism in the intron 1 (+ 281A > G) was linked to a much more pronounced inflammatory response with a positive correlation with high specificity C reactive protein (hsCRP) and IL-6 in patients under RRT [127]. It has been also speculated that PTX3 loss-of-function mutations could promote post ischemic AKI and subsequent CKD [93] even if, according to renal I/R injury models, there is not actually a unique definition of this mechanism [128]
PTX3 interacts with several complement molecules such as C1q, ficolins and mannose binding leptin (MBL) thus promoting complement-dependent opsonization and phagocytosis of microbes and apoptotic cells but, at the same time, interacts with regulatory factors of the complement cascade, such as Factor H (FH) and Complement-4 Binding Protein (C4BP), in order to modulate complement-mediated inflammatory response [4]. All these elements contribute to the activation of the Lectin and Alternative pathway of complement but also, through C3b and C5b opsonizing fragment, to neutrophil upregulation and activation with consequent leukocyte adhesion, inflammatory response and activation of the coagulation cascade. The ending product of complement activation, the membrane attack complex (MAC-C5b-9) complex, exerts consequent endothelial damage [129,130].
One of the leading mechanism for complement activation in HD is binding of Ficolin-2 to the dialyzer surface thus inducing lectin pathway activation with a consequent inflammatory stimulus that [131] enhances, in the short term, a pro-thrombotic milieu and in the long term appearance of cardiovascular events [132]. It’s also established the relationship between PTX3 levels and consequent complement activation: PTX3 enhances innate immune recognition and complement deposition [133] and its C-terminal domain has been reported to bind to C1q with consequent complement activation [134]. Particularly, PTX3 activates the classical complement cascade when interacting with surface-bound C1qand consequent C3 and C4 deposition. Otherwise, in solution, PTX3 downregulates complement cascade blocking interaction sites [135].
7. Conclusions
Patients affected by ESRD are worldwide increasing in absolute number and clinical complexity. Their phenotypic uraemic frailty, in accordance with the development and consequent susceptibility to life threatening comorbidities such as CVD, malignancies or infections, is related to accelerated ageing phenomena generally referred to as “Inflammaging” [138].
In this setting, PTX3 encompasses, better than classic inflammatory biomarkers, the role of “Inflammaging” biomarker, particularly in ESRD and in HD patients. In this patients’ population, PTX3 role could be crucial in order to evaluate not only the most biocompatible membranes or suitable nanostructure but also influence anticoagulation strategies and even dialytic strategies for personalized therapies.
More to date, being more specific than hsCRP and according to its mechanism of release and production, PTX3 defines specific pattern of immune-mediated damage in life threatening conditions such as acute phase GN, vasculitis, TMA and acute rejection providing not only a well-time diagnostic recognition but also a consequent, specific treatment.
Because of the heterogeneity of data collected from literature, particularly in the field of kidney disease, more studies and researches are needed in order to translate in the clinical setting the possible role of PTX3 as a valid biomarker in the diagnosis and progression of the renal damage. Finally, the the knowledge acquired in progression and diagnosis of kidney diseases could help physicians to better understand the molecular mechanism underline the particular phenomena known as accelerated senescence.
Author Contributions
GS and PP: conceptualization, supervision, project administration and funding acquisition; BI: supervision and methodology; SS, SR, SM and AM: writing—original draft preparation; VL and DT: writing—review and editing; VL and SS: image development. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Images created with Biorender™.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garlanda, C.; Bottazzi, B.; Bastone, A.; Mantovani, A. Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu. Rev. Immunol. 2005, 23, 337–366. [Google Scholar] [CrossRef]
- Nauta, A.J.; De Haij, S.; Bottazzi, B.; Mantovani, A.; Borrias, M.C.; Aten, J.; Rastaldi, M.P.; Daha, M.R.; Van Kooten, C.; Roos, A. Human renal epithelial cells produce the long pentraxin PTX3. Kidney Int. 2005, 67, 543–553. [Google Scholar] [CrossRef]
- Chen, J.; Matzuk, M.M.; Zhou, X.J.; Lu, C.Y. Endothelial pentraxin 3 contributes to murine ischemic acute kidney injury. Kidney Int. 2012, 82, 1195–1207. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C. Humoral Innate Immunity and Acute-Phase Proteins. N Engl. J. Med. 2023, 388, 439–452. [Google Scholar] [CrossRef]
- Inforzato, A.; Doni, A.; Barajon, I.; Leone, R.; Garlanda, C.; Bottazzi, B.; Mantovani, A. PTX3 as a paradigm for the interaction of pentraxins with the Complement system. Semin. Immunol. 2013, 25, 79–85. [Google Scholar] [CrossRef]
- Daigo, K.; Inforzato, A.; Barajon, I.; Garlanda, C.; Bottazzi, B.; Meri, S.; Mantovani, A. Pentraxins in the activation and regulation of innate immunity. Immunol. Rev. 2016, 274, 202–217. [Google Scholar] [CrossRef]
- Noone, D.P.; Dijkstra, D.J.; van der Klugt, T.T.; van Veelen, P.A.; de Ru, A.H.; Hensbergen, P.J.; Trouw, L.A.; Sharp, T.H. PTX3 structure determination using a hybrid cryo electron microscopy and Alpha Fold approach offers insights into ligand binding and complement activation. Proc Natl AcadSci USA 2022, 119, e2208144119. [Google Scholar] [CrossRef]
- Kunes, P.; Holubcova, Z.; Kolackova, M.; Krejsek, J. Pentraxin 3(PTX 3): An Endogenous Modulator of the Inflammatory Response. Mediat. Inflamm. 2012, 2012, 920517. [Google Scholar] [CrossRef]
- Yamasaki, K.; Kurimura, M.; Kasai, T.; Sagara, M.; Kodama, T.; Inoue, K. Determination of physiological plasma pentraxin 3 (PTX3) levels in healthy populations. Clin Chem Lab Med. 2009, 47, 471–477. [Google Scholar] [CrossRef]
- Tong, M.; Carrero, J.J.; Qureshi, A.R.; Anderstam, B.; Heimbürger, O.; Bárány, P.; Axelsson, J.; Alvestrand, A.; Stenvinkel, P.; Lindholm, B.; et al. Plasma pentraxin 3 in patients with chronic kidney disease: associations with renal function, protein-energy wasting, cardiovascular disease, and mortality. Clin J Am Soc Nephrol. 2007, 2, 889–897. [Google Scholar] [CrossRef]
