
Submitted:
17 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
Background. The treatment for patients with high-grade gliomas includes radiation therapy and temozolomide. However, some patients do not respond to temozolomide because they have a methylation reversal mechanism through the enzyme O6-methylguanine-DNA-methyltransferase. This biomarker has been used as a prognostic factor in patients receiving treatment with temozolomide. However, not all patients respond in the same way, which suggests the existence of other genes involved in resistance to temozolomide. Materials and Methods. A group of 31 patients with high-grade gliomas was recruited and were clinically, image pattern, and pathologically characterized. The sequencing of 324 genes related to different types of cancer was performed to detect mutations. Subsequently, a statistical analysis was conducted to determine the mutated genes that were most related to resistance to treatment. Results. The genes related to the second relapse of patients with high-grade glioma after the use of temozolomide according to Stupp protocol and metronomic dose were PIK3C2B, KIT, ERBB3, and MLH1. Conclusions. Considering the results obtained, we suggest that the mutations in the four genes and the methylation of the gene promoter that codes for MGMT protein could be related to the clinical evolution of patients with high-grade gliomas and its response to treatment with temozolomide.
Keywords:
gliomas
; temozolomide
; resistance
; genes
; PIK3C2B
; ERBB3
; KIT
; MLH1
; MGMT
1. Introduction
According to the data provided by Cancer Today 2020, the incidence of brain tumors worldwide is 3.5 per 100,000 inhabitants, with a mortality rate of 2.8 per 100,000 inhabitants [1]. The incidence (standardized rate) of cancer in the brain and central nervous system in Colombia ranged from 3.5–4.2 per 100,000 inhabitants, according to data obtained between 2003 and 2016. While mortality ranged between 2.1 and 2.5 per 100,000 inhabitants between 1997 and 2020 (https://infocancer.co/) [2], according to the Brain Tumor Registry (CBTRUS, 2014–2018), the incidence in the United States for primary central nervous system (CNS) tumors was 24 per 100,000, with a mortality rate of 4.3 per 100,000 [3]. Concerning the cases reported in the United States, these data reflect an underreporting of new cases in the rest of the world, including Colombia. Additionally, the CBTRUS estimates that for the United States, the incidence of gliomas, in general, is 5.95, and for the subgroup considered high-grade gliomas, that is, glioblastoma, it is 3.23, anaplastic astrocytoma is 0.41, and anaplastic oligodendroglioma is 0.11 per 100,000 inhabitants [3]. The most abundant cells in the CNS are neurons and glial cells. The glial cells are composed of astrocytes, oligodendrocytes, ependymal cells, and microglia cells. Additionally, meningeal, and pituitary cells are also part of the CNS [4]. When cells have genetic and/or epigenetic alterations, mechanisms such as replication, repair, and senescence are modified, resulting in an uncontrolled proliferation of cells with aggressive characteristics that allow them to invade neighboring tissues and sometimes give rise to metastasis, even in isolated and immune privileged sites such as the brain parenchyma [5]. Tumors originating from astrocytes are called astrocytomas; tumors from oligodendrocytes are oligodendrogliomas; and tumors from ependymal cells are called ependymomas. Additionally, these types of malignant brain tumors are classified according to their degree of differentiation into low-grade and high-grade [6]. High-grade gliomas, now named according to the 2021 WHO classification of tumors of the central nervous system, include IDH wild-type glioblastoma (GBsilv-IDH), IDH-mutated astrocytoma without 1p/19q codeletion, grades 3 and 4 (Amut-IDH, sin-codel-1p/19q, G3-4), and oligodendroglioma mutated for IDH with codeletion 1p/19q grade 3 (Omut-IDH, codel-1p/19q, G3) [7]. Regarding high-grade glioma treatment, the protocol developed by Stupp et al. (2005) is used as standard treatment. This protocol added temozolomide to treatment with radiotherapy, reporting an increase in patient survival (HR not adjusted: 0.63, 95% CI: 0.52–0.75, p < 0.001) and minimal toxicity [8]. However, the treatment was not effective for all patients, so it was necessary to explore at the genomic level which gene was related to resistance to temozolomide [9], finding that promoter methylation of the gene that codes for the MGMT protein had an independent prognostic value of treatment (HR: 0.45, 95%, CI: 0.32–0.61, p < 0.001) and decreased the probability of death (HR: 0.51, 95% CI: 0.31–0.84) in patients in whom temozolomide was added to radiotherapy, the same did not occur in patients who did not have the gene promoter methylated (HR: 0.69, 95% CI: 0.47–1.02) [10], which indicates that this mechanism of resistance to temozolomide does not explain the response to treatment in 100% of the patients. Accordingly, there is a gap in our knowledge about other mechanisms or proteins that are involved in the process of resistance to treatment with temozolomide in patients with high-grade gliomas. Therefore, the objective of our investigation was to identify additional genetic alterations to the promoter's methylation of the gene that codes for the MGMT protein, which helps relate the clinical evolution of patients with high-grade gliomas and their response to treatment with temozolomide.
2. Materials and Methods
2.1. Study population
The study population consisted of oncological patients who attend the Cancer Institute (Las Américas-AUNA Clinic) for management with surgery, radiotherapy, chemotherapy, and oncological support. The study was conducted by open invitation, recruiting patients who met the following inclusion criteria: patients over 18 years of age, diagnosed with a primary malignant brain tumor, who underwent tumor resection surgery or biopsy, and who should have a histological diagnosis of glioblastoma. A total of 31 patients diagnosed with high-grade glioma were included. According to the 2021 WHO classification of tumors of the central nervous system, the patients were classified as 21 glioblastomas, five anaplastic astrocytomas, and five anaplastic oligoastrocytoma. This study is classified as observational, cohort, and ambispective.
2.2. Clinical information
The following data were extracted from clinical history: age, sex, clinical diagnosis, imaging diagnosis, pathology results, molecular biology tests, treatment, and outcomes.
2.3. Sample collection for molecular biology analysis
Twenty-six samples of cancerous brain tissue embedded in paraffin blocks and 5 samples of liquid biopsies were used to conduct DNA extraction, new generation sequencing, and genetic profiling of the 31 patients.
2.4. Sequencing 324 cancer-associated genes
Genomic sequencing was performed using the FoundationOne®CDx (F1CDx) and FoundationONE Liquid CDx (F1LCDx) panels, which are an in vitro diagnostic methodology based on next-generation sequencing for the detection of mutations such as substitutions, insertions, and copy number alterations (CNAs) in 324 genes. Additionally, gene rearrangements and genomic fingerprinting, including microsatellite instability (MSI), tumor mutational burden (TMB), and loss of heterozygosity (LOH), are detected [11].
2.5. Mutated genes identification related with temozolomide resistance mechanisms
A univariate analysis was performed, in which summary measures, including measures of central tendency, were calculated for quantitative variables, along with their respective measures of dispersion, according to the distribution of the variable (Shapiro-Wilk test). For qualitative variables, absolute and relative frequencies were calculated. The results were presented through graphs and tables. To perform the bivariate analysis, the Logrank test, simple Cox regression models, Kaplan-Meier analysis, and survival curves were used. The results were presented in tables and a survival curve graph. To perform the multivariate model, a multiple Cox regression model was performed. For the entry of the variables into the final model, the behavior of the bivariate analysis was considered; that is, those variables that met the following criteria were entered into the model: statistical significance with p < 0.05, Hosmer Lemechow criterion with p < 0.25, and according to the criteria of the investigator (biological plausibility). Variables were entered progressively, and interaction analysis was performed for each model until reaching the final model, considering the principle of parsimony. Statistical analyses were performed using STATA (version 14) and SPSS (version 28) statistical programs.
2.6. Bioinformatic analysis of mutations effect on the proteins
The chemical properties of the proteins were identified using the ProtParam tool web server (https://web.expasy.org/protparam/). The identification of the chemical properties of the amino acids was conducted by means of the GPMAW web server (https://www.alphalyse.com/customer-support/gpmaw-lite-bioinformatics-tool/start-gpmaw-lite/) and the color protein sequence web server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_color.html). The effect of the mutations on the proteins was determined by the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.bii.a-star.edu.sg/) web servers. The signal peptide of the proteins was identified using the SignalP-6 web server (https://dtu.biolib.com/SignalP-6). The InterProScan web server was used to identify the domains, biological processes, molecular function, and cellular component of the four proteins (https://www.ebi.ac.uk/interpro). Subsequently, using the String Interaction Network web server (STRING) and Genecards database, the interactions and metabolic pathways in which the proteins participate were identified (https://string-db.org/ and https://www.genecards.org). The three-dimensional structure of the proteins was modeled using the Swiss-Model web server (https://swissmodel.expasy.org), and the structural change caused by the mutations was determined using the Swiss-pdb-viewer bioinformatics program (http://spdbv.unil.ch), to establish how the structure and function of the protein were affected. Finally, all the diseases that have been associated with mutations in the four genes were identified using the OMIM database (https://www.omim.org).
3. Results
3.1. Study population
A total of 31 patients, who met the inclusion criteria and agreed to enter the study by signing the informed consent, participated in the present study. In relation to the clinical data, the mean age at diagnosis of high-grade glioma was 47 years old (SD 14.52). Regarding gender, 11 patients were male (35.49%) and 20 were female (64.51%).
3.2. Clinical information
The Karnofsky scale was applied to the 31 patients included in the study to assess their functional status at the time of diagnosis, finding values equal to or greater than 70% in all patients, which indicated that all were fit to be taken to surgery. All patients underwent contrasted preoperative magnetic resonance imaging of the brain, which was recorded in the patients' medical records. Without discriminating by which cerebral hemisphere was injured and considering that sometimes the lesions are not confined to a single lobe, it was found that 32% of the patients had the lesion in the frontal lobe, 16% in the parietal, 22% in the temporal, 26% in the occipital, and only one patient had a lesion in the basal ganglia. A grossly complete resection was performed on 45% of the patients, and a partial resection or biopsy was performed on the remainder (55%). According to the pathology report, based on hematoxylin-eosin (H-E) and immunohistochemistry (IHC), it was found that 21 (68%) of the patients had glioblastomas, five (16%) had anaplastic astrocytomas, and five (16%) had oligodendroglial lineage tumors. The latter group was made up of four patients with anaplastic oligoastrocytomas and one patient with an anaplastic oligodendroglioma; see Figure 1.
