
Submitted:
24 April 2023
Posted:
25 April 2023
You are already at the latest version
Abstract
Curcuma longa is only a rich source of curcumin (1) and its major analogues demethoxycurcumin (2) and bisdemethoxycurcumin (3) among the Curcuma species. The content ratio of these three curcumins in turmeric is depended on the varieties and growing environment. Recently, curcumin (1) has been reported as potential inhibitor of hepatic and enzymatic protein tyrosine phosphatase 1B (PTP1B) and its related disorders such as hypertriglyceridemia, hyperlipidemia and liver steatosis. Thus, we further purified curcumins (1–3) from C. longa and investigated their inhibitory effects against PTP1B and α-glucosidase enzymes. As the result, curcumins (1–3) exhibited potential PTP1B inhibition with IC50 values of 37.8 ± 1.4, 45.3 ± 0.7, and 72.6 ± 1.1 μM, respectively, and α-glucosidase inhibition with similar manner (IC50 values of 78.2 ± 0.2, 82.4 ± 0.6, and 90.6 ± 1.0 μM, respectively). These results reveal a key role of methoxylation in the variation of PTP1B and α-glucosidase inhibitory activity for these curcumins. In addition, density functional theory (DFT) was used accompanying with molecular docking (MD) to analyze the ligand stability and the interaction of curcumins (1–3) with PTP1B and glucoside hydrolase proteins. Assay-based results and the MD data obtained are highly correlation suggesting that the deterioration of the enzyme activity caused by the distortion of structural conformation of PTP1B and glucoside hydrolase may be related to the arrangement of amino acids in protein structure. This reported for the first time that inhibitory effects of curcumins (1–3) against PTP1B and glucoside hydrolase have been examined in vitro and in silico as well.
Keywords:
Curcuma longa
; curcumins
; PTP1B
; anti-diabetes
; α-glucosidase
; turmeric
; molecular docking simulation.
1. Introduction
There were about 537 million peoples in the age from 20 to 79 years old having diabetes in 2021 according to the World Diabetes Association (IDF) report. This number may be increased to 643 and 783 million by 2030 and 2045, respectively. Diabetes causes 6.7 million deaths with approximately 541 million adults at risk of type 2 diabetes [1,2,3]. Up to date, the common therapeutic approaches focus on simultaneously inhibit insulin- and glucose-based enzyme. PTP1B is an enzyme founding as a member in the family of the protein tyrosine phosphatase (PTP) [4,5], this enzyme is non-receptor type 1 of tyrosine-protein phosphatase, and it negatively regulates the insulin signaling pathway [6].
Over activation of PTP1B lead to a decrease phosphorylating insulin receptors, mutations in the PTP1B gene lead to the development of diabetes [7,8]. Second, α-glucosidase has now been a well-known drug target to treat diabetes due to its important role in hydrolyzing carbohydrate for absorption in the intestinal track. Thus, inhibition of this enzyme could significantly reduce postprandial hyperglycemia and possibly utilized to control diabetes mellitus [9]. In addition, oligo-1,6-glucosidase protein, a debranching endoenzyme, was previously reported to acts on oligosaccharides with an α-1,6 linkage to produce sugar with an α-configuration as the main product [10]. The malfunction of this enzyme could also play an important role in control diabetes mellitus. Therefore, the study of multiple inhibitor of PTP1B and glucoside hydrolase in the treatment of diabetes could represent an interesting therapeutic approach that promoting the attention of scientists in the recent years [7,8].
Curcuma longa is a favorite medicinal food all over the world, belongs to the family Zingiberaceae, and native to the tropics of Tamil Nadu, Southeast India [11]. The species is one of the oldest plants used as daily spice, grown in Southeast Asian countries with the main chemical component as curcumins. Turmeric was utilized as a medicinal drug to treat various chronic diseases including liver-fat reduction in non-alcoholic fatty liver patients, and anticancer as well [12,13]. Curcumin extracted from C. longa has potential effects in the treatment of psoriasis, antibacterial, antiviral and anti-fungal. In particular, it has antidiabetic effects, the ethanolic extracts of turmeric (containing curcumin, demethoxycurcumin and bisdemethoxycurcumin) were confirmed to reduce blood glucose in rats and prevent hyperglycemia [14,15]. Previous research study reported that curcumin had affected the enzyme PTP1B by increasing the sensitivity of hepatic leptin and insulin of rat liver [14,15]. Curcumin also inhibited PTP1B enzyme action that leads to improve hepatic leptin and insulin signaling in rats fed with fructose [16,17,18,19,20]. In addition, curcumin remarkably reduced the podocyte injury and proteinuria induced by fructose, the expression of PTP1B, and cut down the activity of insulin receptor and insulin receptor substrate 1 of fructose-fed rats [21].
In this study, curcumins (1–3) were purified from C. longa using a rapidly method and structurally identified by spectroscopic techniques. The in vitro inhibitory effects of compounds (1–3) on PTP1B and glucoside hydrolase were investigated by assay-based experiments and their mechanism of action was further elucidated using molecular docking method.
2. Results and Discussion
2.1. Experimental Results
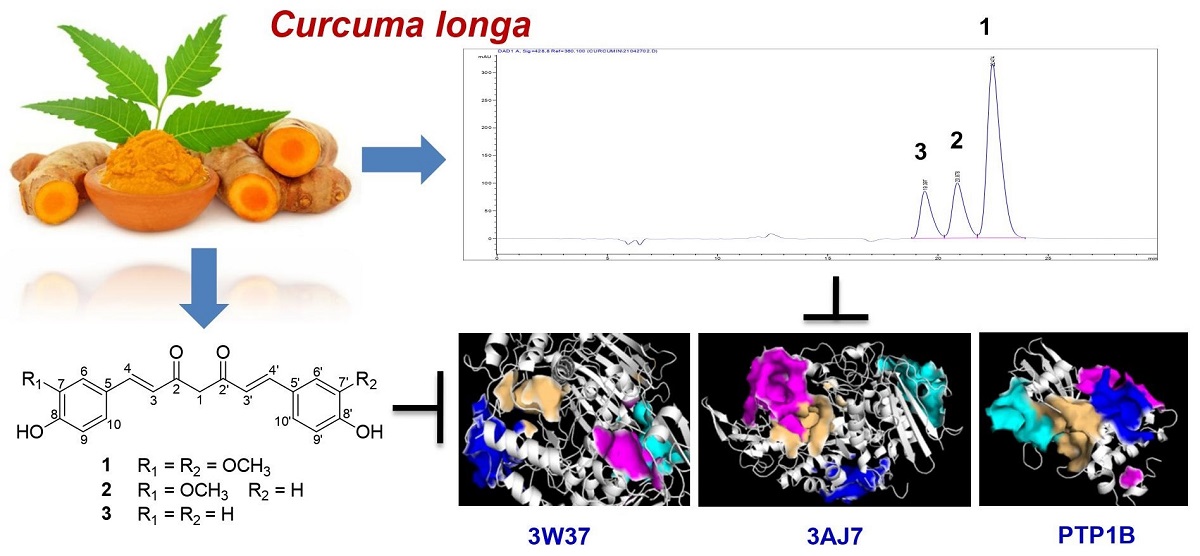
An HPLC method was applied to purified compounds (1–3) from C. longa extract using Agilent 1260 HPLC system and RP-C18 column (Figure 1).
Chemical structures of curcumin (1), demethoxycurcumin (2), and bisdemethoxycurcumin (3) were characterized by analyses of NMR spectral data and comparing with literature (Figure 2).
Compound 1 was obtained as a yellow crystal. The 1H NMR spectrum (Table 1) showed two sets of ABX aromatic protons at [δH 7.28 (2H, d, J = 1.8 Hz, H-6/H-6′), 6.87 (2H, d, J = 7.8 Hz, H-9/H-9′), and 7.14 (2H, dd, J = 1.8, 7.8 Hz, H-10/H-10′], which characterized for the 1,3,4-trisubstituted benzene ring, and two methoxy group [δH 3.89 (6H, s, 7-OCH3/7′-OCH3]. Two pairs of trans-olefinic protons [δH 6.68 (2H, d, J = 16.2 Hz, H-3/H-3′) and 7.58 (2H, d, J = 16.2 Hz, H-4/H-4′], and a methylene group at δH 5.98 (2H, s, H-1) were also observed in the 1H NMR (Figure 2 and Table 1). Consistent with the above 1H NMR analysis, the 13C NMR and DEPT spectra displayed signals of two methoxy groups at δC 56.2 (7-OCH3/7′-OCH3), four olefinic carbons [δC 122.1 (C-3/C-3′) and 141.4 (C-4/C-4′), two conjugated ketones at δC 184.4 (C-2/C-2′), a methylene carbon at δC 101.6 (C-1), and 12 carbons ranging from δC 111.6 to 150.1 ppm corresponding to two aromatic rings (Figure 2 and Table 1). The arrangement of proton-carbon groups were characterized by the aid of HMQC, revealing the structural skeleton of a curcuminoid [13]. The HMBC showed correlations between H-6 and H-10 to C-4 and C-8; H-6′ and H-10′ to C-4′ and C-8′; H-4 to C-2, C-6, and C-10; H-4′ to C-2′, C-6′, and C-10′; H-3 to C-1 and C-5; H-3′ to C-1 and C-5′; as well as H-1 to C-2, C-3, C-2′, and C-3′ (Figure 3). The two methoxy groups were found attaching to C-7 and C-7′ due to the correlations of the methoxy protons with carbon C-7 and C-7′ in the HMBC spectrum (Figure 3). The ESI-MS spectrum of 1 displayed an ion peak at m/z 369.1 [M+H]+, revealing a molecular formula of C21H20O6 for compound 1. Thus, compound 1 was characterized as 1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione [28,29] with a trivial name curcumin.
Compound 2 appeared similar morphology with 1 as yellow crystal. The molecular formula of compound 2 was deduced as C20H18O5 due to the presence of an ion peak at m/z 339.2 [M+H]+ on its ESI-MS. 1D-NMR spectra of compound 2 were quite similar to those of 1, showing an A2B2 aromatic spin system [δH 7.52 (2H, dd, J = 1.8, 6.6 Hz, H-6′/H-10′), 6.87 (2H, dd, J = 1.8, 6.6 Hz, H-7′/H-9′)] for one benzene ring and one ABX aromatic spin for one another (Figure 2 and Table 1). A difference with 1 that compound 2 possessed only one methoxy group at C-7′ [δH 3.89 (3H, s) and δC 56.3]. Thus, compound 2 was determined as 1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-1,6-diene-3,5-dione with a trivial name demethoxycurcumin [30].
