
Submitted:
29 April 2023
Posted:
30 April 2023
You are already at the latest version
Abstract
Fibrosis is an important health problem and its pathogenetic activation is still largely unknown. It can develop either spontaneously or, more frequently, as a consequence of various underlying diseases, such as chronic inflammatory autoimmune diseases. Fibrotic tissue is always characterized by mononuclear immune cells infiltration. The cytokine profile of these cells shows clear proinflammatory and profibrotic characteristics. Furthermore, the production of inflammatory mediators by non-immune cells, in response to several stimuli, can be involved in the fibrotic process. It is now established that defects in the abilities of non-immune cells to mediate immune regulation may be involved in the pathogenicity of a series of inflammatory diseases. The convergence of several, not yet well identified, factors results in the aberrant activation of non-immune cells, such as epithelial cells, endothelial cells and fibroblasts, that producing pro-inflammatory molecules, exacerbate the inflammatory condition leading to the excessive and chaotic secretion of extracellular matrix proteins. However, the precise cellular mechanisms involved in this process have not yet been fully elucidated. In this review we explore the latest discoveries on the mechanisms that initiate and perpetuate the vicious circle of abnormal communications between immune and non-immune cells, responsible for fibrotic evolution of inflammatory autoimmune diseases.
Keywords:
autoimmunity
; inflammation
; fibrosis
1. Introduction
Fibrotic autoimmune disorders are a group of chronic pathologies characterized by a damage in self-tolerance to a broad variety of autoantigens in which fibrosis develops as the end-result of a chronic inflammatory process [1]. The pathogenesis of autoimmunity involves dysfunction of the entire immune system, including neutrophils among the innate immune cells, B and T cells of the adaptive immunity, dendritic cells, and macrophages [2]. Within the various cell types related to fibrotic autoimmune diseases’ pathogenesis, also non-immune cells, as epithelial cells, endothelial cells and fibroblasts, are considered to be key players in the occurrence and progression of these diseases [3]. Based on these premises, immune and non-immune inflammatory cells are considered to be accountable for tissue failure in a wide range of fibrotic autoimmune disorders as like rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), primary sjögren's syndrome (pSS) and systemic sclerosis (SSc) [4]. Indeed, a plethora of recent advances has documented the functional role of inflammatory cells as therapeutic targets in autoimmune disorders [5]. However, major questions and controversies in the field remain and the comprehension of the different mechanisms that trigger fibrosis in autoimmune diseases is a challenge for many researchers. This review collects the latest advances in understanding how an alteration in the delicate balance between immune and non-immune cells is at the basis of the fibrotic evolution that is observed in various autoimmune diseases.
2. The role of immune and non-immune inflammatory cells in fibrotic autoimmune diseases: new discoveries
Inflammatory process is considered to be one of the main steps leading to fibrosis in autoimmune diseases [6]. Numerous studies have demonstrated that the pathophysiology of fibrosis in autoimmune diseases involves an aberrant interplay between the immune and non-immune systems [7]. Both immune and non-immune responses play an essential role in the early events of fibrosis. Dysregulation of these processes comprises inflammatory changes, including proliferation of ECM-producing cells and the occurrence of mononuclear cells inflammatory infiltrates. In this context, both immune and non-immune cells have been implicated as important active participants in inflammatory processes involving in the fibrotic autoimmune diseases [8]. This section will review new insights on the role of immune and non-immune inflammatory cell types in fibrotic autoimmune diseases.
2.1. Current understanding of the involvement of immune cells in fibrotic autoimmune diseases
Both innate and adaptive immunity are involved in fibrogenesis of autoimmune diseases and, interestingly, altered orchestration of immune system might be an early event of fibrosis [7]. Dysregulation of these processes results in autoimmune responses triggered by T lymphocytes, macrophages or dendritic cells [2]. These activated immune cells highly express factors that modulate inflammatory process and rapidly promote progressive fibrosis, involving the activation of resident fibroblasts and their transformation in myofibroblasts [2,7,8]. The following paragraphs report the recent discoveries on the role of immune cells in the fibrotic evolution of autoimmune pathologies.
2.1.1. Update on the correlated pro-fibrotic role of CD4+ and CD8+ T cells
Traditionally, B lymphocytes and CD4+ T lymphocytes are considered to be key cells in the immunopathogenesis of autoimmune diseases and they have already been widely studied and are well recognized [9]. However, more recently, studies have demonstrated the increasing evidence that CD8+ T cells, infiltrating inflamed tissues, cooperate to induce tissue fibrosis in autoimmune diseases [10]. Emerging studies reported that CD8+ T cells infiltrate the lesioned skin of patients with SSc, predominantly in the early stage of the disease and exert a pro-inflammatory and pro-fibrotic activity through the induction of tissue damage [11,12]. Of particular note, key pro-fibrotic mediators, as interleukin (IL)-6, through their signal, activate CD8+ T cells and promote their interactions with fibroblasts leading to the deposition of extracellular matrix (ECM), contributing to perpetuate the fibrotic process in SSc patients [13,14]. High levels of the profibrotic type 2 cytokine IL-13 were produced following activation of peripheral blood effector CD8+ T cells from patients with SSc as compared with healthy controls or with patients with RA. In contrast, CD4+ T cells showed a lower and more variable level of IL-13 production. This abnormality was correlated with the extent of fibrosis and with a high grade of cutaneous involvement [Fuschiotti 2018]. The role of CD4+ resulted controversial because, recently, Sakkas and collaborators demonstrated that, in SSc, a great number of T cells of TH2 type is detected, producing pro-fibrotic IL-4, IL-13, and IL-31; in addition, CD4+ cytotoxic T lymphocytes are increased in skin lesions, and cause fibrosis and endothelial cell apoptosis [15].
A key role for CD8+T cells was also demonstrated in SLE nephritis; Zhang and colleagues showed that tubule-interstitial CD8+T cells correlate with clinic-histologic kidney impairment in SLE nephritis, determining an evident progression of interstitial fibrosis and thus tubular organ atrophy [16]. Also, the expression of cytotoxic T cells is increased and the inactivation of CD4+ T cells induces fibrosis and injury of the liver tissue in patients affected by autoimmune hepatis [17]. Autoimmune hepatitis is a progressive inflammatory liver disease characterized by chronic inflammation of the liver, circulating autoantibodies, hypergammaglobulinemia, and progressive liver fibrosis [18]. CD8+ T lymphocytes may have a significant influence on liver fibrosis and intravascular effects. After activation, CD8+ T cells usually differentiate into cytotoxic T lymphocytes, which represent effector cells that destroy tumour cells and infected cells. Actually, the function of CD8+ T lymphocytes in hepatic fibrosis needs further investigation, because their role is unclear. In the liver, the activity of immune surveillance of the CD8+ T cells against virus-infected cells seems to be reduced in mice with liver fibrosis caused by HBV infection [19]. Additionally, in an experimental mice model of carbon tetrachloride-induced liver fibrosis, the transfer of splenic CD8+ T cells into the mice had the effect to exacerbate fibrosis, a process that can be prevented by IL-10 treatment [20]. On the contrary, a reduction of the number of CD8+ T cells had little effect on the progression of hepatic fibrosis in carbon tetrachloride-treated animals [21]. Given that spleen-derived CD8+ T cells induce liver fibrosis and that hepatic CD8+ T-cell depletion has no effect on liver fibrosis, probably, various subtypes of CD8+ T-cell may be distributed differently in the spleen and liver of mice, playing distinct roles in liver fibrosis [22], (Figure 1).
CD8+ T lymphocytes are crucial players also in the mechanism of exocrine gland injury in pSS [23,24]. In fact, CD8+ T lymphocytes contribute to acinar injury in the salivary glands triggering worsening fibrotic event in pSS [12,23]. Joachims et al. [25] showed that expanded clones of memory CD4 + T cells in the salivary glands displayed sequence similarity both within expanded clones of the same individual and among different patients, indicating that these cells are able to recognize shared antigens. They also observed that an increased frequency of expanded clones in salivary glands was correlated with decreased salivary secretion and increased fibrosis. Although CD4 + cells are the majority of T cells within the glandular infiltrates of pSS patients, CD8 + T cells are also present. A percentage of these CD8+ T cells show an activated phenotype, as shown by a higher expression levels of Human Leukocyte Antigen–DR isotype (HLA-DR, an MHC class II cell surface receptor). Increased proportions of HLA-DR+ T cells were associated with higher disease severity [26]. Also, in the blood of pSS patients with anti-SSA positivity, the increased frequencies of HLA-DR-expressing activated CD4 + and CD8+ T cells in blood resulted correlated with the EULAR Sjögren's syndrome (SS) disease activity index (ESSDAI) scores [26]. Furthermore, the proportion of activated CD8+ T cells in blood was established by a multi-omic study based on whole blood transcriptomes, serum proteomes and peripheral immunophenotyping which identified pSS disease signatures dysregulated in widespread epigenomes, mRNAs and proteins. [27]. For example, the expression of the chemokine receptor CXCR3 by activated CD8 + T cells in pSS patients may be important for their migration to the inflamed salivary glands and, as demonstrated in mice, the recruitment of activated CD8+ T cells to salivary gland tissue was dependent on CXCR3 [28]. We speculate that chronic antigen stimulation leading to systemic inflammation, reflected as higher ESSDAI scores, results in the activation of CD8 + T cells in secondary lymphoid organs, such as spleen, CXCR3 upregulation and consequent migration to the salivary glands [29]. Whether CD8 + T cells, in turn, contribute to glandular dysfunction and fibrotic evolution or systemic disease activity is unknown (Figure 2).