All patients were given concomitant treatment with temozolomide and radiotherapy, followed by adjuvant chemotherapy four to six weeks after surgery. In 15 patients, it was possible to prevent tumor growth with six cycles of temozolomide as recommended by the Stupp Protocol (SP). However, five patients received fewer cycles due to clinical deterioration and tumor growth (progression). While in the remaining 11 patients it was necessary to administer more than six cycles of temozolomide to prevent tumor growth; see Figure 2.
Patients were followed clinically and with imaging, until tumor growth was observed again (first relapse). All patients were given metronomic temozolomide; that is, a dose of 75 mg/mt2 every day to try to avoid the effect of MGMT as this enzyme has a very short half-life. The response of patients to treatment with metronomic temozolomide was variable, ranging from patients who reached the second relapse in the second treatment cycle to patients who reached the second relapse in the 21 cycles of temozolomide treatment at metronomic doses; see Figure 3.
3.3. Sample collection for molecular biology analysis
To classify high-grade gliomas, the following molecular analyses were performed on the samples obtained from the first surgery: a search of the 1p/19q codeletion by FISH in three patients (10% of the sample), which helped to classify them as anaplastic oligoastrocytoma. Immunohistochemistry (IHC) for IDH1 was performed on 19 patients (61%), reporting positive results in 4/10 glioblastomas (40%), 3/4 anaplastic astrocytomas (75%), and in 5/5 oligodendroglial tumors (100%). The methylation status of the promoter of the gene that codes for the MGMT protein was searched by polymerase chain reaction (PCR) in 18 patients, finding methylation in 3/10 glioblastomas (30%), 2/3 anaplastic astrocytomas (66%), and 4/4 tumors of the oligodendroglial lineage (100%); see Supplementary Material 1.
3.4. Sequencing 324 cancer-associated genes
The genetic material was extracted from the 31 samples, and 324 cancer-related genes were sequenced and analyzed according to the protocol described by the FoundationOne®CDx (F1CDx) and FoundationONE Liquid CDx (F1LCDx) panels. An amplification, sequencing, and mutation detection was conducted on each of the genes. According to the results obtained in the gene profiling of the 31 patients with high-grade gliomas, 370 types of mutations were found in 185 genes. The ten most frequently mutated genes, in order of frequency, for the 21 patients initially diagnosed with glioblastomas were EGFR (62%), TERT (62%), PDGFA (57%), TP53 43%, CDKN2A/B 29%), NF1 (24%), ALK (19%), EP300 (19%), KDR (19%), and MLL2 (19%). The most frequently mutated genes in the five patients diagnosed with anaplastic astrocytomas were IDH1 (40%), TP53 (40%), CCN2 (40%), KEAP1 (40%), KIT 40%, MAF (40%), PDGFRA (40%), SPEN (40%), TERT 40%), and TYRO3 (40%). The most frequently mutated genes in the group of five patients with anaplastic oligoastrocytomas were TSC1 (75%), SPEN (50%), IDH (25%), ALK (25%), ARID1A (25%), CIC (25%), MAP3K1 (25%), MLL2 (25%), NF1 (25%), and POLE (25%); see supplementary material 2.
3.5. Mutated genes identification related with temozolomide resistance mechanisms
The simple Cox's regression analysis presents the comparison between the dependent variable "second relapse" (at 22 months) and the presence of mutated genes. Seventy-four mutated genes were found in patients who presented a second relapse. The genes with the greatest statistical significance found were PIK3C2B HR 13.81 (95%, CI: 2.25–84.45, p = 0.004), NOTCH3 HR 6.02 (95%, CI: 1.74–20.84, p = 0.005), KIT HR 3.98 (95%, CI: 1.20–13.18, p = 0.024), ERBB3 HR 3.87 (95%, CI: 1.06–14.04, p = 0.04), and MLH1 HR 3.52 (95%, CI: 0.95–13.09, p = 0.06); see Table 2.
As a second analysis, a multivariate analysis was performed to determine which mutated genes were related to the second relapse, reducing the number to four genes: PIK3C2B with an HR of 82.37 (95%, CI: 8.36–111.67, p = 0.000), KIT with an HR 10.24 (95%, CI: 2.42–43.34, p = 0.002), ERBB3 with an HR 13.20 (95%, CI: 2.77–62.77, p = 0.001), and MLH1 with an HR 8.50 (95%, CI: 1.83–39.45, p = 0.006); see Table 3.
As a third analysis, a survival curve was performed using the Kaplan-Meier method in patients who had mutations in the four genes (PIK3C2B, ERBB3, KIT, and MLH1). This analysis was performed to estimate the time it takes for patients to reach a second relapse after treatment with metronomic temozolomide compared to patients without mutations in these genes. When analyzing the results of the survival curves of the patients who had mutations, it was observed that they presented the second relapse in a shorter time compared to the patients who did not have mutations in these four genes, as can be seen in Figure 4.
3.6. Bioinformatic analysis of mutations effect on the proteins
Based on the PIK3C2B, ERBB3, KIT, and MLH1 mutated genes that were found to be related to the second relapse after the use of temozolomide according to the Stupp protocol and the administration of a metronomic dose, an in-silico bioinformatics analysis was performed. As a first analysis, the functional domains in the four proteins encoded by mutated genes were detected, and whether the different mutations found affected them. Finding that for the PIK3C2B protein, the domain (PI3K_Ras-bd_dom) was affected; for the ERBB3 protein, the domain (Rcp_L) was affected; for the KIT protein, no mutations were found affecting functional domains; and for the MLH1 protein, two domains were affected (DNA_mismatch_repair_N and DNA_mismatch_S5_2-like). Additionally, the biological process, molecular function, and cellular components of the proteins encoded by the four mutated genes are described in Table 4.
In parallel, the interactions and metabolic pathways of the proteins were identified. The PIK3C2B protein interacts with five proteins (RICTOR, EGFR, GRB2, ANKRD32, and SBF2) and participates in five metabolic pathways (DNA damage response, focal adhesion: PI3K-Akt-mTOR-signaling pathway, glioblastoma signaling pathways, phosphoinositide metabolism, and the regulation of actin cytoskeleton). The ERBB3 protein interacts with five proteins (ERBB2, EGFR, NNRG1, GRB2, and SHC1) and participates in five metabolic pathways (apoptosis-related network due to altered Notch3 in ovarian cancer, EGFR tyrosine kinase inhibitor resistance, the ErbB signaling pathway, glioblastoma signaling pathways, and heart development). The KIT protein interacts with five proteins (PIK3R1, PLCG1, KITLG, GRB2, and CRK) and participates in five metabolic pathways (the breast cancer pathway, cardiac progenitor differentiation, focal adhesion: PI3K-Akt-mTOR-signaling pathway, gastrin signaling pathway, and Hippo signaling regulation pathways). The MLH1 protein interacts with five proteins (MSH2, PMS2, PMS1, BLM, and EXO1) and participates in five metabolic pathways (chromosomal and microsatellite instability in colorectal cancer, DNA IR-damage and cellular response via ATR, DNA mismatch repair, DNA repair pathways, and ovarian infertility); see Table 5.
Subsequently, the three-dimensional structure of each of the four proteins was modeled, and the structural effect caused by each of the mutations was determined. The PIK3C2B protein is made up of 1634 amino acids, has a molecular weight of 184739.79 Da, a theoretical isoelectric point of 6.87, 194 negatively charged residues (Asp + Glu), 189 positively charged residues (Arg + Lys), an atomic composition of 8246 carbon, 12875 hydrogen, 2281 nitrogen, 2414 oxygen, and 67 sulfur atoms, an estimated half-life of 30 hours (mammalian reticulocytes, in vitro). Its instability index is computed to be 46.22 and classified as unstable. The R458Q mutation was detected in the protein. The wild-type amino acid is an arginine (R) that is classified as a basic, which is replaced by a glutamine (Q) that is hydrophilic. According to the results of PolyPhen-2 (prediction of functional effects of human nsSNPs), this mutation is predicted to be probably damaging with a score of 0.979 (sensitivity: 0.76; specificity: 0.96). According to the results of SIFT (Sorting Intolerant From Tolerant), substitution at position 458 from R to Q is predicted to be tolerated with a score of 0.45. No signal peptide was detected using the SignalP bioinformatics program (version 6.0). The effect of the mutation on the structure of the protein leads to the shortening of the radical, reducing the Van der Waals forces, which possibly affects the attraction between atoms, molecules, and surfaces. At the structural level, the mutation is located between the amino acids (457–464) that make up a beta sheet, generating an elongation of the beta sheet in the mutated protein. All these alterations can lead to instability and loss of functionality of the protein; see Table 6.
The ERBB3 protein is made up of 1342 amino acids, has a molecular weight of 148098.19 Da, a theoretical isoelectric point of 6.11, 154 negatively charged residues (Asp + Glu), 132 positively charged residues (Arg + Lys), an atomic composition of 6447 carbon, 10126 hydrogen, 1852 nitrogen, 1969 oxygen, and 94 sulfur atoms, an estimated half-life of 30 hours (mammalian reticulocytes, in vitro).Its instability index is computed to be 49.61 and classifies as unstable. The R164K mutation was detected in the protein. The wild-type amino acid is an arginine (R) that is classified as a basic, which is replaced by a lysine (K) that is hydrophilic. According to the results of PolyPhen-2, this mutation is predicted to be probably benign with a score of 0.017 (sensitivity: 0.95; specificity: 0.80). According to the results of SIFT substitution at position 164 from R to K, it is predicted to be tolerated with a score of 0.64. No signal peptide was detected using the SignalP bioinformatics program (version 6.0). At the structural level, an amino acid change is observed that leads to an alteration in the orientation of the radical group that alters the spatial distribution of Van der Waals forces, which possibly affects the attractions between atoms, molecules, and surfaces; see Table 6.
The KIT protein is made up of 977 amino acids, has a molecular weight of 109992.70 Da, a theoretical isoelectric point of 6.54, 111 negatively charged residues (Asp + Glu), 106 positively charged residues (Arg + Lys), an atomic composition of 4925 carbon, 7657 hydrogen, 1301n, 1460 oxygen, and 48 sulfur atoms, and an estimated half-life of 30 hours (mammalian reticulocytes, in vitro). Its instability index is computed to be 39.43 and classified as stable. No signal peptide was detected using the SignalP bioinformatics program (version 6.0). This protein did not present a structural alteration but did show an increase in the number of copies of the KIT gene in the patient's genome. This genetic phenomenon is common in cancer cells, which produce several copies of one or more genes in response to signals from other cells or the environment.