Compound 3 also appeared as a yellow crystal with a molecular formula of C19H16O4 deduced by an ion peak at m/z 309.1 [M+H]+ in its ESI-MS. There was no methoxy group found on its 1D NMR. While two A2B2 spin system of two benzene rings [δH 7.53 (4H, dd, J = 1.8, 7.8 Hz, H-6/H-10/H-6′/H-10′), 6.88 (4H, dd, J = 1.8, 7.8 Hz, H-7/H-9/H-7′/H-9′)] were observed. Detailed analyses of the 1H and 13C NMR data of compound 3 and compare with those of compounds 1 and 2 lead to the structural identification of compound 3 to be 1,7-bis(4-hydroxyphenyl)-1,6-heptadiene-3,5-dione (bisdemethoxycurcumin) [30].
Inhibitory effects of curcumins 1‒3 on protein tyrosine phosphatase 1B (PTP1B) enzyme activity were tested in vitro using a triterpene (ursolic acid) and a quinone emodin as positive control [31]. As shown in Table 2, all compounds potential inhibited this enzyme action with IC50 values of 37.8 ± 1.4 (curcumin 1), 45.3 ± 0.7 (demethoxycurcumin 2), and 72.6 ± 1.1 µM (bisdemethoxycurcumin 3). Emodin possessed potential inhibition with an IC50 value of 7.6 ± 0.3 µM. Among these curcumins, curcumin (1) bearing two methoxy moieties at C-7 and C-7′ (Figure 2), displayed the most potential inhibition against PTP1B (IC50 value of 37.8 ± 1.4 µM), demethoxycurcumin (2) bearing one methoxy group at C-7 (loss of one methoxygroup at C-7′) exhibited less inhibitory activity (IC50 value of 45.3 ± 0.7 µM) than 1. Bisdemethoxycurcumin (3) without methoxy moiety in its structure showed moderate inhibition (IC50 value of 72.6 ± 1.1 µM). This experimental result suggests that substitution of methoxy moiety at C-7 and/or C-7′ in curcuminoid compounds may play a key role for enhancing their inhibitory effect on PTP1B enzyme activity.
Li et al. reported that curcumin (1) possessed inhibition on PTP1B action and affected on the liver of rats with fructose-fed. At 60 mg/kg, curcumin showed similar effect with pioglitazone on liver PTP1B expression, but showed less effect against PTP1B activity than pioglitazone at a concentration of 10 mg/kg [16]. Recently, Kostrzewa et al. demonstrated curcumin showing antidiabetic and anticancer potential via inhibition of PTP1B [17]. Curcumin also reduced the enzymatic activity of PTP1B phosphatase and MCF7 cell viability by 50% inhibition at a concentration of 100 μM. In our in vitro study, curcumin (1) showed more potent inhibition with 50% inhibition at a concentration of 37.8 µM on PTP1B action. This assay has reported for the first time the inhibitory activities of demethoxycurcumin (2) and bisdemethoxycurcumin (3) against PTP1B.
Regarding to α-glucosidase inhibitory experimental assay, acarbose and curcumins (1‒3) were tested using α-glucosidase enzyme from rat intestinal tract. In this assay, acarbose displayed a similar inhibitory IC50 value (147.2 ± 1.0 μM) toward α-glucosidase [22]. Furthermore, curcumin (1) potentially inhibited the action of α-glucosidase with an IC50 value of 78.2 ± 0.2 μM, almost two times stronger than acarbose. Demethoxycurcumin (2) showed an IC50 value of 82.4 ± 0.8 μM and bisdemethoxycurcumin (3) showed less activity than 1 and 2, with an IC50 value of 90.6 ± 1.0 μM. Similar aspect with PTP1B enzyme, the inhibitory effect of curcumins 1‒3 against α-glucosidase was also decreased by the demethoxylation in the curcuminoid structure. This leads to suggest that methylation or methoxyl substitution in the design and synthesis of curcumin analogs may be a key for the search and development of α-glucosidase inhibitory agents.
A published result reported that curcumin (1) exhibited a mild inhibition effect on α-glucosidase through a competitive inhibition mechanism with its IC50 value of 20.54 ± 1.02 mg/mL [32]. Du et al. studied the inhibition of natural curcumins and the synthetic curcumin analogs on α-glucosidase enzyme. However, only natural bisdemethoxycurcumin (3) showed inhibitory activity with an IC50 value of 23.0 μM against α-glucosidase [33], while the inhibitory effects of curcumin (1) and demethoxycurcumin (2) were ambiguously mentioned. Another report demonstrated the inhibition of curcumin (1) and other curcuminoids on enzymatic α-glucosidase activity. But, their inhibitory effects were moderate at 50 μg/mL and at mM level [34,35]. In our experimental assay, α-glucosidase enzyme was obtained from rat intestine and the positive control acarbose showed a stable IC50 value [22]. Consequently, the result obtained in this study was accurate and reachable. And that the inhibitory activity of demethoxycurcumin (2) on α-glucosidase enzyme has first reported.
2.2. Computational Studies
In this study, DFT and MD were the two utilized techniqs used to investigate the interaction between curcumins (1–3) and the targeted protein (3W37, 3AJ7 and PTP1B). Figure 4 shows the geometrically optimized structures of compounds 1–3 and Figure 5 shows the HOMO and LUMO frontier molecular orbitals, Table 3 listed the related quantum parameters for these compounds. These compound structures are identical, compounds 1, 2, and 3 contains -OH and C=O functional groups, the C=O bonds are ~1.21Å, whereas C-OH are 1.35 Å. These were reported to be highly conducive to polarity and solubility of the host compound [36,37], thus implying promising inhibitability towards protein molecules based on polar interactions with highly polarized amino acids. Moreover, these compounds are suitable for intermolecular inhibition suggested by bonding analysis on frontier molecular orbitals. As shown in Figure 5, the large lobes of the HOMO and LUMO are both mainly located on the aromatic ring, with O (C=O bonds), O (-OH group), C (C=C-C bonds). This is indicated that to uphold electron-transferring interactability, the molecules are able to initiate intermolecular inhibition from certain approaching manners. In addition, the parameter values which calculated for these curcumins are not significantly differences. Particularly, the energy gaps (DEGAP) of curcumins (1–3) are -5.962, -5.724 and -5.717 eV, respectively. These obtained values are considered to be low values which may lead to chemical reactivity and inhibitory stability [38,39].
The reason is thought to be that electrons of inhibitory molecules are easily activated and transferred to their surface, ready for intermolecular activities. Furthermore, electronegativity (c), or the chemical potential (m) in a negative value, could be considered as a reliable inhibition indicator since it presents an electron-attracting tendency. In principle, a higher electronegativity implies a stronger attraction of electrons towards the host molecule. Thus, these curcumins (1–3) seem promising for docking study.
As the X-ray structure of PTP1B was not well characterized, we built a three-dimensional structure of PTP1B by homology modeling on the Swiss-Model webserver. It is commonly assumed that a good quality model is expected to have a score over 90% in the most favored regions [35]. Obtained data from Ramachandran plot showed that 98.27% amino acid residues of the PTP1B model located in the most favored regions (Figure 6A,B). In addition, the modeled structure of PTP1B was superimposed with three previously published crystal structure of PTP1B (Figure 6C). The sequence alignment between these models exhibit significant high identity (at least 96.98%), thus, this model could be considered as a liable model for further docking studies.
Amongst various docking softwares, AutoDock4 (AD4) is a non-commercial package that is recognized and used during the last ten years with about 6000 publicaitons. This is an useful tool to rapidly predict the binding affinity of ligands towards a specific protein/ targeted enzyme [41,42], thus, we chose AD4 to perform docking simulation. According to the ranking criteria of Autodock4, the negative value of docking energy express the binding affinity of the compound towards targeted receptor, this means that the value becomes more negative showing better binding affinity [41,43].
The binding site of targeted proteins are investigated using 3DLigandSite webserver (https://www.wass-michaelislab.org/3dlig) in which they are marked as site 1 (yellow), site 2 (cyan), site 3 (magenta), site 4 (blue) (Figure 7). Particularly, the key residues of each site are presented in Table 4. Emodin, ursolic acid and acarbose were selected as reference ligands.
Obtained data showed that all the binding sites are constituted from a larger number of different amino acids, thus, they are assumed as highly conductive to peripheral interactions. Considering α-glucosidase protein (PDB ID: 3W37), a total of 21 amino acids were detected in site 2 binding region which is significantly higher than the other sites. The docking score also proved this is the most suitable site for curcumin compounds given the lowest value of binding free energy (varying from -6.99 to -12.36 kcal/mol) (Table 6). In terms of 3AJ7, site 1 and 3 also expected to be favorable for inhibitors to form interaction based on their dominant in the numbers of constitute residues. Docking analysis has pointed out that site 3 provide the highest binding affinity towards studied ligands, especially curcumin compounds (varying from -9.99 to -10.65 kcal/mol). Regarding protein PTP1B, all the detected binding sites showed no noticeable differences in the number of in-pose amino acids. In this case, docking score revealed that site 3 is the most preferential region for inhibitors to bind with (varying from -8.09 to -12.75 kcal/mol).

Table 5.
