
Submitted:
31 March 2023
Posted:
03 April 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
In the course of SARS-CoV-2 infection, the 3CL or nsp5 protease plays a pivotal role as the most important viral protease required for the maturation of viral proteins during host infection. Herein, we simulated for 500 ns 3CLproWT, 3CLproH41A, , 3CLproBeta, and 3CLproOmicron, in complex with the substrates nsp 4|5 and nsp 5|6. Our results show that mutations in the 3CLpro present in the SARS-CoV-2 variant of concerns (VOCs) did not lead to significant conformational changes or changes in substrate binding affinities. However, significantly high cleavage rates for the boundary between nsp4 and nsp5 were obtained for 3CLproBeta and 3CLproOmicron and may play a key role in the viral replication and virus fitness gain. Our molecular dynamics data suggest that the cleavage rate of nsp4|5 may be related to the increased amount of viral load observed for these VOCs, releasing more nsp4 than other non-structural proteins. Based on our hydrogen bonding analyses, we also discovered that Gly143 and Glu166 are key residues in substrate recognition, suggesting that these residues may be incorporated as pharmacophoric centers for Beta and Omicron variants in drug design. Our results suggest that Gly143 and Glu166 are essential residues to interact with Gln6 of the different substrates and, therefore, are potential broad-spectrum pharmacophoric centers of SARS-CoV-2 3CLpro.
Keywords:
Molecular Dynamics
; SARS-CoV-2 variants of concern
; 3CLpro
; viral load
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused the current pandemic, the largest of the 21st century [1]. According to the World Health Organization (WHO), the novel coronavirus infected more than 759 million individuals and killed more than 6.8 million people worldwide [2]. Previous studies performed with SARS-CoV (referring to the coronavirus responsible for the 2002/2003 epidemic) were fundamental in the development of vaccines and studies of SARS-CoV-2 inhibitors of the virus life cycle in the host cell [3,4]. Vaccination was essential to drastically reduce the number of infections and deaths caused by the virus [5,6]. Although more than 12.4 billion doses of vaccines were applied and more than 5 billion people are fully vaccinated, the vaccination does not prevent the transmission of the virus due to emerging novel SARS-CoV-2 Variants of Concerns (VOCs) that are resistant to neutralizing antibodies [5,6]. The emergence of SARS-CoV-2 VOCs resistant to vaccines has concerned the scientific community [1]. In 2020 December, the Alpha variant (or lineage B.1.1.7, or Clade 20I) emerged and was soon recognized as both more transmissible and more lethal than the ancestral strain [7]. In 2020 September, the Beta variant (or B.1.351 lineage or Clade 20H) emerged in South Africa, showing greater transmissibility and lethality than the Alpha variant [8]. Next, the Gamma variant (or lineage P.1, or Clade 20J) originated in Brazil and Japan, becoming predominant in these countries and spreading to neighboring countries [9]. In 2020 September, the Delta variant (or lineage B.1.617.2 or Clades 21A, 21I, 21J) emerged in India, presenting several mutations that allowed its predominance worldwide [10]. In November 2021, the Omicron variant (or B.1.529 lineage) emerged and, in less than 3 months, became predominant over the Delta variant worldwide [1,11,12]. The lineage B.1.529 (Clade 21M) gave rise to its sub-lineages, BA.1 (Clade 21K), BA.2 (Clade 21L), BA.2.12.1 (Clade 22C), BA.4 (Clade 22A), and BA.5 (Clade 22B). In 2023 March, the proportions of the Omicron sub-variants represented more than 80%, when compared to other VOCs, being XBB.1 and XBB.1.5 more than 55% prevalence in the reported cases of COVID-19 [13].
Among the molecular targets, the Spike protein and the 3-chymotrypsin-like protease (3CLpro or Mpro, SARS-CoV-2 main protease) play key roles in vaccine and drug designs, respectively [14,15,16,17,18,19]. In particular, the 3CLpro cleaves 13 sites of two polyproteins, pp1a and pp1ab, which are results from translation of the genomic mRNA. The active form of the 3CLpro is a homodimer, in which each protomer has domains I, II, and III. Domains I (residues 10-99) and II (residues 100-182) are formed by six antiparallel β-strands, where the catalytic site is located between them [20]. Domain III (residues 198-303) is composed of five α-helices that are involved in the regulation of 3CLpro dimerization through salt interactions between residue E290 of one protomer and residue R4 of the neighboring protomer [21]. Dimerization of 3CLpro is a critical process for catalytic activity because the N-finger of each of the protomers interacts with E166 of the other protomer, adjusting the S1 subsite to bind the substrate [14,22]. The interface between domains I and II is composed of the side chains of V13, L115, F150, P122 of one protomer and P9 of the other protomer, together they form strong hydrophobic interactions [23]. Any substitution of these residues for larger or more polar ones causes a reduction in catalytic activity, suggesting that they are residues important for inter-domain contact [23]. Conversely, at the dimer interface, between the domain I of one protomer and the domain I of another protomer, it is reported that there is an extensive network of hydrogen bonds from residues S10, G11, and E14 of a protomer with residues S10, G11, and E14 from the other protomer [23]. This interaction network is thus expected to play a key role in maintaining SARS-CoV-2 3CLpro dimer [23].
The main catalytic residues of 3CLpro are dyad H41 and C145. Cysteine is conserved among the family Coronaviridae [24] and acts as a nucleophile in the proteolytic process [25,26]. The active site is composed of four subsites, S1, S1', S2 and S4 [26], following the Schecter-Berger nomenclature for proteases [27]. The Sis and Si's represent the subsites of the enzyme and the Pis and Pi's are notations for the substrate, in which the substrate is cleaved between P1 and P1'. Structural studies have shown that the 3CLpro substrate may occupy mainly four subsites, S1', S1, S2, and S4. The S1 subsite is composed of the residues F140, C145, S144, and H163. Specifically, the side chain of F140 makes π-π interaction with H163, interacting with the N-finger of the neighboring protomer. Such interaction networks play an important role for the structural stability of the active site and molecular recognition of the substrate [26]. The S1' subsite is more exposed to the aqueous environment than other subsites and it is composed of residues T25, T26, L27, and H41. This sub-site has a function of accommodating residues that present small bulk side chains [26]. The S2 subsite is highly hydrophobic, composed of residues M49, Y54, and M165 and the alkyl part of the side chain of D187 [26]. Finally, the S4 subsite consists of residues M165, L167, F185, Q192, and Q189 [26]. A multiple alignment of the amino acid sequences in the vicinity of the cleavage sites has shown that 3CLpro recognizes the consensus sequence X-(L/F/M)-Q↓(G/A/S)-X (where X = any amino acid, the arrow ↓ being the substrate cleavage site) [18,28]. Thus, the molecular recognition of 3CLpro by the substrate depends exclusively on the physicochemical features of the specific residues present in the target sequence of each substrate. Therefore, insights into the nature of the interactions involved in substrate recognition by 3CLpro are essential to allow an understanding of how structural information relates to the biological response of the virus, and thus are also essential in drug design of novel anti-SARS-CoV-2 drugs [18,29,30,31,32].
Herein, we simulated 3CLproWT, 3CLproH41A, 3CLproBeta, and 3CLproOmicron in complex with substrates nsp4|5 and nsp5|6 for 500 ns. Our results reveal that nsp4|5 showed good conformational stability in the active site, but was significantly more stable in 3CLproWT and 3CLproOmicron. Furthermore, our root-mean-square fluctuation (RMSF) analysis showed that mutations in 3CLproBeta and 3CLproOmicron make both variants more stable, i.e. diminishes their intrinsic mobility, thus making these proteins more prone to maintain longer interactions with the nsp4|5 and nsp5|6 substrates. However, 3CLproWT, 3CLproBeta, and 3CLproOmicron did not show significant differences in the estimated KD values for the aforementioned substrates. Still, our molecular dynamics data suggest that the cleavage rate of nsp4|5 is higher for the 3CLproOmicron variant and could be related to the higher viral load of these VOC, as the efficient release of nsp4 is essential for replication and assembly of replicative structures of the virus [33]. Therefore, our results suggest that higher viral load of VOCs may be related with enhanced enzymatic activity of the main protease of SARS-CoV-2 [1,34].