- Suliman, M.; Qureshi, A.; Carrero, J.; Barany, P.; Yilmaz, M.; Snaedal-Jonsdottir, S.; Alvestrand, A.; Heimburger, O.; Lindholm, B.; Stenvinkel, P. The long pentraxin PTX-3 in prevalent hemodialysis patients: associations with comorbidities and mortality. Qjm: Int. J. Med. 2008, 101, 397–405. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, J.; Zhu, M.; Lu, R.; Wang, Y.; Ni, Z. Plasma Pentraxin 3 Is Closely Associated with Peripheral Arterial Disease in Hemodialysis Patients and Predicts Clinical Outcome: A 6-Year Follow-Up. Blood Purif. 2015, 39, 266–273, Erratum in: Blood Purif. 2015;40(1):98. [Google Scholar] [CrossRef]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef]
- Betjes, M.G.H. Immune cell dysfunction and inflammation in end-stage renal disease. Nat. Rev. Nephrol. 2013, 9, 255–265. [Google Scholar] [CrossRef]
- Crépin, T.; Legendre, M.; Courivaud, C.; Vauchy, C.; Laheurte, C.; Rebibou, J.M.; Saas, P.; Ducloux, D.; Bamoulid, J. Maladie rénale chronique et immunosenescence prématurée :donné es et perspectives [Premature immune senescence and chronic kidney disease: Update and perspectives]. Nephrol Ther. 2020, 16, 9–18. [Google Scholar] [CrossRef]
- Ikizler, T.A.; Cano, N.J.; Franch, H.; Fouque, D.; Himmelfarb, J.; Kalantar-Zadeh, K.; Kuhlmann, M.K.; Stenvinkel, P.; TerWee, P.; Teta, D.; et al. Prevention and treatment of protein energy wasting in chronic kidney disease patients: a consensus statement by the International Society of Renal Nutrition and Metabolism. Kidney Int. 2013, 84, 1096–1107. [Google Scholar] [CrossRef]
- Alfieri, C.; Malvica, S.; Cesari, M.; Vettoretti, S.; Benedetti, M.; Cicero, E.; Miglio, R.; Caldiroli, L.; Perna, A.; Cervesato, A.; et al. Frailty in kidney transplantation: a review on its evaluation, variation and long-term impact. Clin. Kidney J. 2022, 15, 2020–2026. [Google Scholar] [CrossRef]
- Sjöberg, B.; Qureshi, A.R.; Heimbürger, O.; Stenvinkel, P.; Lind, L.; Larsson, A.; Bárány, P.; Ärnlöv, J. Association between levels of pentraxin 3 and incidence of chronic kidney disease in the elderly. J. Intern. Med. 2016, 279, 173–179. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C.; Bottazzi, B.; Peri, G.; Doni, A.; de la Torre, Y.M.; Latini, R. The long pentraxin PTX3 in vascular pathology. Vasc. Pharmacol. 2006, 45, 326–330. [Google Scholar] [CrossRef]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.-Q. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef]
- Pasterkamp, G.; Van Keulen, J.K.; De Kleijn, D.P.V. Role of Toll-like receptor 4 in the initiation and progression of atherosclerotic disease. Eur. J. Clin. Investig. 2004, 34, 328–334. [Google Scholar] [CrossRef]
- Martin-Rodriguez, S.; Caballo, C.; Gutierrez, G.; Vera, M.; Cruzado, J.M.; Cases, A.; Escolar, G.; Diaz-Ricart, M. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur. J. Clin. Investig. 2015, 45, 160–169. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Lee, R.; Shin, M.-H.; Kim, H.-N.; Lee, Y.-H.; Choi, S.-W.; Ahn, H.-R.; Kweon, S.-S. Relationship between plasma pentraxin 3 level and risk of chronic kidney disease in the Korean elderly: the Dong-gu study. Int. Urol. Nephrol. 2017, 49, 2027–2033. [Google Scholar] [CrossRef]
- Estébanez, B.; Rodriguez, A.L.; Visavadiya, N.P.; Whitehurst, M.; Cuevas, M.J.; González-Gallego, J.; Huang, C.J. Aerobic Training Down-Regulates Pentraxin 3 and Pentraxin 3/Toll-Like Receptor 4 Ratio, Irrespective of Oxidative Stress Response, in Elderly Subjects. Antioxidants (Basel). 2020, 9, 110. [Google Scholar] [CrossRef]
- Nauta, AJ, de Haij, S, Bottazzi, B, Mantovani, A, Borrias, MC, Aten, J, Rastaldi, MP, Daha, MR, van Kooten C, Roos A: Human renal epithelial cells produce the long pentraxin PTX3. Kidney Int 2005, 67, 543–553. [CrossRef]
- Hung, T.-W.; Tsai, J.-P.; Lin, S.-H.; Lee, C.-H.; Hsieh, Y.-H.; Chang, H.-R. Pentraxin 3 Activates JNK Signaling and Regulates the Epithelial-To-Mesenchymal Transition in Renal Fibrosis. Cell. Physiol. Biochem. 2016, 40, 1029–1038. [Google Scholar] [CrossRef]
- Lin CH, Wan C, Liu WS, Wang HH. PM2.5 Induces Early Epithelial Mesenchymal Transition in Human Proximal Tubular Epithelial Cells through Activation of IL-6/STAT3 Pathway. Int J Mol Sci. 2021, 22, 12734. [Google Scholar] [CrossRef]
- Lin, C.-H.; Liu, W.-S.; Wan, C.; Wang, H.-H. Pentraxin 3 mediates early inflammatory response and EMT process in human tubule epithelial cells induced by PM2.5. Int. Immunopharmacol. 2022, 112, 109258. [Google Scholar] [CrossRef]
- Miljković M, Stefanović A, Bogavac-Stanojević N, Simić-Ogrizović S, DumićJ, Černe D, Jelić-Ivanović Z, Kotur-Stevuljević J. Association of Pentraxin-3, Galectin-3 and Matrix Metalloproteinase-9/Timp-1 with Cardiovascular Risk in Renal Disease Patients. Acta Clin Croat. 2017, 56, 673–680. [Google Scholar]
- Dai, L.; Golembiewska, E.; Lindholm, B.; Stenvinkel, P. End-Stage Renal Disease, Inflammation and Cardiovascular Outcomes. Contrib. Nephrol. 2017, 191, 32–43. [Google Scholar] [CrossRef]
- Parlak, A.; Aydogan, U.; Iyisoy, A.; Dikililer, M.A.; Kut, A.; Cakir, E.; Saglam, K. Elevated pentraxin-3 levels are related to blood pressure levels in hypertensive patients: an observational study. Anadolu Kardiyol. Dergisi/The Anatol. J. Cardiol. 2012, 12, 298–304. [Google Scholar] [CrossRef]
- Carrizzo, A.; Lenzi, P.; Procaccini, C.; Damato, A.; Biagioni, F.; Ambrosio, M.; Amodio, G.; Remondelli, P.; Del Giudice, C.; Izzo, R.; et al. Pentraxin 3 Induces Vascular Endothelial Dysfunction Through a P-selectin/Matrix Metalloproteinase-1 Pathway. Circulation 2015, 131, 1495–1505. [Google Scholar] [CrossRef]
- Lech M, Rommele C, Anders HJ (2012) Pentraxins in nephrology: C-reactive protein, serum amyloid P and pentraxin-3. Nephrol Dial Transplant 0:1–8.
- Banfi, C.; Brioschi, M.; Vicentini, L.M.; Cattaneo, M.G. The Effects of Silencing PTX3 on the Proteome of Human Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 13487. [Google Scholar] [CrossRef]
- Yilmaz MI, Carrero JJ, Martín-Ventura JL, Sonmez A, Saglam M, Celik T, Yaman H, Yenicesu M, Eyileten T, Moreno JA, Egido J, Blanco-Colio LM. Combined therapy with renin-angiotensin system and calcium channel blockers in type 2 diabetic hypertensive patients with proteinuria: effects on soluble TWEAK, PTX3, and flow-mediated dilation. Clin J Am Soc Nephrol. 2010 Jul.
