
Submitted:
06 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
Diseases of the lung account for more than 5 million deaths worldwide and are a burden to healthcare. Improving clinical outcomes including mortality and quality of life involves a holistic understanding the etiopathogenesis, which can be provided by multi-omics integration of lung data. An enhanced understanding of large comprehensive datasets provides opportunities to mine those datasets for features that contribute to prevention and amelioration of disease. In this review, we evaluate lung disease models including animal models, organoids and single cell lines as mechanisms to study multiomics in lung health and disease. We provide examples of lung diseases where multi-omics investigations have provided a deeper insight into pathogenesis that has resulted in improved preventive and therapeutic interventions.
Keywords:
multiomics
; lung
; respiratory
; models
; integration
; outcomes
Introduction
Respiratory diseases account for over 5 million deaths yearly and are a huge burden to health-care systems worldwide [1]. Recent advances in high-throughput technologies have provided access to multiomics biological data including genomics, epigenomics, transcriptomics, proteomics, metabolomics and immunomics and provide a holistic view of pathophysiology in lung disease [2]. Biological insights gained from multi-omics can be integrated with clinical and social data and applied in the clinical setting for improved health outcomes. Single omics is limited by the only providing associations whereas multiomic integrations result in a clearer overall mechanistic picture-based overview and thus generates testable hypotheses. State-of-the-art machine-learning methods can integrate these datasets resulting in the ability to predict short- and long-term health trajectories and enable early timely interventions that alters the health course towards better outcomes.
High dimensional data from multiple sources can be integrated using machine learning tools including deep learning and neural networks to yield reliable holistic predictive models to predict mortality, morbidity or other complications in lung disease. Large datasets such as the omics dataset rely on ‘deep learning’ based on neural networks loosely modeled after neurons of the brain. The insights gained by deep learning of multi-omic datasets lead to personalized healthcare decision making (precision medicine) and biomarker discovery.
The NIH defines precision medicine as ‘an innovative approach that takes into account individual differences in patients’ genes, environments, and lifestyles’ [3] (https://www.nih.gov/about-nih/what-we-do/nih-turning-discovery-into-health/promise-precision-medicine). There is an urgent need to shift our current thinking on traditional reactive medicine based on prior literature/data to a more proactive precision medicine (PM) based approach where the trajectory towards health and disease can be predicted in advance, so interventions to improve survival or decrease morbidity can be instituted earlier to improve survival and decrease morbidity. Machine learning tools have already been enabled in a holistic, systems biology approach in oncology fields for prediction of survival, disease severity and biomarker development. This proactive approach need to be adapted in other fields and disciplines. In this review, we will assess the multi-omics strategy as it is integrated into humans, animal and organoid models to provide insight into lung health and disease.
Insights into cell biology using multi-omics
Quantitative omics technologies enable cost-effective and high-throughput profiling of numerous properties of cell biology. Genomics can be profiled using whole-exome or whole-genome sequencing. Transcriptomics is assessed using RNA-Sequencing (RNA-Seq). Protein expression in its total form or as post-translational modifications is measured using mass-spectrometry or antibody-based proteomics assays. Measurable epigenomic properties of the cells span DNA methylation, assessed via whole-genome bisulfite sequencing or probe-based micro-arrays, microRNAs, measured using smallRNA-Seq, histone modifications, measured using Chromatin Immunoprecipitation and sequencing (ChIP-Seq), and open chromatin, measured using assay for transposase-accessible chromatin with sequencing (ATAC-Seq). Getting closer to cell biology, metabolomics and lipidomics can be measured using mass-spectrometry based techniques. Appreciating that humans live in symbiosis with a rich microbial and fungal community, microbiome or mycobiome can be measured using whole genome shotgun sequencing.
The advent of single cell technologies, including single cell RNA-Seq (scRNA-Seq), single cell ATAC-Seq (scATAC-Seq) have provided further insight into cell biology in the last decade. A single-cell multi-omics study CoV2 employed single nuclei RNA-Seq and single nuclei ATAC-Seq in phenotypically healthy lungs of donors with ages of 30 week gestation, 3 years, and 30 years[4]. It aimed to decipher the cell specific landscape of expression and candidate cis-regulatory elements (cCREs) of key host entry genes for SARS-CoV2 infection, including ACE2 and TMPRSS2, and to further explore the changes with age, given the age-associated documented risks of SARS-CoV2 infection and outcome. ACE2 transcript was found in under 100 cells, with almost half of them in AT2 cells. TMPRSS2 expression was detected particularly in AT1, AT2, club, ciliated, and goblet cells. ATAC signal, eg accessible chromatin, was found primarily in gene bodies for ACE2 and TMPRSS2 in AT1, AT2, club, ciliated, and basal cells. cCREs are areas of ATAC peaks, determined within each cell type; cCRE association with a nearby gene is inferred by co-accessibility with promoters of nearby genes. Using the Cicero software, 15 cCREs co-accessible with the promoter were found for ACE2, whereas 73 were found for TMPRSS2[5]. Given the dramatic changes in SARS-CoV2 infection, symptoms, and outcome risk across the age spectrum, the study quantified the expression and cCREs for ACE2 and TMPRSS2 across neonate, infant, and adult lung. A larger proportion of AT2 cells expressed ACE2 and TMPRSS2 in adults compared to neonates and infants. Using the ATAC data, two cCRE clusters for TMPRSS2 were in AT2 cells, comprising nine cCREs, showed enhanced accessibility with age. These clusters associated with genes involved in response to viral infection, immune response, and injury repair, and also overlapped with genes discovered in mouse models to associate with lung epithelial necrosis and chronic inflammation. Overall, this map of epigenomic/transcriptomic at SARS-CoV2 host genes can serve as a reference for studies using lungs from donors with SARS-CoV2 or animal models.
A study utilizing single cell RNA-Seq, single cell ATAC-Seq, and spatial transcriptomics generated an atlas of human fetal lungs spanning 5-22 post conception weeks (PCW)[6]. This study identified 144 cell types. Overall, cell clusters are grouped by age groups, into 5-6, 9-11, and 15-22 PCW. Many of the fetal cells were matched to their adult lung counterpart, such as fetal airway progenitors with adult secretory club cells, and proximal secretory fetal cells with adult goblet cells. Interestingly, fetal AT1 and AT2 cells had highly concordant transcriptomic profiles with the adult cells. This study identified multiple cell types specific to the developing lung, such as progenitor of secretory cells and transition populations. Progenitor cells were further spatially localized. Epithelial progenitor cells were stratified into tip, stalk, airway progenitor, and proximal secretory progenitor, and using the spatial transcriptomics they were spatially localized and assigned on a trajectory of differentiation. The software CellPhoneDB was utilized to elucidate cell-cell communication in distinct lung niches[7]. The airway niche was comprised of airway fibroblasts, late airway SMCs, and airway epithelial cells. Cell-cell communication analysis between airway fibroblasts and airway epithelium included known signaling, such as via TGFB and BMP4, but also novel ones, such as FGF7/18 to FGFR2/3, and non-canonical WNT5A to FZD/ROR. These results were validated via tissue staining, but also by using distal tip based lung organoids; when grown in media with FGF, those organoids showed robust airway differentiation into secretory, basal, and ciliated cells. Using scATAC-Seq, transcription regulation was assessed in each cell type; analysis recapitulated known results, such TCF21 in fibroblasts, KLF in secretory and AT1/AT2 cells, and TP63 in basal cells. A novel observation was of TCF4 enriched in pulmonary neuro-endocrine cells. Overall, this study showed how multi-omic lung profiling can provide rich references for diseases models, used for integration with and interpretation of data generated from human, in-vitro, or in-vivo models of lung disease.