2.1.2. Autoimmune Treg pro-fibrotic role
Recently, an intriguing role identified for a functional T cell subset named regulatory T lymphocytes (Treg) in tissue fibrosis has also begun to emerge [30]. Treg are crucial keepers of the immune system shaping the development of fibrosis and causing lethal organ dysfunction [31]. Although, some investigations have highlighted a controversial role for the Treg cells, depending on the disease model, in recent years, the majority of reports demonstrated an increase in the number of Treg in patients at the early phase of SSc [32]. Treg seem to be able to secrete transforming growth factor- β (TGF-β), the major pro-fibrotic factor, which induces myofibroblast activation and fibrosis [33]. In addition, the dysfunction of Treg cells in the early phase of SSc leads to autoimmunity and inflammation [34]. Notably, Treg cells have the capacity to differentiate in T-helper17 (Th17) cells under inflammatory conditions. Th17 cells secrete IL-17A, which could also promote myofibroblast transformation and fibrosis and was related to vasculopathy by promoting endothelial inflammation. A transcriptomic comparison between the early and late phases of SSc revealed a differentiated gene expression exclusively in Treg cells. Using an RNA-seq analysis to compare early SSc vs. late SSc patients, it was also reported that, in the early phase of SSc, enhancement of the oxidative phosphorylation pathway was observed that represents a metabolic sign of differentiation of Treg to Th17 cells [34]. Therefore, an imbalance between Treg and Th17 cells seems to be implicated in the pathogenesis of the early SSc. The contribution of Treg cells to the pathophysiology of SSc has been explained by several mechanisms, sometimes conflicting. In a normal function, Treg cells release inhibitory cytokines, such as IL-10, TGF-β, and IL-35 which function as immunosuppressive factors [35]. First, in SSc the suppressive effect of Treg cells is limited, causing an altered immune response and leading to chronic inflammation, and fibrosis. The decreased inhibitory ability of Treg cells in SSc patients is attributed to the decreased production of TGF-β and IL-10 [36]. On the other hand, it is now established the promotion of fibrosis by pro-fibrotic cytokines produced by Treg cells. For example, TGF-β contributes to fibrotic pathology through the proliferation of fibroblasts, promoting collagen production and ECM secretion, and also induces the epithelial-mesenchymal transition (EMT). In addition, Treg cells seems to be able to differentiate into Th2-like cells in SSc, and to promote fibrosis through the production of IL-4 and IL-13 [36]. By these mechanisms, Treg cells are thought to be associated with several aspects of immune dysregulation and fibrosis during SSc pathogenesis.
Recent findings, based on the study of the dysfunction and imbalance of Treg cells in pSS, have demonstrated a significantly lower frequency of Treg positive for pSTAT5 in pSS patients after IL-2 stimulation, compared with healthy controls [37]. No differences were demonstrated in other T-cell populations, indicating a specific impact of Tregs in pSS pathogenesis which, of course, will need to be clarified [37] (Figure 2). A decreased number of Treg cells was also demonstrated, specifically, in the patients with SLE, psoriatic arthritis, juvenile idiopathic arthritis and autoimmune liver disease [31,38]. In the liver, a dual role of Tregs in fibrogenesis was detected, because they are responsible for fibrosis promotion or immunosuppression [39]. In fact, a large number of Tregs are revealed in the fibrotic microenvironment in patients with hepatocellular carcinoma in which it was observed that a reduction in Tregs promoted the regression of fibrosis [40]. Conversely, in autoimmune hepatitis, hepatic stellate cells (HSCs) were activated, whose function is to produce and accumulate ECM, a pivotal event in liver fibrosis. Simultaneously, HSCs selectively promote the survival and the activity of Tregs in an IL-2–dependent manner. Tregs can both protect HSCs from NK cell attack and, on the contrary, exert an inhibitory effect on HSCs, confirming the dual role of Tregs in liver fibrogenesis and the importance of equilibrium; the balance between Tregs which could convert to Th17 cells, seems, once again, fundamental in maintaining homoeostasis and immunoregulation; this mechanism, for reasons that are still unclear, can deregulate and leads to the production of pro-inflammatory cytokine by Th17, such as IL-17 and IL-22 [39]. Based on these data, the precise mechanism underlying the immune reaction in fibrogenesis mediated by Tregs, probably depending on different immune microenvironments and molecular pathways, is still unclear and will require further investigation.
2.1.3. Macrophages, dendritic cells, mast cells
In the complexity of the immune scenario, macrophages, key cells that classically initiate and sustain chronic inflammation in a simultaneous or parallel manner, are now recognized as capable of secreting fibrotic factors once activated [41]. Monocytes/macrophages activation, due to the plasticity of these cells, could be an important step for the transition from the inflammatory to the fibrotic phase in SSc pathology. Through the release of fibro-proliferative factors macrophages trigger the fibrotic process determining, for example, skin and lung SSc-related tissue fibrosis [41,42]. Consequently, an autocrine loop begins in which the release of fibrotic factors by macrophages drives the transformation of more monocyte/macrophages into cells with pro-fibrotic phenotype [41,42,43]. This cellular cross-talk occurs, clearly, in autoimmune hepatitis; hepatic resident macrophages, have been shown to exert an intricate role in the initiation of inflammatory responses causing liver injury, and can acquire a pro-fibrogenic phenotype that leads to aberrant tissue remodelling culminating in liver and fibrosis and failure [44]. Interestingly, in line with this concept, studies have highlighted that also dendritic cells display high plasticity after injury, driving pro-fibrotic inflammatory mechanisms in autoimmune diseases. Functional alterations of dendritic cells assist the immune processes favoring the altered T cell polarization and pro-fibrotic inflammation in the SSc [45]. Mast cells are immune cells mainly found in connective tissues with a well-established role in allergy and anaphylaxis. However, a great deal of evidence underlines their active role in tissue healing, angiogenesis, and exacerbation of chronic inflammation that characterizes autoimmune diseases. [46]. Leehan and collaborators have, recently, investigated the role of mast cells in salivary gland fibrosis which is a pathological feature of pSS, positively correlates with high focus scores, but not with the age of the patients [47]. They demonstrated that mast cells are strongly associated with fibrosis and fatty infiltration of salivary glands that represents a biological response to gland injury. It is hypothesized that they promote fibrosis by interacting with local fibroblasts and producing enzymes responsible for cleavage and activation of metalloproteinases, which are important mediators of tissue injury and repair [Leehan]. A schematic comprehensive overview of the involvement of immune cells in autoimmune-related fibrosis is reported in Figure 3, (Figure 3).
2.2. Non-immune cells in fibrotic autoimmune diseases
Recently, it has been proposed that also the non-immune cells, such as epithelial cells, endothelial cells and fibroblasts have been implicated in contributing to inflammation, autoimmunity, as well as fibrosis. Non-immune cells, when damaged or activated, release molecules involved in the regulation of several types of immune responses. Furthermore, the de novo production of bioactive factors by non-immune cells, in response to several stimuli, can influence immunological processes. Therefore, defects in the abilities of non-immune cells to mediate immune regulation may be involved in the pathogenicity of a series of inflammatory autoimmune diseases which, often, show a fibrotic organ evolution. This section will review the main non-immune cell types involved in fibrotic autoimmune diseases.
2.2.1. Epithelial cells
The epithelium includes various highly specialized cells that play critical roles in almost all biological processes and they are considered essential to maintain tissue homeostasis in many organs. In this context, several studies have begun to examine the active role of epithelial cells in several autoimmune disorders characterized by fibrosis. The EMT program, under pathological conditions, can lead to the reduction of normal epithelial cells, destroying tissue architecture, inducing pathogenic activation of fibroblasts and driving organ failure [8]. The knowledge of the molecular mechanisms that occur in EMT program has demonstrated that the epithelial state of the cells initially considered immutable, can undergo important changes in gene expression and post-translational regulation leading to the repression of the epithelial characteristics and to the acquisition of mesenchymal characteristics displaying fibroblast-like morphology and cytoarchitecture [48]. Recently, considerable attention has been paid to chronic inflammatory disorders pSS in which the inflammatory status is often associated with pathological EMT-dependent salivary gland fibrosis [49]. Emerging evidences suggest that epithelial cells are also an important source of myofibroblasts in organ fibrosis [50] and this trans-differentiation is evaluated as a tightly specialized system of the EMT process that may be a central event in the salivary gland fibrosis [49]. The implications of these findings were very important and the recent explosion of knowledge in the biology of cellular differentiation has highlighted, for example, that differentiated cell type, such as a tubular or acinar salivary gland epithelial cell in pSS, with a wide set of glandular characteristics such as secretion and transport, could radically change their transcriptional process, transcribing genes characteristic of mesenchymal cell type [49,50,51,52]. Supporting this opinion, recent evidence highlights that salivary gland epithelial cells derived from healthy biopsies, when exposed to TGF-β1 stimulation, acquired a more fibroblast-like morphology [49,53,54]. Also, in SSc, recent studies have demonstrated anomalous phenotypes of the skin epithelium [55]. Indeed, phenotypically altered epithelial cells possibly explain the selective organ fibrosis in the skin, oesophagus and lung that occur in SSc [55]. In this context, several studies have begun to examine the functional role of tubular epithelial cells in the pathogenesis of lupus nephritis [56]. Renal tubular epithelial cells actively participate in the tubulointerstitial pathology of lupus nephritis through the expression of cytokines, chemokines, and pro-fibrotic factors, and playing a crucial crosstalk with infiltrating cells of the immune system [56,57]. Findings suggest that anti-dsDNA antibodies that bind to the surface of renal tubular epithelial cells, but without cellular uptake and cytoplasmic/nuclear translocation, can promote tubule interstitial fibrosis and subsequently kidney dysfunction [58]. Yung et al. reported that anti-dsDNA antibodies derived from lupus nephritis patients, induce a significant increase in the fibronectin expression in human renal tubular epithelial cells, a process dependent, in part, on the secretion of fibrogenic factors as TGF-β [58]. These data suggest that fibrosis development in lupus nephritis is initiated and amplified via complex signalling pathways involving anti-dsDNA antibodies, fibronectin, and TGF-β in renal tubular epithelial cells [56]. A recent study has identified a key role for IL-23 as pro-fibrotic molecule in RA-associated interstitial lung disease through the induction of EMT-dependent transformation of somatic alveolar type I epithelial cells in fibroblast like cells. The acquisition of a mesenchymal phenotype induced by IL-23 included increased deposition of ECM, the acquisition of invasiveness, and resistance to apoptosis, all events of which may contribute to the formation of fibroblastic foci in fibrotic ILD, especially in the context of autoimmune pathology such as RA [59]. (Figure 4).
