
Submitted:
14 September 2023
Posted:
15 September 2023
You are already at the latest version
Abstract
Viruses frequently contain overlapping genes, which encode functionally unrelated proteins from the same DNA or RNA region but in different reading frames. Yet overlapping genes are often overlooked during genome annotation, in particular in DNA viruses. Here we looked for the presence of overlapping genes likely to encode a functional protein in human parvovirus B19 (genus erythroparvovirus), using an experimentally validated software, Synplot2. Synplot2 detected an open reading frame, X, conserved in all erythroparvoviruses, which overlaps the VP1 capsid gene, and is under highly significant selection pressure. In a related virus, human parvovirus (genus tetraparvovirus), Synplot2 also detected an open reading frame under highly significant selection pressure, ARF1, which overlaps the VP1 gene. X and ARF1 have exactly the same location (both overlap the region of VP1 encoding the phospholipase A2 domain), and encode proteins with similar predicted properties, such as a transmembrane region, strongly suggesting that they are homologous. These findings provide compelling evidence that the X protein must be expressed and functional. It is probably translated either from a polycistronic mRNA by a non-canonical mechanism, or from an unmapped monocistronic mRNA. Finally, we also discovered proteins predicted to be expressed from a frame overlapping VP1 in other species related to parvovirus B19: porcine parvovirus 2 (Z protein) and bovine parvovirus 3 (X-like protein).
Keywords:
Overlapping genes
; overlapping coding sequences
; overlapping reading frames
; 9 kDa protein
; erythroparvovirus
; tetraparvovirus
; Y region
; Y coding sequence
; PLA2
1. Introduction
Parvoviruses are small, non-enveloped viruses (for reviews, see [3,4,5]). Here we focus on two in particular: human parvovirus B19 (B19V) and human parvovirus 4 (PARV4). B19V causes several diseases in humans, such as fifth disease in children, cardiomyopathy, and persistent anemia in immunocompromised persons [6]. PARV4 is not formally associated to any disease, despite suspicions that it may cause encephalitis or accelerate HIV progression [7]. B19V and PARV4 respectively belong to the genera erythroparvovirus and tetraparvovirus, which are closely related [4]; other species in these genera infect a variety of mammals (see Figure 1).
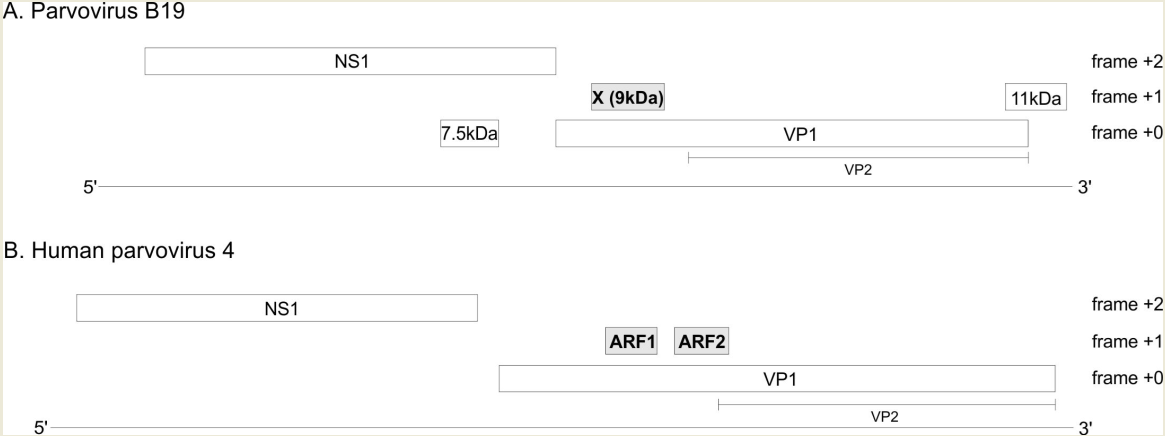
The genome of every erythro- and tetraparvovirus encodes at least two proteins: the replicase NS1 and the capsid protein, of which at least two isoforms are made: VP1 and VP2 (Figure 2). In B19V, three additional ORFs (open reading frames) have been reported (Figure 2A): the 7.5 kDa ORF overlaps the NS1 ORF; the X ORF (which has the potential to code for a 9 kDa protein) overlaps the VP1 ORF; and the 11 kDa ORFpartially overlaps the 3′ region of the VP1 ORF. The expression of the 7.5 kDa protein [8] and of the 11 kDa protein [9,10] have been proven experimentally. In contrast, the expression of the X protein has never been confirmed in infected cells. A substitution meant to knock out the expression of the X ORF caused no discernable change in viral replication or infectivity [11], raising doubts on the expression or functionality of the X protein.
Likewise, in PARV4, two ORFs overlapping the VP1 ORF have been noticed, but never confirmed experimentally [12]: ARF1 and ARF2 (ARF stands for “Alternative Reading Frame) (Figure 2B).
Overlapping ORFs are frequently overlooked in viral genomes [13]. It is possible, in principle, to predict merely from sequence analyses whether a protein is expressed from an overlapping ORF, provided that the protein confers a beneficial function to the virus [14]. In that case, the additional selection pressure caused on the sequence of the reading frame that it overlaps results in a lower rate of synonymous codon substitution in that second frame [15,16].
Surveys of the B19V and PARV4 genomes reported such a lower rate in the region of VP1 corresponding to the X ORF [17], as well as in the region corresponding to ARF1 and ARF2 [12], but did not provide an estimate of the statistical significance of this reduction. In contrast, the dedicated software Synplot2 [18] can quantify the probability that an ORF with a reduced synonymous codon substitution rate is expressed and functional. Synplot2 has been extensively validated on experimentally proven overlaps and since its release was used to detect over 15 overlapping ORFs that were then confirmed experimentally (e.g., [19,20,21]). Interestingly, Synplot2 can also detect non-coding functional elements [18].
We thus chose to use Synplot2 to analyze the VP1 coding sequences of B19V and PARV4. Synplot2 detected several regions that may correspond either to protein-coding ORFs (including that of the X protein and of ARF1) or to non-coding functional elements. We compared the sequence properties of the erythroparvovirus X protein with that of tetraparvovirus ARF1 and determined that they were homologous.
2. Materials and Methods
2.1. Sequence collection
We collected the coding sequences of VP1 for all isolates of viral species investigated here by using Blastn [50] against Genbank (30th July 2019) on the reference sequence of each species. We retained sequences with >75% nucleotide similarity over 90% of the length of the query (i.e., 90% coverage). We removed duplicate sequences, sequences containing insertions or deletions longer than 50 nucleotides with respect to the reference sequence, or those marked as “synthetic” sequences.
2.2. Nucleotide sequence alignment and analysis
To generate codon-respecting alignments based on the coding sequence of VP1, we used the program TranslatorX [31] with the “Muscle” option. The resulting codon-based alignments are in the Alignments S1-S4. Alignment S5 contains the potential start codons of the X ORF in all erythro- and tetraparvoviruses.
2.3. Detection of regions with lower synonymous substitution rate
We used Synplot2 [18] to identify overlapping functional elements, with two sizes of sliding window: 25 and 45 codons. A window of 25 codons provides better specificity, which helped us identify how many regions have a decreased synonymous substitution rate; whereas a window of 45 codons provides better sensitivity, which helped us map the precise boundaries of the regions identified. We present Synplot2 plots computed with a window of either 25 or 45 codons, depending on which window size better shows the regions identified. The boundaries of these regions were always mapped with a window of 45 codons.
2.4. Protein sequence alignment and domain identification
All protein sequence alignments are presented using Jalview [51] with the ClustalX colouring scheme [52]. We carried out phylogenetic analyses using phylogeny.fr [53] with default options. To add unaligned sequences into a reference alignment, we used MAFFT with the --add option [54]. The Alignment S6 contains the sequence alignment of all X proteins in text format. We used HHpred [33] to identify protein domains such as PLA2.
2.5. Prediction of protein structural features
We used MetaDisorder [55] to predict disordered regions, in accordance with the principles described in [56], and DeepCoil [57] to predict coiled-coil regions.
We used two complementary methods to reliably predict transmembrane segments, as explained in [58]. First, we compared the predictions of several transmembrane prediction programs on a single protein, for each protein (“vertical approach”), by using CCTOP [59]. Second, we compared the prediction of a single program (TM-Coffee [60]) on several homologs (“horizontal” approach).
3. Results
3.1. The VP1 gene of B19V contains 3 regions with significantly increased synonymous conservation, among which the X ORF
To determine whether the VP1 gene of B19V might encode other proteins in overlapping reading frames, we collected the VP1 coding sequences (CDS) of all genotypes of B19V available in GenBank, translated them, aligned their amino acid sequences, and back-translated them to yield a nucleotide sequence alignment (see Methods). Next, we determined whether the alignment contained regions with a reduced variability of substitutions at synonymous sites, using Synplot2 [18].
Synplot2 identified 3 regions with a statistically significant decrease in the variability of synonymous substitutions (Figure 3B). These regions are visible as peaks in Figure 3B, since Synplot2 actually plots the increase in conservation of synonymous substitutions instead of their decrease in variability.
The first region spans codons 58-163 of VP1 (see Table 2), and corresponds to the hypothetical X ORF (see Introduction). This ORF is devoid of stop codons in frame +1 relative to VP1 (Figure 3C) in all B19V sequences. A potential AUG start codon overlaps codon 84 of VP1 and is conserved in all B19V sequences, confirming that the X ORF has the potential to code for a protein. As Figure 3A shows, the X ORF is entirely embedded within the region encoding VP1u (the N-terminus of the capsid protein, found in VP1 but not in VP2), and partially overlaps the region encoding the Phospholipase A2 (PLA2) domain of VP1 [22,23].
Note that the reduction in synonymous variability starts quite upstream of the putative AUG start codon of the X ORF (codon 58 and 84 of the VP1 gene, respectively, see Figure 3B and Table 2). This might indicate the presence of a regulatory region that enhances the translation of the X ORF, between codons 58 and 84, underlined in Figure 3B (see Discussion).
The X ORF is found in all other erythroparvoviruses (Table 1 lists the accession numbers of all GenBank reference genome sequences); see below for the special case of bovine parvovirus 3. An X ORF is also found in two erythroparvovirus-like sequences: one thought to be an endogenous virus [24], Indri indri endogenous parvovirus, and one of unknown status, Hyaena hyaena parvovirus-like sequence (Alignment S6).
The second region detected by Synplot2 spans codons 185-239 of VP1 (Figure 3B and Table 2), and has not been described yet, to our knowledge. We called it “Y region”. Other erythroparvoviruses do not contain an equivalent region. The Y region overlaps the region of VP1 located downstream of the PLA2 domain and extends slightly into VP2 (Figure 3A). It is devoid of stop codons in frame +2 relative to VP1 in all B19V sequences (Figure 3C) and thus constitutes a potential CDS of 46aas. However, it lacks a potential AUG start codon. It might thus either correspond to a non-coding functional element, or code for a protein expressed through a non-canonical mechanism.