Docking score of studied compounds on 3W37, 3AJ7 and PTP1B proteins.
| Compounds | 3W37 | 3AJ7 | PTP1B | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Site 1 | Site 2 | Site 3 | Site 4 | Site 1 | Site 2 | Site 3 | Site 4 | Site 1 | Site 2 | Site 3 | Site 4 | |
| 1 | -4.49 | -12.36 | -8.38 | -7.42 | -9.98 | -9.09 | -10.65 | -8.51 | -7.47 | -8.06 | -9.57 | -8.12 |
| 2 | -5.29 | -12.16 | -7.50 | -7.42 | -9.80 | -9.06 | -9.99 | -9.58 | -6.85 | -7.87 | -8.93 | -7.49 |
| 3 | -6.43 | -11.14 | -7.63 | -6.42 | -9.04 | -7.76 | -10.60 | -9.90 | -6.47 | -8.27 | -8.09 | -7.52 |
| Emodin | -4.65 | -6.99 | -6.25 | -5.83 | -7.53 | -7.16 | -7.90 | -7.74 | -6.03 | -7.07 | -11.31 | -5.51 |
| Ursolic acid | -5.27 | -7.36 | -3.70 | -6.20 | -8.81 | -8.01 | -7.68 | -8.80 | -7.43 | -7.28 | -12.37 | -5.98 |
| Acarbose | -6.61 | -12.22 | -6.06 | -12.78 | -13.15 | -12.46 | -12.72 | -13.24 | -12.22 | -8.60 | -12.75 | -10.59 |
The detailed docking simulation interaction of curcuminoid compounds (1–3) are summarized in Table 7. Overall, all curcumins (1–3) exhibit the most inhibitory effect toward α-glucosiase (PDB ID: 3W37) than the others. The most negative values of binding free energies were recorded for curcumin (1), demethoxycurcumin (2) and bisdemethoxycurcumin (3) as -12.36, -12.16 and -11.14 kcal/mol, respectively. The reference ligand, acarbose, was recorded to exhibit high affinity toward this protein with dock score of -12.22 kcal/mol. This ranking were highly correlate with experimental data since the IC50 of tested compounds were: compound 1 (78.2 ± 0.2 µM), compound 2 (82.4 ± 0.8 µM), compound 3 (90.6 ± 1.0 µM), acarbose (147.2 ± 1.0 µM). As reported in previous study [22], Arg676 was assumed to display important role in the functional of α-glucosidase (3W37). Interaction formed with this residue seem to induce sevear conformational changes on the enzyme PTP1B, thus, causing loss of its normal functionality. Obtained data proved that, all three studied ligands form H-bond with Arg676, particularly, the number of hydrogen bonds formed between 3W37 and curcumin compounds (1–3) were 3, 5 and 4, respectively. Additionaly, their interaction were further strengthen through Van der Waals bonding (Figure 8). In terms of oligo-1,6-glucosidase protein (PDB ID: 3AJ7), although the experiments are not included in this study, docking simulation were conducted to gain better insight regarding mechanism of action of studied compounds towards this protein since it is a well-known drug target for diabetes treatment. Initially, docking conformation analysis revealed that residue Asn317 is assumed to play an important role in the fuction of this protein. This hypotheses would need further experiment to validate the inhibitory mechanism of action. Regarding PTP1B protein, the dock score ranking of studied ligands (ursolic acid > emodin > 1 > 2 > 3) showed high correlation with experiment-based assay result on PTP1B inhibition, in which the IC50 value are: ursolic acid (4.3 ± 0.4 µM), emodin (7.6 ± 0.1 µM), compound 1 (37.8 ± 1.4 µM), compound 2 (45.3 ± 0.7 µM) and compound 3 (72.6 ± 1.1 µM). Dock pose analysis showed that amino acid Leu204 participated in forming hydrogen bonds with all the studied curcumin compounds within the site 3 binding region. Therefore, this residue might play a key role in the development of potential compounds to inhibit the function of this protein.
3. Materials and Methods
3.1. General Experimental Procedures
1H-NMR (600 MHz) and 13C NMR (150 MHz) were measured on a Bruker AVANCE 600 spectrometer. MS was obtained from Single Quadrupole LC/MS system of Agilent. Silica gel (Si 60 F254, 230-400 mesh, Merck) was used for open column. Thin layer chromatography was used for analytical purposes using silica gel and RP 60 F254 plates. HPLC was carried out using an Agilent 1260 HPLC system with a DAD detector and an XDB-C18 column and all purified solvents with analytical grade were from Fisher Scientific Korea Ltd.
3.2. Plant Material
The root parts of C. longa were collected in 2019 at Bac Kan province, Vietnam. The sample was identified by Dr. Nguyen Quoc Binh (Vietnam National Museum of Nature, VAST, 18-Hoang Quoc Viet, Cau Giay, and Hanoi, Vietnam). A voucher specimen (CL-BK) was deposited at the Vietnam National Museum of Nature, VAST.
3.3. Extraction and Isolation
The root parts of C. longa (1.4 kg) was cut into small pieces and dried before extracted with MeOH (5 L x 3 time) at 45ºC using sonication for 10 h. After removing the solvent under reduced pressure, the residue (65.4 g) was excessively fractionated with n-hexane and EtOAc to give the n-hexane fraction (16 g) and EtOAc (11 g), respectively. The EtOAc fraction (10 g) was directly chromatographed on an open silica gel column (5.0 × 80 cm; 63−200 mm particle size, Merck) using a stepwise gradient of n-hexane/EtOAc (from 20:1 to 0:1) to give twelve combined fractions (CL-1 to CL-12) according to their TLC profiles. Fraction CL-3 was directly purified by Agilent 1260 HPLC system using an Optima Pak_C18 column (10 × 250 mm, 10 μm particle size), eluted with an isocratic solvent system of ACN:MeOH (25:35%, v/v) in H2O + 0.1% formic acid (flow rate 2 mL/min) over 30 min, UV detection at 428 nm, resulted in the isolation of compounds 1 (88.4 mg; tR = 19.34 min), 2 (21.5 mg; tR = 20.88 min), and compound 3 (28.3 mg; tR = 22.43 min), respectively.
3.4. Spectral and Physical Data of Isolates
Curcumin (1): Yellow crystal; ESI-MS m/z 369.1 [M+H]+ (C21H21O6); 1H NMR (600 MHz) and 13C NMR (150 MHz) spectroscopic data measured in acetone-d6, see Table 1.
Demethoxycurcumin (2): Yellow crystal; ESI-MS m/z 339.2 [M+H]+ (C20H19O5); 1H NMR (600 MHz) and 13C NMR (150 MHz) spectroscopic data measured in acetone-d6, see Table 1.
Bisdemethoxycurcumin (3): Yellow crystal; ESI-MS m/z 309.1 [M+H]+ (C19H17O4); 1H NMR (600 MHz) and 13C NMR (150 MHz) spectroscopic data measured in acetone-d6, see Table 1.
3.5. Inhibitory Assay on PTP1B
PTP1B enzyme was obtained from Biomol International LP, Plymouth Meeting, Pennsylvania, PA, USA, and the experimental assay was performed similar with previous method [22].
3.6. Inhibitory Assay on α-Glucosidase
The α-glucosidase enzyme was from rat intestine and the detailed assay procedure was performed with the same method as described previously [22].
3.7. Molecular Docking Studies
MarvinSketch version 19.27.0 and PyMOL version 1.3r1 were used for preparation of 3D structure of curcumins (1−3) and quantum properties were analyzed by DFT. The Gaussian 09, revision E.01 program was used for all calculations [39]. Geometry optimization and frequency calculations of all structures, transition states (TSs), intermediate species, and products were conducted at the M06-2X functional and 6-311+G(d,p) basis sets without symmetry constraint [40,41]. All single-point calculations were performed at the same level of theory. The PDB entries chosen for docking studies 3W37 (alpha-glucosidase protein) [42], 3AJ7 (oligo-1,6-glucosidase protein) [43] were downloaded from the Protein Data Bank archive. The amino acid sequence of PTP1B enzyme was determined and its information was published at UniProtKB, archived under entry ID: UniProtKB-A0A0U1XP67. The crystal structure of PTP1B was built and validated using Swiss-Model webserver (http://swissmodel.expasy.org). The Graphical User Interface program named Autodock Tools 1.5.6 (ADT) was employed to set up input data. The location and dimensions of the grid box for each protein were chosen such that they incorporate the amino acid domain involved in binding with the reference compound, which was enclosed in a box with the number of grid points in x × y × z directions and a grid spacing of 0.375 Å. The detailed coordinate of grid box parameters are presented in Table 8.
4. Conclusions
A fast and accurate HPLC method was built to purify three curcuminoids [curcumin (1), demethoxycurcumin (2), and bisdemethoxycurcumin (3)] from C. longa. Their structure were characterized by NMR spectra analyses and comparing with published values. Curcumins 1–3 potential inhibited PTP1B enzyme with IC50 values from 37.8 to 72.6 µM, and α-glucosidase enzyme (IC50 values from 78.2 to 90.6 µM). Compound 1 bearing two methoxy groups possessed the most inhibition on both enzymatic assay (IC50 of 37.8 ± 1.4 and 78.2 ± 0.2 µM against PTP1B and α-glucosidase, respectively), while compound 2, with loss of one methoxy group in its structure, showed less inhibition than 1 with IC50 of 45.3 ± 0.7 (PTP1B) and 82.4 ± 0.6 (α-glucosidase) µM. Finally, compound 3 with bisdemethoxylation moderately exhibited the IC50 values of 72.6 and 90.6 µM, respectively. This result may suggest that the demethoxylation in curcuminoid structure may be responsible for the decrease of inhibitory activities against PTP1B and α-glucosidase of these compounds.
In addition, docking simulation and experimental assay data are highly correlation showing the most negative values of binding free energies were recorded for curcumins (1–3) as -12.36, -12.16 and -11.14 kcal/mol, respectively toward PDB ID: 3W37 protein. In case of PTP1B protein, the dock score ranking of studied ligands (ursolic acid > emodin > 1 > 2 > 3) showed high correlate with experiment result, in which the IC50 value are: ursolic acid (4.3 ± 0.4 µM), emodin (7.6 ± 0.1 µM), compound 1 (37.8 ± 1.4 µM), compound 2 (45.3 ± 0.7 µM) and compound 3 (72.6 ± 1.1 µM). Dock pose analysis showed that amino acid Leu204 might play a key role in the development of potential compounds to inhibit the function of this protein due to the forming hydrogen bonds by Leu204 with all the studied curcumins (1–3) at the site 3 binding region. To date, curcumin (1), demethoxycurcumin (2), and bisdemethoxycurcumin (3) have been investigated both in vitro and in silico for their inhibition on PTP1B and glucoside hydrolase enzymes for the first time.
