Submitted:
31 January 2023
Posted:
31 January 2023
You are already at the latest version
Abstract
In this paper we propose a computational framework that merges mechanistic modeling with deep neural networks obeying the Systems Biology Markup Language (SBML) standard. Over the last 20 years, the systems biology community has developed a large number of mechanistic models in SBML that are currently stored in public databases. With the proposed framework, existing SBML mechanistic models may be upgraded to hybrid systems through the incorporation of deep neural networks into the model core, using a freely available python tool. The so-formed hybrid mechanistic/neural network models are trained with a deep learning algorithm based on the adaptive moment estimation method (ADAM), stochastic regularization and semidirect sensitivity equations. The trained hybrid models are encoded in SBML and stored back in model databases, where they can be further analyzed as regular SBML models. The application of this approach is illustrated with three well-known case studies: the threonine synthesis model in Escherichia coli, the P58IPK signal transduction model, and the Yeast glycolytic oscillations model. The proposed framework is expected to greatly facilitate the widespread use of hybrid modeling techniques for systems biology applications.
Keywords:
hybrid modeling
; deep neural networks
; deep learning
; SBML
; systems biology
; computational modeling
1. Introduction
The combination of biological mechanisms with machine learning (ML) in hybrid models is a topic with growing awareness in the systems biology community. In a recent review, Antonakoudis et al. [1] highlighted the potential of combining GEnome-scale Modeling (GEM) and ML for systems biology applications. Hybrid modeling is, however, a well-established topic in process systems engineering (e.g. review by von Stosch et al., [2]). Psichogios and Ungar [3] described one of the first biologic applications of hybrid modeling. The proposed hybrid model combined material balance equations of biochemical species with a shallow feedforward neural network in a common mathematical structure. Thompson and Kramer [4] framed this problem as hybrid semiparametric modeling, as such models merge parametric functions (stemming from knowledge) with nonparametric functions (stemming from data) in the same mathematical structure. Schubert et al. [5] presented the first industrial application of hybrid modeling (material balance equations merged with neural networks) to a Baker’s yeast process. Many studies followed covering a wide array of microbial, animal cells, mixed microbial and enzyme biocatalysis problems in different industries such as wastewater treatment, clean energy, biopolymers and biopharmaceutical manufacturing (e.g. review by Agharafeie et al. [6]). Hybrid models have been mainly applied for predictive modeling/process analysis, process monitoring/software sensors, open- and closed-loop control, batch-to-batch control, model predictive control, intensified design of experiments, process analytical technology, quality-by-design and more recently digital twins mostly for upstream processing [2,6].
Conversely to process systems engineering, the application of hybrid modeling to systems biology has a considerable lag. Antonakoudis et al. [1] recently reviewed the efforts to integrate GEMs with supervised and unsupervised ML. Kim et al. [7] reviewed ML applications in construction and simulation of GEMs and ML applications in use of GEM derived information. The integration of GEMs and ML may be realized through a hybrid pipeline of activities where both frameworks participate to solve particular sub-tasks. Alternatively, GEMs and ML may be merged together in a common mathematical structure, e.g. through hybrid semiparametric modeling, which is the focus of the present paper. Hybrid metabolic flux analysis, combining metabolic networks and principal component analysis (PCA) in the same linear model, has been addressed by Carinhas et al. [8] and Isidro et al. [9]. Hybrid metabolic modeling combining metabolic networks and partial least squares has been proposed by Ferreira et al. [10] and Teixeira et al. [11]. The combination of systems of ODEs with neural networks (hybrid ODEs formalism) has been addressed by von Stosch et al. [12] for modeling biochemical networks with intrinsic time delays. The merge of elementary flux models (EMs) and PCA for hybrid metabolic pathway analysis has been addressed by Folch-Fortuny et al. [13] and von Stosch et al. [14]. Hybrid dynamic modeling that combines ODEs, PCA and EMs has been addressed by Folch-Fortuny et al. [15]. Yang et al. [16] developed a white-box machine learning approach, leveraging carefully curated biological network models to mechanistically link input and output data, for revealing metabolic mechanisms of antibiotic lethality. Lewis and Kemp [17] applied genome-scale Flux Balance Analysis (FBA) to generate data to train ML classifiers to predict tumor radiosensitivity. Vijayakumar et al. [18] developed a hybrid pipeline combining multi-omic MLs with genome-scale FBA to analyze the phenotypic potential of cyanobacterium. Recently, Ramos et al. [19] proposed a hybrid FBA technique that merges GEMs and PCA in a common linear program with mechanistic decision variables (fluxes) simultaneously with empirical decision variables (scores of principal components).
A large number of systems biology models, including GEMs, have been developed and stored in databases (e.g. BioModels [20], JWS online [21], and KiMoSys [22]) in the Systems Biology Markup Language (SBML) format [23]. Previous hybrid modeling efforts did not comply with the SBML format. This significantly hinders the interlink between both modeling approaches. Here, we propose a hybrid modeling framework that is SBML compatible. A previously published python package, SBML2HYB, is used to convert existing systems biology models into hybrid models and vice-versa [24]. The so-formed hybrid models are trained with a deep learning algorithm based on ADAM, stochastic regularization and semidirect sensitivity equations [25]. The final trained hybrid models are uploaded in SBML databases, where they may be further analyzed as regular SBML models. This procedure was applied to three well-known cases studies: the E. coli threonine pathway model in Escherichia coli [26], the P58IPK signal transduction pathway model [27] and the yeast glycolytic oscillations model [28].
2. Materials and methods
2.1. General SBML hybrid model
SBML models are organized in j=1,..,n compartments with size . Each compartment contains species with concentration vector . The species are interlinked though reactions with stoichiometry and reaction kinetics . SBML models also contain parameters, , with given initial values (parameters may be local to compartments or global; for simplicity we assume global). The parameter values may change over time according to predefined algebraic rules. The compartment size may also change over time according to predefined compartment rate rules (other rate rules were not considered here for simplicity). External time dependent stimuli may be defined through events, giving rise to a vector of exogenous input variables, , that may change over time. With these elements, the dynamics of biochemical species in a generic compartment may be described by the following ODEs model:
Equation (1a) is a conservation law of mass assuming a perfectly mixed compartment. Equation (1b) represents a generic compartment rate rule in case the compartment size changes over time. Equation (1c) represents generic algebraic rules to compute model parameters over time. Equations (1a-c) are of parametric nature with fixed structure stemming from prior knowledge (e.g. mass conservation laws, reaction stoichiometry and enzyme kinetics). Some variables may however lack a mechanistic basis (e.g. unknown reaction kinetics mechanism, unknown physicochemical properties of some molecular species such as charge or glycosylation pattern). In the general SBML hybrid model, such variables are defined as loose nonparametric functions, , without fixed structure. Such variables lacking a mechanistic basis are computed by a deep feedforward neural network (FFNN) with hidden layers as function of species concentrations, exogenous inputs, and other relevant variables:
A non-linear pre-processing function, , may be used to compute the FFNN inputs to improve the training. The represents the nodes transfer function in the hidden layers (in this study always the tangent hyperbolic function). The nodes connection weights and are calculated during the training of the model, for which a representative data set is needed. Equations (1)-(2) are a particular form of the generic bioreactor hybrid model published by Pinto et al. [25]. The training of the hybrid models in the present study were performed with the same Octave/Matlab tool for deep learning of hybrid models described by Pinto et al. [25]. More specifically, the adaptive moment estimation algorithm (ADAM) with stochastic minibatch and weights dropout regularization and semidirect sensitivity equations were applied in the present study. For further details, the reader is referred to [25].