Functional RNA elements often have a conserved secondary structure, but we could not detect the signature of such a structure in the Y region (using RNAz [1,2]). We could not either identify by sequence analysis potential sites that would direct non-canonical translation of the Y region, such as an IRES (Internal Ribosome Entry Site) [25] or a ribosomal frameshift [26], though such sites remain difficult to predict. Another possibility for expressing the Y region would be polymerase slippage [27], for which there is no consensus site. Note that all these non-canonical mechanisms would result in a chimeric protein composed of the N-terminus of VP1 fused to the Y coding sequence.
The third region detected by Synplot2 is located at the very C-terminus of the VP1 CDS (codons 771-781) (Figure 3B). It corresponds to the N-terminus of the 11 kDa protein (Figure 3A), known to be expressed in the +1 frame relative to VP1 from an AUG that overlaps codon 756 of VP1 [9,10].
Altogether, these data show that the VP1 gene of B19V encodes a protein (the X protein) in the +1 frame, conserved in all erythroparvoviruses. It also contains another region, Y, which corresponds either to a non-coding functional element or to the coding sequence of a protein in the +2 frame, which might be expressed as a fusion with the N-terminus of VP1.

Table 1.
Nucleotide sequences of virus species analyzed in this work.
| Genus | Species | Common name(s) [Abbreviation] | Genbank genome accession number | Boundaries of the X ORF in the genome sequence (in nucleotides) |
|
|---|---|---|---|---|---|
| Erythroparvovirus | Primate erythroparvovirus 1 |
Parvovirus B19 [B19V] |
NC_000883 | 2874-3119 | |
| Erythroparvovirus | Primate erythroparvovirus 2 | Simian parvovirus | U26342.1 | 2718-2963 | |
| Erythroparvovirus | Primate erythroparvovirus 3 | Rhesus macaque parvovirus | AF221122.1 | 2841 | 3080 |
| Erythroparvovirus | Primate erythroparvovirus 4 |
Pig-tailed macaque parvovirus | AF221123.1 | 2563 | 2802 |
| Erythroparvovirus | Rodent erythroparvovirus 1 | Chipmunk parvovirus | GQ200736.1 | 3031 | 3228 |
| Erythroparvovirus | Seal parvovirus | Seal parvovirus | KF373759.1 | 2789 | 3100 |
| Erythroparvovirus (*) |
Ungulate erythroparvovirus 1 | Bovine parvovirus 3 [bPARV3] | NC_037053 | 2627-2926 | |
| Tetraparvovirus | Chiropteran tetraparvovirus 1 | Eidolon helvum parvovirus | NC_016744.1 | 2829-3062 | |
| Tetraparvovirus | Primate tetraparvovirus 1 |
Human parvovirus 4 [PARV4] |
NC_007018.1 | 2937-3140 | |
| Tetraparvovirus | Ungulate tetraparvovirus 1 | Bovine hokovirus 1 |
NC_028136 | 2857-3111 | |
| Tetraparvovirus | Ungulate tetraparvovirus 2 | Porcine hokovirus | EU200677.1 | 2808 | 3062 |
| Tetraparvovirus | Ungulate tetraparvovirus 5 | Deer tetraparvovirus | NC_031670.1 | 2766-3020 | |
| Tetraparvovirus (*) | Ungulate tetraparvovirus 3 | Porcine parvovirus 2 [pPARV2]; Porcine cnvirus; Parvovirus YX | NC_035180 | No X ORF; boundaries of the Z ORF are 2817-3098 | |
| Tetraparvovirus | Ungulate tetraparvovirus 4 | Ovine hokovirus | JF504699.1 | 2855-3112 | |
| Tetraparvovirus | - | Opossum parvovirus | MG745671.1 | 2862-3092 | |
| Tetraparvovirus | - | Rodent parvovirus | MG745669.1 | 2960-3217 | |
The main species analyzed here are in bold. (*) The taxonomic classification of these species might need a revision in view of our analyses.
Table 2.
Boundaries of VP1 regions with significantly lower synonymous codon variability (identified by Synplot2) encompassing potential protein-coding ORFs.
Table 2.
Boundaries of VP1 regions with significantly lower synonymous codon variability (identified by Synplot2) encompassing potential protein-coding ORFs.
| Virus name | Region | Boundaries of the region with lower synonymous codon variability in the VP1 CDS | Boundaries of the corresponding ORF in the VP1 CDS |
|---|---|---|---|
| Parvovirus B19 | X ORF | Codons 58-163 (nucleotides 172-489) |
Codons 84-166 (Nucleotides 251-496) |
| Parvovirus B19 | Y region(*) | Codons 185-239 (nucleotides 553-715) |
Codons 185-230(*) (nucleotides 553-688) |
| Human parvovirus 4 | ARF1 ORF (=X ORF) |
Codons 180-263 (nucleotides 538-789) |
Codons 187-255 (nucleotides 560-763) |
| Human parvovirus 4 | ARF2 ORF | Codons 294-397 (nucleotides 880-1189) |
Codons 295-379 (nucleotides 884-1135) |
| Bovine parvovirus 3 | X-like ORF | Codons 205-306 (nucleotides 614-916) |
Codons 215-315 (nucleotides 644-943) |
| Porcine parvovirus 2 | Z ORF | Codons 193-309 (nucleotides 577-927) |
Codons 193-285 (nucleotides 578-854) |
(*): this region contains an ORF devoid of stop codon, but lacks a potential AUG start codon, and might not code for a protein.
3.2. The VP1 gene of PARV4 contains 2 regions with significantly reduced synonymous variability, corresponding to ARF1 and ARF2
To determine whether the VP1 gene of PARV4 encodes other proteins in overlapping reading frames, we analyzed the VP1 coding sequence of all strains of PARV4 by using Synplot2, as described above for B19V. Figure 4B shows that two regions have a highly significant increase in the conservation of synonymous sites (Table 2):
The first region spans codons 180-263 of VP1 (Table 2), which corresponds to the ARF1 ORF [12] (see Introduction). ARF1 is devoid of stop codons in frame +1 relative to VP1 (Figure 4B) in all PARV4 sequences. It has a potential AUG start codon conserved in all PARV4 sequences, overlapping codon 187 of VP1. ARF1 is embedded within the VP1u region, and partially overlaps the PLA2 domain (Figure 4A). An ORF similar to ARF1 is found in all other tetraparvoviruses, with the exception of porcine parvovirus 2 (see below).
The second region detected by Synplot2 spans spanning codons 294-397, and corresponds to the ARF2 ORF [12] (see Introduction). ARF2 is devoid of stop codons in frame +1 relative to VP1 (Figure 4C). It has a potential AUG start codon conserved in all PARV4 sequences, overlapping codon 294 of VP1. ARF2 overlaps the region of VP1 immediately downstream the PLA2 domain, and extends slightly into VP2 (Figure 4A). Note that PARV4 ARF2 and the putative Y protein of B19V are not homologous, since they are encoded in different frames relative to VP1 (respectively +1 and 2, compare Figure 4A and Figure 3A).
An ORF similar to ARF2 is found only in tetraparvoviruses closely related to PARV4: hokoviruses (porcine, bovine and ovine), and deer tetraparvovirus. We present their aa sequence in Figure S1. ARF2 has a predicted transmembrane segment near its N-terminus.
Altogether, these data show that the VP1 gene of PARV4 encodes a protein, ARF1 in the +1 frame, conserved in all tetraparvoviruses. It also encodes a second protein, ARF2, in the +1 frame, conserved in tetraparvoviruses closely related to PARV4.
3.3. The X protein and ARF1 are homologous
3.3.1. The B19V X protein and PARV4 ARF1 protein have similar predicted features, in particular a central transmembrane segment
Next, we examined the predicted sequence features of B19V X protein and of PARV4 ARF1. Figure 5 presents multiple sequence alignments of the erythroparvovirus X protein (Figure 5A) and of tetraparvovirus ARF1 (Figure 5B). The erythroparvovirus X protein contains a predicted central transmembrane segment (Figure 5A), followed by a positively charged region, predicted to be inside the cytosol (“positive-inside rule” [29]). In B19V and the three closely related primate erythroparvoviruses, the C-terminus of the X protein is predicted to form a second transmembrane segment (boxed in Figure 5A). It is unusual that closely related proteins would vary in the number of transmembrane proteins they contain, and therefore these predictions should be taken with caution.
Tetraparvovirus ARF1 has a size and predicted organization similar to that of the X protein (compare Figure 5B,A), composed of an extra-cytosolic N-terminus, a central transmembrane segment, and a positively charged, intra-cytosolic region.
3.3.2. The X protein of erythroparvoviruses and the ARF1 protein of tetraparvoviruses are homologous
3 lines of evidence suggest that the erythroparvovirus X protein of and the tetraparvovirus ARF1 protein might be homologous, i.e., share a common origin:
1) they overlap a similar region of the VP1 gene (encoding the PLA2 domain, indicated above the alignments in Figure 5);
2) they are both in the +1 frame relative to VP1 (see Figure 3A and 4A);
3) they have similar sequence features, as shown above.
However, the presence of a transmembrane segment could be explained by convergent evolution [30]. Therefore, to check whether X and ARF1 are homologous, we examined how their sequences align when based on the much more reliable alignment of VP1, and in particular of its PLA2 domain. Indeed, PLA2 contains numerous strictly conserved amino acids (aas) [22,23], which makes its sequence alignment highly reliable.
To generate a reliable alignment of erythroparvovirus X proteins and tetraparvovirus ARF1 based on the alignment of VP1, we:
a) converted the aa alignment of the VP1 proteins into an alignment of nucleotide sequences by using TranslatorX [31];
b) translated this alignment in the reading frame of X and ARF1, i.e., +1 relative to VP1. This procedure is described graphically in a previous article [32].
The resulting alignment of X and ARF1 is shown in Figure 6A, while the reference alignment of VP1 is shown below, in Figure 6B. (Only the PLA2 domain of VP1 is shown, because the region upstream is not well conserved). As Figure 6 A shows, the central transmembrane segments of X and ARF1 align together perfectly. Three aa positions are strictly conserved between X and ARF1, and one position is semi-conserved (aromatic: Y, W or F). They are indicated above the alignment in Figure 6A.
Taken together, this high degree of conservation, coupled to the fact that erythro- and tetraparvoviruses are closely related genera [4], indicates that X and ARF1 are most probably homologous.
