
Submitted:
18 April 2023
Posted:
19 April 2023
You are already at the latest version
Abstract
The purpose of this study is to develop and evaluate a self-microemulsifying drug delivery system (SMEDDS) to improve the oral absorption of poorly water-soluble Olaparib. Through the solubility test of Olaparib in various oils, surfactants and co-surfactants, pharmaceutical excipients for use in SMEDDS manufacturing were selected. Self-emulsifying regions were identified by mixing the selected materials at various ratios, and a pseudoternary phase diagram was constructed by synthesizing these results. Various physicochemical properties of microemulsion incorporating Olaparib were confirmed by investigating the particle morphology, particle size, zeta potential, drug content and stability. In addition, the improved dissolution and oral absorption of Olaparib were also confirmed through a dissolution test and a pharmacokinetic study. An optimal microemulsion was generated in the formulation of Capmul MCM EP/NF 10%, Labrasol 80% and PEG 400 10%. Fabricated nano-sized microemulsions were well-dispersed in aqueous solutions, and it was also confirmed that it was maintained well without any problems of either physical or chemical stability. The 5 min initial dissolution of Olaparib in fabricated formulations was improved by 10.6 times (pH1.2), 14.8 times (pH6.8) and 14.8 times (distilled water) compared to the values of Olaparib powder. The dissolution at 120min was 45.4±4.0%(pH1.2), 53.5±1.1% (pH6.8) and 58.1±1.00% (distilled water) in Olaparib powder but has been significantly improved by 1.7 times (74.8±1.9%), 1.6 times (84.2±0.3%) and 1.5 times (81.2±0.7%) in SMEDDS. Associated with the high dissolutions of Olaparib in fabricated microemulsions, the pharmacokinetic parameters were also greatly improved. The Cmax and AUCinf of Olaparib in the prepared microemulsion were increased 4.2- and 3.2-fold, respectively, compared to powder. Taken together with the results mentioned above, the microemulsion system could be an effective tool as an oral delivery formulation for Olaparib. Additionally, our results demonstrate that the SMEDDS systems comprising Capmul MCM, Labrasol and PEG 400 could be a useful option for improvement of several other poorly water-soluble drugs with physico-chemical properties similar to Olaparib.
Keywords:
Olaparib
; self-microemulsifying drug delivery system
; microemulsion
; solubility
; dissolution
; oral absorption
1. Introduction
The Biopharmaceutics Classification System (BCS) classifies drugs into 4 groups based on the solubility and gastro-intestinal membrane permeability in aqueous [1,2]. Among these, the drugs with low solubility but high permeability are classified as Class II, while other drugs with low solubility and permeability are classified as Class IV [1,2]. About 40% of currently marketed drugs and about 70% of new drug candidates under development are known as poorly water-soluble drugs corresponding to Classes II and IV [3,4,5]. In general, poorly water-soluble drugs have low oral absorption and low bioavailability due to their low solubility, which results in increased administered dose and reduced patient compliance [5,6]. Therefore, how to improve the solubility of poorly water-soluble drugs is an important challenge in oral dosage development [7,8].
In an effort to improve the oral absorption of poorly soluble drugs, various solubilization technologies, including solid dispersion, inclusion complex, salt formation, particle size reduction, co-solvent/co-solvency and lipid-based formulations have been studied [9,10,11,12]. Of these, the lipid-based formulation is pre-dissolving drugs in lipid excipients, so it can effectively avoid the dissolution step and a potentially rate limiting dissolution step in the GI tract, and eventually has the advantage of improving bioavailability [13,14]. Liposome, solid lipid nano-carrier, nano-structured lipid carrier, emulsion and self-emulsifying drug delivery system (SMEDDS) correspond to this lipid-based formulation [15,16,17,18]. SMEDDS is an isotropic mixture of oil, surfactant and co-surfactant, and is a pharmaceutical formulation in which nano-sized emulsions are generated through spontaneous emulsification in the GI tract [19]. SMEDDS has the advantage of increased drug solubility and rapid oral absorption of the drug by the action of fine emulsion droplets as its components, such as oil and surfactants, play a role of solubilizer [19,20]. In addition, solubilizers frequently used in SMEDDS, such as Labrasol and Tween 80, act as effective intestinal permeation enhancers and are known to be effective in increasing oral absorption of Class IV drugs [21,22]. In general, since SMEDDS is prepared in a solid dosage form, such as a gelatin capsule, the improved patient compliance is high and both manufacture and scale-up are easy, making it advantageous for commercial production [19,23]. Owing to these advantages, SMEDDS formulations have been studied to improve the in vivo bioavailability of poorly water-soluble drugs such as fenofibrate, cyclosporine and paclitaxel [24,25]. Moreover, SMEDDS has been applied to cyclosporine, ritonavir and saquinavir, and has been developed and sold as commercial products of Neoral® (Norvatis), Norvir® (AbbVie) and Fortovase® (Roche) [26].
Olaparib (OLA), a poly ADP-ribose polymerase inhibitor (PARP inhibitor), has been known to have therapeutic effects for curing cancers associated with impaired DNA repair capability, especially those with deficiencies in homologous recombination repair pathway [27]. OLA, a potent cytotoxic anticancer drug, is administrated for treatments of patients with advanced, recurrent ovarian cancer who have mutations of breast cancer BRCA1 or breast cancer BRCA2 [28].
Despite the high therapeutic efficacies of OLA, OLA shows low oral bioavailability due to its low solubility and low permeability, which leads to increased administrated dose and frequency [29,30]. The daily dose of Lynparza® capsules is 800 mg, so it is obliged to endure the inconvenience of taking 16 capsules containing 50 mg OLA every day [31,32]. This high-dose administration reduces patient compliance and triggers undesirable side effects such as hematological toxicity, nausea, anemia, vomiting and fatigue [30,33,34]. Therefore, there is a need for a pharmaceutical formulation that can ultimately improve patient compliance and side effects by increasing oral absorption of OLA [30].
In this research, we aimed to prepare a SMEDDS system containing OLA for improving solubility and to evaluate the obtained formulations in vitro and in vivo. Based on results of the solubility test and the pseudoternary phase diagram, the optimal SMEDDS formulations were selected and evaluated on various physico-chemical properties, such as morphologies, particle distributions, zeta potentials, dissolution profiles, stability and in vivo pharmacokinetic profiles. In addition, the optimal SMEDDS formulation was compared with Lynparza® (commercialized product), because our research is the first application of a microemulsion system to OLA.