2.1. Interfacing with SBML databases and SBML modeling tools
The SBML2HYB python package was adopted to read SBML models, redesign to hybrid models and to store in model databases [24]. This freely available python package converts existing systems biology models stored in databases in SBML format into hybrid models that combine mechanistic equations and deep neural networks (currently limited to FFNNs). SBML is not a common format to encode machine learning. An intermediate HMOD format supports the conversion process. The HMOD format is a text-based file (ASCII) with the list of properties (species, reactions, parameters, rates and rules) defining the model that make it easy to parse. This HMOD format organizes the hybrid model components in a similar manner to SBML, by considering any number of species with a certain initial concentration distributed among any number of compartments. These species are then interlinked through a list of reactions and rate rules. The user inputs the information of the deep neural network into the HMOD format either manually or by a pre-configured neural network in Python keras. The resulting hybrid model in HMOD format is reconverted to SBML and stored back in model databases. In this step, the FFNN equations (2a-c) are mapped to assignment rules in the SBML format. The resulting hybrid models in SBML format can be simulated, analyzed, trained with existing tools such as MATLAB and COPASI [29] or special purpose tools with training algorithms for hybrid models that are able to read SBML files. For further details the reader is referred to [24].
2.3. Case studies
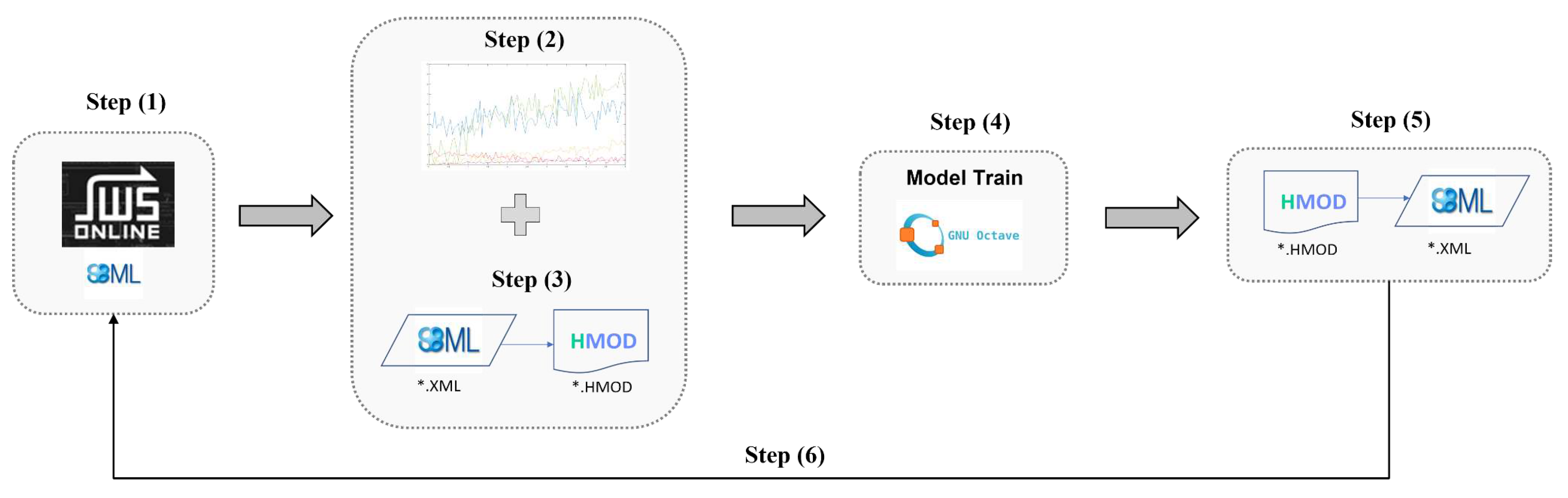
The SBML compatible hybrid modeling framework was applied to three systems biology case studies freely available in the JWS Online database (https://jjj.bio.vu.nl/models/) [21] with the accession ID given in Table 1. The first case study is the metabolic network describing the synthesis of threonine in E. coli proposed by Chassagnole et al. [26]. The second case study is the P58IPK signal transduction network to study Influenza infection dynamics proposed by Goodman et al. [27]. The third case study is the reduced yeast glycolytic model with preserved limit cycle stability proposed by Dano et al. [28]. In order to upgrade the original mechanistic models in hybrid mechanistic/neural network versions, the following pipeline of activities (Figure 1) was applied to each of the case studies:
- Step 1: The original systems biology model was retrieved from the JWS database in SBML format. The respective files are provided as supplementary material
- Step 2: A synthetic time series dataset was generated by simulating the original model in the JWS platform. The resulting data set is provided as supplementary material. This data is needed to train the hybrid model as proof-of-concept. No experimental data was used in this step. More details are provided in the results section.
- Step 3: A feedforward neural network (FFNN) was inserted in the mechanistic model and converted to the HMOD format using the SBML2HYB python tool, freely available in Pinto et al. [24]. The size of the FFNN and interface with the mechanistic model depended on the case study. More details are given in the results section.
- Step 4: the hybrid mechanistic/FFNN ensemble encoded in HMOD format was trained using a Octave/Matlab tool by applying the deep learning approach described by Pinto et al. [25] and the dataset generated in step 2. The ADAM algorithm with stochastic regularization and semidirect sensitivity equations was employed. Implementation details varied in the case studies (more to this in the results section). It should be noted that developing a robust hybrid model may require a deeper analysis than the one performed in this study. Factors such as the size/depth of the FFNN, data partitioning, weights initialization, sensitivity equations and ADAM parameters were investigated elsewhere [25]. The concern here was the proof-of-concept that such hybrid models may be efficiently trained to a comparable performance of the original mechanistic models. The final trained hybrid model with the updated FFNN weights was saved in HMOD format.
- Step 5: The trained hybrid model in HMOD format was reconverted to SBML using the SBML2HYB tool. In this step, the FFNN information is mapped as assignment rules in the SBML file. The hybrid model structure encoded in SBML was visualized using the freely available Cytoscape cy3sbml tool [30]. The respective hybrid model SBML files are provided as supplementary material.
- Step 6: The final trained hybrid model in SBML format is now freely available for the community to analyze. For proof-of-concept, the original SBML model (step 1) and the final trained SBML hybrid model (step 5) were simulated and compared using the JWS online simulator (https://jjj.bio.vu.nl/models/experiments/) showing that their output is practically coincident.

Table 1.
Summary of the three SBML models that were redesigned to hybrid mechanistic/neural network models in the present study.
Table 1.
Summary of the three SBML models that were redesigned to hybrid mechanistic/neural network models in the present study.
| Case Study | Number of species | Number of reactions | Number of parameters | JWS Online ID | Reference |
|---|---|---|---|---|---|
| E. coli threonine synthesis pathway | 11 | 7 | 47 | chassagnole1 | [26] |
| P58IPK signal transduction pathway | 9 (4 fixed) | 9 | 10 | goodman | [27] |
| Yeast glycolytic oscillations | 7 (1 fixed) | 11 | 31 | dano1 | [28] |
Figure 1.
Workflow for redesigning existing SBML models stored in databases into hybrid mechanistic/neural network models.
Figure 1.
Workflow for redesigning existing SBML models stored in databases into hybrid mechanistic/neural network models.
