
Submitted:
10 December 2025
Posted:
19 December 2025
You are already at the latest version
Abstract
Background: Hereditary hypoparathyroidism (hypoPT) is a rare endocrine disorder caused by absent or insufficient parathyroid hormone secretion. Genetic forms are uncommon and frequently underdiagnosed, particularly when clinical onset is atypical.Purpose: We present three cases of genetic hypoPT caused by CASR, GNA11, and GATA3 mutations, highlighting diagnostic challenges and management approaches.Methods: Clinical data, biochemical results, and genetic findings were collected for three female patients with early-onset, nonsurgical hypoPT. Each case was evaluated for presenting symptoms, comorbidities, treatment responses, and outcomes. Relevant literature and guidelines were reviewed for context and management recommendations.Results: All patients exhibited chronic hypocalcemia, hyperphosphatemia, and low PTH without prior neck surgery. Initial manifestations were atypical, delaying diagnosis until adolescence or adulthood. Case 1 (CASR mutation) presented with seizure-like episodes misdiagnosed as epilepsy, and partial response to calcium–vitamin D therapy. Case 2 (GNA11 mutation) showed Raynaud’s phenomenon, dermatologic symptoms, and treatment intolerance without nephrocalcinosis. Case 3 (GATA3 mutation) had early deafness, later renal impairment, and fluctuating calcium levels. Genetic testing confirmed pathogenic variants, and individualized management with active vitamin D analogs achieved partial biochemical control. All patients remain under multidisciplinary follow-up with stabilization of neurological complications and monitoring of renal status. Conclusion: Genetic causes of hypoPT should be suspected in patients with early-onset or atypical presentations in the absence of neck surgery. Timely genetic testing confirms the diagnosis and determines personalized therapy. Management requires careful titration of calcium and active vitamin D to avoid complications and may benefit from emerging treatments (long-acting PTH analogs). Early recognition and tailored interventions can improve patient outcomes and quality of life.
Keywords:
hypoparathyroidism
; CASR
; GNA11
; GATA3
; autosomal dominant hypocalcemia
; Barakat syndrome
Introduction
Hypoparathyroidism (hypoPT) is a rare endocrine pathology defined by absent or insufficient parathyroid hormone (PTH) secretion, hypocalcemia and hyperphosphatemia. The predominant etiology is neck surgery with parathyroid gland injury or excision, whereas other less common cases include autoimmune dysfunction, magnesium imbalance and genetic mutations [1].
Chronic hypoPT poses lifelong management challenges, with risks of neuromuscular irritability, ectopic calcifications and renal complications.
Genetic forms of hypoPT are rare and often underdiagnosed until severe complications (e.g., refractory hypocalcemia, basal ganglia calcification) manifest. Familial isolated hypoPT demonstrates heterogeneous inheritance patterns, including autosomal dominant (AD), autosomal recessive (AR), and X-linked transmission with pathogenic variants identified in PTH, GCM2, CASR, GNA11 and GATA3 among others [2].
Understanding the genetic basis of hypoPT is crucial for accurate diagnosis and management particularly in cases with atypical presentation or early-onset. Advances in genetic testing have facilitated the identification of these mutations, allowing for personalized treatment approaches.
We present a case series of three patients with rare genetic forms of hypoPT due to mutations in the CASR, GNA11, and GATA3 genes, highlighting clinical manifestations, diagnostic challenges and treatment strategies.
Case 1: Autosomal Dominant Hypocalcemia Type 1 (CASR Mutation)
A 28-year-old woman with a decade-long history of episodic loss of consciousness (LOC) since adolescence reported new-onset lower limb cramps, recurrent headaches, and progressive generalized weakness. She was previously managed as atonic epilepsy (on valproate and levetiracetam seizure-free for years). However her last LOC episode coincided with severe menstrual pain, raising suspicion of an alternative etiology. Family history revealed a sibling with similar symptoms, later confirmed to have hypocalcemia and hyperphosphatemia, while parents showed no evidence of mineral metabolism disorders. This atypical presentation (seizure-like episodes without classic hypocalcemic tetany) delayed the diagnosis of hypoPT until early adulthood.