The MLH1 protein is made up of 756 amino acids, has a molecular weight of 84600 Da, a theoretical isoelectric point of 5.51, 104 negatively charged residues (Asp + Glu), 83 positively charged residues (Arg + Lys), an atomic composition of 3740 carbon, 5947 hydrogen, 1017 nitrogen, 1165 oxygen, and 25 sulfur atoms, and an estimated half-life of 30 hours (mammalian reticulocytes, in vitro). Its instability index is computed to be 51.29 and classified as unstable. The F261fs*31 mutation was detected in the protein. No signal peptide was detected using the SignalP bioinformatics program (version 6.0). The deletion of a cytokine at position 783 of MLH1 gene (783delC), leads to structural change in the MLH1 protein (p.Phe261fs*7). This sequence change creates a premature translational stop signal of 7 amino acids after phenylalanine (Frameshift). It is expected to result in an absent or disrupted protein product (this could lead to the production of a truncated protein of 268 amino acids); see Table 6.
Finally, the diseases associated with each of the four mutated genes are identified using the OMIM database (An Online Catalog of Human Genes and Genetic Disorders). Mutations in the gene that codes for the PIK3C2B protein have been associated with the following diseases: high-grade glioma, Maffucci, and Hepatitis C. Mutations in the gene that codes for ERBB3 protein have been associated with the following diseases: high grade-glioma, breast cancer, frontal myelination, enteric nervous system, and diabetes type 1. Mutations in the gene that codes for KIT protein have been associated with the following diseases: high grade glioma, piebaldism, gastrointestinal stromal tumor (GIST), mastocytosis, liver cell membrane autoantibody (LMA), melanoma, and germ cell tumors. Mutations in the gene that codes for MLH1 protein have been associated with the following diseases: high grade glioma, hereditary nonpolyposis colorectal cancer (Lynch), and Turcot syndrome type I; see Table 7.
4. Discussion
According to the Central Brain Tumor Registry of the United States (CBTRUS), gliomas represent 25% of primary central nervous system tumors in adults and are more prevalent in men than in women. In the age range between 15 and 39 years, the oligodendrogliomas and astrocytomas predominate. For those over 40 years of age, the most frequent are glioblastomas [3]. These results are consistent with our patient population, in which 21/31 patients presented with glioblastomas. The 31 patients included in this study had a mean age of 47 (SD: 14.5), and women predominated (65.4%). This result is due to the small number of patients, which is not representative of the population. Traditionally, patients with glial tumors were classified using H-E, and in some cases, the search for the mutation in the gene that codes for Isocitrate Dehydrogenase (IDH) is performed by immunohistochemistry (IHC) according to the WHO 2016 criteria [12]. Based on these two analyses, we obtained the following results in the 31 patients with high-grade gliomas: 21 glioblastomas, five anaplastic astrocytomas, and five anaplastic oligoastrocytoma; see Supplementary Material 1. Currently, the diagnosis of high-grade gliomas is made not only with H-E and IHC but also includes the search for genetic mutations, epigenetic changes, and chromosomal alterations [13]. Microvascular proliferation and necrosis are evaluated using H-E, atypia, mitotic activity, and increased cell density [14]. Meanwhile, IHC makes it possible to verify, among other things, that tumor cells are derived from glia [15]. As noted above, molecular biology studies are the cornerstones of the new 2021 WHO classification of tumors of the central nervous system and allow the classification of high-grade gliomas such as glioblastoma (GBsilv-IDH), in which mutations in the TERT promoter, EGFR promoter, chromosome 7 trisomy, and monosomy chromosome 10 are frequent; Astrocytoma (Amut-IDH, sin-codel-1p/19q, G3-4), in which mutations in the ATRX, TP53 and CDKN2A/B genes can be found; and Oligodendroglioma (Omut-IDH, codel-1p/19q, G3), in which TERT, CIC, FUBP1, and NOTCH1 promoter mutations are common [7]. Therefore, the group of 31 patients was re-evaluated, and patients with high-grade gliomas were classified as 21 previously diagnosed as glioblastoma, of which 19 of them remained GBwt-IDH, and two changed to Amut-IDH, without codel 1p/19q, G-3, 4. Of the five patients with anaplastic astrocytoma, four retained the diagnosis, and one switched to Omut-IDH, codel 1p/19q, G-3. Of the 5 patients with anaplastic oligoastrocytoma, four retained the diagnosis, and one switched to Amut-IDH, codel 1p/19q, G-3, 4, which highlights the importance of molecular methods for a correct classification of high-grade gliomas; see Supplementary Material 1. Performing an accurate diagnosis on these patients would help us provide adequate treatment, which consists of the widest surgical cytoreduction [16], followed by the Stupp Protocol [17], except for the Omut-IDH, codel -1p/19q, G3, in whom the tendency is to give only postoperative temozolomide, postponing radiotherapy [18,19].
Additionally, if an accurate diagnosis is made, we could explain the patient’s life expectancy to them and their family with greater certainty. In our group of patients, at the cut-off point of the study (two years), those with glioblastoma had a median survival of 29 months, not including two long surviving patients. Only 2/5 patients with anaplastic astrocytomas had died (one at 14.5 months and the other at 19 months), and all the patients with anaplastic oligoastrocytoma were still alive. Survival, based on the new 2021 WHO classification, has not yet been calculated for GBsilv-IDH, Amut-IDH, sin-codel-1p/19q, G3-4, or Omut-IDH, codel-1p/ 19q, G3. The perception that patients treated at the Cancer Institute (Las Américas-AUNA clinic) had a better response to treatment can be partly explained by the erroneous diagnosis made. The two patients diagnosed with glioblastomas were expected to survive about two years (14.8% patients) and ten years (2.6%), but they really had an Amut-IDH, sin-codel-1p/19q, G3-4, and these patients were expected to survive about two years (43.3%) and ten years (19% patients) [20]. The patient diagnosed with anaplastic astrocytoma were expected to survive about two years (43.3%) and ten years (19%), but they really had an Omut-IDH, codel-1p/19q, G3, and these patients are expected to survive about two years (68.6% patients) and ten years (39.3% patients) [20]. Finally, the patient diagnosed with anaplastic oligoastrocytoma were expected to survive about two years (68.6%) and ten years (39.3%), but they really had an Amut-IDH, sin-codel-1p/19q, G3-4, and these patients are expected to survive between two years (43.3%) and ten years (19%) [20], which explains its worse prognosis; see Supplementary Material 1. The previous results highlight the importance of making a good diagnosis of the type of high-grade glioma to give accurate information to the patient and their relatives about the outcome, that is, the overall survival (OS) and the progression-free survival time (PFS).
Temozolomide´s mechanism of action is to methylate the O6 position of guanines, which results in a mismatch between guanines and cytosines (O6 guanine pairing with a thymine), which leads to activation of the MMR system [21,22]. This only repairs the chain that contains thymine, accumulating the chains with methylated guanines [23]. Therefore, intra and inter covalent bonds occur, forming hairpins that prevent cell replication and lead to apoptosis [24]. However, cancer cells have a mechanism to reverse the effect of temozolomide through the transcription and subsequent translation of the MGMT protein, which, through a sulfhydryl group, removes the methyl group from O6 methyl-guanine, restoring guanine to its original form [25]. This is reflected when analyzing how an increase in survival was observed (HR: 0.51, 95%, CI: 0.31–0.84) in patients whose tumor had the MGMT gene promoter methylated, and who received radiotherapy and temozolomide, compared to those who received radiotherapy only; while the patients who did not have promoter methylation and who received radiotherapy and temozolomide did not observe a significant increase in survival (HR: 0.69, 95%, CI: 0.47–1.02), when compared with those who only received radiotherapy [10]. In practice, the search for methylation of the MGMT gene only has prognostic value and does not predict a response to treatment [26], so all patients with high-grade gliomas receive temozolomide [27]. Theoretically, patients with MGMT promoter methylation should be expected to respond 100% to treatment, but only a 51% decrease in risk of death has been observed. Additionally, patients without MGMT promoter methylation would be expected to have no response to treatment, but a 31% decreased risk of death has been observed [10]. This highlights the need to find alterations in new genes to improve the prognosis and treatment response of the patients with high-grade gliomas. Promoter methylation was identified in 17 of the 31 patients (10 glioblastomas, three anaplastic astrocytomas, and four anaplastic oligodendrogliomas); see Supplementary Material 1. The methylation data obtained in this study are consistent with what has been reported in the literature according to different types of high-grade gliomas since methylation of the MGMT promoter is found in 50% of glioblastomas, in 75% of anaplastic astrocytomas, and in almost all anaplastic oligodendrogliomas [28]. Overall Survival (OS) in patients with glioblastoma promoter methylation in our study was 59.2 months, compared to 24.6 months for those without promoter methylation. Overall Survival cannot be calculated in patients with anaplastic astrocytomas and anaplastic oligoastrocytomas because, at 24 months (the end of the investigation), 5/10 patients were still alive; therefore, progression-free survival (PFS) was analyzed. In the present investigation, we found that PFS at first and second relapse was higher in patients with gliomas who had the methylated promoter of the gene that codes for MGMT protein compared to those who did not; see Supplementary Material 1. These results are consistent with those reported in the literature [28]. However, new biomarkers are required, which, in association with the methylation of the MGMT promoter, make it possible to predict the response of patients with high-grade glioma to the use of temozolomide.