2.2.2. EMT: new player regulating the interplay between the immunity and fibrosis
In the last years, epithelial to mesenchymal transition (EMT) has been extensively studied as a possible therapeutic target for fibrosis [53,54,60] and therefore a brief refresher in this area is needed. A better understanding of the crosstalk between chronic inflammation, autoimmunity, fibrosis and EMT may represent an opportunity for the development of a broadly effective anti-fibrotic therapy in autoimmune diseases. Cells of multicellular organisms hire several phenotypes that have different functions, morphologies, and gene expression patterns, and, drastically, can undergo specific changes when subjected to determinants stimuli and microenvironments [61]. The inflammatory cells secrete crucial regulatory proteins, such as pro-fibrotic cytokines, chemokines, and growth factors, which can trigger the EMT process [62]. EMT is a highly dynamic process that often gives rise to a series of intermediate phenotypic states in which the cells progressively acquire mesenchymal markers without a concomitant complete loss of epithelial markers [63]. The expression of both mesenchymal and epithelial markers reflects the plasticity of cells depending on their environment [64]. Importantly, EMT leads to the early development of pathological organ fibrosis through paracrine signalling from the epithelium to potential fibroblasts [65]. The fibrotic process affects a variety of organs and tissues, through the activation of specific molecular pathways [65]. However, two common hallmarks are evidenced: the critical role of the TGF-β and the implication of the inflammatory process, which is essential for initiating the fibrotic degeneration [53,60]. EMT is tightly related to fibrosis development in several organs and the fibrogenesis represents the common response of organs and tissues to virtually all chronic repetitive injuries in multiple autoimmune disorders [53,54,60]. During chronic autoimmune diseases, inflammatory and epithelial cells produce fibrogenic mediators. In this context, TGF-β1 emerged as a crucial factor regulating interactions between epithelial and mesenchymal cells, and fibroblasts proliferation [66]. One of the hallmarks of excessive pathological fibrogenesis is the acquisition by resident fibroblasts of a myofibroblasts contractile phenotype expressing high levels of α-Smooth muscle actin (α-SMA). Additional immune cells are recruited into the fibrotic tissue amplifying the fibrotic response, by the secretion of chemokines, cytokines, and growth factors responsible for the differentiation of other myofibroblasts implicated in ECM deposition [67]. The principal EMT pathway is mediated by Smad and we can indicate it as TGF-β1/SMAD/Snail pathway; it is a particularly interesting system active in the EMT-dependent fibrotic process in a number of diseases [53,68]. Alternatively, or parallel to Smads pathways, TGF-β1 also utilizes a multitude of intracellular non-canonical, non-Smads TGF-β-mediated cascade triggered by the binding of ligands different from TGF-β family members to tyrosine kinase receptors [69,70]. This may suggest that the therapeutic use of TGF-β signalling inhibitors, actually used in cancer, may be hypothetically extended also to the treatment of inflammatory autoimmune disorders, but future investigations are needed to prove this hypothesis.
2.2.3. Endothelial cell
Dysregulation of endothelial cell function is proposed as a crucial start event leading to vascular remodelling linked to fibroproliferative vasculopathy. Impaired angiogenesis may be induced by the massive proliferation of fibroblasts observed in some autoimmune diseases characterized by intense pathological fibrosis. New insights have evidenced that myofibroblasts involved in tissue fibrosis can still derive from endothelial cells through a process known as EndoMT [71,72]. It is a non-malignant phenomenon of cellular trans-differentiation by which endothelial cells undergo a phenotypical change where they lose vascular epithelial factors and acquire mesenchymal cell markers [73]. Among systemic autoimmune diseases, endothelial dysfunction has been extensively studied in SLE. In SLE patients, endothelial dysfunction is the main actor of vascular aging and pre-clinical atherosclerosis that led to vascular fibrosis contributing to the early onset of cardiovascular disease and cardiovascular mortality [74]. Interesting studies have highlighted as the endothelial dysfunction occurs also in patients with pSS. Recent epidemiologic data indicate an increase in cardiovascular risk in patients with pSS and endothelial dysregulation may cause vascular fibrosis, leading to arterial stiffness, which precedes the development of high blood pressure [75]. This study demonstrates that patients with pSS, without clinically evident cardiovascular disease or without concomitant cardiovascular risk factors, have an altered endothelial function and a massive proliferation of fibroblasts which suggest a higher susceptibility to the development of vascular fibrosis [75]. Therefore, induction of pro-inflammatory cytokines, such as TNFα and IL-6, involved in atherosclerotic damage, in combination with IFNγ and IL-17 reduces the number of smooth muscle cells, increases collagen production and, favour fibrosis development, with subsequent formation of fibrous atherosclerotic plaque [76]. In this intriguing scenario, the evidence that circulating biomarkers of inflammation predict future cardiovascular events in patients with pSS further reinforces the strict interplay between chronic inflammation and atherosclerosis. Furthermore, subclinical cardiovascular involvement is directly related to elevated inflammatory injury, postulating that inflammation and disease activity are cardio vascular disease risk factors in patients with pSS [75]. In the case of SSc, recent reports have evidenced that this disease was characterized by a massive accumulation of fibroblasts and myofibroblasts and by an abnormal production of interstitial collagens and extracellular matrix components and the dysregulation of endothelial cell activity was identified as a pivotal event that contribute to vasculopathy in SSc [73,77]. (Figure 5).
2.2.4. Fibroblasts
Traditionally, fibroblasts were considered the main contributory cells to the structural integrity of tissues; only recently, they have been recognized as cells that exhibit a dynamic role in physiological or pathological processes [78,79] and are considered active producers of inflammatory cytokines and chemokines. An emerging concept, derived from experimental research on fibroblasts in inflammatory and fibrotic diseases, is that their differentiation is maintained by extrinsic and intrinsic danger signals and local microenvironment-derived morphogens [80,81]. Moreover, fibroblasts can initiate the early molecular processes leading to inflammatory events [81] and, consequently, can be involved with a prominent role in the pathogenesis of fibrotic autoimmune conditions [81].
An interesting paper by Wang, W. et al. [82] demonstrated an incisive role of fibroblasts in systemic sclerosis. In this study, was discovered an altered expression of A20 gene in fibroblasts, isolated from skin and lung systemic sclerosis biopsies; A20 is a gene strongly linked with disease susceptibility and fibrotic manifestations [82]. According to some reports it was demonstrated that A20 expression in fibroblasts can inhibit the fibrotic process, whereas its negative transcriptional regulator, called DREAM (downstream regulatory element antagonist modulator), promotes fibrotic process [83]. The authors proposed that the upregulation of DREAM in systemic sclerosis fibroblasts underlies suppression of A20, which in turn contributes to unchecked pro-fibrotic signalling in stimulated fibroblasts [82]. Interestingly, targeting the A20-DREAM regulatory network could represent a novel therapeutic approach in systemic sclerosis [83].
New reports have documented that the immunomodulatory role of the fibroblasts derived from salivary glands was discovered in a primary site affected by the pSS [84]. Interestingly, these specific clusters of fibroblasts constitute the formation of tertiary lymphoid structures, which are linked to severe disease and can determine a risk factor for the development of lymphoma in pSS [84]. Recent advances in single-cell profiling techniques have demonstrated the presence of fibroblasts in inflamed salivary glands tissue providing evidence of the existence of inflammation-associated fibroblasts in chronically inflamed tissues [85]. Clusters of fibroblasts were identified as key players in the development of renal fibrosis and in particular in lupus nephritis [86].
New discoveries have highlighted as myofibroblasts are the main actors involved in renal fibrogenesis. Interesting, the differentiation of fibroblasts to myofibroblasts is a key cellular event in many autoimmune fibrotic disorders [87].