2. Materials and methods
2.1. Site-directed mutation in silico
We build enzyme/substrate complexes from PDB IDs 7DVP (3CLproH41A/nsp4|5), 7DVW (3CLproH41A/nsp5|6), and 7DW6 (3CLproH41A/nsp14|15) [26]. To obtain WT, Beta and Omicron, we changed the corresponding residues using the Maestro program (academic version v. 2021-4, by Schrödinger) [35]. After modeling the corresponding structures, the generated PDB files were used as inputs in molecular dynamics studies. The protonation states of ionizable residues in an implicitly aqueous environment at pH 7.0 were initially determined using the PROPKA module implemented in the Maestro program (academic version v. 2021-4, by Schrödinger) [35]. Thus, (1) all glutamic and aspartic residues were represented as unprotonated; (2) arginine and lysine residues were assumed to have a positive charge; (3) at the N- and C-terminal regions, the amino and carboxyl groups were converted into charged groups; (4) histidines were protonated according to PROPKA prediction and manual inspection, such as: 3CLproWT, δ-tautomer (all) and ε-tautomer (none); 3CLproBeta, δ-tautomer (all) and ε-tautomer (none); 3CLproOmicron, δ-tautomer (all except H132).
All molecular dynamics simulations were performed using GROMACS, v. 5.1.4 [36,37,38,39], using the OPLS-AA force field [40]. All peptide substrates were parametrized using the OPLS-AA force field implemented in GROMACS. All systems were then explicitly solvated with TIP3P water models in a cubic box and neutralized, maintaining the concentration of 150 mM NaCl and minimized until reaching a maximum force of 10.0 kJ/mol or a maximum number of steps at 50,000. The systems were consecutively equilibrated in isothermal-isochoric (NVT) ensembles for 3 ns (number of steps and intervals of 1,500,000 and 2 fs, respectively) and isothermal-isobaric (NPT) (1 bar, 310 K) for 3 ns ( number of steps and intervals of 1,500,000 and 2 fs, respectively).
All simulations were then performed in a periodic cubic box considering a minimum distance of 1.2 nm between any protein atom and the walls of the box. Molecular dynamics runs were performed in isothermal-isobaric (1 bar, 310 K, NpT) for 500 ns (number of steps and intervals of 250,000,000 and 2 fs, respectively) in the NpT system. The leap-frog algorithm was used to integrate Newton's equations. LINCS (LINEar Constraint Solver) was selected to satisfy the holonomic constraints, whose number of iterations and order were 1 and 4, respectively. Neighbor search grid cells were used (Verlet clipping scheme, frequency to update neighbor list of 20 steps and clipping distance for short range neighbor list of 12 Å). In the van der Waals parameters, the forces were smoothly shifted to zero between 10 and 12 Å. In Coulomb electrostatics, fast and smooth Ewald Particle-Mesh electrostatics (PME) was used for long range electrostatics. In addition, the distance for the Coulomb cut to 12 Å, interpolation order for PME to value 4 (cubic interpolation) and grid spacing for Fast Fourier Transform (FFT) to 1.6 Å. In the temperature coupling, velocity rescheduling with a stochastic term (modified Berendsen thermostat) was used. After obtaining two coupling groups (protein and water/ions), the time constant (0.1 ps) and 310 K were considered as the reference temperature. In the pressure coupling (NpT sets), a Parrinello-Rahman barostat was used (isotropic type, time constant of 2 ps, reference pressure of 1 bar and isothermal compressibility of 4.5x10-5 bar-1). In molecular dynamics calculations, periodic boundary conditions in xyz coordinates (3D space) were used. Then, the percent hydrogen bond occupancy (all frames, considering the cut-off distance and angle of 4 Å and 20°, respectively), root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were calculated using the modules GROMACS and VMD [41].
2.2. Thermodynamic integration
In order to investigate the binding affinities between the different substrates with 3CLpro of WT, Beta and Omicron, we calculated an estimator for ln KD using binding free energies and experimental data of the binding affinities of 3CLproH41A for substrates nsp4|5, nsp5|6 and nsp14|15. This estimator was used to estimate ln KD of these substrates when interacting with the main protease of WT, Beta and Omicron. Thus, we calculated binding free energies of the 3CLproH41A, the binding free energy was calculated using the method of thermodynamic integration. The substrate/3CLpro complexes were placed in a cubic periodic boundary condition (PBC) box. The NVT and NpT procedures were equally applied, keeping the same conditions in our MD simulations. During free energy of perturbation (FEP) simulations, the coupling parameter λ, which varies from 0 to 1, was used to evaluate the change in free energy ΔG of modifying the system from the fully interacting state (λ = 0) to the non-interacting state (λ = 1) by changing the Hamiltonian system. The change in the state of interaction of the ligand from a state of complete interaction to a state of non-interaction with neighboring molecules is known as the process of ligand annihilation. Ten values of λcoul (0.00; 0.10; 0.20; 0.30; 0.40; 0.50; 0.60; 0.70; 0.80; 0.90; 1.00) and ten values of λvdW (0.00; 0.10; 0.20; 0.30; 0.40; 0.50; 0.60; 0.70; 0.80; 0.90; 1.00) were used to change Coulomb and van de Waals interactions, respectively. Twenty simulations were performed considering different values of λ for the substrate in a complex with different 3CLpro. The total energy change of the substrate annihilation process was then summed via Bennet's acceptance ratio (BAR) method. Finally, the binding affinity was estimated using a linear regression model associating the experimental data of the binding affinities of 3CLproH41A for nsp4|5, nsp5|6 and nsp14|15 substrates with the free energy data of the interaction determined by BAR. Finally, we determined a linear regression model using binding free energies (in kJ.mol-1) as independent variables and ln KD as dependent variables.
3. Results and Discussion
3.1. Molecular and structural relationships of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron
Our analysis of multiple sequence alignment of the 3CLpro amino acid sequences of the VOCs showed that the Beta and Omicron variants have substitutions at K90R (K3353R) and P132H (P3395H), respectively (Figure S1). Analogously, other coronaviruses also share mutations in 3CLpro that could impact substrate binding [34,42]. In this context, previous studies have shown that 3CLpro homologs from MERS-CoV, SARS-CoV, and SARS-CoV-2 have different binding affinities to known protease inhibitors [43], raising the possibility that these drugs might also show variable degrees of effectiveness against VOCs. Therefore, structural studies are an essential tool to gather relevant information that could be the basis for drug design and the development of broad-spectrum antivirals. 3CLpro cleaves the nsp4/5 and nsp5/6 sites, as well as other sites on the pp1ab polyprotein [44]. In our molecular dynamics studies, all 3Clpro variants (3CLproH41A, 3CLproWT, 3CLproBeta , and 3CLproOmicron) showed different profiles of RMSD and RMSF while interacting with nsp4|5 and nsp5|6 (Figure 1 and Figure 2). RMSDH41A showed large structural changes throughout the molecular dynamics simulation, ranging from 2.0 to 5.8 Å RMSD. We observed that the backbone RMSD of the four 3CLpro variants showed different profiles in the simulations. The 3CLproH41A presented more structural changes than 3CLproWT, 3CLproBeta and 3CLproOmicron, in which backbone RMSDH41A changed from ~2.7 to ~4.1 Å. Interestingly, the 3CLproWT showed few conformational changes when compared with 3CLproBeta and 3CLproOmicron, with an RMSDWT of ~2.6 Å. Our results are in agreement with Avti and coauthors, in which the Cα RMSD of 3CLproWT in complex with lopinavir and ritonavir converged to ~2.5 Å [45]. We considered that 3CLproBeta and 3CLproOmicron also showed good conformational stabilities, presenting RMSDBeta and RMSDOmicron of ~2.7 and ~2.2 Å, respectively. In general, backbone RMSD of the substrate nsp4|5 presented few structural changes when interacting with 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron. RMSDH41A, RMSDWT, RMSDBeta, and RMSDOmicron values were ~2.1, ~1.7, ~1.3 and ~2.4 Å, respectively.