Therefore, in the present study, the detection and analysis of mutations was performed on 324 cancer-related genes in a group of 31 patients with high-grade gliomas to detect the genes that are related to the patients’ prognoses and resistance to temozolomide, finding 185 mutated genes with 370 different mutations. As a first statistical analysis, a bivariate analysis was conducted (Cox’s regression models, Kaplan-Meyer analysis, and survival curves), through which the relationship of each mutated gene with the second relapse was evaluated after the use of temozolomide as part of the Stupp protocol and metronomic dose, finding 71 genes related to second relapse; however, only the first five genes had significant values of p-value and Hazard Ratio; see Table 2. As a second statistical analysis, a multivariate analysis was performed, and Cox’s regression models were used to determine the mutated genes that were related to the second relapse. The genes with statistically significant values were PIK3C2B with a crude HR of 13.81 (95%, CI: 2.25–84.45, p = 0.004), KIT with a HR of 3.98 (95%, CI: 1.20–13.18, p = 0.024), ERBB3 with a HR of 3.87 (95%, CI: 1.06–14.04, p = 0.04), and MLH1 with a HR of 3.52 (95%, CI: 0.95–13.09, p = 0.06); see Table 3. Additionally, Progression-Free Survival (PFS) was assessed at first and second relapse among patients with and without mutations in the PIK3C2B, ERBB3, KIT, and MLH1 genes. At the first relapse, the patients who had the mutations in the genes presented a lower PFS (8.85 months) than those who lacked the mutations (27.55 months). Also, PFS was compared for the second relapse and patients with mutations in these four genes had lower PFS (19.3 months) than those without mutations (38.17 months). Besides, it is observed that the patients who have mutations in the PIK3C2B gene had the lowest survival free progression. Therefore, presenting mutations in any of these four genes increases the probability of relapse in the patients; see Supplementary Material 1.
In the study we found that PIK3C2B, ERBB3, KIT, and MLH1 genes had different types of mutations, which could affect the structure and functional domains of the proteins. Additionally, these alterations could be affecting different interactions and metabolic pathways, which could help us to hypothesize the mechanisms used by cancer cells to proliferate and acquire resistance to temozolomide treatment. After evaluating the PIK3C2B gene, a mutation, amplification, and rearrangement were found. PIK3C2B protein is involved in the biosynthetic and signaling process of phosphatidylinositol phosphatase called PIK3C2B (phosphatidylinositol-4-phosphate 3-kinase C2), which is part of a family of enzymes capable of phosphorylating the hydroxyl group at the 3' position of the inositol ring of molecules, collectively called phosphatidylinositol, which convert phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3), and then to phosphorylate AKT, among others [29,30,31]. Regarding the ERBB3 gene, two mutations were found: R164K, which affects the domain (Rcp_L) and L1177l, which does not affect any functional domain, but it affects the structure of the protein. This gene encodes a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. This membrane-bound protein has a neuregulin binding domain but not an active kinase domain. Therefore, it can bind this ligand but not convey the signal into the cell through protein phosphorylation. However, it does form heterodimers with other EGF receptor family members which do have kinase activity. Heterodimerization leads to the activation of pathways, which lead to cell proliferation or differentiation. Amplification of this gene and/or overexpression of its protein have been reported in numerous cancers, including prostate, bladder, and breast tumors [32,33]. Regarding the KIT gene, an amplification and equivocal amplification were found. This gene encodes a receptor tyrosine kinase. It was initially identified as a homolog of the feline sarcoma viral oncogene v-kit and is often referred to as proto-oncogene c-Kit. The canonical form of this glycosylated transmembrane protein has an N-terminal extracellular region with five immunoglobulin-like domains, a transmembrane region, and an intracellular tyrosine kinase domain at the C-terminus. Upon activation by its cytokine ligand, stem cell factor (SCF), this protein phosphorylates multiple intracellular proteins that play a role in in the proliferation, differentiation, migration, and apoptosis of many cell types and thereby plays an important role in hematopoiesis, stem cell maintenance, gametogenesis, melanogenesis, and in mast cell development, migration, and function. This protein can be a membrane-bound or soluble protein [34,45]. Regarding the MLH1 gene, the F261fs*7 mutation was found. The protein encoded by this gene can heterodimerize with mismatch repair endonuclease PMS2 to form MutL alpha, part of the DNA mismatch repair system. When MutL alpha is bound by MutS beta and some accessory proteins, the PMS2 subunit of MutL alpha introduces a single strand break near DNA mismatches, providing an entry point for exonuclease degradation. This protein is also involved in DNA damage signaling and can heterodimerize with DNA mismatch repair protein MLH3 to form MutL gamma, which is involved in meiosis [36].
According to the previous biological functions, two possible mechanisms of resistance to temozolomide are postulated, in which these four genes could be involved. Regarding the first resistance mechanism, the catalytically inactive human growth factor receptor 3 (ERBB3) alone can serve as the kinase domain activating partner in a heterodimer in association with ERBB2 (Her2) [33]. Its subsequently transactivated carboxyl-terminal tail domain binds to the SH2 domain of all three PI3K regulatory subunits [37]. The KIT proto-oncogene transcribes the homologous receptor of the feline viral sarcoma v-kit, also called CD117 (c-Kit), which, with its binding to the physiological ligand, produces dimerization, which leads to transphosphorylation. This, in turn, reorients the domain intracellular membrane, freeing it from the autoinhibitory conformation that it acquires in an inactive state, and facilitating its catalytic function [34]. Like the previous receptor, some of these phosphorylation sites in the inner membrane domain of c-Kit bind with SH2 domains, forming docking sites for signaling and activation of the pathway (PI3K, Akt, TSC1/2, mTOR1/2, and transcription factor) [38]. Under physiological conditions, growth factor stimulation, in this case, neuregulin for ERBB3 [39] and stem cell factor for c-kit [40], phosphorylates enzyme-specific kinases that make up PI3K through sequential association with adapter molecules (GRB2-SOS-RAS cascade); activated PI3K phosphorylates and converts PIP2 to PIP3, located on the mid surface of the plasma membrane. Activated AKT phosphorylates and inactivates the TSC1/2 complex. This activates mTORC1 by inhibiting mTORC1 suppression mediated by the Ras homolog of GTP-binding protein enriched in the brain (RHEB) to stimulate cell proliferation and survival [29]. In the RTK-PI3K-mTOR2 pathway, the latter binds to ribosomes and activates AGC subfamily kinases (glucocorticoid/serum-induced Akt kinases and PKCα), which stimulates cell migration [41]. In relation to kinase enzymes, which are part of the PI3K family, and from which phosphorylate inositol phospholipids (PI)—concentrated on the cytosolic surface of membranes—are synthesized, especially in the endoplasmic reticulum, they have more than a structural function; they have important roles in cell signaling [42]. Eighteen percent of the glioblastomas have a somatic mutation of the PIK3CA and PIK3R1 genes, which encode the p85 subunit and the p110 subunit of PI3K, respectively [43]. Most PIK3CA and PIK3R1 mutations are frequently in the domain required for the interaction of the p85 subunit with the p110 subunit [42]. Inhibition of p110 subunit activity by the p85 subunit is lost, and PI3K mutants become constitutively active, resulting in sustained AKT activity [43]. PIK3C2B is a catalytic enzyme of the PI3K family, whose functions are to phosphorylate the hydroxyl group at the 3' position of the inositol ring in the plasma membrane and to generate important second messengers such as PIP2 and PIP3 [44]. PIK3C2B amplification [45] has been described, associated with MDM4 by 1q32.1 amplification, and corresponding to 7.7% of the amplifications detected in glioblastomas [46,47]. PIK3C2B has a dual function, since it can stimulate or repress, depending on how it is programmed [48]. It is involved in the AKT activation of neurons [49], in the repression of mTORC1 [50], in the epithelial-mesenchymal transition [50], in the migration of cancer cells [51], in promoting invasion [52], and in the resistance to cisplatin [53], docetaxel [54], and erlotinib in patients who have the EGFR mutation [55]. In relation to the role of this mutation in the possible resistance to temozolomide, it would possibly be by stimulating the transcription factor NFKB and, in turn, MDR1 [23].
Regarding the second resistance mechanism, alterations in genes that are part of the DNA mismatch repair system have been reported in recurrent glioblastomas after the use of temozolomide [56]. Analysis of the Cancer Genome Atlas (TCGA) revealed a hypermutator phenotype, with mutations in at least one of the MMR genes (MLH1, MSH2, MSH6, or PMS2), suggesting either an escape from MGMT methylation or the selection of MMR mutated clones [57]. Felsberg et al. reported changes in promoter methylation and expression of the MGMT, MLH1, MSH2, MSH6, and PMS2 genes after relapse in 80 patients with glioblastomas, finding that only four patients (6.25%) had a loss or decreased methylation of the MGMT promoter at recurrence, and although none of the four genes that are part of MMR had promoter hypermethylation, they did have mutations, which was confirmed by IHC [58]. DNA double-strand breaks by temozolomide activate the homologous repair and non-homologous end-splicing systems, which, through the ataxia telangiectasia (ATM)-checkpoint kinase 2 (CHK2) pathways, lead to p53 damage repair caused by temozolomide or trigger apoptosis [59]. Other mechanisms of resistance to temozolomide consist of the proper functioning or overactivation of some of the BER components, such as APGN, which would repair methylation at N7 of guanine and N3 of adenine [60]. Resistance to temozolomide can develop due to the overactivation of MDM2, which increases the X-linked inhibitor of the apoptosis protein (XIAP). This key regulator of both intrinsic and extrinsic programmed cell death signaling functions by suppressing the activation of caspases 3, 7, and 9, triggering their degradation mediated by ubiquitination or by the improper functioning of p53; therefore, it is unable to activate the Bcl2 family and the activation of the DR5 receptor [61].
5. Conclusion
Considering the results obtained, we suggest that the mutations in the PIK3C2B, ERBB3, KIT, MLH1 genes, and the methylation of the promoter of the gene that codes for MGMT could be related to clinical evolution of patients with high-grade gliomas and their response to treatment with temozolomide.
Author Contributions
All the authors participated in the formulation, execution, and analysis of the results of the project. In addition, all the authors contributed to the writing, revision, and submission of the manuscript (LDOG, HJCM, PAF, DGP, and RGPS).
Funding
This research was funded by CES University and Americas-AUNA clinic.
Institutional Review Board Statement
The project, entitled "Comprehensive genomic profiling and its usefulness as a prognostic factor for response in patients with high-grade gliomas at second relapse," was conducted according to the guidelines of the Declaration of Helsinki and approved by the ethics committees of the CES University (session 122 of August 5, 2018) and the Cancer Institute of the Americas-AUNA Clinic.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors.
Acknowledgments
The authors thank the Graduate School of the CES University for the training of doctoral students (LDOG) and Américas-AUNA clinic for allowing us to conduct this research.
Conflicts of Interest
This research does not present a conflict of interest.
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021 May;71(3):209–49. [CrossRef]
- Gómez Vega JC, Ocampo Navia MI, De Vries E, Feo Lee O. Sobrevida de los tumores cerebrales primarios en Colombia. Univ Médica [Internet]. 2020 May 7 [cited 2022 Jul 31];61(3).