At the age of 26-year-old, chronic hypoPT was confirmed: PTH 12 pg/mL (15-65), calcium (Ca) total 1.72 mmol/L (2.15-2.55); Ca ionized 0.86 mmol/L (1.0-1.3)). Brain MRI revealed extensive bilateral calcifications in the basal ganglia and cerebellum, consistent with Fahr syndrome. The patient was prescribed Ca carbonate 1500 mg/day, alfacalcidol 1 μg/day, and cholecalciferol 5000 IU/day. However, serum albumin-adjusted Ca (Caadj.) remained low 1.68-1.79 mmol/L with persistent high phosphorus (P) 1.77 mmol/L (0.74-1.52), indicating suboptimal response to conventional therapy.
The patient was admitted to the parathyroid pathology and mineral disorders (PPMD) department (Endocrinology Research Centre) at the age 28-year-old due to worsening symptoms following therapy discontinuation. Physical examination revealed no dysmorphic features. Laboratory results confirmed decompensation of chronic hypoPT: Caadj. 1.68-1.79 mmol/L (2.15-2.55), P 1.96 mmol/L (0.74-1.52), magnesium (Mg) 0.67 mmol/L (0.70-1.05), daily urine Ca 4.08 mmol/day (2.5-8.0).
Genetic testing (next-generation sequencing (NGS)) identified a heterozygous pathogenic variant in the CASR gene (OMIM: 601199, c.382T>C, chr3:122257277T>C), confirming AD hypocalcemia type 1.
Initially, the administration of alfacalcidol (3 μg/day), Ca carbonate (3000 mg/day) in high doses, and Mg supplements (300 mg/day) did not lead to adequate disease compensation. Therefore, we replaced alfacalcidol with calcitriol, and reduced doses of calcitriol and Ca carbonate to prevent hypercalciuria. The patient continued the cholecalciferol therapy (15,000 IU/week).
Despite suboptimal biochemical control due to mild hypercalciuria 9.0 mmol/day, we did not prescribed recombinant human PTH (rhPTH) replacement due to limited availability/access. The main strategy focused on optimizing minimal effective doses of calcitriol and Ca medications.
Neurological status remained stable with no progression of Fahr syndrome on brain imaging (Figure 1). Annual monitoring showed preserved kidney function (eGFR 112.3 mL/min/1.73 m²), with no ultrasound-signs of nephrocalcinosis, nephrolithiasis, and cardiac calcification. Ophthalmologic evaluation revealed only mild myopia without lens pathology. Additional findings included erosive gastritis (positive parietal cell antibodies) and normal bone mineral density (BMD). The patient continues regular check-ups in our Centre.
Case 2: Autosomal Dominant Hypocalcemia Type 2 (GNA11 Mutation)
A 19-year-old female presented with painful hand muscle cramps, acral paresthesias (numbness and cold fingers) and marked asthenia, accompanied by significant weight loss (6 kg over 6 months) with loss of appetite. The clinical picture was further complicated by dermatological manifestations (diffuse alopecia and xerosis) and reproductive dysfunction (10 months of secondary amenorrhea). Family history was unremarkable for mineral metabolism disorders.
At the age of 18-year-old, the patient developed subconjunctival hemorrhages. Brain CT imaging demonstrated extensive bilateral calcifications in subcortical regions (frontal/parietal lobes), basal ganglia and thalamus, confirming Fahr syndrome. Laboratory evaluation revealed inappropriately low PTH 10.8 pg/mL (15-65) with Caadj.1.79 mmol/L (2.15-2.55) and P 2.2 mmol/L (0.74-1.52), establishing hypoPT.
Initial therapy with alfacalcidol 1 μg/day and Ca carbonate 500 mg/day was discontinued due to clinical deterioration, including worsening skin rash and leg pain. Staphylococcal skin infection was identified, requiring antibiotic prescription. Subsequent rheumatologic workup revealed weakly positive antinuclear antibodies (ANA) 1:160 with normal rheumatoid factor and C-reactive protein levels, leading to a diagnosis of Raynaud’s syndrome; however, vasodilator therapy provided slight clinical improvement. These findings suggested complex pathophysiology involving both metabolic and vascular components.