Integration of Multiomics Data
Whereas substantial knowledge can be derived from applying a single omics at scale, in either human samples or model organisms, additional and refined insight can be obtained via multi-omics integration. Multi-omics data may be integrated via early, intermediate and late integration approaches (Figure 1)[8,9]. Early integration or early concatenation although not complicated, may have problems with vast number of features while the number of available data points is low, known as the "curse of dimensionality"[10]. Multi-omics datasets may contain > 50,000 features when the genome, transcriptome, and proteome are combined but available number of patient samples may be relatively small (hundreds or less). The heterogeneity of omics datasets may be a serious issue as omic data sets can have different distributions (e.g., numerical, categorical, continuous, discrete) and differ significantly in size (number of features). A necessary step in multi-omics analysis frequently is dimensionality reduction, which is the process of reducing the number of variables in order to decrease the dimensionality and noise of a dataset. It is an optional simplification step but some (early and iintermediate integration) often require prior dimensionality reduction to be more effective. The intermediate integration strategy works on transforming each omics dataset independently into a simpler representation, thus overcoming some issues with the early integration strategy. Transformation converts the data set to a less dimensional and less noisy one, which decreases heterogeneity, and facilitates integration and analysis. Late integration involves combination of the results from each omics layer or each omics dataset by machine learning tools (or manually) and the predictions combined at the end[8,11]. Since each omics dataset is analyzed by omic-specific machine learning tools, the problems of noise and heterogeneity found in other strategies are not present. However, the downside of the late integration strategy is that it cannot capture inter-omics interactions and the different machine learning models (for the different omics datasets) do not share knowledge or utilize the complementarity information between omics[11]. Combining predictions is simply not enough to accurately exploit multi-omics data and understand the underlying biological mechanisms of diseases.
The potential applications are endless; we will enumerate a subset of them that have been reported in the literature in lung-related disease models. Based on lessons from The Cancer Genome Atlas, molecular disease endotypes can be inferred for lung diseases. Disease drivers, disease presence or response to treatment biomarkers can be refined using multi-omics. Further, new therapeutic vulnerabilities can be determined and exploited by drug repurposing. Finally, multi-omics can be extended to surrogate sites, such as blood, skin, gut, saliva, or nasal cavity. In addition to access to numerous technologies, researchers have access to a trove of public data, using either reference or disease model datasets, including repositories such as LungMap, ENCODE, NIH Epigenome Roadmap, or NCBI Gene Expression Omnibus (GEO), or the Clinical Proteomic Tumor Analysis Consortium (CPTAC)[12,13,14,15].
Lung multiomics Models
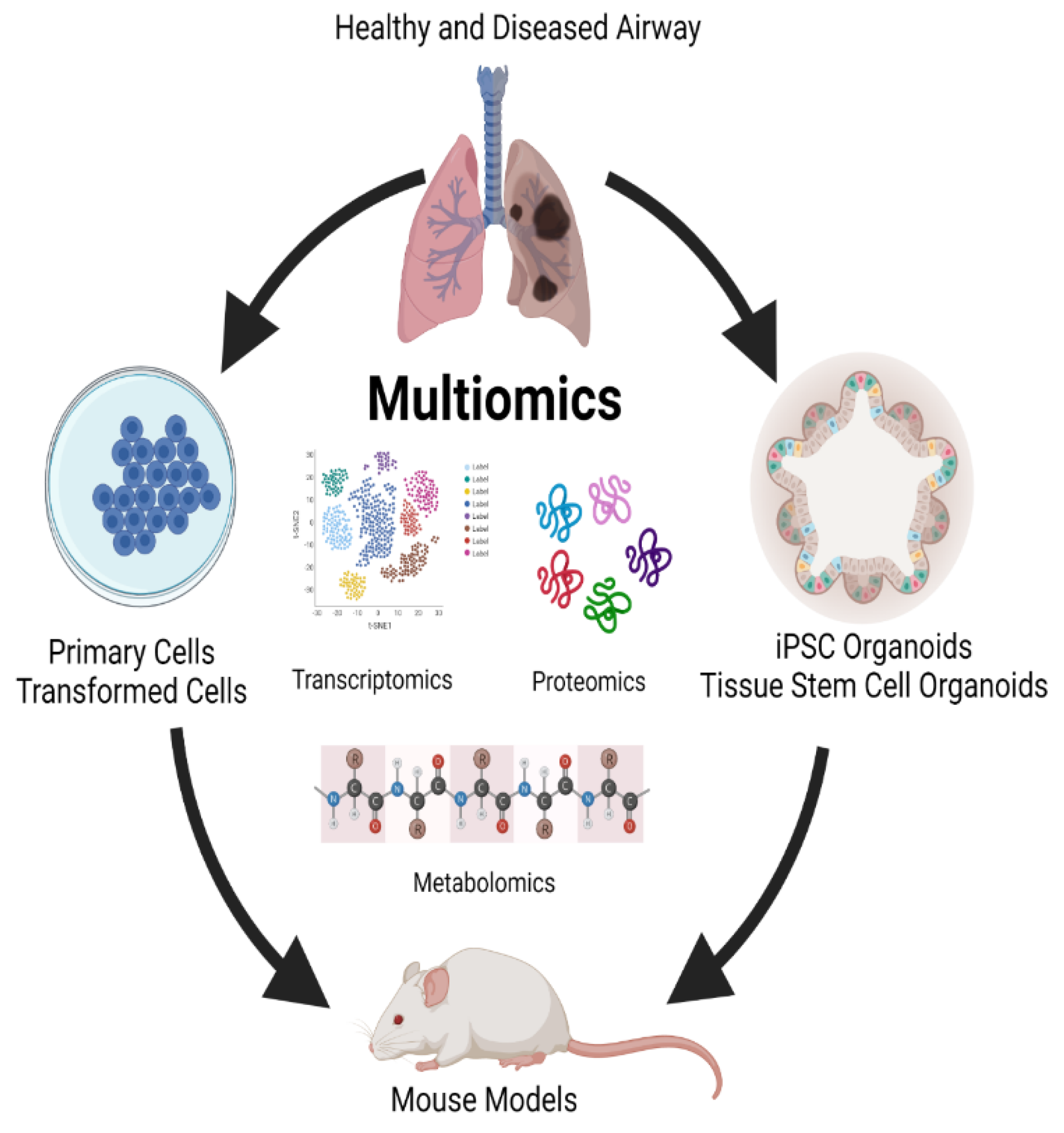
Respiratory disease is a common cause of morbidity and mortality worldwide[16]. With the global ongoing pandemic due to SARS-Covid19[17], respiratory diseases remain a leading cause of death and disability. In almost all respiratory diseases, the epithelium, a monolayer of cells, which comprises the conducting and respiratory airways is damaged which in turn results in functional effects on the proximal airways’ ability to warm, humidify, and cleans inhaled air and on the distal airway to facilitate gas exchange. As a result, health and quality of life are severely impacted by the impaired lung function that occurs in respiratory disease. Human models, such as primary cells and organoids, and animal studies involving integrated multi-omics will allow differences in markers and biologic processes between disease and non-disease models to be elucidated (Figure 2). These differences will provide insight into lung disease including pathways that result in regeneration and repair. Understanding these pathways will be a critical factor in the development of preventive treatments and therapeutic modalities to treat lung diseases and can eventually be harnessed to develop a personalized approach to treating respiratory diseases.
Primary cells and transformed or tumor cells lines have been used for the last half century to understand lung diseases. The cells in these models retain many of the donor tissue characteristics and recapitulate markers and functions that are present in vivo [18,19,20]. These models have the advantage that they are amenable to genetic engineering allowing the dissection of the role of individual molecules and pathways in disease[21]. Additionally, the ease of genetic engineering in these systems has allowed testing function via inducible gene expression[22]. Because of their wide use, many of these cell lines are well characterized, providing a foundation that can only enhance multi-omics studies in which they are used. Studies in cell lines are well suited for high throughput drug screening and evaluation of drug response[23] and are particularly valuable in studying lung cancer[24,25]. However, these models are not without limitations. First and foremost, they fall short at replicating the complex nature of many respiratory diseases. Many lack the multiple cell types and cellular polarity that are present in the proximal and distal respiratory epithelium and exhibit an absence of morphology and structural features that play a significant role in lung biology. Additionally, these cell lines also lack an immune cell component which plays a significant role in the etiology of many lung diseases[26]. Coupled with questions that now have arisen as to the relevance of findings using these models, technical issues including a requirement for tissue donors, a finite lifespan, and limited expansion capacity have contributed to a reduced focus on using multi-omics in these models to study many respiratory diseases.
