
Submitted:
17 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
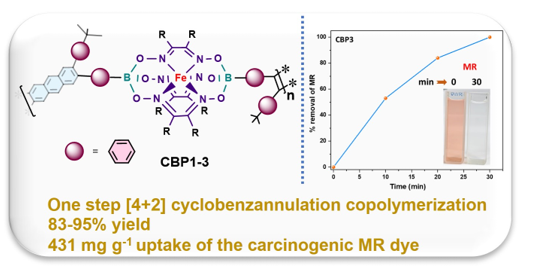
A novel synthetic strategy is disclosed to prepare a new class of metalorganic copolymers which contain iron(II) clathrochelate building blocks by employing a mild and cost-effective copper-catalyzed [4 + 2] cyclobenzannulation reaction, using three specially designed diethynyl iron(II) clathrochelate synthons. The target copolymers CBP1-3 were isolated in high purity and excellent yields as proven by their structural and photophysical characterization, namely, Fourier transform infrared (FTIR), X-ray photoelectron spectroscopy (XPS) and UV-vis absorption and emission spectroscopies. Thermogravimetric analysis (TGA) of CBP1-3 reveals excellent chemical stability. Investigation of the adsorption properties of the target copolymers towards the carcinogenic methyl red dye from aqueous solution reveals a quantitative uptake in 30 minutes. Isothermal adsorption studies disclose that methyl red uptake from aqueous solution follows Langmuir model for all the target copolymers reaching a maximum adsorption capacity (qm) of 431 mg g‒1, whereas kinetic investigation reveals that the adsorption follows pseudo first-order with an equilibrium adsorption capacity (qe,cal) of 79.35 mg g‒1 and whose sorption property was sustained even after its reuse several times.
Keywords:
Metalorganic copolymer
; [4+2] cycloaddition
; cyclobenzannulation
; dye removal
; Environmental Remediation
1. Introduction
Dyes are predominantly utilized as coloring agents in textile industry and also employed in myriad products, such as, pharmaceuticals, food and beverage, leather, plastics, cosmetics, and paper [1,2]. Dyes, whose global annual production is estimated to 7 × 107 tons, [2,3] present several advantages, namely, the variety of their color palette, ease of application on various types of materials, structural diversity, and low energy consumption. [4,5] There are several methods to classify synthetic dyes, notably that based on the chemical structure of their chromophore, hence, they could be grouped as acidic, basic, azoic, nitro, sulphur, etc. [6,7]. It is noteworthy that azo dyes, which are characterized by the presence of one or more azo groups (-N=N-), are widely employed in industry with a production rate that exceeds half that of the total dyes synthesized annually [8,9] Despite its undeniable importance and contribution to economic development[5,10], textile industry is one of the largest global polluters as it consumes large amounts of fuels and chemical reagents [11,12,13]. Additionally, textile industry uses massive quantities of freshwater in the various operations required for its production chain, such as, washing, bleaching, and dyeing, [14], thus, producing effluents which contain high concentrations of pollutants [15]. Inappropriate discharge practices of azo dyes waste lead to harmful environmental effects and severely damage the aquatic life [16,17]. On top of that, azo dyes cause health complications as they are considered carcinogenic, mutagenic, and teratogenic [18,19,20] while their high solubility in water allow them to access the food chain, consequently, causing many health problem, mainly, fever, renal damage, and cramps [5]. Methyl red (MR) is an anionic mono-azo dye (c.f. table S1 in the supporting information file) which is used in paper printing and textile dyeing. Nevertheless, MR is classified as carcinogenic and mutagenic besides being considered as eye and skin irritant and causes damage to the aquatic life if present in excess [19,21].
Various technologies have been tested to remove azo dyes from wastewater, ranging from physicochemical methods, like, oxidation and irradiation, to electrochemical techniques, such as, coagulation-flocculation, photodegradation, ozone treatment, electro Fenton’s and hypochlorite usage [22,23,24] These technologies suffer from several disadvantages, among others, the high dosage required and production of large amounts of sludge, which make them economically unattractive [10]. Nevertheless, the use of adsorbents proved to be promising due to its high efficiency, cost-effectiveness, simplicity, and possibility to be used under ambient conditions [5].
Metal clathrochelates derivatives, which are known for more than five decades, consist of three-dimensional octahedral organometallic complexes with an encapsulated central metal-ion[25,26,27]. Among these metalorganic complexes, myriad of iron(II) clathrochelate derivatives doubly capped with aryl borate groups were synthesized disclosing several advantages, notably, their versatile synthesis from environmentally friendly synthons, modular chemistry allowing for their functionalization, high chemical stability, intricate structure, and cost-effectiveness[28,29,30,31]. Interestingly, iron(II) clathrochelates revealed remarkable properties for applications in biosensors, catalysts for hydrogen generation, materials for electronic transport, organogels, and building blocks to make supramolecular cages and metalorganic frameworks (MOFs) [32,33,34,35,36]. Several adsorbents derived from iron(II) clathrochelate were made and tested against various gases and dyes disclosing remarkable properties[37,38,39,40]. This work discloses the synthesis of three new metalorganic copolymers CBP1-3 in high yield and under mild reaction conditions using a copper-catalyzed [4 + 2] cyclobenzannulation reaction of specially designed diethynyl iron(II) clathrochelate synthons CM1-3, which were made in one step, with 2,5-bis(phenylethynyl)terephthalaldehyde 6. The resulting metalorganic copolymers CBP1-3 were investigated as adsorbents of the carcinogenic Methyl Red dye from aqueous solution, revealing excellent adsorption of the latter.
2. Materials and Methods
All the reactions were carried out under an inert atmosphere of dry argon. Unless otherwise specified, the chemical reagents were used without further purification as purchased from Merck, Aldrich, Alfa Aesar, Honeywell, Loba Chemie, and HiMedia. Decane-5,6-dione dioxime 1, 2-((4-(tert-butyl)phenyl)ethynyl)benzaldehyde 5, and 2,5-bis(phenylethynyl)terephthalaldehyde 6 were synthesized following the literature [34,41]. Synthon 4 was synthesized following a reported procedure[42,43] and its structure was confirmed by 1H- and 13C- nuclear magnetic resonance (NMR), and electrospray ionization mass spectrometry (ESI-MS) (c.f. synthetic procedure (i) and figures S1, S6, and S11 in the supporting information file). Anhydrous solvents, namely, hexane, tetrahydrofuran (THF), dichloroethane (DCE), dichloromethane (DCM), chloroform (CHCl3) methanol, and diisopropylamine (iPr2NH) were further dried over molecular sieves and deoxygenated by bubbling with dry argon gas for 30 minutes. Thin layer chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 and revealed using a UV lamp. NMR spectra (1H: 600 MHz, 13C: 150 MHz) were recorded using a JEOL resonance ECZ600R spectrometer at 25 °C, using CDCl3 and CD2Cl2 as a solvent with the chemical shifts (δ) given in ppm and referenced to tetramethylsilane (TMS). Electro-spray ionization mass spectra (ESI-MS) were recorded on waters QTOF Micro YA263 using a single quadrapole detector-2 (SQD-2). UV-Vis spectra were recorded using a Shimadzu UV1800 spectrophotometer. FT-IR spectra were recorded on PerkinElmer G spectrophotometer using a KBr matrix. Thermogravimetric Analysis (TGA) was recorded on Mettler Toledo Star SW 8.10 system (model number TGA/SDTA851e) analyzer and was used to measure the thermal stability of composites from room temperature to 800 °C with a heating rate of 10 °C/min under an inert atmosphere. X-ray Photoelectron Spectroscopy (XPS) data were recorded with a Thermo scientific using a monochromatic Al K-radiation source (1486.6 eV) with the spot size of samples 10 x 10 × 5 mm. Spectra acquisition and processing were carried out using the software Thermo Advantage Version 4.87. The base pressure in the XPS analysis chamber was in the range 10‒10 to 10‒9 torr. The analyzer was operated with a pass energy of 20 eV, dwell time of 50 min and with a step size of 0.1 eV. Brunauer–Emmett–Teller (BET) surface area were performed at 77 K using a liquid nitrogen bath (77 K) on a Quantachrome Quadrasorb automatic volumetric instrument. All the samples were outgassed for 12 h at 120 °C under vacuum prior to the gas adsorption studies. The surface areas were evaluated using the Brunauer-Emmett-Teller (BET) model applied between P/P0 values of 0.05 and 0.3 for samples. The pore size distributions were calculated using the non-localized density functional theory (NLDFT) method. The surface area of each of the sample has been measured multiple times and then averaged out for proper comparison.