Conventions are as in Figure 5 A) Alignment of the X protein of erythro- and tetraparvoviruses, derived from the reference alignment of VP1 presented in panel B. The X alignment was generated from the VP1 alignment by using TranslatorX [31] (see text). Strictly- or semi-conserved aas are indicated. Predicted transmembrane regions are underlined in the sequence of B19V X and PARV4 ARF1.
B) Reference alignment of VP1. Only the reliably aligned N-terminal part of the PLA2 domain is shown. Thin vertical lines show the correspondence between aas encoded by overlapping codons in the X frame (panel A) and in the VP1 frame (panel B). Aas that overlap conserved positions of the X protein are boxed. Other conserved aas involved in functional elements of PLA2 are also indicated.
3.3.3. Conserved features of the X protein mostly but not exclusively correspond to conserved motifs of the PLA2 domain of VP1
Since the X ORF partially overlaps the PLA2 domain of VP1 (Figure 3A,B), we asked whether conserved sequence features of the X protein are imposed by conserved sequence motifs of PLA2. As Figure 6B shows, the region of PLA2 overlapped by the X protein contains two conserved features: 1) the putative calcium (Ca2+)-binding loop (aa 130-134 in B19V); and 2) a region involved in the catalytic network, containing 3 strictly conserved aas (H153, D154 and Y157 in B19V) [22,23].
The conserved features of the X protein do correspond to these conserved features of PLA2. First, the transmembrane segment of the X protein overlaps the Ca2+-binding loop. Second, strictly conserved positions of the X protein (P43, L50, G73 in B19V, boxed in Figure 6A) overlap strictly conserved positions of PLA2, boxed in Figure 6B: P126 and P133 (both within the Ca2+-binding loop), and R156, close to conserved aas of the catalytic network. Likewise, the semi-conserved position of the X protein (Y54 in B19V) corresponds to a strictly conserved position of VP1 (L137 in B19V).
Clearly the PLA2 enzyme is under stringent selection pressure to conserve aas responsible for its catalytic activity. Therefore, one might assume that the sequence conservation within the X protein is dictated by PLA2. However, the sequence of strictly conserved aas of X is not completely imposed by PLA2. For example, consider the strictly conserved P133 and G134 in PLA2, which overlap the aa L50 strictly conserved in the X frame (Figure 6). Conservation of this Leucine is not imposed by the conservation of P133 and G134, since the dipeptide PG (Proline-Glycine) can be encoded by the nucleotides CCNGGN, in which N is any nucleotide. The first corresponding codon in the +1 frame relative to PLA2, CNG, can encode not only Leucine (CTG), but also Proline (CCG), Glutamine (CAG), or Arginine (CGG). Likewise, none of the conserved positions of the X protein are completely imposed by conservation of PLA2.
Altogether, these data show that conserved features of the X protein mostly but not exclusively correspond to conserved motifs of the PLA2 domain of VP1.
3.4. The VP1 gene of Bovine parvovirus 3 and porcine parvovirus 2 differs from that of other erythro- and tetraparvoviruses
3.4.1. Bovine parvovirus 3 VP1 gene encodes an X-like ORF, despite not encoding a PLA2 domain
We noticed that in one species, ungulate erythroparvovirus 1, VP1 completely lacks the sequence signature of the PLA2 domain found in all other erythro- and tetraparvoviruses. This species is also commonly called bovine parvovirus 3 (bPARV3) [34], and is basal to the erythroparvovirus phylogeny [34] (Figure 1).
Synplot2 detected in the VP1 CDS of bPARV3 a region with significantly reduced synonymous variability, slightly upstream of the VP1/VP2 boundary (Figure 7B). This region corresponds almost exactly to an ORF conserved in all strains of bPARV3, in frame +1 relative to VP1 (Figure 7C). We called it “X-like” ORF, since it has a similar location as the X ORF of erythro- and tetraparvoviruses and has the potential to code for a protein with a similar length (99 aas) and organization (central transmembrane segment) as the X protein. The sequence of the bPARV3 X-like protein is shown in Figure 5C.
These data show that the bPARV3 VP1 gene encodes an X-like protein in the +1 frame despite not encoding a PLA2 domain (see also the Discussion).
3.4.2. Porcine parvovirus 2 does not encode an X ORF, but encodes a “Z ORF” overlapping VP1
As mentioned above, there is no X-like ORF in porcine parvovirus 2 (pPARV2) (also called cnvirus [35]), which belongs to the species Ungulate tetraparvovirus 3, and is basal to the tetraparvovirus phylogeny [35] (Figure 1). We examined its VP1 coding sequence with Synplot2. Three regions have a significant increase in the conservation of synonymous sites (Figure 8B):
The first region spans codons 1-57. It is interrupted by stop codons both in +1 and +2 frames relative to VP1 (Figure 8C) and may thus correspond to a non-coding functional element.
The second region spans codons 193-309. It is devoid of stop codons in frame +1 relative to VP1 (Figure 8C) in all sequences of pPARV2, except one (accession number MK378188). It contains a potential AUG start codon overlapping codon 193 of VP1, conserved in all sequences. Thus, this region probably encodes a protein, which we called “Z protein”. The Z ORF overlaps the region of VP1 upstream the PLA2 domain and slightly extends into the N-terminus of PLA2 (Figure 8A). The sequence of the Z protein is shown in Figure S2. It has a rather low sequence complexity, and its N-and C-termini are predicted to be structurally disordered.
The third region spans codons 355-449. It is interrupted by stop codons both in frames +1 and +2 relative to VP1 (Figure 8C) and may thus correspond to a non-coding functional element.
Altogether, these data show that the pPARV2 VP1 gene encodes a protein (the Z protein), unrelated to the X protein of other erythro- and tetraparvoviruses, in the +1 frame. It also encodes two probable non-coding functional elements.
4. Discussion
4.1. Sequence analyses provide compelling evidence that the X protein must be expressed and have a crucial function
The X ORF was noticed as early as 1986 [36], but has lived up to its name, since no experimental support has ever been provided for its translation or essentiality in infected cells. Indeed, substituting its presumed start codon by a stop codon had no effect on replication, infectivity, or capsid production in cells permissive for B19 [11].
An earlier sequence analysis provided hints that the product of the X ORF was functional, by detecting a decrease in synonymous codon variability in the region of overlap with VP1 [17], but could not determine whether this decrease was significant. Here we show that it is highly significant, using a dedicated software extensively validated on experimentally proven overlaps, Synplot2. In addition, we show that the X ORF is conserved not only in all erythroparvoviruses but also in tetraparvoviruses (in which it was called ARF1 [12]). Given the high rate of evolution of viruses, the conservation of the X ORF in two genera provides compelling evidence that it must be expressed and play a function essential for the viral life cycle.
4.1.1. The X protein could be translated either by a non-conventional mechanism or from an overlooked mRNA
The X ORF has a potential AUG start codon in all erythro- and tetraparvoviruses, but cannot be encoded in a monocistronic fashion by any known viral mRNA (see Figure S3). These observations suggest that the X protein is translated either 1) by a non-canonical mechanism, or 2) from a currently unmapped mRNA. We briefly discuss both hypotheses, which we present only as a starting point to guide experimental approaches.
1) Translation of the X ORF through a non-canonical mechanism
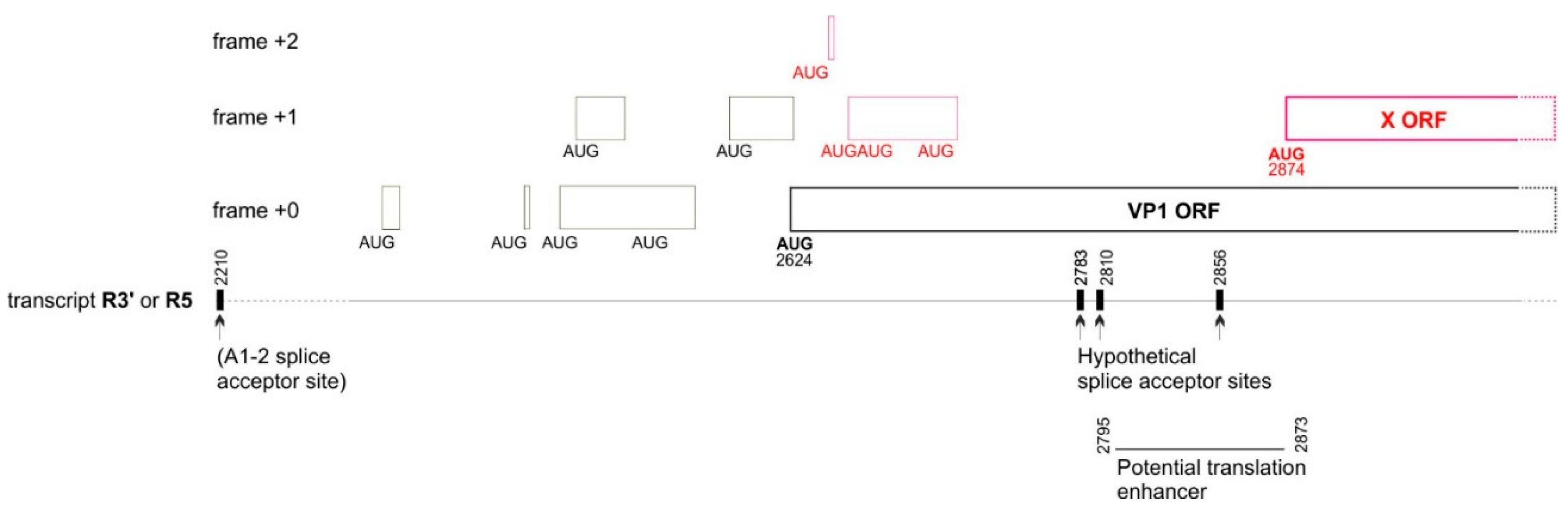
In vertebrates, two main factors influence canonical translation: 1) the strength of the “Kozak sequence” surrounding the initiator AUG codon [38]; and 2) the position of the AUG codon in the mRNA. In general, translation initiates at the first AUG with an optimal Kozak sequence, but many exceptions are known (for a review, see [39]). For example, a downstream AUG can sometimes initiate translation even if it is separated from the first optimal AUG by intervening AUGs, thanks to a mechanism called “re-initiation” (for a review, [40]). For example, in B19V, the VP1 AUG codon is preceded by 7 upstream AUG codons that form mini-ORFs (Figure 9), and is accessed by re-initiation after having first initiated translation at some of these mini-ORFs [41]. Note that the presence of these 7 upstream AUGs severely decreases the translation level of VP1 [41].
In principle, the B19V X ORF might also be translated from the VP1 mRNA by re-initiation, since it is separated from the VP1 AUG start codon by 4 AUGs (Figure 9). However, the efficiency of translation would presumably be very low [40]. Interestingly, in B19V, the 77 nucleotides upstream of the presumed AUG start codon of the X OR have a significantly reduced variability in synonymous codons (nt 172-250 of the VP1 CDS, see Figure 3B and Table 2, corresponding to nucleotides 2795-2873 of the genome, see Figure 9, bottom right) It is tempting to speculate that this region corresponds to a translation enhancer, i.e., a regulatory element that would enhance the translation efficiency of the X ORF.
