
Submitted:
09 February 2023
Posted:
09 February 2023
You are already at the latest version
Abstract
While genetic testing is becoming mainstream in the management of patients with potentially inherited cardiovascular disease, the prevalence of uncertain results severely limits its utility. One promising approach is to generate variant effect maps that report the function of all possible variants in a gene prospectively. The proactive clinical application of these maps is nascent, and it requires careful integration with current American College of Medical Genetics guidelines for variant interpretation. Here, we describe three pediatric cases of cardiac arrest or sudden cardiac death with variants of uncertain significance in calmodulin genes. We demonstrate the prospective clinical utility of a calmodulin variant effect map to inform variant interpretation, and therefore diagnosis and family care, in each case. This study was approved by the Stanford University and Vanderbilt University Medical Center IRBs. Consent was waived based on low risk of de-identified retrospective data collection per the IRB.
Keywords:
variant effect maps
; cardiac arrest
; cardiovascular genetics
Genetic testing is becoming mainstream in the management of cardiovascular disease. However, up to 80% of variants reported on genetic testing are classified as variants of uncertain significance (VUS), which cannot be acted upon with high confidence in the clinic [1]. Conventionally, VUS are functionally investigated reactively, after they have been identified in a patient. This time- and labor-intensive approach represents a significant barrier to patient care. Here, we describe a first implementation of proactive variant effect mapping in clinical cardiovascular genetics. We show that prospective determination of the functions of large numbers of variants in calmodulin genes (CALM1, CALM2, and CALM3) allows rapid integration of functional data in clinical decision-making in pediatric cardiac arrest cases, illustrating the promise of this strategy in overcoming the VUS barrier toward widespread implementation of genomic medicine.
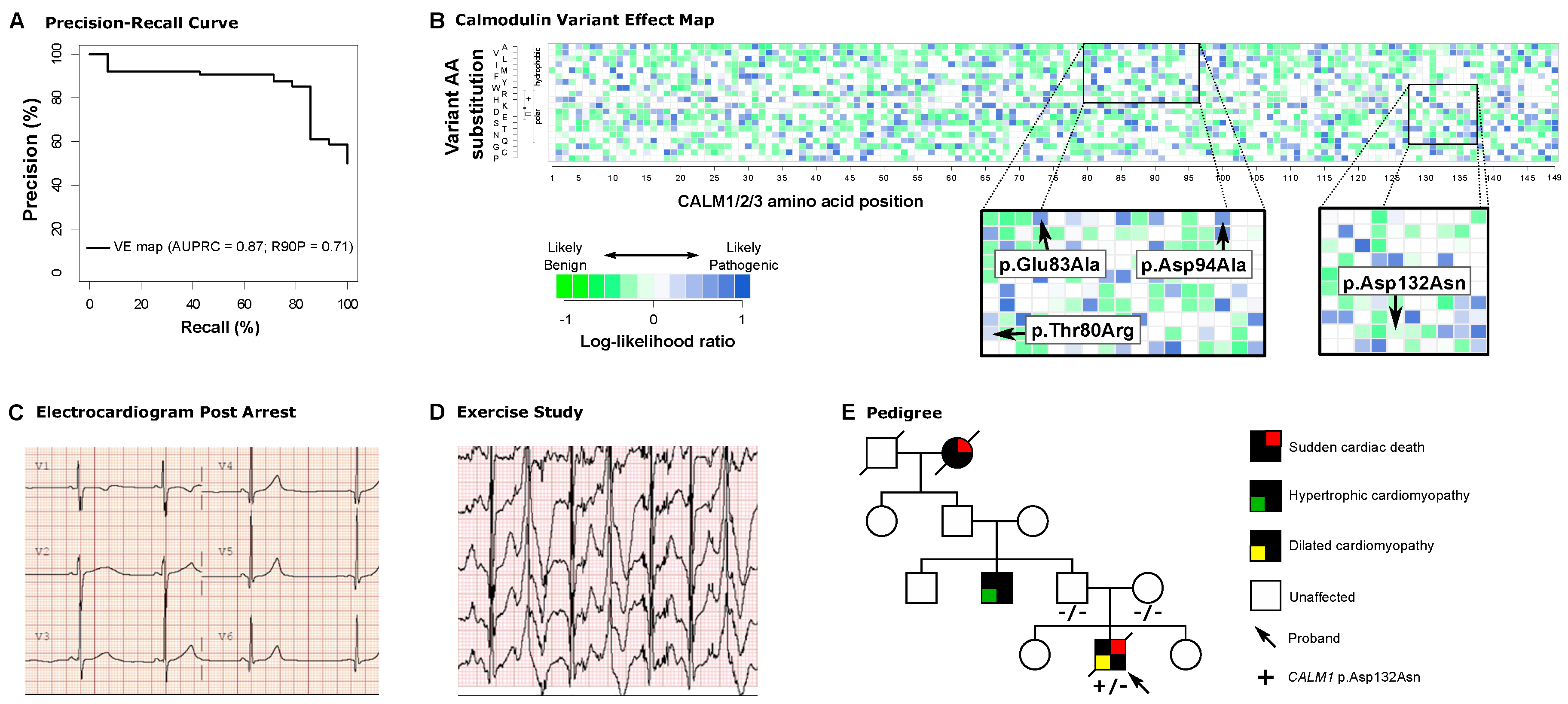
In 3 pediatric cardiac arrest cases, each involving a VUS in 1 of the 3 genes encoding the CALM (calmodulin) protein, we incorporated data from a previously published proactive variant effect map based on the ability of human calmodulin to complement loss of its ortholog in yeast [2,3]. This map was previously validated by its ability to distinguish variants common in gnomAD from pathogenic variants in ClinVar, out-performing existing prediction tools (SIFT2, PROVEAN) [2]. We performed precision recall analysis for known likely pathogenic variants using these data and showed that the area under the curve from the variant effect map is 0.87 (P<10-5 versus randomly permuted orderings, Figure 1A). Here, to relate each map score to a corresponding likelihood of variant pathogenicity, we compared each variant’s fitness score with those of known pathogenic and benign variants to derive a log10-likelihood ratio (LLR) score representing the strength of evidence the map provides for or against pathogenicity (Figure 1B, see Appendix for additional methods, and raw data are available at https://mavedb.org/scoreset/urn:mavedb:00000001-c-2/). Putatively pathogenic (Positive) benchmark cases were curated from ClinVar using known pathogenic and likely pathogenic variants in CALM1, CALM2, and CALM3 with the provision of assertion criteria (at least 1-star review