More recently organoid models have come to the forefront of models in which the use of multi-omics to understand respiratory disorders are being used. Organoids can be derived from either induced pluripotent stem cells or embryonic stem cells (hereafter referred to as iPSC organoids) or established from tissue derived multipotent stem cells (referred to as ASC organoids)[27]. Differentiation of iPSC organoids occurs in a multistep process that involves a definitive endoderm stage, anterior foregut stage, and then into NKX2-1+ lung epithelial progenitors[28,29]. ASC organoids are established following mechanical and enzymatic isolation of either conducting airway or respiratory epithelium stem cells from lavage, small amounts of native tissue, or biopsy specimen[30,31,32,33]. Both iPSC and ASC organoids rely heavily on manipulation of exogenously added growth factors to induce differentiation of the mature polarized airway epithelium and the presence of extracellular matrix such as Matrigel®, synthetic matrices, or decellularized tissue scaffoldings. Both organoid models require cultivation on transwells under air-liquid interface conditions (ALI) where the basal side of the epithelium is in contact with media and the apical side is exposed to air to achieve maximum differentiation potential[32,34]. iPSC and ASC organoids can give rise to alveolar organoid models that recapitulate respiratory epithelium, nasal, trachea, or bronchial organoids that recapitulate conducting airway epithelium, and lung organoids that are mixture[35,36,37]. Like primary and transformed cell models, organoids are amenable to genetic engineering and can be established from donors with genetic disorders that cause lung disease[38]. Organoids are well suited for drug screening and as models for infectious disease research[39,40,41,42] and recapitulate many aspects of other chronic lung diseases such as idiopathic pulmonary fibrosis [43] and cancer[32]. However, iPSC derived airway cells do not seem to achieve the maturation levels observed in human lung[44] and although ASC organoids seem to contain mature epithelial cells, they lack stromal components such as the immune system that play a significant role in most lung diseases. However, the increased cellular complexity and the modeling of human epithelium combined with a forward-thinking multi-omics approach provide an area for advancement in information surrounding respiratory illness and significant translational potential.
There are many animal models of lung diseases including chronic diseases such as cystic fibrosis [45], idiopathic pulmonary fibrosis[46,47], viral and bacterial infections[48], and cancer [49]. Animal models of respiratory diseases offer several advantages including reproducibility, control of environmental factors, unlimited numbers of replicates, genetic phenotyping, and accessibility to lung tissue. Multi-omics approaches can be easily used to provide insight into the relationship between environmental stressors and the effect of the stressor on respiratory disease. Information gained can lead to detailed physiologic and pathologic pathways that contribute to disease pathogenesis. Animal models of lung disease are particularly useful in assessing the predisposition of genetic mutations in causing a specific disease and provide a model in which interactions between components of the whole system can also be examined[50]. Animal models are limited by the fact that in most cases, there are significant differences between human lung tissue and animal lung tissue[12,51]. In addition, many human respiratory diseases are not recapitulated in animal models and clinical manifestations are difficult to assess. However, comparisons between human and animal multi-omics analyses can provide validation for animal models and together multi-omic based approaches combining data collected from both human in vitro and animal in vivo models will provide robustness, rigor, and reproducibility to support drawn conclusions.
Multiomics Insight Into Clinical Disease
a. Cystic fibrosis
Cystic fibrosis is a disease due to a mutation in the chloride transporter gene leading to thickened airway secretions [52,53]. The hallmark of the disease is chronic obstructive airway disease with infection and chronic inflammation[52,54]. However, persistent and heightened inflammation is seen without infection[55,56].
Microbiome studies have pointed to the presence of complex polymicrobial communities in the airways and the lungs of patients with cystic fibrosis [57,58,59,60,61,62,63]. Although microbiome evaluations using 16S ribosomal RNA (rRNA) sequencing have pointed out bacterial communities in the airways (upper and lower and shifts with time[60,64,65,66,67], it does not provide information on the community functionalities or metabolism. Whole genome sequencing of the microbiome enhances our understanding of the metabolic potential of the bacterial communities[59]. The availability of high throughput multi-omics technology will help us understand the microbe-microbe and microbe- host interplay that may determine disease severity and progression [2]. Identification and profiling of the metabolites both host and microbial by metabolomics will provide a view of the metabolic landscape and may identify biomarkers for disease diagnosis and prognosis[68].
Samples that have been evaluated in CF include upper airway secretions, sputum and bronchoalveolar fluid (BAL)[57,59,63] and exhaled breath[69,70]. Metabolites have been identified in BAL associated with inflammation and disease pathogenesis in CF [71,72,73]. O’Connor et al compared CF patients’ BAL with control subjects to draw comparisons by network analysis[63].
A holistic approach integrating multi-omics that include microbiome WGS, metabolome and the proteome and epigenome may provide a deep insight into the pathogenesis of the disease and aid in management and improve clinical outcomes in CF patients[59,74]. Sputum evaluations using multiomics [57,59,61,62,75] and found to have correlations between presence of pathogenic bacteria, metabolites and inflammation[58,75]. Twomey et al identified strong correlations identified between the presence of strict anaerobes in sputum and the abundance of putrescine, pyruvate, and lactate[57].
a. Chronic obstructive pulmonary disease (COPD)
COPD is one of the leading causes of death in the United States and could benefit from a multi-omics approach to decipher complex pathways and networks of potential biomarkers[76]. Yan et al report airway microbe-host interactions in a study of 99 patients with COPD compared to 36 controls from China[77] for 2 endotypes, neutrophilic or eosinophilic inflammation. In neutrophil dominant COPD, altered tryptophan metabolism leads to decreased indole-3-acetic acid (IAA), which affects host interleukin-22 signaling and epithelial cell apoptosis pathways. Yan et al observed that airway microbiome-derived IAA mitigates neutrophilic inflammation, apoptosis, emphysema and lung function decline, via macrophage-epithelial cell cross-talk mediated by interleukin-22. Also, intranasal inoculation of two airway Lactobacilli restored IAA and recapitulated its protective effects in mice [77]. Multi-omic analysis in COPD also can help us understand the pathogenesis of pulmonary hypertension in COPD as it relates to heterogeneity and response to therapy[78], that may lead us to precision medicine- individualizing therapy and prognosis. Wang et al, using a multi-omic meta-analysis approach, used public COPD datasets (1640 16S evaluations and 26 samples from metagenomic sequencing) [79]. The investigators identified microbial shifts and established a global classifier for COPD using 12 microbial genera. Metabolic potentials of the airway microbiome was inferred and linked to host targets. 29.6% of differentially expressed human pathways were predicted to be targeted by microbiome metabolism[79].
b. SARS-CoV-2 infection
The Coronavirus Disease 2019 (COVID-19)is caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and as a pandemic has pervaded our work, productivity and socio-economic development. Xu et al in a multi-omics analysis showed that epithelial cells activated strong innate immune response, including interferon and inflammatory responses[80]. Ubiquitinomics showed SARS-CoV-2 proteins were ubiquitinated during infection despite the fact that SARS-CoV-2 itself didn't code any E3 ligase, and that ubiquitination at three sites on the Spike protein could significantly enhance viral infection. Therefore SARS-CoV-2 not only modulates innate immunity, but also promotes viral infection, by hijacking ubiquitination-specific processes, highlighting potential antiviral and anti-inflammation targets[80]. Unterman et al in a single cell multiomics analysis, in patients with COVID 19 assessed the immunology (T and B cell responses) and their response to tocilizumab, report desynchrony of the innate and adaptive immune interactions in progressive COVID 19[81]. In a review Li et al summarize the multiomics integration-based molecular characterizations of COVID-19, which to date include the integration of transcriptomics, proteomics, genomics, lipidomics, immunomics and metabolomics to explore virus targets and developing suitable therapeutic solutions through systems biology tools[82]. Wu et al suing multi-omic analysis report putative genes for covid severity (COVID-19 HGI using complementary CMO and S-PrediXcan methods), namely XCR1, CCR2, SACM1L, OAS3, NSF, WNT3, NAPSA, and IFNAR2 at 5 genomic loci[83]. Cantwell et al, in a hamster model infected with human SARS-CoV2 report the dynamic changes in gene transcription and protein expression over the course of the infection in a multi-organ kinetic analysis[84]. In a multiomics study involving transcriptomic, epigenomic, and proteomic analyses, report dysfunction in innate immunity in sever and fatal COVID19 infection that included hyperactivation signatures in neutrophils and NK cells[85].
c. Lung cancer
Lung diseases cover a large spectrum; for this review we will focus on non-small cell lung cancer including Lung Adenocarcinoma (LUAD) and Lung Squamous Carcinoma (LUSC), Idiopathic Pulmonary Fibrosis (IPF), Chronic obstructive pulmonary disease (COPD), Broncho-pulmonary dysplasia (BPD), and tuberculosis (TB).