At the age of 19-year-old, the patient was admitted to the PPMD department with severe metabolic disturbances and clinical manifestations including marked acrocyanosis, xerosis cutis (Figure 2), and significant underweight (body weight 34 kg and height 149 sm, BMI 15.3 kg/m²). Biochemical evaluation showed extremely low Caadj. 1.57 mmol/L (2.15-2.55), high P 2.5 mmol/L (0.74-1.52), and daily urine Ca 2.16 mmol/day. There was no evidence of Mg deficiency (0.78 mmol/l – RR 0.66-1.05). Ca supplements and vitamin D analogs in tablets triggered allergic reactions, thus loratadine 10 mg/day administration with reduced-dose oil solution of alfacalcidol 0.5-0.75 μg/day were prescribed. Despite subjective improvement in well-being, biochemical parameters at discharge remained non-optimal (Caadj. 1.63 mmol/L, P 2.4 mmol/L).rhPTH therapy was not initiated because of limited therapy access and patient’s avoidance due to polyvalent drug allergy.
The patient developed bilateral cataracts as a complication of chronic hypoPT, while renal and cardiac evaluations were excluded. Significant underweight was associated with hypogonadotropic hypogonadism (low estradiol, luteinizing hormone, and follicle-stimulating hormone levels), consistent with functional hypothalamic amenorrhea, prompting combined psychiatric and nutritional interventions. Given the early disease onset and absence of obvious causes of hypoPT we suggested genetic form of the disease. NGS analysis revealed a pathogenic heterozygous GNA11 variant (c.179G>T, p.Arg60Leu; rs587777707), confirming AD hypocalcemia type 2 (OMIM: 139313.0007).
Case 3: Hypoparathyroidism, Sensorineural Deafness and Renal Disease Syndrome (HDR Syndrome; Barakat Syndrome (GATA3 Mutation)
A 29-year-old female patient was admitted to the PPMD department with complaints of recurrent headaches, skin eruptions on the face, chest and back, redness and flaking of the scalp and forehead, and progressive worsening of pre-existing grade III mixed (sensorineural and conductive) hearing loss since childhood. We revealed patient’s mild facial dysmorphism (wide forehead, small low-set ears) and widespread dermatological manifestations, including numerous papules, pustules, comedones, and areas of hyperemia with desquamation. Hereditary anamnesis was unremarkable, with no known mineral metabolism or endocrine disorders in her relatives.
The patient developed first symptoms at the age of 12 (episodic paresthesias and subjective «loss of body control», dental enamel defects). Laboratory blood tests revealed low Ca total 1.81 mmol/L (2.15-2.55), Ca ionized 0.79 mmol/L (1.03-1.29). Brain MRI demonstrated calcifications in the basal ganglia and right frontal lobe (Fahr syndrome). These findings were initially interpreted as symptomatic frontal lobe epilepsy, prompting anticonvulsant therapy; however, subsequent evaluations failed to verify epileptiform activity. She did not prescribed other medications.
At the age of 23, the patient presented with recurrent episodes of limb numbness and weakness in the postpartum period. Laboratory tests showed persistent hypocalcemia (Caadj. 1.76 mmol/L), hypocalciuria 1.22 mmol/day (2.5-8.0) and an inappropriately low PTH 7.3 pg/mL (15-65), confirming hypoPT. Treatment with Ca 2500 mg/day and active vitamin D 2.0 µg/day supplementation was initiated.
Despite conventional therapy, there was no hypoPT compensation with episodes of both hypocalcemia, severe hypercalcemia (Caadj. up to 3.65 mmol/L), and progressive renal impairment (eGFR (CKD-EPI) 17 mL/min/1.73 m²).
At the age of 29, she developed acute tubulointerstitial nephritis and was treated with methylprednisolone. Hypercalcemia necessitated withdrawal of Ca and vitamin D supplements, in 5 days therapy was resumed (Ca carbonate 1500 mg/day, alfacalcidol (2.0 µg/day) because of hypocalcemia (Caadj. 1.9 mmol/L). When the patient was admitted to the PPMD department in some months laboratory test showed hypercalcemia again, so we cancelled all supplements and closely monitored her Ca-P metabolism parameters. HypoPT therapy was resumed in 5 days with alfacalcidol (0.5 µg/day) and Ca carbonate (500 mg/day), achieving Caadj. 2.4 mmol/L and P 1.28 mmol/L. Subsequent evaluation demonstrated nephrocalcinosis, bilateral renal microlithiasis, bilateral cataracts, and intracranial calcifications. Additionally, markedly increased BMD was observed, with Z-scores up to 5.4 SD in the lumbar spine and 3.6 SD in the left femoral neck.