2. Materials and Methods
2.1. Materials
OLA was received by ScinoPharm Taiwan Ltd (Shan-Hua, Taiwan). Labrafil M 1944 CS, Labrafac PG, Labrasol, Transcutol HP and Plurol Oleique CC 497 were supplied by Gattefosse (St. Priest Cedextran, France). Cotton seed oil, Span 80, isopropyl myristate and PEG 400 were obtained from Daejung Co. Ltd (Siheung, Korea). oleic acid and Tween 80 were purchased from Duksan Co. Ltd (Ansan, Korea). Mineral oil, Capmul MCM EP/NF and Kolliphor EL (Cremophor EL) were obtained from Samchun Chemicals Co. (Pyeongtaek, Korea), ABITEC Corp. (Columbus, USA) and BASF (Ludwigshafen, Germany), respectively. All chemicals and solvents used were of reagent or HPLC grade.
2.2. Solubility Test
The solubilities of OLA were quantitatively analyzed in various aqueous solutions (distilled water, pH1.2 and pH6.8 solution), oils (Capmul MCM EP/NF, Cotton seed oil, Mineral oil, Labrafil M 1944 CS, Labrafac PG and oleic acid) and surfactants (Kolliphor EL, Tween 80, Span 80, Labrasol, Transcutol HP, Plurol Oleique CC 497, isopropyl myristate and PEG 400). An excess amount of OLA was poured into each microtube containing 1.0mL of aqueous solution, oil or surfactant. Using a DAIHAN RT-10 rotary mixer (Seoul, Korea), each of the blends of materials were shaken for 72 hours at room temperature. Then, the tubes were centrifuged at 10,000 rpm for 15 min using a DAIHAN CF-10 microcentrifuge (Seoul, Korea) to separate undissolved OLA. The obtained supernatants were filtered using a 0.45 μm nylon membrane and quantitatively evaluated by a high performance liquid chromatography system. Quantitative analysis of OLA in each filtered solution was conducted using a Hitachi Chromaster® HPLC system (Tokyo, Japan) equipped with UV detector and LB-SCIENCE Supersil 120 ODS II C18 column (4.6×250 mm, 5μm; Dalian, China). The mobile phase comprised a mixture solution (acetonitrile: distilled water, 50:50 (v/v)). The flow rate of the mobile phase solution was 1.0mL/min, and the column temperature was maintained at 30℃. Detection UV wavelength was set at 200nm.
2.3. Self-emulsification and Pseudoternary Phase diagram
Considering the solubility results of OLA in the solubility test, Capmul MCM EP/NF, Labrasol and PEG 400 were selected as oil, surfactant and co-surfactant, respectively. To identify the self-emulsifying region of SMEDDS, the self-emulsification of a variety of compositions was meticulously observed. We added 50 μL of a mixture of Capmul MCM EP/NF, Labrasol, and PEG 400 to 50 mL distilled water, gently mixed them using a magnetic stirrer, and observed its self-emulsification with naked eyes. If we obtained a nearly transparent homogenous emulsion without phase separation, it was judged that the microemulsion was successful. On the contrary, if we observed a turbid mixture or no emulsification progress, it was judged that the microemulsion ended up as a failure. Based on the obtained self-emulsification observations, a pseudoternary phase diagram was constructed.
2.4. Preparation of SMEDDS
The SMEDDS formulations were selected based on the identification of a self-emulsifying region in the pseudoternary phase diagram. The formulations were prepared by mixing a certain amount of OLA, oil, surfactant and co-surfactant. 100mg of OLA was dissolved in a 3mL mixture solution of Capmul MCM EP/NF, Labrasol and PEG 400, and mildly stirred until the mixture was clear. The obtained solutions were stored at room temperature until physico-chemical characterizations.
2.5. Morphological analysis
The morphologies of fine emulsion droplets of the prepared SMEDDS formulation were observed using a Philips CM120 Transmission Electron Microscope (Eindhoven, Netherlands). After dilution of the sample solution with distilled water, the samples were deposited on the carbon-coated grid and dried at room temperature. Then the dried samples were negatively stained with 1% (w/v) uranyl acetate for about 10 mins prior to observation. The pre-treated samples were observed at 34,000 x (0.32nm/pixel) and 100kV acceleration voltage using an UltraScan 1000CCD camera (Gatan, USA).
2.6. Droplet size and zeta potential analysis
The droplet size distribution and zeta potential of the prepared microemulsions were evaluated by an Otsuka ELSZ-100 particle size analyzer (Tokyo, Japan). 5μL of the prepared microemulsion is added to 50 mL of distilled water and the diluted sample was analyzed at room temperature (n=3).
2.7. Stability test
To evaluate the storage stability of the SMEDDS formulation, samples were tightly sealed in 5 mL glass vials. According to ICH guideline (Q1), the samples were stored for 3 months in a long-term storage condition (25±2℃, 60±5%RH) and an accelerated storage condition (40±2℃, 75±5%RH). A storage stability test was conducted on the physical application, emulsification, droplet distribution, zeta potential and OLA contents at 1 and 3 months from the initial time. Each sample was evaluated three times and data were processed.
2.8. In vitro dissolution test
An in vitro dissolution study of OLA was conducted using a Kukje KDT-600 dissolution tester (Goyang, Korea) under USP apparatus II (paddle method). The dissolution media were HCl buffer (pH1.2), phosphate buffer (pH6.8) and distilled water. The paddles were rotated at 100 rpm and the temperature was 37℃. The SMEDDS system (equivalent of OLA 10mg) was filled into capsules (hard gelatin) prior to the dissolution test, and compared with OLA powder. During the dissolution test, each dissolution medium (3mL) was taken at predetermined times (5, 10, 15, 30, 45, 60, 90 and 120min) and filtered through a 0.45μm syringe filter. The obtained aliquots were quantitatively analyzed using the HPLC system as described in section 2.2. After taking dissolution samples, an equal volume of dissolution medium was replaced at each time point and all experiments were conducted three times. The statistical significances between the results of OLA powder and SMEDDS formulation were confirmed by the student's t-test method using Jandel scientific Sigmaplot Ver 12.0 (CA, USA).
2.9. In vivo pharmacokinetic study
To confirm the improvement of oral absorption of OLA contained in the SMEDDS formulation, a rat pharmacokinetic study of OLA powder, SMEDDS formulation and Lynparza® tablet was conducted. The rats used in this experiment were fasted overnight, but water was freely consumed. Blood samples were taken from the jugular vein using a 1.0mL syringe treated with heparin at predetermined times (0, 30, 60, 120, 240, 480 and 1440 mins) after oral administration of each formulation. The blood samples were centrifuged at 7000rpm and 4°C for 10mins, and the obtained plasma was stored at -80°C until quantitive analysis.