Disease endotypes can be determined using transcriptomics then characterized with several omics. The TCGA LUSC study identified 4 endotypes, classical, primitive, basal, and secretory, with DNA hypermethylation associated with the classical expression endotype[86]. The TCGA LUAD project identified 3 endotypes: the proximal proliferative, with high rates of KRAS mutations, proximal inflammatory, with co-mutations of NF1 and TP53, and terminal respiratory unit, characterized by EGFR mutations and better prognosis[87]. In TB, transcriptomic profiling of the blood identified two major endotypes, further characterized using proteomics as hyper- and hypo-inflammatory, with the hyper-inflammatory exhibiting worse clinical outcomes; drug repurposing analysis indicated that HDAC inhibitors might have opposite effects depending on the patient endotype[88].
microRNA/mRNA networks identify a subset of transcriptomic response that can be attributed to one mechanism of epigenomic dysregulation. microRNA targets can be inferred using several prediction engines, including TargetScan, mirDB, DIANA-TarBase; miRNA/mRNA networks [89,90,91]can be inferred using algorithm such as SigTerms. miRNA/mRNA[89] were identified in LUAD, [92]including hub miRNAs such as miR-539-5p, miR-656-3p, miR-2110, let-7b-5p, and miR-92b-3p; interestingly, other studies found further associations between LUAD, LUSC, miRNAs such as hsa-miR-195, hsa-miR-26b, and hsa-miR-126, and exercise[93]. An analysis of PanSCC cancers identified over-expressed miRNAs miR-205-5p and miR-944 across multiple squamous cancers including LUSC, and further determined an association of their targets with epithelial-mesenchymal transition (EMT)[94]. A study using RNA-Seq transcriptomics and Mass-Spectrometry metabolomics in LUAD derived a 28-gene signature with prognosis capability and also led to the identification of a novel lung cancer drug, AZD-6482, a PI3Kβ inhibitor. [95]
COPD is a risk factor for lung cancer and Sandri et al report multi-omic analysis of the lung stroma in patients with COPD who developed cancer compared to those who did not, testing the hypothesis that lung stroma in COPD has upregulated molecular mechanisms that support carcinogenesis[96]. Predictive variables for cancer compared to adjacent stroma, were mainly represented in the transcriptomic data, whereas predictive variables associated with adjacent tissue, compared to controls. Pathway analysis revealed extracellular matrix and phosphatidylinositol-4,5-bisphosphate 3-kinase-protein kinase B signaling pathways as important signals in the tumor adjacent stroma[96].
Ho et al in combined mass cytometry, immunohistochemistry, and RNA sequencing identified the tumor microenvironment (TME) of lung metastases of pancreatic ductal adenocarcinomas (PDAC)[97]. The investigators report that the lung TME exhibits higher levels of immune infiltration, immune activation, and pro-immune signaling pathways, whereas multiple immune-suppressive pathways are emphasized in the liver TME. Sun 2021 et al report integration of extensive multiomics data sources, utilizing a total of 40 genome-wide functional annotations to prioritize and characterize single nucleotide polymorphisms (SNPs) that increase risk of squamous cell lung cancer through the inflammatory and immune responses[98] including reanalysis of the ILCCO data. Their work highlights SNPs of genes associated with nuclear factor-κB signaling pathway genes and major histocompatibility complex mediated variation in immune responses[98]. In a multi-omics investigation involving whole exome sequencing (WES), RNA sequencing, methylation microarray, and immunohistochemistry (IHC) on 8 pairs of non-small cell lung cancer (NSCLC) primary tumors and matched distant metastases [99] suggests that metastasis is a molecularly late event, and immunosuppression driven by different molecular events, including somatic copy number aberration, may be a common characteristic of tumors with metastatic plasticity [99].
d. Bronchopulmonary dysplasia in preterm infants
Bronchopulmonary dysplasia (BPD) is lung disease in preterm infants defined by oxygen requirements at 36 weeks post menstrual age. Jensen et al reported that the best BPD definition out of 18 prespecified evaluated definitions to predict death or serious respiratory morbidity through 18 to 26 months of corrected age was based on the mode of respiratory support administered at 36 weeks PMA, regardless of whether supplemental oxygen was used [100]. The conclusions were based on evaluation of a prospective study from the National Institute of Child Health and Development (NICHD) network of 2677 preterm infants (GA <32 weeks, 90 percent of the cohort were extremely preterm (EPT) with GA <27 weeks) born between 2011 and 2015. BPD results from multi-factorial etiology including disordered alveolar and microvascular development and inflammation leading to airway injury, inflammation and parenchymal fibrosis. Multiomics may provide a holistic view on the molecular changes in BPD and may provide clues towards prevention or amelioration of the disease. The development of a molecular atlas of the developing lung (LungMAP) has been funded by the National Heart, Lung, and Blood Institute (NHLBI)[12,101]. This proposal will endeavor to create a molecular atlas of the developing lung (LungMAP), which will aid research and public education. Using multi-omics and other resources the research will study lung development including interactive gene networks and dynamic cross talk among multiple cell types to control and coordinate lineage specification, cell proliferation, differentiation, migration, morphogenesis, and injury repair. The information will benefit preterm infants with appropriate interventions to improve clinical outcomes including survival and neurodevelopment.
Lal et al, in a study evaluating the functional metagenome of tracheal aspirates in preterm infants from 16srDNA data and metabolomics, reported differences in preterm infants who develop BPD[102]. The airway metabolome was enriched for metabolites involved in fatty acid activation and androgen and estrogen biosynthesis in BPD infants[102]. The role of exosomal microRNAs (miRNA) in BPD has been investigated and found that BPD susceptible infants had reduced miR-876-3p in their tracheal aspirates[103]. A gain of function of miR-876-3p restores lung architecture in an animal model of BPD. Addition of lipopolysaccharide (LPS) in animal models leads to a decrease in miR-876-3p[103]. These results in combination of the finding of increased abundances of Proteobacteria in tracheal aspirates of BPD outlines the importance of lung dysbiosis and inflammation in the etiopathogenesis of BPD[104,105]. Hyperoxia exposure of newborn mice serves as a model for BPD development in premature born human babies; integration of RNA-Seq transcriptomics with miRNA targets revealed miR-30a as a potential driver for the reported sex disparity between males and females with respect to BPD risk [106,107]. In a mouse model of BPD, a study of lung microbiome and metabolome in 1-14 days old mice in response to exposure to hyperoxia and lipopolysaccharide (LPS) revealed that hyperoxia increased Intestinimonas abundance, whereas LPS decreased Clostridiales, Dorea, and Intestinimonas; further integration with a published lung transcriptomics signature of hyperoxia derived a gene signature with biomarkers potential for risk of BPD development[108]. The importance and relevance of lung T-cell multi-omic interactions with the genome, epigenome and microbiome has been reviewed in the context of BPD in preterm infants[109].
e. Pulmonary hypertension
Chen 2022 et al studied association between gut microbiota composition and host metabolome signatures in a left pulmonary artery ligation (LPAL)-induced PH rat model and report significant gut dysbiosis in LPAL-PH rats, characterized by altered gut microbiota composition, in association with specific changes in gut and lung metabolome profiles[110]. In another multiomic study Konigsberg 2021 et al studied molecular signatures in an idiopathic pulmonary fibrosis model[111] and Titz et al investigated the multiomics of toxic effects of aerosols in mouse models[112]. Hong et al performed co-expression analysis by RNA sequencing 96 disease and 52 control, samples from the lung biobank[113]. The investigators report a co-expression module of 266 genes that were associated with PAH severity such as increased PVR and intimal thickness, but also with compensated PAH such as lower number of hospitalizations, WHO functional class and NT-proBNP. A pharmaco-transcriptomic screen discovered ubiquitin-specific peptidases (USPs) as potential therapeutic targets[113].

Table 1.
Omics Definitions and Descriptions (adapted from Pammi et al[114].
Table 1.