Given the triad of hypoPT, early-onset sensorineural hearing loss, and renal impairment, HDR syndrome was suspected. NGS identified a previously unreported heterozygous variant in the GATA3 gene (c.1061C>A, p.Pro354His), confirming the diagnosis. Follow-up tests showed normal Ca (2.05 mmol/L) and P (1/17 mmol/L) levels, with eGFR improvement (57 mL/min/1.73 m²).
Key information on the described patients with hereditary hypoPT is presented in the Table 1.
Discussion
All three young female patients exhibited chronic hypocalcemia, varying degrees of hyperphosphatemia, and low or undetectable PTH levels, consistent with chronic hypoPT. They had no prior neck surgery or features of autoimmune polyglandular syndrome. All patients demonstrated neurological complications (basal ganglia calcifications and neuromuscular symptoms) secondary to chronic hypocalcemia and hyperphosphatemia.
The initial presentation of all cases was atypical (neurologic, dermatologic, reproductive, or renal-first), which contributed to delayed diagnosis of hypoPT in adulthood.
While different gene mutations share similar biochemical profiles of mineral metabolism, each exhibits distinct clinical features: ADH-1 demonstrates relative resistance to conventional therapy and rapid onset of hypercalciuria; ADH-2 is associated with autoimmune pathology; and HDR syndrome presents with deafness and renal dysfunction.
Gain-of-function mutations in the CASR gene (causing ADH-1) lower the calcium-sensing receptor’s (CaSR) set point, resulting in inappropriate PTH suppression despite hypocalcemia. ADH-1 typically manifests with hypocalcemia, hyperphosphatemia, hypercalciuria and basal ganglia calcifications. Patients frequently develop neuromuscular symptoms including muscle cramps, paresthesias, and seizures, and may exhibit cognitive or psychiatric disorders. Interestingly, while hypercalciuria represents a hallmark feature, our patient demonstrated normal urinary Ca excretion at disease onset, potentially attributable to severe Ca deficiency [3,4,5].
GNA11 mutations (ADH-2) impair CaSR downstream signaling. In contrast to ADH-1, ADH-2 typically demonstrates milder hypercalciuria and is infrequently connected with hypomagnesemia. This could be due to the fact that CASR couples with proteins other than alfa subunit of G11 in the kidneys. Patients with ADH-2 tend to exhibit shorter height, suggesting that GNA11 may influence skeletal development, as well as calcifications in the basal ganglia. However, our patient exhibited an expanded phenotype including autoimmune manifestations and treatment intolerance that has not been previously described in this disease [6]. While both ADH-1 and ADH-2 share hypocalcemia with low PTH, their renal and neurological involvement differs markedly. Notably, our patient lacked nephrocalcinosis – a frequent finding in reported cases [3]. The autoimmune and other features of impaired immune function in our ADH-2 patient may be a random combination of diseases or caused by PTH deficiency [7].
GATA3 mutations (HDR syndrome) result in haploinsufficiency of the GATA3 transcription factor, leading to impaired development of the parathyroid glands, kidneys, and auditory system. While patients usually exhibited the classic diagnostic triad, our case also presented with atypical manifestations including dermatological features, which can be associated with immune system dysregulation [8,9,10].
According to current clinical guidelines, genetic testing is indicated in early-onset hypoPT after excluding surgery, autoimmune or other obvious factors. Identification of CASR, GNA11, or GATA3 mutations enables accurate diagnosis, prognosis assessment, and family counseling [11].