Next, we investigated backbone RMSD of the substrate nsp 5|6 while interacting with 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron. Such substrates presented low binding affinity by 3CLproH41A [26]. Thus, both substrate and 3CLpro variants are expected to show large conformational changes in molecular dynamics simulations. Indeed, both substrate nsp 5|6 and the enzyme variants showed large variability in the RMSD values and, in each case, a different structural behavior was observed, even maintaining the same experimental conditions. Although RMSD of nsp 5|6 presented convergence in ~280 ns, the mutation from H41 to A41 caused significant structural changes of the 3CLproH41A, mainly while interacting with this low binding affinity substrate. Conversely, we observed that nsp 5|6 presented less structural variability, mainly after ~250 ns. The 3CLproBeta and 3CLproOmicron showed different backbone RMSD profiles in the simulation (Figure 2 c-d). Note that the substrate nsp5|6 exhibited similar variability of the backbone RMSD while interacting with these variants. However, 3CLproOmicron showed better structural stability than 3CLproBeta while interacting with this substrate. Thus, changes in structural stability of nsp 4|5 and nsp 5|6 in the active site of the 3CLpro variants play a key role in the substrate/enzyme complex interaction. Strengthening of these interactions increases the binding affinity of these molecules to the active site, an effect often deployed as a basic principle in drug design. Indeed, previous molecular dynamics studies showed that 3CLpro in complexes with inhibitors of high binding affinities have stable active sites [46]. Therefore, the stability of both substrates in 3CLpro of VOCs demonstrate the effectiveness of accommodation of high and low affinity substrates in the active site, in order to facilitate the cleavage process.
Next, we investigated how structural flexibility of each amino acid residue is affected in relation to mean movement throughout the molecular dynamics simulation. Thus, we analyzed the backbone RMSF of different 3CLpro (Figure 3). Comparing the RMSF of the 3CLpro variants interacting with the substrate nsp4|5. The 3CLproBeta and 3CLproH41A showed significantly higher RSMF values than 3CLproWT and 3CLproOmicron, suggesting that 3CLproBeta and 3CLproH41A are more flexible when bound to nsp4|5 than 3CLproWT and 3CLproOmicron. In general, all four 3CLpro variants, when interacting with substrates nsp4|5 and nsp5|6, presented high RMSF values (RMSF > 3Å) at residues 45-55 (domain I), 90-200 (domain II), and 230-300 (domain III). Our results are in agreement with Muralidharan and coauthors, who showed that free enzyme and the enzyme in complex with different inhibitors presented the same RMSF profile [46]. Therefore, our results suggest that the mutations identified in the 3CLproBeta and 3CLproOmicron do not significantly affect structural flexibility of the amino acid residues of these variants when interacting with substrates nsp 4|5 and nsp 5|6.
3.2. Predicted binding affinity of different substrates interacting with 3CLproH41A, 3CLproWT, 3CLproBeta and 3CLproOmicron
Next, we developed a linear regression model using our binding free energy results from three different substrates and the experimental data from Zhao et. al [26]. Assuming that the active site is kept conserved in the 3CLpro variants, this linear regression model was used to infer the binding affinities of these substrates in the active site of the 3CLproWT, 3CLproBeta, and 3CLproOmicron in complex with the substrates nsp 4|5, nsp 5|6, and nsp 14|15. Table 1 shows the binding free energies predicted by free energy of perturbation (FEP). Note that the FEP-based binding free energies values of the different 3CLpro variants interacting with the same substrate showed the same order of magnitude: (1) nsp 4|5 in complex with 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron were, respectively, 3057.57, 3017.41, 3016.58 and 3026 kJ.mol-1; (2) nsp 5|6 in complex with 3CLproH41A, 3CLproWT, 3CLproBeta and 3CLproOmicron were, respectively, 3428.13, 3418.68, 3402.94, and 3412.27 kJ.mol-1; (3) nsp 14|15 in complex with 3CLproH41A, 3CLproWT, 3CLproBeta and 3CLproOmicron were, respectively, 3290.29, 3276.84, 3292.97, and 3265.98 kJ.mol-1. Therefore, these results reveal that corresponding mutations in these 3CLpro variants are not affecting free binding affinities by different substrates. Using these FEP-based energies, we determined the linear regression model:
where is the binding free energy obtained by the FEP, R is the gas constant, T is the absolute temperature, and is a dissociation constant in μM. In Table 2, we show the estimated KD values calculated using this linear regression model at T = 310 K. For the substrate nsp4|5, estimated KD values varied between 22.1 and 36.9 μM. In substrate nsp5|6, estimated KD values were from 3129.3 to 4326.4 μM. Finally, for substrate nsp 14|15, estimated KD values varied between 537.7 and 760.8 μM. Therefore, these results suggest that the mutations did not significantly affect the binding affinity of the corresponding substrates in 3CLproH41A, 3CLproWT, 3CLproBeta and 3CLproOmicron.
Our previous study showed that significant variations in viral load may be associated with other genes, in addition to Spike, that influence viral load [1]. This suggests the hypothesis that the activity of these genes may probably influence other stages of the viral replication cycle. In the other words, our hypothesis suggests that viral load may be related to cell invasion and protein maturation or the inhibition of innate cell response [1], supported by experimental studies [1,34] that showed VOCs have higher viral load when compared to the wild type. Since the structural changes of 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron were not affected by mutations, we investigated other kinetic parameters that could be related with viral load. We therefore considered the mechanism of action, where the transfer of protons must occur from C145 to H41, making the cysteine active for a possible nucleophilic attack on the carbonyl carbon of the substrate glutamine. To evaluate the feasibility of this transfer, we measured the distance between the hydrogen of the catalytic cysteine (C145-H) and delta 1 nitrogen (H41-Nδ1) of H41. Furthermore, we also calculated the distance between the sulfur of C145 and the carbonyl carbon of glutamine present in the substrate. We considered that proteolytic cleavage may only occur if the catalytic residues, the nucleophile (Cys145) and the point of attack on the substrate are close enough and in the right geometry. We thus assumed that a distance of 4.5 Å between these residues would be enough for catalysis and then quantified the substrate cleavage rate in the different molecular dynamics simulations (Table 3). As shown in Table 3, under these assumptions, substrate nsp4|5 may be cleaved 154, 9941 and 555 times, respectively, by 3CLproWT, 3CLproBeta, and 3CLproOmicron. However, the substrate nsp 5|6 may be cleaved 1025, 70 and 445 times by 3CLproWT, 3CLproBeta, and 3CLproOmicron. Although these results are still preliminary, the much higher number of conformations where both the Omicron and the Beta variants have the opportunity to cleave the peptide bond at the nsp4|5 boundaries might help explain how these VOCs achieve higher viral loads. Indeed, expression of nsp4 is essential for replication and assembly of virus replicative structures [33] and cleavage between nsp4 and nsp5 may be critical for virus particle production, which may explain the increased viral loads [1,34].
At the same time, a smaller number of conformations capable of catalyzing the cleavage of the nsp5|6 is visited by 3CLpro from the Beta and, to a lesser extent, by Omicron variants. Since nsp5 is the catalytic core of 3CLpro and is known to act on cellular proteins that regulate innate immunity and inflammation, a reduction in the intracellular concentrations of mature nsp5 could impact the capacity of these VOCs to inhibit the innate immunity and stress response pathways [47,48]. Still, as seen by the number of expected cleavage events, the Omicron variant might still be very effective at attacking the nsp5|6 and reports actually suggest the Beta variant is more effective than the wild-type in evading humoral responses [49,50]. It is therefore possible that the lower number of cleavage events leading to maturation of Nsp5 does not significantly affect the attack of innate immunity regulators by Nsp5.