3. Results and discussion
Case study 1: threonine synthesis pathway in E. coli
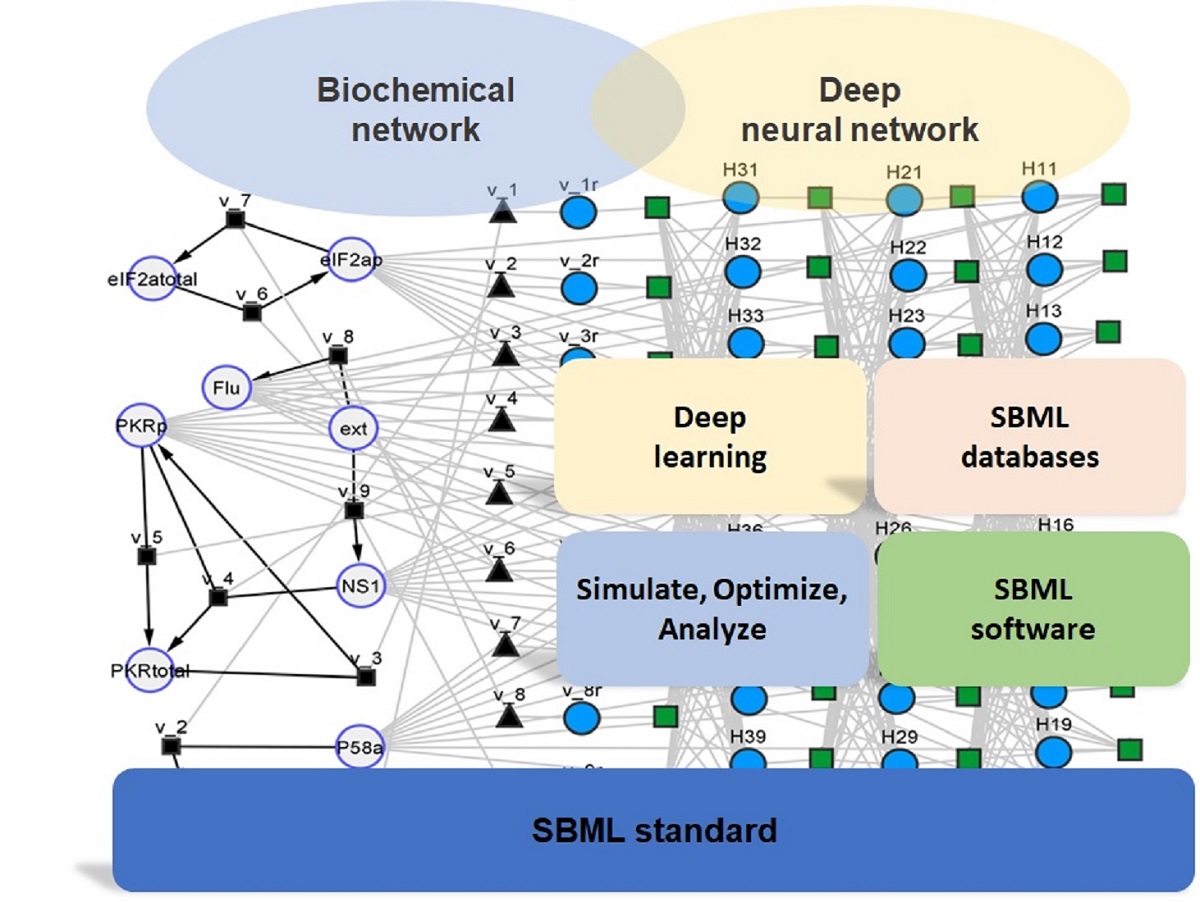
The first case study is the metabolic model proposed by Chassagnole et al. [26] describing the threonine synthesis pathway in E. coli (Table 1). This model dynamically simulates the time course of 11 species (adp, asa, asp, aspp, atp, hs, hsp, nadp, naph, phos and thr) in a single compartment corresponding to 11 ODEs. It has 7 reactions (with rates vak, vasd, vatpase, vhdh, vhk, vnadph_endo and vtsy) and 47 kinetic parameters (the names of variables were kept the same as the original SBML model to facilitate cross-reference; for details on variables the reader is referred to the original SBML model stored in the JWS Online database with accession ID ‘chassagnole’). A hybrid model was then created by combining a deep FFNN with the original mechanistic model following the previously described steps (Figure 1). The FFNN had the configuration 11×5×5×7 with a total number of 132 weights. The inputs to the FFNN were the concentrations of the 11 species (adp, asa, asp, aspp, atp, hs, hsp, nadp, naph, phos, thr), followed by 2 hidden layers (5×5) with tanh activation function, and 7 outputs corresponding to the maximum reaction rate vales of the 7 metabolic reactions. The kinetic law equations of the original SBML model are thus fully retained in the hybrid model. The job of the FFNN is thus to model the maximum reaction rate parameters as a function of species concentrations. The resulting hybrid model in SBML format can be visualized using the Cytoscape cy3sbml tool (Figure 2). The SBML hybrid model is represented as an heterogenous (hybrid) network composed of nodes and edges of different nature. On the biochemical network (left) side, the large circles represent the molecular species, which have a physical concentration associated. The small black squares and respective edges represent biochemical reactions with well-defined stoichiometry. The black triangles (with equal number as black squares) are the reaction kinetic rates. On the neural network side, the blue circles are neural network nodes with a numerical value defining the node strength. The green squares and respective edges represent signal propagation between nodes. The interlink between the two sides of the network is set by the black triangles, which for the present case study represent the maximum reaction rate parameters to be applied in the kinetic law equations. An interesting analogy may be established between the neural network part and the cell nucleus with associated signal transduction networks and gene regulatory networks, with the job to control the cellular metabolic processes.
The hybrid model was afterwards trained with a synthetic data set as proof-of-concept following the steps of Figure 1. A time series dataset was created by simulating the original SBML model directly in the JWS platform. A 2-factor central composite design of experiments (CC-DOE) was carried out to the initial concentrations of atp between 5 and 15 and of asp between 1 and 3 resulting in 9 experiments. The data for each experiment was simulated directly in the JWS Online platform and recorded as a time series with 100 data points and a sampling time of 1 (au) for each experiment. Finally, 10% Gaussian noise was added to concentrations of species thereby simulating experimental error. This synthetic dataset is available in the supplementary material (Simulation_data.xlsx; chassagnole_data sheet). From the 9 experiments, 8 were used for training whereas the 9th (the center point of the CC-DOE) was used for testing. The training was performed with ADAM with 0.001 learning rate and 5000 iterations, semidirect sensitivity equations and stochastic regularization with minibatch size of 0.78 and weights dropout of 0.22. Figure 3 shows the training and test Mean-Squared-Error (MSE) decay over training iteration. The training and test error reached a minimum after 2000 iterations. The final training and test MSE values are practically coincident (1.11 and 1.10 respectively) and very close to the noise MSE (0.93), denoting a successful training without overfitting.
The final hybrid model can be simulated and analyzed in any systems biology platform complying with the SBML standard. As proof-of-concept, the trained hybrid model in SBML format was uploaded to the JWS online platform. Figure 4 shows the JWS online simulation of the original model and of the trained hybrid model for the test experiment. The results show that the hybrid model perfectly mimicked the dynamics of the original mechanistic model.