The clinical presentation of hypoPT demonstrates variability, even within the same genetic subtype, necessitating personalized therapeutic strategies. In ADH-1 patients, treatment requires careful titration to avoid hypercalcemia while minimizing renal complications such as hypercalciuria and nephrolithiasis/nephrocalcinosis. Our patient with ADH-2 developed recurring allergic reactions to standard Ca and vitamin D supplements, thus we adjusted both the dosage and forms of the medications. Similarly, HDR syndrome with progressive renal impairment required regular assessment of GFR, serum electrolyte levels, and long-term treatment safety. These cases underscore the complexity of managing rare genetic forms of hypoPT and the importance of individualized therapeutic approaches.
Current guidelines support considering rhPTH therapy in chronic hypoPT when conventional treatment fails to achieve biochemical targets, leads to complications (e.g., persistent hyperphosphatemia, hypercalciuria, renal insufficiency), or does not sufficiently improve quality of life. In our cases, especially in the patient with ADH-1, rhPTH could have been an optimal therapeutic option, but it was not administered due to the unavailability of the medication.
Recent therapeutic developments offer promising alternatives for patients with suboptimal control. Long-acting PTH analogs, such as palopegteriparatide (TransCon PTH), have demonstrated sustained normocalcemia, independence from active vitamin D, and reduced daily urinary Ca over 52 weeks in Phase 3 trials (PaTHway). Moreover preliminary results show improvements in quality of life and renal outcomes. Similarly, eneboparatide (AZP-3601), a PTH1R agonist, normalized serum Ca compared with placebo in a Phase 3 trial (topline results) and showed favorable pharmacodynamic profiles in early-phase studies. While data specific to CASR-, GNA11-, or GATA3-related hypoPT remain limited, mechanism-based benefits may be expected [12,13,14,15,16,17].
Conclusions
Genetic forms of hypoPT are rare but should be suspected in patients with early-onset disease, absence of prior neck surgery and/or atypical features. Genetic testing allows to confirm the correct diagnosis associated with pathogenic variants (e.g., in CASR, GNA11, or GATA3 genes) and define personalized management. The presented cases illustrate clinical heterogeneity among these molecular subtypes, complicating timely diagnosis. Effective management requires a multidisciplinary team, including endocrinologist, nephrologist, genetics, and other specialists. Long-term management must include careful monitoring of Ca-P parameters, renal and neurological complications, and treatment response. Early diagnosis and interventions may improve outcomes and quality of life.
Funding
The research was carried out within the state assignment of Ministry of Health of the Russian Federation (theme No. 123021300096-3).
Author contributions
All authors contributed to the conception and design of the study, data collection, analysis, and interpretation. The manuscript was drafted and critically revised by all authors. All authors read and approved the final version of the manuscript.
Institutional Review Board Statement
The study was conducted in accordance with the principles of the Declaration of Helsinki. Ethical approval was obtained from the Local Ethics Committee of the National Medical Research Center for Endocrinology, Ministry of Health of the Russian Federation (protocol No. 6, approved on 23 March 2022). Written informed consent to participate was obtained from all patients.
Consent for Publication
Written informed consent for publication of anonymized clinical data was obtained from all patients