The nomenclature of transcripts and splice sites is as in [6]. Thin boxes represent mini-ORFs. The mini-ORFs in black influence the translation of VP1 [40], and might also influence that of the X protein. The mini-ORFs in red are expected to influence the translation of the X protein but presumably not that of VP1. The region immediately upstream of the X ORF has a decreased synonymous variability (see Table 2 and Figure 3B), suggesting it has a regulatory function and might act as a translation enhancer.
2) Translation of the X ORF from a currently unmapped mRNA
A second mechanism might in principle ensure translation of the X ORF: the existence of an unmapped mRNA, generated by an overlooked splice acceptor site. Two conditions would be required for a splice acceptor site to generate a monocistronic transcript that encodes the X protein of B19V: 1) this site should be conserved in all isolates of B19V; 2) it should be located in the region between the VP1 start codon and the presumed start codon of the X protein (nt 251-253 of the VP1 CDS).
We found 3 such potential sites (having the canonical sequence (C/U)AG preceded by a region rich in pyrimidines (C/U) [42]), at nucleotides 158-160, 185-187, and 231-233 of the VP1 CDS. (The respective coordinates of the acceptor G in the genomic sequence of B19V are 2783, 2810 and 2856, see Figure 9). Each acceptor site would yield a monocistronic transcript that encodes the X ORF, by splicing out both the VP1 AUG start codon and the 4 following AUG codons located upstream of the presumed AUG start codon of the X protein (in red in Figure 9). Interestingly, these potential splice acceptor sites are located in or near in the potential regulatory region immediately upstream of the X ORF (Figure 9, bottom right), which has a decreased synonymous variability (Figure 3B and Table 2).
The X ORF most probably originated by overprinting the VP1 ORF
Most overlapping gene pairs originate by overprinting, a process in which substitutions in an ancestral reading frame enable the expression of a second reading frame (the novel frame), while preserving the expression of the first frame [43,44]. The ancestral frame can be identified by its phylogenetic distribution (the ORF with the widest distribution is most probably the ancestral one) [43,45], or by their codon usage [46] if both frames have the same phylogenetic distribution [14].
The phylogenetic distribution of X and of VP1 indicates that VP1 is necessarily the ancestral reading frame, since a PLA2 domain is found not only in most Parvoviridae, but also in a wide variety of metazoans and plants [47], whereas the X protein is found only in erythro- and tetraparvoviruses. Therefore, the X protein must have originated by overprinting the region of the the VP1 frame encoding PLA2, in the putative common ancestor of erythro- and tetraparvoviruses.
An intriguing observation is that the VP1 gene of bPARV3, which is basal to the erythroparvovirus phylogeny, contains an X-like ORF despite lacking a PLA2 domain (Figure 7). This raises two hypotheses–either:
1) the X-like ORF of bPARV3 arose independently from the X ORF of erythro- and tetraparvoviruses (i.e., their similarity is coincidental–they are not homologous); or
2) the X-like ORF of bPARV3 is homologous to the X ORF of erythro- and tetraparvoviruses, and the PLA2 domain was lost in bPARV3. In that case, the X-like ORF would constitute a “genetic palimpsest” (a palimpsest is a manuscript that has been erased and written on again), i.e., the X-like ORF would have been overprinted (“written over”) on a now “erased” PLA2 domain.
In the absence of intermediate sequences to reconstruct the evolution of bPARV3, it is not yet possible to settle the issue.
The X protein is not homologous to the protoparvovirus SAT protein
An earlier work [12] hypothesized that the ARF1/X protein of PARV4 was homologous to the SAT protein, another short, transmembrane protein encoded in the +1 frame of the VP1 gene in the genus protoparvovirus [48]. However, SAT and X cannot have a common origin, since SAT is encoded by the N-terminus of VP2, downstream of the region encoding the PLA2 domain (our observations), unlike the X protein, which overlaps PLA2 (see Figs 3 and 4).
5. Conclusion
While a systematic effort has been made to discover overlapping genes in RNA viruses by sequence analyses [18], this has not yet been the case in DNA viruses. Our findings confirm (if that were needed) that overlapping genes remain to be discovered in DNA viruses (we know of at least another case already flagged by sequence analyses, in human bocavirus [49]). We encourage all virologists who sequence genomes to look for overlapping genes using the simple tools and strategies presented here. This is perfectly feasible if you are a bench virologist lacking programming skills (like the author), since all analyses were done using web-based, relatively user-friendly programs (see Methods) on a standard laptop computer.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org: Figure S1: Multiple sequence alignment of tetraparvovirus ARF2 ORFs; Figure S2: Sequence of the Z protein of porcine parvovirus 2. Conventions are as in Figure 5. N-terminal Methionines that could correspond to an AUG start codon are in bold. In more distant tetraparvoviruses (not shown) the ARF2 ORF is interrupted by stop codons.Figure S3: All known transcripts that could express the X protein are polycistronic; A. Numbering refers to the B19V reference genome. Transcripts most likely to encode the X protein are marked by an asterisk (*). B. Numbering refers to the PARV4 reference genome. Color coding is different from panel A.Alignment S1: Codon alignment of all B19V VP1 coding sequences; Alignment S2: Codon alignment of all PARV4 VP1 coding sequences; Alignment S3: Codon alignment of all bPARV3 VP1 coding sequences; Alignment S4: Codon alignment of all pPARV2 VP1 coding sequences; Alignment S5: The X ORF has a potential AUG start codon in all erythro- and tetraparvoviruses; Alignment S6: Alignment of the X protein of erythroparvoviruses, tetraparvoviruses, and reported endogenous erythroparvoviruses.
Funding Information: This research received no external funding.
Acknowledgments
I gratefully acknowledge AE Firth for useful advice on using Synplot2 and for help with preparing the Synplot2 figures, K McNair for PRfect predictions, and CW Nelson for preliminary analyses using OLgenie. I thank S. Courtès, J Qiu, G. Gallinella, R Gifford, and two anonymous reviewers of a previous version for commenting on the manuscript. I thank all the authors of the user-friendly, web-based software without whom this work would not have been possible. I thank the Marie Skłodowska-Curie European programme for not supporting my projects and thereby allow me to pursue research as a rewarding hobby.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gruber AR, Neubock R, Hofacker IL, Washietl S. The RNAz web server: prediction of thermodynamically stable and evolutionarily conserved RNA structures. Nucleic Acids Research. 2007, 35, W335–W338. [CrossRef]
- Washietl S, L. Hofacker I. Identifying Structural Noncoding RNAs Using RNAz. In: Baxevanis AD, Davison DB, Page RDM, Petsko GA, Stein LD, Stormo GD, editors. Current Protocols in Bioinformatics. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2007. p. bi1207s19. [CrossRef]
- Söderlund-Venermo, M. Emerging Human Parvoviruses: The Rocky Road to Fame. Annu Rev Virol. 2019, 6. [Google Scholar] [CrossRef]
- Kailasan S, Agbandje-McKenna M, Parrish CR. Parvovirus Family Conundrum: What Makes a Killer? Annu Rev Virol. 2015, 2, 425–450. [CrossRef]
- Cotmore SF, Tattersall P. Parvoviruses: Small Does Not Mean Simple. Annu Rev Virol. 2014, 1, 517–537. [CrossRef]
- Ganaie SS, Qiu J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front Cell Infect Microbiol. 2018, 8, 166. [CrossRef]
- Matthews PC, Sharp C, Simmonds P, Klenerman P. Human parvovirus 4 ‘PARV4′ remains elusive despite a decade of study. F1000Res. 2017, 6, 82. [CrossRef]
- Luo W, Astell CR. A Novel Protein Encoded by Small RNAs of Parvovirus B19. Virology. 1993, 195, 448–455. [CrossRef]
- St Amand J, Beard C, Humphries K, Astell CR. Analysis of splice junctions and in vitro and in vivo translation potential of the small, abundant B19 parvovirus RNAs. Virology. 1991, 183, 133–142. [CrossRef]
- St Amand J, Astell CR. Identification and characterization of a family of 11-kDa proteins encoded by the human parvovirus B19. Virology. 1993, 192, 121–131. [CrossRef]
- Zhi N, Mills IP, Lu J, Wong S, Filippone C, Brown KE. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J Virol. 2006, 80, 5941–5950. [CrossRef]
- Simmonds P, Douglas J, Bestetti G, Longhi E, Antinori S, Parravicini C, et al. A third genotype of the human parvovirus PARV4 in sub-Saharan Africa. J Gen Virol. 2008, 89, 2299–2302. [CrossRef]
- Pavesi A, Vianelli A, Chirico N, Bao Y, Blinkova O, Belshaw R, et al. Overlapping genes and the proteins they encode differ significantly in their sequence composition from non-overlapping genes. PLoS ONE. 2018, 13, e0202513. [CrossRef]
- Pavesi, A. Computational methods for inferring location and genealogy of overlapping genes in virus genomes: approaches and applications. Current Opinion in Virology. 2022, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Firth AE, Brown CM. Detecting overlapping coding sequences with pairwise alignments. Bioinformatics. 2005, 21, 282–292. [CrossRef]
- Sabath N, Landan G, Graur D. A method for the simultaneous estimation of selection intensities in overlapping genes. PLoS ONE. 2008, 3, e3996. [CrossRef]
- Norja P, Eis-Hübinger AM, Söderlund-Venermo M, Hedman K, Simmonds P. Rapid sequence change and geographical spread of human parvovirus B19: comparison of B19 virus evolution in acute and persistent infections. J Virol. 2008, 82, 6427–6433. [CrossRef]
- Firth, AE. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res. 2014, 42, 12425–12439. [Google Scholar] [CrossRef]
- Chung BY-W, Miller WA, Atkins JF, Firth AE. An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci USA. 2008, 105, 5897–5902. [CrossRef]
- Jagger BW, Wise HM, Kash JC, Walters K-A, Wills NM, Xiao Y-L, et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science. 2012, 337, 199–204. [CrossRef]
- Ratinier M, Caporale M, Golder M, Franzoni G, Allan K, Nunes SF, et al. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 2011, 7, e1002477. [CrossRef]
- Zádori Z, Szelei J, Lacoste MC, Li Y, Gariépy S, Raymond P, et al. A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell. 2001, 1, 291–302.