Author Contributions
Methodology, P.-H.N. and M.-Q.P.; software, M.-Q.P. and M.-H.T.; validation, P.-H.N., N.-Q.D. and T.-D.N.; formal analysis, P.-H.N., A.-T.N., D.-C.T. and M.-Q.P; investigation, T.-T.D., A.-T.N., P.-H.N. and M.-Q.P; resources, T.-H.N. and H.-T.N.; data curation, T.-T.D., and A.-T.N.; writing—original draft preparation, P.-H.N., D.-C.T., M.-H.T. and M.-Q.P.; writing—review and editing, P.-H.N., D.-C.T., M.-H.T. and M.-Q.P.; supervision, P.-H.N. and N.-Q.D.; funding acquisition, P.-H.N. and T.-D.N.
Funding
The study was supported by Vietnam Academy of Science and Technology with a grant number: QTRU02.04/20-21.
Acknowledgments
T.-D.N thanks for the support of Thai Nguyen University of Agriculture and Forestry (TUAF) under a research program funded by the Ministry of Education and Training (MOET) with the project code number: CT 2020.03.
Conflicts of Interest
no confliction.
Sample Availability
Curcumins (1–3) and their NMR data can be supported by the authors.
References
- Preethi, K.A.; Selvakumar, S.C.; Sekar, D. Diagnostic and Therapeutic Application of Exosomal microRNAs Inducing Inflammation in Type 2 Diabetes Mellitus. Crit. Rev. Immunol. 2022, 42, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Muntean, C.; Starcea, I.M.; Banescu, C. Diabetic kidney disease in pediatric patients: A current review. World J. Diabetes 2022, 13, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Yeh, J.M.; Reddy, C.L.; Gomber, A.; Ross, C.; Rittiphairoj, T.; Manne-Goehler, J.; Abdalla, A.T.; Abdullah, M.A.; Ahmed, A.; Ankotche, A.; Azad, K.; Bahendeka, S.; Baldé, N.; Jain, S.M.; Kalobu, J.C.; Karekezi, C.; Kol, H.; Prasannakumar, K.M.; Leik, S.K.; Mbanya, J.C.; Mbaye, M.N.; Niang, B.; Paturi, V.R.; Raghupathy, P.; Ramaiya, K.; Sethi, B.; Zabeen, B.; Atun, R. Estimating the total incidence of type 1 diabetes in children and adolescents aged 0-19 years from 1990 to 2050: a global simulation-based analysis. Lancet Diabetes Endocrinol. 2022, 10, 848–858. [Google Scholar] [CrossRef]
- Liu, R.; Mathieu, C.; Berthelet, J.; Zhang, W.; Dupret, J.M.; Rodrigues Lima, F. Human Protein Tyrosine Phosphatase 1B (PTP1B): From Structure to Clinical Inhibitor Perspectives. Int. J. Mol. Sci. 2022, 23, 7027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Gupta, A.; Sehgal, A.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Najmi, A.; Bhatia, S.; Bungau, S. Exploring protein tyrosine phosphatases (PTP) and PTP-1B inhibitors in management of diabetes mellitus. Biomed. Pharmacother. 2022, 153, 113405. [Google Scholar] [CrossRef]
- Verma, M.; Gupta, S.J.; Chaudhary, A.; Garg, V.K. Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents - A brief review. Bioorg. Chem. 2017, 70, 267–283. [Google Scholar] [CrossRef]
- Nandi, S.; Saxena, M. Potential Inhibitors of Protein Tyrosine Phosphatase (PTP1B) Enzyme: Promising Target for Type-II Diabetes Mellitus. Curr. Top. Med. Chem. 2020, 20, 2692–2707. [Google Scholar] [CrossRef]
- Hirsh, A.J.; Yao, S.Y.; Young, J.D.; Cheeseman, C.I. Inhibition of glucose absorption in the rat jejunum: A novel action of alpha-D-glucosidase inhibitors. Gastroenterology 1997, 113, 205–211. [Google Scholar] [CrossRef]
- Taniguchi, H.; Honnda, Y. Amylases. In Encyclopedia of Microbiology, 3rd ed.; Academic Press, 2009; pp. 159–173. [Google Scholar] [CrossRef]
- Liu, X.; Machado, G.C.; Eyles, J.P.; Ravi, V.; Hunter, D.J. Dietary supplements for treating osteoarthritis: a systematic review and meta-analysis. Br. J. Sports Med. 2018, 52, 167–175. [Google Scholar] [CrossRef]
- Bich, D.H.; Chung, D.Q.; Chuong, B.X.; Dong, N.T.; Dam, D.T.; Hien, P.V.; Lo, V.N.; Mai, P.D.; Man, P.K.; Nhu, D.T.; Tap, N.; Toan, T. The medicinal plants and medicinal animals in Vietnam, 1st ed.; Science and Technology Publishing House: Hanoi, Vietnam, 2006; pp. 383–391. [Google Scholar]
- Tung, B.T.; Nham, D.T.; Hai, N.T.; Thu, D.K. Curcuma longa, the polyphenolic curcumin compound and pharmacological effects on liver. Dietary Interventions in Liver Disease; Academic Press: London, England, 2019. [Google Scholar]
- Soleimani, V.; Sahebkar, A.; Hosseinzadeh, H. Turmeric (Curcuma longa) and its major constituent (curcumin) as nontoxic and safe substances. Phytother. Res. 2018, 32, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Karłowicz-Bodalska, K.; Han, S.; Freier, J.; Smolenski, M.; Bodalska, A. Curcuma longa as medicinal herb in the treatment of diabetic complications. Acta Pol. Pharm. 2017, 74, 605–610. [Google Scholar]
- Li, J.M.; Li, Y.C.; Kong, L.D.; Hu, Q.H. Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology. 2010, 51, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, T.; Przychodzen, P.; Gorska-Ponikowska, M.; Kuban-Jankowska, A. Curcumin and Cinnamaldehyde as PTP1B Inhibitors With Antidiabetic and Anticancer Potential. Anticancer Res. 2019, 39, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, T.; Wołosewicz, K.; Jamrozik, M.; Drzeżdżon, J.; Siemińska, J.; Jacewicz, D.; Górska-Ponikowska, M.; Kołaczkowski, M.; Łaźny, R.; Kuban-Jankowska, A. Curcumin and Its New Derivatives: Correlation between Cytotoxicity against Breast Cancer Cell Lines, Degradation of PTP1B Phosphatase and ROS Generation. Int J Mol Sci. 2021, 22, 10368. [Google Scholar] [CrossRef]
- Bozkurt, O.; Kocaadam-Bozkurt, B.; Yildiran, H. Effects of curcumin, a bioactive component of turmeric, on type 2 diabetes mellitus and its complications: an updated review. Food Funct. 2022, 13, 11999–12010. [Google Scholar] [CrossRef]
- Duan, J.; Yang, M.; Liu, Y.; Xiao, S.; Zhang, X. Curcumin protects islet beta cells from streptozotocin-induced type 2 diabetes mellitus injury via its antioxidative effects. Endokrynol Pol. 2022, 73, 942–946. [Google Scholar] [CrossRef]
- Ding, X.Q.; Gu, T.T.; Wang, W.; Song, L.; Chen, T.Y.; Xue, Q.C.; Zhou, F.; Li, J.M.; Kong, L.D. Curcumin protects against fructose-induced podocyte insulin signaling impairment through upregulation of miR-206. Mol Nutr Food Res. 2015, 59, 2355–2370. [Google Scholar] [CrossRef]
- To, D.C.; Bui, T.Q.; Nhung, N.T.A.; Tran, Q.T.; Do, T.T.; Tran, M.H.; Hien, P.P.; Ngu, T.N.; Quy, P.T.; Nguyen, T.H.; Nguyen, H.T.; Nguyen, T.D.; Nguyen, P.H. On the Inhibitability of Natural Products Isolated from Tetradium ruticarpum towards Tyrosine Phosphatase 1B (PTP1B) and α-glucosidase (3W37): an in vitro and in silico Study. Molecules 2021, 26, 3691. [Google Scholar] [CrossRef]
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Free radicals induced oxidative stress at a molecular level: the current status, challenges and perspectives of computational chemistry based protocols. J. Mex. Chem. Soc. 2015, 7, 335–352. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. Kinetics of Radical-Molecule Reactions in Aqueous Solution: A Benchmark Study of the Performance of Density Functional Methods. J. Comput. Chem. 2014, 35, 2019–2026. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A Computational Methodology for Accurate Predictions of Rate Constants in Solution: Application to the Assessment of Primary Antioxidant Activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Yamashita, K.; Okuyama, M.; Mori, H.; Yao, M.; Kimura, A. Molecular basis for the recognition of long-chain substrates by plant & alpha-glucosidase. J. Biol. Chem. 2013, 288, 19296–19303. [Google Scholar] [PubMed]
- Yamamot, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, S.; Feng, J.; Xiao, Y.; Xue, X.; Zhang, H.; Wang, Y.; Liang, X. Structure elucidation and NMR assignments for curcuminoids from the rhizomes of Curcuma longa. Magn. Reson. Chem. 2009, 47, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Payton, F.; Sandusky, P.; Alworth, W.L. NMR Study of the Solution Structure of Curcumin. J. Nat. Prod. 2007, 70, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakasha, G.K.; Rao, L.J.M.; Sakariah, K.K. Improved HPLC Method for the Determination of Curcumin, Demethoxycurcumin, and Bisdemethoxycurcumin, J. Agric. Food Chem. 2002, 50, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Le, H.L.; To, D.C.; Tran, M.H.; Do, T.T.; Nguyen, P.H. Natural PTP1B Inhibitors from Polygonum cuspidatum and Their 2-NBDG Uptake Stimulation. Nat Prod. Commun. 2020, 15, 1–7. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, J.; Yu, J.; Chen, X.; Zhang, S.; Cai, Y.X.; Li, L. Curcumin as a mild natural α-glucosidase inhibitor: a study on its mechanism in vitro. International Journal of Food Science and Technology 2021, 57, 2689–2700. [Google Scholar] [CrossRef]
- Du, Z.Y.; Liu, R.R.; Shao, W.Y.; Mao, X.P.; Ma, L.; Gu, L.Q.; Huang, Z.S.; Chan, A.S.C. α-Glucosidase inhibition of natural curcuminoids and curcumin analogs. European Journal of Medicinal Chemistry 2006, 41, 213–218. [Google Scholar] [CrossRef]
- Al-Lahham, S.; Jaradat, N.; Hamayel, A.; Assaassa, A.; Hammad, F.; Mosa, A.; Nafaa, F.; Chanim, M.; Dwikat, M.; Alqub, M.; Rahim, A.A.; Barquawi, A. Hexane extract of Curcuma longa L. inhibits the activities of key enzymes and pro- inflammatory adipokines linked to obesity. European Journal of Integrative Medicine 2021, 48, 101400–101408. [Google Scholar] [CrossRef]
- Cao, W.X.; Chen, X.; Chin, Y.X.; Zheng, J.K.; Lim, P.E.; Xue, C.H.; Tang, Q.J. Identification of curcumin as a potential α-glucosidase And dipeptidyl-peptidase 4 inhibitor: Molecular docking study, in vitro and in vivo biological evaluation. J. Food Biochem. 2021, 00, e13686. [Google Scholar] [CrossRef]
- Thomas, G. Medicinal Chemistry: An Introduction; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Sessler, J.L.; Gale, P.A.; Cho, W.S. Anion Receptor Chemistry; Royal Society of Chemistry: London, UK, 2006. [Google Scholar]
- Carvalho, A.A.; Andrade, L.N.; Batista, É.; Sousa, V.; De Sousa, D.P. Antitumor Phenylpropanoids Found in Essential Oils. BioMed Res. Int. 2015, 2015, 392674. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; De Morais, M.C. The Dual Antioxidant/Prooxidant Effect of Eugenol and Its Action in Cancer Development and Treatment. Nutrients 2017, 9, 1367. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.; Rullmann, J.A.; MacArthur, M.; Kaptein,R. ; Thornton, J. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Hai, N.T.; Han, P.T.N.; Huy, N.T.; Bay, M.V.; Quan, P.M.; Nam, P.C.; Vu, V.V.; Tung, N.S. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model. 2019, 60, 204–211. [Google Scholar] [CrossRef]
- Dan, N.T.; Quang, H.D.; Van, V.T.; Nghi, D.H.; Cuong, N.M.; Cuong, T.D.; Toan, T.Q.; Giang, B.L.; Anh, N.T.H.; Mai, N.T.; Lan, N.T.; Chinh, L.V.; Quan, P.M. Design, synthesis, structure, in vitro cytotoxic activity evaluation and docking studies on target enzyme GSK-3β of new indirubin-3ʹ-oxime derivatives. Sci. Rep. 2020, 10, 11429. [Google Scholar] [CrossRef]
- Ngo, Q.A.; Hang, N.T.T.; Quan, P.M.; Delfino, D.; Thao, D.T. Antiproliferative and antiinflammatory coxib–combretastatin hybrids suppress cell cycle progression and induce apoptosis of MCF7 breast cancer cells. Mol. Divers. 2020, 25, 2307–2319. [Google Scholar] [CrossRef]
Figure 1.