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Clarke, B.L., Vokes, T.J., Bilezikian, J.P., Shoback, D.M., Lagast, H., Mannstadt, M.: Effects of parathyroid hormone replacement therapy on hypoparathyroidism: a randomized double-blind placebo-controlled study (REPLACE). Endocrine 55, 273–282 (2017). [CrossRef]
- Ding, C., Buckingham, B., Levine, M.A.: Familial isolated hypoparathyroidism caused by a mutation in the gene for the transcription factor GCMB. J. Clin. Invest. 108, 1215–1220 (2001). [CrossRef]
- Roszko, K.L., Stapleton Smith, L.M., Sridhar, A.V., Roberts, M.S., Hartley, I.R., Gafni, R.I., Collins, M.T., Fox, J.C., Nemeth, E.F.: Autosomal dominant hypocalcemia type 1: a systematic review. J. Bone Miner. Res. 37, 1926–1935 (2022). [CrossRef]
- Roszko, K.L., Bi, R.D., Mannstadt, M.: Autosomal dominant hypocalcemia (hypoparathyroidism) types 1 and 2. Front. Physiol. 7, 458 (2016). [CrossRef]
- De Coster, T., David, K., Breckpot, J., Decallonne, B.: Genetics of hypoparathyroidism: an updated review. J. Endocrinol. Invest. 48, 831–844 (2025). [CrossRef]
- Li, D., Opas, E.E., Tuluc, F., Metzger, D.L., Hou, C., Hakonarson, H., Levine, M.A.: Autosomal dominant hypoparathyroidism caused by germline mutation in GNA11: phenotypic and molecular characterization. J. Clin. Endocrinol. Metab. 99, E1774–E1783 (2014). [CrossRef]
- Puliani, G., Hasenmajer, V., Sciarra, F., Barbagallo, F., Sbardella, E., Pofi, R., Gianfrilli, D., Romagnoli, E., Venneri, M.A., Isidori, A.M.: Hypoparathyroidism and associated autoimmune comorbidities: insights from clinical practice. J. Clin. Endocrinol. Metab. 106, e2215–e2227 (2021). [CrossRef]
- Valenciaga, A., Brock, P., O’Donnell, B., Ing, S.W.: HDR syndrome due to GATA3 mutation: a case report. J. Clin. Endocrinol. Metab. Case Rep. 3, luae246 (2025). [CrossRef]
- Prabhu, P.P., Ballal, S., Augustine, R., Shetty, M.: Hypoparathyroidism–deafness–renal disease (HDR) syndrome: a case report. Indian J. Nephrol. 33, 377–380 (2023). [CrossRef]
- Tao, Y., Yang, L., Han, D., Zhao, C., Song, W., Li, M., Zhang, X., Li, G.: Novel GATA3 mutation in HDR syndrome with atypical features. Front. Genet. 14, 1254556 (2023). [CrossRef]
- Mannstadt, M., Cianferotti, L., Gafni, R.I., Giusti, F., Kemp, E.H., Koch, C.A., et al.: Hypoparathyroidism: clinical management guidelines of the European Society of Endocrinology. J. Bone Miner. Res. 37, 2615–2629 (2022). [CrossRef]
- Clarke, B.L.: Advances in the management of hypoparathyroidism. Arch. Endocrinol. Metab. 66, 604–610 (2022). [CrossRef]
- Khan, A.A., Rubin, M.R., Schwarz, P., Vokes, T., Shoback, D.M., Gagnon, C., et al.: Long-term safety and efficacy of recombinant parathyroid hormone therapy in hypoparathyroidism. J. Bone Miner. Res. 38, 14–25 (2023). [CrossRef]
- Clarke, B.L., Khan, A.A., Rubin, M.R., Schwarz, P., Vokes, T., Shoback, D.M., et al.: Efficacy and safety of TransCon PTH in adults with hypoparathyroidism: 52-week results from the PaTHway trial. J. Clin. Endocrinol. Metab. 110, 951–960 (2025). [CrossRef]
- Rejnmark, L., Gosmanova, E.O., Khan, A.A., Makita, N., Imanishi, Y., Takeuchi, Y., et al.: Eneboparatide in hypoparathyroidism: results from a phase 2 study. Adv. Ther. 41, 2500–2518 (2024). [CrossRef]
- Ovize, M., Allas, S., Culler, M.D., Milano, S., Ouldrouis, T., Sumeray, M., et al.: Eneboparatide (AZP-3601) for hypoparathyroidism: phase 3 trial results. Endocr. Connect. 14, e240464 (2025). [CrossRef]
- ClinicalTrials.gov: Phase 3 Study of Eneboparatide (AZP-3601) in Chronic Hypoparathyroidism (CALYPSO trial). NCT05778071. Accessed 30 August 2025.
Figure 1.
MRI scan of patient`s brain (Fahr syndrome).