3.3. Pharmacophoric hypothesis based on molecular dynamics simulations
In rational drug design, one way to predict a potential inhibitor of a target protein is to know the pharmacophoric elements of bioactive molecules and/or main amino acid residues of the active site. Such pharmacophoric elements are defined as basic requirements for a substrate and/or ligand to make the molecular recognition at the active site of the target protein. It has been reported that residues His41, Met165, Asp187, Arg188, and Gln189 are important for the interaction of 3CLpro with its inhibitors [14,51]. In general, His41 may favor interaction with aromatic groups of inhibitors [51]. Rhamnocitrin, Isokaempferide, and kaempferol may make important interactions into the active site with amino acid residues His41, Cys145, Asn142, and Glu166 [52]. The α-ketoamide-derived inhibitor interacts with 3CLpro, specifically with residues His41, His164, and Cys145 [20]. Three drug-like peptides were identified and shown to interact with His41, Gly143, and Glu166 of the 3CLpro [19]. The Glu166 is a potential pharmacophore for drug design and plays a key role in preventing 3CLpro dimerization [14]. Catalytic residues H41 and Cys145 are essential for the substrate proteolytic cleavage process [53], being fundamental for the rational design of 3CLpro inhibitors [14]. In this regard, we compare our molecular dynamics simulation results with these previous studies and propose a pharmacophore for the rational design of inhibitors for 3CLproWT and its variants 3CLproBeta and 3CLproOmicron.
In general, pharmacophoric models have been proposed based on chemical structures of inhibitors, crystal structure of 3CLpro, and molecular dynamics simulation studies. Previous studies of X-ray crystallography and computational data showed that His41, Asn142, Cys145, Glu166, and Gln189 may be potential pharmacophoric centers for 3CLpro inhibitors [14,18,54,55,56]. Considering the conformational diversity of the 3CLpro active site, Pathak and collaborators identified six pharmacophoric clusters for SARS-CoV-2 main protease [57]. These clusters allowed identifying the fundamental residues to interact with different chemical groups of inhibitors, allowing mapping of pharmacophoric subsites of 3CLpro [57]. Thus, the six pharmacophoric groups presented important residues that interact with different inhibitors in the subsites S1 (Phe140, Leu141, His163, Met165, Glu166, His172), S1' (Thr25, Thr26, Leu27, Asn142, Gly143, Ser144, Cys145, His164), S2 (His41, Met49, Asp187, Arg188, Gln189) and S4 (Leu167, Phe168, Thr190, Ala191, Gln192) [57]. Wang and coauthors constructed a 3D pharmacophoric model with four components, being one hydrophobic, one hydrogen bond donor and two hydrogen bond acceptors, in which corresponding residues are hypothesized to be His41, Met49, Phe140, Ser144 e Met165, Glu166 e Gln189 [58]. In our previous study, we built models of artificial neural networks, partial least squares regression model and sequential minimal optimization regression, and simulated twelve potential inhibitors into the 3CLpro active site [14]. These combined results led us to propose a pharmacophoric model with few components that included the residues His41, Asn142, Cys145, Glu166, and Gln189 [14].
In order to investigate potential pharmacophoric centers, we calculated hydrogen bonding (HB) occupancies of the subsite on the enzymatic cavity of each 3CLpro variant. In Table 4 and Table 5 we show the main hydrogen bonds, considering values of hydrogen bond occupancies above 20%. We classify the hydrogen bonds as weak interaction (HB occupancy between 20 and 30%), medium interaction (HB occupancy between 30.1 and 50%), and strong interaction (HB occupancy above 50%). Gln6 is conserved among the SARS-CoV-2 polyprotein cleavage sites. Our results show that Gln6 is essential for making strong hydrogen bonding interactions with different residues in the enzymatic cavity, as observed between pairs SCGln6nsp4|5/SCGlu166H41A, MCGln6nsp4|5/MCHis164WT, MCGln6nsp4|5/MCGly143WT, SCGln6nsp4|5/SCHis163Omicron., and MCGln6nsp4|5/MCGly143Omicron. Notably, the nsp5|6 also contains medium and strong hydrogen bonding interactions between SCGln6nsp5|6/MCGly143H41A, SCGln6nsp5|6/SCGlu166WT, SCGln6nsp5|6/SCGlu166Beta, SCGln6nsp5|6/SCGlu166Omicron, MCGln6nsp5|6/MCGly143WT, MCGln6nsp5|6/MCGly143Beta, MCGln6nsp5|6/MCGly143Omicron. Gln6 seems essential to maintain strong or medium hydrogen bonding interactions with the residues Gly143 (in the S1 subsite) or Glu166 (S4 subsite), suggesting that these residues are fundamental for the enzyme/substrate recognition.
Other positions of both nsp4|5 and nsp5|6 were found to be involved in interactions in different 3CLpro variants. The MCAla3nsp4|5 makes strong hydrogen bonding interactions only with MCThr190WT (71% hydrogen bond occupancy). The MCVal4nsp4|5 is key to make strong and medium hydrogen bonding interactions with Glu166 in all 3CLpro. The MCLeu5nsp4|5 forms a strong hydrogen bonding interaction with SCGln189WT, this interaction being weak in the other variants of 3CLpro. Considering nsp5|6, the MCThr4 makes strong hydrogen bonding with the main chain of Glu166 in all 3CLpro while MCSer7 forms strong hydrogen interactions with only the main chain of the MCTHR26Beta. The 3CLproWT inhibitors may interact with other residues such as His41, Asn142, Ser144, Cys145, His164, Arg188, and Gln192 (Table S1). However, considering that Gly143 and Glu166 are key in substrate recognition, we suggest that these residues be incorporated as pharmacophoric centers of Beta and Omicron variants in drug design. Our analysis shows that Gly143 and Glu166 are essential residues to interact with substrate Gln6 and, therefore, are potential broad-spectrum pharmacophoric centers of SARS-CoV-2 3CLpro VOCs.
4. Conclusion
The 3CLpro mutations may be related to the high viral load observed for SARS-CoV-2 VOCs. RMSD values of the substrate nsp4|5 interacting with 3CLpro from all variants analyzed indicates conformational stability in the active site, with significantly higher stability being observed for 3CLproWT and 3CLproOmicron. The 3CLpro variants interacting with nsp4|5 and nsp 5|6 showed similar profiles of RMSF, suggesting little variation in overall flexibility. Furthermore, the 3CLproWT, 3CLproBeta and 3CLproOmicron did not present significant differences in the estimated KD values in their interactions with the substrates, presenting the same order of magnitude for their binding affinities. However, the cleavage rate of nsp 4|5 may be related to the increased viral load of these VOCs because the releasing nsp4 is essential for replication and assembly of replicative structures of the virus [33]. Our results have limitations due to the use of a simplified system for performing the molecular dynamics calculations. Kinetic studies measuring the catalytic efficiency (kcat/KM) of the substrates in these 3CLpro variants are therefore required to confirm our computational data. Importantly, despite these limitations, our simulations allowed us to suggest that Gly143 and Glu166 are key in substrate recognition and may be incorporated as pharmacophoric centers of Beta and Omicron variants in drug design. Our conclusion that Gly143 and Glu166 are essential residues to interact with the substrate’s Gln6 residue implies that these residues have great potential to be used as broad-spectrum pharmacophoric centers of SARS-CoV-2 3CLpro.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The authors acknowledge the National Council for Scientific and Technological Development (CNPq), the Coordination for the Improvement of Higher Education Personnel (CAPES, grant 88887.374931/2019-00, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Finance Code 01), and the São Paulo Research Foundation (FAPESP, grants 2019/00195-2, 2020/04680-0, 2016/09047-8), Rede Virus MCTI (grant FINEP 0459/20), Brazil, for financial support. The authors also acknowledge Geoambiente Sensoriamento Remoto LTDA for their generous support and for providing access to the Google Cloud services supported by grant FAPESP 2021/00070-5.