Single-cell sequencing has demonstrated that myofibroblasts have different gene expression profiles with dynamic changes in fibrosis of different organs [88]. Myofibroblasts, armed with myosin and smooth muscle actin (α-SMA) to connect physically to their environment, secreting TGFβ, VEGF, CTGF, IL-1, IL-6, and IL-8 [89]
It has been suggested that myofibroblasts localized in renal fibrotic tissue may derive from different precursor resident cells, including fibroblasts and epithelial cells [90]. Moreover, myofibroblasts not only contribute to deposition of ECM, but they can produce radical oxygen species and through their intrinsic contractile properties, can alter renal tissue architecture [90]. Their pathogenic role in renal fibrosis has been discovered in different murine models in which the removal of myofibroblasts can reduce fibrogenesis [79].Moreover, myofibroblasts are considered as one of the principal participants in the final point of EMT. After an acute insult, a temporary activation of the EMT process is considered of fundamental importance in renal repair [86,91]. These studies challenge the preconceived notion that fibroblasts can provide new insights into progression of the inflammatory pathology and the identification of key morphogen signals that regulate fibroblast differentiation in diseases could provide a therapeutic opportunity to block differentiation that is required to drive pathology (Figure 6).
2.3. Conclusion
The fibrotic consequences of various primary autoimmune diseases, characterized by tissue damage resulting from chronic inflammatory conditions, remain a major unsolved diagnostic and therapeutic challenge. From experimental experience it seems that all fibrotic tissues derived from autoimmune patients display signs of a chronic immunologically-mediated inflammation during the earliest periods of fibrosis. In these initial stages of fibrotic evolution, a predominant role is certainly played by immune cells, although some questions remain open about the specificity of lymphocyte subtypes occurring in fibrotic tissue as well as about a possible imbalance of pro- and anti-fibrotic factors produced by components of the immune cells infiltrate. Actually, there is accumulating evidence showing that non-immune cells, such as epithelial cells, endothelial cells and fibroblasts are cells with important immunomodulatory properties, playing a pivotal role in the switch to chronic inflammation. Determining the exact contribution of these mechanisms remains a challenge, as they are at the cross-point of multiple regulatory networks also involving immune and non-immune cells, and this, in an autoimmune condition in which the immune system works in an altered way, makes the scenario even more complex. For example, whether EMT activation may interfere with the crosstalk between epithelial cells, mesenchymal cells and immune cells, stimulating fibrotic evolution, remains elusive. Since valid biomarkers for the diagnosis and staging of autoimmune-related fibrosis are not yet available, more detailed knowledge on the cellular and molecular basis of fibrogenesis is urgently needed. From this point of view, a better knowledge of the non-immune cells contribution to the autoimmune fibrosis should help to appreciate the reasons underlying the actual clinical failures and design more effective therapies.
Author Contributions
all authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version for publication. M.S. and S.L. had full access to the data collected in the review, take responsibility for their integrity and performed a critical reading. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
the authors declare no conflict of interest.
References
- Duan, L.; Rao, X.; Sigdel, K.R. Regulation of Inflammation in Autoimmune Disease. J. Immunol. Res. 2019, 2019, 7403796. [Google Scholar] [CrossRef] [PubMed]
- Frizinsky, S.; Haj-Yahia, S.; Machnes Maayan, D.; Lifshitz, Y.; Maoz-Segal, R.; Offengenden, I.; Kidon, M.; Agmon-Levin, N. The innate immune perspective of autoimmune and autoinflammatory conditions. Rheumatology (Oxford). 2019, 58, vi1–vi8. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wei, X.; Ye, T.; Hang, L.; Mou, L.; Su, J. Interrelation Between Fibroblasts and T Cells in Fibrosing Interstitial Lung Diseases. Front. Immunol. 2021, 12, 747335. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liu, H.; Huang, G.; Dai, S.S. The emerging role of neutrophils in autoimmune-associated disorders: Effector, predictor, and therapeutic targets. Med. Comm. 2021, 2, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Kim, W.U. Targeted Immunotherapy for Autoimmune Disease. Immune Netw. 2022, 22, e9. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.W.; Gyorfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Konigshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New insights into fibrosis from the ECM degradation perspective: The macrophage-MMP-ECM interaction. Cell Biosci. 2022, 12, 117. [Google Scholar]
- Raphael, I.; Joern, R.R.; Forsthuber, T.G. Memory CD4+ T Cells in Immunity and Autoimmune Diseases. Cells. 2020, 9, 531. [Google Scholar] [CrossRef]
- Deng, Q.; Luo, Y.; Chang, C.; Wu, H.; Ding, Y.; Xiao, R. The Emerging Epigenetic Role of CD8+T Cells in Autoimmune Diseases: A Systematic Review. Front. Immunol. 2019, 10, 856. [Google Scholar] [CrossRef]
- Fuschiotti, P. Current perspectives on the role of CD8+ T cells in systemic sclerosis. Immunol. Lett. 2018, 195, 55–60. [Google Scholar] [CrossRef]
- Kaneko, N.; Chen, H.; Perugino, C.A.; Maehara, T.; Munemura, R.; Yokomizo, S.; Sameshima, J.; Diefenbach, T.J.; Premo, K.R.; Chinju, A.; et al. Cytotoxic CD8+ T cells may be drivers of tissue destruction in Sjögren's syndrome. Sci. Rep. 2022, 12, 15427. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y. Cytokines Involved in the Pathogenesis of SSc and Problems in the Devel-opment of Anti-Cytokine Therapy. Cells. 2021, 10, 1104. [Google Scholar] [CrossRef]
- Valenzi, E.; Tabib, T.; Papazoglou, A.; Sembrat, J.; Trejo Bittar, H.E.; Rojas, M.; Lafyatis, R. Disparate Interferon Signaling and Shared Aberrant Basaloid Cells in Single-Cell Profiling of Idiopathic Pulmonary Fibrosis and Systemic Sclerosis-Associated Interstitial Lung Disease. Front. Immunol. 2021, 12, 595811. [Google Scholar] [CrossRef]
- Sakkas, L.I.; Bogdanos, D.P. The Role of T Cells in SSc: An Update. Immuno 2022, 2, 534–547. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, M.; Zhang, J.; Feng, X.; Liu, Z.; Cheng, Z. Association between tubulointerstitial CD8+T cells and renal prognosis in lupus nephritis. Int. Immunopharmacol. 2021, 99, 107877. [Google Scholar] [CrossRef] [PubMed]
- Vuerich, M.; Wang, N.; Kalbasi, A.; Graham, J.J.; Longhi, M.S. Dysfunctional Immune Regulation in Autoimmune Hepatitis: From Pathogenesis to Novel Therapies. Front. Immunol. 2021, 12, 746436. [Google Scholar] [CrossRef] [PubMed]
- Covelli, C.; Sacchi, D.; Sarcognato, S.; Cazzagon, N.; Grillo, F.; Baciorri, F.; Fanni, D.; Cacciatore, M.; Maffeis, V.; Guido, M. Pathology of autoimmune hepatitis. Pathologica 2021, 113, 185–193. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8(+) T. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef]
- Safadi, R.; Ohta, M.; Alvarez, C.E.; Fiel, M.I.; Bansal, M.; Mehal, W.Z.; Friedman, S.L. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology 2004, 127, 870–882. [Google Scholar] [CrossRef]
- Novobrantseva, T.I.; Majeau, G.R.; Amatucci, A.; Kogan, S.; Brenner, I.; Casola, S.; Shlomchik, M.J.; Koteliansky, V.; Hochman, P.S.; Ibraghimov, A. Attenuated liver fibrosis in the absence of B cells. J. Clin. Invest. 2005, 115, 3072–3082. [Google Scholar] [CrossRef]
- Sun, R.; Xiang, Z.; Wu, B. T cells and liver fibrosis. Port Hypertens Cirrhos. 2022, 1, 125–132. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, J.; Tian, J.; Wang, S. CD8+ T Lymphocytes: Crucial Players in Sjögren's Syndrome. Front. Immunol. 2021, 11, 602823. [Google Scholar] [CrossRef]
- Chihaby, N.; Orliaguet, M.; Le Pottier, L.; Pers, J.O.; Boisramé, S. Treatment of Sjögren's Syndrome with Mesenchymal Stem Cells: A Systematic Review. Int J Mol Sci. 2021, 22, 10474. [Google Scholar] [CrossRef]
- Joachims, M.L.; Leehan, K.M.; Lawrence, C.; Pelikan, R.C.; Moore, J.S.; Pan, Z.; Rasmussen, A.; Radfar, L.; Lewis, D.M.; Grundahl, K.M.; et al. Single-cell analysis of glandular T cell receptors in Sjögren's syndrome. JCI Insight 2016, e85609. [Google Scholar] [CrossRef] [PubMed]
- Mingueneau, M.; Boudaoud, S.; Haskett, S.; Reynolds, T.L.; Nocturne, G.; Norton, E.; Zhang, X.; Constant, M.; Park, D.; Wang, W.; et al. Cytometry by time-of-flight immunophenotyping identifies a blood Sjögren's signature correlating with disease activity and glandular inflammation. J Allergy Clin Immunol. 2016, 137, 1809–1821.e12. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, S.; Suzuki, K.; Nishikawa, A.; Kassai, Y.; Takiguchi, M.; Kurisu, R.; Okuzono, Y.; Miyazaki, T.; Takeshita, M.; Yoshimoto, K.; et al. Multiomic disease signatures converge to cytotoxic CD8 T cells in primary Sjögren's syndrome. Ann Rheum Dis. 2017, 76, 1458–1466. [Google Scholar] [CrossRef]