The pipeline presented in Figure 1 may result in mathematical structures that are more detailed mechanistically and much more complex than previously published hybrid models. This may raise concerns about the training feasibility of embedded FFNNs. Pinto et al. [25] compared traditional shallow hybrid modeling (using the Levenberg-Marquardt algorithm coupled with the indirect sensitivity equations, cross-validation and tanh activation function) with deep hybrid modeling (using ADAM, semidirect sensitivity equations, stochastic regularization and multiple hidden layers). A clear advantage of adopting hybrid deep learning both in terms of predictive power and computational cost was shown. However, all experiments addressed had a simplistic mechanistic core. Here, case study 1 (which retained the original kinetic law equations in the hybrid model core) results suggest that the previously published deep learning approach is able to efficiently train hybrid models with complex parametric functions in its core.
3.1. Case study 2: P58IPK signal transduction pathway
The second case study was based on the viral infection model proposed by Goodman et al. [27] (Table 1). This study focused on the dynamics of the P58IPK signal transduction pathway during Influenza viral infection. A mathematical model was developed to evaluate the effect of protein P58a activation on the P58IPK pathway dynamics, particularly on the activation of the PKR kinase and on the phosphorylation of eIF2, which control viral protein expression. This model comprehends 9 species (Flu, NS1, P58a, P58total, PKRp, PKRtotal and eIF2ap, eIF2atotal, ext) in a single compartment, of which 4 were fixed (P48total, PKRtotal, eIF2atotal and ext) thus translated to 5 ODEs. The model also has 9 reactions and 10 parameters. This model is freely available in SBML at the JWS Online database (http://www.jjj.bio.vu.nl) under accession ID ‘goodman’. The names of variables were kept the same and are explained in the database. As in the previous example, a SBML hybrid model was created by combining a FFNN with the original mechanistic model following the pipeline of Figure 1. Figure 5 shows the resulting hybrid ensemble, with the right side representing the original mechanistic signal transduction network and the left side the added FFNN. The FFNN has 5 inputs corresponding to the concentrations of the 5 dynamical species (Flu, NS1, P58a, PKRp and EIF2ap), 3 hidden layers (10×10×10) with tanh activation function, and 9 outputs corresponding to the kinetic rates (v_1r, v_2r, v_3r, v_4r, v_5r, v_6r, v_7r, v_8r, v_9r as they are named in the original SBML implementation). In this case study, the FFNN completely replaced the kinetic laws of the original model, which were deleted in the hybrid model. This network may be interpreted as a hybrid signal transduction network with a physical part composed by proteins and an artificial part composed by neural network nodes.
The hybrid SBML model was afterward trained with a synthetic data set. A time series dataset was created by simulations of the original SBML model directly in the JWS platform following a similar procedure to case study 1. A 2-factor CC-DOE was carried out to the initial amount of Flu (overall level of infection within the host cell) between 2 and 6 and the initial amount of PKRp (phosphorylated PKR protein) between 0 and 2. The data for each experiment was simulated directly in the JWS Online platform as a time series with 100 points and sampling time of 0.05 (au). This resulted in a total of 9 experiments with 100 time points each, to which an additional 10% Gaussian noise was added to concentrations of species. As in the previous example, 8 experiments were used for training and a single experiment (the center point of the CC-DOE) for testing. This synthetic dataset is available as supplementary material (Simulation_data.xlsx; goodman_data sheet). The training was performed using ADAM with 0.001 learning rate and 5000 iterations, semidirect sensitivity equations and stochastic regularization (minibatch size of 0.78 and weight dropout of 0.22). The training converged after approximately 3000 iterations to a final MSE error of 1.00 and 1.03 for training and testing respectively (results not shown). These errors are slightly above the noise MSE of 0.92, denoting a successful training without overfitting. Finally, the trained hybrid model in SBML format was uploaded to the JWS online platform and simulated comparatively to the original mechanistic model (Figure 6). As in the previous case study, results show that the hybrid model was able to perfectly mimic the dynamics of the original mechanistic model.
3.1. Case study 3: yeast glycolytic oscillations
The third case study consisted in the reduced dynamical model of yeast glycolysis proposed by Dano et al. [28]. The proposed model is thus a reduced version of a more detailed yeast glycolysis model with preserved dynamical properties. More specifically, both the original and reduced models exhibit limit cycle stability under certain conditions, with a certain number of species showing stable oscillations over time. This model comprehends 8 species (ADP, AMP, ATP, BPG, DHAP, FBP, GAP, sink) in a single compartment. The species ‘sink’ was the only fixed, thus translating to a system of 7 ODEs. The model further comprehends 11 metabolic reactions and 31 parameters. This model is freely available in SBML format at the JWS Online database (http://www.jjj.bio.vu.nl) with accession ID ‘dano1’. The reader is referred to the database for further details on model variables. As in the previous case studies, a SBML hybrid model was created by combining a FFNN with the original mechanistic model (Figure 7). The right side represents the original metabolic network and the left side the incorporated FFNN. The FFNN has in this case 7 inputs corresponding to the concentrations of the 7 dynamical species (ADP, AMP, ATP, BPG, DHAP, FBP, GAP), 3 hidden layers (10×10×10) with tanh activation function, and 11 outputs corresponding to the kinetic rates (v_1r, v_2r, v_3r, v_4r, v_5r, v_6r, v_7r, v_8r, v_9r, v_10r, v_11r as they are named in the original SBML model). The FFNN configuration was thus (7×10×10×10×11) with 421 weights.
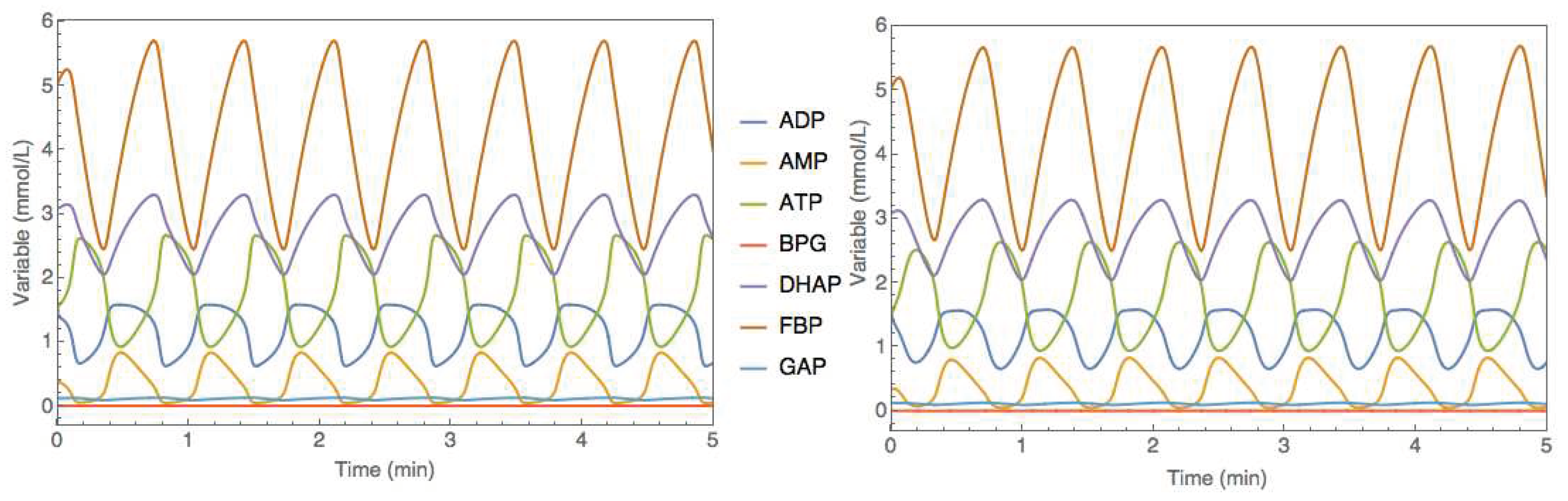
The hybrid SBML model was afterward trained with a synthetic dataset following a similar process to the previous case studies. A 2-factor CC-DOE was carried out by varying the amount of initial ADP concentration between 1 and 2 and the initial ATP concentration between 1 and 2 resulting in 9 experiments. Each experiment was simulated directly in the JWS Online platform with the resulting time series recorded with a sampling time of 0.05 (au) (100 time points). A 10% Gaussian noise was added to the concentrations of species. As before, 8 experiments were used for training and one experiment (the CC-DOE center point) used for testing. The synthetic data is available as supplementary material (Simulation_data.xlsx; dano1_data sheet). The hybrid model was then trained with this data using the same method as before (ADAM with 0.001 learning rate, 20000 iterations, semidirect sensitivity equations, stochastic regularization with minibatch size of 0.78 and weight dropout of 0.22). The training converged to a final MSE of 1.00 and 1.21 for the train and test partitions. The training error is slightly higher than the noise MSE of 0.93 denoting a successful training without overfitting. The test error is slightly higher than the train error due to the complex oscillatory dynamics. Unsurprisingly, limit cycle stability is a more challenging problem to be addressed with hybrid modeling. The trained hybrid model in SBML format was uploaded to the JWS online platform and simulated comparatively to the original metabolic model (Figure 8). Despite the higher test error, the hybrid model was able to reproduce very faithfully the oscillatory behavior as the original metabolic model.