Conflicts of Interest
The authors declare no conflict of interest.
References
- de Souza, A.S.; de Freitas Amorim, V.M.; Guardia, G.D.A.; Dos Santos, F.F.; Ulrich, H.; Galante, P.A.F.; de Souza, R.F.; Guzzo, C.R. Severe Acute Respiratory Syndrome Coronavirus 2 Variants of Concern: A Perspective for Emerging More Transmissible and Vaccine-Resistant Strains. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- WHO. Available online: https://covid19.who.int/ (accessed on 3 August 2022).
- Milman, O.; Yelin, I.; Aharony, N.; Katz, R.; Herzel, E.; Ben-Tov, A.; Kuint, J.; Gazit, S.; Chodick, G.; Patalon, T.; et al. Community-Level Evidence for SARS-CoV-2 Vaccine Protection of Unvaccinated Individuals. Nat. Med. 2021, 27, 1367–1369. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Colaneri, M.; Di Filippo, A.; Blanchini, F.; Bolzern, P.; De Nicolao, G.; Sacchi, P.; Colaneri, P.; Bruno, R. Modeling Vaccination Rollouts, SARS-CoV-2 Variants and the Requirement for Non-Pharmaceutical Interventions in Italy. Nat. Med. 2021, 27, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A.; Mulholland, K. Effectiveness of an Inactivated SARS-CoV-2 Vaccine. New England Journal of Medicine 2021, 385, 946–948. [Google Scholar] [CrossRef] [PubMed]
- Halley, J.M.; Vokou, D.; Pappas, G.; Sainis, I. SARS-CoV-2 Mutational Cascades and the Risk of Hyper-Exponential Growth. Microb. Pathog. 2021, 161, 105237. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Corado, A.L.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- Li, M.; Lou, F.; Fan, H. SARS-CoV-2 Variants of Concern Delta: A Great Challenge to Prevention and Control of COVID-19. Signal Transduction and Targeted Therapy 2021, 6. [Google Scholar] [CrossRef]
- Barnard, R.C.; Davies, N.G.; Pearson, C.A.B.; Jit, M.; John Edmunds, W. Projected Epidemiological Consequences of the Omicron SARS-CoV-2 Variant in England, December 2021 to April 2022 2021.
- Khare, S.; GISAID Global Data Science Initiative (GISAID); Munich; Germany; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; et al. GISAID’s Role in Pandemic Response. China CDC Weekly 2021, 3, 1049–1051.
- GISAID - NextStrain. Available online: https://gisaid.org/phylodynamics/global/nextstrain/ (accessed on 30 March 2023).
- de Souza, A.S.; de Souza, R.F.; Guzzo, C.R. Quantitative Structure-Activity Relationships, Molecular Docking and Molecular Dynamics Simulations Reveal Drug Repurposing Candidates as Potent SARS-CoV-2 Main Protease Inhibitors. J. Biomol. Struct. Dyn. 2021, 1–18. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.S.; de Freitas Amorim, V.M.; Guardia, G.D.A.; Dos Santos, F.R.C.; Dos Santos, F.F.; de Souza, R.F.; de Araujo Juvenal, G.; Huang, Y.; Ge, P.; Jiang, Y.; et al. Molecular Dynamics Analysis of Fast-Spreading Severe Acute Respiratory Syndrome Coronavirus 2 Variants and Their Effects on the Interaction with Human Angiotensin-Converting Enzyme 2. ACS Omega 2022, 7, 30700–30709. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.S.; de Amorim, V.M.F.; de Souza, R.F.; Guzzo, C.R. Molecular Dynamics Simulations of the Spike Trimeric Ectodomain of the SARS-CoV-2 Omicron Variant: Structural Relationships with Infectivity, Evasion to Immune System and Transmissibility. J. Biomol. Struct. Dyn. 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.S.; Rivera, J.D.; Almeida, V.M.; Ge, P.; de Souza, R.F.; Farah, C.S.; Ulrich, H.; Marana, S.R.; Salinas, R.K.; Guzzo, C.R. Molecular Dynamics Reveals Complex Compensatory Effects of Ionic Strength on the Severe Acute Respiratory Syndrome Coronavirus 2 Spike/Human Angiotensin-Converting Enzyme 2 Interaction. J. Phys. Chem. Lett. 2020, 11, 10446–10453. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun Biol 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of Key Interactions between SARS-CoV-2 Main Protease and Inhibitor Drug Candidates. Sci. Rep. 2020, 10, 12493. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Shi, J.; Song, J. The Catalysis of the SARS 3C-like Protease Is under Extensive Regulation by Its Extra Domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of Coronavirus Main Proteinase Reveals Combination of a Chymotrypsin Fold with an Extra Alpha-Helical Domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Iketani, S.; Hong, S.J.; Sheng, J.; Bahari, F.; Culbertson, B.; Atanaki, F.F.; Aditham, A.K.; Kratz, A.F.; Luck, M.I.; Tian, R.; et al. Functional Map of SARS-CoV-2 3CL Protease Reveals Tolerant and Immutable Sites. Cell Host Microbe 2022. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro Proteinase Cleavage Sites: Possible Relevance to SARS Virus Pathology. BMC Bioinformatics 2004, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Grottesi, A.; Bešker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, Y.; Liu, X.; Jin, Z.; Duan, Y.; Zhang, Q.; Wu, C.; Feng, L.; Du, X.; Zhao, J.; et al. Structural Basis for Replicase Polyprotein Cleavage and Substrate Specificity of Main Protease from SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2117142119. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined Drug Repurposing and Virtual Screening Strategies with Molecular Dynamics Simulation Identified Potent Inhibitors for SARS-CoV-2 Main Protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef] [PubMed]
- Jade, D.; Ayyamperumal, S.; Tallapaneni, V.; Nanjan, C.M.J.; Barge, S.; Mohan, S.; Nanjan, M.J. Virtual High Throughput Screening: Potential Inhibitors for SARS-CoV-2 PLPRO and 3CLPRO Proteases. European Journal of Pharmacology 2021, 901, 174082. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.-S.; Shin, D.H. Flavonoids with Inhibitory Activity against SARS-CoV-2 3CLpro. J. Enzyme Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef]
- Molavi, Z.; Razi, S.; Mirmotalebisohi, S.A.; Adibi, A.; Sameni, M.; Karami, F.; Niazi, V.; Niknam, Z.; Aliashrafi, M.; Taheri, M.; et al. Identification of FDA Approved Drugs against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) and 3-Chymotrypsin-like Protease (3CLpro), Drug Repurposing Approach. Biomed. Pharmacother. 2021, 138, 111544. [Google Scholar] [CrossRef]
- Oostra, M.; te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.M.; de Haan, C.A.M. Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J. Virol. 2007, 81, 12323–12336. [Google Scholar] [CrossRef]
- Puhach, O.; Adea, K.; Hulo, N.; Sattonnet, P.; Genecand, C.; Iten, A.; Jacquérioz, F.; Kaiser, L.; Vetter, P.; Eckerle, I.; et al. Infectious Viral Load in Unvaccinated and Vaccinated Individuals Infected with Ancestral, Delta or Omicron SARS-CoV-2. Nat. Med. 2022, 28, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger Maestro. Available online: https://www.schrodinger.com/maestro.