- Fassett, R.G.; Robertson, I.K.; Ball, M.J.; Geraghty, D.P.; Coombes, J.S. Effects of atorvastatin on biomarkers of inflammation in chronic kidney disease. Clin. Nephrol. 2014, 81, 75–85. [Google Scholar] [CrossRef]
- Ristagno, G.; Fumagalli, F.; Bottazzi, B.; Mantovani, A.; Olivari, D.; Novelli, D.; Latini, R. Pentraxin 3 in Cardiovascular Disease. Front. Immunol. 2019, 10, 823. [Google Scholar] [CrossRef]
- Zanetti, M.; Bosutti, A.; Ferreira, C.; Vinci, P.; Biolo, G.; Fonda, M.; Valente, M.; Cattin, L.; Guarnieri, G.; Barazzoni, R. Circulating pentraxin 3 levels are higher in metabolic syndrome with subclinical atherosclerosis: evidence for association with atherogenic lipid profile. Clin. Exp. Med. 2009, 9, 243–248. [Google Scholar] [CrossRef]
- Jaillon, S.; Peri, G.; Delneste, Y.; Frémaux, I.; Doni, A.; Moalli, F.; Garlanda, C.; Romani, L.; Gascan, H.; Bellocchio, S.; et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 2007, 204, 793–804. [Google Scholar] [CrossRef]
- Norata, G.D.; Marchesi, P.; Venu, V.K.P.; Pasqualini, F.; Anselmo, A.; Moalli, F.; Pizzitola, I.; Garlanda, C.; Mantovani, A.; Catapano, A.L. Deficiency of the Long Pentraxin PTX3 Promotes Vascular Inflammation and Atherosclerosis. Circulation. 2009, 120, 699–708. [Google Scholar] [CrossRef]
- Jenny NS, Arnold AM, Kuller LH, Tracy RP, Psaty BM. Associations of pentraxin 3 with cardiovascular disease and all-cause death: the cardiovascular health study. Arterioscler Thromb Vasc Biol 2009, 29, 594–599. [Google Scholar] [CrossRef]
- Peri, G.; Introna, M.; Corradi, D.; Iacuitti, G.; Signorini, S.; Avanzini, F.; Pizzetti, F.; Maggioni, A.P.; Moccetti, T.; Metra, M.; et al. PTX3, A Prototypical Long Pentraxin, Is an Early Indicator of Acute Myocardial Infarction in Humans. Circ. 2000, 102, 636–641. [Google Scholar] [CrossRef]
- Suzuki, S.; Takeishi, Y.; Niizeki, T.; Koyama, Y.; Kitahara, T.; Sasaki, T.; Sagara, M.; Kubota, I. Pentraxin 3, a new marker for vascular inflammation, predicts adverse clinical outcomes in patients with heart failure. Am. Hear. J. 2008, 155, 75–81. [Google Scholar] [CrossRef]
- Matsui, S.; Ishii, J.; Kitagawa, F.; Kuno, A.; Hattori, K.; Ishikawa, M.; Okumura, M.; Kan, S.; Nakano, T.; Naruse, H.; et al. Pentraxin 3 in unstable angina and non-ST-segment elevation myocardial infarction. Atherosclerosis 2010, 210, 220–225. [Google Scholar] [CrossRef]
- Roberto Latini, MD; Aldo P. Maggioni, MD; Giuseppe Peri, BS; Lucio Gonzini, BS; Donata Lucci, BS; Paolo Mocarelli, MD; Luca Vago, MD; Fabio Pasqualini, BS; Stefano Signorini, MD; Dario Soldateschi, BS; Lorenzo Tarli, BS; Carlo Schweiger, MD; Claudio Fresco, MD; Rossana Cecere, BS; Gianni Tognoni, MD; Alberto Mantovani, MD; on behalf of the Lipid Assessment Trial Italian Network (LATIN) Investigators: Prognostic significance of the long pentraxin ptx3 in acute myocardial infarction. Circulation 2004, 110, 2349–2354. [Google Scholar]
- Blanco-Colio LM, Martin-Ventura JL, Munoz-Garcia B, Orbe J, Pa’ramo JA, Michel JB, Ortiz A, Meilhac O, Egido J: Identification of soluble tumor necrosis factor-like weak inducer of apoptosis (sTWEAK) as a possible biomarker of subclinical atherosclerosis. Arterioscler Thromb Vasc Biol 2007, 27, 916–922. [CrossRef]
- Yilmaz MI, Carrero JJ, Ortiz A, Martín-Ventura JL, Sonmez A, Saglam M, Yaman H, Yenicesu M, Egido J, Blanco-Colio LM: Soluble TWEAK plasma levels as a novel biomarker of endothelial function in patients with chronic kidney disease. ClinJ Am SocNephrol 2009, 4, 1716–1723. [CrossRef]
- Yilmaz MI, Sonmez A, Ortiz A, Saglam M, Kilic S, Eyileten T, Caglar K, Oguz Y, Vural A, Çakar M, Egido J, Altun B, Yenicesu M, Blanco-Colio LM, Carrero JJ. Soluble TWEAK and PTX3 in non-dialysis CKD patients: impact on endothelial dysfunction and cardiovascular outcomes. Clin J Am Soc Nephrol. 2011, 6, 785. [Google Scholar] [CrossRef]
- Jager KJ, Kovesdy C, Langham R, Rosenberg M, Jha V, Zoccali C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrol Dial Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Anders, H.-J.; Kitching, A.R.; Leung, N.; Romagnani, P. Glomerulonephritis: immunopathogenesis and immunotherapy. Nat. Rev. Immunol. 2023, 23, 453–471. [Google Scholar] [CrossRef]
- Bussolati, B.; Peri, G.; Salvidio, G.; Verzola, D.; Mantovani, A.; Camussi, G. The Long Pentraxin Ptx3 Is Synthesized in IgA Glomerulonephritis and Activates Mesangial Cells. J. Immunol. 2003, 170, 1466–1472. [Google Scholar] [CrossRef]
- Lech, M.; Rommele, C.; Anders, H.-J. Pentraxins in nephrology: C-reactive protein, serum amyloid P and pentraxin-3. Nephrol. Dial. Transplant. 2013, 28, 803–811. [Google Scholar] [CrossRef]
- Bassi, N.; Ghirardello, A.; Blank, M.; Zampieri, S.; Sarzi-Puttini, P.; Mantovani, A.; Shoenfeld, Y.; Doria, A. IgG anti-pentraxin 3 antibodies in systemic lupus erythematosus. Ann. Rheum. Dis. 2010, 69, 1704–1710. [Google Scholar] [CrossRef]
- Inforzato, A.; Jaillon, S.; Moalli, F.; Barbati, E.; Bonavita, E.; Bottazzi, B.; Mantovani, A.; Garlanda, C. The long pentraxin PTX3 at the crossroads between innate immunity and tissue remodelling. Tissue Antigens 2011, 77, 271–282. [Google Scholar] [CrossRef]
- Presta, M.; Camozzi, M.; Salvatori, G.; Rusnati, M. Role of the soluble pattern recognition receptor PTX3 in vascular biology. J. Cell. Mol. Med. 2007, 11, 723–738. [Google Scholar] [CrossRef]