4. Conclusions
Hybrid models combining mechanisms with machine learning is a topic with 3 decades of history in process systems engineering. The application of this approach to systems biology has however a considerable lag. With few exceptions, previously published hybrid modeling studies are limited to relatively simple mechanistic models (mechanistic scale-gap) and to relatively simple machine learning components (machine learning scale-gap). Here we propose a methodology for SBML compatible hybrid modeling that may significantly narrow the mechanistic scale-gap. It is shown with three examples how publicly available SBML models may be upgraded to hybrid mechanistic/neural network models still obeying to the SBML standard. Such hybrid models may be trained with state-of-the-art deep learning algorithms to either mimic, improve or extend existing SBML models. They can be further uploaded, trained and analyzed in SBML compatible software tools. All in all, we expect this framework to greatly facilitate the application of hybrid modeling techniques to systems biology problems.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization, methodology, software, data curation, investigation, writing, original draft preparation – José Pinto; Writing, review and editing - João Ramos; Conceptualization, methodology, writing, review, editing - Rafael S. Costa; Conceptualization, methodology, writing, review, editing and supervision - Rui Oliveira; All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Associate Laboratory for Green Chemistry - LAQV which is financed by national funds from FCT/MCTES (UIDB/50006/2020 and UIDP/50006/2020). This work has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement number 870292 (BioICEP project). JP acknowledges PhD grant (SFRD/BD14610472019), Fundação para a Ciência e Tecnologia (FCT) and RSC the contract CEECIND/01399/2017.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Antonakoudis, A.; Barbosa, R.; Kotidis, P.; Kontoravdi, C. The era of big data: Genome-scale modelling meets machine learning. Comput Struct Biotec 2020, 18, 3287–3300. [Google Scholar] [CrossRef] [PubMed]
- von Stosch, M.; Oliveira, R.; Peres, J.; de Azevedo, S.F. Hybrid semi-parametric modeling in process systems engineering: Past, present and future. Computers & Chemical Engineering 2014, 60, 86–101. [Google Scholar] [CrossRef]
- Psichogios, D.C.; Ungar, L.H. A Hybrid Neural Network-1st Principles Approach to Process Modeling. Aiche Journal 1992, 38, 1499–1511. [Google Scholar] [CrossRef]
- Thompson, M.L.; Kramer, M.A. Modeling Chemical Processes Using Prior Knowledge and Neural Networks. Aiche Journal 1994, 40, 1328–1340. [Google Scholar] [CrossRef]
- Schubert, J.; Simutis, R.; Dors, M.; Havlik, I.; Lubbert, A. Hybrid Modeling of Yeast Production Processes - Combination of a-Priori Knowledge on Different Levels of Sophistication. Chem Eng Technol 1994, 17, 10–20. [Google Scholar] [CrossRef]
- Agharafeie, R.; Oliveira, R.; Ramos, J.; Mendes, J. Application of Hybrid Neural Models to Bioprocesses: A Systematic Literature Review. 2023. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, G.B.; Lee, S.Y. Machine learning applications in genome-scale metabolic modeling. Current Opinion in Systems Biology 2021, 25. [Google Scholar] [CrossRef]
- Carinhas, N.; Bernal, V.; Teixeira, A.P.; Carrondo, M.J.T.; Alves, P.M.; Oliveira, R. Hybrid metabolic flux analysis: combining stoichiometric and statistical constraints to model the formation of complex recombinant products. Bmc Systems Biology 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Isidro, I.A.; Portela, R.M.; Clemente, J.J.; Cunha, A.E.; Oliveira, R. Hybrid metabolic flux analysis and recombinant protein prediction in Pichia pastoris X-33 cultures expressing a singlechain antibody fragment. Bioprocess and Biosystems Engineering 2016, 39, 1351–1363. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Dias, J.M.L.; Teixeira, A.P.; Carinhas, N.; Portela, R.M.C.; Isidro, I.A.; von Stosch, M.; Oliveira, R. Projection to latent pathways (PLP): a constrained projection to latent variables (PLS) method for elementary flux modes discrimination. Bmc Systems Biology 2011, 5. [Google Scholar] [CrossRef]
- Teixeira, A.P.; Dias, J.M.L.; Carinhas, N.; Sousa, M.; Clemente, J.J.; Cunha, A.E.; von Stosch, M.; Alves, P.M.; Carrondo, M.J.T.; Oliveira, R. Cell functional enviromics: Unravelling the function of environmental factors. Bmc Systems Biology 2011, 5. [Google Scholar] [CrossRef]
- von Stosch, M.; Peres, J.; de Azevedo, S.F.; Oliveira, R. Modelling biochemical networks with intrinsic time delays: a hybrid semi-parametric approach. Bmc Systems Biology 2010, 4. [Google Scholar] [CrossRef] [PubMed]