- Dorsch S, Liebisch G, Kaufmann B, von Landenberg P, Hoffmann JH, Drobnik W, et al. The VP1 unique region of parvovirus B19 and its constituent phospholipase A2-like activity. J Virol. 2002, 76, 2014–2018. [CrossRef]
- Campbell MA, Loncar S, Kotin RM, Gifford RJ. Comparative analysis reveals the long-term coevolutionary history of parvoviruses and vertebrates. Quental TB, editor. PLoS Biol. 2022, 20, e3001867. [CrossRef]
- Wang J, Gribskov M. IRESpy: an XGBoost model for prediction of internal ribosome entry sites. BMC Bioinformatics. 2019, 20, 409. [CrossRef]
- McNair K, Salamon P, Edwards RA, Segall AM. PRFect: A tool to predict programmed ribosomal frameshifts in prokaryotic and viral genomes. Res Sq. 2023; rs.3.rs-2997217. [CrossRef]
- Atkins JF, Loughran G, Bhatt PR, Firth AE, Baranov PV. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [CrossRef]
- Leisi R, Di Tommaso C, Kempf C, Ros C. The Receptor-Binding Domain in the VP1u Region of Parvovirus B19. Viruses. 2016, 8, 61. [CrossRef]
- Baker JA, Wong W-C, Eisenhaber B, Warwicker J, Eisenhaber F. Charged residues next to transmembrane regions revisited: “Positive-inside rule” is complemented by the “negative inside depletion/outside enrichment rule.” BMC Biol. 2017, 15, 66. [CrossRef]
- Wong W-C, Maurer-Stroh S, Eisenhaber F. Not all transmembrane helices are born equal: Towards the extension of the sequence homology concept to membrane proteins. Biol Direct. 2011, 6, 57. [CrossRef]
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–13. [CrossRef]
- Lo MK, Søgaard TM, Karlin DG. Evolution and structural organization of the C proteins of paramyxovirinae. PLoS ONE. 2014, 9, e90003. [CrossRef]
- Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–248. [CrossRef]
- Allander T, Emerson SU, Engle RE, Purcell RH, Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc Natl Acad Sci USA. 2001, 98, 11609–11614. [CrossRef]
- Wang F, Wei Y, Zhu C, Huang X, Xu Y, Yu L, et al. Novel parvovirus sublineage in the family of Parvoviridae. Virus Genes. 2010, 41, 305–308. [CrossRef]
- Shade RO, Blundell MC, Cotmore SF, Tattersall P, Astell CR. Nucleotide sequence and genome organization of human parvovirus B19 isolated from the serum of a child during aplastic crisis. J Virol. 1986, 58, 921–936.
- Filippone C, Zhi N, Wong S, Lu J, Kajigaya S, Gallinella G, et al. VP1u phospholipase activity is critical for infectivity of full-length parvovirus B19 genomic clones. Virology. 2008, 374, 444–452. [CrossRef]
- Hernández G, Osnaya VG, Pérez-Martínez X. Conservation and Variability of the AUG Initiation Codon Context in Eukaryotes. Trends in Biochemical Sciences. 2019; S096800041930146X. [CrossRef]
- Firth AE, Brierley I. Non-canonical translation in RNA viruses. Journal of General Virology. 2012, 93, 1385–1409. [CrossRef]
- Gupta A, Bansal M. RNA-mediated translation regulation in viral genomes: computational advances in the recognition of sequences and structures. Briefings in Bioinformatics. 2019; bbz054. [CrossRef]
- Ozawa K, Ayub J, Young N. Translational regulation of B19 parvovirus capsid protein production by multiple upstream AUG triplets. J Biol Chem. 1988, 263, 10922–10926.
- Baralle M, Baralle FE. The splicing code. Biosystems. 2018, 164, 39–48. [CrossRef]
- Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol. 2009, 83, 10719–10736. [CrossRef]
- Keese PK, Gibbs A. Origins of genes: “big bang” or continuous creation? Proc Natl Acad Sci USA. 1992, 89, 9489–9493. [CrossRef]
- Sabath N, Wagner A, Karlin D. Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol. 2012, 29, 3767–3780. [CrossRef]
- Pavesi A, Magiorkinis G, Karlin DG. Viral proteins originated de novo by overprinting can be identified by codon usage: application to the “gene nursery” of Deltaretroviruses. PLoS Comput Biol. 2013, 9, e1003162. [CrossRef]
- Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochimica et Biophysica Acta (BBA)–Molecular and Cell Biology of Lipids. 2006, 1761, 1246–1259. [CrossRef]
- Zádori Z, Szelei J, Tijssen P. SAT: a late NS protein of porcine parvovirus. J Virol. 2005, 79, 13129–13138. [CrossRef]
- Sealfon RS, Lin MF, Jungreis I, Wolf MY, Kellis M, Sabeti PC. FRESCo: finding regions of excess synonymous constraint in diverse viruses. Genome Biol. 2015, 16, 38. [CrossRef]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [CrossRef]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009, 25, 1189–1191. [CrossRef]
- Procter JB, Thompson J, Letunic I, Creevey C, Jossinet F, Barton GJ. Visualization of multiple alignments, phylogenies and gene family evolution. Nat Methods. 2010, 7, S16–25. [CrossRef]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–469. [CrossRef]
- Katoh K, Frith MC. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012, 28, 3144–3146. [CrossRef]
- Kozlowski LP, Bujnicki JM. MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics. 2012, 13, 111. [CrossRef]
- Ferron F, Longhi S, Canard B, Karlin D. A practical overview of protein disorder prediction methods. Proteins. 2006, 65, 1–14. [CrossRef]
- Ludwiczak J, Winski A, Szczepaniak K, Alva V, Dunin-Horkawicz S. DeepCoil-a fast and accurate prediction of coiled-coil domains in protein sequences. Bioinformatics. 2019, 35, 2790–2795. [CrossRef]
- Kuchibhatla DB, Sherman WA, Chung BYW, Cook S, Schneider G, Eisenhaber B, et al. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J Virol. 2014, 88, 10–20. [CrossRef]
- Dobson L, Reményi I, Tusnády GE. CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res. 2015, 43, W408–412. [CrossRef]
- Floden EW, Tommaso PD, Chatzou M, Magis C, Notredame C, Chang J-M. PSI/TM-Coffee: a web server for fast and accurate multiple sequence alignments of regular and transmembrane proteins using homology extension on reduced databases. Nucleic Acids Res. 2016, 44, W339–343. [CrossRef]
Figure 1.
Cladogram of the VP1 proteins of erythro- and tetraparvoviruses.
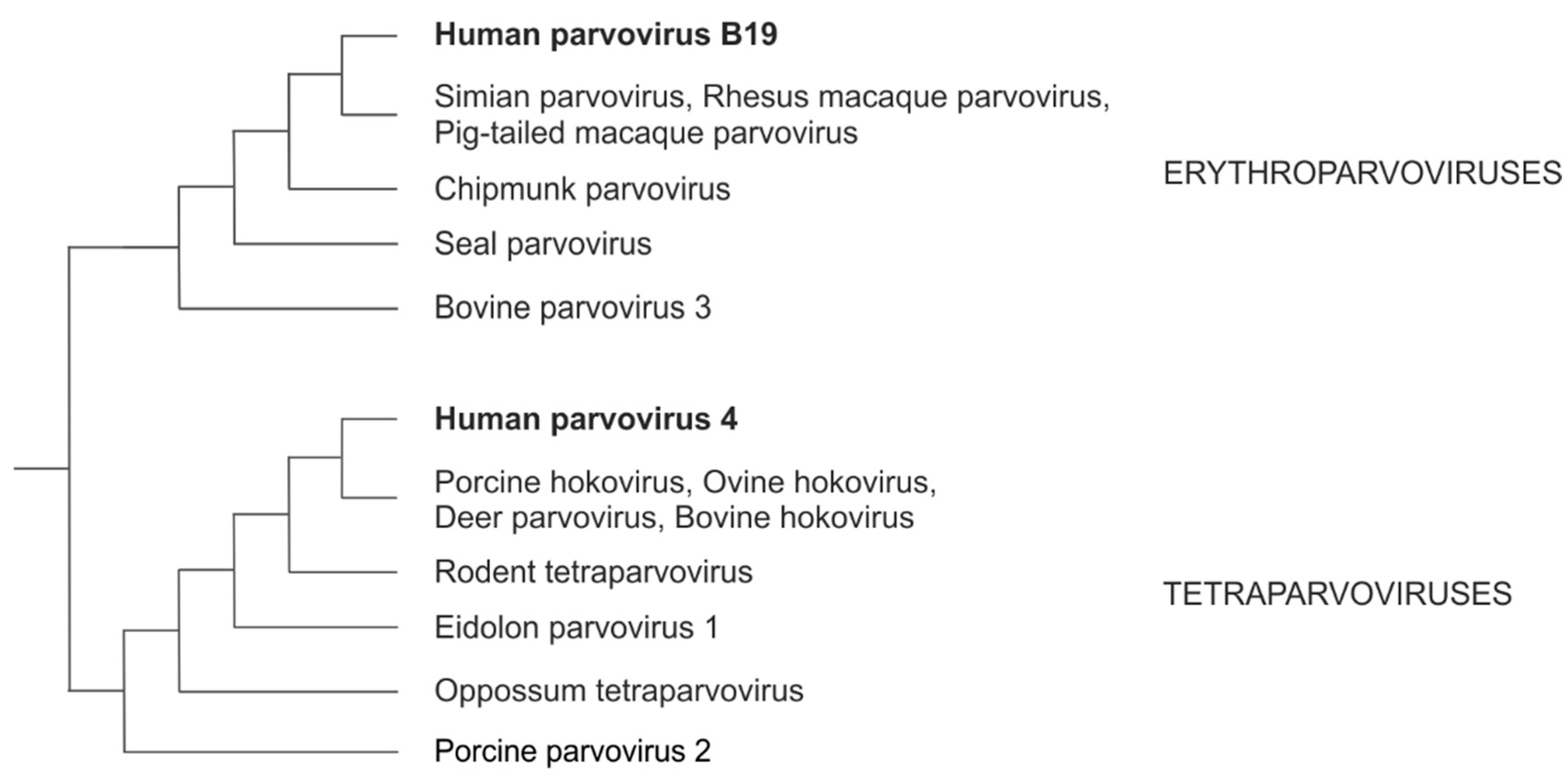
Figure 2.
B19V and PARV4 encode three suspected protein-coding ORFs. Long, horizontal lines represent the viral genomes. Boxes represent ORFs (Open reading frames). The three ORFs suspected to code for a protein are in grey. The VP2 isoform of VP1 is represented under VP1.
Figure 2.
B19V and PARV4 encode three suspected protein-coding ORFs. Long, horizontal lines represent the viral genomes. Boxes represent ORFs (Open reading frames). The three ORFs suspected to code for a protein are in grey. The VP2 isoform of VP1 is represented under VP1.