- Fazzini, F.; Peri, G.; Doni, A.; Dell'Antonio, G.; Cin, E.D.; Bozzolo, E.; D'Auria, F.; Praderio, L.; Ciboddo, G.; Sabbadini, M.G.; et al. PTX3 in small-vessel vasculitides: An independent indicator of disease activity produced at sites of inflammation. Arthritis Rheum. 2001, 44, 2841–2850. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Jaillon, S.; Peri, G.; Delneste, Y.; Frémaux, I.; Doni, A.; Moalli, F.; Garlanda, C.; Romani, L.; Gascan, H.; Bellocchio, S.; et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 2007, 204, 793–804. [Google Scholar] [CrossRef]
- Ishida, R.; Nakai, K.; Fujii, H.; Goto, S.; Hara, S.; Imai, N.; Nishi, S. Elevated Expression of Pentraxin 3 in Anti-neutrophil Cytoplasmic Antibody-associated Glomerulonephritis with Normal Serum C-reactive Protein. Intern. Med. 2015, 54, 1369–1373. [Google Scholar] [CrossRef]
- Chen, X.; Luo, J.; Wu, M.; Pan, Z.; Xie, Y.; Wang, H.; Chen, B.; Zhu, H. Study on Association of Pentraxin 3 and Diabetic Nephropathy in a Rat Model. J. Diabetes Res. 2018, 2018, 8968573. [Google Scholar] [CrossRef]
- Cieślik, P.; Hrycek, A. Pentraxin 3 as a biomarker of local inflammatory response to vascular injury in systemic lupus erythematosus. Autoimmunity 2014, 48, 242–250. [Google Scholar] [CrossRef]
- Pang Y, Tan Y, Li Y, Zhang J, Guo Y, Guo Z, Zhang C, Yu F, Zhao MH. Pentraxin 3 Is Closely Associated with Tubulointerstitial Injury in Lupus Nephritis: A Large Multicenter Cross-Sectional Study. Medicine (Baltimore). 2016, 95, e2520. [Google Scholar] [CrossRef]
- Mistry, P.; Kaplan, M.J. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin. Immunol. 2017, 185, 59–73. [Google Scholar] [CrossRef]
- Kang, S.; Rogers, J.L.; Monteith, A.J.; Jiang, C.; Schmitz, J.; Clarke, S.H.; Tarrant, T.K.; Truong, Y.K.; Diaz, M.; Fedoriw, Y.; et al. Apoptotic Debris Accumulates on Hematopoietic Cells and Promotes Disease in Murine and Human Systemic Lupus Erythematosus. J. Immunol. 2016, 196, 4030–4039. [Google Scholar] [CrossRef]
- Bassi, N.; Del Prete, D.; Ghirardello, A.; Gatto, M.; Ceol, M.; Zen, M.; Bettio, S.; Mantovani, A.; Iaccarino, L.; Punzi, L.; et al. PTX3, Anti-PTX3, and Anti-C1q Autoantibodies in Lupus Glomerulonephritis. Clin. Rev. Allergy Immunol. 2015, 49, 217–226. [Google Scholar] [CrossRef]
- Augusto, J.-F.; Onno, C.; Blanchard, S.; Dubuquoi, S.; Mantovani, A.; Chevailler, A.; Jeannin, P.; Subra, J.-F. Detection of anti-PTX3 autoantibodies in systemic lupus erythematosus. Rheumatology 2009, 48, 442–444. [Google Scholar] [CrossRef]
- Yuan, M.; Tan, Y.; Pang, Y.; Li, Y.-Z.; Song, Y.; Yu, F.; Zhao, M.-H. Anti-pentraxin 3 auto-antibodies might be protective in lupus nephritis: a large cohort study. Ren. Fail. 2017, 39, 465–473. [Google Scholar] [CrossRef]
- Ma K, Li J, Wang X. TLR4+CXCR4+ plasma cells drive nephritis development in systemic lupus erythematosus. Ann Rheum Dis. 2018, 77, 1498–1506. [Google Scholar] [CrossRef]
- Gatto, M.; Wiedemann, A.; Nomovi, N.; Reiter, K.; Schrezenmeier, E.; Rose, T.; Szelinski, F.; Lino, A.C.; Valentino, S.; Ghirardello, A.; et al. Circulating Pentraxin3-Specific B Cells Are Decreased in Lupus Nephritis. Front. Immunol. 2019, 10, 29. [Google Scholar] [CrossRef]
- Gatto, M.; Radu, C.M.; Luisetto, R.; Ghirardello, A.; Bonsembiante, F.; Trez, D.; Valentino, S.; Bottazzi, B.; Simioni, P.; Cavicchioli, L.; et al. Immunization with Pentraxin3 prevents transition from subclinical to clinical lupus nephritis in lupus-prone mice: Insights from renal ultrastructural findings. J. Autoimmun. 2020, 111, 102443. [Google Scholar] [CrossRef]
- Yoshida, Y.; Kato, H.; Ikeda, Y.; Nangaku, M. Pathogenesis of Atypical Hemolytic Uremic Syndrome. J. Atheroscler. Thromb. 2019, 26, 99–110. [Google Scholar] [CrossRef]
- Raina, R.; Grewal, M.K.; Radhakrishnan, Y.; Tatineni, V.; DeCoy, M.; Burke, L.L.; Bagga, A. Optimal management of atypical hemolytic uremic disease: challenges and solutions. Int. J. Nephrol. Renov. Dis. 2019, ume 12, 183–204. [Google Scholar] [CrossRef]
- Deban, L.; Jaillon, S.; Garlanda, C.; Bottazzi, B.; Mantovani, A. Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res. 2011, 343, 237–249. [Google Scholar] [CrossRef]
- Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, Spreafico F, Pausa M, D’Ettorre C, Gianazza E, Tagliabue A, Salmona M, Tedesco F, Introna M, Mantovani A. Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem. 1997, 272, 32817–23. [Google Scholar] [CrossRef]
- Nauta, A.J.; Bottazzi, B.; Mantovani, A.; Salvatori, G.; Kishore, U.; Schwaeble, W.J.; Gingras, A.R.; Tzima, S.; Vivanco, F.; Egido, J.; et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur. J. Immunol. 2003, 33, 465–473. [Google Scholar] [CrossRef]
- Deban, L.; Jarva, H.; Lehtinen, M.J.; Bottazzi, B.; Bastone, A.; Doni, A.; Jokiranta, T.S.; Mantovani, A.; Meri, S. Binding of the Long Pentraxin PTX3 to Factor H: Interacting Domains and Function in the Regulation of Complement Activation. J. Immunol. 2008, 181, 8433–8440. [Google Scholar] [CrossRef]
- Doni, A.; Garlanda, C.; Bottazzi, B.; Meri, S.; Garred, P.; Mantovani, A. Interactions of the humoral pattern recognition molecule PTX3 with the complement system. Immunobiology 2012, 217, 1122–1128. [Google Scholar] [CrossRef]
- Braunschweig, A.; Józsi, M. Human Pentraxin 3 Binds to the Complement Regulator C4b-Binding Protein. PLOS ONE 2011, 6, e23991. [Google Scholar] [CrossRef]
- Trojnar, E.; Józsi, M.; Szabó, Z.; Réti, M.; Farkas, P.; Kelen, K.; Reusz, G.S.; Szabó, A.J.; Garam, N.; Mikes, B.; et al. Elevated Systemic Pentraxin-3 Is Associated With Complement Consumption in the Acute Phase of Thrombotic Microangiopathies. Front. Immunol. 2019, 10, 240. [Google Scholar] [CrossRef]
- Haapasalo, K.; Meri, S. Regulation of the Complement System by Pentraxins. Front. Immunol. 2019, 10, 1750. [Google Scholar] [CrossRef]