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. Journal of Molecular Modeling 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. Journal of Molecular Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lyngse, F.P.; Mølbak, K.; Skov, R.L.; Christiansen, L.E.; Mortensen, L.H.; Albertsen, M.; Møller, C.H.; Krause, T.G.; Rasmussen, M.; Michaelsen, T.Y.; et al. Increased Transmissibility of SARS-CoV-2 Lineage B.1.1.7 by Age and Viral Load. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef]
- Astuti, I. ; Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An Overview of Viral Structure and Host Response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Avti, P.; Chauhan, A.; Shekhar, N.; Prajapat, M.; Sarma, P.; Kaur, H.; Bhattacharyya, A.; Kumar, S.; Prakash, A.; Sharma, S.; et al. Computational Basis of SARS-CoV 2 Main Protease Inhibition: An Insight from Molecular Dynamics Simulation Based Findings. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, N.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. Computational Studies of Drug Repurposing and Synergism of Lopinavir, Oseltamivir and Ritonavir Binding with SARS-CoV-2 Protease against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2673–2678. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.-W.; Xu, Y.; Zhang, J.; Nan, M.-L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N Protein Counteract the RIG-I Signaling Pathway by Suppressing the Formation of Stress Granules. Signal Transduct Target Ther 2022, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Barrett, B.S.; Morrison, J.H.; Mickens, K.L.; Vladar, E.K.; Hasenkrug, K.J.; Poeschla, E.M.; Santiago, M.L. Interferon Resistance of Emerging SARS-CoV-2 Variants. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2203760119. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Flury, P.; Krüger, N.; Su, H.; Schäkel, L.; Barbosa Da Silva, E.; Eppler, O.; Kronenberger, T.; Nie, T.; Luedtke, S.; et al. Small-Molecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure-Activity Relationships, Antiviral Activity, and X-Ray Structure Determination. J. Med. Chem. 2022, 65, 9376–9395. [Google Scholar] [CrossRef]
- Johnson, T.O.; Adegboyega, A.E.; Ojo, O.A.; Yusuf, A.J.; Iwaloye, O.; Ugwah-Oguejiofor, C.J.; Asomadu, R.O.; Chukwuma, I.F.; Ejembi, S.A.; Ugwuja, E.I.; et al. A Computational Approach to Elucidate the Interactions of Chemicals From Targeted Toward SARS-CoV-2 Main Protease Inhibition for COVID-19 Treatment. Front. Med. 2022, 9, 907583. [Google Scholar] [CrossRef]
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Tuñón, I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir Ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and Molecular Dynamics Simulation Studies Reveal Potential Covalent and FDA-Approved Inhibitors of SARS-CoV-2 Main Protease 3CL. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef]
- Khan, A.; Heng, W.; Wang, Y.; Qiu, J.; Wei, X.; Peng, S.; Saleem, S.; Khan, M.; Ali, S.S.; Wei, D.-Q. In Silico and in Vitro Evaluation of Kaempferol as a Potential Inhibitor of the SARS-CoV-2 Main Protease (3CLpro). Phytother. Res. 2021, 35, 2841–2845. [Google Scholar] [CrossRef] [PubMed]
- Sk, M.F.; Roy, R.; Jonniya, N.A.; Poddar, S.; Kar, P. Elucidating Biophysical Basis of Binding of Inhibitors to SARS-CoV-2 Main Protease by Using Molecular Dynamics Simulations and Free Energy Calculations. J. Biomol. Struct. Dyn. 2021, 39, 3649–3661. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.; Chen, Y.-T.; Hsu, Y.-C.; Hsu, N.-Y.; Kuo, C.-J.; Tsai, H.P.; Kang, J.-J.; Huang, C.-H.; Chang, S.-Y.; Chang, Y.-H.; et al. Uncovering Flexible Active Site Conformations of SARS-CoV-2 3CL Proteases through Protease Pharmacophore Clusters and COVID-19 Drug Repurposing. ACS Nano 2021, 15, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, Y.; Wu, Y.; Yu, H.; Wang, Z.; Ma, Y. Pharmacophore-Based Virtual Screening of Potential SARS-CoV-2 Main Protease Inhibitors from Library of Natural Products. Nat. Prod. Commun. 2022, 17, 1934578X2211436. [Google Scholar] [CrossRef]
Figure 1.
Structural changes of different 3CLpro variants while interacting with nsp 4|5 substrate. Backbone root-mean-square deviation (RMSD) of a) 3CLproH41A, b) 3CLproWT, c) 3CLproBeta and d) 3CLproOmicron calculated using molecular dynamics simulations for 500 ns.
Figure 1.
Structural changes of different 3CLpro variants while interacting with nsp 4|5 substrate. Backbone root-mean-square deviation (RMSD) of a) 3CLproH41A, b) 3CLproWT, c) 3CLproBeta and d) 3CLproOmicron calculated using molecular dynamics simulations for 500 ns.
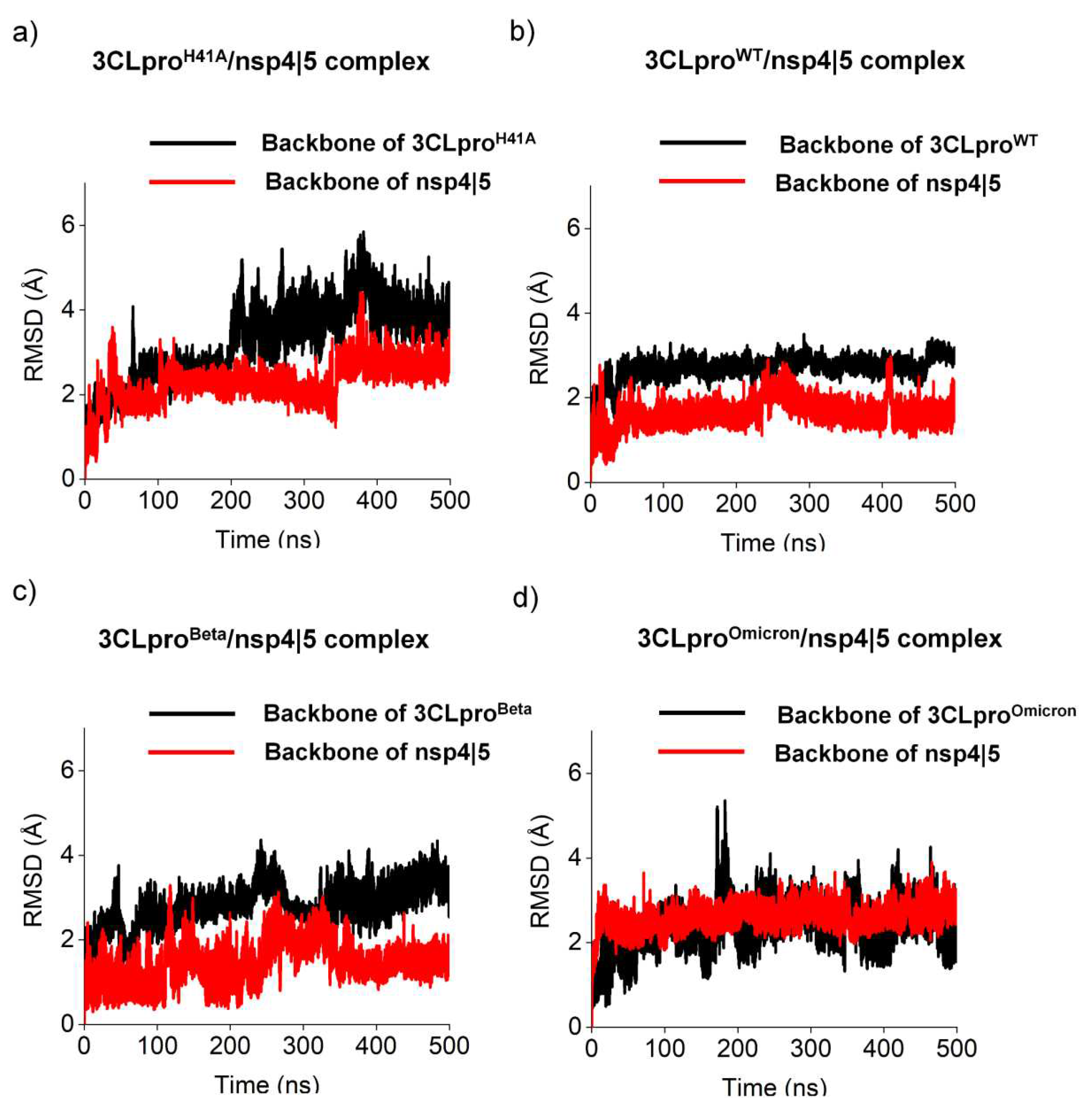
Figure 2.