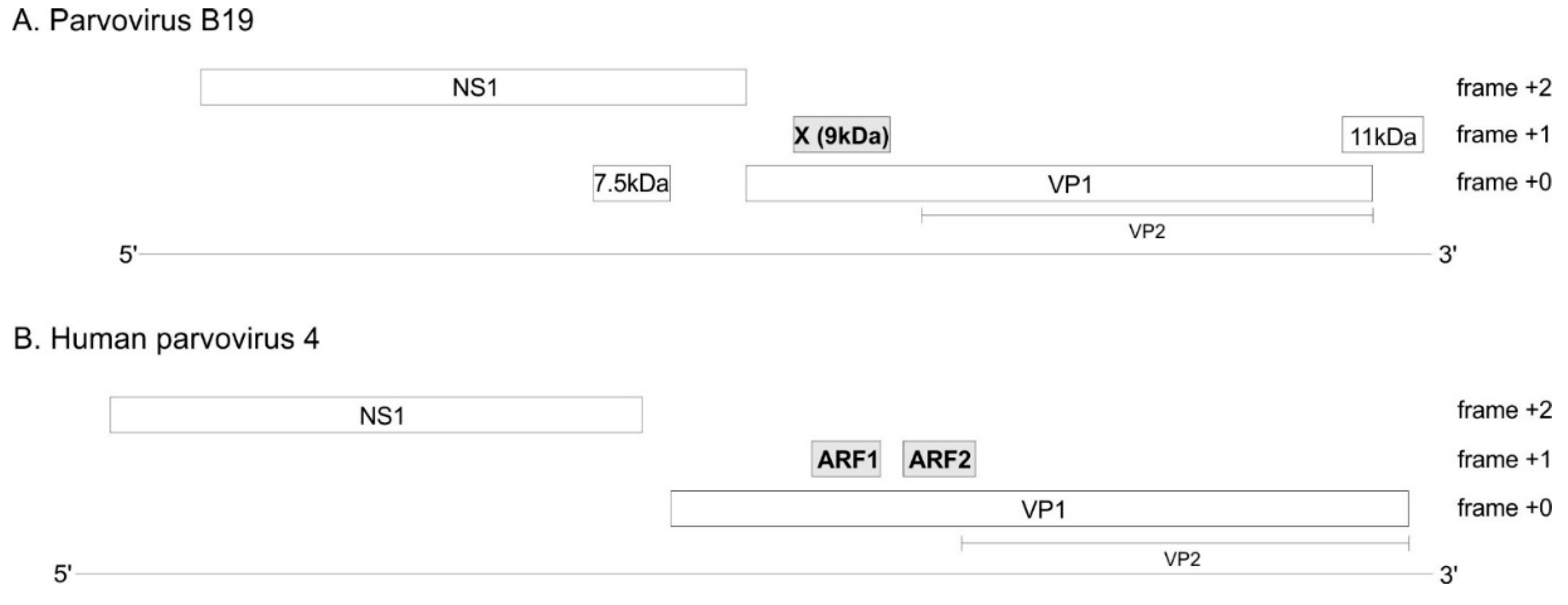
Figure 3.
Synplot2 detects 3 regions with significantly lower synonymous variability in the VP1 coding sequence of B19V, among which the X ORF. (A.) The VP1 gene and of its overlapping elements (coding sequences or functional elements). PLA2: Phospholipase A2 domain. RBD: receptor-binding domain [28]. VP1u: Vp1-unique region.(B.) Sequence conservation at synonymous sites in an alignment of coding sequences of B19V VP1 (121 sequences ranging from 87% to 99% nucleotide identity), using a 25-codon sliding window. The plot corresponds to the P-value calculated by Synplot2 based on the number of substitutions observed and the number expected under a null model (in which synonymous sites evolve neutrally). Regions in which synonymous substitutions are significantly decreased are indicated. The horizontal dotted line shows the significance cut-off value (10−3). Note that the first region with reduced synonymous variability starts markedly before the potential AUG start codon of the X protein (in green). This region, indicated by a thick line, might correspond to a functional element, which might facilitate the non-canonical translation of the X protein or the splicing of an X-specific RNA transcript (see text and Discussion). (C.) Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 121 sequences.
Figure 3.
Synplot2 detects 3 regions with significantly lower synonymous variability in the VP1 coding sequence of B19V, among which the X ORF. (A.) The VP1 gene and of its overlapping elements (coding sequences or functional elements). PLA2: Phospholipase A2 domain. RBD: receptor-binding domain [28]. VP1u: Vp1-unique region.(B.) Sequence conservation at synonymous sites in an alignment of coding sequences of B19V VP1 (121 sequences ranging from 87% to 99% nucleotide identity), using a 25-codon sliding window. The plot corresponds to the P-value calculated by Synplot2 based on the number of substitutions observed and the number expected under a null model (in which synonymous sites evolve neutrally). Regions in which synonymous substitutions are significantly decreased are indicated. The horizontal dotted line shows the significance cut-off value (10−3). Note that the first region with reduced synonymous variability starts markedly before the potential AUG start codon of the X protein (in green). This region, indicated by a thick line, might correspond to a functional element, which might facilitate the non-canonical translation of the X protein or the splicing of an X-specific RNA transcript (see text and Discussion). (C.) Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 121 sequences.
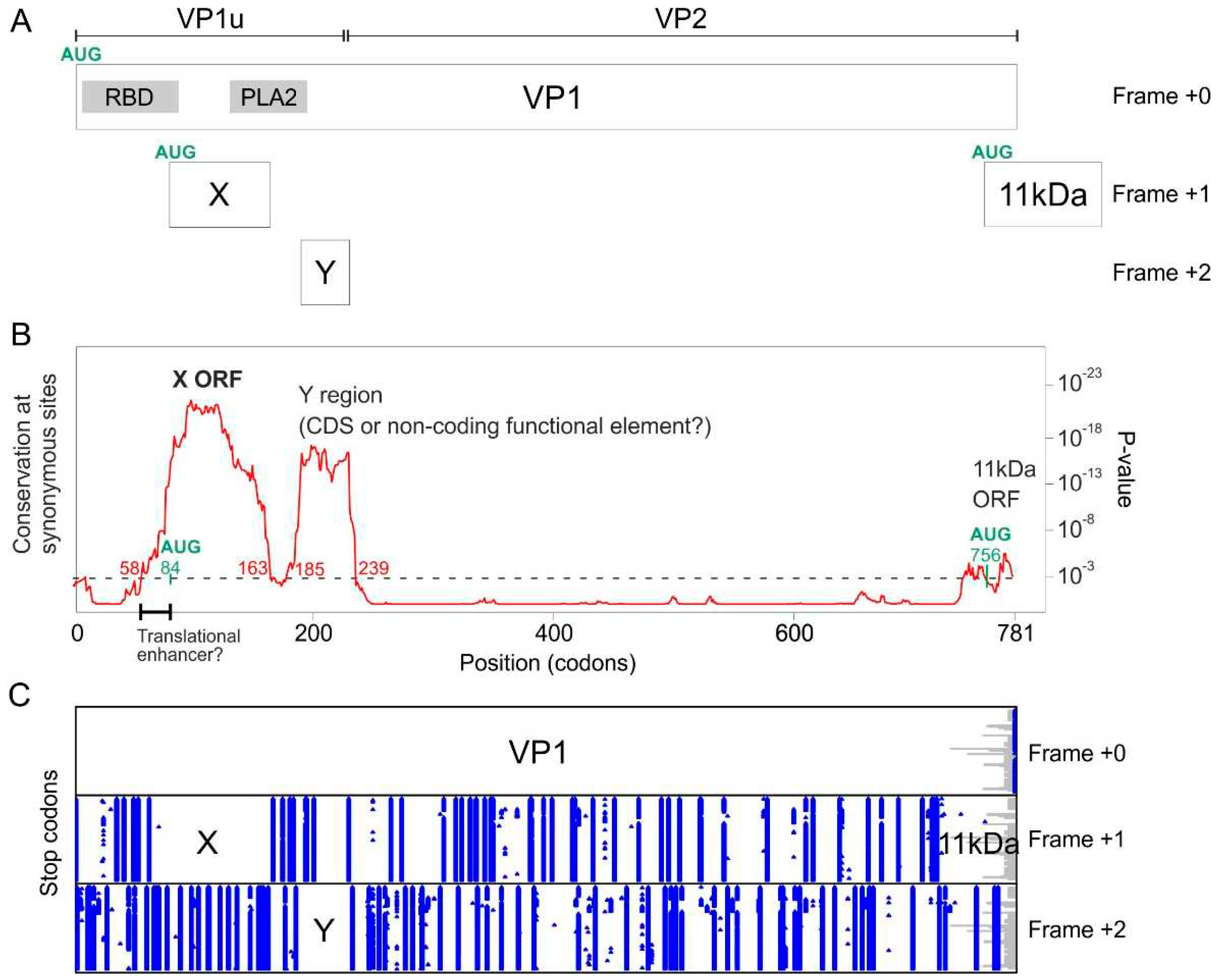
Figure 4.
Synplot2 detects 2 regions with significantly lower synonymous-site variability in the VP1 coding sequence of PARV4. A. Conventions are as in Figure 3. B. Conservation at synonymous sites in an alignment of coding sequences of PARV4 VP1 (21 sequences ranging from 93% to 99% identity), using a 25-codon sliding window in Synplot2. C. Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 21 sequences.
Figure 4.
Synplot2 detects 2 regions with significantly lower synonymous-site variability in the VP1 coding sequence of PARV4. A. Conventions are as in Figure 3. B. Conservation at synonymous sites in an alignment of coding sequences of PARV4 VP1 (21 sequences ranging from 93% to 99% identity), using a 25-codon sliding window in Synplot2. C. Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 21 sequences.