- Kopp, A.; Strobel, S.; Tortajada, A.; de Córdoba, S.R.; Sánchez-Corral, P.; Prohászka, Z.; López-Trascasa, M.; Józsi, M. Atypical Hemolytic Uremic Syndrome-Associated Variants and Autoantibodies Impair Binding of Factor H and Factor H-Related Protein 1 to Pentraxin 3. J. Immunol. 2012, 189, 1858–1867. [Google Scholar] [CrossRef]
- Csincsi. I.; Kopp, A.; Zöldi, M.; Bánlaki, Z.; Uzonyi, B.; Hebecker, M.; Caesar, J.J.E.; Pickering, M.C.; Daigo, K.; Hamakubo, T.; et al. Factor H–Related Protein 5 Interacts with Pentraxin 3 and the Extracellular Matrix and Modulates Complement Activation. J. Immunol. 2015, 194, 4963–4973. [Google Scholar] [CrossRef] [PubMed]
- Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C; ADQI XIII Work Group. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J Am Soc Nephrol. 2016, 27, 687–97. [Google Scholar] [CrossRef]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Matzuk, M.M.; Zhou, X.J.; Lu, C.Y. Endothelial pentraxin 3 contributes to murine ischemic acute kidney injury. Kidney Int. 2012, 82, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Divella, C.; Stasi, A.; Franzin, R.; Rossini, M.; Pontrelli, P.; Sallustio, F.; Netti, G.S.; Ranieri, E.; Lacitignola, L.; Staffieri, F.; et al. Pentraxin-3-mediated complement activation in a swine model of renal ischemia/reperfusion injury. Aging 2021, 13, 10920–10933. [Google Scholar] [CrossRef]
- Giuseppe Castellano, Rita Melchiorre, Antonia Loverre, Pasquale Ditonno, Vincenzo Montinaro, Michele Rossini, Chiara Divella, Michele Battaglia, Giuseppe Lucarelli, Gennaro Annunziata, Silvano Palazzo, Francesco Paolo Selvaggi, Francesco Staffieri, Antonio Crovace, Mohamed R. Daha, Maurice Mannesse, Sandra van Wetering, Francesco Paolo Schena, Giuseppe Grandaliano Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010, 176, 1648–1659. [Google Scholar] [CrossRef]
- Delpech, P.-O.; Thuillier, R.; SaintYves, T.; Danion, J.; Le Pape, S.; van Amersfoort, E.S.; Oortwijn, B.; Blancho, G.; Hauet, T. Inhibition of complement improves graft outcome in a pig model of kidney autotransplantation. J. Transl. Med. 2016, 14, 1–13. [Google Scholar] [CrossRef]
- Kurzhagen, J.T.; Dellepiane, S.; Cantaluppi, V.; Rabb, H. AKI: an increasingly recognized risk factor for CKD development and progression. J. Nephrol. 2020, 33, 1171–1187. [Google Scholar] [CrossRef]
- Xiao Y, Yang N, Zhang Q, Wang Y, Yang S, Liu Z. Pentraxin 3 inhibits acute renal injury-induced interstitial fibrosis through suppression of IL-6/Stat3pathway. Inflammation. 2014, 37, 1895–901. [Google Scholar] [CrossRef]
- Lech M, Römmele C, Gröbmayr R, Eka Susanti H, Kulkarni OP, Wang S, Gröne HJ, Uhl B, Reichel C, Krombach F, Garlanda C, Mantovani A, Anders HJ. Endogenous and exogenous pentraxin-3 limits post-ischemic acute and chronic kidney injury. Kidney Int. 2013, 83, 647–61. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, J.; Gomez, H.; A Kellum, J.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Kim, S.Y.; Na, J.C.; Yoon, Y.E.; Han, W.K. Exogenous pentraxin-3 inhibits the reactive oxygen species-mitochondrial and apoptosis pathway in acute kidney injury. PLOS ONE 2018, 13, e0195758. [Google Scholar] [CrossRef]
- Turkmen, K.; Tonbul, H.Z.; Toker, A.; Gaipov, A.; Erdur, F.M.; Cicekler, H.; Anil, M.; Ozbek, O.; Selcuk, N.Y.; Yeksan, M.; et al. The Relationship between Oxidative Stress, Inflammation, and Atherosclerosis in Renal Transplant and End-Stage Renal Disease Patients. Ren. Fail. 2012, 34, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Witasp, A.; Rydén, M.; Carrero, J.J.; Qureshi, A.R.; Nordfors, L.; Näslund, E.; Hammarqvist, F.; Arefin, S.; Kublickiene, K.; Stenvinkel, P. Elevated Circulating Levels and Tissue Expression of Pentraxin 3 in Uremia: A Reflection of Endothelial Dysfunction. PLOS ONE 2013, 8, e63493. [Google Scholar] [CrossRef] [PubMed]
- Gursu M, Celik K, Ozturk S, Turkmen A, Gorcin S, Kocak B, Sari S, Koldas M,Feyizoglu H, Kazancioglu R. Pentraxin 3 and C-reactive protein as inflammatory markers after a kidney transplant. Exp Clin Transplant. 2014, 12, 295–299. [Google Scholar]
- Imai, N.; Nishi, S.; Yoshita, K.; Ito, Y.; Osawa, Y.; Takahashi, K.; Nakagawa, Y.; Saito, K.; Takahashi, K.; Narita, I. Pentraxin-3 expression in acute renal allograft rejection. Clin. Transplant. 2012, 26, 25–31. [Google Scholar] [CrossRef]
- Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. 2003, 349, 2326–2333. [Google Scholar] [CrossRef]
- Su, L.; Tam, N.; Deng, R.; Chen, P.; Li, H.; Wu, L. Everolimus-based calcineurin-inhibitor sparing regimens for kidney transplant recipients: a systematic review and meta-analysis. Int. Urol. Nephrol. 2014, 46, 2035–2044. [Google Scholar] [CrossRef]
- Paoletti, E.; Project, E.; Citterio, F.; Corsini, A.; Potena, L.; Rigotti, P.; Sandrini, S.; Bussalino, E.; Stallone, G. Everolimus in kidney transplant recipients at high cardiovascular risk: a narrative review. J. Nephrol. 2019, 33, 69–82. [Google Scholar] [CrossRef]
- Infante, B.; Bellanti, F.; Correale, M.; Pontrelli, P.; Franzin, R.; Leo, S.; Calvaruso, M.; Mercuri, S.; Netti, G.S.; Ranieri, E.; et al. mTOR inhibition improves mitochondria function/biogenesis and delays cardiovascular aging in kidney transplant recipients with chronic graft dysfunction. Aging 2021, 13, 8026–8039. [Google Scholar] [CrossRef] [PubMed]