A 50 μL of plasma samples was added to 150 μL of an internal standard solution (carbamazepine 200 ng/ml in acetonitrile), then mixed on a vortex mixer for three min. After centrifugation for 10 min at 13,000 rpm at 4°C, the supernatants were transferred into new LC vials and analyzed by injecting 5 μL of the supernatants into an LC-MS/MS system. The autosampler was set at 10°C during analysis. The chromatographic separation of OLA was conducted onto a Gemini-NX C18 column (50 × 2.0 mm, 3μ particle size, 110 Å; Phenomenex Inc., Torrance, CA, USA) using an isocratic elution condition of acetonitrile and 10 mM ammonium formate buffer (80:20, v/v) at a flow rate of 0.3 ml/min and a column oven temperature of 40°C using Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA). The method of mass spectrometry known as multiple reaction monitoring (MRM) was utilized for analytical procedure, and the following parameters were used: an ion-spray voltage of +5500 V at 550°C; a decluttering potential of 111 V; an entrance potential of 11 V; a collision energy of 37 V; and a mass transition m/z of 435.1>281.3 for OLA.
The following pharmacokinetic parameters were calculated using the WinNonlin program ver 5.0 (CA, USA): the time to reach the maximum plasma concentration (Tmax), the maximum plasma concentration (Cmax); the area under the plasma concentration-time curve to the last time point or infinity time (AUClast or AUCinf,); and the half-life (T1/2). Additionally, the relative oral bioavailability (BA) was calculated by dividing the AUC for the SMEDDS formulation or the Lynparza® tablet by that of OLA. All data were expressed means ±S.D. The student’s t-test was conducted to determine statistically significant differences (p<0.05) among the tested groups.
3. Results and Discussion
3.1. Solubility test of OLA
Solubility tests of OLA in various aqueous solutions, oils and surfactants were performed (Table 1). The aqueous solubility of OLA was 0.073±0.25mg/ml in distilled water, 0.063±0.57mg/ml at pH1.2 and 0.059±0.42mg/ml at pH6.8, consistently very low regardless of pH. These results show that, even after oral administration of OLA with a daily dose of 800 mg, most of the OLA is not dissolved in the GI tract and is precipitated in an insoluble state, which may lead to very low oral absorption. In general, it is known that the lower the solubility of a drug, the lower the bioavailability, which increases the administered dose as well as side effects [30].
Improving the drug solubility using an appropriate solubilizer may increase oral absorption, thereby decreasing the administrated dose. As a result, this study conducted a screening test of excipients frequently used in the pharmaceutical field to search for substances that can improve the solubility of OLA. Among various oils, the solubility of Capmul MCM EP/NF was about 574 times that of distilled water (41.68±0.02mg/ml vs 0.07±0.25mg/ml). In the case of surfactants, the solubilities of PEG 400 and Labrasol showed 47.66 ± 0.38 mg/ml and 36.60 ± 0.07 mg/ml, respectively, which were improvements of about 657.3 times and 504.8 times, respectively, compared to distilled water. Through the excipient solubility screening test described above, Capmul MCM EP/NF, PEG 400 and Labrasol were selected as oil, surfactant and co-surfactant to be used in the production of OLA SMEDDS.
3.2. Construction of a pseudoternary phase diagram
A self-emulsification assessment was performed using the selected oil (Capmul MCM EP/NF) and surfactant (PEG 400 or Labrasol). Various SMEDDS mixtures were prepared by diversifying the ratio of the selected substances to proceed with various evaluations. By constructing a pseudoternary phase diagram based on the obtained results, self-emulsifying regions could be identified. The region shown on Figure 1 represents a stable self-emulsifying region, either clear or slightly bluish. When the oil ratio was 5-30%, an excellent microemulsion was observed.
It was confirmed that the higher the oil ratio, the larger the droplet size of the emulsion. When checking the pseudoternary phase diagram, the higher ratio of Labrasol or PEG 400 that was used as surfactant rather than Capmul MCM EP/NF, the better the emulsions were generated. This phenomenon is mentioned in several literatures, revealing the higher proportion of surfactant and co-surfactant increases, the more stabilized and condensed interface of the emulsion [35]. Based on the pseudoternary phase diagram, Capmul MCM EP/NF 10%, Labrasol 80%, PEG 400 10% was selected as the optimal SMEDDS production ratio of OLA. Considering the solubility of OLA in each substance, a SMEDDS formulation containing 100 mg of OLA in a mixture of Capmul MCM EP/NF 0.3mL, Labrasol 2.4mL and PEG 400 0.3mL was obtained. Using this resultant formulation, several characterizations were investigated.
3.3. Characterization of SMEDDS
The morphology of prepared SMEDDS was observed using TEM. Figure 2 shows that the microemulsion droplets are well dispersed. The mean droplet size was 141.1±0.2 nm, and the polydispersity index was very small, 0.19. In general, having a PDI value of 0.2 or less in a dispersed system means that the particle size distribution is quite narrow [36]. The zeta potential was measured at -15.8±1.1 mV, and it has been reported that nano-sized dispersed systems with a zeta potential lower than 10 mV can maintain physical stability and improve drug efficacy by improving in vivo absorption [37]. Judging from the obtained test results and various literatures, it was expected that the manufactured OLA SMEDDS system could show high physical stability and oral absorption of OLA because it had appropriate physical properties.
3.4. Stability study of SMEDDS
To evaluate the stability of the obtained SMEDDS formulation, the physico-chemical properties were checked while storing the fabricated samples under the long-term stability test condition (25±2℃, 60±5% RH) and accelerated stability test condition (40±2℃, 75±5% RH) (Table 2). During the storage period, the formulation’s physical stability was confirmed as it maintained its clear liquid state without drug precipitation and phase separation. Also, when the SMEDDS formulation was dispersed in distilled water, self-emulsification was also confirmed to have maintained its initial state. For 3 months, the droplet size increased from 141.1±0.2 nm to 166.1±2.7 nm, and the zeta potential slightly increased from -13.5±0.8 mV to -18.8±0.4 mV, but the nano-sized dispersion still remained stable. Drug contents were 101.0±0.2% and 98.5±0.2% in long term storage and accelerated storage, respectively, not significantly different from the initial value (101.1±0.5%). Because the SMEDDS system does not contain water in the pharmaceutical formulation, it is generally known to have relatively high stability compared to other liquid-type dosage formulations [23]. Taken together, these results show that self-emulsification behavior was maintained and the physical properties of the SMEDDS formulation did not change significantly.