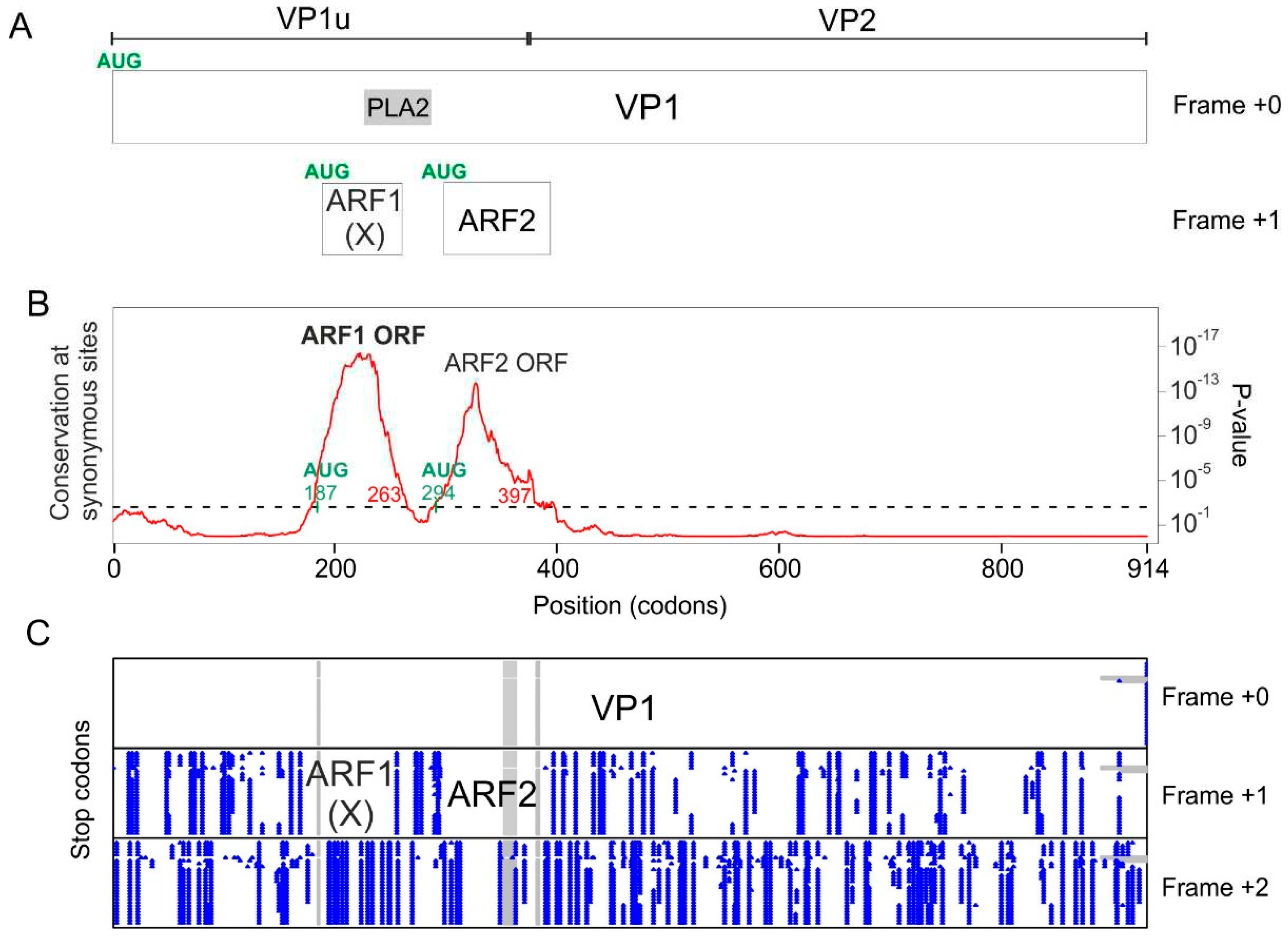
Figure 5.
Similar organization of the erythroparvovirus X protein, tetraparvovirus ARF1, and bovine parvovirus 3 X-like protein.A. Multiple sequence alignment of the erythroparvovirus X proteins. Numbering above the alignment corresponds to B19V. The sequences presented assume that the first AUG of each X ORF is used to initiate translation. PLA2: Phospholipase A2 domain.B. Alignment of the tetraparvovirus ARF1. Numbering corresponds to PARV4C. Sequence of the X-like protein of bovine parvovirus 3.
Figure 5.
Similar organization of the erythroparvovirus X protein, tetraparvovirus ARF1, and bovine parvovirus 3 X-like protein.A. Multiple sequence alignment of the erythroparvovirus X proteins. Numbering above the alignment corresponds to B19V. The sequences presented assume that the first AUG of each X ORF is used to initiate translation. PLA2: Phospholipase A2 domain.B. Alignment of the tetraparvovirus ARF1. Numbering corresponds to PARV4C. Sequence of the X-like protein of bovine parvovirus 3.
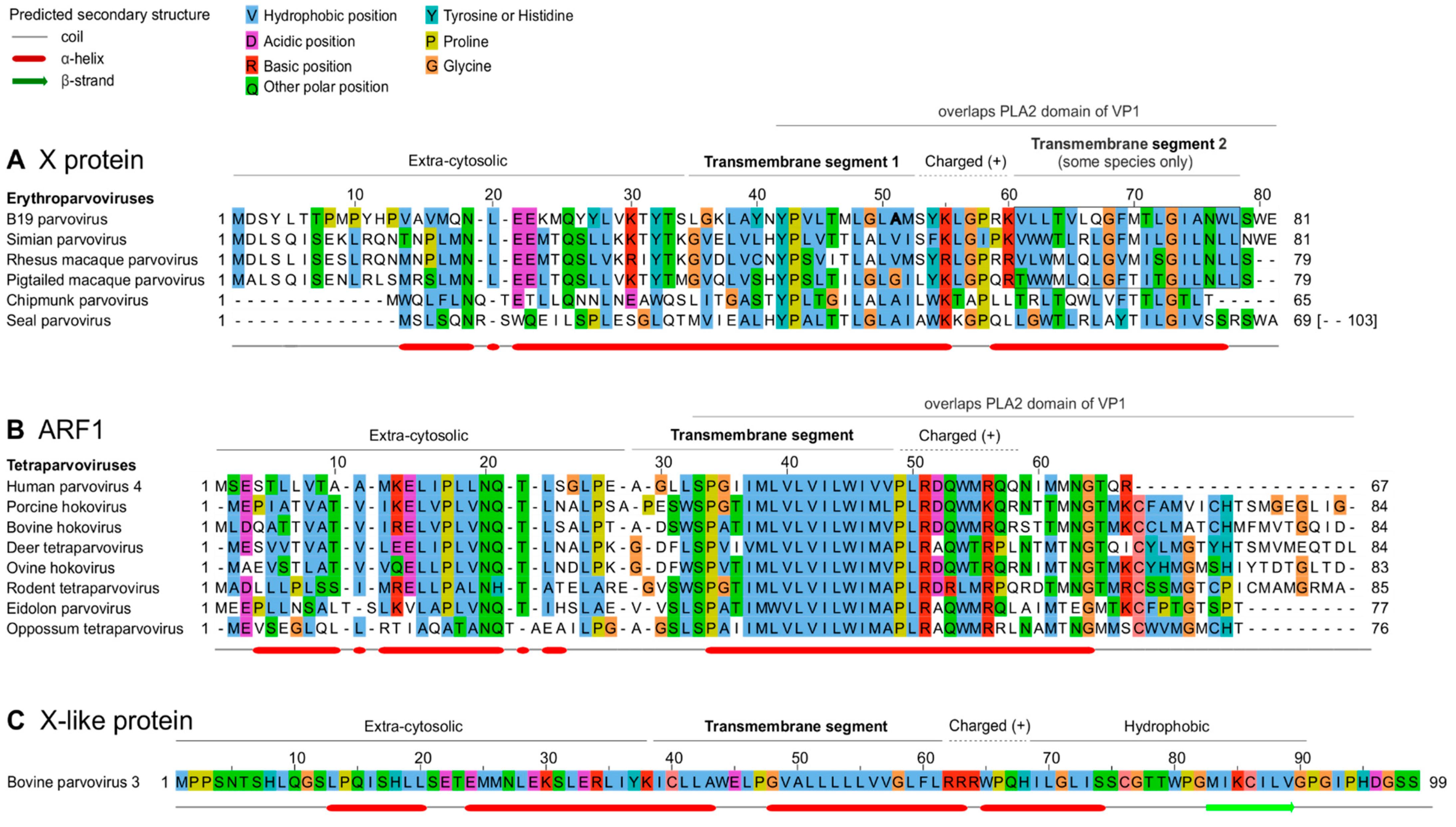
Figure 6.
Alignment of all X and ARF1 proteins based on the reference alignment of the PLA2 domain of VP1.
Figure 6.
Alignment of all X and ARF1 proteins based on the reference alignment of the PLA2 domain of VP1.
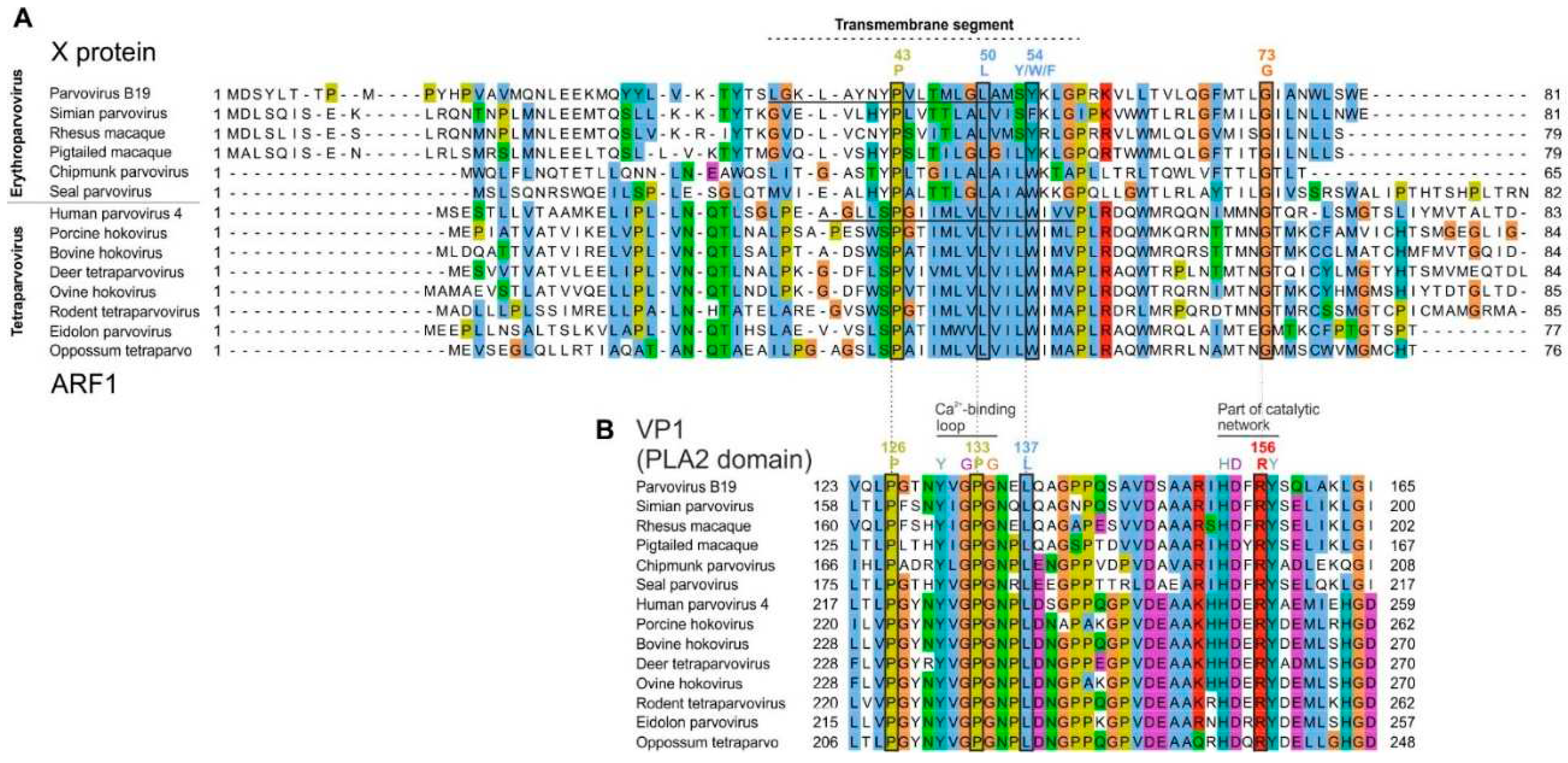
Figure 7.