2.1. Synthesis
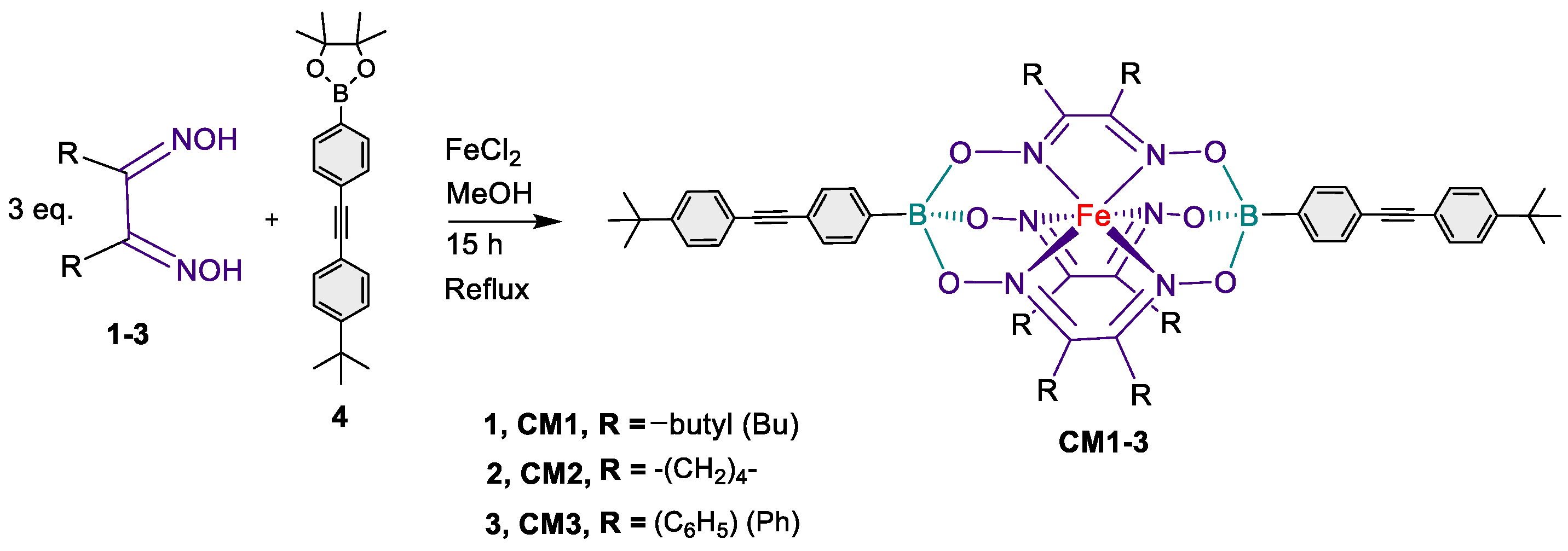
2.1.1. Synthesis of CM1 (Procedure A).
2-(4-((4-(tert-butyl)phenyl)ethynyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 4 (540 mg, 1.5 mmol, 2.3 eq.), decane-5,6-dione dioxime 1 (405 mg, 2.0 mmol, 3 eq.) and iron(II) chloride (FeCl2, 83 mg 0.66 mmol, 1 eq.) in 6 mL of degassed methanol was refluxed under argon for 24 hours. The solvent was evaporated under reduced pressure and the orange red solid was precipitated from methanol. The resulting orange red precipitate was filtered and washed with methanol, hexane followed by diethyl ether. Yield: (691 mg, 93 %). 1H-NMR (CDCl3, 600 MHz, ppm): δ 7.70 (d, J = 7.7 Hz, 4H, ArH), 7.55 (d, J = 8.0 Hz, 4H, ArH), 7.48 (d, J = 8.3 Hz, 4H, ArH), 7.38 (d, J = 8.1 Hz, 4H, ArH), 2.83 (t, J = 7.7 Hz, 12H, N=C-CH2), 1.58 (m, 12H, CH2), 1.36–1.33 (m, 30H, CH2 & CH3), 0.90 (t, 18H, CH3); 13C-NMR (CDCl3, 150 MHz, ppm): δ 157.14 (C=N), 151.24, 131.68, 131.38, 130.59, 125.37, 122.39, 120.86, 89.91, 88.91, 34.84, 31.29, 29.28, 27.19, 22.52, 13.93; ESI-MS; Calcd. for M•+ C66H88B2FeN6O6: 1138.63; found 1138.63 FTIR (KBr, cm−1): 2958 (Aliphatic C-H str), 2211 (CΞC str), 1468 (Aliphatic C-H ben) 1396 (B-O str), 1184 (B-C str), 828 (Ar-C-H ben).
2.1.2. Synthesis of CM2
CM2 was prepared following procedure A with: 2-(4-((4-(tert-butyl)phenyl)ethynyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 4 (280 mg, 0.77 mmol, 2.3 eq.), 1,2-Cyclohexanedione dioxime 2 (147 mg, 1.0 mmol, 3 eq.), and iron(II) chloride (FeCl2, 43 mg 0.34 mmol, 1 eq.) in 5 mL of degassed methanol. Brick-red solid (298 mg, 91%). 1H-NMR (600 MHz, CD2Cl2, ppm): δ 7.69 (d, J = 8.1 Hz, 6H, ArH), 7.53-7.51 (brm, 6H, ArH), 7.45 (d, J = 8.4 Hz, 4H, ArH), 2.97 (brs, 12H, CH2), 1.86 (brs, 12H, CH2), 1.38 (brs, 18H, CH3); 13C-NMR (150 MHz, CD2Cl2, ppm): δ 152.66 (C=N), 142.14, 135.11, 132.25, 131.74, 130.93, 126.00, 123.02, 121.00, 90.00, 89.48, 35.26, 31.48, 26.80, 22.17; ESI-MS; Calcd. for M•+ C54H58B2FeN6O6: 964.39; found 964.39 FTIR (KBr, cm−1): 2947 (Aliphatic C-H str), 2208 (CΞC str), 1431 (Aliphatic C-H ben) 1399 (B-O str), 1194 (B-C str), 829 (Ar-C-H ben).
2.1.3. Synthesis of CM3
CM3 was prepared following procedure A with: 2-(4-((4-(tert-butyl)phenyl)ethynyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 4 (270 mg, 0.75 mmol, 2.3 eq.), anti-diphenylglyoxime 3 (243 mg, 1.0 mmol, 3 eq.), and iron(II) chloride (FeCl2, 42 mg 0.33 mmol, 1 eq.) in 5 mL of degassed methanol. Brick-red solid (382 mg, 94%). 1H-NMR (600 MHz, CDCl3, ppm): δ 7.77 (d, J = 8.0 Hz, 4H, ArH), 7.51 (d, J = 8.0 Hz, 4H, ArH), 7.46 (m, 14H, ArH), 7.37 (d, J = 8.3 Hz, 6H, ArH), 7.32-7.28 (m, 18H, ArH), 1.35 (brs, 18H, CH3); 13C-NMR (150 MHz, CDCl3, ppm): δ 155.89 (C=N), 151.76, 134.65, 131.81, 131.47, 131.02, 130.81, 130.27, 129.84, 127.74, 126.31, 125.44, 120.21, 90.98, 89.00, 34.88, 31.25; FTIR (KBr, cm−1): 2973 (Aliphatic C-H str), 2203 (CΞC str), 1450 (Aliphatic C-H ben) 1396 (B-O str), 1144 (B-C str), 868 (Ar-C-H ben).
2.1.4. Synthesis of CBM
A Schlenk tube was charged under argon with 2-((4-(tert-butyl)phenyl)ethynyl) benzaldehyde 5 (27 mg, 0.10 mmol, 2.1 eq.), CM2 (48 mg, 0.05 mmol, 1 eq.), copper(II) triflate Cu(OTf)2 (3.6 mg, 0.01 mmol, 0.2 eq.), and trifluoroacetic acid TFA (15 μL, 0.2 mmol) in 4 mL of deoxygenated dichloroethane. The solution was heated at 100 °C overnight and the solvent was evaporated under reduced pressure. The resulting mixture was dissolved in DCM and extracted with a saturated solution of NaHCO3 (50 mL × 2). The combined organic layer was washed with deionized water (100 mL × 3), concentrated, and the desired product was isolated using silica gel column chromatography with DCM/hexane (50:50 v/v) as the eluent. Brick-red solid (55 mg, 95%). 1H-NMR (600 MHz, CDCl3, ppm): δ 7.81-7.78 (brm, 4H, ArH), 7.57 (t, J = 7.9 Hz, 2H, ArH), 7.48 (d, J = 7.9 Hz, 2H, ArH) 7.43-7.39 (m, 8H, ArH), 7.32-7.26 (m, 4H, ArH), 7.20-7.11 (m, 8H, ArH), 2.84 (brs, 12H, CH2), 1.73 (brs, 12H, CH2), 1.25 (brs, 18H, CH3); 13C-NMR (150 MHz, CDCl3, ppm): δ 152.48, 150.17, 139.47, 133.19, 132.25, 131.83, 131.74, 130.93, 130.25, 130.10, 129.51, 128.19, 126.64, 126.00, 125.74, 125.58, 125.40, 125.05, 34.39, 31.68, 26.79, 22.17. ESI-MS; Calcd. for M•+ C70H70B2FeN6O6: 1168.48; found 1168.48 FTIR (KBr, cm−1): 2947 (Aliphatic C-H str), 1435 (Aliphatic C-H ben), 1391 (B-O str), 1194 (B-C str), 833 (Ar-C-H ben).
2.1.5. Synthesis of copolymer CBP1 (Procedure B)
2,5-bis(phenylethynyl) terephthalaldehyde 6 (73 mg, 0.22 mmol, 1 eq.), CM1 (250 mg, 0.22 mmol, 1 eq.), Cu(OTf)2 (32 mg, 0.09 mmol, 0.4 eq.), and TFA (136 μL, 1.76 mmol) were refluxed in 35 mL of deoxygenated dichloroethane in a Schlenk tube under argon. After 2 days of reaction, the precipitate was filtered and washed exhaustively with a sequence of the following solvents 50 mL of DCM, 50 mL of THF, 50 mL of acetone, 50 mL of water, and 50 mL of diethyl ether affording a brown solid (235 mg, 83%). FTIR (KBr, cm−1): 2955 (Aliphatic C-H str), 1461 (Aliphatic C-H ben), 1396 (B-O str), 1180 (B-C str), 820 (Ar-C-H ben). UV-vis: (THF, 10−6 M), λmax [nm] = 313, 452, fluorescence: (THF, 10−6 M), λmax [nm] = 440.
2.1.6. Synthesis of CBP2
CBP2 was prepared following procedure B with: 2,5-bis(phenylethynyl) terephthalaldehyde 6 (63 mg, 0.19 mmol, 1 eq.), CM2 (183 mg, 0.19 mmol, 1 eq.), Cu(OTf)2 (27 mg, 0.08 mmol, 0.4 eq.), and TFA (117 μL, 1.52 mmol) in 30 mL of degassed dichloroethane. Brown solid (200 mg, 95%); FTIR (KBr, cm−1): 2948 (Aliphatic C-H str), 1450 (Aliphatic C-H ben), 1392 (B-O str), 1190 (B-C str), 830 (Ar-C-H ben). UV-vis: (THF, 10−6 M), λmax [nm] = 312, 450, fluorescence: (THF, 10−6 M), λmax [nm] = 440
2.1.7. Synthesis of CBP3
CBP3 was prepared following procedure B with: 2,5-bis(phenylethynyl) terephthalaldehyde 6 (60 mg, 0.18 mmol, 1 eq.), CM3 (226 mg, 0.18 mmol, 1 eq.), Cu(OTf)2 (26 mg, 0.07 mmol, 0.4 eq.), and TFA (110 μL, 1.44 mmol) in 30 mL of degassed dichloroethane. Brown solid (228 mg, 90%); FTIR (KBr, cm−1): 2965 (Aliphatic C-H str), 1449 (Aliphatic C-H ben), 1370 (B-O str), 1200 (B-C str), 890 (Ar-C-H ben); UV-vis: (THF, 10−6 M), λmax [nm] = 297, 481, fluorescence: (THF, 10−6 M), λmax [nm] = 451.
3. Results and discussion
3.1. Synthesis
Scheme 1 discloses the reaction conditions to synthesize comonomers CM1-3 from a one-pot complexation reaction[31,34] of iron(II) chloride (FeCl2, 0.44 eq.) with a dioxime derivative 1-3 (1.35 eq.) in refluxing methanol overnight and in presence of the diboronic acid pinacol ester capping reagent 4 (1.0 eq.), thus, affording the desired building blocks in excellent yields. The structures of the desired comonomers CM1-3 were confirmed by 1H- and 13C- nuclear magnetic resonance (NMR), electrospray ionization mass spectra (ESI-MS), and FTIR spectroscopy (figures S2-4, S7-9, and S12-13 in the supporting information file).