3.5. In vitro dissolution study
In vitro dissolution profiles of OLA from SMEDDS formulation were monitored and compared with OLA powder under different pH conditions (pH1.2, pH6.8 and distilled water) for 2 hours (Figure 3). At pH 1.2 solution, dissolutions of OLA powder were only 6.09±0.27% dissolution within 5 min and 45.39±3.97% at 120 min. On the other hand, that of OLA in SMEDDS formulation was 64.38±0.48% within 5 min, showing a very rapid dissolution profile, followed by a final dissolution of 74.81 ± 1.87% at 120 min. Similar results to those at pH 1.2 were also obtained at pH 6.8 and distilled water. In pH 6.8 and distilled water, the dissolution values of OLA in the SMEDDS system were 84.20±0.31% and 67.93±0.64%, respectively, for the initial five minutes. In contrast, those of OLA powders were very low at 5.70±0.59% and 4.58±0.14%, respectively. According to the solubility results, the solubility of OLA is independent of pH conditions. Therefore, it is estimated that dissolution profiles in pH1.2, pH6.8 and distilled water are all similar. The dissolutions of OLA contained in SMEDDS at initial 5 min were improved 10.57 times (pH1.2), 14.78 times (pH6.8) and 14.83 times (distilled water) compared to OLA powder, and the final dissolution rates (at 120 min) were improved 1.65 times (pH1.2), 1.57 times (pH6.8) and 1.52 times (distilled water). The improved dissolution rates of drugs contained in SMEDDS formulations has been reported in several documents. These results showed that the improved dissolutions of the SMEDDS formulation containing OLA were because of increased specific surface area of nano-sized microemulsion droplets and solubilization effects of optimal oils and surfactants. As such, the SMEDDS formulation showed an improved dissolution profile compared to the OLA powder, which would lead to enhanced oral adsorption of OLA.
3.6. In vivo pharmacokinetic study
In vivo pharmacokinetic behavior of OLA with SMEDDS, marketed product (Lynparza® tablet) and OLA powder was investigated in rats. The mean plasma concentration profiles of OLA were plotted as a function of time, as presented in Figure 4. The mean pharmacokinetic parameters of OLA absorption are summarized in Table 3. Cmax and AUClast of OLA powder were 83.1±49.6ng/mL and 384.1±136.2ng⦁hr/mL, respectively. Cmax and AUClast of the Lynparza® tablet were 335.7±100.2ng/mL and 518.2±154.9ng⦁hr/mL, respectively. On the other hand, Cmax and AUClast following SMEDDS administration were 351.2±173.3ng/mL and 1242.7±420.3ng⦁hr/mL, a significant increase by 4.2- and 3.2-, respectively compared to OLA powder. When compared with the Lynparza® tablet, Cmax was similar, but AUClast was significantly increased by 2.4-fold. AUCinf of OLA powder and the Lynparza® tablet were 634.0±437.2ng⦁hr/mL and 522.0±155.9ng⦁hr/mL, respectively. AUCinf of SMEDDS was 1427.3±460.3ng⦁hr/mL, which was increased by 2.25- from OLA powder (634.0±437.2ng⦁hr/mL) and by 2.73-fold from the Lynparza® tablet (522.0±155.9ng⦁hr/mL), respectively. Tmax of SMEDDS was slightly delayed compared to the Lynparza® tablet, but T1/2 showed was no significant difference among formulations.
In most cases, the toxicity of anticancer drugs increases in proportion to the administered dose [38]. In particular, it is known that high-dose administration of OLA can cause side effects such as gastrointestinal toxicities, fatigue and anemia, and therefore, dose reduction through formulation improvement can reduce toxicity [39]. As a result, when analyzing the results of the pharmacokinetic study conducted in this study comprehensively, the SMEDDS formulation is expected to significantly reduce the dosage and toxicity compared to the marketed formulation.
4. Conclusions
The optimized SMEDDS containing OLA was developed using Capmul MCM EP/NF, Labrasol and PEG 400. This formulation system formed a stable microemulsion system at least for 3 months under long-term storage (25±2℃, 60±5%RH) and accelerated storage (40±2℃, 75±5%RH). In addition, it showed higher dissolution patterns than OLA powder and even the commercial formulation. In vivo pharmacokinetic results also showed improvement of OLA oral absorption. In conclusion, this system suggests the potential use of SMEDDS for the oral administration of OLA or other hydrophobic drugs.
Author Contributions
Conceptualization, D.J. Jang, H.G. Choi, Y.H. Kim and S.B. Kim; Methodology, S.H. Choi and T.T.L. N; Software, S.H. Ahn and K.S. Moon; Validation, K.H. Cho and T.Y. Sim; Formal Analysis, S.H. Choi, T.T.L. N and K.S. Moon; Investigation, S.H. Choi and Y.H. Kim; Data Curation, S.H. Choi and Y.H. Kim; Writing, S.H. Choi, D.J. Jang and Y.H. Kim; Writing–Original Draft Preparation, S.H. Choi, D.J. Jang and Y.H. Kim; Writing–Review & Editing, S.B. Kim, S.T. Kim, H.G. Choi and D.J. Jang; Supervision, H.G. Choi and D.J. Jang; Project Administration, S.B. Kim and D.J. Jang; Funding Acquisition, D.J. Jang.
Funding
This research was supported by 2022 Research Grant from Kangwon National University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Amidon, G. L. Lennernäs, H., Shah, V. P., & Crison, J. R. (1995). A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharmaceutical research, 12(3), 413-420.
- Dahan, A.; Miller, J.M.; Amidon, G.L. Prediction of Solubility and Permeability Class Membership: Provisional BCS Classification of the World’s Top Oral Drugs. AAPS J. 2009, 11, 740–746. [Google Scholar] [CrossRef]
- Karashima, M.; Kimoto, K.; Yamamoto, K.; Kojima, T.; Ikeda, Y. A novel solubilization technique for poorly soluble drugs through the integration of nanocrystal and cocrystal technologies. Eur. J. Pharm. Biopharm. 2016, 107, 142–150. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.T.; Kim, H.; Cho, J.M.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.Y.K.; Ibisogly, A.; Bauer-Brandl, A. Solubility enhancement of BCS Class II drug by solid phospholipid dispersions: Spray drying versus freeze-drying. Int. J. Pharm. 2015, 496, 382–391. [Google Scholar] [CrossRef]
- Savjani, K. T. , Gajjar, A. K., & Savjani, J. K. (2012). Drug solubility: importance and enhancement techniques. International Scholarly Research Notices. 2012. [Google Scholar]
- Chaudhary, A. , Nagaich, U., Gulati, N., Sharma, V. K., Khosa, R. L., & Partapur, M. U. (2012). Enhancement of solubilization and bioavailability of poorly soluble drugs by physical and chemical modifications: A recent review. J Adv Pharm Educ Res, 2(1), 32-67.
- Sikarra, D. , Shukla, V., Kharia, A. A., & Chatterjee, D. P. (2012). Techniques for solubility enhancement of poorly soluble drugs: an overview. JMPAS, 1, 1-22. 1.