- Caldeira-Dantas, S.; Furmanak, T.; Smith, C.; Quinn, M.; Teos, L.Y.; Ertel, A.; Kurup, D.; Tandon, M.; Alevizos, I.; Snyder, C.M. The Chemokine Receptor CXCR3 Promotes CD8+ T Cell Accumulation in Uninfected Salivary Glands but Is Not Necessary after Murine Cytomegalovirus Infection. J Immunol. 2018, 200, 1133–1145. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Kroese, F.G.M.; Bootsma, H. T cells in primary Sjögren’s syndrome: Targets for early intervention. Rheumatology 2021, 60, 3088–3098. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.; Min, B. Tissue Resident Foxp3+ Regulatory T Cells: Sentinels and Saboteurs in Health and Disease. Front. Immunol. 2022, 13, 865593. [Google Scholar] [CrossRef]
- Rajendeeran, A.; Tenbrock, K. Regulatory T cell function in autoimmune disease. J. Transl Autoimmun. 2021, 4, 100130. [Google Scholar] [CrossRef]
- Miao, M.; Hao, Z.; Guo, Y.; Zhang, X.; Zhang, S.; Luo, J.; Zhao, X.; Zhang, C.; Liu, X.; Wu, X.; et al. Short-term and low-dose IL-2 therapy restores the Th17/Treg balance in the peripheral blood of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2018, 77, 1838–1840. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Auffray, C.; Avouac, J.; Allanore, Y. Regulatory T Cells in Systemic Sclerosis. Front Immunol. 2018, 9, 2356. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Nagafuchi, Y.; Shoda, H.; Fujio, K. The Pathophysiological Roles of Regulatory T Cells in the Early Phase of SSc. Front Immunol. 2022, 13, 900638. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat Rev Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- MacDonald, K.G.; Dawson, N.A.J.; Huang, Q.; Dunne, J.V.; Levings, M.K.; Broady, R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol. 2015, 135, 946–955.e9. [Google Scholar] [CrossRef]
- Keindl, M.; Davies, R.; Bergum, B.; Brun, J.G.; Hammenfors, D.; Jonsson, R.; Lyssenko, V.; Appel, S. Impaired activation of STAT5 upon IL-2 stimulation in Tregs and elevated sIL-2R in Sjögren's syndrome. Arthritis Res. Ther. 2022, 24, 101. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. Recent progress on the Roles of Regulatory T Cells in IgG4-Related Disease. Immuno 2022, 2, 430–442. [Google Scholar] [CrossRef]
- Wu, K.J.; Qian, Q.F.; Zhou, J.R.; Sun, D.L.; Duan, Y.F.; Zhu, X.; Sartorius, K.; Lu, Y.J. Regulatory T cells (Tregs) in liver fibrosis. Cell Death Discov. 2023, 9, 53. [Google Scholar] [CrossRef]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef]
- Al-Adwi, Y.; Westra, J.; van Goor, H.; Burgess, J.K.; Denton, C.P.; Mulder, D.J. Macrophages as determinants and regulators of fibrosis in systemic sclerosis. Rheumatology 2023, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Hoeft, K.; Schaefer, G.J.L.; Kim, H.; Schumacher, D.; Bleckwehl, T.; Long, Q.; Klinkhammer, B.M.; Peisker, F.; Koch, L.; Nagai, J.; et al. Platelet-instructed SPP1+ macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner. Cell Rep. 2023, 42, 112131. [Google Scholar] [CrossRef]
- Cheng, D.; Chai, J.; Wang, H.; Fu, L.; Peng, S.; Ni, X. Hepatic macrophages: Key players in the development and progression of liver fibrosis. Liver Int. 2021, 41, 2279–2294. [Google Scholar] [CrossRef] [PubMed]
- Choreño-Parra, J.A.; Cervantes-Rosete, D.; Jiménez-Alvarez, L.A.; Ramírez-Martínez, G.; Márquez-García, J.E.; Cruz-Lagunas, A.; Magaña-Sanchez, A.Y.; Lima, G.; López-Maldonado, H.; Gaytán-Guzmán, E.; et al. Dendritic cells drive profibrotic inflammation and aberrant T cell polarization in systemic sclerosis. Rheumatology 2022, 489. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Stellin, L.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjogren’s Syndrome: Promising Inhibitory Effect of IL-37. Int. J. Mol. Sci. 2020, 21, 4297. [Google Scholar] [CrossRef] [PubMed]
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjogren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 36, 80–88. [Google Scholar]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Sisto, M.; Ribatti, D.; Lisi, S. Sjögren’s Syndrome-Related Organs Fibrosis: Hypotheses and Realities. J. Clin. Med. 2022, 11, 3551. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nat. Cell Biol. 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFβ-Dependent EMT. Biomolecules. 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Ribatti, D.; Lisi, S. Molecular Mechanisms Linking Inflammation to Autoimmunity in Sjögren's Syndrome: Identification of New Targets. Int. J. Mol. Sci. 2022, 23, 13229. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Takahashi, T.; Saigusa, R. Systemic sclerosis: Is the epithelium a missing piece of the pathogenic puzzle? J. Dermatol. Sci. 2019, 94, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Healy, H.; Kassianos, A.J. The Emerging Role of Renal Tubular Epithelial Cells in the Immunological Pathophysiology of Lupus Nephritis. Front. Immunol. 2020, 11, 578952. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Yung, S.; Ng, C.Y.; Ho, S.K.; Cheung, K.F.; Chan, K.W.; Zhang, Q. Anti-dsDNA antibody induces soluble fibronectin secretion by proximal renal tubular epithelial cells and downstream increase of TGF-beta1 and collagen synthesis. J. Autoimmun. 2015, 58, 111–122. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, S.; Lau, J.; Roden, A.C.; Matteson, E.L.; Sun, J.; Luo, F.; Tschumperlin, D.J.; Vassallo, R. IL-23 amplifies the epithelial-mesenchymal transition of mechanically conditioned alveolar epithelial cells in rheumatoid arthritis-associated interstitial lung disease through mTOR/S6 signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L1006–L1022. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef]
- Wang, W.; Poe, D.; Yang, Y.; Hyatt, T.; Xing, J. Epithelial-to-mesenchymal transition proceeds through directional destabilization of multidimensional attractor. Elife. 2022, 11, e74866. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells. 2021, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dai, F.; Feng, L.; Zou, H.; Feng, L.; Xu, M. Communication Between Epithelial-Mesenchymal Plasticity and Cancer Stem Cells: New Insights Into Cancer Progression. Front. Oncol. 2021, 11, 617597. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S. Epithelial-to-Mesenchymal Transition in Fibrosis: Concepts and Targeting Strategies. Front. Pharmacol. 2021, 12, 737570. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 signaling and tissue fibrosis. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Jimenez, S.A. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol 2013, 2013, 835948. [Google Scholar] [CrossRef] [PubMed]
- Ebmeier, S.; Horsley, V. Origin of fibrosing cells in systemic sclerosis. Curr. Opin. Rheumatol 2015, 27, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Vomero, M.; Navarini, L.; Dolo, V.; Cipriani, P.; Giacomelli, R. Endothelial-to-mesenchymal transition in systemic sclerosis. Clin. Exp. Immunol. 2021, 205, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, L.; Piantoni, S.; Vizzardi, E.; Sciatti, E.; Riccardi, M.; Franceschini, F.; Cavazzana, I. Endothelial Dysfunction in Systemic Lupus Erythematosus and Systemic Sclerosis: A Common Trigger for Different Microvascular Diseases. Front. Med. 2022, 9, 849086. [Google Scholar] [CrossRef] [PubMed]
- Łuczak, A.; Małecki, R.; Kulus, M.; Madej, M.; Szahidewicz-Krupska, E.; Doroszko, A. Cardiovascular Risk and Endothelial Dysfunction in Primary Sjogren Syndrome Is Related to the Disease Activity. Nutrients. 2021, 13, 2072. [Google Scholar] [CrossRef]
- Robert, M.; Miossec, P. Effects of Interleukin 17 on the cardiovascular system. Autoimmun Rev. 2017, 16, 984–991. [Google Scholar] [CrossRef]
- Mostmans, Y.; Cutolo, M.; Giddelo, C.; Decuman, S.; Melsens, K.; Declercq, H.; Vandecasteele, E.; De Keyser, F.; Distler, O.; Gutermuth, J.; et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: A systematic review. Autoimmun. Rev. 2017, 16, 774–786. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and Function of Myofibroblasts in Kidney Fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Wei, K.; Korsunsky, I.; Marshall, J.L.; Gao, A.; Watts, G.F.M.; Major, T.; Croft, A.P.; Watts, J.; Blazar, P.E.; Lange, J.K.; et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature. 2020, 582, 259–264. [Google Scholar] [CrossRef]
- Smith, T.J. Insights into the role of fibroblasts in human autoimmune diseases. Clin. Exp. Immunol. 2005, 141, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bale, S.; Wei, J.; Yalavarthi, B.; Bhattacharyya, D.; Yan, J.J.; Abdala-Valencia, H.; Xu, D.; Sun, H.; Marangoni, R.G.; et al. Fibroblast A20 governs fibrosis susceptibility and its repression by DREAM promotes fibrosis in multiple organs. Nat.Commun. 2022, 13, 6358. [Google Scholar] [CrossRef] [PubMed]
- Onuora, S. Fibroblast A20 and its suppressor DREAM regulate fibrosis in SSc. Nat Rev Rheumatol, 2023, 19, 1. [Google Scholar] [CrossRef]
- Klein, K. Fibroblasts in Sjögren’s Syndrome, Fibroblasts-Advances in Inflammation, Autoimmunity and Cancer. FROM THE EDITED VOLUME Fibroblasts Edited by Mojca Frank Bertoncelj and Katja Lakota, Published: July 16th, 2021.