3.1.1. Synthesis of the prototypical monomer CBM
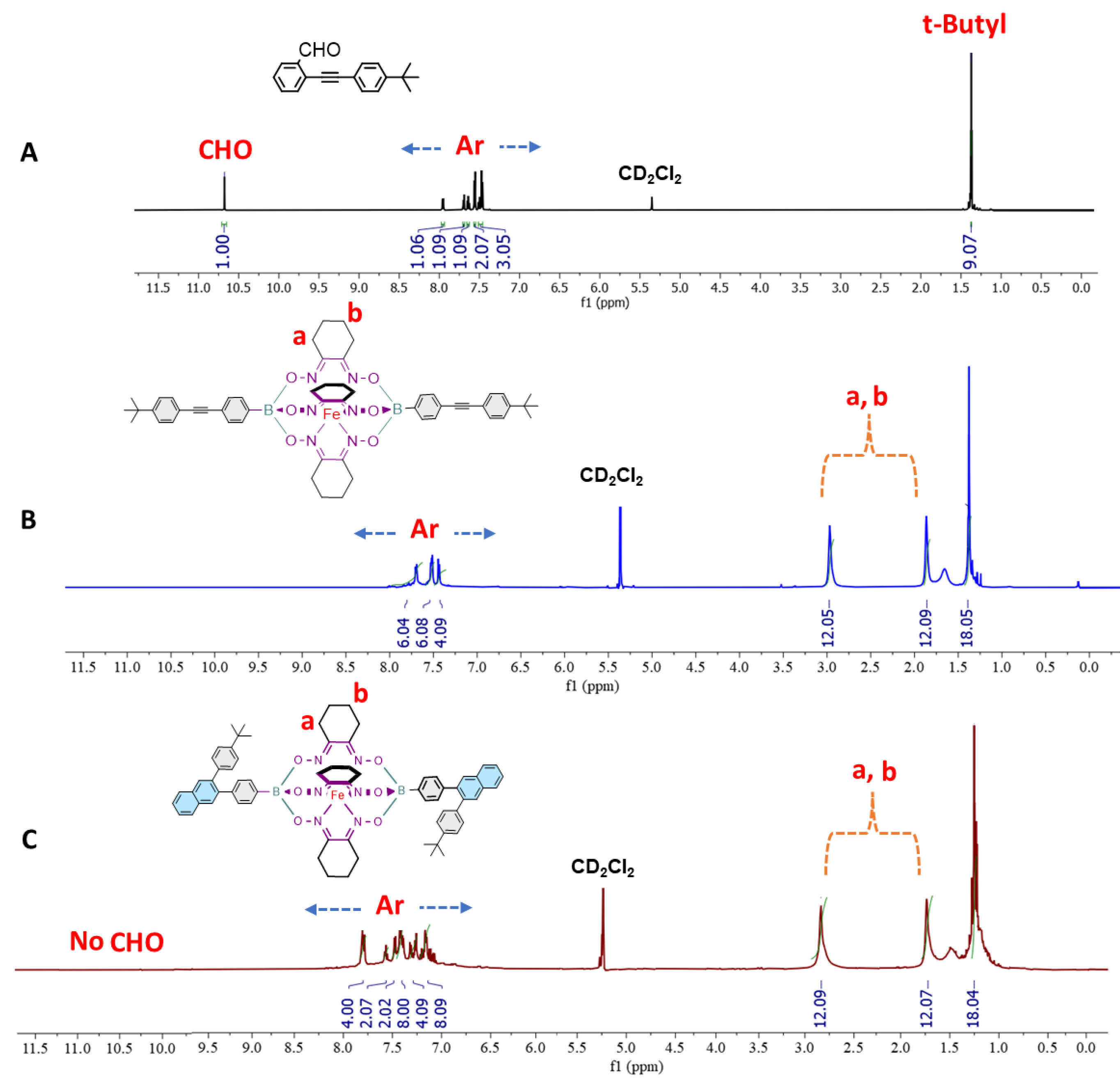
As a proof of concept, the prototypical iron(II) clathrochelate monomer CBM was prepared using the copper-catalyzed [4 + 2] cyclobenzannulation reaction conditions where the diethynyl-containing iron(II) clathrochelate synthon CM2 was reacted with two equivalents of 2-((4-(tert-butyl)phenyl)ethynyl) benzaldehyde 5 in presence of copper(II) triflate and trifluoracetic acid (TFA) in refluxing dichloroethane overnight affording CBM in quantitative yield (Scheme 2). The structure of the latter was confirmed by 1H- and 13C- nuclear magnetic resonance (NMR), ESI-MS, and FTIR spectroscopy (Figure 1 and figures S5, S10, S14 and S18 in the supporting information file).
Figure 1 portrays the comparative 1H-NMR spectra of the starting materials 5 and CM2 with CBM, where the spectrum of the latter clearly confirms the presence of all the desired peaks and the disappearance of those which could be attributed to synthon 5, namely, the fingerprint chemical shifts of the carbaldehyde group at 10.66 ppm and the t-butyl group at 1.37 ppm. The chemical shifts in the 1H-NMR spectrum of CBM ranging from 7.1 ppm to 7.8 ppm are assigned to the aromatic protons while those detected at 2.8 ppm, 1.73 ppm are attributed to the characteristic methylene (-CH2-) protons of the cyclohexyl groups (c.f. peaks labeled a, b in Figure 1). Likewise, the chemical shifts observed at 1.25 ppm are assigned to the methyl (-CH3) protons of the tertiary butyl group (Figure 1). 13C-NMR spectrum of CBM displays all the expected aromatic peaks in the range of 152.4–125.0 ppm in addition to the chemical shifts of the aliphatic carbons of both the cyclohexyl and tertiary butyl groups at 34.9 ppm, 31.6 ppm, 26.7 ppm, and 22.1 ppm (figures S10 in the supporting information file). In addition, the high purity of CBM was confirmed by electrospray ionization mass spectrometry (ESI-MS, figure S14 in the supporting information file).
3.1.2. Synthesis of copolymers CBP1-3
Synthesis of target copolymers CBP1-3 (Scheme 3) was carried out using reaction conditions similar to those employed to make the prototypical monomer CBM described in Scheme 2. The copper-catalyzed [4 + 2] cyclobenzannulation reaction of the diethynyl iron(II) clathrochelate derivatives CM1-3 and 2,5-bis(phenylethynyl)terephthalaldehyde 6 afforded the target copolymers CBP1-3 in excellent yields in the range of 83-95% (Scheme 3). Table 1 summarizes the attempts carried out to optimize the copolymerization reaction conditions: when a 2.5 x 10−2 M solution of 2,5-bis(phenylethynyl)terephthalaldehyde 6 and an equimolar amount of CM1 are reacted in refluxing dichloroethane (DCE) in presence of Cu(OTf)2 and TFA for 48 hours, CBP1 was isolated as an insoluble solid in 48% yield (Table 1, entry 1). Thus, to improve the reaction yield, the concentration of comonomers CM1 and 6 was diluted to a molar concentration of 1.25 x 10−2 M which afforded CBP1 in 65% (Table 1, entry 2). Further dilution of the comonomers to a concentration of 6 x 10−3 M resulted in the improvement of the reaction yield affording CBP1 in 83% (Table 1, entry 3).
Similar reaction conditions were employed in the copolymerization of 6 in presence of an equimolar amount of either CM2 or CM3, thus, affording CBP2 and CBP3 in 95% and 90% yields, respectively (Table 1 entry 4 and 5).
CBP1-3 were characterization by various techniques, namely, FTIR, XPS, TGA, UV-vis absorption, and emission spectroscopies (Figure 2-5 and figures S19-S21 and S22-S26 in the supporting information file). Nevertheless, the target copolymers were found to be insoluble in common organic solvents, such as THF, DCM, DMSO, methanol, acetone, and chloroform, which prevented their molar masses determination.
Figure 2 portrays the comparative FTIR absorption spectra for comonomer CM1 and its corresponding target copolymer CBP1. The characteristic stretching vibrations of the ethynyl (CΞC) group were detected at ~2211 cm−1 [44]. for CM1 which disappear from the spectrum of copolymer CBP1. It is noteworthy that the absorption bands identified at ~2955 cm−1 and ~1461 cm−1 correspond to the distinctive aliphatic C-H stretching and bending vibrations, respectively, which clearly indicates the presence of the butyl groups in CBP1 [45,46]. In addition, the fingerprint stretching vibrations were confirmed for each of the B-O (1396 cm−1), B-C (1180 cm−1) and aromatic C-H (820 cm−1) bending vibration peaks which further supports the formation of target copolymer CBP1[34,47]. Likewise, FTIR absorption spectra of target copolymers CBP2,3 reveal their distinctive stretching and bending vibration peaks which corroborates their successful formation (Figure 2 and figure S19-21 in the supporting information file).
X-ray Photoelectron spectroscopy (XPS) survey-scan spectra of CBP1-3 confirm the presence of all their constituting elements. The C1s, O1s, and N1s binding energies were detected in the range of ~284.8-284.7 eV, 532.2 eV, and 400.5-400.1 eV, respectively, whereas those for B1s and Fe2p were revealed in the range of 191.1-191.0 eV and 708.9-722.1 eV, respectively (Figure 3 and S22-23 in the supporting information file)[34].
Figure 4 illustrates the thermogravimetric analysis (TGA) of copolymers CBP1-3 depicting their 10% weight loss temperatures in the range of 200-310°C, which indicates their relatively high thermal stability.
Interestingly, UV–Vis absorption and emission spectra of the target copolymers CBP1-3 display similar features. The butyl- and cyclohexyl- containing iron(II) clathrochelate cyclobenzannulated copolymers CBP1,2 reveals a similar UV absorption band at ~312 whereas the one with phenyl side groups i.e., CBP3 displays a strong absorption band at 297 (Figure 5). The emission spectra of CBP1,2 portray a broad peak with an intensity maximum at 442 nm while the phenyl-containing copolymer CBP3 discloses an emission maximum at 451 nm (Figure 5 and S24 in the supporting information file).
The porosity of the target copolymers CBP1-3 was investigated using nitrogen adsorption-desorption experiments at 77 K and low relative pressure (figures S25-27 in the supporting information file). Brunauer-Emmett-Teller (BET) method revealed a surface area of 74.0 m2g‒1 for the cyclohexyl-containing copolymer CBP2 whereas those with butyl- and phenyl- side groups, i.e., CBP1 and CBP3, respectively show lower BET surface areas of 7.0 m2g‒1 and 35.0 m2g‒1, respectively. The pore volumes of CBP1-3 derived from these isotherms disclose values of 0.014 cm3 g‒1, 0.081 cm3 g‒1, and 0.030 cm3 g‒1 respectively.
3.2. Methyl red adsorption studies
Copolymers CBP1-3 were tested as adsorbents of the cancerogenic and mutagenic azo dye methyl red (MR). The uptake capacity was evaluated by soaking an aliquot of CBP1-3 in aqueous solution of MR (Figure 6 and figure S28-S29 in the supporting information file). The removal efficiency of MR by copolymers CBP1-3 was investigated by recording the UV-visible absorbance spectra of the dye’s aqueous solutions at different time intervals. The dye adsorption experiments were carried out by stirring a 5 mg sample of a given copolymer in a 5 mL aqueous solution of MR (20 mg L−1) at ambient temperature. The adsorption efficiency, E (%), and amount of dye adsorbed by the copolymer, qe (mg g‒1), were calculated using the following equations:[25]
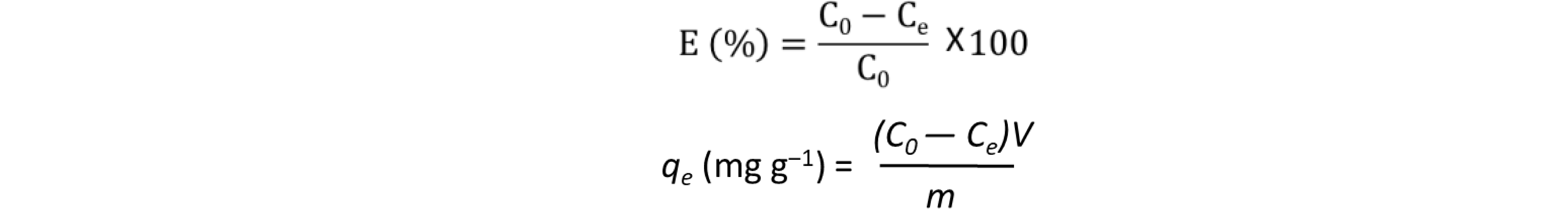
with C0 and Ce are the initial and equilibrium dye concentrations (mg L‒1), respectively, m (g) is the quantity of the adsorbent used, and V (L) is the volume of dye solution.