- Folch-Fortuny, A.; Marques, R.; Isidro, I.A.; Oliveira, R.; Ferrer, A. Principal elementary mode analysis (PEMA). Mol Biosyst 2016, 12, 737–746. [Google Scholar] [CrossRef]
- von Stosch, M.; Hamelink, J.M.; Oliveira, R. Hybrid modeling as a QbD/PAT tool in process development: an industrial E-coli case study. Bioprocess and Biosystems Engineering 2016, 39, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Folch-Fortuny, A.; Teusink, B.; Hoefsloot, H.C.J.; Smilde, A.K.; Ferrer, A. Dynamic elementary mode modelling of non-steady state flux data. Bmc Systems Biology 2018, 12. [Google Scholar] [CrossRef]
- Yang, J.H.; Wright, S.N.; Hamblin, M.; McCloskey, D.; Alcantar, M.A.; Schrubbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661. [Google Scholar] [CrossRef]
- Lewis, J.E.; Kemp, M.L. Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance. Nat Commun 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, S.; Rahman, P.K.S.M.; Angione, C. A Hybrid Flux Balance Analysis and Machine Learning Pipeline Elucidates Metabolic Adaptation in Cyanobacteria. Iscience 2020, 23. [Google Scholar] [CrossRef]
- Ramos, J.R.C.; Oliveira, G.P.; Dumas, P.; Oliveira, R. Genome-scale modeling of Chinese hamster ovary cells by hybrid semi-parametric flux balance analysis. Bioprocess Biosyst Eng 2022, 45, 1889–1904. [Google Scholar] [CrossRef]
- Le Novere, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Research 2006, 34, D689–D691. [Google Scholar] [CrossRef]
- Olivier, B.G.; Snoep, J.L. Web-based kinetic modelling using JWS Online. Bioinformatics 2004, 20, 2143–2144. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.S.; Verissimo, A.; Vinga, S. KiMoSys: a web-based repository of experimental data for KInetic MOdels of biological SYStems. BMC Syst Biol 2014, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Fineey, A.; Sauro, H.M.; al., e. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [CrossRef]
- Pinto, J.; Costa, R.S.; Alexandre, L.; Ramos, J.; Oliveira, R. SBML2HYB: a Python interface for SBML compatible hybrid modelling. Bioinformatics 2023. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Mestre, M.; Ramos, J.; Costa, R.S.; Striedner, G.; Oliveira, R. A general deep hybrid model for bioreactor systems: Combining first principles with deep neural networks. Computers & Chemical Engineering 2022, 165. [Google Scholar] [CrossRef]
- Chassagnole, C.; Fell, D.A.; Rais, B.; Kudla, B.; Mazat, J.P. Control of the threonine-synthesis pathway in Escherichia coli: a theoretical and experimental approach. Biochemical Journal 2001, 356, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.G.; Tanner, B.C.W.; Chang, S.T.; Esteban, M.; Katze, M.G. Virus infection rapidly activates the P58(IPK) pathway, delaying peak kinase activation to enhance viral replication. Virology 2011, 417, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Dano, S.; Madsen, M.F.; Schmidt, H.; Cedersund, G. Reduction of a biochemical model with preservation of its basic dynamic properties. Febs Journal 2006, 273, 4862–4877. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI — a COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef]
- Konig, M.; Drager, A.; Holzhutter, H.G. CySBML: a Cytoscape plugin for SBML. Bioinformatics 2012, 28, 2402–2403. [Google Scholar] [CrossRef]
Figure 2.
Hybrid model structure for the threonine synthesis pathway (case study 1) visualized in the cy3sbml tool [30]. Large circles represent biochemical species. Black squares and black edges represent biochemical reactions. Black triangles represent kinetic laws. Small blue circles represent neural network nodes. Green squares and gray edges represent signal propagation between neural network nodes.
Figure 2.
Hybrid model structure for the threonine synthesis pathway (case study 1) visualized in the cy3sbml tool [30]. Large circles represent biochemical species. Black squares and black edges represent biochemical reactions. Black triangles represent kinetic laws. Small blue circles represent neural network nodes. Green squares and gray edges represent signal propagation between neural network nodes.
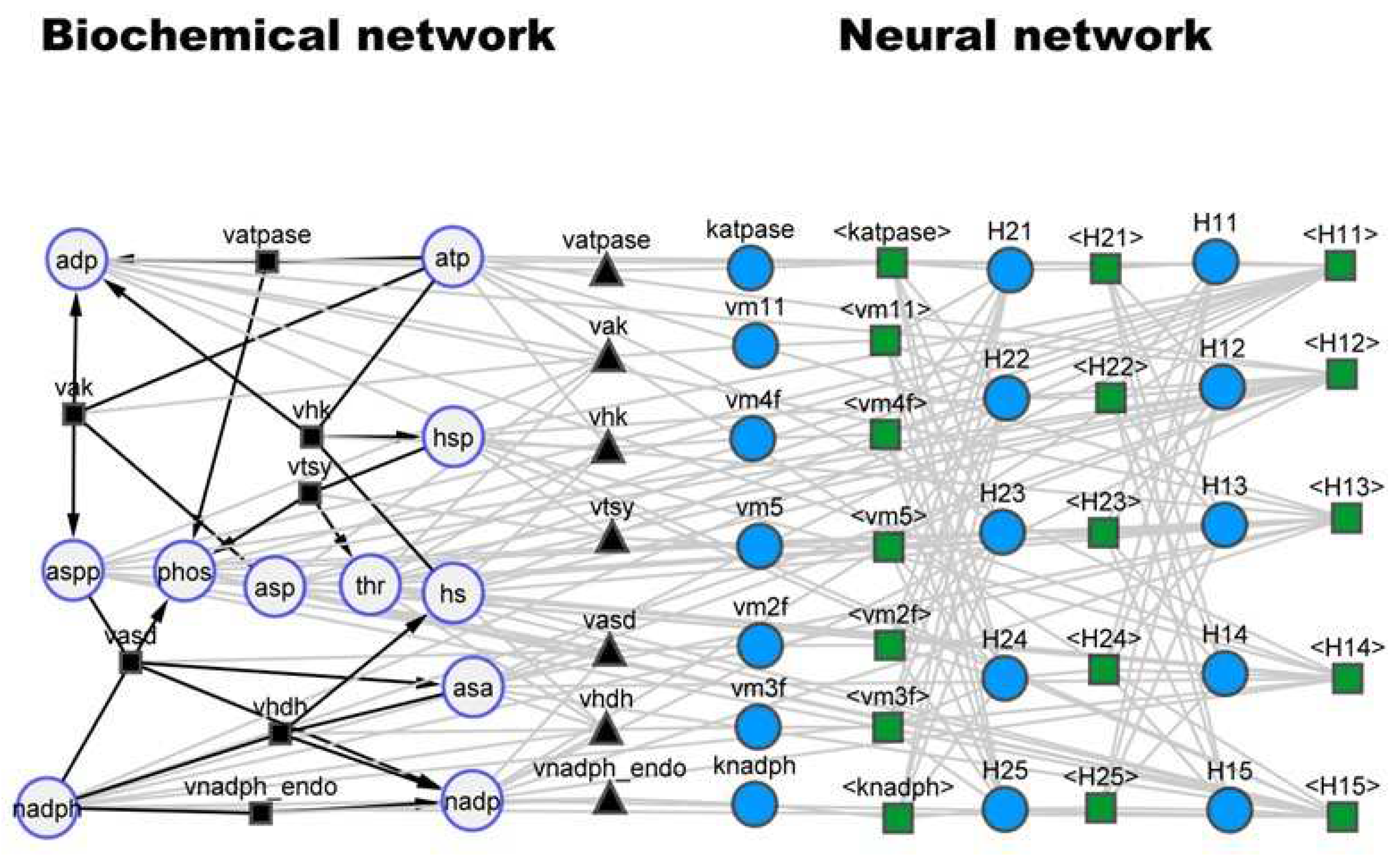
Figure 3.
Mean-Squared-Error (MSE) for the training and testing partitions over training iteration for case study 1.
Figure 3.
Mean-Squared-Error (MSE) for the training and testing partitions over training iteration for case study 1.

Figure 4.
Comparison between original and hybrid SBML models dynamics simulated in the JWS Online platform for case study 1 - threonine synthesis pathway in E. coli. Full lines represent species concentrations over time. left panel: original SBML model simulation. right panel: trained SBML hybrid model simulation.
Figure 4.