Structural changes of different 3CLpro while interacting with nsp 5|6 substrate. Backbone root-mean-square deviation (RMSD) of a) 3CLproH41A, b) 3CLproWT, c) 3CLproBeta and d) 3CLproOmicron.
Figure 2.
Structural changes of different 3CLpro while interacting with nsp 5|6 substrate. Backbone root-mean-square deviation (RMSD) of a) 3CLproH41A, b) 3CLproWT, c) 3CLproBeta and d) 3CLproOmicron.
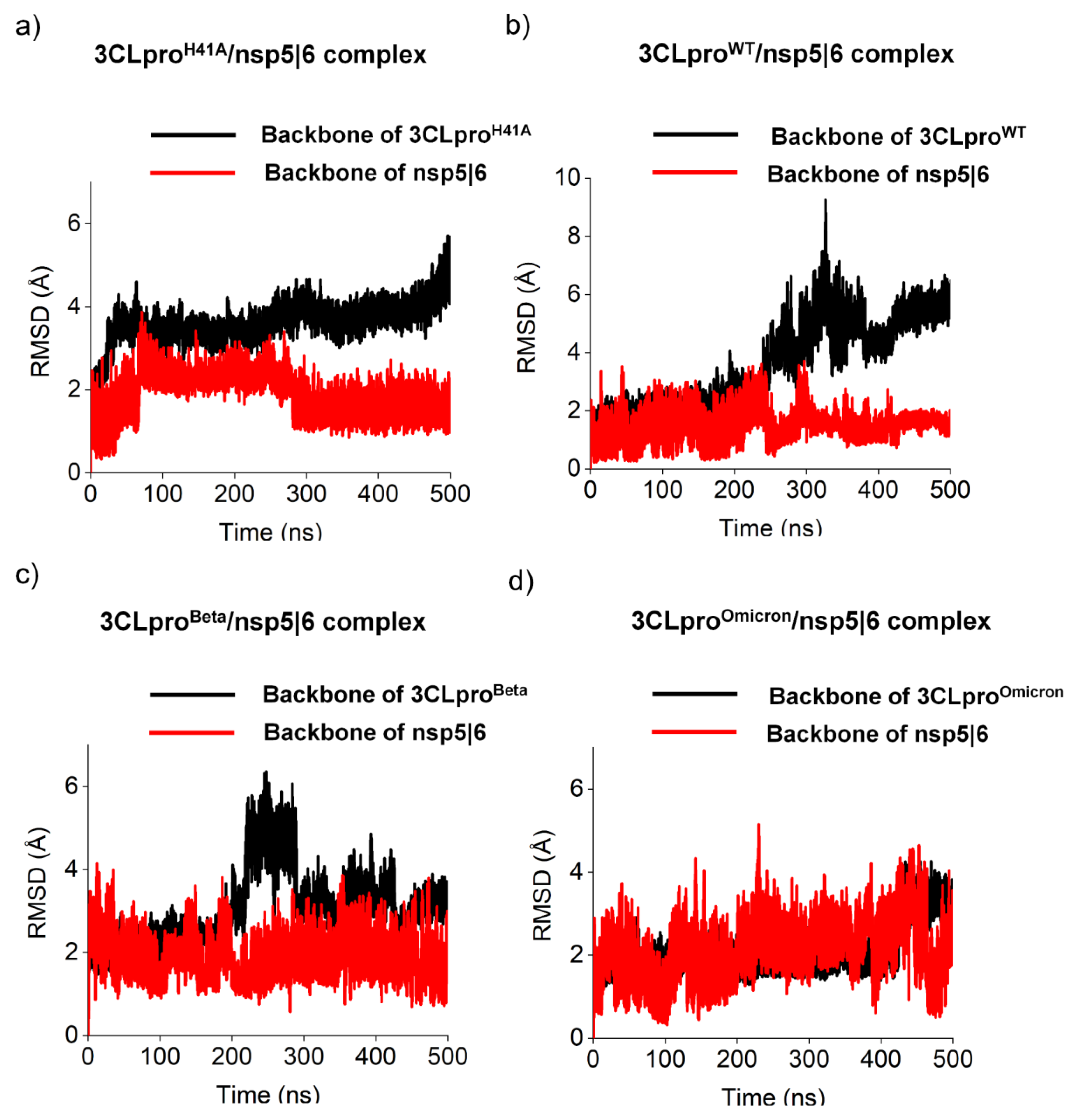
Figure 3.
Backbone root-mean-square fluctuation (RMSF) of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron. a) Backbone RMSF of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron while interacting with nsp 4|5. b) Backbone RMSF of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron while interacting with nsp 5|6.
Figure 3.
Backbone root-mean-square fluctuation (RMSF) of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron. a) Backbone RMSF of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron while interacting with nsp 4|5. b) Backbone RMSF of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron while interacting with nsp 5|6.
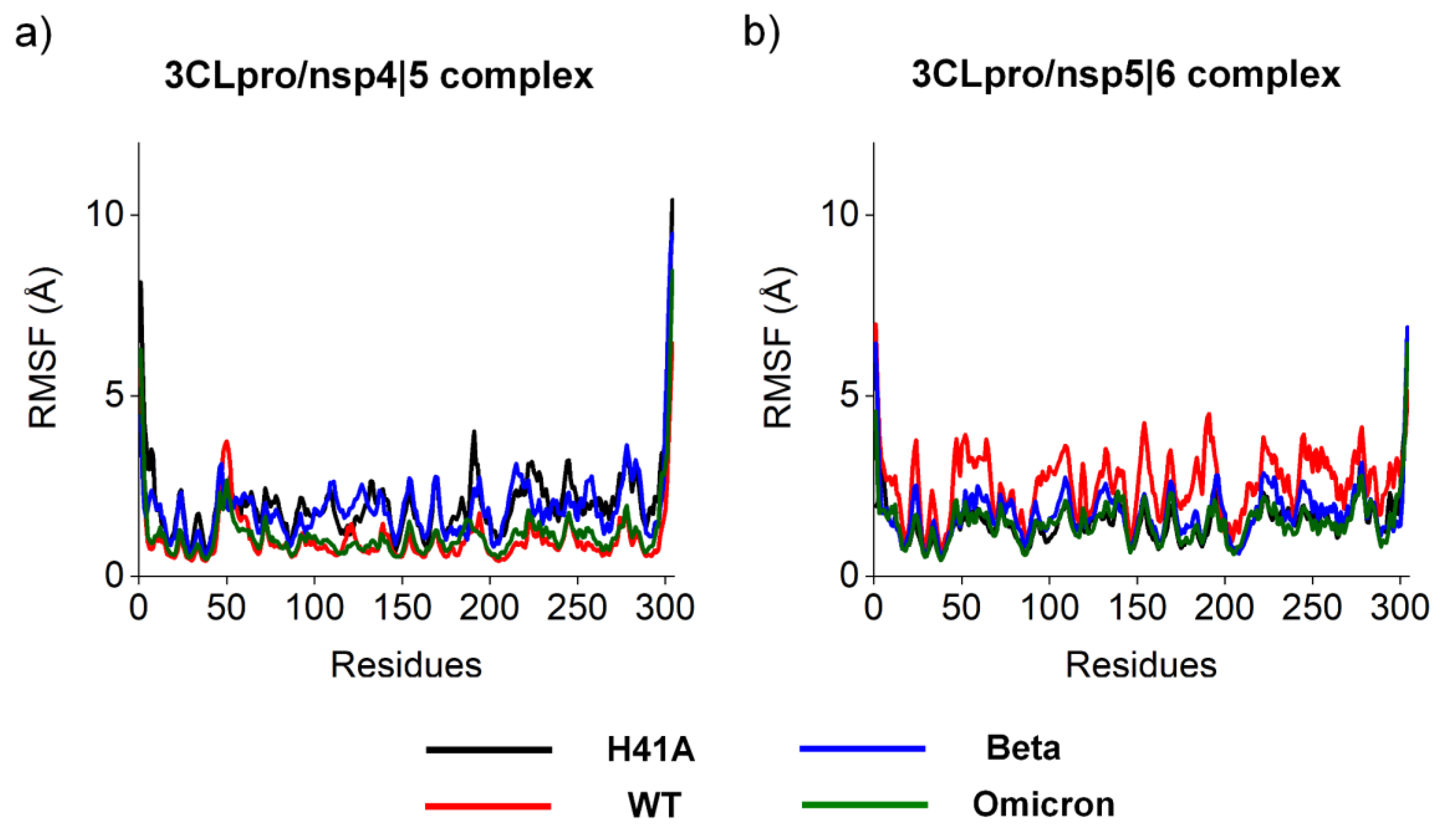

Table 1.