Synonymous-site variability in the VP1 coding sequence of bovine parvovirus 3 (bPARV3). A. Conventions are as in Figure 3. The location of the VP1/VP2 boundary is only inferred. bARV3 VP1 contains no PLA2 domain, unlike all other erythro- and tetraparvoviruses (see text). B. Conservation at synonymous sites in an alignment of the coding sequences of bPARV3 VP1 (13 sequences ranging from 92% to 99% identity), using a 45-codon sliding window in Synplot2. C. Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 13 sequences.
Figure 7.
Synonymous-site variability in the VP1 coding sequence of bovine parvovirus 3 (bPARV3). A. Conventions are as in Figure 3. The location of the VP1/VP2 boundary is only inferred. bARV3 VP1 contains no PLA2 domain, unlike all other erythro- and tetraparvoviruses (see text). B. Conservation at synonymous sites in an alignment of the coding sequences of bPARV3 VP1 (13 sequences ranging from 92% to 99% identity), using a 45-codon sliding window in Synplot2. C. Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 13 sequences.
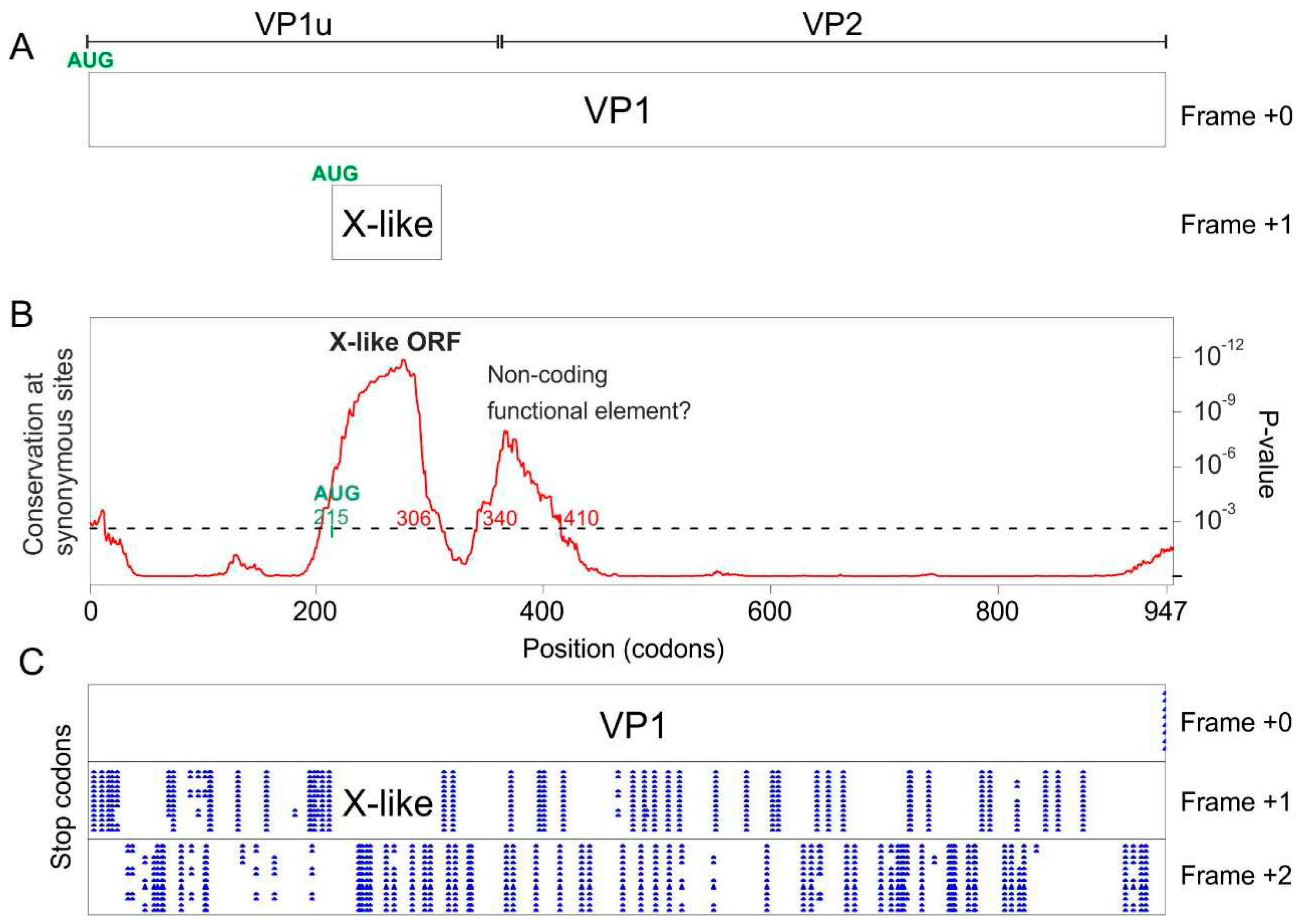
Figure 8.
Synonymous-site variability in the VP1 coding sequence of porcine parvovirus 2. A. Conventions are as in Figure 3. The location of the VP1/VP2 boundary is only inferred. B. Conservation at synonymous sites in an alignment of the coding sequences of pPARV2 VP1 (90 sequences ranging from 93% to 99% identity), using a 45-codon sliding window in Synplot2. C. Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 90 sequences.
Figure 8.
Synonymous-site variability in the VP1 coding sequence of porcine parvovirus 2. A. Conventions are as in Figure 3. The location of the VP1/VP2 boundary is only inferred. B. Conservation at synonymous sites in an alignment of the coding sequences of pPARV2 VP1 (90 sequences ranging from 93% to 99% identity), using a 45-codon sliding window in Synplot2. C. Position of stop codons (blue) in the 3 potential frames, and gaps in alignment (gray) in the 90 sequences.
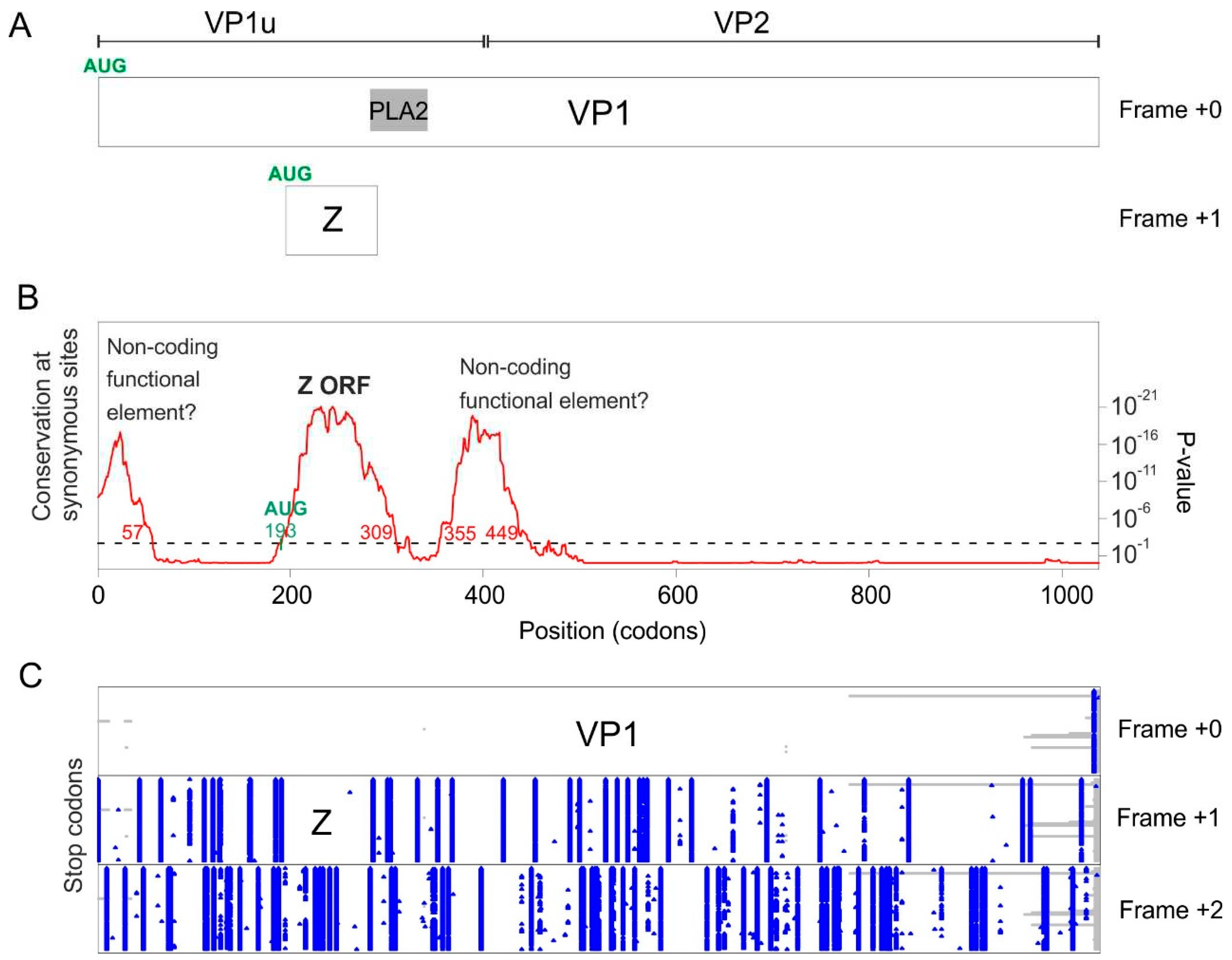
Figure 9.
Elements that could influence translation of the VP1 and X ORFs: upstream mini-ORFs, potential splice acceptors, and potential translation enhancer.
Figure 9.
Elements that could influence translation of the VP1 and X ORFs: upstream mini-ORFs, potential splice acceptors, and potential translation enhancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