Comparison between original and hybrid SBML models dynamics simulated in the JWS Online platform for case study 1 - threonine synthesis pathway in E. coli. Full lines represent species concentrations over time. left panel: original SBML model simulation. right panel: trained SBML hybrid model simulation.
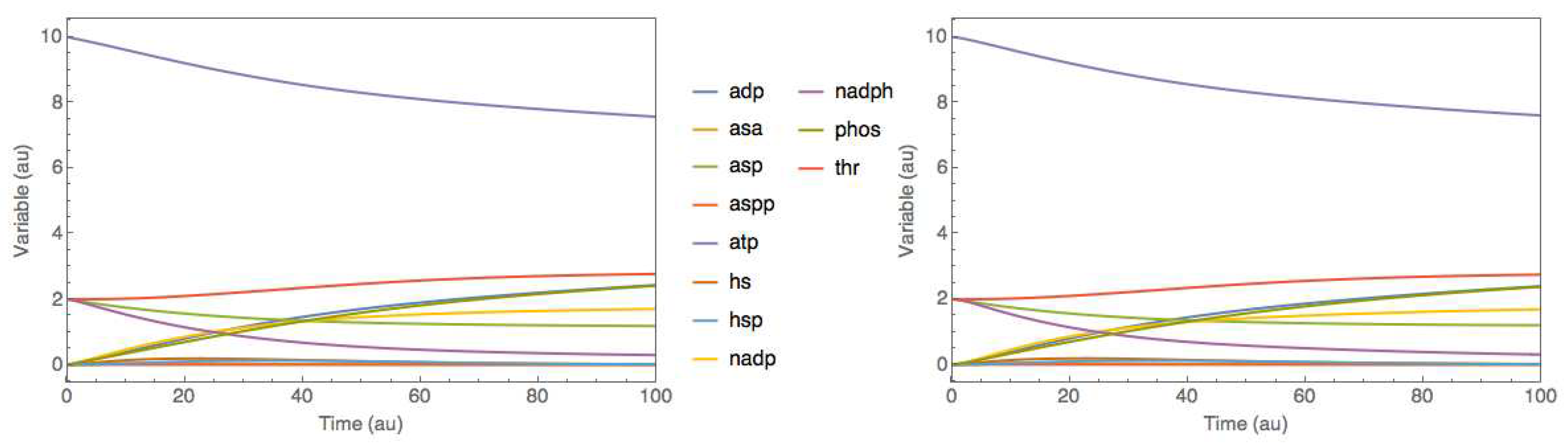
Figure 5.
Hybrid SBML model of the P58IPK signal transduction pathway visualized with the cy3sbml tool [30]. Large circles represent biochemical species. Black squares and black edges represent biochemical reactions. Black triangles represent kinetic laws. Small blue circles represent neural network nodes. Green squares and gray edges represent signal propagation between nodes.
Figure 5.
Hybrid SBML model of the P58IPK signal transduction pathway visualized with the cy3sbml tool [30]. Large circles represent biochemical species. Black squares and black edges represent biochemical reactions. Black triangles represent kinetic laws. Small blue circles represent neural network nodes. Green squares and gray edges represent signal propagation between nodes.
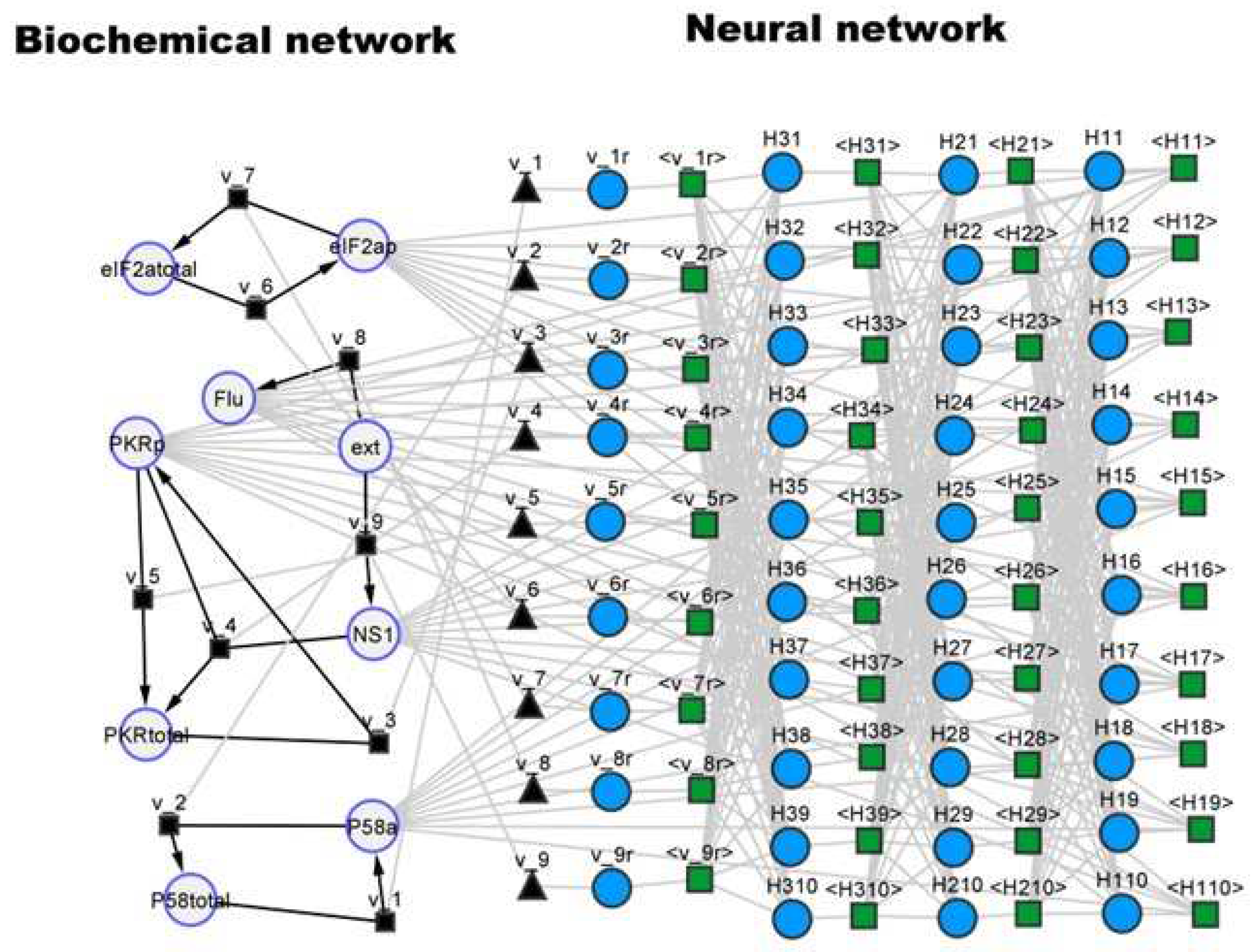
Figure 6.
Comparison between original and hybrid model simulations in the JWS Online platform for case study 2. Full lines represent species concentrations over time. Right panel - original mechanistic P58IPK signal transduction pathway. Left panel - hybrid P58IK pathway represented in Figure 5.
Figure 6.
Comparison between original and hybrid model simulations in the JWS Online platform for case study 2. Full lines represent species concentrations over time. Right panel - original mechanistic P58IPK signal transduction pathway. Left panel - hybrid P58IK pathway represented in Figure 5.