Perturbation free energy (FEP) estimated by molecular dynamics. Binding free energies (ΔG in kJ.mol-1) between different substrates and different 3CLpro.
Table 1.
Perturbation free energy (FEP) estimated by molecular dynamics. Binding free energies (ΔG in kJ.mol-1) between different substrates and different 3CLpro.
| ΔG (kJ.mol-1) | ||||
|---|---|---|---|---|
| 3CLproH41A | 3CLproWT | 3CLproBeta | 3CLproOmicron | |
| nsp 4|5 | 3057,57 | 3017,41 | 3016,58 | 3026 |
| nsp 5|6 | 3428,13 | 3418,68 | 3402,94 | 3412,27 |
| nsp 14|15 | 3290,29 | 3276,84 | 3292,97 | 3265,98 |
Table 2.
Relationship between the affinity rates obtained experimentally with the rates obtained by prediction. Prediction rates were described using a regression model and compared with experimental KD data from Zhao and co-authors [26].
Table 2.
Relationship between the affinity rates obtained experimentally with the rates obtained by prediction. Prediction rates were described using a regression model and compared with experimental KD data from Zhao and co-authors [26].
| KD (μM) | |||||
|---|---|---|---|---|---|
| Experimental | Prediction | ||||
| 3CLproH41A | 3CLproH41A | 3CLproWT | 3CLproBeta | 3CLproOmicron | |
| nsp 4|5 | 28 ± 3 [26] | 36.9 | 22,0 | 21,8 | 24,6 |
| nsp 5|6 | 2,730 ± 900 [26] | 4,326.4 | 3,831.3 | 3,129.3 | 3,528.2 |
| nsp 14|15 | 1,530 ± 350 [26] | 735.0 | 618.3 | 760.8 | 537.7 |
Table 3.
Cleavage rates of different 3CLpro. To calculate the cleavage rate, the frequency at which the catalytic dyad of the 3CLpro protein approached the substrate was taken into account, taking into account a cutoff of 4.5 Å.
Table 3.
Cleavage rates of different 3CLpro. To calculate the cleavage rate, the frequency at which the catalytic dyad of the 3CLpro protein approached the substrate was taken into account, taking into account a cutoff of 4.5 Å.
| Cleavage rate (cut of 4.5 Å) | |||
|---|---|---|---|
| 3CLproWT | 3CLproBeta | 3CLproOmicron | |
| nsp 4|5 | 154 | 9941 | 555 |
| nsp 5|6 | 1025 | 70 | 445 |
Table 4.
Hydrogen bonding occupancies of substrate nsp 4|5 interacting with 3CLpro variants. The hydrogen bonding occupancies considered relevant were at least 20%. Note that MC = main chain; SC = Side chain; HBA = Hydrogen bonding acceptor; HBD = Hydrogen bonding receptor.
Table 4.
Hydrogen bonding occupancies of substrate nsp 4|5 interacting with 3CLpro variants. The hydrogen bonding occupancies considered relevant were at least 20%. Note that MC = main chain; SC = Side chain; HBA = Hydrogen bonding acceptor; HBD = Hydrogen bonding receptor.
| Amino acid sequence of 3CLpro variants | ||||
|---|---|---|---|---|
| nsp 4|5 | H41A | WT | Beta | Omicron |
| MCAla3 |
SCGln189HBD (31.6) SCGln189HBA (24.9) |
MCThr190HBA (71.0) SCPro168HBD (22.0) |
MCThr190HBA (26.8) | - |
| MCVal4 |
MCGlu166HBA (62.1) MCGlu166HBD (54.4) |
MCGlu166HBA (41.9) MCGlu166HBD (75.6) SCGln189HBA (20.2) |
MCGlu166HBA (38.6) MCGlu166HBD (41.9) |
MCGlu166HBA (56.3) MCGlu166HBD (49.0) |
| MCLeu5 | - | SCGln189HBA (50.6) | SCGln189HBA (28.7) | - |
| SCGln6 |
SCGlu166HBA (72.7) MCLeu141HBA (21.9) |
SCGlu166HBA (20.4) |
SCGlu166HBA (26.3) |
SCHis163HBA (70.3) MCPhe140HBA (47.9) |
| MCGln6 | - |
MCHis164HBA (50.6) SCCys145HBD (23.4) MCGly143HBD (68.2) |
- |
SCCys145HBD (37.4) MCGly143HBD (81.6) |
| SCSer7 | - | SCHis41HBA (29.0) | SCThr25HBD (21.7) | - |
| MCGly8 |
MCThr26HBA (30.0) SCAsn142HBD (20.8) |
- | SCAsn142HBD (36.2) | SCAsn142HBD (34.4) |
Table 5.
Hydrogen bonding occupancies of substrate nsp 5|6 interacting with 3CLpro variants. The hydrogen bonding occupancies considered relevant are at least 20%. Note that MC = main chain; SC = Side chain; HBA = Hydrogen bonding acceptor; HBD = Hydrogen bonding receptor.
Table 5.
Hydrogen bonding occupancies of substrate nsp 5|6 interacting with 3CLpro variants. The hydrogen bonding occupancies considered relevant are at least 20%. Note that MC = main chain; SC = Side chain; HBA = Hydrogen bonding acceptor; HBD = Hydrogen bonding receptor.
| Amino acid sequence of 3CLpro variants | ||||
|---|---|---|---|---|
| nsp 5|6 | H41A | WT | Beta | Omicron |
| MCVal3 |
MCThr190HBA (30.4) SCGln189HBD (20.9) |
SCGln189HBD (26.3) | SCGln189HBD (23.4) | SCGln189HBD (27.3) |
| MCThr4 |
MCGlu166HBA (58.3) MCGlu166HBD (55.9) |
MCGlu166HBA (59.6) MCGlu166HBD (74.9) |
MCGlu166HBA (52.5) MCGlu166HBD (70.5) |
MCGlu166HBA (59.0) MCGlu166HBD (62.6) |
| SCGln6 |
MCGly143HBD (66.3) MCSer144HBD (22.4) |
SCGlu166HBD (36.4) | SCGlu166HBD (49.9) | SCGlu166HBA (42.5) |
| MCGln6 | - | MCGly143HBD (31.6) | MCGly143HBD (36.6) |
MCGly143HBD (42.9) SCCys145HBD (25.4) mCHis164HBA (22.7) |
| SCSer7 | MCAla41HBA (30.1) | SCHis41HBA (22.2) | SCHis41HBA (27.1) | SCHis41HBA (22.2) |
| MCSer7 | MCTHR26HBA (30.9) | - | MCTHR26HBD (53.1) | - |
| MCAla8 | - | - | - | SCAsn142HBA (21.3) |
| MCVal9 | MCSER46HBA (30.1) | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