- Singh, A.; Worku, Z.A.; Mooter, G.V.D.; Bpharm; Mpharm; D, P. Oral formulation strategies to improve solubility of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1361–1378. [Google Scholar] [CrossRef]
- Fahr, A.; Liu, X.; D, P. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 2007, 4, 403–416. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef]
- Jannin, V.; Chevrier, S.; Michenaud, M.; Dumont, C.; Belotti, S.; Chavant, Y.; Demarne, F. Development of self emulsifying lipid formulations of BCS class II drugs with low to medium lipophilicity. Int. J. Pharm. 2015, 495, 385–392. [Google Scholar] [CrossRef]
- Kalepu, S. , Manthina, M., & Padavala, V. (2013). Oral lipid-based drug delivery systems–an overview. Acta Pharmaceutica Sinica B, 3(6), 361-372.
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128. [Google Scholar] [CrossRef]
- Göke, K.; Lorenz, T.; Repanas, A.; Schneider, F.; Steiner, D.; Baumann, K.; Bunjes, H.; Dietzel, A.; Finke, J.H.; Glasmacher, B.; et al. Novel strategies for the formulation and processing of poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2018, 126, 40–56. [Google Scholar] [CrossRef]
- Kim, H.; Kim, Y.; Lee, J. Liposomal formulations for enhanced lymphatic drug delivery. Asian J. Pharm. Sci. 2013, 8, 96–103. [Google Scholar] [CrossRef]
- Patel, D.; Sawant, K.K. Self Micro-Emulsifying Drug Delivery System: Formulation Development and Biopharmaceutical Evaluation of Lipophilic Drugs. Curr. Drug Deliv. 2009, 6, 419–424. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- McCartney, F.; Jannin, V.; Chevrier, S.; Boulghobra, H.; Hristov, D.R.; Ritter, N.; Miolane, C.; Chavant, Y.; Demarne, F.; Brayden, D.J. Labrasol® is an efficacious intestinal permeation enhancer across rat intestine: Ex vivo and in vivo rat studies. J. Control. Release 2019, 310, 115–126. [Google Scholar] [CrossRef]
- Buyukozturk, F.; Benneyan, J.C.; Carrier, R.L. Impact of emulsion-based drug delivery systems on intestinal permeability and drug release kinetics. J. Control. Release 2010, 142, 22–30. [Google Scholar] [CrossRef]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS) – challenges and road ahead. Drug Deliv. 2014, 22, 675–690. [Google Scholar] [CrossRef]
- Reddy, M.S.; Gurram, A.; Deshpande, P.; Kar, S.; Nayak, U.; Udupa, N. Role of components in the formation of self-microemulsifying drug delivery systems. Indian J. Pharm. Sci. 2015, 77, 249–57. [Google Scholar] [CrossRef]
- Kyatanwar, A. U. , Jadhav, K. R., & Kadam, V. J. (2010). Self micro-emulsifying drug delivery system (SMEDDS). Journal of Pharmacy Research, 3(2), 75-83.
- Narang, A.S.; Delmarre, D.; Gao, D. Stable drug encapsulation in micelles and microemulsions. Int. J. Pharm. 2007, 345, 9–25. [Google Scholar] [CrossRef]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 1–12. [Google Scholar] [CrossRef]
- Weil, M.K.; Chen, A.P. PARP Inhibitor Treatment in Ovarian and Breast Cancer. Curr. Probl. Cancer 2011, 35, 7–50. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L. Patent Review of Manufacturing Routes to Recently Approved PARP Inhibitors: Olaparib, Rucaparib, and Niraparib. Org. Process. Res. Dev. 2017, 21, 1227–1244. [Google Scholar] [CrossRef]
- Pathade, A.D.; Kommineni, N.; Bulbake, U.; Thummar, M.M.; Samanthula, G.; Khan, W. Preparation and Comparison of Oral Bioavailability for Different Nano-formulations of Olaparib. AAPS PharmSciTech 2019, 20, 276. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Banerjee, S.; Mileshkin, L.; Scott, C.; Shannon, C.; Goh, J. Practical guidance on the use of olaparib capsules as maintenance therapy for women with BRCA mutations and platinum-sensitive recurrent ovarian cancer. Asia-Pacific J. Clin. Oncol. 2016, 12, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Li, J.; Bui, K.; Learoyd, M.; Berges, A.; Milenkova, T.; Al-Huniti, N.; Tomkinson, H.; Xu, H. Bridging Olaparib Capsule and Tablet Formulations Using Population Pharmacokinetic Meta-analysis in Oncology Patients. Clin. Pharmacokinet. 2018, 58, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaurasiya, A.; Singh, M.; Upadhyay, S.C.; Mukherjee, R.; Khar, R.K. Exemestane Loaded Self-Microemulsifying Drug Delivery System (SMEDDS): Development and Optimization. Aaps Pharmscitech 2008, 9, 628–634. [Google Scholar] [CrossRef]
- Chen, M.; Liu, X.; Fahr, A. Skin penetration and deposition of carboxyfluorescein and temoporfin from different lipid vesicular systems: In vitro study with finite and infinite dosage application. Int. J. Pharm. 2011, 408, 223–234. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Feng, Y.; Firempong, C.K.; Zhu, Y.; Omari-Siaw, E.; Zheng, Y.; Pu, Z.; Xu, X.; Yu, J. Enhanced oral bioavailability of [6]-Gingerol-SMEDDS: Preparation, in vitro and in vivo evaluation. J. Funct. Foods 2016, 27, 703–710. [Google Scholar] [CrossRef]
- Powis, G. Dose-Dependent Metabolism, Therapeutic Effect, and Toxicity of Anticancer Drugs in Man. Drug Metab. Rev. 1983, 14, 1145–1163. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Moreno, V.; Gupta, A.; Kaye, S.B.; Dean, E.; Middleton, M.R.; Friedlander, M.; Gourley, C.; Plummer, R.; Rustin, G.; et al. An Adaptive Study to Determine the Optimal Dose of the Tablet Formulation of the PARP Inhibitor Olaparib. Target. Oncol. 2016, 11, 401–415. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pseudoternary phase diagram of oil, surfactant, and co-surfactant. (Capmul MCM EP/NF, Labrasol, and PEG 400, respectively).
Figure 1.
Pseudoternary phase diagram of oil, surfactant, and co-surfactant. (Capmul MCM EP/NF, Labrasol, and PEG 400, respectively).