The absorbance maximum peak intensity of MR detected at 430 nm noticeably decreased upon the addition of the target polymers CBP1-3 to the solution, which confirms the latter to be very good adsorbents. Interestingly, all the copolymers reached 100% adsorption capacity of MR, but at different time intervals, with CBP3 being the fastest by quantitatively removing MR from the aqueous solution in 30 minutes at room temperature (Figure 6 and figure S28-S29 in the supporting information file).
To better comprehend the adsorption behavior of copolymers CBP1-3, the adsorption isotherm of MR removal experiments were obtained by preparing different aqueous solutions of MR with initial concentrations ranging from 50 to 600 mg L‒1 where Langmuir and Freundlich linear isotherm models were employed to fit the adsorption isotherm data. In case of Langmuir isotherm model, the following linear equation was utilized [25]:
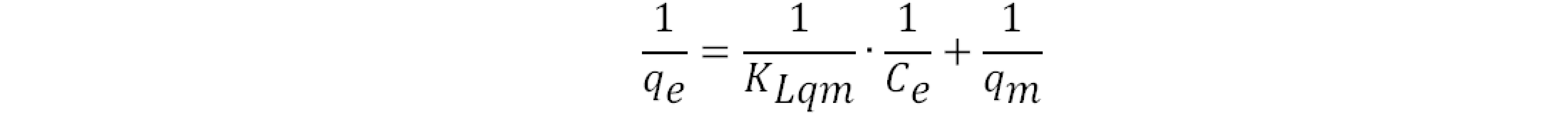
On the other hand, the linear equation below was employed for Freundlich isotherm model:[25]
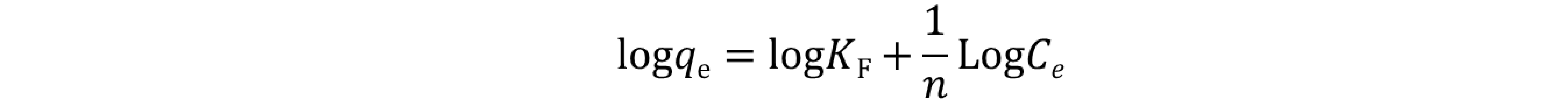 where qe (mg g‒1) denotes the equilibrium adsorption capacity, Ce (mg L‒1) represents the equilibrium dye concentration, and qm (mg g‒1) indicates the maximum adsorption capacity. KL is the Langmuir constant whereas KF and n are Freundlich constants correlated to the sorption capacity and sorption intensity, respectively (figures S30-S32 in the supporting information file). The Langmuir parameters were obtained by plotting the graph of 1/qe versus 1/Ce and those for Freundlich were derived from the plot of log qe versus log Ce (figures S30-S32 in the supporting information file). Both models were used to fit the equilibrium data obtained for MR adsorption. It is worthwhile to note that the correlation coefficient (R2) derived from the linear equation using the Langmuir model was found to be higher than that computed for the Freundlich isotherm model of MR (figure S32), thus, implying that the Langmuir isotherm is a more favorable model to illustrate the equilibrium data, and which suggests a homogenous adsorption and formation of monolayers of MR dye on the adsorbates CBP1-3. Additionally, the maximum adsorption capacity (qm) derived from the Langmuir model was found to be 199.20 mg g‒1 and 219.8 mg g‒1 for CBP2 and CBP1, respectively and it reaches 431.03 mg g‒1 for the phenyl-bearing iron(II) clathrochelate cyclobenzannulated copolymer CBP3, which to the best of our knowledge, is superior to the adsorption capacity values for most of the materials reported in the literature [5,24,48].
where qe (mg g‒1) denotes the equilibrium adsorption capacity, Ce (mg L‒1) represents the equilibrium dye concentration, and qm (mg g‒1) indicates the maximum adsorption capacity. KL is the Langmuir constant whereas KF and n are Freundlich constants correlated to the sorption capacity and sorption intensity, respectively (figures S30-S32 in the supporting information file). The Langmuir parameters were obtained by plotting the graph of 1/qe versus 1/Ce and those for Freundlich were derived from the plot of log qe versus log Ce (figures S30-S32 in the supporting information file). Both models were used to fit the equilibrium data obtained for MR adsorption. It is worthwhile to note that the correlation coefficient (R2) derived from the linear equation using the Langmuir model was found to be higher than that computed for the Freundlich isotherm model of MR (figure S32), thus, implying that the Langmuir isotherm is a more favorable model to illustrate the equilibrium data, and which suggests a homogenous adsorption and formation of monolayers of MR dye on the adsorbates CBP1-3. Additionally, the maximum adsorption capacity (qm) derived from the Langmuir model was found to be 199.20 mg g‒1 and 219.8 mg g‒1 for CBP2 and CBP1, respectively and it reaches 431.03 mg g‒1 for the phenyl-bearing iron(II) clathrochelate cyclobenzannulated copolymer CBP3, which to the best of our knowledge, is superior to the adsorption capacity values for most of the materials reported in the literature [5,24,48].Pseudo-first- and pseudo-second- order kinetic experiments were carried out to better understand the adsorption mechanism of MR on CBP3 using an initial concentration of 500 mg L−1 of MR dye at different time intervals every 15 minutes up to 150 minutes (Figure 7).
The linear equation that is detailed below was employed to investigate the pseudo-first-order model:[34]
ln (qe ‒ qt) = ln qe ‒ K1t
Alternatively, the pseudo-second-order model is expressed by:[34]
where qe (mg g‒1) and qt (mg g‒1) are the adsorption capacities at equilibrium and time t (min), respectively. k1 is the rate constant of the pseudo first-order model whereas k2 is the rate constant of the pseudo second-order model.
t/qt = t/qe + 1/k2qe2
As shown in Table 2, the calculated adsorption capacity at equilibrium, qe,cal, was derived from the pseudo-first-order model by plotting ln(qe – qt) versus t, whereas the same was computed from the plot of t/qt versus t from the pseudo-second-order model. Interestingly, Table 2 also discloses a correlation coefficient, R2 = 0.9821, derived from the linear equation using the pseudo-first-order model which is higher than the one derived from the pseudo-second-order model (R2 = 0.6559). Moreover, the comparison of the experimental capacity at equilibrium, qe,exp = 76 mg g‒1 with that calculated, qe,cal, = 76 mg g‒1, clearly reveal a better agreement between these values when derived from the pseudo-first-order model [49]. This strongly suggests that that the adsorption of MR by CBP3 follows a pseudo-first-order kinetic model, therefore, suggesting that the adsorption process is most likely physisorption.
Reusability experiments were carried out in order to test the adsorbing performance of copolymer CBP3 towards MR after several adsorption–desorption cycles. Thus, a used sample of CBP3 copolymer loaded with MR was ultrasonicated in deionized water for 10 min followed by its isolation by vacuum filtration over a membrane filter before adding the regenerated copolymer sample to a freshly prepared aqueous solution of MR. This procedure was repeated for several cycles proving a removal efficiency of 90.4% for CBP3 even after eight cycles (Figure 8).
4. Conclusions
We report the synthesis of a new class of three metalorganic copolymers CBP1-3 bearing iron(II) clathrochelate unit and interlinked by anthracene groups via a typical copper-catalyzed [4 + 2] cycloaddition polymerization reaction conditions. The target copolymers were isolated in excellent yields and revealed excellent removal capacities of the carcinogenic dye methyl red from aqueous medium, especially CBP3 which disclosed ultrafast and superior adsorption efficiency up to 100% in 30 minutes and exhibited maximum adsorption capacity (qm) of 431 mg g‒1 with the possibility to regenerate the hitherto mentioned polymer for several cycles. The novel iron (II) clathrochelate-based copolymers presented herein confer several advantages, particularly, a versatile synthesis methodology, low cost, superior stability and excellent adsorption capacity. Therefore, these materials are prominent candidates for environmental remediation applications, specifically, as adsorbents of the hazardous azo dyes.
Data Availability Statement
The following supporting information can be downloaded at: Data Availability Statement: The raw/processed data required to reproduce these findings can be shared upon demand.
Acknowledgements
The project was partially supported by Kuwait Foundation for the Advancement of Sciences (KFAS) under project code PN18-14SC-03.