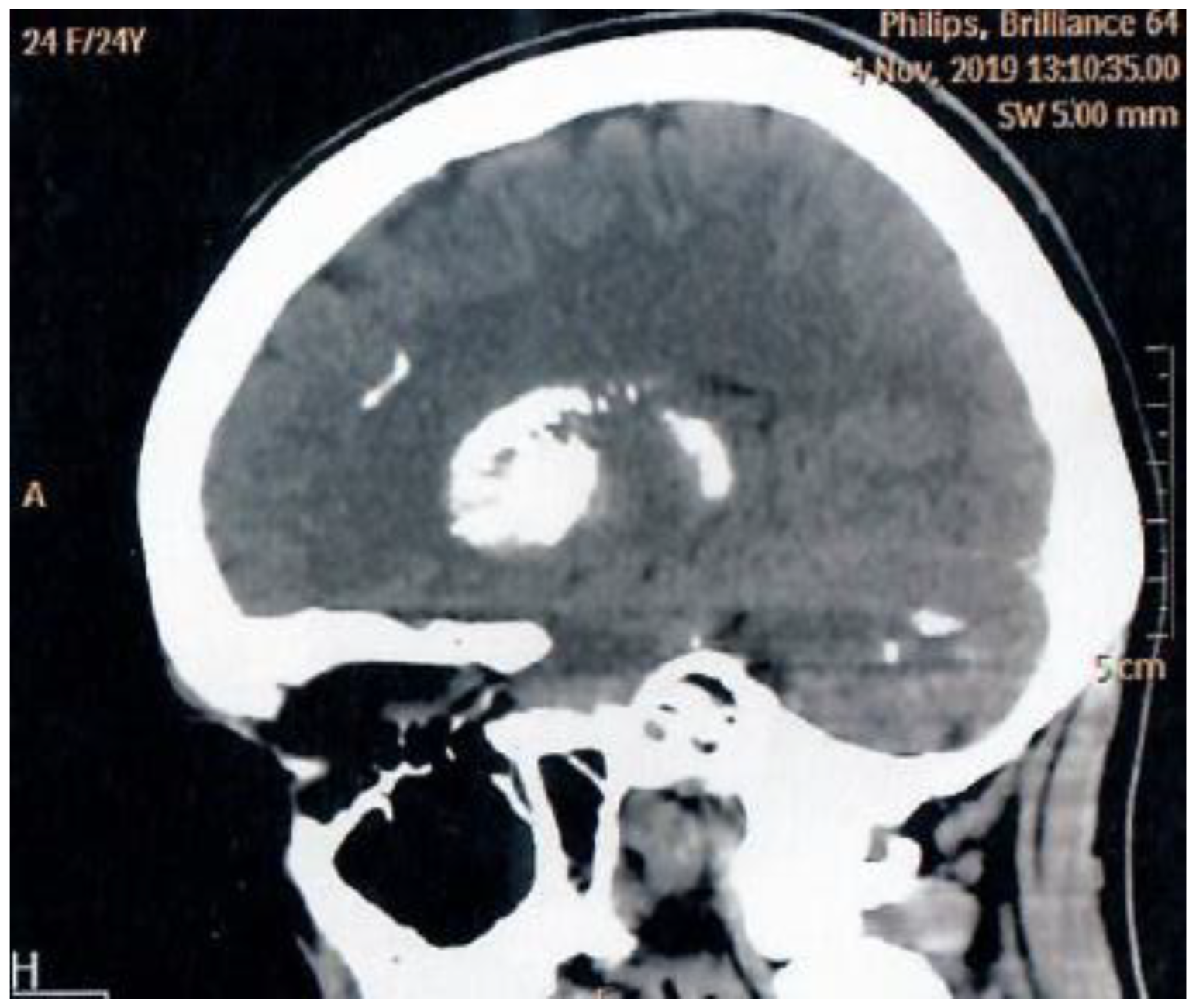
Figure 2.
Raynaud’s syndrome`s signs.