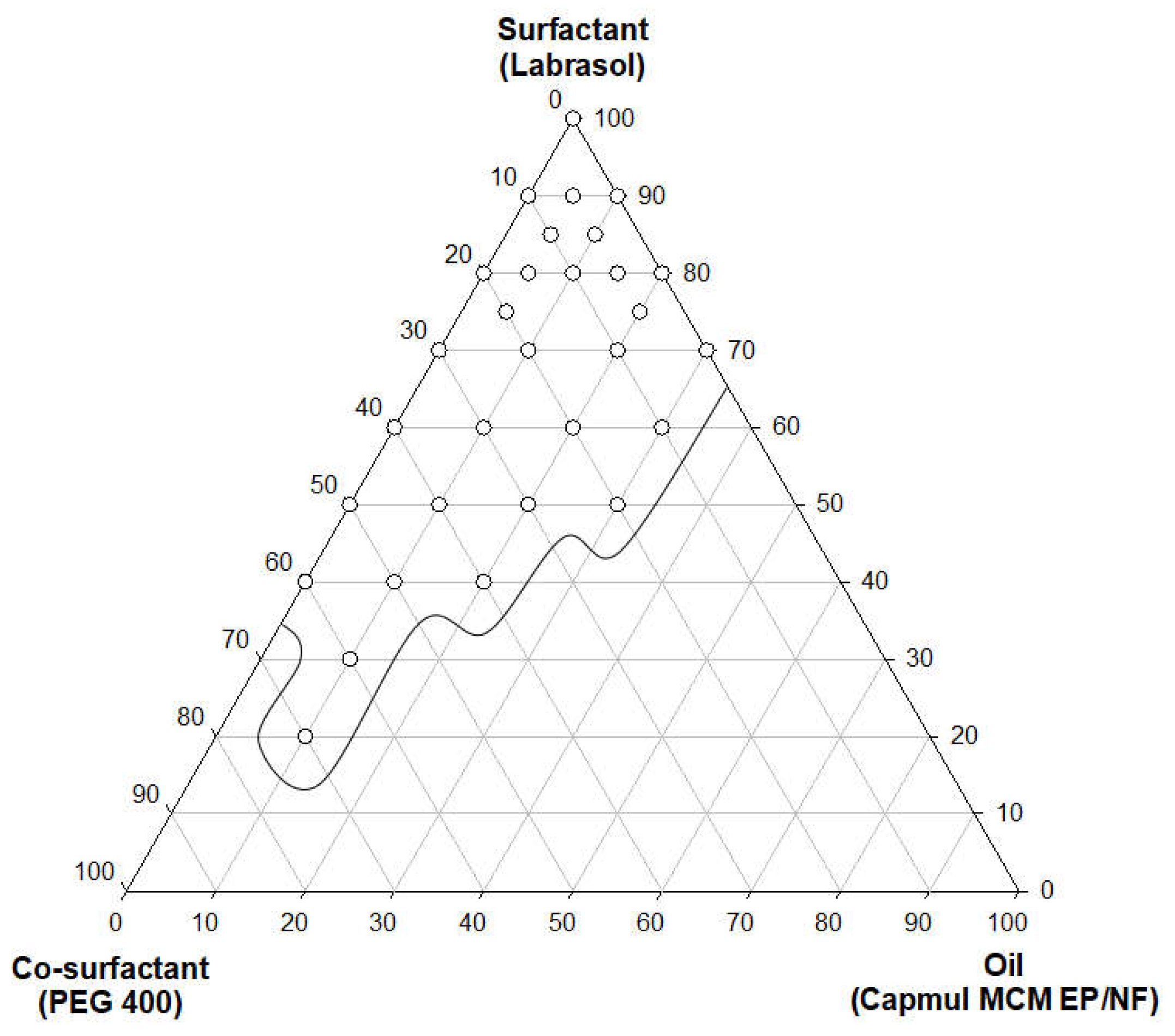
Figure 2.
Transmission electron microscopy image of OLA SMEDDS.

Figure 3.
In vitro dissolution study of SMEDDS and OLA powder. Dissolution profile in (a) HCl buffer (pH 1.2); (b) phosphate buffer (pH 6.8); (c) distilled water; **p < 0.01 compared with OLA.
Figure 3.
In vitro dissolution study of SMEDDS and OLA powder. Dissolution profile in (a) HCl buffer (pH 1.2); (b) phosphate buffer (pH 6.8); (c) distilled water; **p < 0.01 compared with OLA.