Omics Definitions and Descriptions (adapted from Pammi et al[114].
| “Omic” technology | Description |
|---|---|
| Genome | The basic template of DNA. Technologies can identify genetic (DNA) variants associated with diseases. |
| Microbiome | Allows for accurate quantitative determination of microbial taxa, their abundance and diversity that can be associated with healthy and diseased states. |
| Transcriptome | Examines RNA levels transcribed from DNA template. A small amount of RNA is transcribed for protein synthesis, a much larger amount is encoded for other purposes, which may be implicated in disease. |
| Proteome | Quantifies peptides which may be used as disease biomarkers. |
| Metabolome | Detects and quantifies small molecules which include carbohydrates, amino and fatty acids, and other products of cellular metabolism. Abnormally high or low levels may predict disease. |
| Epigenome | Characterizes modifications of DNA or DNA associated proteins. |
Summary
The availability and accessibility of high-throughput multi-omics technologies including microbiome evaluation of polymicrobial communities in the airway and the lung, transcriptomics, metabolomics, proteomics and genome wide evaluation approaches have increased our understanding in systems biology approach. These technologies are contributing to a better understanding of etiopathogenesis of many respiratory diseases. Ultimately, this knowledge will open avenues for novel preventative and therapeutic strategies to treat airway diseases and contribute to novel and innovative research areas that continue to improve human health and well-being.
References
- Humbert, M.V.; Spalluto, C.M.; Bell, J.; Blume, C.; Conforti, F.; Davies, E.R.; Dean, L.S.N.; Elkington, P.; Haitchi, H.M.; Jackson, C.; et al. Towards an artificial human lung: modelling organ-like complexity to aid mechanistic understanding. Eur Respir J 2022. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Einarsson, G.G.; Gilpin, D.F.; Tunney, M.M. Multi-Omics Approaches: The Key to Improving Respiratory Health in People With Cystic Fibrosis? Front Pharmacol 2020, 11, 569821. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. NIH's 'precision nutrition' bet aims for individualized diets. Science 2021, 371, 552. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Chiou, J.; Poirion, O.B.; Buchanan, J.; Valdez, M.J.; Verheyden, J.M.; Hou, X.; Kudtarkar, P.; Narendra, S.; Newsome, J.M.; et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell 2018, 71, 858–871.e858. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Lim, K.; Sun, D.; Pett, J.P.; Jeng, Q.; Polanski, K.; Dong, Z.; Bolt, L.; Richardson, L.; Mamanova, L.; et al. A human fetal lung cell atlas uncovers proximal-distal gradients of differentiation and key regulators of epithelial fates. Cell 2022, 185, 4841–4860. [Google Scholar] [CrossRef] [PubMed]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine Learning for Integrating Data in Biology and Medicine: Principles, Practice, and Opportunities. Inf Fusion 2019, 50, 71–91. [Google Scholar] [CrossRef]
- Bellman, R. Dynamic programming. Science 1966, 153, 34–37. [Google Scholar] [CrossRef]
- Picard, M.; Scott-Boyer, M.P.; Bodein, A.; Périn, O.; Droit, A. Integration strategies of multi-omics data for machine learning analysis. Comput Struct Biotechnol J 2021, 19, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Ardini-Poleske, M.E.; Clark, R.F.; Ansong, C.; Carson, J.P.; Corley, R.A.; Deutsch, G.H.; Hagood, J.S.; Kaminski, N.; Mariani, T.J.; Potter, S.S.; et al. LungMAP: The Molecular Atlas of Lung Development Program. Am J Physiol Lung Cell Mol Physiol 2017, 313, L733–l740. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res 2020, 48, D882–d889. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J Proteome Res 2015, 14, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med 2020, 8, 585–596. [CrossRef]
- Atzrodt, C.L.; Maknojia, I.; McCarthy, R.D.P.; Oldfield, T.M.; Po, J.; Ta, K.T.L.; Stepp, H.E.; Clements, T.P. A Guide to COVID-19: a global pandemic caused by the novel coronavirus SARS-CoV-2. Febs j 2020, 287, 3633–3650. [Google Scholar] [CrossRef]
- Gruenert, D.C.; Willems, M.; Cassiman, J.J.; Frizzell, R.A. Established cell lines used in cystic fibrosis research. J Cyst Fibros 2004, 3 Suppl 2, 191–196. [Google Scholar] [CrossRef]
- Ren, H.; Birch, N.P.; Suresh, V. An Optimised Human Cell Culture Model for Alveolar Epithelial Transport. PLoS One 2016, 11, e0165225. [Google Scholar] [CrossRef]
- Hermanns, M.I.; Unger, R.E.; Kehe, K.; Peters, K.; Kirkpatrick, C.J. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: development of an alveolo-capillary barrier in vitro. Lab Invest 2004, 84, 736–752. [Google Scholar] [CrossRef]
- Kiełbus, M.; Czapiński, J.; Kałafut, J.; Woś, J.; Stepulak, A.; Rivero-Müller, A. Genetically Engineered Lung Cancer Cells for Analyzing Epithelial-Mesenchymal Transition. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Barisic, M.; Jäättelä, M.; Liu, B. How to Choose the Right Inducible Gene Expression System for Mammalian Studies? Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Ling, A.; Gruener, R.F.; Fessler, J.; Huang, R.S. More than fishing for a cure: The promises and pitfalls of high throughput cancer cell line screens. Pharmacol Ther 2018, 191, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front Bioeng Biotechnol 2020, 8, 322. [Google Scholar] [CrossRef]
- Wong, A.H.; Li, H.; Jia, Y.; Mak, P.I.; Martins, R.; Liu, Y.; Vong, C.M.; Wong, H.C.; Wong, P.K.; Wang, H.; et al. Drug screening of cancer cell lines and human primary tumors using droplet microfluidics. Sci Rep 2017, 7, 9109. [Google Scholar] [CrossRef]
- The lungs at the frontlines of immunity. Nat Immunol 2015, 16, 17. [CrossRef]
- van der Vaart, J.; Clevers, H. Airway organoids as models of human disease. J Intern Med 2021, 289, 604–613. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell 2017, 20, 844–857. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Kotton, D.N. Derivation of Epithelial-Only Airway Organoids from Human Pluripotent Stem Cells. Curr Protoc Stem Cell Biol 2018, 45, e51. [Google Scholar] [CrossRef]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef]