- Argani, H.; Ghorbanihaghjo, A.; Panahi, G.; Rashtchizadeh, N.; Safa, J.; Meimand, S.M. Serum Fetuin-A and Pentraxin3 in hemodialysis and renal transplant patients. Clin. Biochem. 2012, 45, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Rollag H, Asberg A, Ueland T, Hartmann A, Jardine AG, Humar A, Pescovitz MD, Bignamini AA, Aukrust P. Treatment of cytomegalovirus disease in solid organ transplant recipients: markers of inflammation as predictors of outcome. Transplantation. 2012, 94, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, K.; Husson, C.; Dondeyne, J.-P.; Vanherweghem, J.-L.; Tielemans, C. Cytotoxicity of Mononuclear Cells as Induced by Peritoneal Dialysis Fluids: Insight into Mechanisms that Regulate Osmotic Stress-Related Apoptosis. 2008, 28, 655–666. [CrossRef]
- Palomar-Fontanet R, Lavin-Gómez BA, Quintanar-Lartundo JA, García-Unzueta MT, Gago-Fraile M, Torrealba-Rodríguez MI, Arias-Rodriguez MA, Gómez-Gerique JA. Markers of inflammation before and during peritoneal dialysis. AdvPerit Dial. 2011, 27, 28–32. [Google Scholar]
- Ishimatsu, N.; Miyamoto, T.; Ueno, H.; Hasegawa, E.; Kuma, A.; Fujimoto, Y.; Bando, K.; Nakamata, J.; Furuno, Y.; Serino, R.; et al. High glucose concentration-induced expression of pentraxin-3 in a rat model of continuous peritoneal dialysis. 2016, 31, 1251–1258. [CrossRef]
- Kanda, R.; Hamada, C.; Kaneko, K.; Nakano, T.; Wakabayashi, K.; Io, H.; Horikoshi, S.; Tomino, Y. Pentraxin 3 as a new biomarker of peritoneal injury in peritoneal dialysis patients. J. Artif. Organs 2013, 16, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Wardoyo, E.Y.; Nainggolan, G.; Hustrini, N.M.; Setiati, S. Factors Associated with Arterial Stiffness in Chronic Hemodialysis Patients in Jakarta: The Role of Hemodialysis Frequency and Pentraxin 3. Acta Med Indones. 2021, 53, 177–183. [Google Scholar]
- Boehme, M.; Kaehne, F.; Kuehne, A.; Bernhardt, W.; Schröder, M.; Pommer, W.; Fischer, C.; Becker, H.; Müller, C.; Schindler, R. Pentraxin 3 is elevated in haemodialysis patients and is associated with cardiovascular disease. Nephrol. Dial. Transplant. 2007, 22, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, X.; Zou, J.; Liu, Z.; Jiang, S.; Xu, S.; Shen, B.; Chen, Y.; Shan, Y.; Cao, X. Plasma Pentraxin 3 is Associated with Cardiovascular Disease in Hemodialysis Patients. Ren. Fail. 2011, 33, 998–1004. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Ou, S.-M.; Wu, C.-C.; Huang, C.-C.; Hsieh, J.-T.; Tseng, P.-Y.; Lee, C.-Y.; Yang, C.-Y.; Tarng, D.-C. Pentraxin 3 Predicts Arteriovenous Fistula Functional Patency Loss and Mortality in Chronic Hemodialysis Patients. Am. J. Nephrol. 2022, 53, 148–156. [Google Scholar] [CrossRef]
- Nishi, K.; Imamura, T.; Kitamura, K.; Ogawa, T.; Fujimoto, S.; Kakitsubata, Y.; Ishikawa, T.; Asada, Y.; Kodama, T. Associations of Plasma Pentraxin 3 and Monocyte Chemoattractant Protein-1 Concentrations with Cardiovascular Disease in Patients with Chronic Kidney Disease. Ren. Fail. 2011, 33, 398–404. [Google Scholar] [CrossRef]
- Cardiovascular disease in dialysis patients. Cozzolino M, Mangano M, Stucchi A, Ciceri P, Conte F and Galassi A. Nephrol Dial Transplant (2018) 33: iii28–iii34. i.
- Valente, M.J.; Rocha, S.; Coimbra, S.; Catarino, C.; Rocha-Pereira, P.; Bronze-Da-Rocha, E.; Oliveira, J.G.; Madureira, J.; Fernandes, J.C.; Sameiro-Faria, M.D.; et al. Long Pentraxin 3 as a Broader Biomarker for Multiple Risk Factors in End-Stage Renal Disease: Association with All-Cause Mortality. Mediat. Inflamm. 2019, 2019, 3295725. [Google Scholar] [CrossRef] [PubMed]
- Foshati, S.; Askari, G.; Bagherniya, M.; Mortazavi, M.; Moeinzadeh, F.; Taheri, S.; Heidari, Z.; Rouhani, M.H. Association between nutritional, inflammatory and oxidative status (NIOS) and risk of adverse outcomes in patients on haemodialysis (HD): the NIOS-HD prospective cohort study protocol. BMJ Open 2022, 12, e064367. [Google Scholar] [CrossRef] [PubMed]
- Calabia J, Arcos E, Carrero JJ, Comas J, Vallés M. Does the obesity survival paradox of dialysis patients differ with age? Blood Purif. 2015, 39, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Naderi, N.; Kleine, C.-E.; Park, C.; Hsiung, J.-T.; Soohoo, M.; Tantisattamo, E.; Streja, E.; Kalantar-Zadeh, K.; Moradi, H. Obesity Paradox in Advanced Kidney Disease: From Bedside to the Bench. Prog. Cardiovasc. Dis. 2018, 61, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Qureshi, A.R.; Heimbürger, O.; Bárány, P.; Carrero, K.; Sjöberg, B.; Lindholm, B.; Stenvinkel, P.; Carrero, J.J. Inverse Relationship between the Inflammatory Marker Pentraxin-3, Fat Body Mass, and Abdominal Obesity in End-Stage Renal Disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Barazzoni, R.; Aleksova, A.; Carriere, C.; Cattin, M.R.; Zanetti, M.; Vinci, P.; Stolfo, D.; Guarnieri, G.; Sinagra, G. Obesity and high waist circumference are associated with low circulating pentraxin-3 in acute coronary syndrome. Cardiovasc. Diabetol. 2013, 12, 167–167. [Google Scholar] [CrossRef]
- Oldani, S.; Finazzi, S.; Bottazzi, B.; Garlanda, C.; Baldassarre, E.; Valaperta, S.; Cuccovillo, I.; Albini, M.; Child, M.; Montanelli, A.; et al. Plasma pentraxin-3 as a marker of bioincompatibility in hemodialysis patients. J. Nephrol. 2012, 25, 120–126. [Google Scholar] [CrossRef]
- Sangeetha Lakshmi B, Harini Devi N, Suchitra MM, Srinivasa Rao PVLN, Siva Kumar V. Changes in the inflammatory and oxidative stress markers during a single hemodialysis session in patients with chronic kidney disease. Ren Fail. 2018, 40, 534–540. [Google Scholar] [CrossRef]
- Tong M, Anderstam B, Sjoberg B, Alvestrand A: Circulating levels of long pentraxin 3 (PTX3) are elevated in hemodialysis patients and increase acutely after a single HD session (abstract). J Am Soc Nephrol 2006, 17, 735.