A representative HPLC chromatography for isolation of curcumin (1), demethoxycurcumin (2), and bisdemethoxycurcumin (3) from C. longa.
Figure 1.
A representative HPLC chromatography for isolation of curcumin (1), demethoxycurcumin (2), and bisdemethoxycurcumin (3) from C. longa.
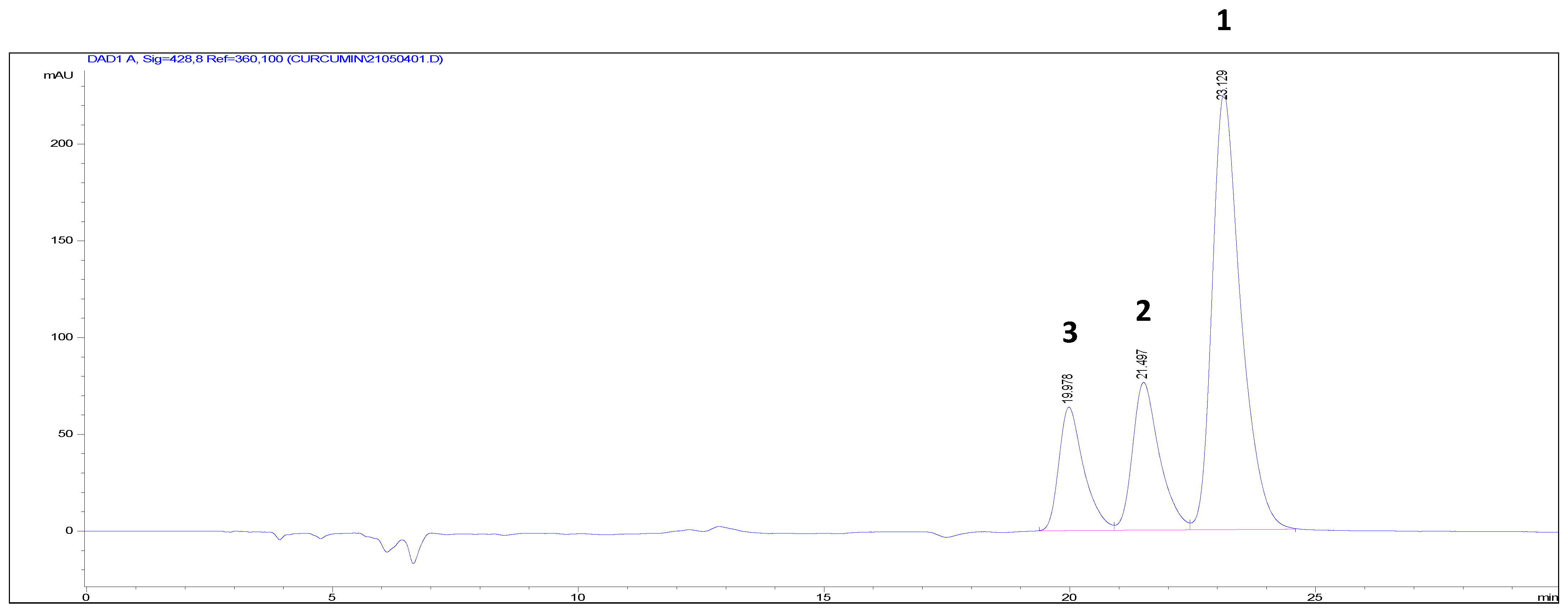
Figure 2.
Chemical structure of compounds (1–3) isolated from C. longa.
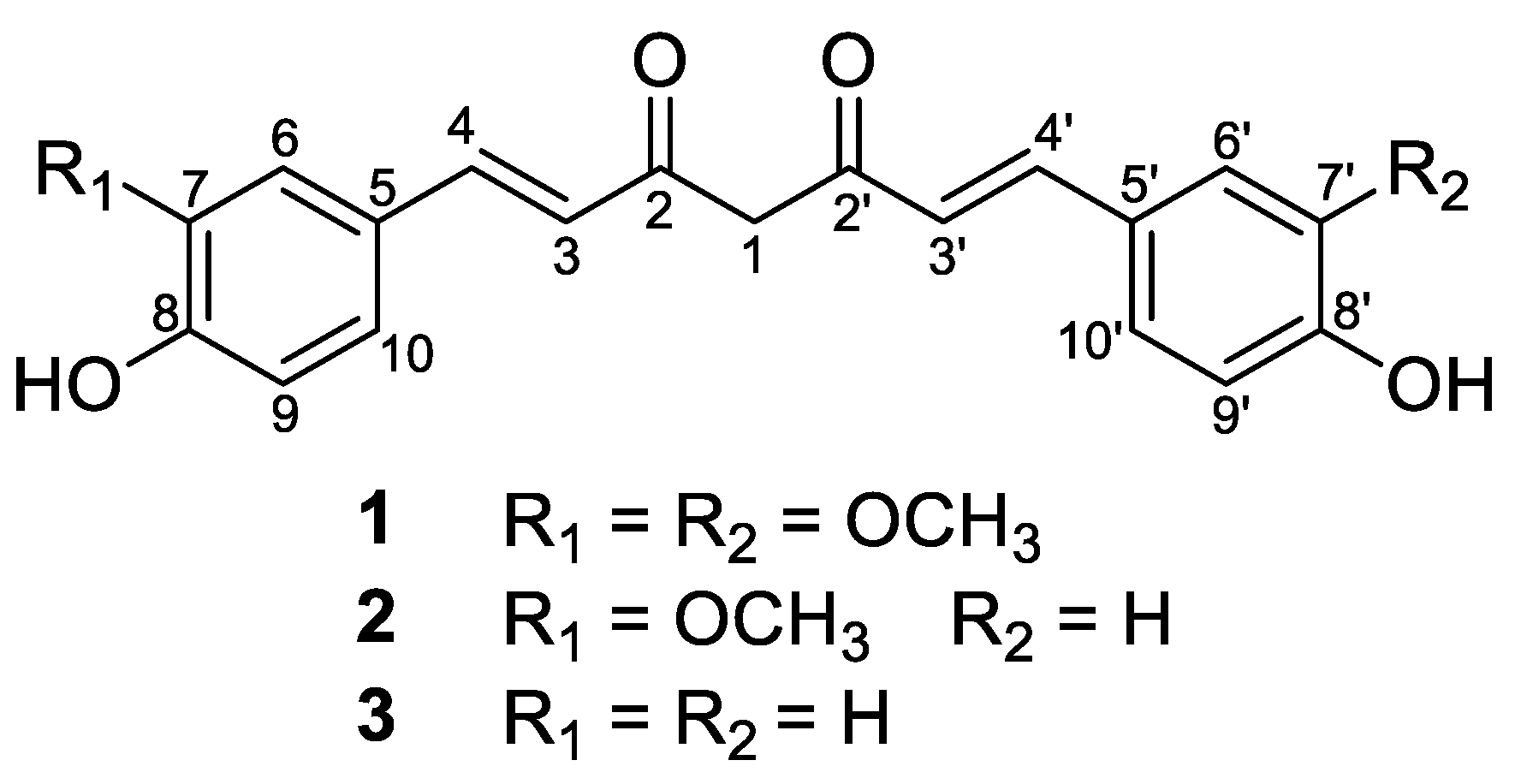
Figure 3.