- Low JT, Ostrom QT, Cioffi G, Neff C, Waite KA, Kruchko C, et al. Primary brain and other central nervous system tumors in the United States (2014-2018): A summary of the CBTRUS statistical report for clinicians. Neuro-Oncol Pract. 2022 May 13;9(3):165–82. [CrossRef]
- Jeans A, Esiri M. Brain histology. Pract Neurol. 2008 Oct 1;8(5):303–10.
- Goryaynov SA, Potapov AA, Ignatenko MA, Zhukov VYu, Protskiy SV, Zakharova NA, et al. Glioblastoma metastases: a literature review and a description of six clinical observations. Vopr Neirokhirurgii Im NN Burdenko. 2015;79(2):33. [CrossRef]
- Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics. 2017 Apr;14(2):284–97. [CrossRef]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-Oncol. 2021 Aug 2;23(8):1231–51. [CrossRef]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005 Mar 10;352(10):987–96. [CrossRef]
- Martinez R, Esteller M. The DNA methylome of glioblastoma multiforme. Neurobiol Dis. 2010 Jul;39(1):40–6. [CrossRef]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N Engl J Med. 2005 Mar 10;352(10):997–1003. [CrossRef]
- Woodhouse R, Li M, Hughes J, Delfosse D, Skoletsky J, Ma P, et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. Ha P, editor. PLOS ONE. 2020 Sep 25;15(9): e0237802. [CrossRef]
- Back, M., Rodriguez, M., Jayamanne, D., Khasraw, M., Lee, A., & Wheeler, H. (2018). Understanding the Revised Fourth Edition of the World Health Organization Classification of Tumours of the Central Nervous System (2016) for Clinical Decision-making: A Guide for Oncologists Managing Patients with Glioma. Clinical oncology, 30(9), 556–562. [CrossRef]
- Komori T. Grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab Invest. 2022 Feb;102(2):126–33. [CrossRef]
- Mikkelsen VE, Solheim O, Salvesen Ø, Torp SH. The histological representativeness of glioblastoma tissue samples. Acta Neurochir (Wien). 2021 Jul;163(7):1911–20. [CrossRef]
- Iwanaga T, Takahashi Y, Fujita T. Immunohistochemistry of neuron-specific and glia-specific proteins. Arch Histol Cytol. 1989;52(Suppl):13–24. [CrossRef]
- Hervey-Jumper SL, Berger MS. Maximizing safe resection of low- and high-grade glioma. J Neurooncol. 2016 Nov;130(2):269–82. [CrossRef]
- Lakomy R, Kazda T, Selingerova I, Poprach A, Pospisil P, Belanova R, et al. Real-World Evidence in Glioblastoma: Stupp’s Regimen After a Decade. Front Oncol. 2020 Jul 3;10:840. [CrossRef]
- Paleologos N, Newton H, editors. Oligodendroglioma: clinical presentation, pathology, molecular biology, imaging, and treatment. San Diego: Elsevier/Academic Press; 2019. 402 p.
- Profyris C, Chen E, Young IM, Chendeb K, Ahsan SA, Briggs RG, et al. Anaplastic Oligodendroglioma – Is Adjuvant Radiotherapy Mandatory following Maximal Surgical Resection? Clin Neurol Neurosurg. 2021 Jan;200:106303. [CrossRef]
- Weller, M., Wick, W., Aldape, K., Brada, M., Berger, M., Pfister, S. M., Nishikawa, R., Rosenthal, M., Wen, P. Y., Stupp, R., & Reifenberger, G. (2015). Glioma. Nature reviews. Disease primers, 1, 15017. [CrossRef]
- Zhang J, F.G. Stevens M, D. Bradshaw T. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr Mol Pharmacol. 2012 Jan 1;5(1):102–14. [CrossRef]
- Wang JYJ, Edelmann W. Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell. 2006 Jun;9(6):417–8. [CrossRef]
- Massoud TF, Paulmurugan R, editors. Glioblastoma resistance to chemotherapy: molecular mechanisms and innovative reversal strategies. 1st ed. San Diego: Academic Press is an imprint of Elsevier; 2021.
- Shen, W., Hu, J. A., & Zheng, J. S. (2014). Mechanism of temozolomide-induced antitumour effects on glioma cells. The Journal of international medical research, 42(1), 164–172. [CrossRef]
- Chen Y, Hu F, Zhou Y, Chen W, Shao H, Zhang Y. MGMT Promoter Methylation and Glioblastoma Prognosis: A Systematic Review and Meta-analysis. Arch Med Res. 2013 May;44(4):281–90. [CrossRef]
- Nabors LB, Portnow J, Ahluwalia M, Baehring J, Brem H, Brem S, et al. Central Nervous System Cancers, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020 Nov;18(11):1537–70. [CrossRef]
- Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010 Jan;6(1):39–51. [CrossRef]
- Binabaj, M. M., Bahrami, A., ShahidSales, S., Joodi, M., Joudi Mashhad, M., Hassanian, S. M., Anvari, K., & Avan, A. (2018). The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. Journal of cellular physiology, 233(1), 378–386. [CrossRef]
- Knobbe, C. B., & Reifenberger, G. (2003). Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3'-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain pathology (Zurich, Switzerland), 13(4), 507–518. [CrossRef]
- Shepherd P. R. (2005). Mechanisms regulating phosphoinositide 3-kinase signalling in insulin-sensitive tissues. Acta physiologica Scandinavica, 183(1), 3–12. [CrossRef]
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017 Mar 9;168(6):960-976. [CrossRef]
- Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, et al. Oncogenic ERBB3 Mutations in Human Cancers. Cancer Cell. 2013 May;23(5):603–17.
- Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014 Jan;79:34–74. [CrossRef]
- Vanajothi R, Rajamanika S, Sudha A, Srinivasan P. Structural and Functional Analysis of KIT Gene Encoding Receptor Tyrosine Kinase and its Interaction with Sunitinib and HDAC Inhibitors: An in silico Approach. Pak J Biol Sci. 2012 Jan 15;15(3):121–31. [CrossRef]
- Pathania S, Pentikäinen OT, Singh PK. A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim Biophys Acta BBA - Rev Cancer. 2021 Dec;1876(2):188631. [CrossRef]
- RR, Li KKW, Zhang ZY, Chan AKY, Wang WW, Chan DTM, et al. Mismatch repair proteins PMS2 and MLH1 can further refine molecular stratification of IDH-mutant lower grade astrocytomas. Clin Neurol Neurosurg. 2021 Sep;208:106882. [CrossRef]
- Harder BG, Peng S, Sereduk CP, Sodoma AM, Kitange GJ, Loftus JC, et al. Inhibition of phosphatidylinositol 3-kinase by PX-866 suppresses temozolomide-induced autophagy and promotes apoptosis in glioblastoma cells. Mol Med. 2019 Dec;25(1):49. [CrossRef]
- Gomes AL, Reis-Filho JS, Lopes JM, Martinho O, Lambros MBK, Martins A, et al. Molecular Alterations of KIT Oncogene in Gliomas. Anal Cell Pathol. 2007 Jan 1;29(5):399–408. [CrossRef]
- Beauchamp RL, Erdin S, Witt L, Jordan JT, Plotkin SR, Gusella JF, et al. mTOR kinase inhibition disrupts neuregulin 1-ERBB3 autocrine signaling and sensitizes NF2-deficient meningioma cellular models to IGF1R inhibition. J Biol Chem. 2021 Jan;296:100157. [CrossRef]
- Botchkareva NV, Khlgatian M, Jack Longley B, Botchkarev VA, Gilchrest BA. SCF/c-kit signaling is required for cyclic regeneration of the hair pigmentation unit. FASEB J. 2001 Mar;15(3):645–58. [CrossRef]
- Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017 Mar;168(6):960–76. [CrossRef]
- Parthasarathy R, Eisenberg F. The inositol phospholipids: a stereochemical view of biological activity. Biochem J. 1986 Apr 15;235(2):313–22. [CrossRef]
- Tanaka S, Batchelor TT, Iafrate AJ, Dias-Santagata D, Borger DR, Ellisen LW, et al. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol Commun. 2019 Dec;7(1):66. [CrossRef]
- Liggett LA, DeGregori J. Changing mutational and adaptive landscapes and the genesis of cancer. Biochim Biophys Acta BBA - Rev Cancer. 2017 Apr;1867(2):84–94. [CrossRef]
- Gozzelino L, Kochlamazashvili G, Baldassari S, Mackintosh AI, Licchetta L, Iovino E, et al. Defective lipid signalling caused by mutations in PIK3C2B underlies focal epilepsy. Brain. 2022 Jul 29;145(7):2313–31. [CrossRef]
- Knobbe CB, Reifenberger G. Genetic Alterations and Aberrant Expression of Genes Related to the Phosphatidyl-lnositol-3′-Kinase/Protein Kinase B (Akt) Signal Transduction Pathway in Glioblastomas. Brain Pathol. 2006 Apr 5;13(4):507–18. [CrossRef]
- Rao SK, Edwards J, Joshi AD, Siu IM, Riggins GJ. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol. 2010 Jan;96(2):169–79. [CrossRef]
- Maini J, Ghasemi M, Yandhuri D, Thakur SS, Brahmachari V. Human PRE-PIK3C2B, an intronic cis-element with dual function of activation and repression. Biochim Biophys Acta BBA - Gene Regul Mech. 2017 Feb;1860(2):196–204. [CrossRef]
- Aung KT, Yoshioka K, Aki S, Ishimaru K, Takuwa N, Takuwa Y. The class II phosphoinositide 3-kinases PI3K-C2α and PI3K-C2β differentially regulate clathrin-dependent pinocytosis in human vascular endothelial cells. J Physiol Sci. 2019 Mar;69(2):263–80. [CrossRef]
- Wang C, Yang Y, Yin L, Wei N, Hong T, Sun Z, et al. Novel Potential Biomarkers Associated With Epithelial to Mesenchymal Transition and Bladder Cancer Prognosis Identified by Integrated Bioinformatic Analysis. Front Oncol. 2020 Jun 30;10:931. [CrossRef]
- Margaria JP, Ratto E, Gozzelino L, Li H, Hirsch E. Class II PI3Ks at the Intersection between Signal Transduction and Membrane Trafficking. Biomolecules. 2019 Mar 15;9(3):104. [CrossRef]
- Liu Z, Li X, Ma J, Li D, Ju H, Liu Y, et al. Integrative Analysis of the IQ Motif-Containing GTPase-Activating Protein Family Indicates That the IQGAP3-PIK3C2B Axis Promotes Invasion in Colon Cancer. OncoTargets Ther. 2020 Aug;Volume 13:8299–311. [CrossRef]
- Liu Z, Sun C, Zhang Y, Ji Z, Yang G. Phosphatidylinositol 3-Kinase-C2β Inhibits Cisplatin-Mediated Apoptosis via the Akt Pathway in Oesophageal Squamous Cell Carcinoma. J Int Med Res. 2011 Aug;39(4):1319–32. [CrossRef]
- Cisse O, Quraishi M, Gulluni F, Guffanti F, Mavrommati I, Suthanthirakumaran M, et al. Downregulation of class II phosphoinositide 3-kinase PI3K-C2β delays cell division and potentiates the effect of docetaxel on cancer cell growth. J Exp Clin Cancer Res. 2019 Dec;38(1):472. [CrossRef]
- Löw S, Vougioukas VI, Hielscher T, Schmidt U, Unterberg A, Halatsch ME. Pathogenetic pathways leading to glioblastoma multiforme: association between gene expressions and resistance to erlotinib. Anticancer Res. 2008 Dec;28(6A):3729–32.