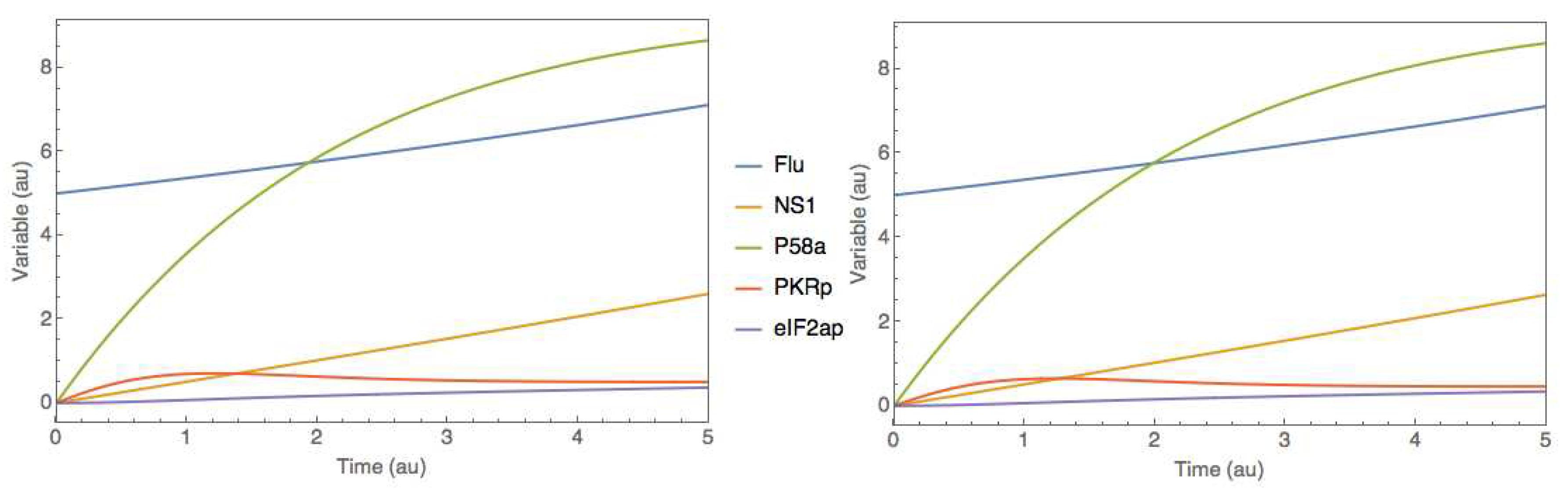
Figure 7.
Hybrid SBML model of the yeast glycolysis pathway visualized with the cy3sbml tool [30]. Large circles represent biochemical species. Black squares and black edges represent biochemical reactions. Black triangles represent kinetic laws. Small blue circles represent neural network nodes. Green squares and gray edges represent signal propagation between neural network nodes.
Figure 7.
Hybrid SBML model of the yeast glycolysis pathway visualized with the cy3sbml tool [30]. Large circles represent biochemical species. Black squares and black edges represent biochemical reactions. Black triangles represent kinetic laws. Small blue circles represent neural network nodes. Green squares and gray edges represent signal propagation between neural network nodes.
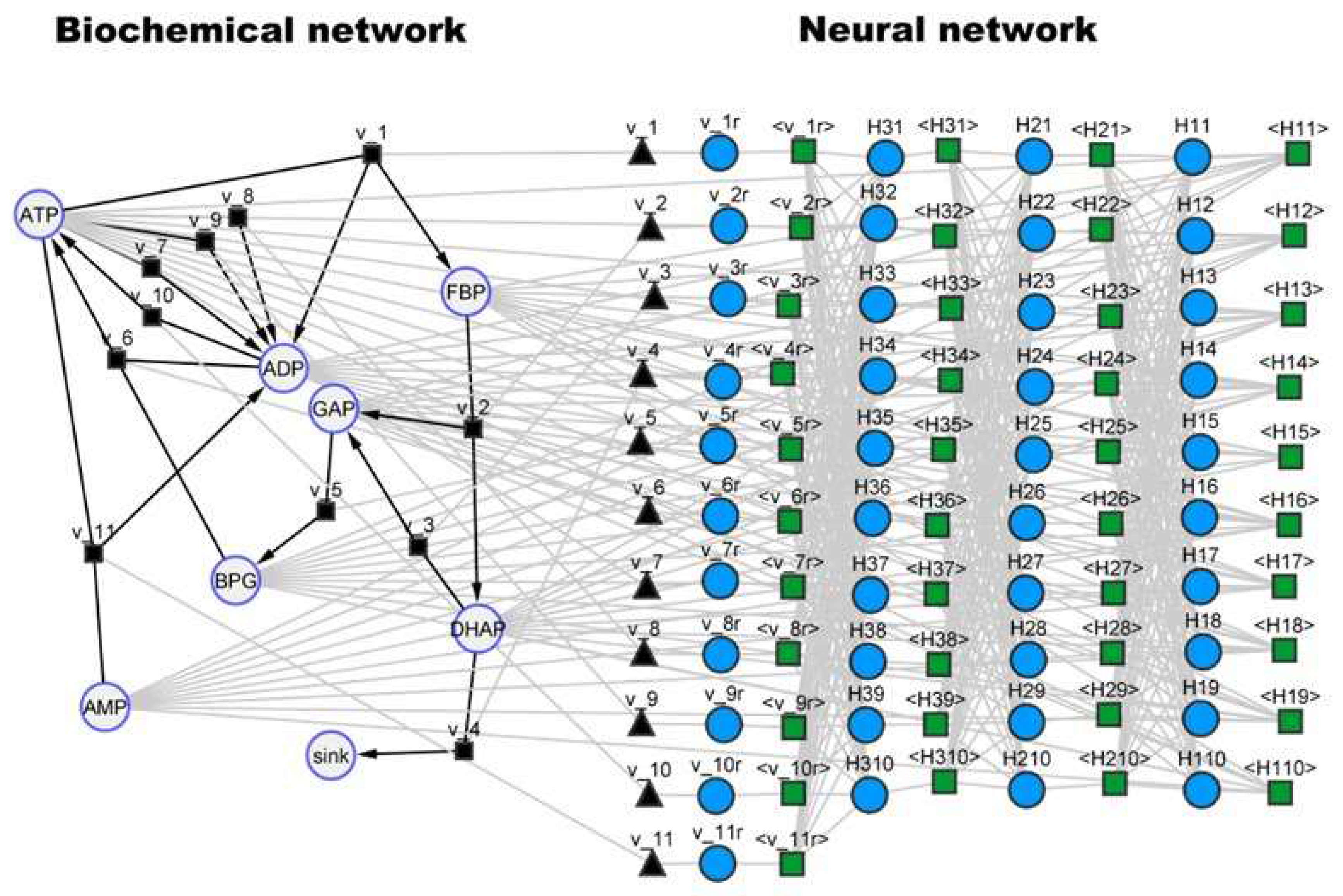
Figure 8.
Comparison between original and hybrid model simulations in the JWS Online platform for the yeast glycolysis model (case study 3). Full lines represent species concentrations over time. left panel - original mechanistic model. right panel - hybrid SBML model represented in Figure 7.
Figure 8.
Comparison between original and hybrid model simulations in the JWS Online platform for the yeast glycolysis model (case study 3). Full lines represent species concentrations over time. left panel - original mechanistic model. right panel - hybrid SBML model represented in Figure 7.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