The key HMBC correlations for curcumins 1–2 from C. longa.
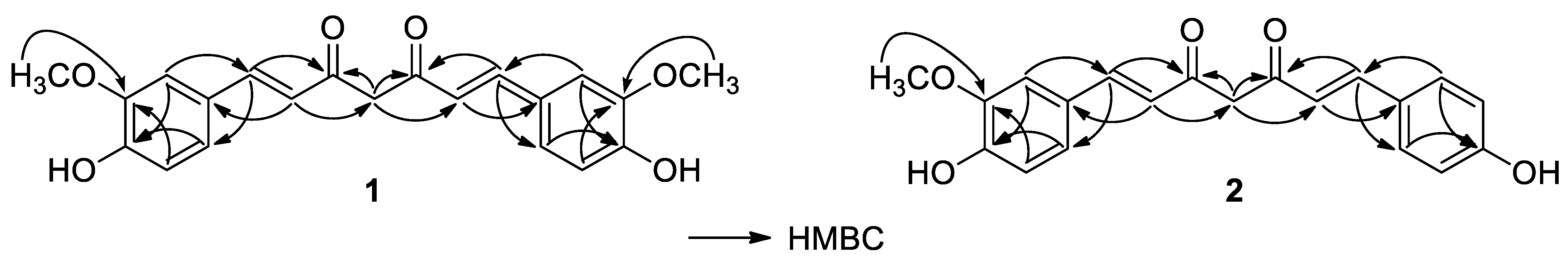
Figure 4.
DFT calculation for structural optimization of curcumins 1–3 using basis M062X/6-311+G(d,p).
Figure 4.
DFT calculation for structural optimization of curcumins 1–3 using basis M062X/6-311+G(d,p).
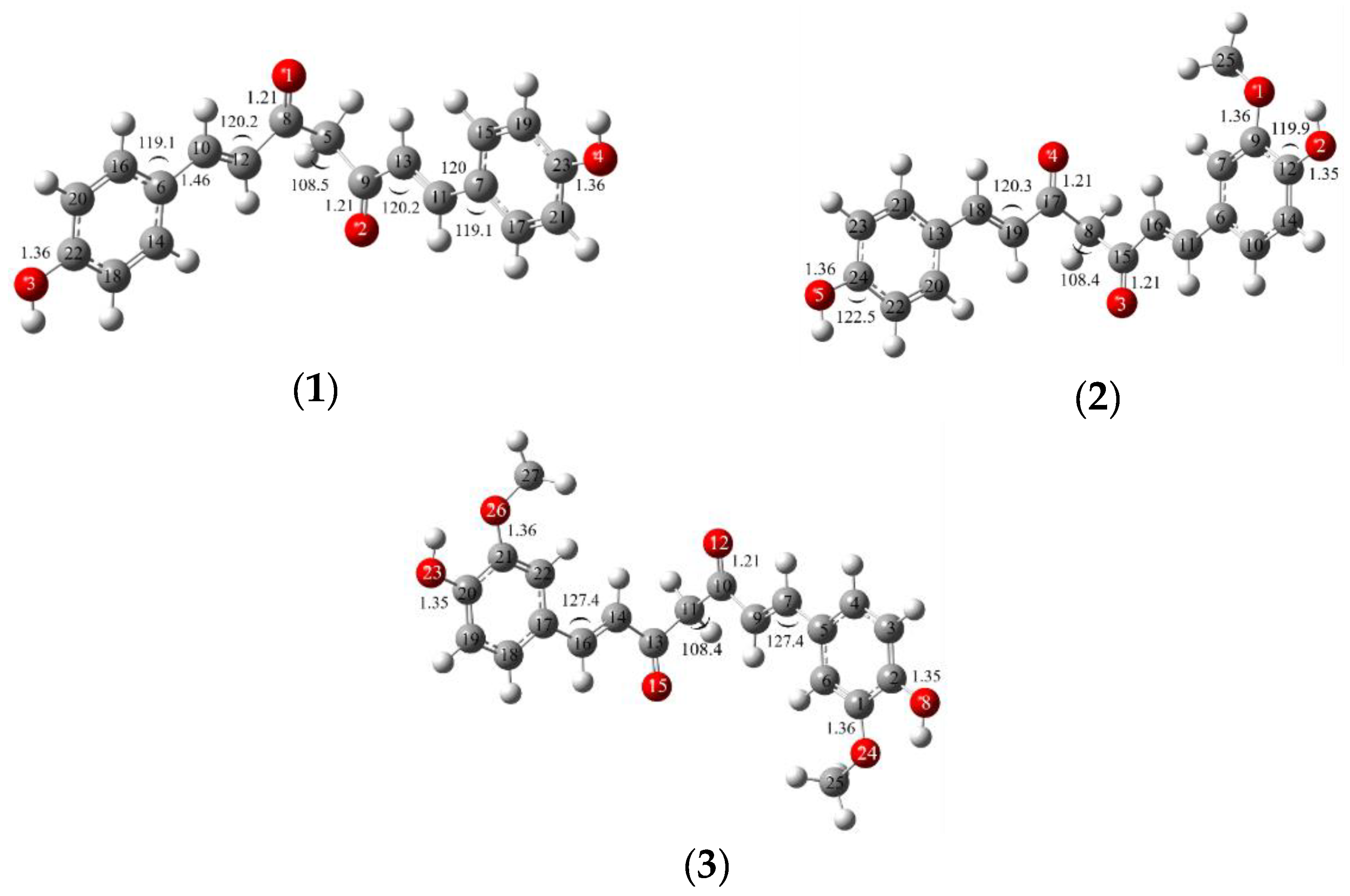
Figure 5.
HOMO and LUMO of curcumins 1–3 calculated by DFT using M062X/6-311+G(d,p). Red and green colours refer to the electron density deformation.
Figure 5.
HOMO and LUMO of curcumins 1–3 calculated by DFT using M062X/6-311+G(d,p). Red and green colours refer to the electron density deformation.
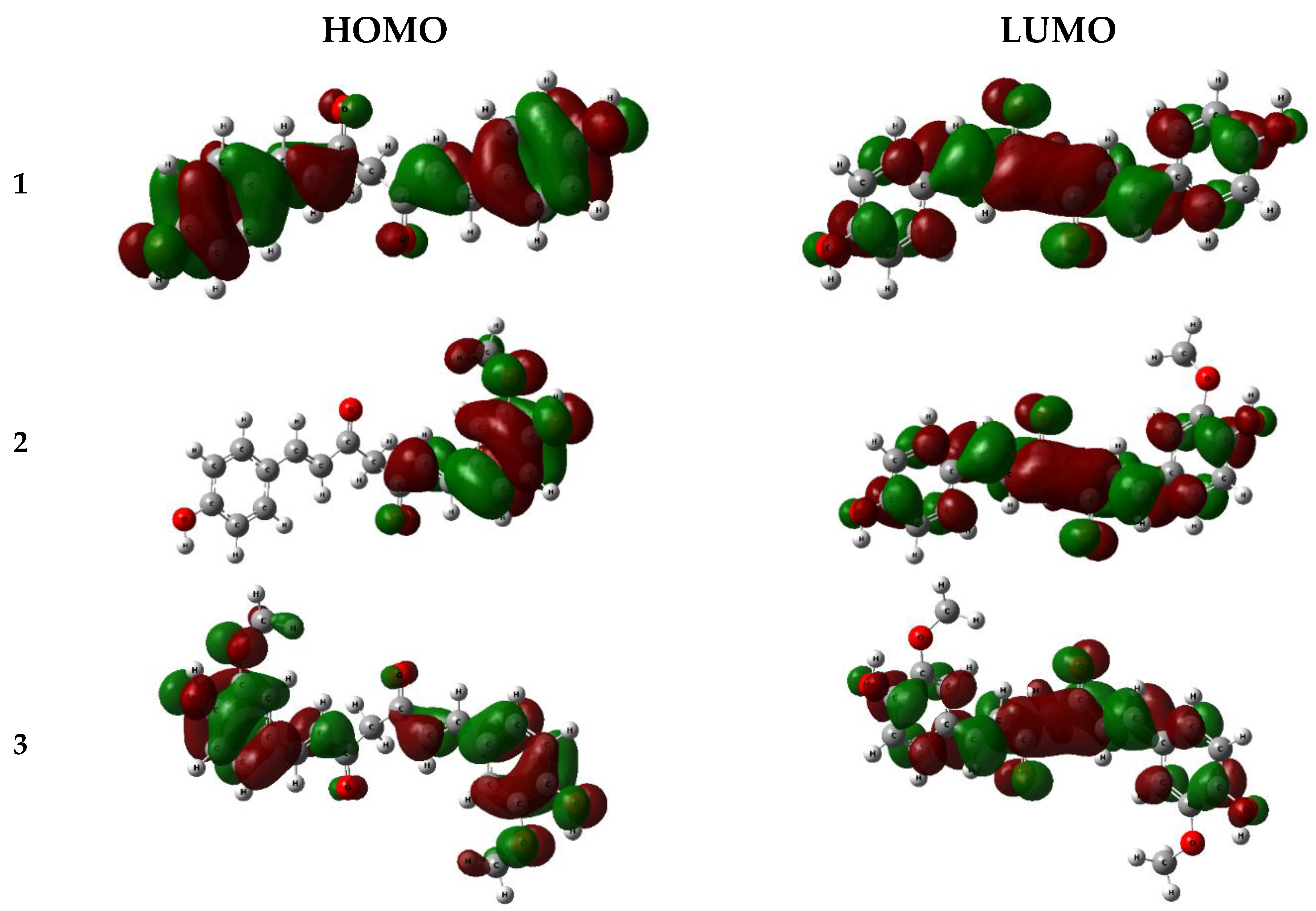
Figure 6.
(A) Swiss-Model of PTP1B; (B) Ramachandran plot analysis of the structure of PTP1B model; (C) An overlay of the ribbon diagrams of the PTP1B homology modeled structure (apricot) with three published crystal structure of PTP1B on Protein Data Bank (PDB entry: 1G7F-cyan; 1PXH-magenta; 4Y14-green, respectively). The images were generated using Chimera 1.16.
Figure 6.