- Ortiz R, Perazzoli G, Cabeza L, Jiménez-Luna C, Luque R, Prados J, et al. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr Neuropharmacol. 2021 Apr;19(4):513–37. [CrossRef]
- Indraccolo S, Lombardi G, Fassan M, Pasqualini L, Giunco S, Marcato R, et al. Genetic, Epigenetic, and Immunologic Profiling of MMR-Deficient Relapsed Glioblastoma. Clin Cancer Res. 2019 Mar 15;25(6):1828–37. [CrossRef]
- Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer. 2011 Aug 1;129(3):659–70. [CrossRef]
- Higuchi F, Nagashima H, Ning J, Koerner MVA, Wakimoto H, Cahill DP. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin Cancer Res. 2020 Apr 1;26(7):1690–9. [CrossRef]
- Yang B, Han N, Sun J, Jiang H, Xu HY. CtIP contributes to non-homologous end joining formation through interacting with ligase IV and promotion of TMZ resistance in glioma cells. Eur Rev Med Pharmacol Sci. 2019 Mar;23(5):2092–102. [CrossRef]
- Zhang P, Wang H, Chen Y, Lodhi AF, Sun C, Sun F, et al. DR5 related autophagy can promote apoptosis in gliomas after irradiation. Biochem Biophys Res Commun. 2020 Feb;522(4):910–6. [CrossRef]
Figure 1.
Type of tumor of patients with high-grade glioma. The figure shows the classification of the 31 patients analyzed according to the type of tumor: Glioblastoma (GB; 21 patients; 67,74%), Anaplastic Astrocytoma (AA; 5 patients; 16,13%), and Anaplastic Oligoastrocytoma (AO; 5 patients; 16,13%). The most frequent tumor was glioblastoma.
Figure 1.
Type of tumor of patients with high-grade glioma. The figure shows the classification of the 31 patients analyzed according to the type of tumor: Glioblastoma (GB; 21 patients; 67,74%), Anaplastic Astrocytoma (AA; 5 patients; 16,13%), and Anaplastic Oligoastrocytoma (AO; 5 patients; 16,13%). The most frequent tumor was glioblastoma.
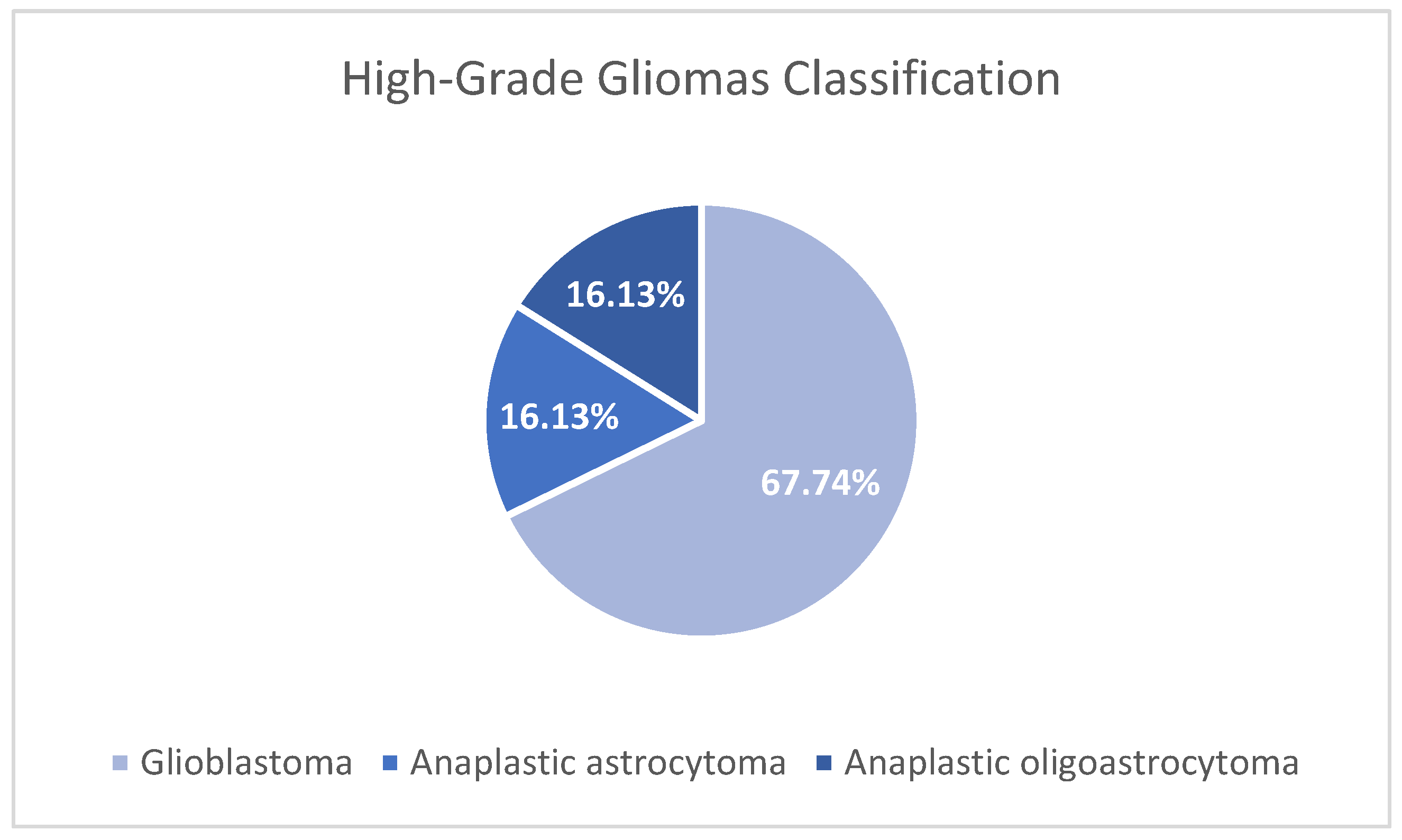
Figure 2.
The figure shows the treatment cycles with adjuvant temozolomide given to the 31 patients with high-grade glioma. Fifteen patients received the six cycles recommended by the Stupp protocol, five patients received fewer cycles due to an early relapse, and 11 patients needed more cycles due to disease persistence.
Figure 2.
The figure shows the treatment cycles with adjuvant temozolomide given to the 31 patients with high-grade glioma. Fifteen patients received the six cycles recommended by the Stupp protocol, five patients received fewer cycles due to an early relapse, and 11 patients needed more cycles due to disease persistence.
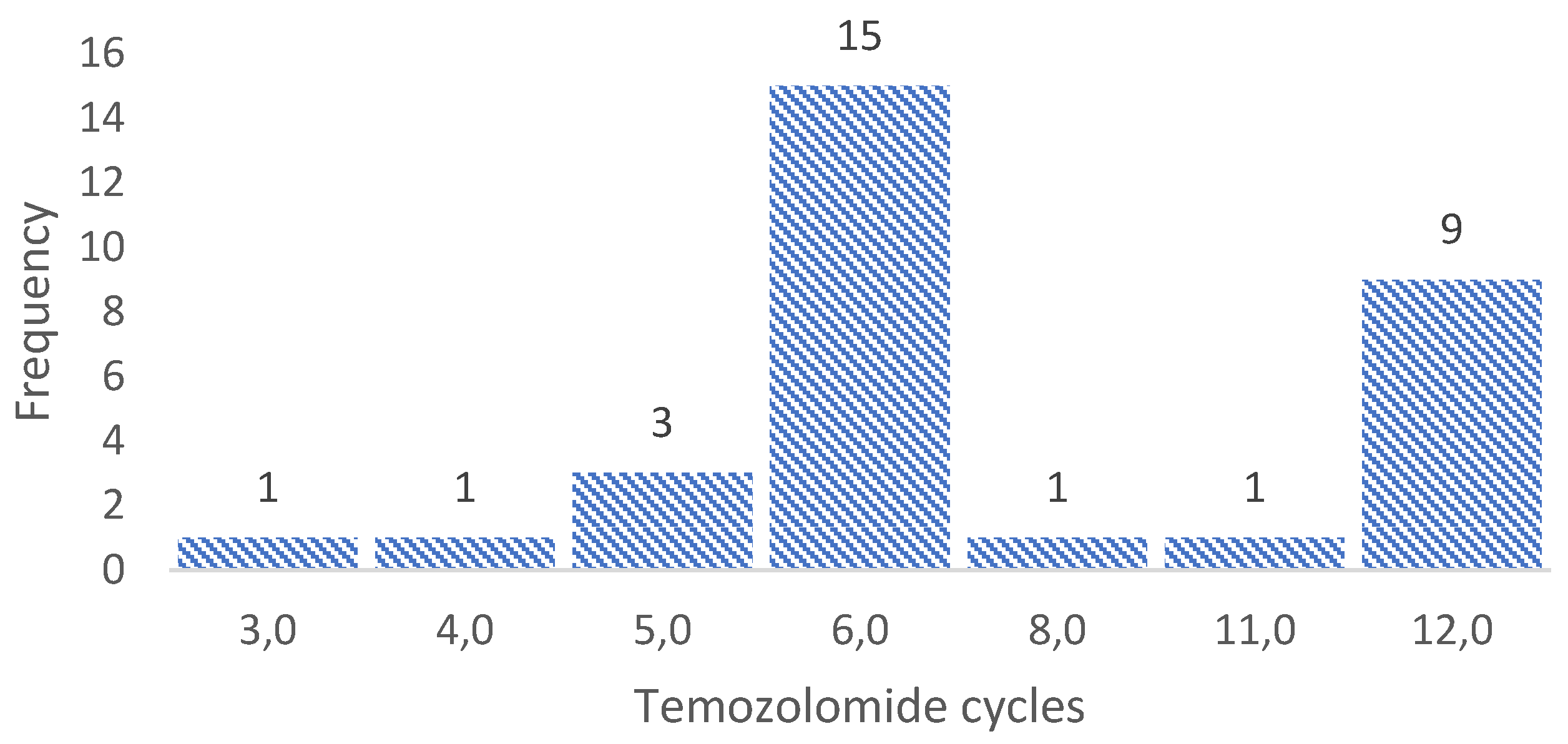
Figure 3.