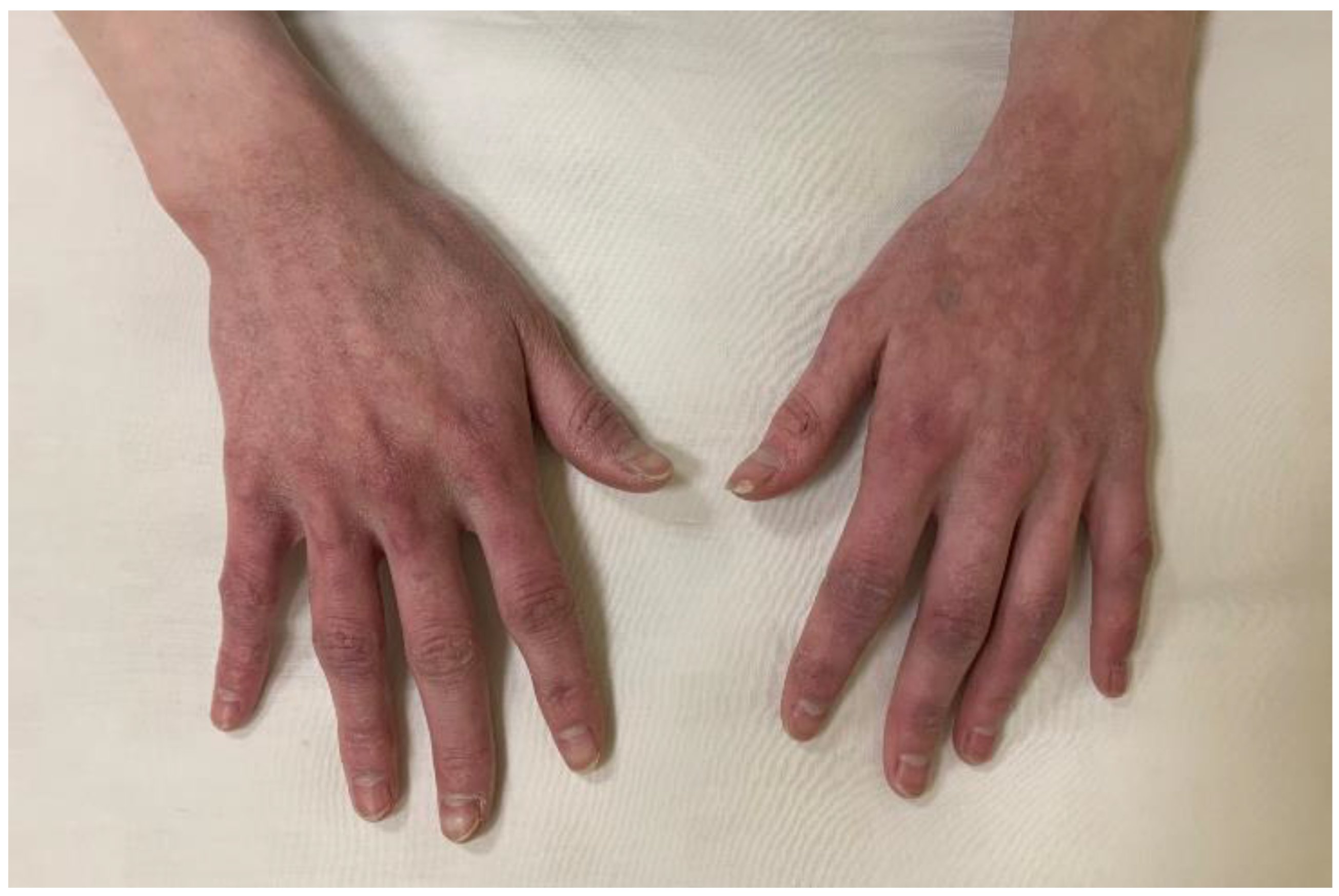

Table 1.
Summary of clinical and genetic characteristics of patients with hereditary hypoPT.
| Patient | Gene | Variant | Inheritance type | Age at hypoPT onset (years) | Age at hypoPT diagnosis (years) | hypoPT complications | Comorbidities |
|---|---|---|---|---|---|---|---|
| 1 | CASR | c.382T>C (p.Phe128Leu) | AD | 9 | 26 | Fahr syndrome | - |
| 2 | GNA11 | c.179G>T (p.Arg60Leu) | AD | 18 | 18 | Fahr syndrome, bilateral cataract | Raynaud’s syndrome, low BMI (15.3 kg/m²), functional hypothalamic amenorrhea |
| 3 | GATA3 | c.1061C>A (p.Pro354His) | AD | 12 | 23 | Fahr syndrome, bilateral nephrocalcinosis, bilateral phacopathy, CKD stage 4, high BMD syndrome | Bilateral sensorineural hearing loss, enamel hypoplasia, psoriasis, nodular goiter, normocytic normochromic anemia, chronic blepharitis, chalazion |
Abbreviations: hypoPT – hypoparathyroidism; BMD – bone mineral density; CKD – chronic kidney disease; BMI – body mass index.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