References
- Etale, A.; Onyianta, A. J.; Turner, S. R.; Eichhorn, S. J. Cellulose: A Review of Water Interactions, Applications in Composites, and Water Treatment. Chemical Reviews 2023, 123(5), 2016–2048. [Google Scholar] [CrossRef] [PubMed]
- Al-Tohamy, R.; Ali, S. S.; Li, F.; Okasha, K. M.; Mahmoud, Y. A. G.; Elsamahy, T.; Jiao, H.; Fu, Y.; Sun, J. A critical review on the treatment of dye-containing wastewater: Ecotoxicological and health concerns of textile dyes and possible remediation approaches for environmental safety. Ecotoxicology and Environmental Safety 2022, 231, 113160. [Google Scholar] [CrossRef]
- Ogugbue, C. J.; Sawidis, T. Bioremediation and Detoxification of Synthetic Wastewater Containing Triarylmethane Dyes by <Aeromonas hydrophila> Isolated from Industrial Effluent. Biotechnology Research International 2011, 2011, 967925. [Google Scholar] [CrossRef]
- Kaur, H.; Devi, N.; Siwal, S. S.; Alsanie, W. F.; Thakur, M. K.; Thakur, V. K. Metal–Organic Framework-Based Materials for Wastewater Treatment: Superior Adsorbent Materials for the Removal of Hazardous Pollutants. ACS Omega 2023, 8(10), 9004–9030. [Google Scholar] [CrossRef] [PubMed]
- Takkar, S.; Tyagi, B.; Kumar, N.; Kumari, T.; Iqbal, K.; Varma, A.; Thakur, I. S.; Mishra, A. Biodegradation of methyl red dye by a novel actinobacterium Zhihengliuella sp. ISTPL4: Kinetic studies, isotherm and biodegradation pathway. Environmental Technology & Innovation 2022, 26, 102348. [Google Scholar] [CrossRef]
- Rojas, S.; Horcajada, P. Metal–Organic Frameworks for the Removal of Emerging Organic Contaminants in Water. Chemical Reviews 2020, 120(16), 8378–8415. [Google Scholar] [CrossRef] [PubMed]
- Lellis, B.; Fávaro-Polonio, C. Z.; Pamphile, J. A.; Polonio, J. C. Effects of textile dyes on health and the environment and bioremediation potential of living organisms. Biotechnology Research and Innovation 2019, 3(2), 275–290. [Google Scholar] [CrossRef]
- Benkhaya, S.; M’Rabet, S.; El Harfi, A. Classifications, properties, recent synthesis and applications of azo dyes. Heliyon 2020, 6(1), e03271. [Google Scholar] [CrossRef]
- Overdahl, K. E.; Gooden, D.; Bobay, B.; Getzinger, G. J.; Stapleton, H. M.; Ferguson, P. L. Characterizing azobenzene disperse dyes in commercial mixtures and children’s polyester clothing. Environmental Pollution 2021, 287, 117299. [Google Scholar] [CrossRef]
- Khan, Z.; Jain, K.; Soni, A.; Madamwar, D. Microaerophilic degradation of sulphonated azo dye—Reactive Red 195 by bacterial consortium AR1 through co-metabolism. International Biodeterioration & Biodegradation 2014, 94, 167–175. [Google Scholar] [CrossRef]
- Leal Filho, W.; Perry, P.; Heim, H.; Dinis, M. A. P.; Moda, H.; Ebhuoma, E.; Paço, A. An overview of the contribution of the textiles sector to climate change. Frontiers in Environmental Science 2022, 10. [Google Scholar] [CrossRef]
- Kishor, R.; Purchase, D.; Saratale, G. D.; Saratale, R. G.; Ferreira, L. F. R.; Bilal, M.; Chandra, R.; Bharagava, R. N. Ecotoxicological and health concerns of persistent coloring pollutants of textile industry wastewater and treatment approaches for environmental safety. Journal of Environmental Chemical Engineering 2021, 9(2), 105012. [Google Scholar] [CrossRef]
- Niinimäki, K.; Peters, G.; Dahlbo, H.; Perry, P.; Rissanen, T.; Gwilt, A. The environmental price of fast fashion. Nature Reviews Earth & Environment 2020, 1 (4), 189-200. [CrossRef]
- Hossain, M. S.; Das, S. C.; Islam, J. M. M.; Al Mamun, M. A.; Khan, M. A. Reuse of textile mill ETP sludge in environmental friendly bricks—effect of gamma radiation. Radiation Physics and Chemistry 2018, 151, 77–83. [Google Scholar] [CrossRef]
- Koulini, G. V.; Laiju, A. R.; Ramesh, S. T.; Gandhimathi, R.; Nidheesh, P. V. Effective degradation of azo dye from textile wastewater by electro-peroxone process. Chemosphere 2022, 289, 133152. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Bankole, P. O.; Sadasivam, S. K. Biodecolorization and degradation of textile azo dyes using Lysinibacillus sphaericus MTCC 9523. Frontiers in Environmental Science 2022, 10. [Google Scholar] [CrossRef]
- Franca, R. D. G.; Oliveira, M. C.; Pinheiro, H. M.; Lourenço, N. D. Biodegradation Products of a Sulfonated Azo Dye in Aerobic Granular Sludge Sequencing Batch Reactors Treating Simulated Textile Wastewater. ACS Sustainable Chemistry & Engineering 2019, 7 (17), 14697-14706. [CrossRef]
- Mishra, A.; Gupta, B.; Kumar, N.; Singh, R.; Varma, A.; Thakur, I. S. Synthesis of calcite-based bio-composite biochar for enhanced biosorption and detoxification of chromium Cr (VI) by Zhihengliuella sp. ISTPL4. Bioresource Technology 2020, 307, 123262. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Gupta, B.; Srivastava, S. K.; Gupta, A. K. Recent advances on the removal of dyes from wastewater using various adsorbents: a critical review. Materials Advances 2021, 2(14), 4497–4531. [Google Scholar] [CrossRef]
- Wu, L.; Liu, X.; Lv, G.; Zhu, R.; Tian, L.; Liu, M.; Li, Y.; Rao, W.; Liu, T.; Liao, L. Study on the adsorption properties of methyl orange by natural one-dimensional nano-mineral materials with different structures. Scientific Reports 2021, 11(1), 10640. [Google Scholar] [CrossRef]
- Maniyam, M. N.; Ibrahim, A. L.; Cass, A. E. G. Decolourization and biodegradation of azo dye methyl red by Rhodococcus strain UCC 0016. Environ Technol 2020, 41(1), 71–85. [Google Scholar] [CrossRef]
- Baena-Baldiris, D.; Montes-Robledo, A.; Baldiris-Avila, R. Franconibacter sp., 1MS: A New Strain in Decolorization and Degradation of Azo Dyes Ponceau S Red and Methyl Orange. ACS Omega 2020, 5 (43), 28146-28157. [CrossRef]
- Ajaz, M.; Rehman, A.; Khan, Z.; Nisar, M. A.; Hussain, S. Degradation of azo dyes by Alcaligenes aquatilis 3c and its potential use in the wastewater treatment. AMB Express 2019, 9(1), 64. [Google Scholar] [CrossRef]
- Muthuraman, G.; Teng, T. T. Extraction of methyl red from industrial wastewater using xylene as an extractant. Progress in Natural Science 2009, 19(10), 1215–1220. [Google Scholar] [CrossRef]
- Shetty, S.; Baig, N.; Al-Mousawi, S.; Alameddine, B. Cover Image, Volume 139, Issue 43. Journal of Applied Polymer Science 2022, 139(43), e51150. [Google Scholar] [CrossRef]
- Losytskyy, M.; Chornenka, N.; Vakarov, S.; Meier-Menches, S. M.; Gerner, C.; Potocki, S.; Arion, V. B.; Gumienna-Kontecka, E.; Voloshin, Y.; Kovalska, V. Sensing of Proteins by ICD Response of Iron(II) Clathrochelates Functionalized by Carboxyalkylsulfide Groups Biomolecules [Online], 2020.
- Jansze, S. M.; Severin, K. Clathrochelate Metalloligands in Supramolecular Chemistry and Materials Science. Accounts of Chemical Research 2018, 51(9), 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Idrees, K. B.; Shetty, S.; Xie, H.; Wasson, M. C.; Gong, W.; Zhang, X.; Alameddine, B.; Farha, O. K. Regulation of Catenation in Metal–Organic Frameworks with Tunable Clathrochelate-Based Building Blocks. Crystal Growth & Design 2021, 21 (12), 6665-6670. [CrossRef]
- Pomadchik, A. L.; Belov, A. S.; Lebed, E. G.; Belaya, I. G.; Vologzhanina, A. V.; Voloshin, Y. Z. Dramatic Effect of A Ring Size of Alicyclic α-Dioximate Ligand Synthons on Kinetics of the Template Synthesis and of the Acidic Decomposition of the Methylboron-Capped Iron(II) Clathrochelates Molecules [Online], 2021.
- Kovalska, V.; Vakarov, S.; Losytskyy, M.; Kuperman, M.; Chornenka, N.; Toporivska, Y.; Gumienna-Kontecka, E.; Voloshin, Y.; Varzatskii, O.; Mokhir, A. Dicarboxyl-terminated iron(ii) clathrochelates as ICD-reporters for globular proteins. RSC Advances 2019, 9(42), 24218–24230. [Google Scholar] [CrossRef] [PubMed]
- Alameddine, B.; Shetty, S.; Baig, N.; Al-Mousawi, S.; Al-Sagheer, F. Synthesis and characterization of metalorganic polymers of intrinsic microporosity based on iron(II) clathrochelate. Polymer 2017, 122, 200–207. [Google Scholar] [CrossRef]
- Shetty, S.; Idrees, K. B.; Xie, H.; Alameddine, B.; Farha, O. K. Synthesis of zirconium-based metal–organic frameworks with iron(ii) clathrochelate ligands. CrystEngComm 2023, 25(10), 1550–1555. [Google Scholar] [CrossRef]
- Gong, W.; Xie, Y.; Pham, T. D.; Shetty, S.; Son, F. A.; Idrees, K. B.; Chen, Z.; Xie, H.; Liu, Y.; Snurr, R. Q.; Chen, B.; Alameddine, B.; Cui, Y.; Farha, O. K. Creating Optimal Pockets in a Clathrochelate-Based Metal–Organic Framework for Gas Adsorption and Separation: Experimental and Computational Studies. Journal of the American Chemical Society 2022, 144(8), 3737–3745. [Google Scholar] [CrossRef]
- Baig, N.; Shetty, S.; Habib, S. S.; Husain, A. A.; Al-Mousawi, S.; Alameddine, B. Synthesis of Iron(II) Clathrochelate-Based Poly(vinylene sulfide) with Tetraphenylbenzene Bridging Units and Their Selective Oxidation into Their Corresponding Poly(vinylene sulfone) Copolymers: Promising Materials for Iodine Capture. Polymers 2022, 14(18), 3727. [Google Scholar] [CrossRef]
- Shetty, S.; Baig, N.; Al-Mousawi, S.; Al-Sagheer, F.; Alameddine, B. Synthesis of secondary arylamine copolymers with Iron(II) clathrochelate units and their functionalization into tertiary Polyarylamines via Buchwald-Hartwig cross-coupling reaction. Polymer 2019, 178, 121606. [Google Scholar] [CrossRef]
- Alameddine, B.; Shetty, S.; Anju, R. S.; Al-Sagheer, F.; Al-Mousawi, S. Highly soluble metal-organic polymers based on iron(II) clathrochelates and their gelation induced by sonication. European Polymer Journal 2017, 95, 566–574. [Google Scholar] [CrossRef]
- Shetty, S.; Baig, N.; Alameddine, B. Synthesis and Iodine Adsorption Properties of Organometallic Copolymers with Propeller-Shaped Fe(II) Clathrochelates Bridged by Different Diaryl Thioether and Their Oxidized Sulfone Derivatives. Polymers 2022, 14(22), 4818. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Baig, N.; Moustafa, M. S.; Al-Mousawi, S.; Alameddine, B. Synthesis of Metalorganic Copolymers Containing Various Contorted Units and Iron(II) Clathrochelates with Lateral Butyl Chains: Conspicuous Adsorbents of Lithium Ions and Methylene Blue. Polymers 2022, 14(16), 3394. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Baig, N.; Hassan, A.; Al-Mousawi, S.; Das, N.; Alameddine, B. Fluorinated Iron(ii) clathrochelate units in metalorganic based copolymers: improved porosity, iodine uptake, and dye adsorption properties. RSC Advances 2021, 11(25), 14986–14995. [Google Scholar] [CrossRef] [PubMed]
- Baig, N.; Shetty, S.; Al-Mousawi, S.; Al-Sagheer, F.; Alameddine, B. Influence of size and nature of the aryl diborate spacer on the intrinsic microporosity of Iron(II) clathrochelate polymers. Polymer 2018, 151, 164–170. [Google Scholar] [CrossRef]
- Baig, N.; Shetty, S.; Al-Mousawi, S.; Alameddine, B. Conjugated microporous polymers using a copper-catalyzed [4 + 2] cyclobenzannulation reaction: promising materials for iodine and dye adsorption. Polymer Chemistry 2021, 12(15), 2282–2292. [Google Scholar] [CrossRef]
- Baig, N.; Shetty, S.; Tiwari, R.; Pramanik, S. K.; Alameddine, B. Aggregation-Induced Emission of Contorted Polycondensed Aromatic Hydrocarbons Made by Edge Extension Using a Palladium-Catalyzed Cyclopentannulation Reaction. ACS Omega 2022, 7(49), 45732–45739. [Google Scholar] [CrossRef] [PubMed]
- Baig, N.; Shetty, S.; Al-Mousawi, S.; Alameddine, B. Synthesis of conjugated polymers via cyclopentannulation reaction: promising materials for iodine adsorption. Polymer Chemistry 2020, 11(17), 3066–3074. [Google Scholar] [CrossRef]
- Baig, N.; Shetty, S.; Al-Mousawi, S.; Al-Sagheer, F.; Alameddine, B. Synthesis of triptycene-derived covalent organic polymer networks and their subsequent in-situ functionalization with 1,2-dicarbonyl substituents. Reactive and Functional Polymers 2019, 139, 153–161. [Google Scholar] [CrossRef]
- Baig, N.; Shetty, S.; Moustafa, M. S.; Al-Mousawi, S.; Alameddine, B. Selective removal of toxic organic dyes using Trӧger base-containing sulfone copolymers made from a metal-free thiol-yne click reaction followed by oxidation. RSC Advances 2021, 11(34), 21170–21178. [Google Scholar] [CrossRef]
- Shetty, S.; Baig, N.; Moustafa, M. S.; Al-Mousawi, S.; Alameddine, B. Sizable iodine uptake of porous copolymer networks bearing Tröger’s base units. Polymer 2021, 229, 123996. [Google Scholar] [CrossRef]
- Baig, N.; Shetty, S.; Pasha, S. S.; Pramanik, S. K.; Alameddine, B. Copolymer networks with contorted units and highly polar groups for ultra-fast selective cationic dye adsorption and iodine uptake. Polymer 2022, 239, 124467. [Google Scholar] [CrossRef]
- Gul, S.; Kanwal, M.; Qazi, R. A.; Gul, H.; Khattak, R.; Khan, M. S.; Khitab, F.; Krauklis, A. E. Efficient Removal of Methyl Red Dye by Using Bark of Hopbush Water [Online], 2022.