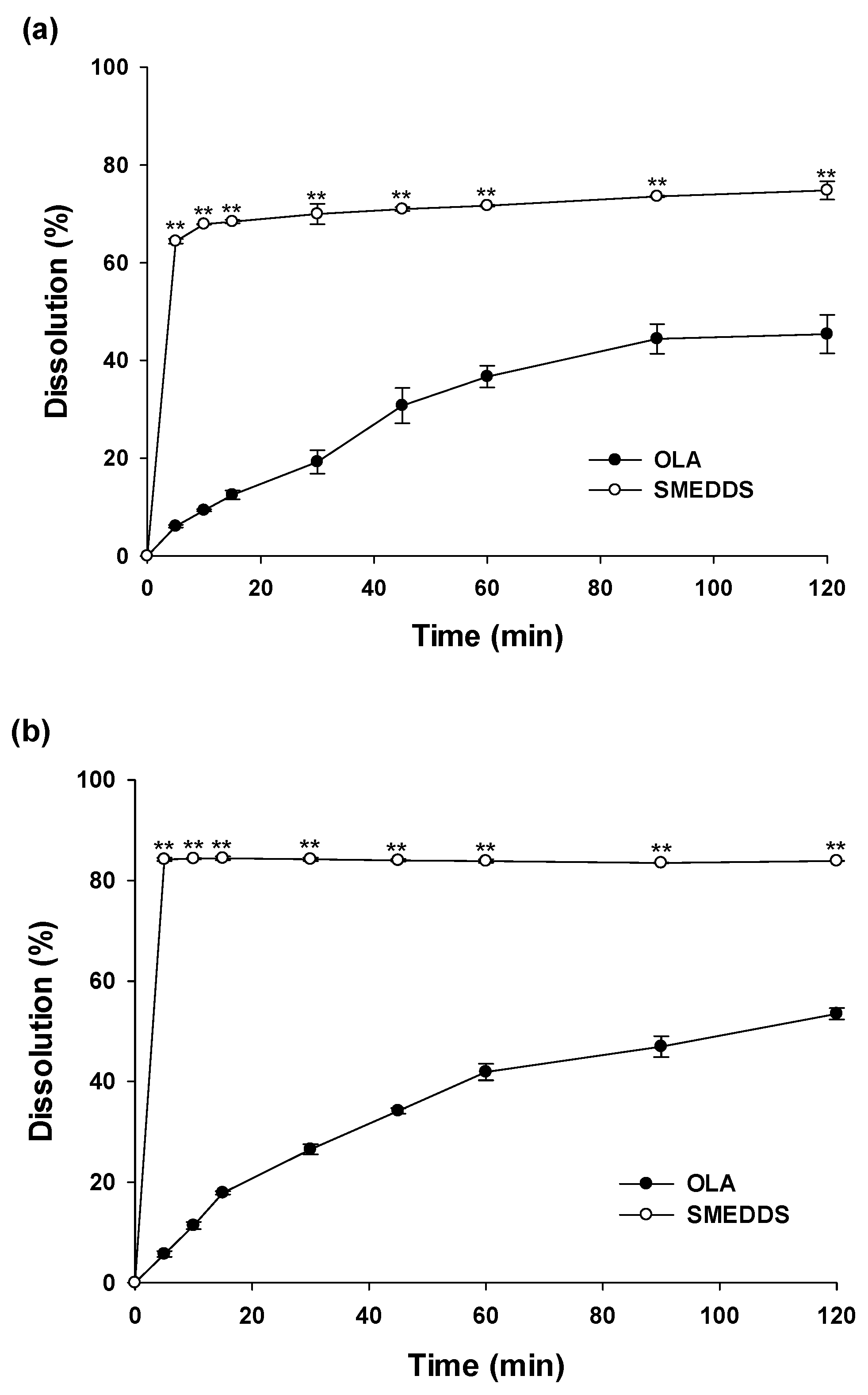
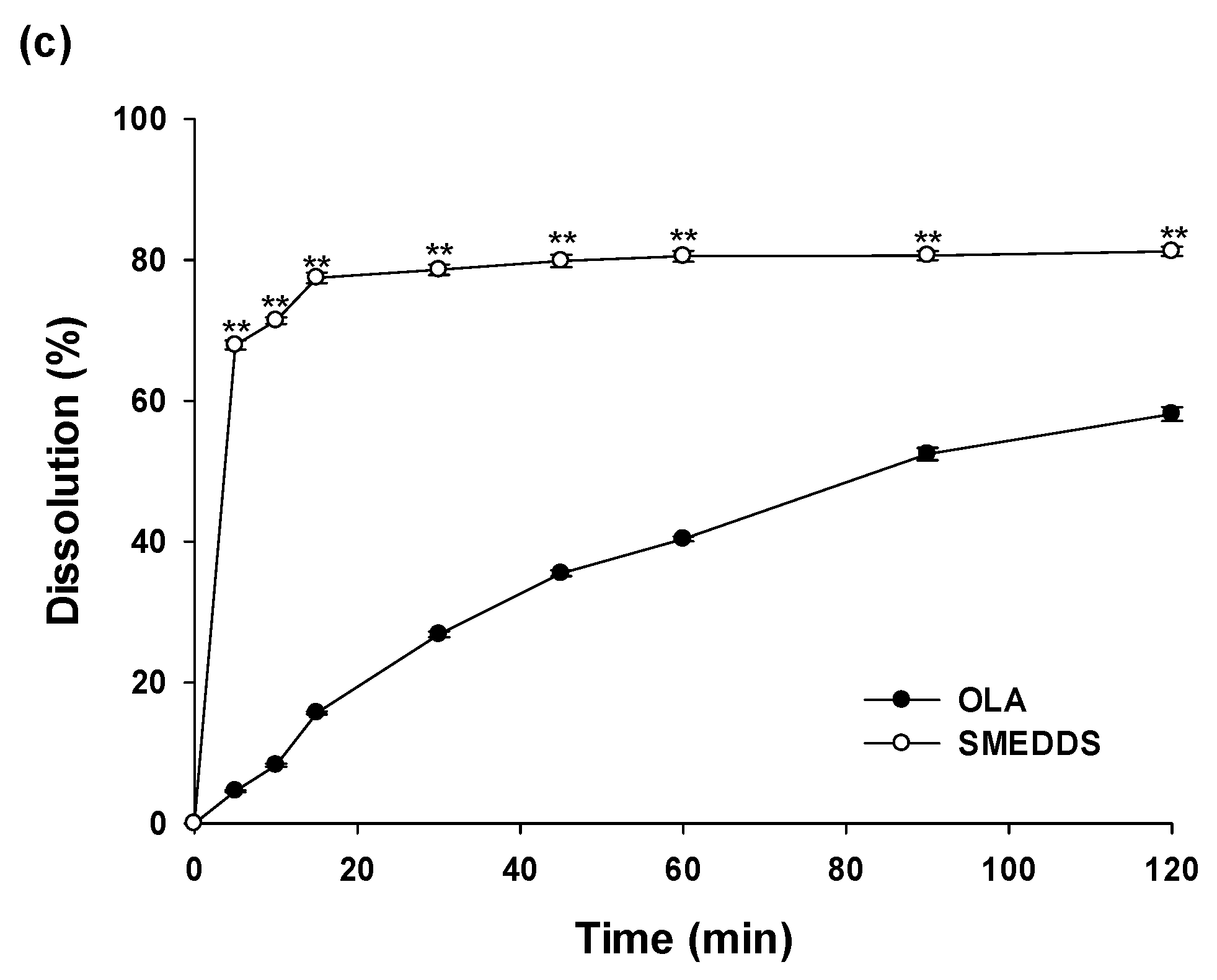
Figure 4.
Plasma concentration-time profiles in rats after oral administration of OLA powder, SMEDDS and Lynparza® tablet; *p < 0.05 compared with OLA; #p < 0.05 compared with Lynparza® tablet.
Figure 4.
Plasma concentration-time profiles in rats after oral administration of OLA powder, SMEDDS and Lynparza® tablet; *p < 0.05 compared with OLA; #p < 0.05 compared with Lynparza® tablet.