- Usui, S.; Shimizu, T.; Kishioka, C.; Fujita, K.; Sakakura, Y. Secretory cell differentiation and mucus secretion in cultures of human nasal epithelial cells: use of a monoclonal antibody to study human nasal mucin. Ann Otol Rhinol Laryngol 2000, 109, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. Embo j 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.C.; Li, C.; Liu, X.; Yu, Y.; Huang, J.; Wan, Z.; Xiao, D.; Chu, H.; Cai, J.P.; Zhou, B.; et al. A bipotential organoid model of respiratory epithelium recapitulates high infectivity of SARS-CoV-2 Omicron variant. Cell Discov 2022, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Bluhmki, T.; Traub, S.; Müller, A.K.; Bitzer, S.; Schruf, E.; Bammert, M.T.; Leist, M.; Gantner, F.; Garnett, J.P.; Heilker, R. Functional human iPSC-derived alveolar-like cells cultured in a miniaturized 96-Transwell air-liquid interface model. Sci Rep 2021, 11, 17028. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, Y.; Shi, F.; Liu, H. Human lung organoid: Models for respiratory biology and diseases. Dev Biol 2023, 494, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Liu, F.; Liang, X.; Duan, L.; Li, Q.; Pan, G.; Ma, C.; Liu, M.; Li, M.; Wang, P.; et al. iPSC-Derived Airway Epithelial Cells: Progress, Promise, and Challenges. Stem Cells 2023, 41, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cao, Y.; Zhao, P.; Shen, S.; Xi, Y. Organoid: a powerful tool to study lung regeneration and disease. Cell Regen 2021, 10, 21. [Google Scholar] [CrossRef]
- Chen, J.; Na, F. Organoid technology and applications in lung diseases: Models, mechanism research and therapy opportunities. Front Bioeng Biotechnol 2022, 10, 1066869. [Google Scholar] [CrossRef]
- van der Sanden, S.M.G.; Sachs, N.; Koekkoek, S.M.; Koen, G.; Pajkrt, D.; Clevers, H.; Wolthers, K.C. Enterovirus 71 infection of human airway organoids reveals VP1-145 as a viral infectivity determinant. Emerg Microbes Infect 2018, 7, 84. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.; Chu, H.; Poon, V.K.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc Natl Acad Sci U S A 2018, 115, 6822–6827. [Google Scholar] [CrossRef]
- Heo, I.; Dutta, D.; Schaefer, D.A.; Iakobachvili, N.; Artegiani, B.; Sachs, N.; Boonekamp, K.E.; Bowden, G.; Hendrickx, A.P.A.; Willems, R.J.L.; et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat Microbiol 2018, 3, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.P.Y.; Ching, R.H.H.; Chan, S.K.H.; Nicholls, J.M.; Sachs, N.; Clevers, H.; Peiris, J.S.M.; Chan, M.C.W. Tropism, replication competence, and innate immune responses of influenza virus: an analysis of human airway organoids and ex-vivo bronchus cultures. Lancet Respir Med 2018, 6, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.C.; Alva-Ornelas, J.A.; Sucre, J.M.; Vijayaraj, P.; Durra, A.; Richardson, W.; Jonas, S.J.; Paul, M.K.; Karumbayaram, S.; Dunn, B.; et al. Development of a Three-Dimensional Bioengineering Technology to Generate Lung Tissue for Personalized Disease Modeling. Stem Cells Transl Med 2017, 6, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat Protoc 2019, 14, 518–540. [Google Scholar] [CrossRef] [PubMed]
- Wilke, M.; Buijs-Offerman, R.M.; Aarbiou, J.; Colledge, W.H.; Sheppard, D.N.; Touqui, L.; Bot, A.; Jorna, H.; de Jonge, H.R.; Scholte, B.J. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros 2011, 10 Suppl 2, S152–171. [Google Scholar] [CrossRef]
- Walters, D.M.; Kleeberger, S.R. Mouse models of bleomycin-induced pulmonary fibrosis. Curr Protoc Pharmacol 2008. Chapter 5, Unit 5.46,. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front Med (Lausanne) 2017, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, J.; Naninck, T.; Delache, B.; Creppy, J.; Huber, P.; Holzapfel, M.; Bouillier, C.; Contreras, V.; Martinon, F.; Kahlaoui, N.; et al. Non-human primate models of human respiratory infections. Mol Immunol 2021, 135, 147–164. [Google Scholar] [CrossRef]
- Kwon, M.C.; Berns, A. Mouse models for lung cancer. Mol Oncol 2013, 7, 165–177. [Google Scholar] [CrossRef]
- Baron, R.M.; Choi, A.J.; Owen, C.A.; Choi, A.M. Genetically manipulated mouse models of lung disease: potential and pitfalls. Am J Physiol Lung Cell Mol Physiol 2012, 302, L485–497. [Google Scholar] [CrossRef]
- Pan, H.; Deutsch, G.H.; Wert, S.E. Comprehensive anatomic ontologies for lung development: A comparison of alveolar formation and maturation within mouse and human lung. J Biomed Semantics 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Burns, J.L.; Ramsey, B.W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003, 168, 918–951. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med 2014, 190, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Chmiel, J.; Berger, M. Chronic inflammation in the cystic fibrosis lung: alterations in inter- and intracellular signaling. Clin Rev Allergy Immunol 2008, 34, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Keiser, N.W.; Birket, S.E.; Evans, I.A.; Tyler, S.R.; Crooke, A.K.; Sun, X.; Zhou, W.; Nellis, J.R.; Stroebele, E.K.; Chu, K.K.; et al. Defective innate immunity and hyperinflammation in newborn cystic fibrosis transmembrane conductance regulator-knockout ferret lungs. Am J Respir Cell Mol Biol 2015, 52, 683–694. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, J.B.; Mottlowitz, M.M.; Wagner, B.D.; Boyne, K.L.; Stevens, M.J.; Robertson, C.E.; Harris, J.K.; Laguna, T.A. Divergence of bacterial communities in the lower airways of CF patients in early childhood. PLoS One 2021, 16, e0257838. [Google Scholar] [CrossRef] [PubMed]
- Twomey, K.B.; Alston, M.; An, S.Q.; O'Connell, O.J.; McCarthy, Y.; Swarbreck, D.; Febrer, M.; Dow, J.M.; Plant, B.J.; Ryan, R.P. Microbiota and metabolite profiling reveal specific alterations in bacterial community structure and environment in the cystic fibrosis airway during exacerbation. PLoS One 2013, 8, e82432. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Wagner, B.D.; Robertson, C.E.; Stevens, M.J.; Szefler, S.J.; Accurso, F.J.; Sagel, S.D.; Harris, J.K. Assessment of airway microbiota and inflammation in cystic fibrosis using multiple sampling methods. Ann Am Thorac Soc 2015, 12, 221–229. [Google Scholar] [CrossRef]
- Quinn, R.A.; Phelan, V.V.; Whiteson, K.L.; Garg, N.; Bailey, B.A.; Lim, Y.W.; Conrad, D.J.; Dorrestein, P.C.; Rohwer, F.L. Microbial, host and xenobiotic diversity in the cystic fibrosis sputum metabolome. Isme j 2016, 10, 1483–1498. [Google Scholar] [CrossRef]
- Jorth, P.; Ehsan, Z.; Rezayat, A.; Caldwell, E.; Pope, C.; Brewington, J.J.; Goss, C.H.; Benscoter, D.; Clancy, J.P.; Singh, P.K. Direct Lung Sampling Indicates That Established Pathogens Dominate Early Infections in Children with Cystic Fibrosis. Cell Rep 2019, 27, 1190–1204. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Vasco, K.; Vázquez-Baeza, Y.; Jiang, L.; Morton, J.T.; Li, D.; Gonzalez, A.; DeRight Goldasich, L.; Humphrey, G.; Ackermann, G.; et al. High-Resolution Longitudinal Dynamics of the Cystic Fibrosis Sputum Microbiome and Metabolome through Antibiotic Therapy. mSystems 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Whiteson, K.; Davis, T.J.; Phan, J.; Sami, I.; Koumbourlis, A.C.; Freishtat, R.J.; Crandall, K.A.; Bean, H.D. Longitudinal Associations of the Cystic Fibrosis Airway Microbiome and Volatile Metabolites: A Case Study. Front Cell Infect Microbiol 2020, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- O'Connor, J.B.; Mottlowitz, M.; Kruk, M.E.; Mickelson, A.; Wagner, B.D.; Harris, J.K.; Wendt, C.H.; Laguna, T.A. Network Analysis to Identify Multi-Omic Correlations in the Lower Airways of Children With Cystic Fibrosis. Front Cell Infect Microbiol 2022, 12, 805170. [Google Scholar] [CrossRef] [PubMed]