- Khan, E. A. ; Shahjahan; Khan, T. A. Adsorption of methyl red on activated carbon derived from custard apple (Annona squamosa) fruit shell: Equilibrium isotherm and kinetic studies. Journal of Molecular Liquids 2018, 249, 1195–1211. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of comonomers CM1-3.
Scheme 2.
Synthesis of CBM.
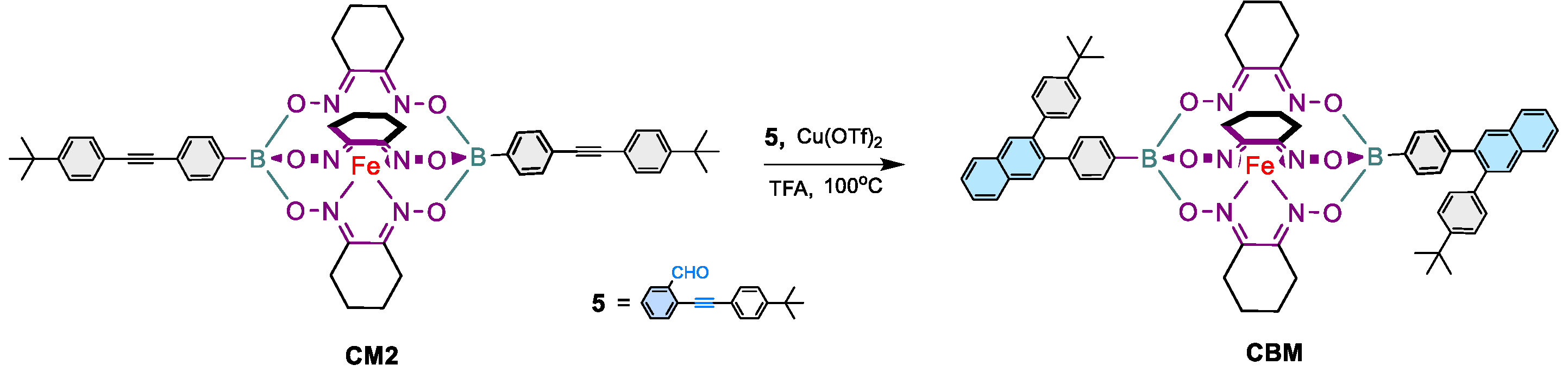
Figure 1.
Comparative 1H-NMR spectra of 5 (A), CM2 (B), and CBM (C).
Scheme 3.
Synthesis of copolymers CBP1-3.
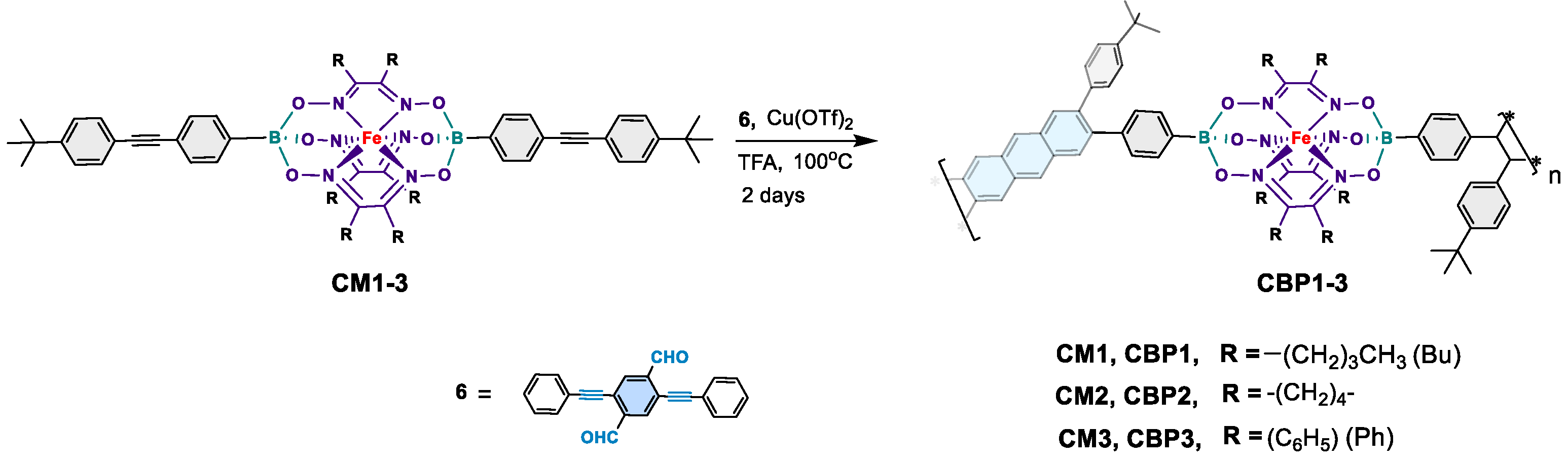
Figure 2.
Comparative FT-IR spectrum of CM1 (red) and CBP1 (blue).
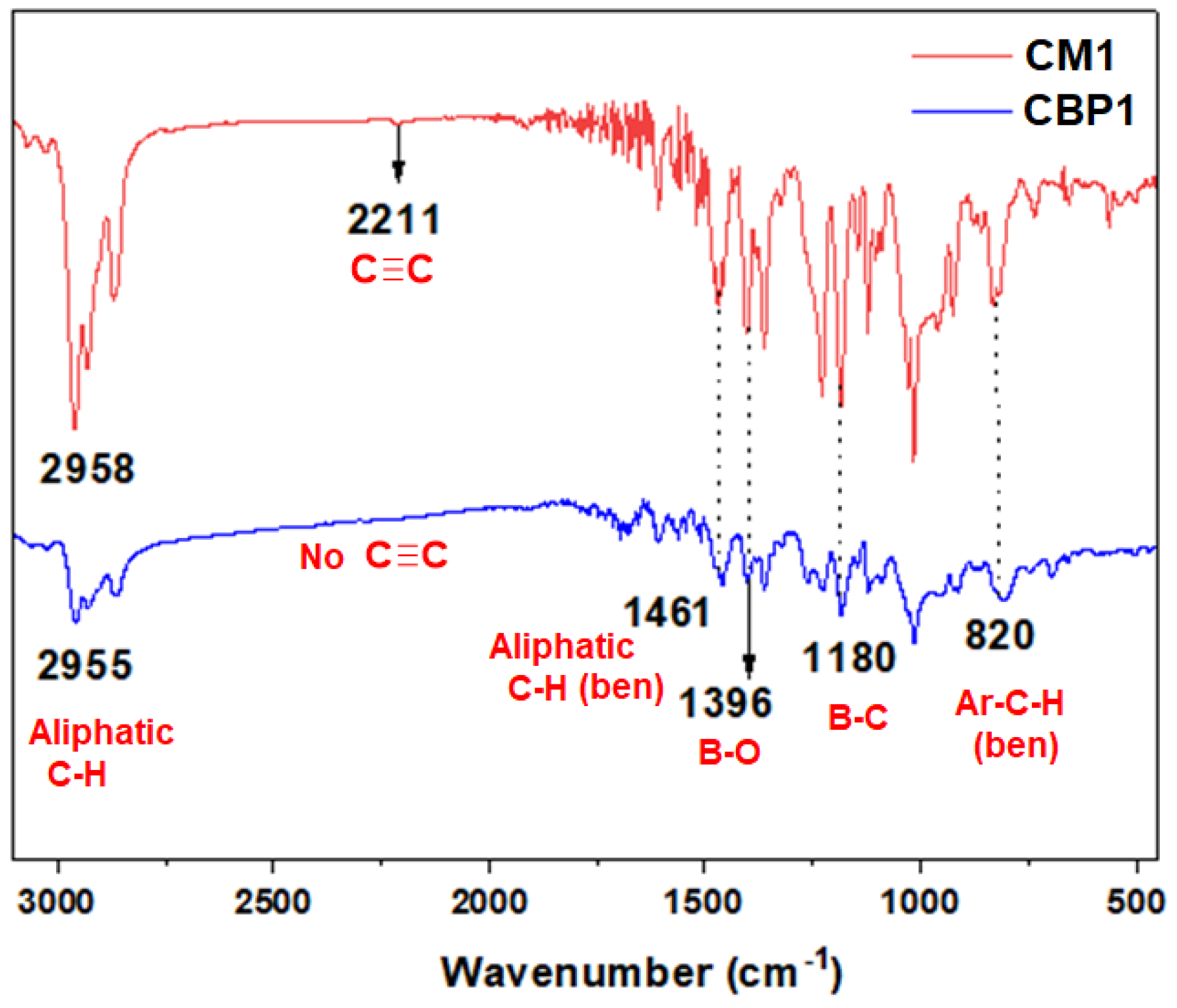
Figure 3.
High-resolution XPS survey scan and spectra of B1s, C1s, N1s, O1s, and Fe2p of copolymer CBP2.
Figure 3.
High-resolution XPS survey scan and spectra of B1s, C1s, N1s, O1s, and Fe2p of copolymer CBP2.