(A) Swiss-Model of PTP1B; (B) Ramachandran plot analysis of the structure of PTP1B model; (C) An overlay of the ribbon diagrams of the PTP1B homology modeled structure (apricot) with three published crystal structure of PTP1B on Protein Data Bank (PDB entry: 1G7F-cyan; 1PXH-magenta; 4Y14-green, respectively). The images were generated using Chimera 1.16.
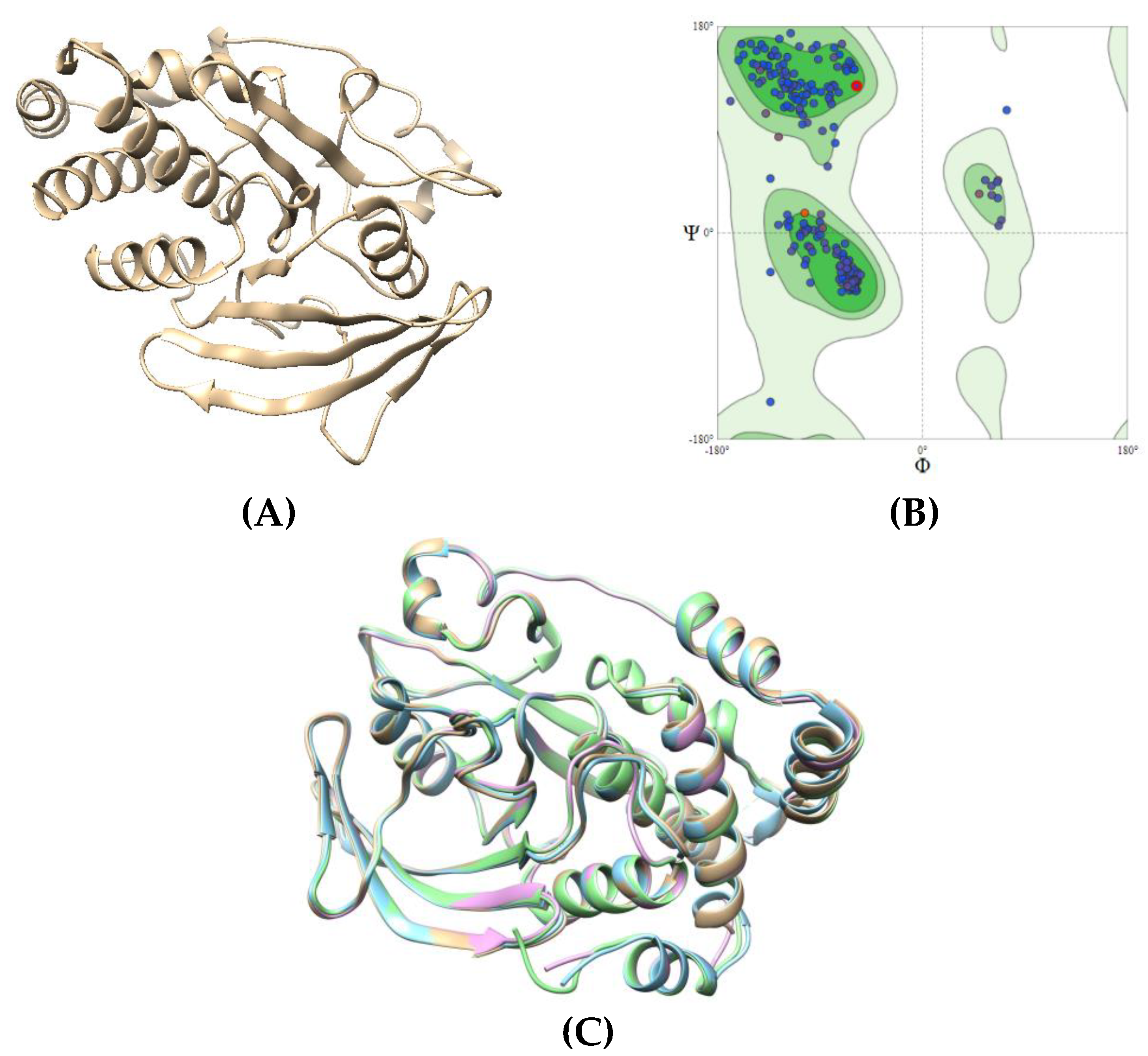
Figure 7.
Crystal structures of proteins 3W37, 3AJ7, and PTP1B with their binding sites by investigated curcumins: yellow (site 1), cyan (site 2), magenta (site 3), and blue (site 4).
Figure 7.
Crystal structures of proteins 3W37, 3AJ7, and PTP1B with their binding sites by investigated curcumins: yellow (site 1), cyan (site 2), magenta (site 3), and blue (site 4).
Figure 8.
Docking pose of compounds 1−3 in the most active binding site of protein targets, suggested by molecular docking studies. (A) Compound 1 dock with 3W37; (B) Compound 2 dock with 3W37; (C) Compound 3 dock with 3W37; (D) Compound 1 dock with 3AJ7; (E) Compound 2 dock with 3AJ7; (F) Compound 3 dock with 3AJ7; (G) Compound 1 dock with PTP1B; (H) Compound 2 dock with PTP1B; (I) Compound 3 dock with PTP1B.
Figure 8.
Docking pose of compounds 1−3 in the most active binding site of protein targets, suggested by molecular docking studies. (A) Compound 1 dock with 3W37; (B) Compound 2 dock with 3W37; (C) Compound 3 dock with 3W37; (D) Compound 1 dock with 3AJ7; (E) Compound 2 dock with 3AJ7; (F) Compound 3 dock with 3AJ7; (G) Compound 1 dock with PTP1B; (H) Compound 2 dock with PTP1B; (I) Compound 3 dock with PTP1B.
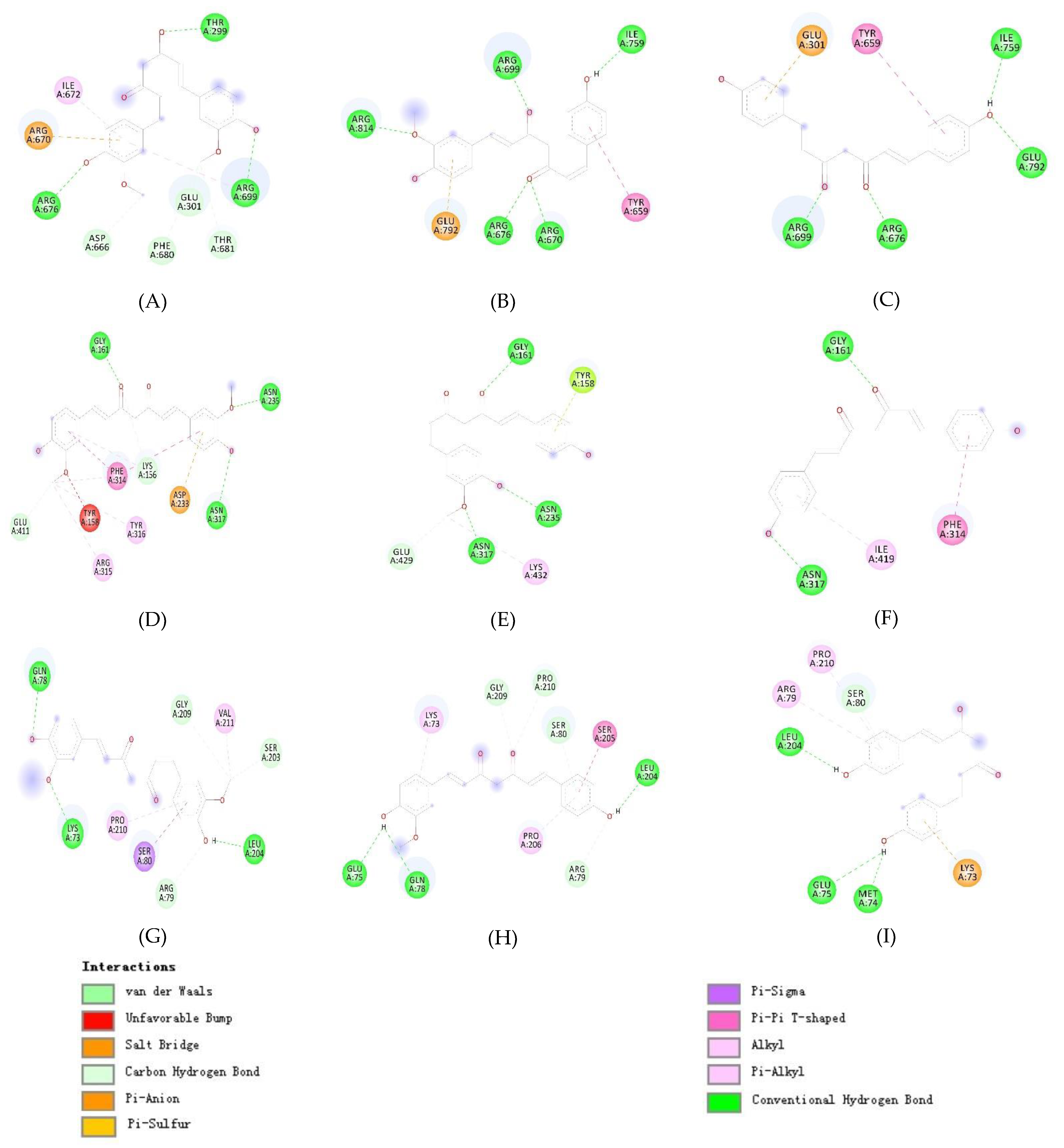
Table 1.
NMR spectroscopic data of compounds 1‒3 measured in acetone-d6 at 600 MHz (1H) and 150 MHz (13C).
Table 1.
NMR spectroscopic data of compounds 1‒3 measured in acetone-d6 at 600 MHz (1H) and 150 MHz (13C).