- Hoen, A.G.; Li, J.; Moulton, L.A.; O'Toole, G.A.; Housman, M.L.; Koestler, D.C.; Guill, M.F.; Moore, J.H.; Hibberd, P.L.; Morrison, H.G.; et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J Pediatr 2015, 167, 138–147.e131-133. [Google Scholar] [CrossRef] [PubMed]
- LiPuma, J.J. Assessing Airway Microbiota in Cystic Fibrosis: What More Should Be Done? J Clin Microbiol 2015, 53, 2006–2007. [Google Scholar] [CrossRef] [PubMed]
- Tracy, M.; Cogen, J.; Hoffman, L.R. The pediatric microbiome and the lung. Curr Opin Pediatr 2015, 27, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Prevaes, S.M.; de Winter-de Groot, K.M.; Janssens, H.M.; de Steenhuijsen Piters, W.A.; Tramper-Stranders, G.A.; Wyllie, A.L.; Hasrat, R.; Tiddens, H.A.; van Westreenen, M.; van der Ent, C.K.; et al. Development of the Nasopharyngeal Microbiota in Infants with Cystic Fibrosis. Am J Respir Crit Care Med 2016, 193, 504–515. [Google Scholar] [CrossRef]
- Serkova, N.J.; Standiford, T.J.; Stringer, K.A. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am J Respir Crit Care Med 2011, 184, 647–655. [Google Scholar] [CrossRef]
- Montuschi, P.; Paris, D.; Melck, D.; Lucidi, V.; Ciabattoni, G.; Raia, V.; Calabrese, C.; Bush, A.; Barnes, P.J.; Motta, A. NMR spectroscopy metabolomic profiling of exhaled breath condensate in patients with stable and unstable cystic fibrosis. Thorax 2012, 67, 222–228. [Google Scholar] [CrossRef]
- Monge, M.E.; Pérez, J.J.; Dwivedi, P.; Zhou, M.; McCarty, N.A.; Stecenko, A.A.; Fernández, F.M. Ion mobility and liquid chromatography/mass spectrometry strategies for exhaled breath condensate glucose quantitation in cystic fibrosis studies. Rapid Commun Mass Spectrom 2013, 27, 2263–2271. [Google Scholar] [CrossRef]
- Wolak, J.E.; Esther, C.R., Jr.; O'Connell, T.M. Metabolomic analysis of bronchoalveolar lavage fluid from cystic fibrosis patients. Biomarkers 2009, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Esther, C.R., Jr.; Coakley, R.D.; Henderson, A.G.; Zhou, Y.H.; Wright, F.A.; Boucher, R.C. Metabolomic Evaluation of Neutrophilic Airway Inflammation in Cystic Fibrosis. Chest 2015, 148, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Esther, C.R., Jr.; Turkovic, L.; Rosenow, T.; Muhlebach, M.S.; Boucher, R.C.; Ranganathan, S.; Stick, S.M. Metabolomic biomarkers predictive of early structural lung disease in cystic fibrosis. Eur Respir J 2016, 48, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.J.; Zhuang, Y.; Russell, P.H.; Hobbs, B.D.; Parker, M.M.; Castaldi, P.J.; Rudra, P.; Vestal, B.; Hersh, C.P.; Saba, L.M.; et al. Unsupervised discovery of phenotype-specific multi-omics networks. Bioinformatics 2019, 35, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.A.; Adem, S.; Mills, R.H.; Comstock, W.; DeRight Goldasich, L.; Humphrey, G.; Aksenov, A.A.; Melnik, A.V.; da Silva, R.; Ackermann, G.; et al. Neutrophilic proteolysis in the cystic fibrosis lung correlates with a pathogenic microbiome. Microbiome 2019, 7, 23. [Google Scholar] [CrossRef]
- Abdel-Hafiz, M.; Najafi, M.; Helmi, S.; Pratte, K.A.; Zhuang, Y.; Liu, W.; Kechris, K.J.; Bowler, R.P.; Lange, L.; Banaei-Kashani, F. Significant Subgraph Detection in Multi-omics Networks for Disease Pathway Identification. Front Big Data 2022, 5, 894632. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Chen, B.; Yang, Y.; Yi, X.; Wei, M.; Ecklu-Mensah, G.; Buschmann, M.M.; Liu, H.; Gao, J.; Liang, W.; et al. Multi-omics analyses of airway host-microbe interactions in chronic obstructive pulmonary disease identify potential therapeutic interventions. Nat Microbiol 2022, 7, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Sweatt, A.J.; Maron, B.A. Harnessing Big Data to Advance Treatment and Understanding of Pulmonary Hypertension. Circ Res 2022, 130, 1423–1444. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.; Yan, Z.; Liu, H.; Chen, B.; Liang, Z.; Wang, F.; Miller, B.E.; Tal-Singer, R.; Yi, X.; et al. Multi-omic meta-analysis identifies functional signatures of airway microbiome in chronic obstructive pulmonary disease. Isme j 2020, 14, 2748–2765. [Google Scholar] [CrossRef]
- Xu, G.; Wu, Y.; Xiao, T.; Qi, F.; Fan, L.; Zhang, S.; Zhou, J.; He, Y.; Gao, X.; Zeng, H.; et al. Multiomics approach reveals the ubiquitination-specific processes hijacked by SARS-CoV-2. Signal Transduct Target Ther 2022, 7, 312. [Google Scholar] [CrossRef]
- Unterman, A.; Sumida, T.S.; Nouri, N.; Yan, X.; Zhao, A.Y.; Gasque, V.; Schupp, J.C.; Asashima, H.; Liu, Y.; Cosme, C., Jr.; et al. Single-cell multi-omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. Nat Commun 2022, 13, 440. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Gao, J.; Zhang, Z.; Chen, L.; Li, X.; Zhou, M.; Wheelock Å, M. Multiomics integration-based molecular characterizations of COVID-19. Brief Bioinform 2022, 23. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, J.; Liu, D.; Sun, Y.; Wu, C. An integrative multiomics analysis identifies putative causal genes for COVID-19 severity. Genet Med 2021, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Cantwell, A.M.; Singh, H.; Platt, M.; Yu, Y.; Lin, Y.H.; Ikeno, Y.; Hubbard, G.; Xiang, Y.; Gonzalez-Juarbe, N.; Dube, P.H. Kinetic Multi-omic Analysis of Responses to SARS-CoV-2 Infection in a Model of Severe COVID-19. J Virol 2021, 95, e0101021. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Lee, M.J.; Wei, B.; Parks, B.; Pi, R.; Martínez-Colón, G.J.; Ranganath, T.; Zhao, N.Q.; Taylor, S.; Becker, W.; et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J Exp Med 2021, 218. [Google Scholar] [CrossRef]
- Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [CrossRef]
- CGARN. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- DiNardo, A.R.; Gandhi, T.; Heyckendorf, J.; Grimm, S.L.; Rajapakshe, K.; Nishiguchi, T.; Reimann, M.; Kirchner, H.L.; Kahari, J.; Dlamini, Q.; et al. Gene expression signatures identify biologically and clinically distinct tuberculosis endotypes. Eur Respir J 2022, 60. [Google Scholar] [CrossRef]