- Lee, B.; Lee, S.H.; Shin, K. Crosstalk between fibroblasts and T cells in immune networks. Front.Immunol. 2023, 13, 1103823. [Google Scholar] [CrossRef]
- Sciascia, S.; Cozzi, M.; Barinotti, A.; Radin, M.; Cecchi, I.; Fenoglio, R.; Mancardi, D.; Wilson Jones, G.; Rossi, D.; Roccatello, D. RenalFibrosis in Lupus Nephritis. Int. J. Mol. Sci. 2022, 23, 14317. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Woods, E.L.; Dally, J.; Kong, D.; Steadman, R.; Moseley, R.; Midgley, A.C. Myofibroblasts: Function, Formation, and Scope of Molecular Therapies for Skin Fibrosis. Biomolecules. 2021, 11, 1095. [Google Scholar] [CrossRef] [PubMed]
- Tabib, T.; Morse, C.; Wang, T.; Chen, W.; Lafyatis, R. SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. J. Invest. Dermatol. 2018, 138, 802–810. [Google Scholar] [CrossRef] [PubMed]
- van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse Origins of the Myofibroblast—Implications for Kidney Fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT-β-Catenin Signalling—A Versatile Player in Kidney Injury and Repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef]
Figure 1.
schematic representation of the involvement of the immune cells in autoimmune hepatitis.
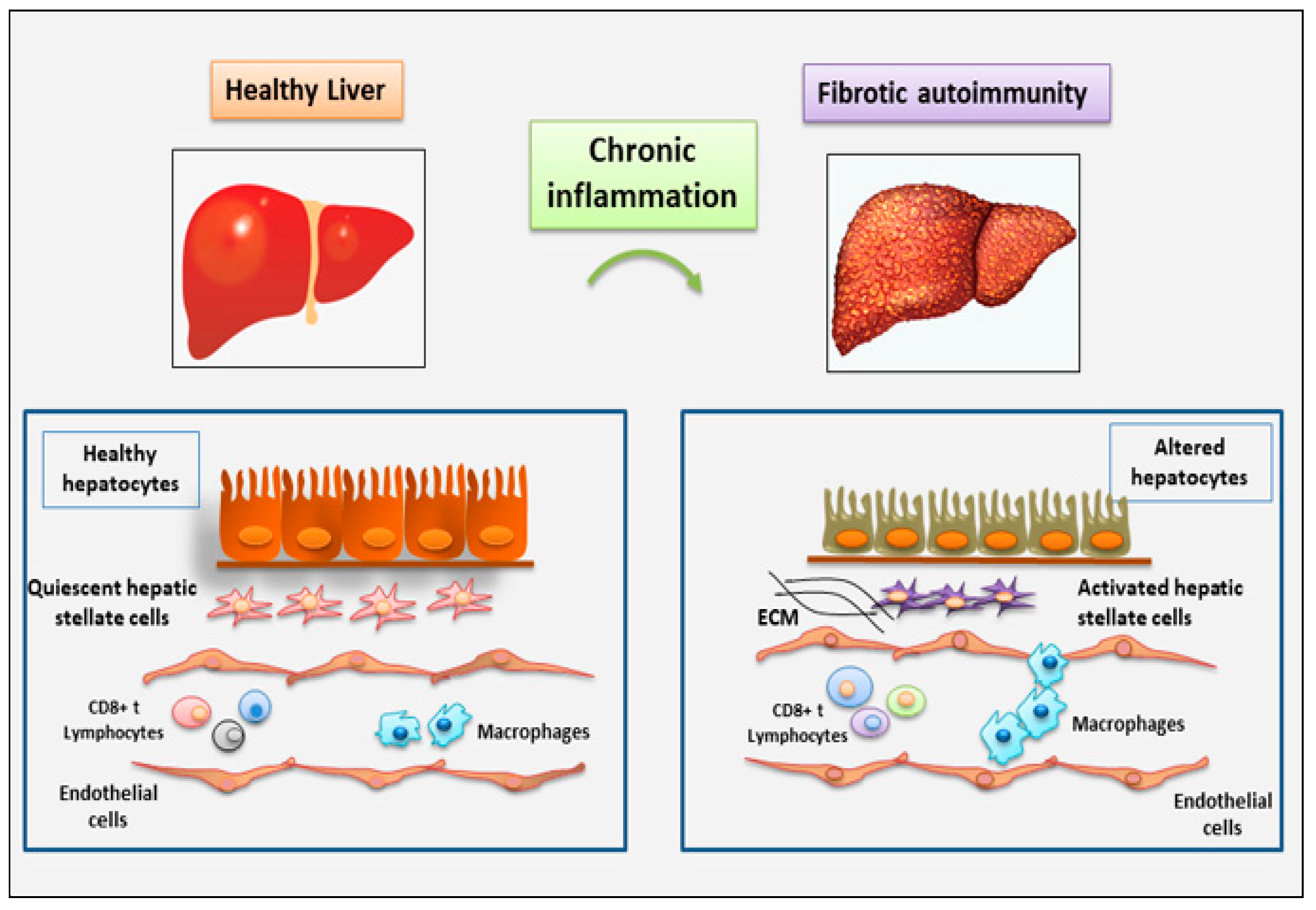
Figure 2.
immune cells involved in the fibrosis of salivary glands observed in the chronic inflammatory autoimmune diseases primary Sjӧgren’s syndrome (pSS), accordingly with the most recent findings. The figure highlights the role of TGF-β as pro-fibrotic factor.
Figure 2.
immune cells involved in the fibrosis of salivary glands observed in the chronic inflammatory autoimmune diseases primary Sjӧgren’s syndrome (pSS), accordingly with the most recent findings. The figure highlights the role of TGF-β as pro-fibrotic factor.
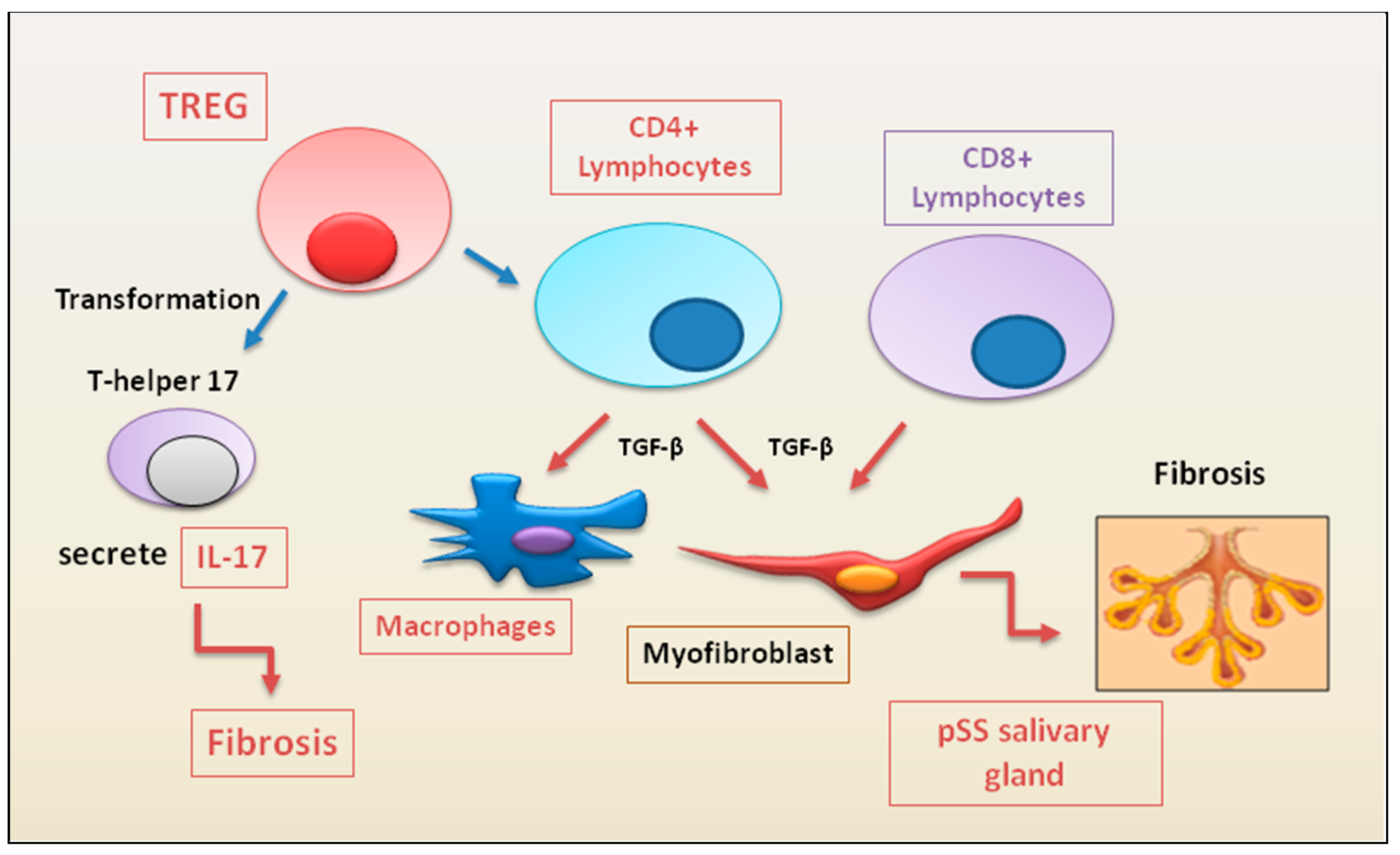
Figure 3.
immune cells recently linked to the fibrotic evolution of autoimmune diseases. In a condition of chronic inflammation, many immune cell populations with diverse functions are activated to produce multiple cytokines that lead to the proliferation and activation of myofibroblasts, directly involved in the development of fibrosis in various autoimmune diseases.