- Dell'Oglio, M.P.; Simone, S.; Ciccone, M.; Corciulo, R.; Gesualdo, M.; Zito, A.; Cortese, F.; Castellano, G.; Gigante, M.; Gesualdo, L.; et al. Neutrophil-dependent pentraxin-3 and reactive oxygen species production modulate endothelial dysfunction in haemodialysis patients. Nephrol. Dial. Transplant. 2017, 32, gfw363–1549. [Google Scholar] [CrossRef]
- Fukushi, T.; Yamamoto, T.; Yoshida, M.; Fujikura, E.; Miyazaki, M.; Nakayama, M. Enhanced neutrophil apoptosis accompanying myeloperoxidase release during hemodialysis. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Valente, M.J.; Coimbra, S.; Catarino, C.; Rocha-Pereira, P.; Oliveira, J.G.; Madureira, J.; Fernandes, J.C.; do Sameiro-Faria, M.; Miranda, V.; et al. Interleukin 6 (rs1800795) and pentraxin 3 (rs2305619) polymorphisms-association with inflammation and all-cause mortality in end-stage-renal disease patients on dialysis. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- de Oliveira, T.H.C.; Souza, D.G.; Teixeira, M.M.; Amaral, F.A. Tissue Dependent Role of PTX3 During Ischemia-Reperfusion Injury. Front. Immunol. 2019, 10, 1461. [Google Scholar] [CrossRef]
- Bowry, S.K.; Kircelli, F.; Himmele, R.; Nigwekar, S.U. Blood-incompatibility in haemodialysis: alleviating inflammation and effects of coagulation. Clin. Kidney J. 2021, 14, i59–i71. [Google Scholar] [CrossRef] [PubMed]
- Song G, Wang S, Barkestani MN. Membrane attack complexes, endothelial cell activation, and direct allorecognition. Front Immunol. 2022, 13, 1020889. [Google Scholar] [CrossRef] [PubMed]
- Poppelaars F, Faria B, Gaya da Costa M, Franssen CFM, van Son WJ, Berger SP, Daha MR, Seelen MA. The Complement System in Dialysis: A Forgotten Story? Front Immunol. 2018, 9, 71. [Google Scholar] [CrossRef]
- Poppelaars, F.; da Costa, M.G.; Faria, B.; Berger, S.P.; Assa, S.; Daha, M.R.; Pestana, J.O.M.; van Son, W.J.; Franssen, C.F.M.; Seelen, M.A. Intradialytic Complement Activation Precedes the Development of Cardiovascular Events in Hemodialysis Patients. Front. Immunol. 2018, 9, 2070. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Doni, A.; Hummelshøj, T.; Honoré, C.; Bastone, A.; Mantovani, A.; Thielens, N.M.; Garred, P. Synergy between Ficolin-2 and Pentraxin 3 Boosts Innate Immune Recognition and Complement Deposition. J. Biol. Chem. 2009, 284, 28263–28275. [Google Scholar] [CrossRef]
- Nauta, A.J.; Bottazzi, B.; Mantovani, A.; Salvatori, G.; Kishore, U.; Schwaeble, W.J.; Gingras, A.R.; Tzima, S.; Vivanco, F.; Egido, J.; et al. Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur. J. Immunol. 2003, 33, 465–473. [Google Scholar] [CrossRef]
- Garlanda C, Bottazzi B, Magrini E. PTX3, a Humoral Pattern Recognition Molecule, in Innate Immunity, Tissue Repair, and Cancer. Physiol Rev. 2018, 98, 623–639. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G.L. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, E33. [Google Scholar] [CrossRef] [PubMed]
- Rosner MH, Reis T, Husain-Syed F, Vanholder R, Hutchison C, Stenvinkel P, Blankestijn PJ, Cozzolino M, Juillard L, Kashani K, Kaushik M, Kawanishi H, Massy Z, Sirich TL, Zuo L, Ronco C. Classification of Uremic Toxins and TheirRole in Kidney Failure. Clin J Am SocNephrol. 2021, 16, 1918–1928. [Google Scholar] [CrossRef]
- Losappio, V.; Franzin, R.; Infante, B.; Godeas, G.; Gesualdo, L.; Fersini, A.; Castellano, G.; Stallone, G. Molecular Mechanisms of Premature Aging in Hemodialysis: The Complex Interplay between Innate and Adaptive Immune Dysfunction. Int. J. Mol. Sci. 2020, 21, 3422. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PTX3 involvement in CKD progression. Expression and production of PTX3 is shown for primary mesangial cells, primary tubular epithelial cells (PTECs) and renal fibroblasts and it is enhanced by IL-1 or TNF-α. Activation of PTECs with IL-17 and CD40L resulted in increased production of PTX3. PTX3 produced by PTECs is able to bind C1q leading to MAC formation, inflammation/infiltration into the tubulo-interstitium, progressive decrease in peritubular capillary (PTC) blood flow, tubulo-interstitial hypoxia and oxidative stress. PTX3 induces cell migration via upregulation of EMT in a JNK-dependent mechanism leading to fibrosis and tubular atrophy.
Figure 1.
PTX3 involvement in CKD progression. Expression and production of PTX3 is shown for primary mesangial cells, primary tubular epithelial cells (PTECs) and renal fibroblasts and it is enhanced by IL-1 or TNF-α. Activation of PTECs with IL-17 and CD40L resulted in increased production of PTX3. PTX3 produced by PTECs is able to bind C1q leading to MAC formation, inflammation/infiltration into the tubulo-interstitium, progressive decrease in peritubular capillary (PTC) blood flow, tubulo-interstitial hypoxia and oxidative stress. PTX3 induces cell migration via upregulation of EMT in a JNK-dependent mechanism leading to fibrosis and tubular atrophy.