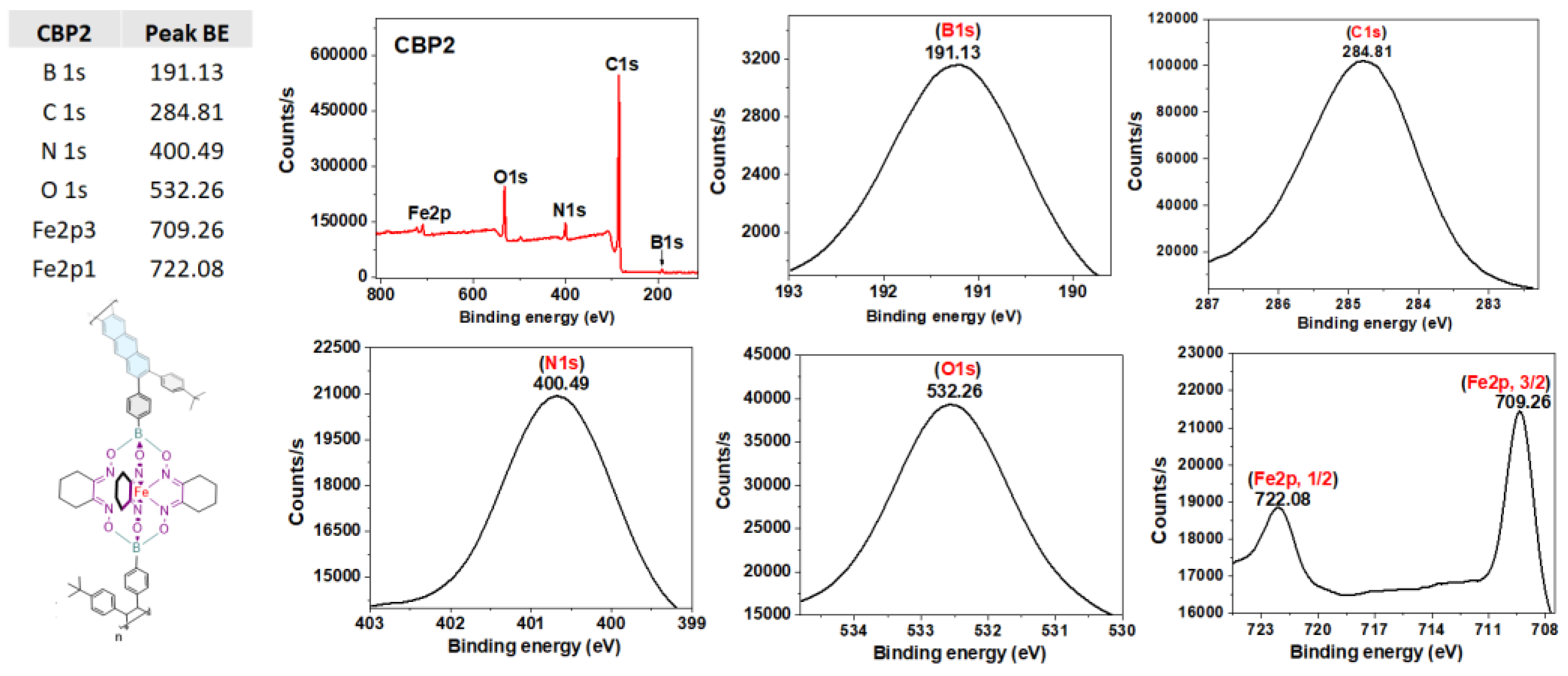
Figure 4.
TGA thermograms of polymers CBP1-3, Td represents the temperature of 10% weight loss.
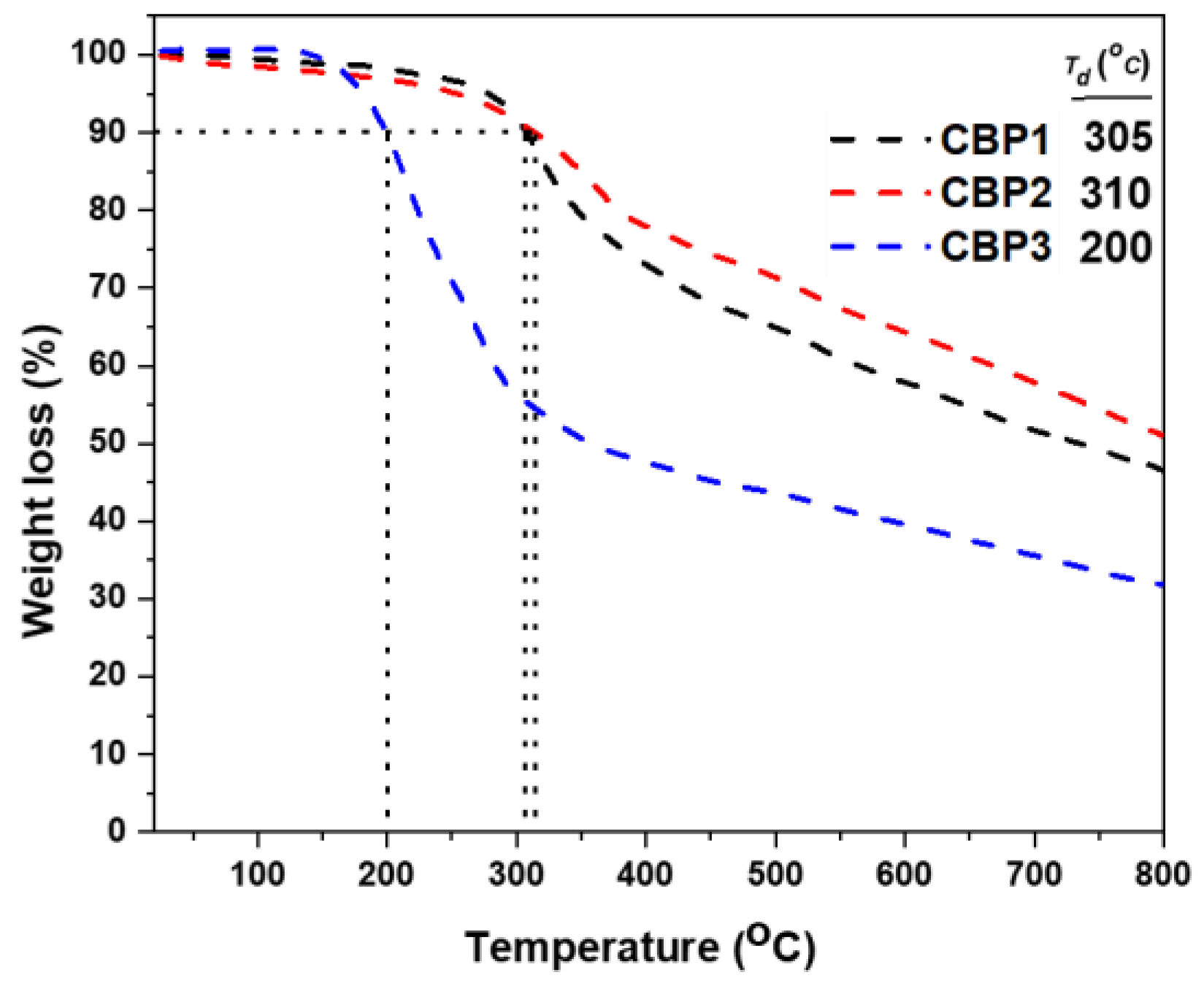
Figure 5.
Normalized UV-VIS absorption (solid lines) and emission (dotted lines) spectra of CBP1-3 (CM = 10−8 M in THF).
Figure 5.
Normalized UV-VIS absorption (solid lines) and emission (dotted lines) spectra of CBP1-3 (CM = 10−8 M in THF).
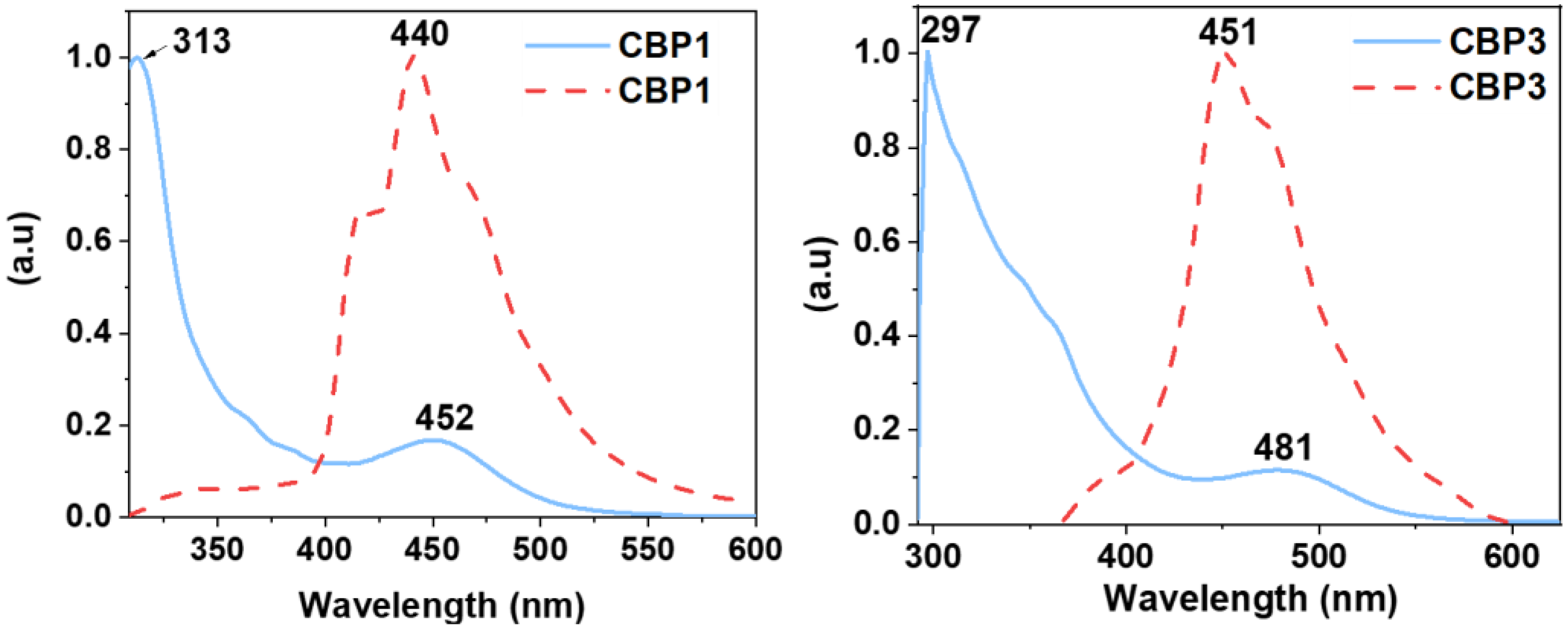
Figure 6.
UV–vis absorption spectra of aqueous solution of MR in the presence CBP3 at various time intervals (inset: Photographs showing the color change upon dye adsorption).
Figure 6.
UV–vis absorption spectra of aqueous solution of MR in the presence CBP3 at various time intervals (inset: Photographs showing the color change upon dye adsorption).