Figure 3.
immune cells recently linked to the fibrotic evolution of autoimmune diseases. In a condition of chronic inflammation, many immune cell populations with diverse functions are activated to produce multiple cytokines that lead to the proliferation and activation of myofibroblasts, directly involved in the development of fibrosis in various autoimmune diseases.
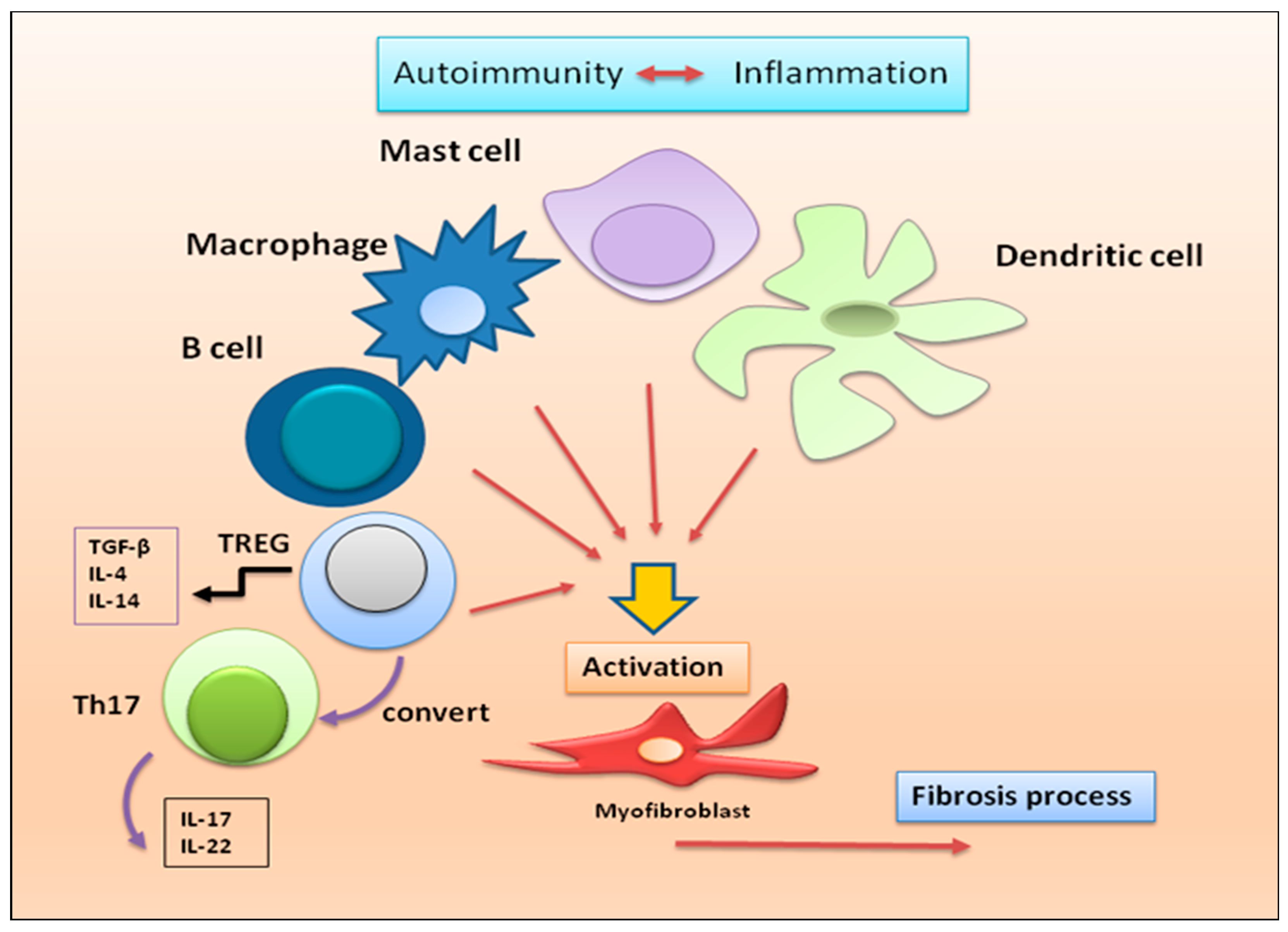
Figure 4.
representation of the hypothetical role of epithelial cells in the activation of the fibrotic program in various autoimmune diseases. (ECM= extracellular matrix; EMT= epithelial to mesenchymal transformation).
Figure 4.
representation of the hypothetical role of epithelial cells in the activation of the fibrotic program in various autoimmune diseases. (ECM= extracellular matrix; EMT= epithelial to mesenchymal transformation).
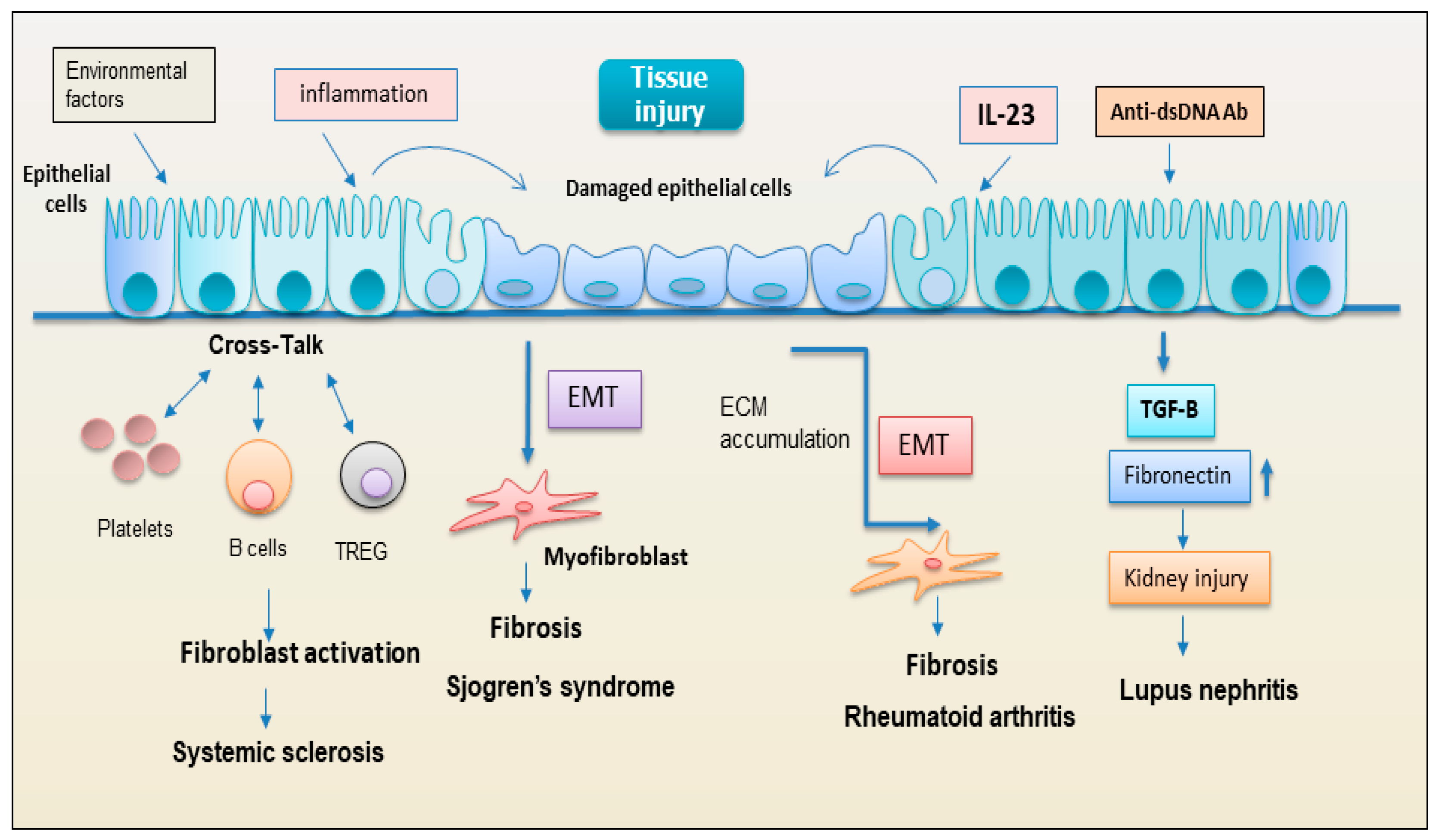
Figure 5.
in chronic autoimmune diseases, endothelial cells are often the primary site of inflammation that triggers the downstream molecular events of fibrosis. The activation of myofibroblasts in various autoimmune diseases such as SLE (Systemic Lupus Erythematosus) and SSc (systemic sclerosis) may result from the phenotypic conversion of endothelial cells into activated mesenchymal cells, a process known as endothelial to mesenchymal transition (EndoMT). .