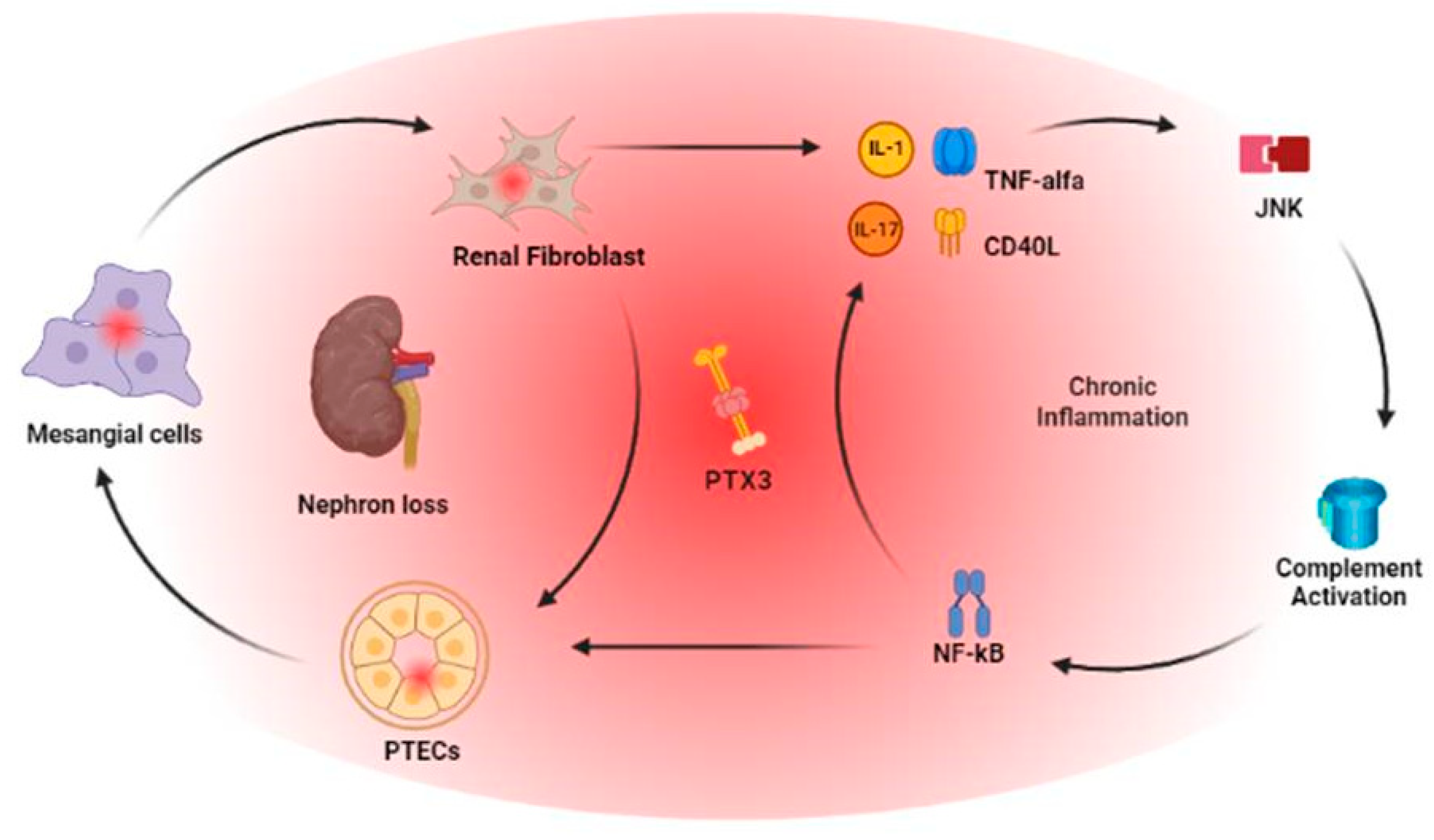
Figure 2.
PTX3 in HD. PTX3 is overexpressed in CKD. Circulating PTX3 interacts with C1q, Ficolins and Mannose binding leptin (MBL) thus determining the over activation of all three complement ways already enhanced during RRT because of the interaction of complement with the dialysis membrane. The upregulation during a single treatment determines: 1. endothelial damage through MAC release and 2. neutrophils activation with consequent ROS release, 3. inflammatory response due to pro-inflammatory cytokines release. More biocompatible membranes could reduce PTX3 production, specific membrane properties/design could target its removal.
Figure 2.
PTX3 in HD. PTX3 is overexpressed in CKD. Circulating PTX3 interacts with C1q, Ficolins and Mannose binding leptin (MBL) thus determining the over activation of all three complement ways already enhanced during RRT because of the interaction of complement with the dialysis membrane. The upregulation during a single treatment determines: 1. endothelial damage through MAC release and 2. neutrophils activation with consequent ROS release, 3. inflammatory response due to pro-inflammatory cytokines release. More biocompatible membranes could reduce PTX3 production, specific membrane properties/design could target its removal.
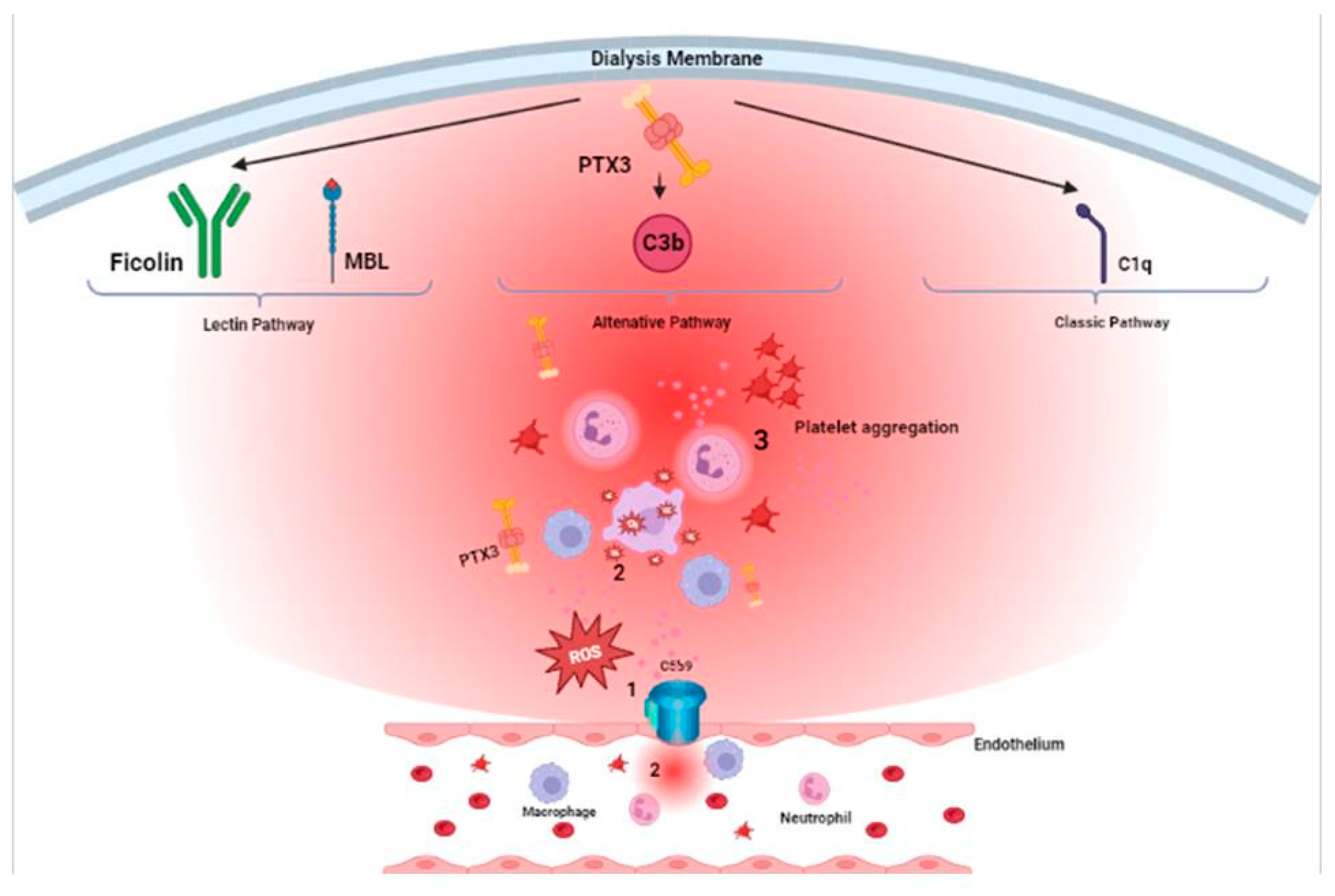
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