status). Putatively benign (Negative) benchmark cases were curated from ClinVar using known benign or likely benign variants in CALM1, CALM2, and CALM3 with at least 1-star review status, and from gnomAD with minor allele frequency >10-6. We estimated the densities of benchmark variant sets of putatively pathogenic and benign variants at given fitness scores. These were in turn used to calculate a LLR of pathogenicity, measuring how much more likely it is to observe a given fitness score for a variant that is pathogenic than for one that is benign. This study was approved by the Stanford University and Vanderbilt University Medical Center Institutional Review Boards (IRBs). Consent was waived based on low risk of de-identified retrospective data collection per the IRB.
Case 1 was an otherwise healthy 11-year-old male patient who presented with aborted ventricular fibrillation arrest preceded by anxiety. Postarrest electrocardiogram (EKG) showed a prolonged QTc interval of 525 milliseconds, and exercise testing provoked ventricular bigeminy (Figure 1C,D). Clinical genetic testing identified an allele with 2 suspicious VUSs in cis in CALM2, c.[239C>G;248A>C], p.[(Thr80Arg);(Glu83Ala)], neither of which were present in population databases. Parental genotypes were unknown. Both CALM2 variants had high LLRs in the CALM variant effect map, indicating a high likelihood of pathogenicity for each variant (Figure 1B, inset). Based on the likelihood of detrimental functional effects of both variants in the variant effect map, this allele was deemed to contribute to the patient’s phenotype. Prior to prospective functional variant effect mapping, it would have taken months to test the functional effects of these variants on calmodulin function.
Case 2 was an 8-year-old female patient with a ventricular fibrillation arrest after falling from monkey bars. Postarrest EKG showed a QTc of 547 milliseconds. Clinical genetic testing identified a heterozygous VUS in CALM3, c.281A>C, p.(Asp94Ala) that had not been observed in population databases. The variant was upgraded to pathogenic only after parental testing confirmed its de novo status as per ACMG criteria [4]. The CALM variant effect map shows a LLR consistent with pathogenicity (Figure 1B, inset), which would have facilitated variant interpretation in real time without the delay of obtaining parental testing results.
Case 3 was a 17-year-old male patient with sudden death during sleep, with cardiac fibrosis on autopsy histology. Molecular autopsy revealed a VUS in CALM1, c.394G>A, p.(Asp132Asn), which was not observed in ClinVar or population databases. No other candidate variants were identified. The family history was significant for hypertrophic cardiomyopathy in a paternal uncle and sudden death in a paternal grandmother (Figure 1E). The variant interpretation was upgraded to pathogenic by the laboratory upon demonstration of de novo status (Figure 1E). However, the variant effect map shows a LLR near 0, in a range where there is an equal prevalence of pathogenic and benign variants (Figure 1B, inset). In the setting of the patient’s cardiac fibrosis, which is not a typical feature of CALM-related disease, and the family history of cardiomyopathy and sudden death, these results serve to underscore the possibility that this de novo variant was not solely responsible for this patient’s sudden cardiac death. Together, these data suggest that another cause of cardiomyopathy may coexist in family members, who should be clinically screened accordingly.