- Creighton, C.J.; Nagaraja, A.K.; Hanash, S.M.; Matzuk, M.M.; Gunaratne, P.H. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. Rna 2008, 14, 2290–2296. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 2020, 48, D127–d131. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res 2015, 43, D153–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Gao, J.; Wang, Z.; Yu, Q. Identification of a Potentially Functional microRNA-mRNA Regulatory Network in Lung Adenocarcinoma Using a Bioinformatics Analysis. Front Cell Dev Biol 2021, 9, 641840. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, L.; Wang, W. Systematic assessment of microRNAs associated with lung cancer and physical exercise. Front Oncol 2022, 12, 917667. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep 2018, 23, 194–212.e196. [Google Scholar] [CrossRef] [PubMed]
- Thaiparambil, J.; Dong, J.; Grimm, S.L.; Perera, D.; Ambati, C.S.R.; Putluri, V.; Robertson, M.J.; Patel, T.D.; Mistretta, B.; Gunaratne, P.H.; et al. Integrative metabolomics and transcriptomics analysis reveals novel therapeutic vulnerabilities in lung cancer. Cancer Med 2023, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Sandri, B.J.; Kaplan, A.; Hodgson, S.W.; Peterson, M.; Avdulov, S.; Higgins, L.; Markowski, T.; Yang, P.; Limper, A.H.; Griffin, T.J.; et al. Multi-omic molecular profiling of lung cancer in COPD. Eur Respir J 2018, 52. [Google Scholar] [CrossRef]
- Ho, W.J.; Erbe, R.; Danilova, L.; Phyo, Z.; Bigelow, E.; Stein-O'Brien, G.; Thomas, D.L., 2nd; Charmsaz, S.; Gross, N.; Woolman, S.; et al. Multi-omic profiling of lung and liver tumor microenvironments of metastatic pancreatic cancer reveals site-specific immune regulatory pathways. Genome Biol 2021, 22, 154. [Google Scholar] [CrossRef]
- Sun, R.; Xu, M.; Li, X.; Gaynor, S.; Zhou, H.; Li, Z.; Bossé, Y.; Lam, S.; Tsao, M.S.; Tardon, A.; et al. Integration of multiomic annotation data to prioritize and characterize inflammation and immune-related risk variants in squamous cell lung cancer. Genet Epidemiol 2021, 45, 99–114. [Google Scholar] [CrossRef]
- Lee, W.C.; Reuben, A.; Hu, X.; McGranahan, N.; Chen, R.; Jalali, A.; Negrao, M.V.; Hubert, S.M.; Tang, C.; Wu, C.C.; et al. Multiomics profiling of primary lung cancers and distant metastases reveals immunosuppression as a common characteristic of tumor cells with metastatic plasticity. Genome Biol 2020, 21, 271. [Google Scholar] [CrossRef]
- Jensen, E.A.; Dysart, K.; Gantz, M.G.; McDonald, S.; Bamat, N.A.; Keszler, M.; Kirpalani, H.; Laughon, M.M.; Poindexter, B.B.; Duncan, A.F.; et al. The Diagnosis of Bronchopulmonary Dysplasia in Very Preterm Infants. An Evidence-based Approach. Am J Respir Crit Care Med 2019, 200, 751–759. [Google Scholar] [CrossRef]
- Sun, X.; Perl, A.K.; Li, R.; Bell, S.M.; Sajti, E.; Kalinichenko, V.V.; Kalin, T.V.; Misra, R.S.; Deshmukh, H.; Clair, G.; et al. A census of the lung: CellCards from LungMAP. Dev Cell 2022, 57, 112–145.e112. [Google Scholar] [CrossRef] [PubMed]
- Lal, C.V.; Kandasamy, J.; Dolma, K.; Ramani, M.; Kumar, R.; Wilson, L.; Aghai, Z.; Barnes, S.; Blalock, J.E.; Gaggar, A.; et al. Early airway microbial metagenomic and metabolomic signatures are associated with development of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2018, 315, L810–l815. [Google Scholar] [CrossRef] [PubMed]
- Lal, C.V.; Olave, N.; Travers, C.; Rezonzew, G.; Dolma, K.; Simpson, A.; Halloran, B.; Aghai, Z.; Das, P.; Sharma, N.; et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Lal, C.V.; Travers, C.; Aghai, Z.H.; Eipers, P.; Jilling, T.; Halloran, B.; Carlo, W.A.; Keeley, J.; Rezonzew, G.; Kumar, R.; et al. The Airway Microbiome at Birth. Sci Rep 2016, 6, 31023. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Lal, C.V.; Wagner, B.D.; Mourani, P.M.; Lohmann, P.; Luna, R.A.; Sisson, A.; Shivanna, B.; Hollister, E.B.; Abman, S.H.; et al. Airway Microbiome and Development of Bronchopulmonary Dysplasia in Preterm Infants: A Systematic Review. J Pediatr 2019, 204, 126–133.e122. [Google Scholar] [CrossRef]
- Zhang, Y.; Coarfa, C.; Dong, X.; Jiang, W.; Hayward-Piatkovskyi, B.; Gleghorn, J.P.; Lingappan, K. MicroRNA-30a as a candidate underlying sex-specific differences in neonatal hyperoxic lung injury: implications for BPD. Am J Physiol Lung Cell Mol Physiol 2019, 316, L144–l156. [Google Scholar] [CrossRef]
- Coarfa, C.; Zhang, Y.; Maity, S.; Perera, D.N.; Jiang, W.; Wang, L.; Couroucli, X.; Moorthy, B.; Lingappan, K. Sexual dimorphism of the pulmonary transcriptome in neonatal hyperoxic lung injury: identification of angiogenesis as a key pathway. Am J Physiol Lung Cell Mol Physiol 2017, 313, L991–l1005. [Google Scholar] [CrossRef]
- El Saie, A.; Fu, C.; Grimm, S.L.; Robertson, M.J.; Hoffman, K.; Putluri, V.; Ambati, C.S.R.; Putluri, N.; Shivanna, B.; Coarfa, C.; et al. Metabolome and microbiome multi-omics integration from a murine lung inflammation model of bronchopulmonary dysplasia. Pediatr Res 2022. [Google Scholar] [CrossRef]
- Toldi, G.; Hummler, H.; Pillay, T. T Lymphocytes, Multi-Omic Interactions and Bronchopulmonary Dysplasia. Front Pediatr 2021, 9, 694034. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, D.; Miao, J.; Zhang, C.; Li, X.; Feng, H.; Xing, Y.; Zhang, Z.; Bao, C.; Lin, Z.; et al. Microbiome and metabolome dysbiosis of the gut-lung axis in pulmonary hypertension. Microbiol Res 2022, 265, 127205. [Google Scholar] [CrossRef]
- Konigsberg, I.R.; Borie, R.; Walts, A.D.; Cardwell, J.; Rojas, M.; Metzger, F.; Hauck, S.M.; Fingerlin, T.E.; Yang, I.V.; Schwartz, D.A. Molecular Signatures of Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol 2021, 65, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Titz, B.; Szostak, J.; Sewer, A.; Phillips, B.; Nury, C.; Schneider, T.; Dijon, S.; Lavrynenko, O.; Elamin, A.; Guedj, E.; et al. Multi-omics systems toxicology study of mouse lung assessing the effects of aerosols from two heat-not-burn tobacco products and cigarette smoke. Comput Struct Biotechnol J 2020, 18, 1056–1073. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wong, B.; Rhodes, C.J.; Kurt, Z.; Schwantes-An, T.H.; Mickler, E.A.; Gräf, S.; Eyries, M.; Lutz, K.A.; Pauciulo, M.W.; et al. Integrative Multiomics to Dissect the Lung Transcriptional Landscape of Pulmonary Arterial Hypertension. bioRxiv 2023. [Google Scholar] [CrossRef]
- Pammi, M.; Aghaeepour, N.; Neu, J. Multiomics, artificial intelligence, and precision medicine in perinatology. Pediatr Res 2023, 93, 308–315. [Google Scholar] [CrossRef]
Figure 1.
-Multi-omics integration.
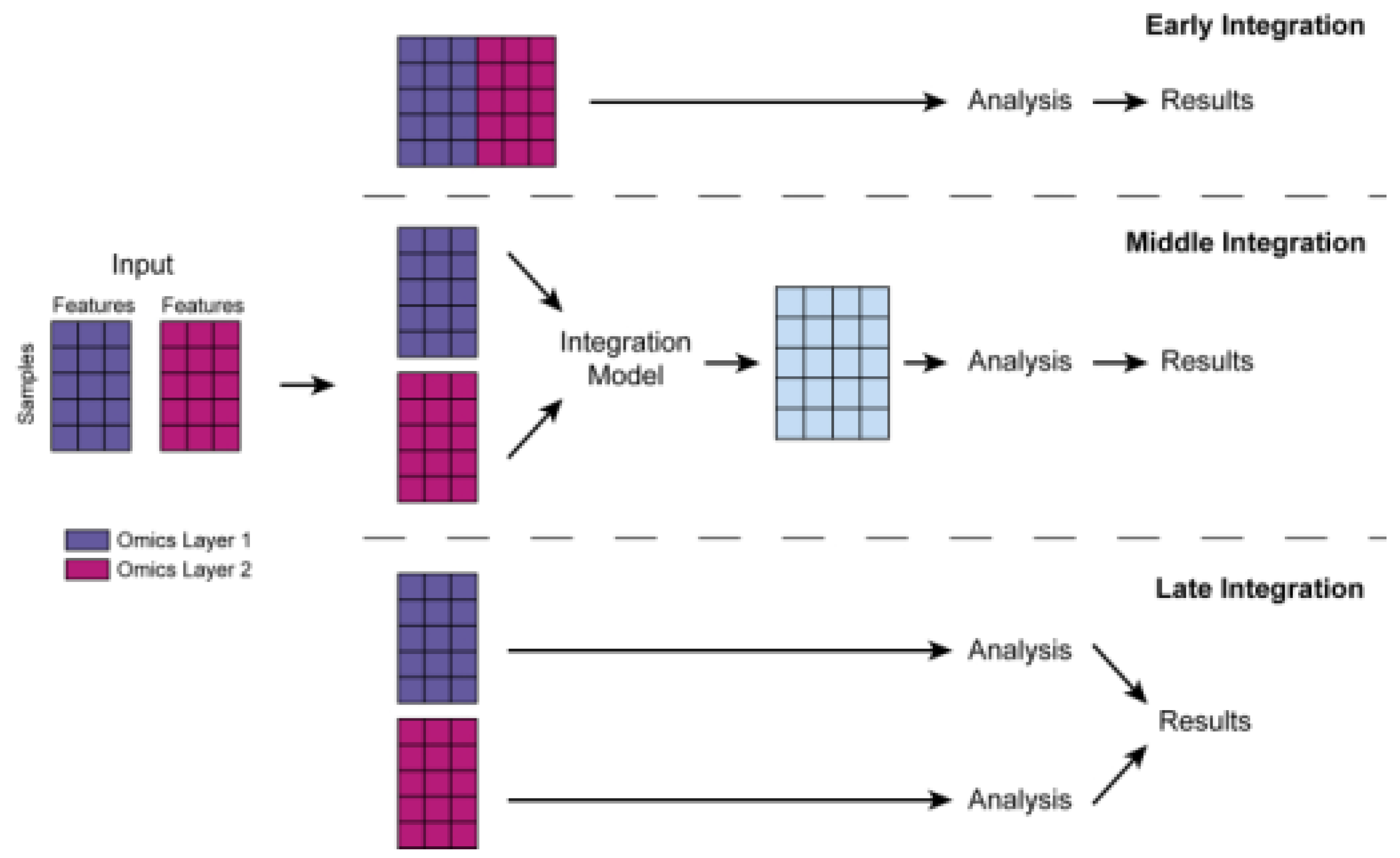
Figure 2.
Modelling multi-omics studies in the study of human lung disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