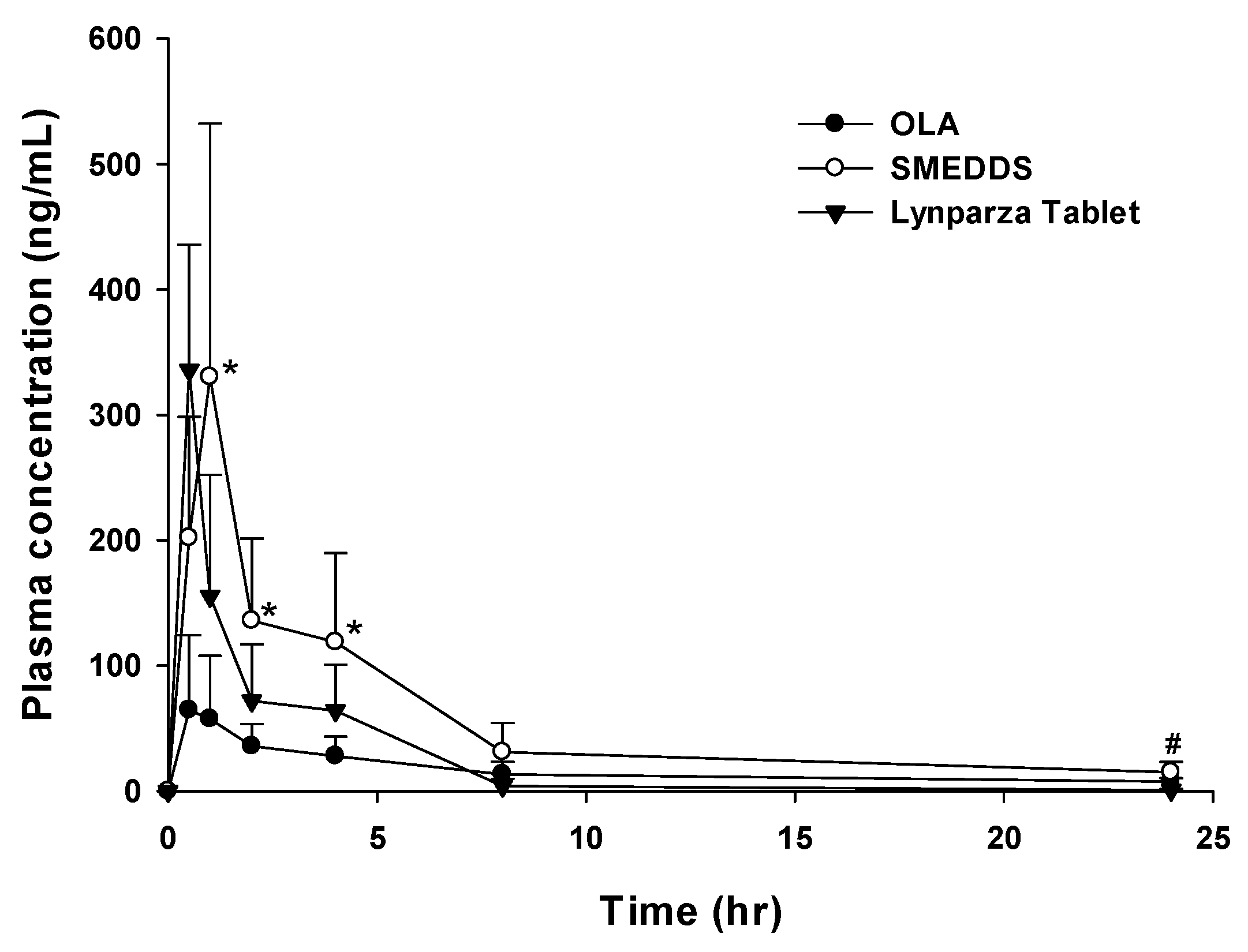

Table 1.
Solubility of OLA in various aqueous solution, oils, surfactants and co-surfactants (mean ± S.D.; n = 3).
Table 1.
Solubility of OLA in various aqueous solution, oils, surfactants and co-surfactants (mean ± S.D.; n = 3).
| Vehicle | Solubility (mg/ml) |
|---|---|
| Aqueous solution | |
| Distilled Water | 0.07 ± 0.25 |
| pH 1.2 | 0.06 ± 0.57 |
| pH 6.8 | 0.06 ± 0.42 |
| Oils | |
| Capmul MCM EP/NF | 41.68 ± 0.02 |
| Cotton Seed Oil | 0.06 ± 0.20 |
| Labrafil M 1944 CS | 0.95 ± 0.71 |
| Labrafac PG | 0.13 ± 0.10 |
| Oleic acid | 3.83 ± 0.22 |
| Surfactants and co-surfactants | |
| Kolliphor EL | 7.83 ± 6.06 |
| Tween 80 | 8.82 ± 0.28 |
| Span 80 | 4.82 ± 3.52 |
| Labrasol | 36.60 ± 0.07 |
| Transcutol HP | 19.69 ± 0.14 |
| Plurol Oleique CC 497 | 4.99 ± 0.09 |
| Isopropyl Myristate | 0.04 ± 0.02 |
| PEG 400 | 47.66 ± 0.38 |
Table 2.
Stability study of SMEDDS at 25 °C, 60% RH and 40 °C, 75% RH (Mean ± S.D.; n = 3).
| Time (month) | Droplet size (nm) | Zeta potential (mV) | Drug content (%) |
|---|---|---|---|
| (a) 25 °C, 60% RH | |||
| 0 | 141.1 ± 0.24 | -15.83 ± 1.09 | 101.05 ± 0.45 |
| 1 | 142.7 ± 0.39 | -18.75 ± 0.43 | 101.09 ± 0.17 |
| 3 | 164.1 ± 0.33 | -16.31 ± 0.65 | 101.01 ± 0.24 |
| (b) 40 °C, 75% RH | |||
| 0 | 141.1 ± 0.24 | -15.83 ± 1.09 | 101. 05 ± 0.45 |
| 1 | 143.0 ± 0.70 | -13.45 ± 0.82 | 99.02 ± 0.14 |
| 3 | 166.1 ± 2.69 | -15.99 ± 0.65 | 98.50 ± 0.15 |
Table 3.
Pharmacokinetic parameters of SMEDDS and OLA at an equivalent dose of 10 mg/kg in rats (mean ± S.D.; n = 5); *p < 0.05 and**p < 0.01 compared with OLA; #p < 0.05 compared with Lynparza® tablet.
Table 3.
Pharmacokinetic parameters of SMEDDS and OLA at an equivalent dose of 10 mg/kg in rats (mean ± S.D.; n = 5); *p < 0.05 and**p < 0.01 compared with OLA; #p < 0.05 compared with Lynparza® tablet.
| PK Parameters | OLA | Lynparza® tablet | SMEDDS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Tmax(hr) | 0.8 | ± | 0.3 | 0.5 | ± | 0.0 | 0.9 | ± | 0.2# |
| Cmax(ng/mL) | 83.1 | ± | 49.6 | 335.7 | ± | 100.2 | 351.2 | ± | 173.3* |
| T1/2(hr) | 9.4 | ± | 0.1 | 4.0 | ± | 0.9 | 7.3 | ± | 3.2 |
| AUClast(ng·hr/mL) | 384.1 | ± | 136.2 | 518.2 | ± | 154.9 | 1242.7 | ± | 420.3**,# |
| AUCinf(ng·hr/mL) | 634.0 | ± | 437.2 | 522.0 | ± | 155.9 | 1427.3 | ± | 460.3*,# |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