The figure shows the number of treatment cycles with metronomic temozolomide that patients received before presenting the second relapse. The largest number of patients presented a second relapse at the sixth treatment cycle (10 patients), but the results were highly variable, with early (first cycle) and late (21 cycles) second relapses.
Figure 3.
The figure shows the number of treatment cycles with metronomic temozolomide that patients received before presenting the second relapse. The largest number of patients presented a second relapse at the sixth treatment cycle (10 patients), but the results were highly variable, with early (first cycle) and late (21 cycles) second relapses.
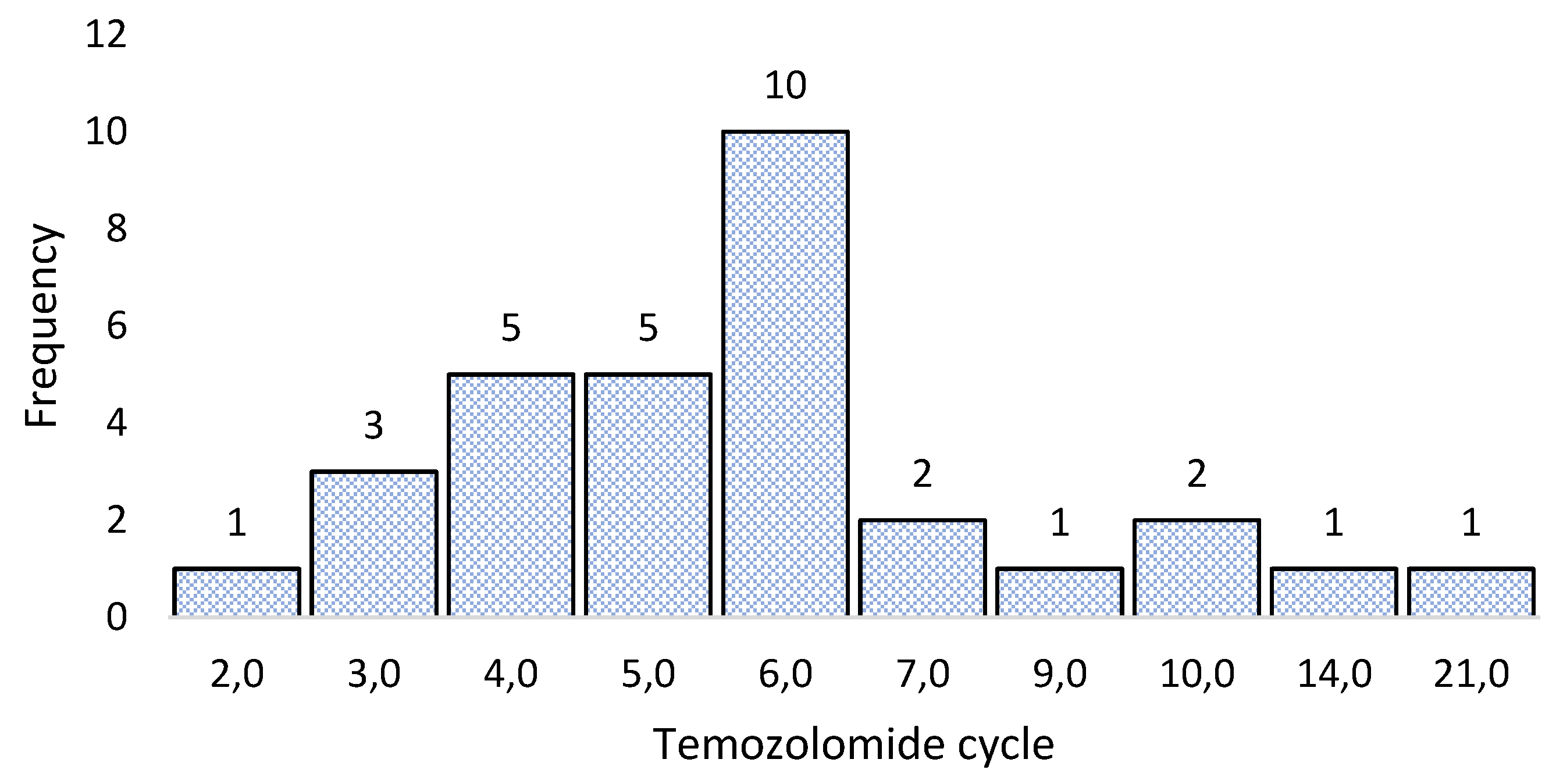
Figure 4.
The figure shows the survival curves according to the Kaplan-Meier method. This analysis was performed to estimate the time it takes for patients with and without mutations in the PIK3C2B, KIT, ERBB3, and MLH1 genes to present a second relapse after treatment with metronomic temozolomide. The graph shows patients without mutations with blue lines, while the red lines represent patients with mutations, observing that patients with mutations present a second relapse sooner. The 0 symbol represents patients without mutations, and the 1 symbol represents patients with mutations. The Y-axis represents the survival rate, and the X-axis shows time in months.
Figure 4.
The figure shows the survival curves according to the Kaplan-Meier method. This analysis was performed to estimate the time it takes for patients with and without mutations in the PIK3C2B, KIT, ERBB3, and MLH1 genes to present a second relapse after treatment with metronomic temozolomide. The graph shows patients without mutations with blue lines, while the red lines represent patients with mutations, observing that patients with mutations present a second relapse sooner. The 0 symbol represents patients without mutations, and the 1 symbol represents patients with mutations. The Y-axis represents the survival rate, and the X-axis shows time in months.
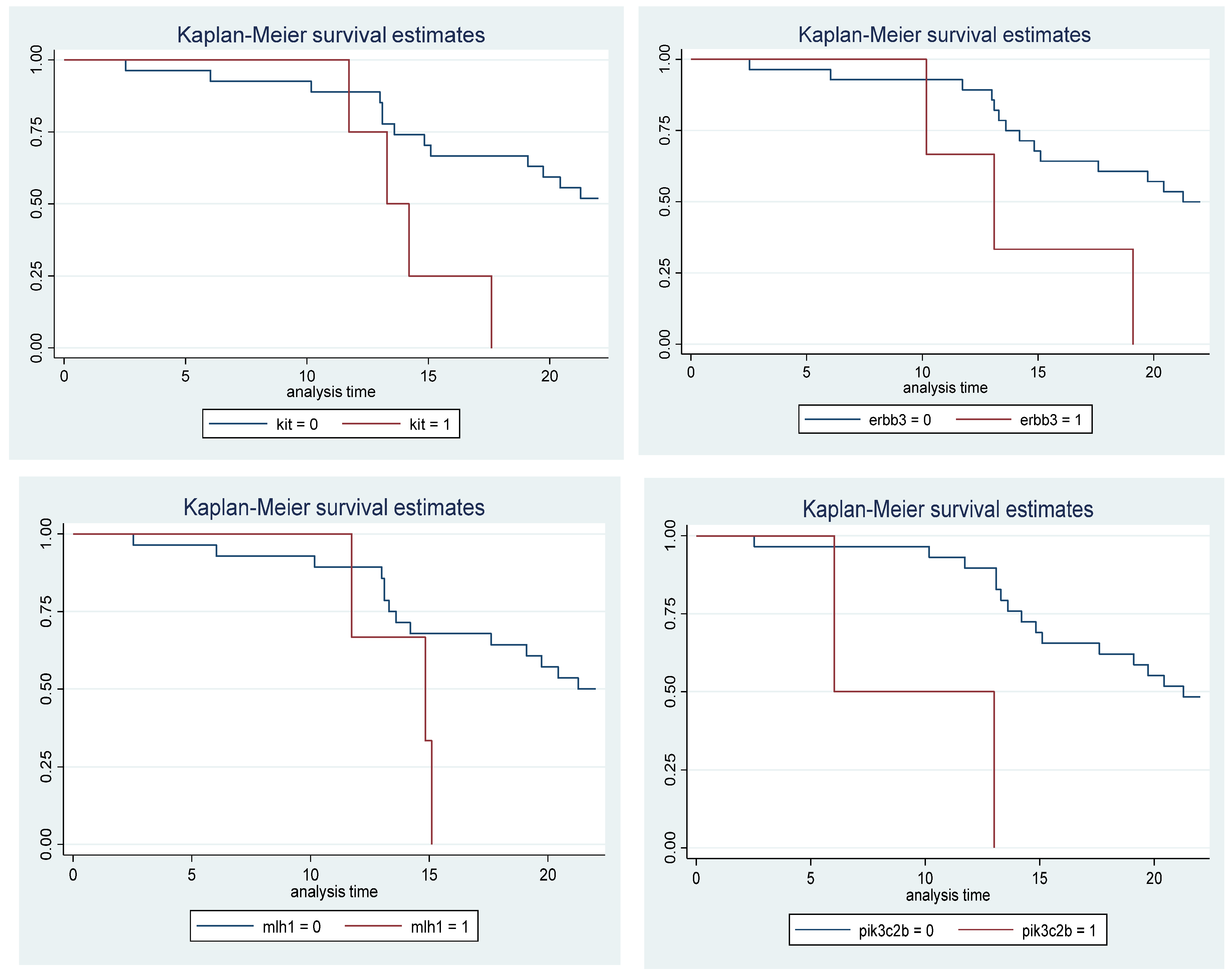

Table 2.