Figure 5.
in chronic autoimmune diseases, endothelial cells are often the primary site of inflammation that triggers the downstream molecular events of fibrosis. The activation of myofibroblasts in various autoimmune diseases such as SLE (Systemic Lupus Erythematosus) and SSc (systemic sclerosis) may result from the phenotypic conversion of endothelial cells into activated mesenchymal cells, a process known as endothelial to mesenchymal transition (EndoMT). .
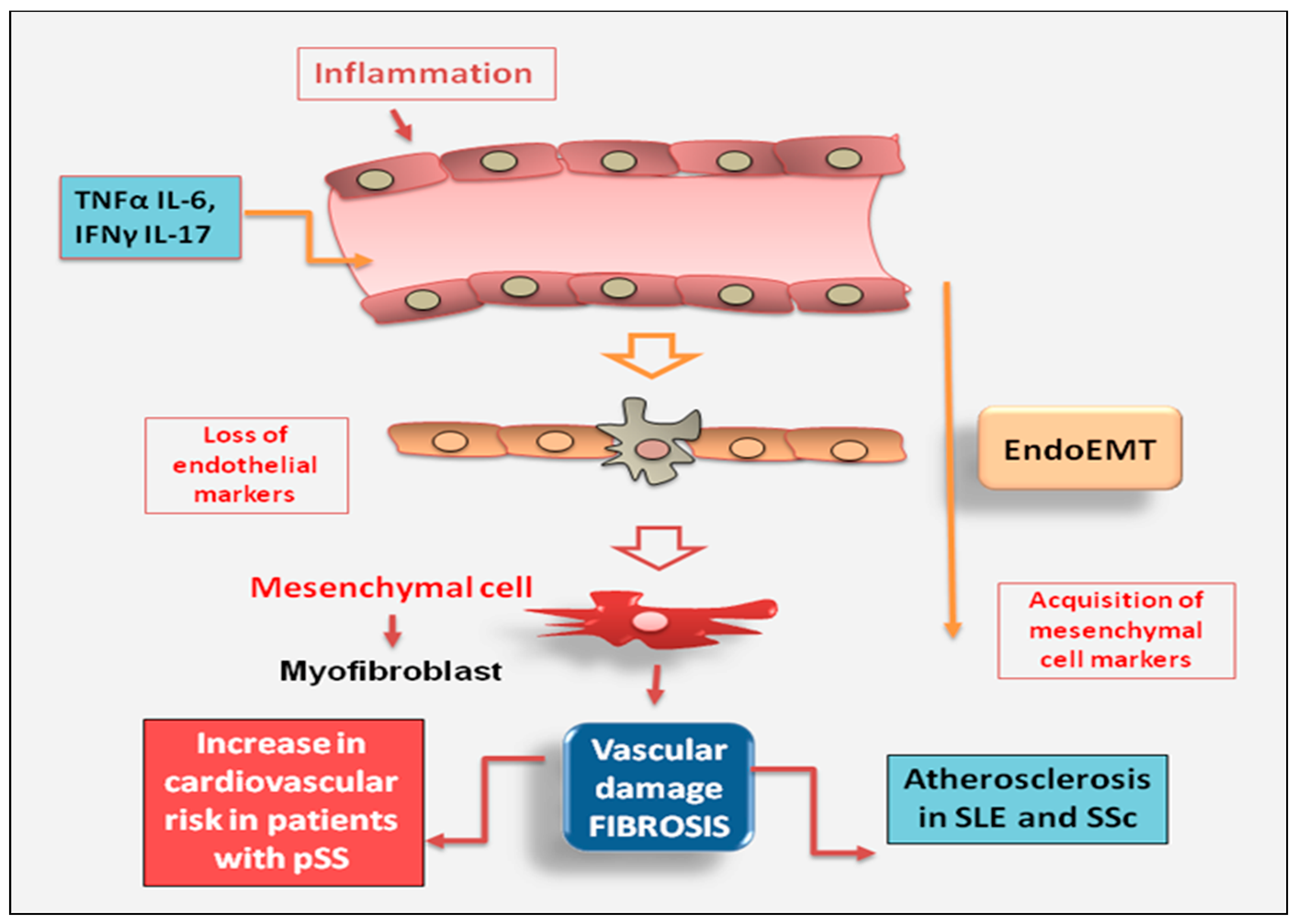
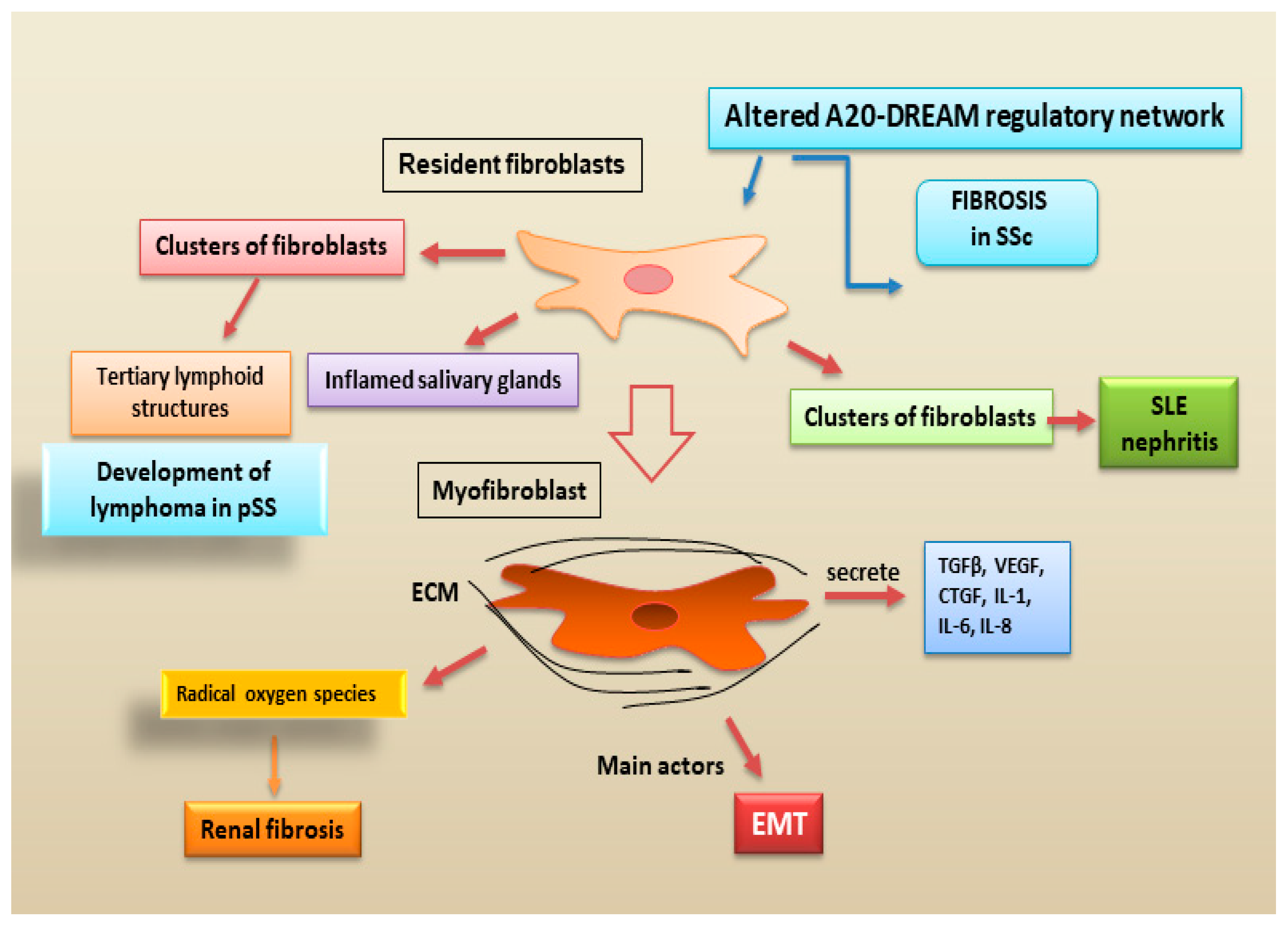
Figure 6.
recent advances in the pro-fibrotic role of fibroblasts in autoimmune diseases. The traditional view that fibroblasts represent purely structural elements has been gradually replaced by the acknowledgement that they are dynamic cells actively involved in the evolution from inflammatory states to fibrosis. SLE = Systemic Lupus Erythematosus; SSc = systemic sclerosis; pSS = primary Sjӧgren’s syndrome; ECM = extracellular matrix; EMT = epithelial to mesenchymal transition).
Figure 6.
recent advances in the pro-fibrotic role of fibroblasts in autoimmune diseases. The traditional view that fibroblasts represent purely structural elements has been gradually replaced by the acknowledgement that they are dynamic cells actively involved in the evolution from inflammatory states to fibrosis. SLE = Systemic Lupus Erythematosus; SSc = systemic sclerosis; pSS = primary Sjӧgren’s syndrome; ECM = extracellular matrix; EMT = epithelial to mesenchymal transition).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