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 5.98 (2H, s) | 101.6 | 5.99 (2H, s) | 101.6 | 5.99 (2H, s) | 101.6 |
| 2 | 184.4 | 184.6 | 184.5 | |||
| 3 | 6.68 (1H, d, 16.2) | 122.1 | 6.69 (1H, d, 15.6) | 122.1 | 6.64 (1H, d, 15.6) | 121.7 |
| 4 | 7.58 (1H, d, 16.2) | 141.4 | 7.59 (1H, d, 15.6) | 141.4 | 7.58 (1H, d, 15.6) | 141.1 |
| 5 | 127.9 | 127.9 | 127.3 | |||
| 6 | 7.28 (1H, d, 1.8) | 111.6 | 7.29 (1H, d, 1.8) | 111.6 | 7.53 (1H, dd, 1.8, 7.8) | 130.8 |
| 7 | 148.9 | 148.9 | 6.88 (1H, dd, 1.8, 7.8) | 116.7 | ||
| 8 | 150.1 | 150.2 | 160.7 | |||
| 9 | 6.87 (1H, d, 7.8) | 116.3 | 6.86 (1H, d, 7.8) | 116.3 | 6.88 (1H, dd, 1.8, 7.8) | 116.7 |
| 10 | 7.14 (1H, dd, 1.8, 7.8) | 123.7 | 7.14 (1H, dd, 1.8, 7.8) | 123.8 | 7.53 (1H, dd, 1.8, 7.8) | 130.8 |
| 2′ | 184.4 | 184.4 | 184.5 | |||
| 3′ | 6.68 (1H, d, 16.2) | 122.1 | 6.64 (1H, d, 15.6) | 121.8 | 6.64 (1H, d, 15.6) | 121.7 |
| 4′ | 7.58 (1H, d, 16.2) | 141.4 | 7.57 (1H, d, 15.6) | 141.2 | 7.58 (1H, d, 15.6) | 141.1 |
| 5′ | 127.9 | 127.3 | 127.3 | |||
| 6′ | 7.28 (1H, d, 1.8) | 111.6 | 7.52 (1H, dd, 1.8, 6.6) | 130.9 | 7.53 (1H, dd, 1.8, 7.8) | 130.8 |
| 7′ | 148.9 | 6.87 (1H, dd, 1.8, 6.6) | 116.8 | 6.88 (1H, dd, 1.8, 7.8) | 116.7 | |
| 8′ | 150.1 | 160.8 | 160.7 | |||
| 9′ | 6.87 (1H, d, 7.8) | 116.3 | 6.87 (1H, dd, 1.8, 6.6) | 116.8 | 6.88 (1H, dd, 1.8, 7.8) | 116.7 |
| 10′ | 7.14 (1H, dd, 1.8, 7.8) | 123.7 | 7.52 (1H, dd, 1.8, 6.6) | 130.9 | 7.53 (1H, dd, 1.8, 7.8) | 130.8 |
| 7-OCH3 | 3.89 (3H, s) | 56.2 | 3.89 (3H, s) | 56.3 | ||
| 7′-OCH3 | 3.89 (3H, s) | 56.2 | ||||
Table 2.
The inhibitory activities of curcumins (1‒3) on PTP1B and α-glucosidase.
| Compounds | PTP1B | α-glucosidase |
|---|---|---|
| IC50, µM | IC50, µM | |
| Curcumin (1) | 37.8 ± 1.4 | 78.2 ± 0.2 |
| Demethoxycurcumin (2) | 45.3 ± 0.7 | 82.4 ± 0.8 |
| Bisdemethoxycurcumin (3) | 72.6 ± 1.1 | 90.6 ± 1.0 |
| Emodina | 7.6 ± 0.1 | - |
| Ursolic acida | 4.3 ± 0.4 | - |
| Acarboseb | - c | 147.2 ± 1.0 |
aThe compounds used as positive controls for PTP1B. bPositive controls for α-glucosidase. cNot detected.
Table 3.
Quantum chemical parameters of củcumins 1–3 calculated by Pop analysis at level M062X/6-311+G(d,p) including HOMO energy (EHOMO), LUMO energy (ELUMO), energy gap (DEGAP); ionization potential (I); electron affinity (A); electronegativity (c); chemical potential (m).
Table 3.
Quantum chemical parameters of củcumins 1–3 calculated by Pop analysis at level M062X/6-311+G(d,p) including HOMO energy (EHOMO), LUMO energy (ELUMO), energy gap (DEGAP); ionization potential (I); electron affinity (A); electronegativity (c); chemical potential (m).
| Compounds | EHOMO (eV) | ELUMO (eV) | ∆EGAP = ELUMO − EHOMO | I=− EHOMO | A = −ELUMO | χ=(I+A)/2 |
μ =−χ = −(∂E/∂N)v® |
|---|---|---|---|---|---|---|---|
| 1 | -7.298 | -1.581 | 5.717 | 7.298 | 1.581 | 4.439 | -4.439 |
| 2 | -7.298 | -1.574 | 5.724 | 7.298 | 1.574 | 4.436 | -4.436 |
| 3 | -7.525 | -1.563 | 5.962 | 7.525 | 1.563 | 4.544 | -4.544 |
Table 4.
Key residues of different binding site of 3W37, 3AJ7, and PTP1B proteins.
| Site | Residues of 3W37 | Residues of 3AJ7 | Residues of PTP1B |
|---|---|---|---|
| 1 | Tyr360, Phe367, Pro395, Ile396, Leu397, Ile454, Phe457, Arg458, Ile463, Ile466 | Asp69, Tyr72, His112, Lys156, Ser157, Tyr158, Phe159, Leu177, Phe178, Gln182, Arg213, Asp307, Thr310, Ser311, Pro312, Leu313, Phe314, Arg315, Tyr316, His351, Asp352, Gln353, Glu411, Ile440, Arg442, Arg446 | Arg24, Ala27, Ser28, Asp29, Phe30, Pro31, Cys32, Lys36, Asp48, Val49, Phe52, Ile219, Arg254, Arg257, Met258, Gly259, GLn262 |
| 2 | Glu301, Tyr659, Thr662, Leu663, Asp666, Arg670, Ile672, Arg676, Ile697, Gly698, Arg699, Gly700, Ile701, Ile754, Asn758, Ile759, Val760, Ala761, Thr790, Gly&91, Glu792 | Val369, Ile370, Lys373, Pro488, Asn489, Ser490, Asn493, Phe494, Glu497, Leu561, Glu562, Phe593, Gly564, Asn565, Tyr566, Pro567, Lys568, Val571 | Ala35, Lys36, Leu37, Pro38, Asn40, Lys41, Asn44, Arg45, Tyr46, Arg47, Asp48, Val49,Ser50 |
| 3 | Tyr319, Pro658, Tyr661, Gln763, Arg773, Phe777, Leu793, Phe794, Leu795, Asp796, Trp841 | Lys156, Ser157, Tyr158, Phe159, Gly160, Gly161, Asp233, Asn235, Ser236, Thr237, Trp238, Ser311, Leu313, Phe314, Asn317, Asn415, Ala418, Ile419, Glu422, His423, Glu428, Glu429, Lys432 | Leu71, Lys73, Met74, Glu75, Ala77, Gln78, Arg79, Ser80, Ser203, Leu204, Ser205, Pro206, His208, Gly209, Pro210, Val211, Leu233, Lys237 |
| 4 | Tys331, Arg332, Asp333, Ile358, Asp359,Tyr360, Met361, Asp362, Ala363, Phe364, Asp370, His373, Phe374, Arg629 | Lys, Trp15, Asn259, Ile262, Glu271, Ile272, Met273, Thr274, Tyr289, Thr290, Ser291, Ala292, Arg294, His295, Glu296, Leu297, Ser298, Asp341, Cys342, Trp343 | Lys73, Met74, Glu75, Glu76, Ala77, Thr230, Leu234, Lys248, Val249, Glu252, Lys255, Phe256 |
Table 6.
Interaction analysis between studied compounds (curcumins 1–3) and 3W37, 3AJ7 and PTP1B targeted proteins.
Table 6.
Interaction analysis between studied compounds (curcumins 1–3) and 3W37, 3AJ7 and PTP1B targeted proteins.
| Compounds | 3W37 | 3AJ7 | PTP1B | |||
|---|---|---|---|---|---|---|
| H-bond | Van der Waals interaction | H-bond | Van der Waals interaction | H-bond | Van der Waals interaction | |
| 1 | Thr299, Arg676, Arg699 | Glu301, Asp666, Arg670, Ile672, Phe680, Thr681 | Gly161, Asn235, Asn317 | Lys156, Tyr158, Asp233, Phe314, Arg315, Tyr316, Glu411 | Lys73, Gln78, Leu204 | Arg79, Ser80, Ser203, Gly209, Pro210, Val211 |
| 2 | Arg670, Arg676, Arg699, Ile759, Arg814 | Tyr659, Glu792 | Gly161, Asn235, Asn317 | Tyr158, Glu429, Lys432 | Glu75, Gln78, Leu204 | Lys73, Arg79, Ser80, Ser205, Pro206, Gly209, Pro210 |
| 3 | Arg676, Arg699, Ile759, Glu792 | Glu301, Tyr659 | Gly161, Asn317 | Phe314, Ile419 | Met74, Glu75, Leu204 | Lys73, Arg79, Ser80, Pro210 |
Table 8.
Detail grid box parameters for each binding site of targeted proteins.
| Site | 3W37 | 3AJ7 | PTP1B | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Grid box center | |||||||||
| x | y | z | x | y | z | x | y | z | |
| 1 | -2.783 | -20.139 | -14.192 | 16.920 | -10.869 | 19.269 | -27.669 | 40.981 | -8.880 |
| 2 | 16.542 | -28.867 | -41.997 | 28.176 | 16.748 | -0.427 | -23.809 | 30.047 | -16.569 |
| 3 | 8.457 | -41.335 | -37.723 | 15.262 | -21.012 | 13.686 | -34.929 | 32.542 | 9.157 |
| 4 | -13.147 | -11.244 | -19.031 | 12.365 | 10.215 | 30.416 | -34.929 | 42.799 | 5.639 |
| Grid box size | |||||||||
| x | y | Z | x | y | Z | x | y | Z | |
| 1 | 50 | 55 | 50 | 74 | 64 | 70 | 66 | 55 | 50 |
| 2 | 68 | 52 | 70 | 50 | 60 | 55 | 60 | 40 | 40 |
| 3 | 55 | 55 | 55 | 60 | 60 | 60 | 60 | 68 | 55 |
| 4 | 55 | 50 | 65 | 60 | 60 | 60 | 55 | 50 | 50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