Bivariate analysis was used to identify the mutated genes associated with the second relapse after the use of temozolomide according to Stupp protocol and the metronomic dose in patients with high-grade gliomas. Seventy-one genes were related to the second relapse; however, only the first five genes with significant values of p-values and Hazard Ratios are shown.
Table 2.
Bivariate analysis was used to identify the mutated genes associated with the second relapse after the use of temozolomide according to Stupp protocol and the metronomic dose in patients with high-grade gliomas. Seventy-one genes were related to the second relapse; however, only the first five genes with significant values of p-values and Hazard Ratios are shown.
| Gene | Gene presence | P-value | Hazard Ratio (CI 95%) | |
|---|---|---|---|---|
| Si | Not | |||
| PIK3C2B | 2(11,7) | 15(88,2) | 0,004 | 13,81 (2,258 - 84,45) |
| NOTCH3 | 4(23,5) | 13(76,4) | 0,005 | 6,026 (1,742 - 20,84) |
| KIT | 4(23,5) | 13(76,4) | 0,024 | 3,985 (1,204 - 13,18) |
| ERBB3 | 3(17,6) | 14(82,3) | 0,04 | 3,871 (1,066 - 14,04) |
| MLH1 | 3(17,6) | 14(82,3) | 0,06 | 3,528 (0,950 - 13,09) |
Table 3.
Multivariate analysis was used to identify the mutated genes associated with the second relapse, after the use of temozolomide, according to the Stupp protocol and metronomic dose, in patients with high-grade gliomas.
Table 3.
Multivariate analysis was used to identify the mutated genes associated with the second relapse, after the use of temozolomide, according to the Stupp protocol and metronomic dose, in patients with high-grade gliomas.
| Gene | P-value | HR Raw value (CI 95%) | P-value | HR Adjusted value (CI 95%) | |
|---|---|---|---|---|---|
| PIK3C2B | 0,004 | 13,81 (2,258 - 84,45) | 0,000 | 82,37 (8,36 - 811,67) | |
| KIT | 0,024 | 3,985 (1,204 - 13,18) | 0,002 | 10,24 (2,42 - 43,34) | |
| ERBB3 | 0,04 | 3,871 (1,066 - 14,04) | 0,001 | 13,20 (2,77 - 62,77) | |
| MLH1 | 0,06 | 3,528 (0,950 - 13,09) | 0,006 | 8,50 (1,83 - 39,45) |
Table 4.
The following table shows the domains, biological processes, molecular function, and cellular components of the four proteins related to the second relapse after using temozolomide according to the Stupp protocol and a metronomic dose in patients with high-grade gliomas.
Table 4.
The following table shows the domains, biological processes, molecular function, and cellular components of the four proteins related to the second relapse after using temozolomide according to the Stupp protocol and a metronomic dose in patients with high-grade gliomas.
| Protein | Domains | Biological processes | Molecular function | Cellular component |
|---|---|---|---|---|
| PIK3C2B | -R458Q mutation affects domain (PI3K_Ras-bd_dom) - Amplification - Rearrangement |
-Phosphatidylinositol-mediated signaling - Phosphatidylinositol phosphatase biosynthetic process |
-Phosphatidylinositol binding -kinase activity |
None |
| ERBB3 | - R164K mutation affects domain (Rcp_L) - L1177l the mutation does not affect any functional domain |
-Membrane receptor protein tyrosine kinase signaling pathway. -Protein phosphorylation |
-Protein kinase activity -Protein tyrosine kinase activity -ATP binding |
Membrane |
| KIT | - Amplification - Wrong amplification |
-Protein phosphorylation -Transmembrane receptor protein tyrosine kinase signaling pathway -Kit signaling pathway -Fc receptor signaling pathway |
-ATP binding -Transmembrane receptor -Protein tyrosine kinase activity -Protein kinase activity -Cytokine binding -Protein tyrosine kinase activity |
None |
| MLH1 | -(F261fs*7) mutation affects domain (DNA_mismatch_repair_N and DNA_mismatch_S5_2-like) | DNA mismatch repair | -Mismatched DNA binding -ATP binding -ATP-dependent DNA damage Sensor activity -ATP hydrolysis activity |
Mismatch Repair Complex |
Table 5.
Identification of the protein-protein interactions and the metabolic pathways of the proteins encoded by the four mutated genes related to the second relapse after the use of temozolomide according to the Stupp protocol and the administration of metronomic doses in patients with high-grade gliomas.
Table 5.
Identification of the protein-protein interactions and the metabolic pathways of the proteins encoded by the four mutated genes related to the second relapse after the use of temozolomide according to the Stupp protocol and the administration of metronomic doses in patients with high-grade gliomas.
| Protein | protein-protein interactions | Metabolic routes |
|---|---|---|
| PIK3C2B | RICTOR, EGFR, GRB2, ANKRD32, SBF2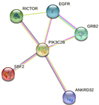
|
-DNA damage response (only ATM dependent) -Focal adhesion: PI3K-Akt-mTOR-signaling pathway -Glioblastoma signaling pathways -Phosphoinositides metabolism -Regulation of actin cytoskeleton |
| ERBB3 | ERBB2, EGFR, NNRG1, GRB2, SHC1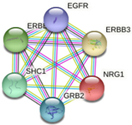
|
-Apoptosis-related network due to altered Notch3 in ovarian cancer -EGFR tyrosine kinase inhibitor resistance -ErbB signaling pathway -Glioblastoma signaling pathways -Heart development |
| KIT | PIK3R1, PLCG1, KITLG, GRB2, CRK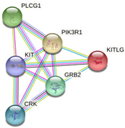
|
-Breast cancer pathway -Cardiac progenitor differentiation -Focal adhesion: PI3K-Akt-mTOR-signaling pathway -Gastrin signaling pathway -Hippo signaling regulation pathways |
| MLH1 | MSH2, PMS2, PMS1, BLM, EXO1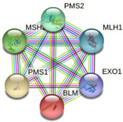
|
-Chromosomal and microsatellite instability in colorectal cancer -DNA IR-damage and cellular response via ATR -DNA mismatch repair -DNA repair pathways, full network -Ovarian infertility |
Table 6.
Structural analysis of the effect of the mutations in the four proteins encoded by mutated genes related to the second relapse after the use of temozolomide according to the Stupp protocol and a metronomic dose in patients with high-grade gliomas. The first column shows the protein name and mutation found. The second column shows the structural modeling of the wild-type protein. The third column shows the effect of the mutations on protein structure.
Table 6.
Structural analysis of the effect of the mutations in the four proteins encoded by mutated genes related to the second relapse after the use of temozolomide according to the Stupp protocol and a metronomic dose in patients with high-grade gliomas. The first column shows the protein name and mutation found. The second column shows the structural modeling of the wild-type protein. The third column shows the effect of the mutations on protein structure.
| Gene | Normal protein | Mutated protein |
|---|---|---|
|
PIK3C2B R458Q Mutation |
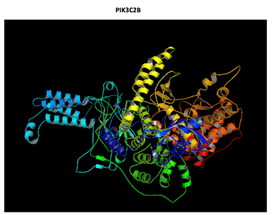 |
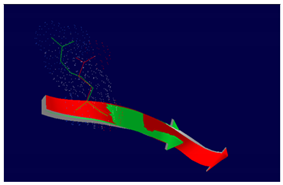 The effect of the mutation on the structure of the protein leads to the shortening of the radical, reducing the Van der Waals forces. At structural level, the mutation is located be-tween the amino acids (457-464) that make up a beta sheet, generating an elongation of the beta sheet in the mutated protein. |
|
ERBB3 R164K Mutation |
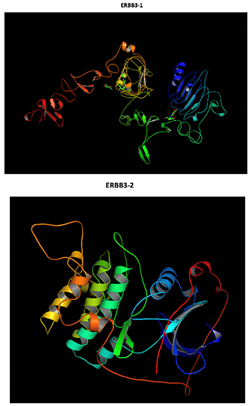 Two-part patterned protein |
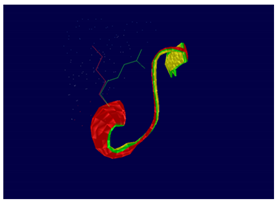 At structural level, an amino acid change is observed which leads to an alteration in the orientation of the radical group that alters the spatial distribution of Van der Waals forces. |
|
MLH1 F261fs*31 Mutation |
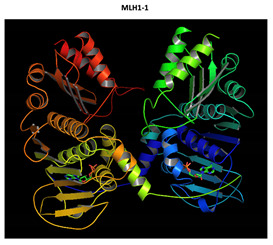 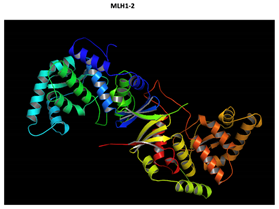 Two-part patterned protein. |
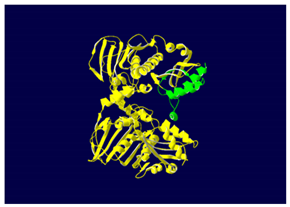 The yellow color shows the common region between the wild-type and mutated protein, the green color shows the region that would be lost in the mutated protein by the occurrence of a premature stop codon. |
Table 7.
Diseases related to the four mutated genes related to second relapse after the use of temozolomide according to the Stupp protocol and the metronomic dose in patients with high-grade gliomas. The first column shows the genes' names. The second column shows the diseases related to mutations in the PIK3C2B, ERBB3, KIT, and MLH1 genes.
Table 7.
Diseases related to the four mutated genes related to second relapse after the use of temozolomide according to the Stupp protocol and the metronomic dose in patients with high-grade gliomas. The first column shows the genes' names. The second column shows the diseases related to mutations in the PIK3C2B, ERBB3, KIT, and MLH1 genes.
| Gene | Associated diseases |
|---|---|
| PIK3C2B | -High grade glioma -Maffucci -Hepatitis C |
| ERBB3 | -High grade glioma -Breast cancer -Frontal myelination -Enteric nervous system -Diabetes type 1 |
| KIT | -High grade glioma -Piebaldism -GIST -Mastocytosis -LMA -Melanoma - Germ cell tumors |
| MLH1 | -High grade glioma -Hereditary nonpolyposis colorectal cancer (Lynch) -Turcot type I |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