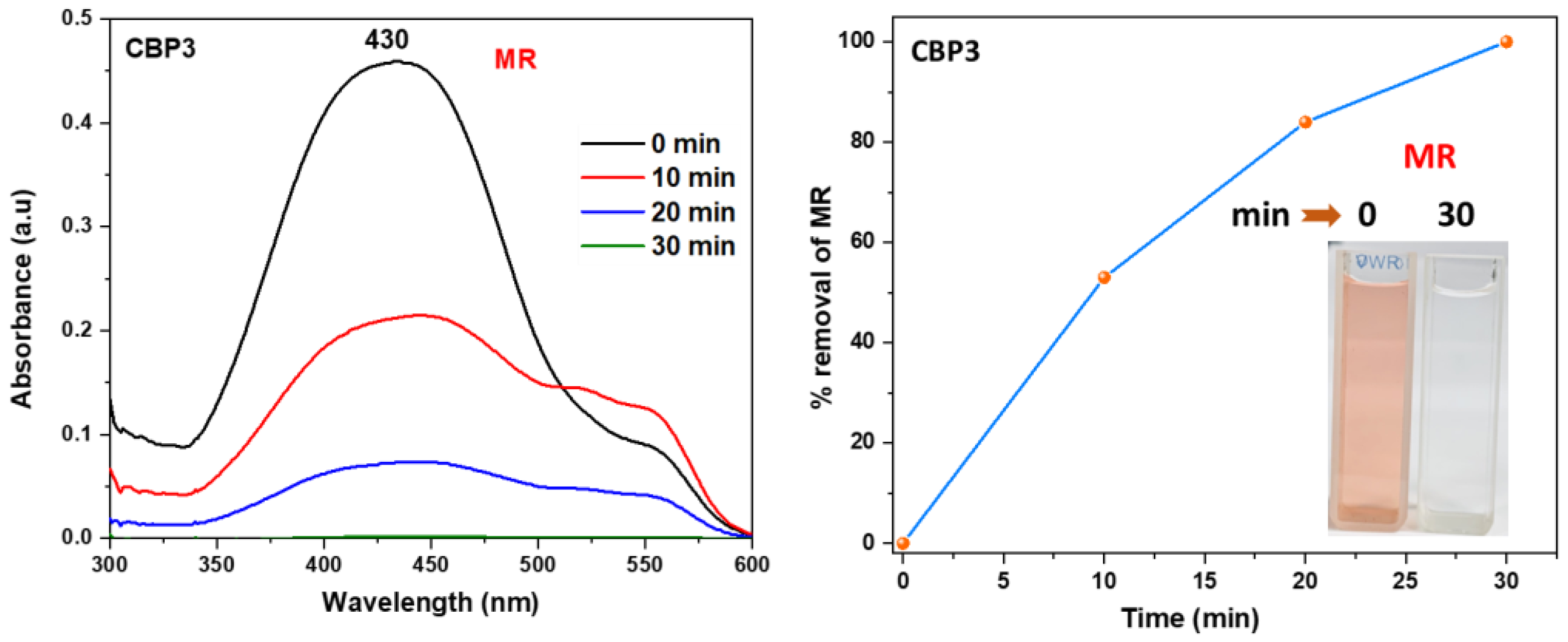
Figure 7.
Kinetic modelling of MR by CBP3 using pseudo-first-order (left) and pseudo-second-order kinetic model (right).
Figure 7.
Kinetic modelling of MR by CBP3 using pseudo-first-order (left) and pseudo-second-order kinetic model (right).
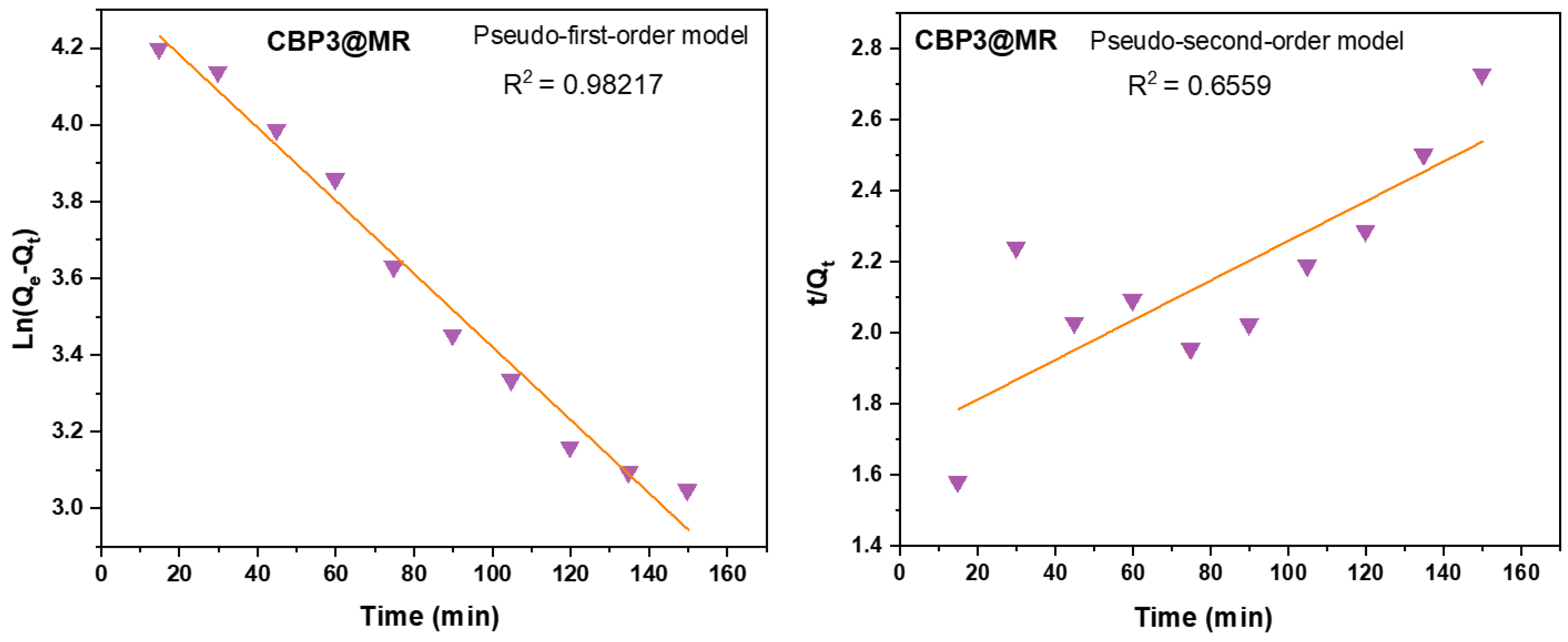
Figure 8.
Graphical representation of MR adsorption recyclability of CBP3.
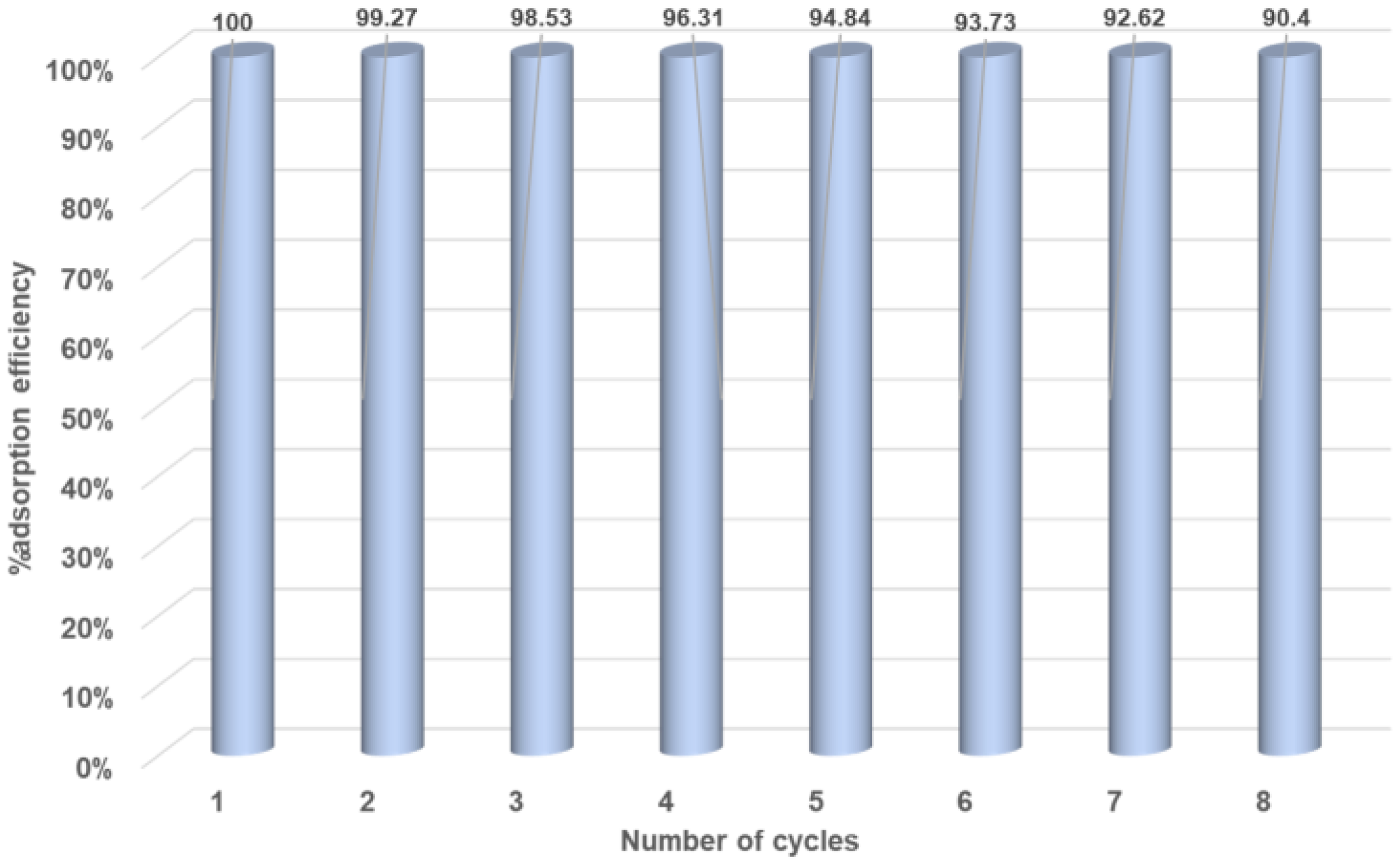

Table 1.
Summary of optimized reaction conditions of copolymers CBP1-3.
| Entry | Copolymer a | Time in days | CM b[M] | Yield (%) |
|---|---|---|---|---|
| 1 | CBP1 | 2 | 2.5x10‒2 | 48 |
| 2 | CBP1 | 2 | 1.25x10‒2 | 65 |
| 3 | CBP1 | 2 | 6.0x10‒3 | 83 |
| 4 | CBP2 | 2 | 6.0x10‒3 | 95 |
| 5 | CBP3 | 2 | 6.0x10‒3 | 90 |
a: Cu(OTf)2 (0.4 eq.), TFA (8.0 eq.), and DCE;b Molar concentration of 6.
Table 2.
pseudo-first-order and pseudo-second-order kinetic model parameters for the adsorption of MR on CBP3.
Table 2.
pseudo-first-order and pseudo-second-order kinetic model parameters for the adsorption of MR on CBP3.
| Dye on CBP3 | Pseudo 1st order model | Pseudo 2nd order model | ||||||
| C0 (mg L‒1) |
qe, exp (mg g−1) |
qe, cal (mg g−1) |
k1 (min−1) |
R2 | qe, cal (mg g−1) |
k2 (min−1) |
R2 | |
| MR | 500 | 76 | 79.35 | -0.00013 | 0.9821 | 32116 | 1.83E-05 | 0.6559 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