These cases reveal the potential of variant effect maps to facilitate patient diagnosis and refine clinical decision-making in cardiovascular genetics. The CALM map provided evidence toward rapid reclassification of VUSs for 2 of 3 patients. In the third case, the map provided functional data that, in concordance with additional clinical data, calls into question the sufficiency of its de novo variant status to clear other family members from future screening. Thus, prospectively generated variant effect maps offer an opportunity to refine current approaches to variant classification, and thereby accelerate the integration of genetic testing into cardiovascular care [5]. Ongoing technology development for variant effect mapping includes the optimization of multiple model systems and multiplexed phenotypic read-outs of variant effect to provide immediately clinically actionable findings, potentially ultimately replacing the slow process of reactive functional testing with a readily available atlas of variant effects. Given the significant burden of genetic disease in cardiovascular medicine, it is critical that we prepare to capitalize on this wealth of immediately clinically actionable knowledge by implementing systems for its direct, proactive incorporation into patient care.
Funding Sources
The authors, as members of the CardioVar Consortium, are supported by NIH grant R01 HL164675. Dr. Parikh is supported by grants from the Sarnoff Cardio- vascular Research Foundation and NHLBI K08 HL143185 and R01 HL144843. Dr. Ashley is supported by NHLBI grant R01 HL144843 and grants from Google LLC, Verily Life Sciences LLC, Bristol Myers Squibb Co, and Takeda Pharmaceuticals International, Inc. Dr. Roth is supported by NIH/NHGRI Center of Excellence in Genomic Science grant RM1HG010461, by the NHGRI Impact of Genomic Variation on Function Initiative (UM1HG011989), by the One Brave Idea Initiative (jointly funded by the American Heart Association, Verily Life Sciences LLC, and Astra-Zeneca, Inc), and by a Canadian Institutes of Health Research Foundation Grant. Dr. MacRae is Supported by NIH grant R24 OD017870. Dr. Glazer is supported by NIH grant R00HG010904.
Disclosure Statement
Dr. Parikh is a scientific advisory board member for and receives research support from BioMarin, Inc. Dr. Ashley is a founder of Personalis, Inc, Deepcell, Inc, Silicon Valley Exercise Analytics, an advisor for Nuevocor Pte Ltd, CathayCapital NA, LLC, Third Rock Ventures, Medical Excellence Capital LLC, Foresite Capital Management LLC, Novartis AG, Apple, Inc, Sequence Bio, Inc, Genome Medical, Inc, Disney, and a member of the AstraZeneca board of directors. Dr. Roth is a shareholder and advisor for SeqWell, Constantiam, BioSymetrics, and a share- holder of Ranomics. Dr. Roth's lab has received research support from Alnylam, Deep Genomics, Beam Therapeutics and Biogen, Inc. Ms. Reuter is a consultant for My Gene Counsel. The other authors report no conflicts.
References
- van Lint FHM, Mook ORF, Alders M, Bikker H, Lekanne Dit Deprez RH, Christiaans I. Large next-generation sequencing gene panels in genetic heart disease: yield of pathogenic variants and variants of unknown significance. Neth Heart J. 2019; 27:304–309. [CrossRef]
- Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC, Wu Y, Pons C, Wong C, van Lieshout N, et al. A framework for exhaustively mapping functional missense variants. Mol Syst Biol. 2017; 13:957. [CrossRef]
- Wu Y, Weile J, Cote AG, Sun S, Knapp J, Verby M, Roth FP. A web application and service for imputing and visualizing missense variant effect maps. Bioinformatics. 2019; 35:3191–3193. [CrossRef]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–423. [CrossRef]
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, Biesecker LG, ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018; 20:1054–1060. [CrossRef]
Figure 1.
Proactive functional genomics for variant interpretation in CALM1, CALM2, and CALM3. (A) Precision recall curve for variant classifications from Weile et al. displays AUC 0.87 and identifies 71% of pathogenic variants at 90% precision. (B) Variant effect map of CALM1, CALM2, and CALM3 in terms of log10-likelihood ratio, with insets indicating likelihood of pathogenicity for variants evaluated in this report. Columns represent each amino acid position with each row representing a different variant at that position. (C) EKG demonstrating long QT (525 milliseconds) in Case 1. (D) Ventricular ectopy consistent with catecholaminergic polymorphic tachycardia at peak exercise in Case 1. (E) Pedigree demonstrating complex family history of cardiomyopathy and sudden death despite de novo status of the CALM1 VUS in Case 3.
Figure 1.
Proactive functional genomics for variant interpretation in CALM1, CALM2, and CALM3. (A) Precision recall curve for variant classifications from Weile et al. displays AUC 0.87 and identifies 71% of pathogenic variants at 90% precision. (B) Variant effect map of CALM1, CALM2, and CALM3 in terms of log10-likelihood ratio, with insets indicating likelihood of pathogenicity for variants evaluated in this report. Columns represent each amino acid position with each row representing a different variant at that position. (C) EKG demonstrating long QT (525 milliseconds) in Case 1. (D) Ventricular ectopy consistent with catecholaminergic polymorphic tachycardia at peak exercise in Case 1. (E) Pedigree demonstrating complex family history of cardiomyopathy and sudden death despite de novo status of the CALM1 VUS in Case 3.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



